

Figure 1.
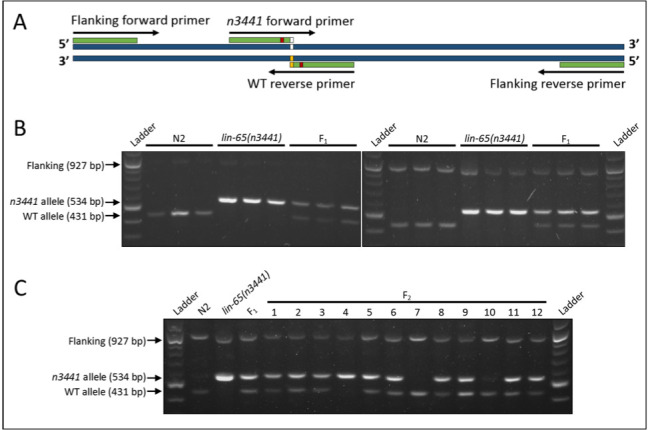
(A) Schematic of tetra-primer ARMS-PCR with lin-65(n3441)SNP indicated in white, WT SNP in yellow, and mismatches in red. (B) Three replicates of single-worm PCR products of C. elegansgene lin-65in N2 strain, a strain homozygous for the lin-65(n3441)SNP allele, and heterozygous (n3441/WT) F1progeny under standard PCR conditions (left) and conditions optimized for amplification of the SNP flanking sequence (right). (C) PCR products of parental, F1, and F2individuals with flanking sequence amplification.
Description
Single nucleotide polymorphisms (SNPs) can be difficult to detect using traditional PCR and gel electrophoresis, especially when no enzyme restriction sites overlap the SNP. Although alleles can be identified through amplification with flanking primers and sequencing, this adds time and cost, and sequencing is typically outsourced. Tetra-primer Amplification-Refractory MutationSystem (ARMS)-PCR allows SNPs to be distinguished through a 3’ primer mismatch at the SNP site for one allele, and an additional mismatch 1 – 3 bases upstream from the 3’ end (Figure 1A), limiting amplification to only the allele with a single mismatch (Yeet al.2001). Each internal primer (one forward and one reverse) is allele-specific, while external primers are different distances from the SNP, producing allele-specific fragments of different sizes in a single PCR reaction that can be distinguished by gel electrophoresis. This is useful for single-worm PCR where separate reactions may not be feasible, such as selection of individuals from a segregating population. Here, we designed primers and optimized PCR conditions to allow clear genotyping of lin-65(n3441), a G->A point mutation, without the need for sequencing. Additionally, we show that minor changes to reaction conditions allow for amplification of the larger flanking fragment (Figure 1B) which can then be gel purified and sequenced. While this may not be necessary for routine genotyping, sequencing the flanking fragment provides confirmation and higher confidence in SNP identification. To demonstrate the accuracy of our protocol, N2 and lin-65(n3441) adult hermaphrodites, a heterozygous F1hermaphrodite, and twelve segregating F2progeny were genotyped by PCR using conditions optimized for amplification of the flanking fragment (Figure 1C). Flanking fragments were then gel purified and sequenced. Results showed the presence of the G allele (WT) in N2, F2-7, and F2-10, the A allele (n3441) in lin-65(n3441) and F2-4, and heterozygous G/A in F1 and the remaining F2samples, confirming the genotypes indicated by PCR. Interestingly, sequencing showed a higher proportion of the G allele in all nine heterozygous samples and the heterozygous control. Those samples, as well as the homozygous n3441 sample and control, were also heterozygous G/A at the -2 base, showing a higher proportion of the A allele; this change corresponds to the introduced G->A mismatch in the n3441-specific internal primer. These two observations suggest the possibility of low level overlapping PCR during amplification, incorporating the primer sequence into a portion of PCR product. Additionally, the higher proportion of the G allele at the SNP site indicates amplification bias of the n3441-specific primer over the flanking forward primer in the presence of the n3441 allele, leaving the two flanking primers with mostly the WT allele for amplification in heterozygotes. This idea is supported by the visibly brighter n3441 bands in all samples where the allele is present.
The current primer sets were chosen following the guidelines from Liu et al. and optimized to give the highest chance of success, though alternative primers were not tested. To overcome bias of n3441 amplification in the larger flanking fragment, an improvement in amplification efficiency of the WT allele could be achieved by redesigning the WT-specific internal primer to be longer. Different mismatches could also be tested from the one chosen here, and at different positions relative to the SNP (-1, -3, or -4). However, we do not feel that the WT-specific primer is amplifying the WT band inefficiently, rather the n3441-specific primer amplifies exceptionally well; only 0.33 µl PCR product was loaded into each lane to avoid smearing due to overloading. An alternative to improving WT amplification is to slightly reduce efficiency of n3441 amplification. This could be accomplished by following the suggestions outlined above and applying them to the n3441-specific primer; a slightly less efficient primer could then be selected. While tetra-primer ARMS-PCR is largely limited by the SNP site, as adjacent upstream and downstream sequence must be amenable to primer design, we have demonstrated here that certain SNPs may be worth pursuing with minimal or no changes to standard PCR protocol.
Detailed Protocol
Primers were developed using Multiple Primer Analyzer (Thermo Fisher Scientific). Internal allele-specific primers were designed following steps outlined by Liu et al. (2012). The wild-type (WT)-specific primer set included a 24-nt forward primer beginning at 412 bases upstream from the SNP, and a 19-nt reverse primer with the WT SNP (G) as its terminal (3’) base. A GC mismatch was introduced into the primer at the -2 base from the SNP site. The n3441-specific primer set consisted of a 20-nt reverse primer beginning 514 bases downstream from the SNP, and a 20-nt forward primer with the n3441 SNP (A) as its terminal base. A TG mismatch was introduced into the primer at the -2 base from the SNP site. The WT and n3441 primer sets were tested individually using single worm PCR across a temperature gradient to determine appropriate annealing temperature. Both primer sets were then combined into a single 25 µl PCR reaction with each primer at 0.2 µM, dNTP (New England BioLabs® Inc., Ipswich, MA) at 0.2 mM, 1x EconoTaq Buffer with Mg (Lucigen Corp., Middleton, WI), and 0.1 µl Taq Polymerase per reaction (http://bio-protocol.org/e-136). Samples were denatured at 94oC for 3 minutes, followed by 30 cycles of denaturation (94oC for 30 seconds), annealing (61oC for 30 seconds), and extension (72oC for 10 seconds), ending with a final extension of 72oC for 10 minutes. To improve amplification of the fragment flanking the SNP, external primer concentrations were increased to 0.4 µM and dNTP raised to 0.3 mM, with a 50 second extension time during the 30 cycles. All other conditions remained the same. Amplification of the WT allele, n3441 allele, and flanking fragment generated products of 431, 534, and 927 bp, respectively.
To develop a test population, N2 males were mated with a single lin-65(n3441) hermaphrodite to produce heterozygous cross-progeny (F1). A single F1hermaphrodite was isolated and allowed to produce self-progeny (F2). After two days of egg laying, the F1hermaphrodite was moved to a new plate for later genotyping.Once F2progeny reached adult stage, single N2 and lin-65(n3441) adult hermaphrodites, the F1 parent, and twelve F2 progeny were PCR amplified under conditions for flanking fragment amplification. 84–100 ng of PCR product per sample were loaded onto a 1% agarose gel and separated by electrophoresis at 100 V for 1.5 hours. Using 5.1 – 6.0 µg of DNA from the same PCR reaction, samples were run through a new gel and the flanking fragments purified using Monarch® DNA Gel Extraction Kit (New England BioLabs® Inc., Ipswich, MA). Purified fragments were sent to GENEWIZ (South Plainfield, NJ) for Sanger sequencing. Trace files confirmed a single peak corresponding to the allele in samples identified as homozygous by PCR. In all samples identified as heterozygous, peaks corresponding to the G and A alleles were both observed, with G peaks being noticeably higher.
Reagents
Strains: N2 (wild-type, var. Bristol); MT13232 [lin-65(n3441)]. Both strains are available at the CGC.
Primer sequences: flanking forward: 5’-attcagatttccgggagaaaaatc-3’; flanking reverse: 5’-tttggtagacgtgtcgaatc-3’; WT reverse: 5’-ggcggtggaattgcttgcc-3’; n3441forward: 5’-cttccggcaacagtcaggta-3’
Acknowledgments
Funding
NIH R15 GM119029
References
- Liu J, Huang S, Sun M, Liu S, Liu Y, Wang W, Zhang X, Wang H, Hua W. An improved allele-specific PCR primer design method for SNP marker analysis and its application. Plant Methods. 2012 Aug 24;8(1):34–34. doi: 10.1186/1746-4811-8-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye S, Dhillon S, Ke X, Collins AR, Day IN. An efficient procedure for genotyping single nucleotide polymorphisms. Nucleic Acids Res. 2001 Sep 01;29(17):E88–E88. doi: 10.1093/nar/29.17.e88. [DOI] [PMC free article] [PubMed] [Google Scholar]