

Abstract
Introduction:
Cxcl12, or stromal-derived factor-1, is a chemokine produced by several hepatic cell types, including hepatocytes, after liver injury and surgical resection. Studies have revealed that Cxcl12 is important for regeneration of the liver after surgical resection and for development of liver fibrosis during chronic liver injury. While the function of Cxcl12 in the liver is well established, the mechanism by which Cxcl12 is upregulated is not fully understood. Because regions of hypoxia develop in the liver following injury, we tested the hypothesis that hypoxia upregulates Cxcl12 in hepatocytes by a hypoxia-inducible factor (HIF)-dependent mechanism.
Methods:
To test this hypothesis, primary mouse hepatocytes were isolated from the livers of HIF-1α-deficient mice or HIF-1α-deficient mice and exposed to 1% oxygen. Cxcl12 expression was increased following exposure of primary mouse hepatocytes to 1% oxygen. Previously we that in addition to HIFs, transforming growth factor-β is required for upregulation of a subset of genes in hypoxic hepatocytes. To examine the role of TGF-β in regulation of Cxcl12 during hypoxia, hepatocytes were pretreated with the TGF-β receptor I inhibitor, SB431542.
Results:
Upregulation of Cxcl12 by hypoxia was partially prevented in hepatocytes from HIF-1α-deficient mice and completely prevented in hepatocytes from HIF-1β-deficient hepatocytes. This suggests that under hypoxic conditions, both HIF-1α and HIF-2α regulate Cxcl12 in hepatocytes. Pretreatment of hepatocytes with SB431542 completely prevented upregulation Cxcl12 by hypoxia. Further, treatment of hepatocytes with recombinant TGF-β1 upregulated Cxcl12 in hepatocytes cultured in room air.
Conclusion:
Collectively, these studies demonstrate that hypoxia upregulates Cxcl12 in primary mouse hepatocytes by a mechanism that involves HIFs and TGF-β.
Keywords: Cxcl12, hypoxia, TGF-β, fibrosis, liver, hepatocytes
Graphical Abstract
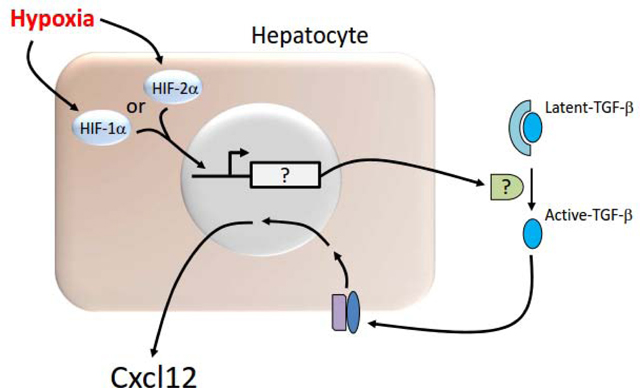
Hypoxia activates HIF-1a and/or HIF-2a in hepatocytes which stimulates their translocation to the nucleus. In the nucleus, they with HIF-1b (not shown) which increases expression of a yet to be identified mediator (indicated by a question mark in the diagram) that is secreted and converts latent TGF-b to active TGF-b. TGF-b then activates its receptor which stimulates upregulation of Cxcl12.
1. Introduction
Cxcl12, or stromal cell-derived factor-1, is a member of the CXC chemokine family. Chemokines, like Cxcl12, are small proteins that play a vital role in the migration of cells throughout the body under homeostatic conditions and also during periods of inflammation and tissue regeneration. In the liver, Cxcl12 is expressed by bile duct epithelial cells, hepatic stellate cells, and sinusoidal endothelial cells (SECs) under various conditions[18]. Cxcl12 is also expressed by hepatocytes during oval cell-mediated liver regeneration[30]. Once released, Cxcl12 activates the G-protein coupled receptors CXCR4 and CXCR7, which are constitutively expressed by several types of hepatic cells, including hepatic stellate cells, oval cells, and hepatocytes[17,18,29].
Several studies have demonstrated a key role for Cxcl12 in liver repair after acute liver injury. Following liver injury from either carbon tetrachloride or acetaminophen, Cxcl12 is released and activates CXCR7 on SECs[11]. Selective deletion of CXCR7 on SECs setting resulted in reduced hepatocyte proliferation and worsened liver injury[11]. Similar to acute injury, Cxcl12 is also important for liver regeneration after surgical resection. In rats subjected to partial hepatectomy, inhibition of Cxcl12 reduced both hepatocyte proliferation and recruitment of bone marrow-derived SEC progenitors[10]. Interestingly, inhibition of hepatocyte proliferation after partial hepatectomy with 2-acetylaminofluorene increased Cxcl12 secretion by hepatocytes which stimulated expansion of hepatic progenitor cells that ultimately differentiated into mature hepatocytes[30].
Activation of the Cxcl12/CXCR4 chemokine axis has also been implicated in promoting fibrosis in cases of chronic liver injury. Activation of CXCR4 by Cxcl12 in SECs during chronic liver injury generates profibrogenic signals that activate hepatic stellate cells leading to extracellular matrix deposition and ultimately liver fibrosis[11,14,24]. In liver fibrosis patients, Cxcl12 protein levels in the liver and plasma correlate with the severity of liver fibrosis consistent with a role for this chemokine in liver fibrosis in humans[28].
Collectively, these studies demonstrate an important role for Cxcl12 in regulation of proregenerative and profibrotic pathways in the liver after acute and chronic injury[17]. The mechanism by which SDF-1 is upregulated in the liver under these conditions, however, is not fully understood. Because hepatocellular hypoxia occurs in the setting of both acute and chronic liver injury, we tested the hypothesis that hypoxia upregulates Cxcl12 in hepatocytes through activation of hypoxia-inducible factor transcription factors (HIF)[5,20]. Our findings reveal that hypoxia and HIFs increase expression of Cxcl12 in hepatocytes, by a complex mechanism involving transforming growth factor-β (TGF-β).
2. MATERIALS AND METHODS
2.1. Mice
The following mice were used in the present studies: C57BL/6 mice (Harlan, Madison, WI), HIF-1α (control and deficient mice), and HIF-1β-(control and deficient mice). The generation of HIF-1α-control mice, HIF-1α-deficient mice, HIF-1β-control mice, and HIF-1β-deficient mice was described in detail by us previously[6].
All mice were maintained using a 12-h light/dark cycle with controlled temperature (18–21 C) and humidity. Both food (Rodent Chow; Harlan-Teklad, Madison, WI) and tap water were allowed ad libitum. procedures on animals were carried out in accordance with the Guide for the Care and Use of Laboratory Animals promulgated by the National Institutes of Health.
2.2. Hepatocyte Isolation
Collagenase perfusion used to isolate cultures of primary hepatocytes from mouse livers as described in detail previously[1]. Primary hepatocytes were cultured in Williams’ medium E supplemented with 10% fetal bovine serum and Penicillin-Streptomycin. Following a 3-hour period of cell attachment, medium with unattached cells were removed, and fresh serum-free Williams’ E medium supplemented with Penicillin-Streptomycin was added. Following a 16 hour incubation, hepatocyte cultures were maintained in an environment containing either room air or 1% oxygen for the indicated times in a NAPCO CO2 7000 cell culture incubator (NAPCO Precision, Winchester, VA). These environments were balanced with nitrogen and contained 5% CO2.
For studies with SB-431542, hepatocytes were treated with either vehicle (dimethyl sulfoxide) or 10 μM SB-431542 (Tocris, Ellisville, MO) for 30 minutes prior to culturing the cells in either room air or 1% oxygen or treating the cells with 5 ng/ml recombinant transforming growth factor-β1 (TGF-β1) (R&D Systems, Minneapolis, MN). For studies with latent TGF-β1, hepatocytes were treated with 20 ng/ml latent TGF-β1 (R&D Systems), 30 minutes prior to hypoxia exposure.
2.3. Real-time PCR
TRI reagent (Sigma Chemical Company, St. Louis, MO) was used to isolate total liver RNA, which was reverse transcribed into cDNA as described by us previously[1]. Real-time PCR was used to quantify and 18S. Real-time PCR was performed on an Applied Biosystems Prism 7300 Real-time PCR Instrument (Applied Biosystems, Foster City, CA) using a SYBR green DNA PCR kit (Applied Biosystems) as described by us previously[1]. The following primer sequences were used for Real-time PCR: 18S Forward: 5’-TTG ACG GAA GGG CAC CAC CAG-3’; 18S Reverse: 5’-GCA CCA CCA CCC ACG GAA TCG-3’; Cxcl12 Forward: 5’-AAA CCA GTC AGC CTG AGC TAC C-3’; SDF-1 Reverse: 5’-GCA CAG TTT GGA GTG TTG AGG A-3’.
2.4. Cxcl12 Protein
Cxcl12 protein was measured by ELISA (R&D Systems, Minneapolis, MN).
2.5. Statistical Analysis
The results are presented as mean + SEM and were analyzed using an Analysis of Variance (ANOVA). For instances in which variances were not homogenous, ANOVAs were performed on log X-transformed data. For comparisons among group means a Student-Newman-Keuls test was used. For all studies, the criterion for significance was p < 0.05.
3. Results
Exposure of primary mouse hepatocytes to hypoxia (1% O2) increased levels of Cxcl12 mRNA and protein by 72 hours after treatment (Fig. 1). To determine whether upregulation of Cxcl12 by hypoxia required HIF transcription factors, hepatocytes were isolated from HIF-1α-deficient mice and HIF-1β-deficient mice. As shown in Figure 2, deletion of HIF-1α in hepatocytes reduced hypoxia-mediated induction of Cxcl12 by approximately 50%, whereas, deletion of HIF-1β completely prevented induction of Cxcl12 by hypoxia. Not all hypoxia-induced genes were increased in a HIF-dependent manner, however. As shown in Supplemental Figure 1, MMP13 was increased by hypoxia in a HIF-independent manner.
Figure 1.

Primary mouse hepatocytes were exposed to room air or 1% oxygen. (A) mRNA levels of Cxcl12 were measured by real-time PCR at the indicated time. (B) Cxcl12 protein levels were measured by ELISA at 72 hours after exposure to hypoxia. n=3 where each n represents cells isolated from different mice. aSignificantly different from cells cultured in room air.
Figure 2.

Primary mouse hepatocytes were isolated from control mice and mice deficient in either (A) HIF-1α or (B) HIF-1β. The cells were exposed to room air or 1% oxygen for 72 hours and mRNA levels of Cxcl12 were measured by real-time PCR. n=3 where each n represents cells isolated from different mice. aSignificantly different from cells cultured in room air. bSignificantly different from control cells cultured in 1% oxygen.
We showed previously that upregulation of a subset of genes in hepatocytes by hypoxia required transforming growth factor-β (TGF-β) receptor signaling[7]. To determine whether upregulation of Cxcl12 by hypoxia occurred by a similar mechanism, primary mouse hepatocytes pretreated with the TGF-β receptor I inhibitor, SB431542. As above, exposure of hepatocytes to hypoxia increased Cxcl12 by 72 hours (Fig. 3). Pretreatment with SB431542, however, completely prevented upregulation of Cxcl12 by hypoxia (Fig. 3). We next determined whether treatment of primary mouse hepatocytes alone with TGF-β increases expression of Cxcl12. As shown in Figure 4A, treatment of hepatocytes with TGF-β1 increased Cxcl12 mRNA levels, an effect that was completely prevented by pretreatment with SB431542 (Fig. 4A). Further, treatment of hepatocytes with TGF-β1 increased Cxcl12 by 24 hours, and Cxcl12 levels remained elevated at 72 hours (Fig. 4B).
Figure 3.

Primary mouse hepatocytes were isolated from mice and treated with either DMSO vehicle or the TGF-β receptor antagonist, SB-431542. The cells were then exposed to room air or 1% oxygen for 72 hours and mRNA levels of Cxcl12 were measured by real-time PCR. n=4 where each n represents cells isolated from different mice. aSignificantly different from cells cultured in room air. bSignificantly different from DMSO-treated cells cultured in 1% oxygen.
Figure 4.

(A) Primary mouse hepatocytes were isolated from mice and treated with either DMSO vehicle or the TGF-β receptor antagonist, SB-431542. The cells were then treated with TGF-β 1 for 72 hours and mRNA levels of Cxcl12 were measured by real-time PCR. (B) Hepatocytes were treated with TGF-β1 for the indicated time and Cxcl12 mRNA levels were measured. n=3 where each n represents cells isolated from different mice. aSignificantly different from cells with control vehicle. bSignificantly different from DMSO-treated cells treated with TGF-β1.
TGF-β1 is secreted as an inactive, latent form that is processed to the active form of TGF-β1 by proteolytic cleavage and by other mechanisms[2]. We previously demonstrated that cultured primary mouse hepatocytes secrete latent-TGF-β that is activated when the hepatocytes are placed in hypoxia[7]. If this mechanism regulates Cxcl12 in hypoxic hepatocytes, then addition of recombinant latent-TGF-β1 should enhance upregulation of Cxcl12 by hypoxia. As shown in Fig. 5, pretreatment of hepatocytes with latent-TGF-β1 enhanced upregulation of Cxcl12 by hypoxia.
Figure 5.

Primary mouse hepatocytes were isolated from mice and treated with either vehicle or 20 ng/ml of latent TGF-β. mRNA levels of Cxcl12 were measured by real-time PCR. n=3 where each n represents cells isolated from different mice. aSignificantly different from cells cultured in room air. bSignificantly different from vehicle-treated cells cultured in 1% oxygen.
4. Discussion
Several studies have established an important role for Cxcl12 in liver repair after injury[17]. The mechanism by which Cxcl12 is upregulated in various cell types, including hepatocytes, after injury, however, remains poorly defined. Several studies have previously shown regions of hypoxia develop in the liver following injury. Furthermore, we have shown that hypoxia develops in the liver following bile duct ligation, a model of obstructive cholestasis and after treatment with the hepatotoxicant, monocrotaline[5,20]. Similarly, others have demonstrated regions of hypoxia develop in the liver following toxicity from acetaminophen or diethylnitrosamine, suggesting that hepatocellular hypoxia is a common feature after liver injury [4,9]. Because of this, we tested the hypothesis that hypoxia stimulates upregulation of Cxcl12 in primary mouse hepatocytes. Our results demonstrate that hypoxia is a stimulus for Cxcl12 induction, and that this process requires transcription factors (Figs. 1 and 2). HIFs are composed of an alpha subunit, HIF-1α or HIF-2α, and a beta subunit, HIF-1β[25]. In the presence of hypoxia, the alpha subunit becomes stabilized and translocates to the nucleus where it heterodimerizes with HIF-1β and regulates expression of genes. We have demonstrated previously that HIF-1α and HIF-2α are both activated in hypoxic hepatocytes[6]. Additionally, we have demonstrated that both of these transcription factors regulate expression of genes in these cells[6]. In the present study, deletion of HIF-1α in hepatocytes partially prevented upregulation of Cxcl12 by hypoxia, whereas deletion of HIF-1β completely prevented upregulation of Cxcl12 in hypoxic hepatocytes. This suggests that both HIF-1α and HIF-2α are important for upregulation of Cxcl12 in hypoxic hepatocytes.
We showed previously that a subset of genes upregulated in hypoxic hepatocytes required HIFs and TGF-β signaling[7]. In these studies, cultured hepatocytes constitutively expressed and secreted latent-TGF-β that was activated when the hepatocytes were placed in hypoxia[7]. Once activated, TGF-β increased expression of several genes associated with epithelial to mesenchymal transition. Similar to this study, hypoxia increased expression of Cxcl12 in hepatocytes by a mechanism that required TGF-β signaling. Further, recombinant, active TGF-β 1 increased expression of Cxcl12 in hepatocytes cultured in room air, and addition of TGF-β1 to hypoxic hepatocytes enhanced upregulation of Cxcl12. This suggests that during hypoxia, HIFs increase expression of a factor that converts latent TGF-β into active TGF-β. TGF-β then increases expression of Although we do not yet know the identity of the factor that activates TGF-β in hepatocytes, several HIF regulated genes are known activators of TGF-β, such as thrombospondin-1 [19.22].
As discussed, Cxcl12 is upregulated in the liver after surgical resection and during chronic liver injury. After surgical resection, deletion of Cxcl12 limits liver regeneration, and deletion of Cxcl12 during chronic liver injury reduces fibrosis[10,11]. Whether hypoxia is important for induction of Cxcl12 in these situations remains to be determined. In support of a role for hypoxia, though, we showed that deletion of HIF-1α in the liver reduces liver fibrosis[20]. Further, others have reported deletion of HIFs reduces liver regeneration after surgical resection[27]. Although the observed effects of HIF deletion are similar to those after deletion of Cxcl12, additional studies are needed to determine the importance of hypoxia, HIFs, and TGF-β for upregulation of Cxcl12 in the liver during regeneration and fibrosis. Overall, though, our studies indicate that hypoxia is a key regulator of Cxcl12 in hepatocytes.
Supplementary Material
Highlights.
The mechanism by which Cxcl12 is upregulated in the liver is poorly understood.
Hypoxia stimulates upregulation of Cxcl12 in a HIF-1α-and HIF-2α-dependent manner.
TGF-β is also required for upregulation of Cxcl12 in hypoxic hepatocytes.
Acknowledgements:
This work was supported by National Institute of Health Grant DK103895 to BLC
Abbreviations
- ANOVA
Analysis of Variance
- HIF
hypoxia-inducible factor
- SECs
sinusoidal endothelial cells
- TGF-β
transforming growth factor-β
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Allen K, Jaeschke H, Copple BL. Bile acids induce inflammatory genes in hepatocytes: a novel mechanism of inflammation during obstructive cholestasis. Am J Pathol 2011; 178(1): 175–86 [DOI: 10.1016/j.ajpath.2010.11.026] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Annes JP, Munger JS, Rifkin DB. Making sense of latent TGFbeta activation. J Cell Sci 2003; 116(Pt2): 217–24 [DOI: 10.1242/jcs.00229] [DOI] [PubMed] [Google Scholar]
- [3].Bertran E, Crosas-Molist E, Sancho P, Caja L, Lopez-Luque J, Navarro E, Ega G, Lastra R, Serrano T, Ramos E, Fabregat I. Overactivation of the TGF-β pathway confers a mesenchymal-like phenotype and CXCR4-dependent migratory properties to liver tumor cells. Hepatology 2013, 58(6): 2032–2044 [DOI: 10.1002/hep26597] [DOI] [PubMed] [Google Scholar]
- [4].Chaudhuri S, McCullough SS, Hennings L, Letzig L, Simpson PM, Hinson JA, James LP. Acetaminophen hepatotoxicity and HIF-1α induction in acetaminophen toxicity in mice occurs without hypoxia. Toxicol Appl 2001; 252(3):211–20 [DOI: 10.1016/j.taap.2011.02.005] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Copple BL, Roth RA, Ganey PE. Anticoagulation and inhibition of nitric oxide synthase influence hepatic hypoxia after monocrotaline exposure. Toxicology 2006; 225(2–3):128–37 [DOI: 10.1016/j.tox.2006.05.016] [DOI] [PubMed] [Google Scholar]
- [6].Copple BL, Bustamante JJ, Welch TP, Kim ND, Moon JO. Hypoxia-inducible factor-dependent production of profibrotic mediators by hypoxic hepatocytes. Liver Int 2009; (7):1010–21 [DOI: 10.1111/j.1478-3231.2009.02015.x] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Copple BL. Hypoxia stimulates hepatocyte epithelial to mesenchymal transition by hypoxia-inducible factor and transforming growth factor-beta-dependent mechanisms. Liver Int 2010; 30(5):669–682 [DOI: 10.1111/j.1478-3231.2010.02205.x] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Copple BL, Bai S, Burgoon LD, Moon JO. Hypoxia-inducible Factor-1α Regulates Expression of Genes in Hypoxic Hepatic Stellate Cells Important for Collagen Deposition and Angiogenesis. Liver International 2011; 31(2): 230–244 [DOI: 10.1111/j.1478-3231.2010.02347.x] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Corpechot C, Barbu V, Wendum D, Chignard N, Housset C, Poupon R, Rosmorduc O. Hepatocyte growth factor and c-Met inhibition by hepatic cell hypoxia: a potential mechanism for liver regeneration failure in experimental cirrhosis. Am J Pathol 2002; 160(2):613–20. [DOI: 10.1016/S0002-9440(10)64881-X] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].DeLeve LD, Wang X, Wang L. VEGF-sdf1 recruitment CXCR7+ bone marrow progenitors of liver sinusoidal endothelial cells promotes rat liver regeneration. Am J Physiol Gastrointest Liver Physiol 2016;310(9):G739–46 [DOI: 10.1152/ajpgi.00056.2016] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Ding BS, Cao Z, Lis R, Nolan P, Simons M, Penfold ME, Shido K, Rabbany SY, Rafii S. Divergent angiocrine signals from niche balance liver regeneration and fibrosis. Nature 2014; 505(7481):97–102 [DOI: 10.1038/nature12681] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Fabregat I, Moreno-Càceres J, Sánchez A, Dooley S, Dewidar B, Giannelli G, ten Dijke P. TGF-β signalling and liver disease. FEBS Journal 2016; 283:2219–2232. [DOI: 10.1111/febs.13665] [DOI] [PubMed] [Google Scholar]
- [13].Hitchon C, Wong K, Ma G, Reed J, Lyttle D, El-Gabalawy H. Hypoxia-induced production of stromal cell-derived factor 1 (CXCL12) and vascular endothelial growth factor by synovial fibroblasts. Arthritis and Rheumatism 2002; 46(10): 2587–2597. [DOI: 10.1002/art.10520] [DOI] [PubMed] [Google Scholar]
- [14].Hong F, Tuyama A, Lee TF, Loke J, Agarwal R, Cheng X, Garg A, Fiel MI, Schwartz M, Walewski J, Branch A, Schecter AD, Bansal MB. Hepatic stellate cells express functional CXCR4: role in stromal cell-derived factor-1alpha-mediated stellate cell activation. Hepatology 2009; 49(6):2055–67 [DOI: 10.1002/hep.22890] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Kojima Y, Acar A, Eaton EN, Mellody KT, Scheel C, Ben-Porath I, Onder TT, Wang ZC, Richardson AL, Weinberg RA, Orimo A. Autocrine TGF-and stromal cell-derived factor-1 (SDF-1) signaling drives the evolution of tumor-promoting mammary stromal myofibroblasts. Proceedings of the National Academy of Sciences 2010; 107(46): 20009–20014 [DOI: 10.1073/pnas.1013805107] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Kollet O, Shivtiel S, Chen YQ, Suriawinata J, Thung SN Dabeva MD, Kahn J, Spiegal A, Dar A, Samira S, Goichberg P, Kalinkovich A, Arenzana-Seisdedos F, Nagler A, Hardan I, Revel M, Shafritz D, Lapidot T. HGF, SDF-1, and MMP-9 are involved in stress-induced human CD34+stem cell recruitment to the liver. The of Experimental Medicine 2003;112(2):160–169 [DOI: 10.1172/JCI17902] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Liepelt A, Tacke F. Stromal cell-derived factor-1 (SDF-1) as a target in liver diseases. American Journal of Physiology-Gastrointestinal and Liver Physiology 2016; 311(2):G203–G209. [DOI: 10.1152/ajpgi.00193.2016] [DOI] [PubMed] [Google Scholar]
- [18].Marra F, Tacke F. Roles for chemokines in liver disease. Gastroenterology 2014; 147(3):577–594 [DOI: 10.1053/j.gastro.2014.06.043] [DOI] [PubMed] [Google Scholar]
- [19].Miyoshi A, Kitajima Y, Ide T, Ohtaka K, Nagasawa H, Uto Y, Hori H, Miyazaki K. Hypoxia accelerates cancer invasion of hepatoma cells by upregulating MMP expression in an HIF-1alpha-independent manner. Int J Oncol 2006; 29(6):1533–9 [DOI: 10.3892/ijo.29.6.1533] [DOI] [PubMed] [Google Scholar]
- [20].Moon JO, Welch TP, Gonzalez FJ, Copple BL. Reduced liver fibrosis in hypoxia-indcuible factor-1alpha-deficient mice. Am J Phyisol Gastrointest Liver Physiol 2009; 296(3):G582–92. [DOI: 10.1152/ajpgi.90368.2008] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Murphy PM. Chemokines and the molecular basis of cancer metastasis. N Engl J Med 2001; 345(11):833–835. [DOI: 10.1056/NEJM200109133451113] [DOI] [PubMed] [Google Scholar]
- [22].Osada-Oka M, Ikeda T, Akiba S, Sato T. Hypoxia stimulates the autocrine regulation of migration of vascular smooth muscle cells via HIF-1alpha-dependent expression of thrombospondin-1. J Cell Biochem 2008; 104(5):1918–26 [DOI: 10.1002/jcb.21759] [DOI] [PubMed] [Google Scholar]
- [23].Schioppa T, Uranchimeg B, Saccani A, Biswas SK, Doni A, Rapisarda A, Bernasconi S, Saccani S, Nebuloni M, Vago L, Mantovani A, Melillo G, Sica A. Regulation of the Chemokine Receptor CXCR4 by Hypoxia. The Journal Medicine 2003; 198(9):1391–1402 [1084/jem.20030267] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Saiman Y, Jiao J, Fiel MI, Friedman SL, Aloman C, Bansal MB. Inhibition of the CXCL12/CXCR4 chemokine axis with AMD3100, a CXCR4 small molecule inhibitor, worsens murine hepatic injury. Hepatol Res. 2015;45(7):794–803 [DOI: 10.1111/hepr.12411] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Schito L, Semenza GL. Hypoxia-Inducible Factors: Master Regulators of Cancer Progression. Trends Cancer 2016; 2(12):758–770. [DOI: 10.1016/j.trecan.2016.10.016] [DOI] [PubMed] [Google Scholar]
- [26].Sun X, Cheng G, Hao M, Zheng J, Zhou X. CXCL12/CXCR4/CXCR7 Chemokine Axis and Cancer Progression. Cancer Metastasis Reviews, 2010; 29(4):709–722 [DOI: 10.1007/s10555-010-9256-x] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Tajima T, Goda N, Fujiki N, Hishiki T, Nishiyama Y, Senoo-Matsuda N, Shimazu M, Soga T, Yoshimura Y, Johnson RS, Suematsu M. HIF-1alpha is necessary to support gluconeogenesis during liver regeneration. Biochem Biophys Res Commun. 2009; 387(4):789–94. [DOI: 10.1016/j.bbrc.2009.07.115] [DOI] [PubMed] [Google Scholar]
- [28].Wald O, Pappo O, Safadi R, Dagan-Berger M, Beider K, Wald H, Franitza S, Weiss I, Avniel S, Boaz P, Hanna J, Zamir G, Eid A, Mandelboim O, Spengler U, Galun E, Peled A. Involvement of the CXCL12/CXCR4 pathway in the advanced liver disease that is associated with hepatitis C virus or hepatitis B virus. Eur J Immunol 2004;34(4):1164–74. [DOI] [PubMed] [Google Scholar]
- [29].Young KC, Torres E, Hatzistergos KE, Hehre D, Suguihara C, and Hare JM. Inhibition of the SDF-1/CXCR4 axis attenuates neonatal hypoxia-induced pulmonary hypertension. Circulation Research, 2009; 104(11):1293–1301 [DOI: 10.1161/CIRCRESAHA.109.197533] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Zheng D, Oh SH, Jung Y, and Petersen BE. Oval cell response in 2-acetylaminofluorene/partial rat is attenuated by short interfering RNA targeted to stromal cell-derived factor-1. American Journal of Pathology 2006; 169(6):2066–2074 [DOI: 10.2353/ajpath.2006.060211] [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.