

Abstract
S100B is a small, dimeric, calcium-binding protein that is implicated in various diseases, most significantly cancer; therefore, there is interest in identifying S100B inhibitors that may have therapeutic value (Bresnick et al. Nat Rev Cancer 15:96–109, 2015; Chong et al. Curr Med Chem 23:1571–1596). Two fiuorescence polarization competition assays (FPCA) are described here for S100B and S100A1 that are amenable to high-throughput screening (HTS) campaigns and can be used to determine the binding affinity (Ki) of the inhibitors. One FPCA is used to identify and characterize inhibitors of S100B with the aim of finding new therapeutics, and the other was developed as a counter-screen to avoid inhibitors of S100A1 due to its role in regulating skeletal and cardiac muscle function. Also outlined are methods for expressing and purifying S100B and S100A1 in quantities needed for performing large HTS campaigns.
Keywords: High-throughput screening, Fluorescence polarization competition assay, S100 protein, Small molecule, Protein-protein interface, Inhibitors, Calcium binding, S100B, S100A1, Cancer, Melanoma
1. Introduction
The calcium-regulated S100B protein is a prognostic marker for poor survival in several cancers including malignant melanoma [1]. In addition to its role as a cancer marker, S100B is involved in promoting tumor growth and metastasis. Specifically, S100B binds and regulates several proteins and enzymes involved in the progression of cancer including RSK, Hdm2, Hdm4, and p53, to name a few, and S100B modulates the tumor microenvironment when elevated in cancer [1, 3–5]. As a result, S100B is considered a therapeutic target for treating cancer, particularly in malignant melanoma where a majority of patients have elevated S100B [1, 2]. With this in mind, it is important to develop S100B inhibitors that are highly specific since there are 21 members of the S100 protein family that have a similar global fold and high levels of sequence homology. Like S100B, S100A1 is involved in a wide array of cellular activities, including regulating calcium signaling in heart and skeletal muscle via interaction(s) with the ryanodine receptor [6–8]. Thus, when developing S100B inhibitors, care must be taken to avoid blocking S100A1, which is 60% homologous to S100B, and could potentiate problems with skeletal and cardiac muscle function. To this end, fiuorescence polarization competition assays (FPCA) were developed to identify highly specific S100B inhibitors that do not block S100A1.
FPCAs developed to block S100-target interactions are well suited for high-throughput screening (HTS) campaigns due to their sensitivity, speed, homogenous format, and the need for relatively simple instrumentation [9–11]. The FPCA takes advantage of the ability to distinguish the fiuorescence polarization of small peptides (<3 kDa) labeled with probes such as fiuorescein isothiocyanate (FITC) or 5,6-carboxytetramethylrhodamine (TAMRA) in the free state versus bound to a S100 homodimer (>20 kDa). Peptide probes for both S100B and S100A1 were identified by testing several peptides derived from full-length S100 targets. Those labeled peptides deemed acceptable had a binding affinity (KD) of 0.1–5 μM, an interpretable stoichiometry (n = 1, per monomeric subunit), and the fiuorescently labeled peptide was shown to bind in the same site as the unlabeled peptide via direct competition experiments. The concept for the FPCA is straightforward; the fiuorescent probe is highly polarized when bound to the S100 protein; however, upon being displaced by the unlabeled peptide, or a small molecule inhibitor, the fiuorescent peptide probe is released, and its fiuorescence polarization (FP) value decreases significantly (see Fig. 1). The final FPCAs developed here are robust due to the stability of the S100 proteins themselves and the relative simplicity of the method. Below is a description of the FPCA for identifying inhibitors of S100B versus S100A1, calculations to determine the inhibitor affinity (Ki), and for the expression and purification of recombinant S100 proteins needed for the assay.
Fig. 1.
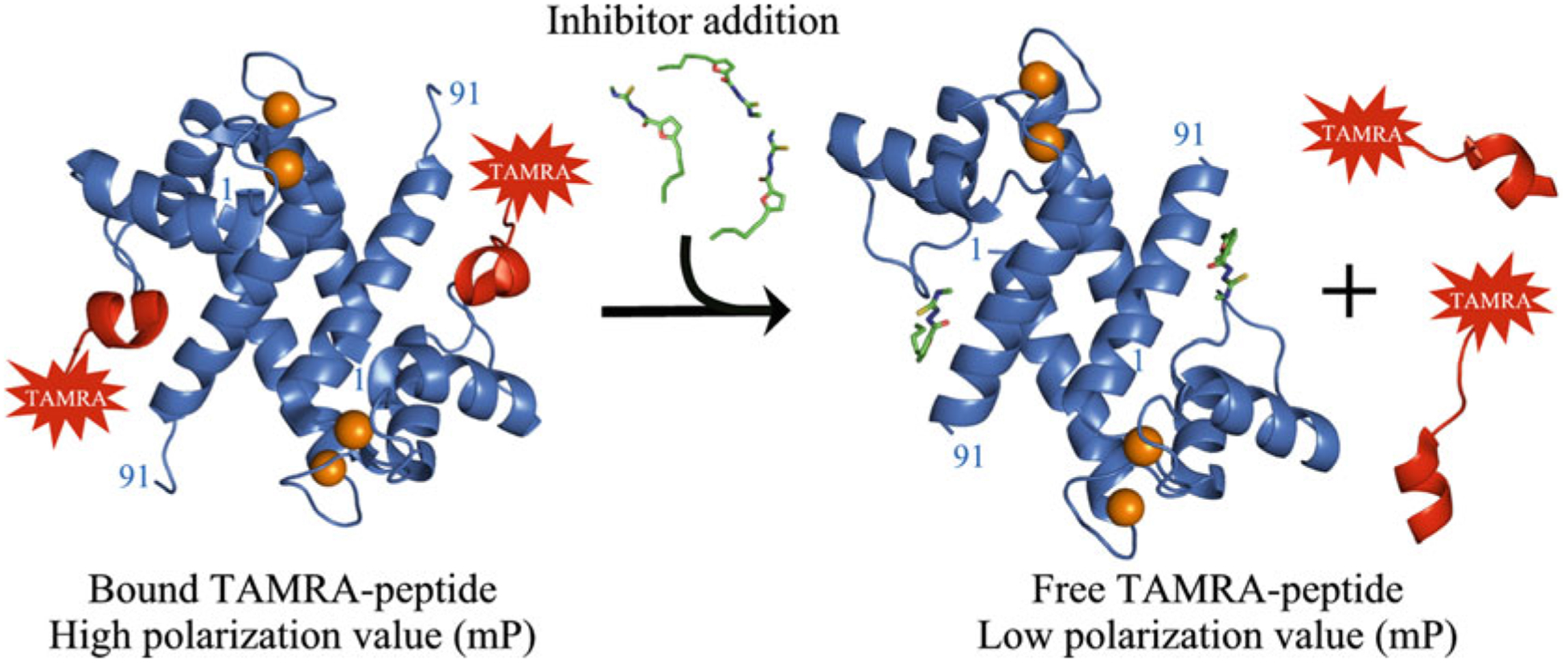
Schematic illustration of the S100B TAMRA-TRTK FPCA
2. Materials
All chemical reagents are American Chemical Society grade or higher unless otherwise indicated, and the solutions need to be prepared using ultrapure water.
2.1. Recombinant S100B and S100A1 Production and Purification
2.1.1. Equipment
Misonix Sonicator 3000 (600 W with 1/2” horn) or equivalent sonicator.
Fraction collector.
UV monitor.
SDS-PAGE electrophoresis equipment.
Sorvall Superspeed refrigerated centrifuge equipped with both a SS-34 and GS-3 fixed angle rotor or equivalent.
Oak Ridge high-speed, polypropylene centrifuge tubes, 50 mL.
Centrifuge bottles, 500 mL, to fit Sorvall GS-3 rotor.
Shaker incubator for 2.5 L culture fiasks.
2.1.2. Common Materials
10 kDa MWCO Amicon Ultra-15 centrifugal filter units.
8–10 kDa MWCO dialysis tubing: Dry dialysis tubing is hydrated and washed to remove metals and glycerin ahead of time by (1) boiling the tubing in 0.4 M EDTA, pH 8.0, for 30 min; (2) boiling the tubing in 0.5 M NaCl for 30 min;(3) boiling the tubing in 0.1 M EDTA, pH 8.0, for 30 min, twice; (4) boiling in water at least three times or until water remains clear; and (5) storing at 4 °C in 0.35 mM NaN3,0.5 mM EDTA. Rinse tubing with high-quality water inside and out before using.
Chelex 100 resin slurry: Clean the resin by (1) hydrating it in water and transferring to a chromatography column or Büchner funnel, (2) washing the resin with two bed volumes of 1 M HCl, (3) washing the resin with five bed volumes of water,(3) washing the resin with two bed volumes of 1 M NaOH,(4) washing the resin with five bed volumes of water, and then(5) washing the resin with five bed volumes of water with0.35 mM NaN3. Make a 50% slurry in the final buffer and store at 4 °C.
Bradford protein assay.
BSA protein standards or S100 standards with known concentrations as determined by amino acid analyses.
2.1.3. Glycerol Stocks, Cell Growth, Protein Expression, and Cell Harvesting
-
1.
LB-Amp: Luria-Bertani broth with 100 μg/mL ampicillin.
-
2.
LB-Amp plates: Luria-Bertani agar plates with 100 μg/mL ampicillin.
-
3.
Ampicillin stock: 100 mg/mL ampicillin water and sterilize by passing thru a 0.2 μm filter.
-
4.
Sterile 80% glycerol: Autoclaved 80% glycerol in water.
-
5.
Isopropyl-1-thio-ß-D-galactopyranoside (IPTG) solution:0.1 g/mL prepared from powder day of use.
-
6.
pET11b S100 protein expression vectors (see Note 1).
-
9.
Competent HMS174(DE3) cells (see Note 2).
-
10.
Tunair full-baffie shake fiask, 2.5 L.
2.1.4. Cell Lysis, Ammonium Sulfate Purification, and Heat Precipitation
Lysis buffer: 0.5 mM AEBSF added to DEAE buffer A right before use.
Lysozyme solution: 10 mg/mL lysozyme in water.
DNase solution: 1 mg/mL DNase I, 5 mM NaOAc, 1 mM CaCl2, 50% glycerol. This can be stored at −20 °C for 3 months.
1 g/mL MgCl2·6H2O.
500 mM EDTA.
10% streptomycin sulfate solution in water.
Ammonium sulfate.
2.1.5. DEAE Anion Exchange
DEAE Sepharose fast-fiow resin in a 2.5-cm-diameter column, ~6 cm bed height, ~30 mL bed volume, or equivalent.
DEAE buffer A: 50 mM Tris–HCl, pH 7.5, 0.5 mM DTT.
DEAE buffer B: 2 M NaCl, 50 mM Tris–HCl, pH 7.5,0.5 mM DTT.
0.10 M NaCl DEAE buffer: 95.00 mL DEAE buffer A + 5.00 mL DEAE buffer B.
0.17 M NaCl DEAE buffer: 27.45 mL DEAE buffer A + 2.55 mL DEAE buffer B.
0.22 M NaCl DEAE buffer: 26.70 mL DEAE buffer A + 3.30 mL DEAE buffer B.
0.35 M NaCl DEAE buffer: 24.75 mL DEAE buffer A + 5.25 mL DEAE buffer B.
0.50 M NaCl DEAE buffer: 45.00 mL DEAE buffer A + 15.00 mL DEAE buffer B.
Resin storage buffer: 20% ethanol, 0.02% NaN3.
2.1.6. G-75 Size Exclusion
Sephadex G-75 medium resin (GE Healthcare 17-0050-01) in a 2.5 cm diameter column, 100 cm length, ~480 mL bed volume, or equivalent.
G-75 column buffer: 10 mM Tris, 50 mM NaCl, 0.25 mM DTT.
Chelex slurry: 50% Chelex 100 in water.
G-75 resin storage buffer: G-75 column buffer, 0.02% NaN3.
2.1.7. Phenyl-Sepharose
Phenyl-Sepharose CL-4B resin in a 2.5-cm diameter column, ~6 cm bed height, ~30 mL bed volume, or equivalent.
Phenyl-Sepharose buffer A: 10 mM Tris, pH 7.5, 500 mM NaCl, 10 mM CaCl2, 0.25 mM DTT.
Phenyl-Sepharose buffer B: 10 mM Tris, pH 7.5, 500 mM NaCl, 10 mM EDTA, 0.25 mM DTT.
2.1.8. G-25 Size Exclusion
Sephadex G-25 medium resin (GE Healthcare 17-0033-01) in a 1.5 cm diameter column, 30–50 cm length, 45–80 mL bed volume, or equivalent.
G-25 column buffer: 10 mM HEPES, pH 7.2, 15 mM NaCl,0.25 mM DTT.
G-25 resin storage buffer: G-25 column buffer, 0.02% NaN3.
2.2. FPCA
2.2.1. Equipment
Microplate reader with fiuorescence polarization measurement capabilities to read 10 nM TAMRA, 20 μL, in a 384-well black microplate from the top (see Note 3).
Origin software (OriginLab Corp., Northampton, MA) or other software for determining the IC50 values from a dose titration plot using nonlinear least squares analysis.
Multichannel pipette.
2.2.2. Common Materials
384-well black polypropylene microplate(s) (see Note 4).
10× FPCA buffer: 0.5 M HEPES, pH 7.2, 1 M KCl, 0.15 M NaCl, 0.1 M CaCl2, 0.1% Triton X-100. Store at 4 °C.
2.2.3. S100A1 Buffers
TAMRA-HDM425–43 peptide: Peptide corresponds to HDM4 residues 25–43 with a single N-terminal 5,6-carboxytetramethylrhodamine (TAMRA) and an amidated C-terminus. Make stock in water up to 1 mM, and immediately adjust to a pH of ~7 (see Note 5).
S100A1 full FPCA buffer: 1X FPCA buffer, 10 nM TAMRA-HDM425–43, 6 μM S100A1 protein. Make at least 0.02 mL per well for 384-well microplate (0.1 mL per well for 96-well microplate) fresh.
S100A1 no S100 control: 1X FPCA buffer, 10 nM TAMRA-HDM425–43. Make at least 0.02 mL per well for 384-well microplate (0.1 mL per well for 96-well microplate) fresh.
2.2.4. S100B Buffers
TAMRA-TRTK: Peptide corresponds to the CapZ protein residues 265–276 [12] with a single N-terminal 5,6-carboxytetramethylrhodamine (TAMRA) and an amidated C-terminus. Make stock in water up to 1 mM, and immediately adjust to a pH of ~7 (see Note 5).
S100B full FPCA buffer: 1× FPCA buffer, 10 nM TAMRA-TRTK, 1 μM S100B protein. Make at least 0.02 mL per well for 384-well microplate (0.1 mL per well for 96-well microplate) fresh.
S100B no S100 control: 1× FPCA buffer, 10 nM TAMRA-TRTK. Make at least 0.02 mL per well for 384-well microplate(0.1 mL per well for 96-well microplate) fresh.
3. Methods
3.1. Recombinant S100B and S100A1 Production and Purification
3.1.1. Glycerol Stocks
Transform competent HMS174(DE3) cells with pET11b S100 protein expression vector construct, and spread plate them on LB-Amp plates overnight at 37 °C (see Note 2).
Inoculate 5 mL LB-Amp medium with a single colony. Repeat this for five to ten unique colonies. Incubate overnight at 37 °C with orbital shaking.
Dilute an aliquot of the overnight cultures (1:100) into 10 mL LB-Amp medium, and incubate at 37 °C with orbital shaking until the A600 is 0.6–0.8. Save the overnight cultures at 4 °C.
Remove 500 μL, sediment the bacteria, suspend the cell pellet in 100 μL SDS-PAGE sample buffer, and store at −20 °C.
Add 20 μL IPTG solution to the remaining culture, and incubate for another 2–4 h at 37 °C with orbital shaking, 150 rpm.
Remove 500 μL, sediment the bacteria, and suspend the cell pellet in 100 μL SDS-PAGE sample buffer.
Run SDS-PAGE analysis on 15 μL of the preinduction (step 4) and post-induction (step 6) cultures, and visually determine the lane with the highest expression of the S100 protein (see Note 6).
Dilute the overnight culture of the highest expression colony (1:100) in 10 mL LB-Amp medium, and incubate at 37 °C with orbital shaking until the A600 is 0.6–0.8.
Aliquot 100 μL sterile 80% glycerol into pre-labeled sterile1.5 mL Eppendorf tubes (three to five tubes).
Add 900 μL of the bacterial culture, mix quickly by pipetting or vortex mixing, and rapidly freeze by placing in crushed dry ice powder, a dry ice ethanol bath, or into liquid nitrogen.
After 5 min transfer to −80 °C for long-term storage (see Note 7).
3.1.2. Cell Growth, Protein Expression, and Cell Harvesting for S100B and S100A1
Inoculate a 5 mL LB-Amp culture with a scraping from the glycerol stock using a sterile pipet tip or inoculating loop and grow at 37 °C with orbital shaking of 150 rpm overnight (see Note 7).
The next morning, inoculate 200 mL sterile LB-Amp (2 L fiask) with 2 mL of the overnight 5 mL LB culture and at 37 °C, 150 rpm, and grow to an A600 of ~0.6.
Inoculate 5 × 1 L cultures in baffied 2.5 L fiask with 30 mL from the 200 mL culture, and grow at 37 °C with 200 rpm orbital shaking.
Start monitoring the A600 of the cultures every hour, and graph read on a semilog plot. Cultures will be ready for induction when A600 = 0.8–1.0. To avoid overshooting, take more frequent measurements when the A600 reaches ~0.6.
During this period prepare 2 mL IPTG solution for each 1 L culture induction by dissolving 0.1 g/mL IPTG in ddH2O for each 6 L fiask in use.
Before induction, take a 15 μL aliquot from each fiask for SDS-PAGE analysis.
Induce cells by adding 2 mL of IPTG solution per 1 L culture.
Incubate 3 h at 37 °C with 200 rpm orbital shaking.
During this time weigh six empty centrifuge bottles and record their total weight.
Record the A600 and take a 15 μL aliquot from each fiask for SDS-PAGE analysis.
Load the centrifuge bottles to their maximum volume, balance, and spin at 6000 × g for 20 min at 4 °C in the GS-3 rotor. Keep remaining culture suspension on ice.
Carefully remove the media from the cell pellet, and add the remaining cell culture suspension to the same centrifuge bottles containing the pellet, balance, and spin at 6000 × g for 20 min at 4 °C in the GS-3 rotor. Repeat until all the cells are pelleted.
After final supernatant is decanted, remove as much media as possible with a pipette, and weigh bottles, subtracting the weight of the empty bottles to determine the pellet weight.
Place centrifuge bottles in −80 °C freezer (see Note 8).
3.1.3. Cell Lysis for S100B and S100A1
If frozen, thaw pellets in water bath at room temperature, and immediately put on ice.
After this point all steps are to be performed at or near 4 °C by using ice bath or cold room unless specified. Use only plastic to avoid introducing metals and losing protein (see Note 9).
Dissolve AEBSF in DEAE buffer A to make lysis buffer.
Suspend the pellets using 1 mL lysis buffer per 1 g of pellet. Work quickly but gently to avoid lather.
Pour cell suspension into 250 mL plastic beaker with stir bar, and add 80 mL lysozyme solution per 1 g of pellet. Stir gently on ice for 20 min. The viscosity should increase due to DNA release.
Add 300 μL DNase solution and 100 μL MgCl2 solution. Continue stirring on ice for 15–20 min. The viscosity should decrease as the DNA is degraded; if not make new DNase solution and repeat.
Add sufficient volume of EDTA solution to make suspension 5 mM in EDTA (i.e., add 0.01 × cell suspension volume of 500 mM EDTA solution).
Remove the stir bar. Set the sonicator to 70% of maximum power. Sonicate 0.5 min/mL solution. Allow 20–30 s in an ice bath after each minute of sonication or as necessary to keep the liquid cool.
Centrifuge the solution for 45 min at 27,000 × g and 4 °C in the SS-34 rotor.
Pour the supernatant into a 100 mL plastic beaker with a stir bar. Slowly add streptomycin solution over the next 30 min in ten aliquots; each aliquot should be 0.01× volume of supernatant.
Continue to stir for an additional 30 min.
Centrifuge the solution for 45 min at 27,000 g and 4 °C in the SS-34 rotor.
For S100B proceed with ammonium sulfate purification (see Subheading 3.1, step 4), and for S100A1 proceed with heat precipitation (see Subheading 3.1, step 5).
3.1.4. Ammonium Sulfate Purification for S100B
Measure the volume of the supernatant and transfer into a 100 mL plastic beaker.
With constant stirring, make the solution 80% (NH4)2SO4 by slow addition over a period of 1 h on ice or in cold room at 4 °C.
Continue stir for an additional 30 min.
Centrifuge the solution for 45 min at 27,000 × g and 4 °C in the SS-34 rotor.
Transfer the supernatant into 8–10 kDa MW cutoff dialysis tubing allowing for threefold volume expansion during dialysis.
Dialyze the supernatant twice against 4 L DEAE buffer A at 4 °C for a minimum of 4 h each with gentle mixing. It can be left overnight.
Proceed with DEAE anion exchange (see Subheading 3.1, step 6).
3.1.5. Heat Precipitation for S100A1
Transfer the supernatant to a new centrifuge tube.
Suspend the uncapped centrifuge tube in boiling water.
Heat until the internal sample temperature reaches 60 °C.
Remove from the boiling water, allow to cool, and cap.
Centrifuge the solution for 45 min at 27,000 × g and 4 °C in the SS-34 rotor, and save the supernatant.
Proceed with DEAE anion exchange (see Subheading 3.1, step 6).
3.1.6. DEAE Anion Exchange for S100B and S100A1
Clean DEAE Sepharose fast-fiow resin with 100 mL DEAE buffer B.
Equilibrate with five column volumes (CV) (~150 mL) DEAE buffer A at ≤5 mL/min (60 cm/h linear fiow rate; maximum for this resin is 750 cm/h).
Load sample, and run through column at a rate not to exceed that of the equilibration step above. Collect and save the eluent.
Follow the sample with 100 mL DEAE buffer A to wash the column. Collect and save the eluent.
Set up fraction collector for 60 5 mL (150-drop) fractions.
Load the buffer solutions in order of increasing salt concentrations making sure to wait until each buffer has run into the resin bed before loading the next one and recording the fractions collected for each.
Add 100 mL of 0.1 M NaCl DEAE buffer.
Add 30 mL of 0.17 M NaCl DEAE buffer.
Add 30 mL of 0.22 M NaCl DEAE buffer.
Add 30 mL of 0.35 M NaCl DEAE buffer.
Add 60 mL of 0.5 M NaCl DEAE buffer.
Wash column with ~60 mL DEAE buffer B, saving the eluent.
Wash column with 60–100 mL of DEAE buffer A, saving the eluent.
Measure the UV absorbance at 280 nm and 260 nm of fractions, and run SDS-PAGE analysis on the samples containing protein based on UV.
Pool fractions containing S100 protein verified by SDS-PAGE. The bulk is released with 0.17 M NaCl and will be in fractions 29–41 but can range from 16 to 47.
After it has been determined that S100B has been eluted, equilibrate column with 150 mL resin storage buffer. Resin should be stored at 4 °C, either in the column or another container.
For S100A1 proceed with Phenyl-Sepharose (see Subheading 3.1, step 7), and for S100B proceed with G-75 size exclusion (see Subheading 3.1, step 8).
3.1.7. Phenyl-Sepharose for S100A1
Dialyze the pooled fractions twice against 4 L Phenyl-Sepharose buffer A at 4 °C for a minimum of 4 h each with gentle mixing. This can be left over overnight.
Wash the Phenyl-Sepharose CL-4B column with five column volumes of 1:9 dilution of Phenyl-Sepharose buffer B in Phenyl-Sepharose buffer A.
Equilibrate the Phenyl-Sepharose CL-4B column with five CV of Phenyl-Sepharose buffer A.
Load S100A1 protein sample and collect the fiow through.
Set up the fraction collector to collect 3 mL (100-drop) fractions.
Wash the column with five CV of Phenyl-Sepharose buffer A, and collect the fiow through in 3 mL fraction.
Elute with three CV buffer B and save in 3 mL fraction.
Run an SDS page gel on all of the fiow through samples and fractions.
Pool pure S100A1 samples, and proceed with G-25 size exclusion (see Subheading 3.1, step 9).
Pool remaining samples with S100A1, and proceed with G-75 size exclusion to further purify (see Subheading 3.1, step 8).
After it has been determined that S100A1 protein has been eluted, equilibrate column with two CV of water followed by two CV of resin storage buffer. Resin should be stored at 4 °C.
3.1.8. G-75 Size Exclusion for S100B and S100A1
Transfer the pooled fractions containing S100 protein into 8–10 kDa MW cutoff dialysis tubing allowing for threefold volume expansion during dialysis.
Dialyze the pooled fractions twice against 4 L G-75 column buffer containing 2 mL Chelex slurry at 4 °C for a minimum of 4 h each with gentle mixing. This can be left overnight.
Concentrate the sample using Amicon Ultra-15 centrifugal filter units 10 kDa MWCO to 0.5–1.0% of the G-75 column bed volume.
Equilibrate G-75 column with at least two CV of G-75 column buffer at 4 °C using no more than 1.5 mL/min (18 cm/h linear fiow rate).
Set up the fraction collector for 175 3 mL (100-drop) fractions.
Load sample, and allow to run through column at a rate not to exceed that of the equilibration step above. Begin the fraction collector as the sample is loaded.
Measure the UV absorbance at 260–280 nm of fractions, and run SDS-PAGE analysis on the samples containing protein based on UV.
Pool fractions containing S100 protein.
After it has been determined that S100 protein (S100B or S100A1) has been eluted, equilibrate column with 500 mL G-75 resin storage buffer. Resin should be stored at 4 °C in the column.
Proceed to G-25 size exclusion (see Subheading 3.1, step 9).
3.1.9. G-25 Size Exclusion S100B and S100A1
Transfer the pooled fractions containing S100 protein into 8–10 kDa MW cutoff dialysis tubing allowing for threefold volume expansion during dialysis.
Dialyze the pooled fractions twice against 4 L G-25 column buffer containing 2 mL Chelex slurry at 4 °C for a minimum of 4 h each with gentle mixing. This can be left overnight.
Concentrate the sample using Amicon Ultra-15 centrifugal filter units 10 kDa MWCO to 0.5–1.0% of the G-25 column bed volume (see Note 10).
Equilibrate with five CV G-25 column buffer at 4 °C using at no more than 2 mL/min.
Set up the fraction collector with enough tubes to collect one bed volume in 30-drop (1 mL) fractions.
Load sample, and allow to run through column at a rate not to exceed that of the equilibration step above. Begin the fraction collector as the sample is loaded.
Measure the UV absorbance at 260–280 nm of fractions, and run SDS-PAGE analysis on the samples containing protein based on UV.
Pool fractions containing S100 protein.
After it has been determined that S100 protein has been eluted, equilibrate column with two column volumes of G-25 resin storage buffer. Resin should be stored at 4 °C in the column.
Concentrate the sample using Amicon Ultra-15 centrifugal filter units 10 kDa MWCO to 0.5–1.0% of the G-25 column bed volume.
Determine an accurate protein concentration of the final concentrated protein using the Bradford protein assay. Freeze protein stock at −20° C at 0.5–10 mM S100 protein (see Note 10).
3.2. Fluorescence Polarization Competition Assay (FPCA)
3.2.1. FPCA for Screening
See Fig. 2 for assay setup in a 384-well microplate.
Add 20 μL no S100 control + DMSO to columns 23–24.
Add 20 μL full FPCA buffer to every other well, except those in columns 23–24.
Add up to 1 μL of the test compounds per well so that the final concentration of DMSO will not exceed 5% (see Note 12).
Add an equivalent amount of DMSO to the control wells so that all of the wells have the same final DMSO concentration.
Take care not to introduce bubbles while pipetting, but the plate can be centrifuged ~5 min to remove bubbles.
Seal the plates with sealing tape.
Incubate the plates for at least 4 h room temperature (25 °C).
Remove the sealing tape carefully as not to introduce bubbles.
For the first assay plate only, calibrate the G-factor for the fiuorescence microplate reader with polarization capabilities on one of the wells containing the no S100 control + DMSO, such as well B23, setting the polarization as 100 mP using 544 nm excitation and 590 nm emission wavelength (see Note 11).
Read the fiuorescence polarization for all the plates using those settings.
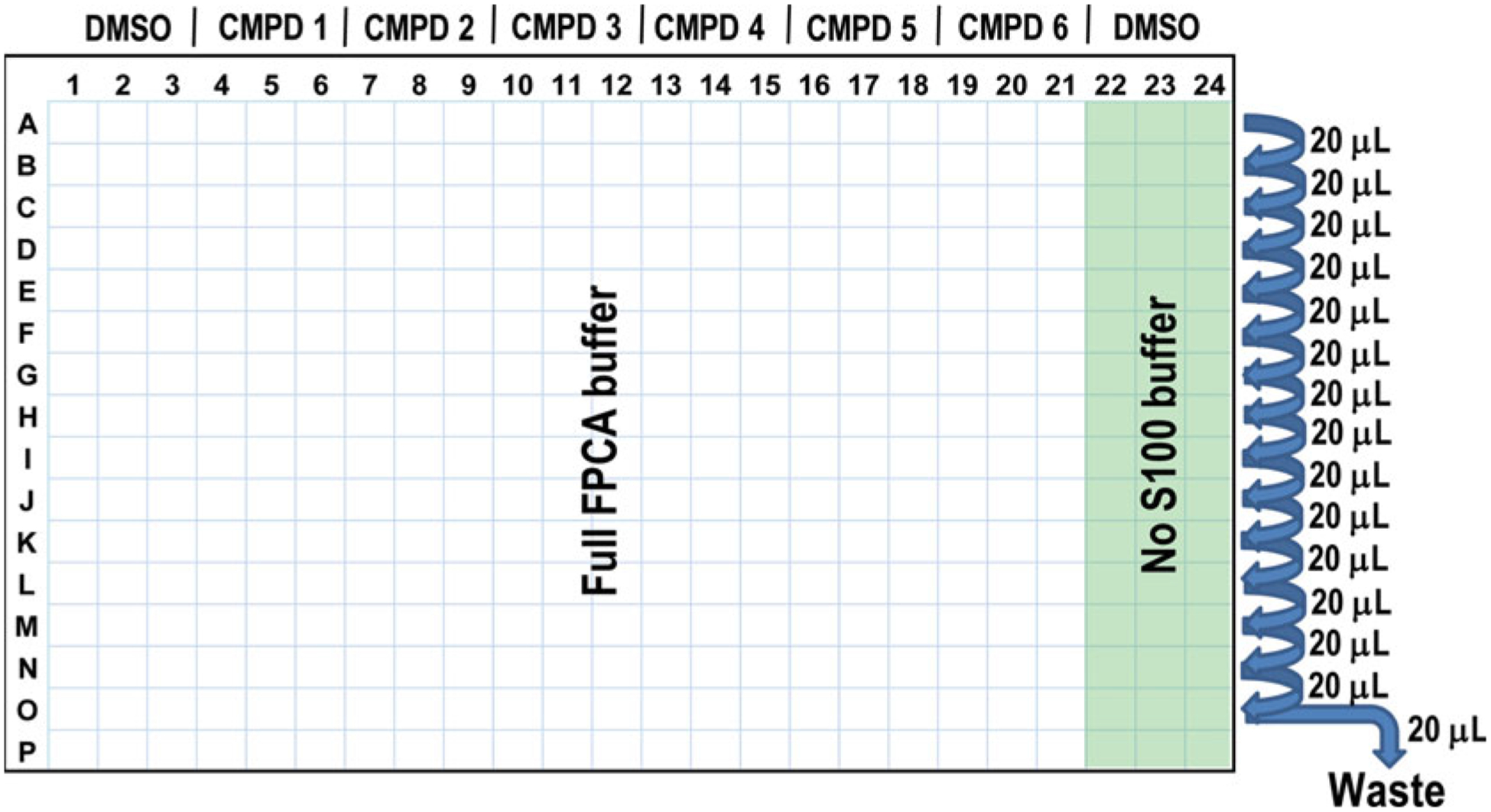
Fig. 2.

Layout of typical HTS assay in a 384-well microplate for screening compound libraries that normally have DMSO on the first and last columns (CMPD, test compound)
3.2.2. Z-Factor
Determine μb: the mean polarization values for all the wells containing full FPCA buffer + DMSO only (bound probe).
Determine μf: the mean polarization values for all the wells containing no S100 control + DMSO only (free probe).
Determine σb: the standard deviation of the polarization values for wells containing full FPCA buffer + DMSO only (bound probe).
Determine σf: the standard deviation of the polarization values for wells containing full FPCA buffer + DMSO only (free probe).
Determine the Z-factor: 1 ((3 × σb + 3 × σf)/(μb−μf))
The Z-factor can be any value ≤1, with a value of 1 being an ideal assay with a value ≥0.5–1.0 being acceptable for screening [9]; however, below 0.5 the assay should be redone.
3.2.3. Hit Identification
Determine the mean (μ) and standard deviation (σ) of the polarization values for all the wells except the no S100 Control + DMSO only.
A “hit” (i.e., perspective inhibitor) is defined to result in a polarization value ≤μ (3 × σ).
3.2.4. FPCA Titration for Determining IC50
See Fig. 3 for assay setup in a 384-well microplate.
Add 40 μL of the highest concentration of the test compound in full FPCA buffer into wells in the first row (row A) of the 384-well microplate plate with each test compound done in triplicate (see Note 13).
Add 40 μL of full FPCA buffer with the equivalent amount of DMSO as the test compound(s) samples in the first row of the first three columns (row A, columns 1–3).
Add 20 μL of the full FPCA buffer in the wells below the compound samples and DMSO control. The same pipette tips can be used repeatedly taking care not to create bubbles.
Add 40 μL of no S100 FPCA buffer with the equivalent amount of DMSO as the test compound(s) samples in the first row of the first three columns (row A, columns 22–24).
Add 20 μL of the no S100 FPCA buffer in the remaining wells in columns 22–24 below DMSO control. The same pipette tips can be used repeatedly taking care not to create bubbles.
Serial dilute the sample by pipetting 20 μL from the first wells (row A) into the next wells (row B) mixing slowly and carefully pipetting up and down two to four times with the existing 20 μL buffer in the well. Be careful not to blow out the sample and introduce bubbles.
Using the same pipette tips, transfer 20 μL from these current wells (row B) to the next wells (row C) mixing slowly and carefully pipetting up and down two to four times with the existing 20 μL buffer in the well. Be careful not to blow out the sample and introduce bubbles.
Repeat step 8 until you are in the next-to-final wells (row O).
Remove 20 μL from the next-to-final well (row O) and dispose it off. The final well should not contain any compound.
Seal the plates with a sealing tape.
Incubate the plates for at least 4 h room temperature (25 °C).
Remove the sealing tape carefully as not to introduce bubbles.
Calibrate the G-factor for the fiuorescence microplate reader with polarization capabilities on one of the wells containing the no S100 control + DMSO, such as well B23, setting the polarization as 100 mP using 544 nm excitation and 590 nm emission wavelength (see Note 11).
Read the fiuorescence polarization for all the plates using those settings.
- Convert the raw polarization values, typically given as mP, into % inhibition:
where:- SAMPLEmP = the polarization value of experimental sample.
- NO S100mP = mean polarization value of the no S100 Control + DMSO wells.
- S100mP = mean polarization value of the S100 Control + DMSO wells.
Determine the IC50 by fitting the data to a single-site binding model using nonlinear least-squares analysis.
Fig. 3.
Layout of typical compound titrations performed in a 384-well microplate where the sample is serial diluted down the plate with row A having the highest, row O the lowest, and row P containing no compound (CMPD, test compound)
3.2.5. Ki Calculation
Determine the Ki from the experimentally determined IC50, the variables in Table 1, using the Nikolovska-Coleska et al. equation as follows (see Note 14):
where:
P = S100 protein.
L = fiuorescence-labeled peptide probe.
I = inhibitor
(see Table 1)
(see Table 1)
KD = dissociation constant of the PL complex (see Table 1)
Table 1.
Values for Ki calculation
| [PT] (μM) | [LT] (μM) | KD (μM) | |
|---|---|---|---|
| S100A1 | 6.0 | 0.01 | 3.67 ± 0.07 |
| S100B | 1.0 | 0.01 | 0.13 ± 0.01 |
3.2.6. Hit Confirmation
It is necessary to have an independent secondary assay to confirm that the interaction of S100 occurs directly with the compound(i.e., one not depending on the same technique). This would rule out the possibility that the compound is a false positive due to fiuorescence overlap or quenching of the fiuorescent probe, interaction with the peptide, or other indirect effects.
4. Notes
The methods described here were performed successfully with human, bovine, and rat S100B and S100A1. The S100A1 and S100B pET11b expression vectors were synthesized using codon optimization for expression in E. coli.
BL21(DE3) or BL21(DE3) Star cells are also suitable E. coli for expression.
The assay can be run on any instrument capable of measuring TAMRA fiuorescence polarization accurately at 10 nM with a 544 nm excitation and 590 nm emission wavelength. The assay could be adapted easily to run in a 96-well microplate or in a cuvette; however the throughput is highly limited using a single cuvette system. Also, since the final sample volume would have to be increased in these lower-throughput methods, more reagents including S100 protein, compound, and fiuorescently-labeled peptide would be needed. For example, just going from the 384-well protocol to a typical 96-well microplate would necessitate increasing the sample from 20 μL to 100 μL and requires fivefold more sample. The use of a BMG PHERAstar FS multimode microplate reader is recommended, but Tecan, BioTek, Molecular Devices, Perki-nElmer, and other manufacturers produce instruments with similar capabilities.
Microplates are manufactured with different materials to either minimize or maximize the binding of proteins or other biomolecules. For the FPCA, minimizing binding of the fiuorescently labeled peptide, S100 proteins, and compounds of interest to the microplate is desired. Untreated polystyrene should not be used. While there are polystyrene microplates with surfaces engineered to have very low binding that can be purchased, the polypropylene microplates are sufficient for this application at a fraction of the cost.
Both peptides were synthesized using solid-state peptide synthesis, and their purity was determined to be >95% by high-pressure liquid chromatography (HPLC) and mass spectrometry. Since the peptides used are not terminal in the full-length proteins, the C- and N-terminus residues are modified to neutralize the charge. A TAMRA label was chosen as the fiuorescent probe since it is readily available, most fiuorimeters are capable of measuring its FP, it is relatively inexpensive compared to other fiuorophores, and its fiuorescence properties are not as susceptible to pH and oxidation as is the case for fiuorescein isothiocyanate (FITC). The concentration of the peptide solutions are determined using the extinction coefficient for TAMRA, ε547 = 65,000 cm−1 M−1 [11]. Most synthetic peptides have TFA counter ions, so the pH must be adjusted to ~pH 7 immediately upon suspending the powder in water, or else peptide degradation can occur. Titrations of the respective S100 proteins were done to determine the respective TAMRA-peptide binding affinity (KD; see Table 1), and the fiuorescently labeled peptide was competed off with the addition of the unlabeled version of the same peptide, as needed to demonstrate that the label does not cause the peptide to bind to an alternative site(s).
A 4–20% gradient gel for SDS-PAGE is recommended, since it readily separates S100A1 and S100B from the buffer front; however, even with a high percent gel, it is necessary to run the buffer front completely to the bottom of the gel to obtain good separation. Be aware that S100B and S100A1 will run at ~6 kDa by the SDS-PAGE rather than the expected ~10.7 kDa (expected monomer size) because this family of proteins are typically highly negatively charged.
Do not thaw the glycerol stocks, and return them to −80 °C immediately after using. If stored and handled correctly, the glycerol stocks can be used for over 10 years.
The cells can be stored at −80 °C for up to 3 months. Even though freeze-thawing the cells helps lyse the cells and release the overexpressed protein of interest, this step can be skipped if the plan includes immediately performing the cell lysis step (see Subheading 3.1, step 3).
S100 proteins bind calcium and in some cases zinc and copper. This protocol will produce mostly S100 proteins in the metal-free state (<1%), which is sufficient since the FPCA assay will typically be run with saturating calcium concentrations to observe peptide binding and the competition experiments. Additional steps can be taken to further lower contaminating calcium for “Ca2+-free” control experiments. Such steps include using only plastic containers when possible, acid washing glass that contacts the sample or buffers, starting with highest-purity metal-free water, Chelex treating all buffers and reagents, and adding 5 mM EGTA to the G-75 column buffer for the G-75 size exclusion step. For full removal of the 5 mM EGTA, it will be necessary to dialyze twice against 4 L of G-75 column buffer +100 mM NaCl before the G-25 size exclusion, step 2. If this is not done, the EGTA can contaminate the final sample. A longer G-25 column, ~80 mL column volume, will more efficiently remove EGTA.
The purified S100 protein stock can be stored in 5–50 mM HEPES, Tris, or other nonfiuorescent buffer at pH 6.8–7.5. An accurate protein concentration is critical for the FPCA particularly if you wish to use the assay to determine the Ki of the compound. A Bradford protein assay can be used to determine the stock concentration. The S100 concentration is calculated for a single subunit since S100 proteins exists typically as a symmetric dimer with a single fiuorescent peptide probe bound per S100 subunit.
Most microplate readers do not determine the absolute polarization, instead the manufacturer instructions say to calibrate the instrument to a fiuorophore sample with a known polarization value such as FITC (27 mP) or TAMRA (50 mP) to determine a grating factor (G-factor). It is recommended to use the “no S100 control” sample set for 100 mP to calibrate the G-factor and to avoid having to make a separate sample. (The absolute values of the free peptides were determined in these assay conditions, 72.9 ± 5.7 mP for TAMRA-HDM425–43 and 134.2 ± 5.1 for TAMRA-TRTK, but the only concern is since with the change in polarization rather than the absolute value, for the FPCA, the 100 mP value is fine.)
There are several sources of small molecule compound collections available commercially and from public sources (NCI) for screening. Most will be supplied in 96- or 384-well plates and will be suspended at 10–20 mM in 100% DMSO. Since both the S100 FPCAs can be performed with ≤5% DMSO, the compounds concentration for the inhibition studies can approach the high μM range. The test compounds can be added via a multichannel pipette; however, the use of auto-mated liquid handlers using 96 or 384 channel pipetting head or pin tools will greatly increase the throughput.
Start with a maximum concentration of compound ~20-fold higher than the expected IC50 as long as the total DMSO does not exceed 5% and the compound is soluble (i.e., does not precipitate). For example, if the primary screen was as 10 μM compounds and the hit decreased the mP by 50% inhibition, then your IC50 is about 10 μM, and then start with a maximum compound concentration of at least 200 μM. It may require multiple trials to get the best concentration range, resulting in a good curve fit to the data.
Once a putative S100 inhibitor is identified in a single-point screening assay, a titration of the compound can be carried out to determine the IC50. From that, the Ki of the inhibitor for the S100 can be calculated. While the IC50 values can be used to compare the potency of inhibitors within the same assay, the Ki is more useful for comparing binding affinities determined in different assay conditions or types. An equation developed by Nikolovska-Coleska et al. [13] is used here to calculate the Ki (i.e., versus the Cheng-Prusoff equation) [14] as FPCA fails to meet the basic assumption of the latter equation. While both equations give estimates of the Ki, the Nikolovska-Coleska et al. equation was shown to be closer to the true Ki. The authors maintain a publicly accessible web-based computer program for determining the Ki using the Nikolovska-Coleska et al. equation at http://sw16.im.med.umich.edu/software/calc_ki/.
References
- 1.Bresnick AR, Weber DJ, Zimmer DB (2015) S100 proteins in cancer. Nat Rev Cancer 15(2):96–109. 10.1038/nrc3893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chong ZZ, Changyaleket B, Xu H, Dull RO, Schwartz DE (2016) Identifying S100B as a biomarker and a therapeutic target for brain injury and multiple diseases. Curr Med Chem 23(15):1571–1596 [DOI] [PubMed] [Google Scholar]
- 3.Wilder PT, Lin J, Bair CL, Charpentier TH, Yang D, Liriano M, Varney KM, Lee A, Oppenheim AB, Adhya S, Carrier F, Weber DJ (2006) Recognition of the tumor suppressor protein p53 and other protein targets by the calcium-binding protein S100B. Biochim Biophys Acta 1763(11):1284–1297. 10.1016/j.bbamcr.2006.08.024 [DOI] [PubMed] [Google Scholar]
- 4.Hartman KG, Vitolo MI, Pierce AD, Fox JM, Shapiro P, Martin SS, Wilder PT, Weber DJ (2014) Complex formation between S100B protein and the p90 ribosomal S6 kinase (RSK) in malignant melanoma is calcium-dependent and inhibits extracellular signal-regulated kinase (ERK)-mediated phosphorylation of RSK. J Biol Chem 289(18):12886–12895. 10.1074/jbc.M114.561613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang H, Zhang L, Zhang IY, Chen X, Da Fonseca A, Wu S, Ren H, Badie S, Sadeghi S, Ouyang M, Warden CD, Badie B (2013) S100B promotes glioma growth through chemoattraction of myeloid-derived macrophages. Clin Cancer Res 19(14):3764–3775. 10.1158/1078-0432.CCR-12-3725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Prosser BL, Wright NT, Hernandez-Ochoa EO, Varney KM, Liu Y, Olojo RO, Zimmer DB, Weber DJ, Schneider MF (2008) S100A1 binds to the calmodulin-binding site of ryanodine receptor and modulates skeletal muscle excitation-contraction coupling. J Biol Chem 283(8):5046–5057. 10.1074/jbc.M709231200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wright NT, Prosser BL, Varney KM, Zimmer DB, Schneider MF, Weber DJ (2008) S100A1 and calmodulin compete for the same binding site on ryanodine receptor. J Biol Chem 283(39):26676–26683. 10.1074/jbc.M804432200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cannon BR, Zimmer DB, Weber DJ (2011) S100A1 (S100 calcium binding protein A1). Atlas Genet Cytogenet Oncol Haematol 15(10):873–876. 10.4267/2042/46035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hall MD, Yasgar A, Peryea T, Braisted JC, Jadhav A, Simeonov A, Coussens NP (2016) Fluorescence polarization assays in high-throughput screening and drug discovery: a review. Methods Appl Fluoresc 4(2):022001 10.1088/2050-6120/4/2/022001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huang X, Aulabaugh A (2016) Application of fiuorescence polarization in HTS assays. Methods Mol Biol 1439:115–130. 10.1007/978-1-4939-3673-1_7 [DOI] [PubMed] [Google Scholar]
- 11.Wilder PT, Charpentier TH, Liriano MA, Gianni K, Varney KM, Pozharski E, Coop A, Toth EA, Mackerell AD, Weber DJ (2010) In vitro screening and structural characterization of inhibitors of the S100B-p53 interaction. Int J High Throughput Screen 2010(1):109–126. 10.2147/IJHTS.S8210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ivanenkov VV, Jamieson GA Jr, Gruenstein E, Dimlich RV (1995) Characterization of S-100b binding epitopes. Identification of a novel target, the actin capping protein, CapZ. J Biol Chem 270(24):14651–14658 [DOI] [PubMed] [Google Scholar]
- 13.Nikolovska-Coleska Z, Wang R, Fang X, Pan H, Tomita Y, Li P, Roller PP, Krajewski K, Saito NG, Stuckey JA, Wang S (2004) Development and optimization of a binding assay for the XIAP BIR3 domain using fiuorescence polarization. Anal Biochem 332(2):261–273. 10.1016/j.ab.2004.05.055 [DOI] [PubMed] [Google Scholar]
- 14.Cheng Y, Prusoff WH (1973) Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem Pharmacol 22(23):3099–3108 [DOI] [PubMed] [Google Scholar]