

Abstract
Interplay between the nervous and immune systems is critical for homeostasis, and its dysfunction underlies pathologies like multiple sclerosis, autism, leukemia, and inflammation. The nematode C. elegans provides an opportunity to define evolutionarily conserved mechanisms of regulation of host innate immunity and inflammation in a genetically tractable whole-animal system. In the past few years, the C. elegans nervous system has emerged as an integral part of host defense against pathogens, acting through diverse mechanisms to repress or induce protective transcriptional responses to infection in distal tissues. In this review, we discuss current knowledge of the mechanisms through which the C. elegans nervous system controls the expression of host defense genes in the intestinal epithelium. Although still incomplete, the insights derived from such work have broad implications for neural regulation of epithelial function at mucosal barriers in higher organisms in health and disease.
Graphical Abstract
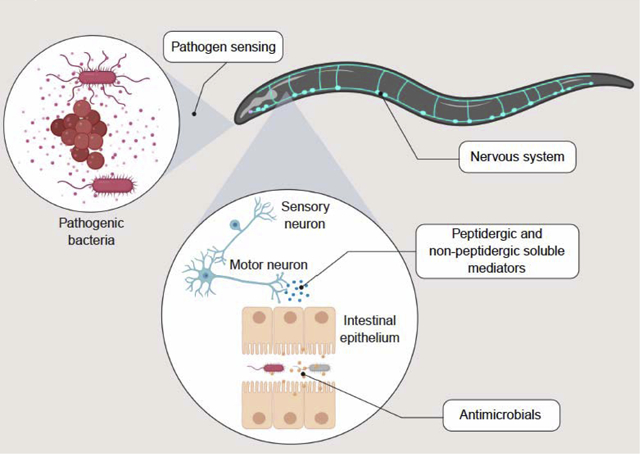
Introduction
Free-living nematodes like Caenorhabditis elegans ingest microbes as a source of nutrition. Their habitats harbor diverse communities of microorganisms, some of which have evolved virulence potential, or the capability to fight back against predation[1]. This poses an evolutionary conundrum to nematodes, not unlike that posed to humans in relation to their microbiota: how to detect and protect against pathogenic microbes, while foraging for nonpathogenic or commensal nutritional sources? Hence the evolution in nematodes of sophisticated molecular sensing and defense mechanisms, the majority of which are shared with mammals, and thus must have existed in their last common ancestor[2,3]. This article reviews the most recent advances in our understanding of the critical role that the nervous system plays in detecting pathogenic microbes and in inducing the expression of antimicrobial host defense genes in the intestine in the model nematode C. elegans. The insights derived from this work have implications for our understanding of the evolution of host defense strategies in animals, and can shed light on yet undiscovered mechanisms of neuro-immune regulation in higher organisms.
C. elegans host defense
Invertebrates possess only innate mechanisms of host defense, which may include pattern recognition receptors such as the Toll-like receptors (TLR), downstream signaling pathways, and transcription factors like NF-κB that induce the expression of antimicrobial peptides upon infection[4]. In addition, patrolling phagocytes in their “blood” contribute to pathogen clearance. In these animals, innate host defense results from interactions between endocrine signals and cellular defenses to enhance survival of infection.
Not so in C. elegans. These nematodes have lost genes encoding NF-κB and several key components of known TLR pathways[3]. Furthermore, although C. elegans does possess 3 pairs of phagocyte-like coelomocytes, these do not patrol and do not appear to ingest bacteria[5]. One might be tempted to think that C. elegans represents a more primordial host defense system in relation to, say, arthropods like Drosophila. However, its phylogenetic position in relation to more basal metazoans like cnidariae, which do carry genes for TLR pathways and patrolling phagocytes, clearly favors the hypothesis that nematodes lost TLR pathways and phagocytes in the course of evolution[3], presumably in favor of alternative mechanisms of defense that increased their fitness. What might these mechanisms of defense be?
Research conducted over the last 15 years, following the pioneering work of Ausubel and Ewbank[6,7], has demonstrated that infections with pathogenic bacteria, fungi, and viruses elicit pathogen-specific programs of gene expression in many tissues throughout the nematode body[8]. In the specific case of ingested pathogenic bacteria, C. elegans rely on their simplified intestinal epithelium to fight infections[3]. Different bacterial infections elicit distinct transcriptional programs in these cells, which include genes that may encode antimicrobial peptides and enzymes, and which enhance host survival of infection[9,10]. Although such activities have been formally demonstrated in only a handful of cases, the majority of these genes have human correlates, if not direct homologs[11,12].
The induction of these putative or demonstrated antimicrobials has been used with remarkable success to identify upstream signaling pathways that regulate their expression. In a relatively short time, forward and reverse genetics approaches have facilitated the discovery and characterization of multiple evolutionarily-conserved signaling pathways, which are engaged in pathogen-specific manners to produce the organismal defense response. Thus, pathways mediated by p38 MAPK[13,14], β-catenin[15], hypoxia-inducible factor[16], heat shock factor[17], transcription factor EB (TFEB)[18], the unfolded protein response[19], ZIP-type transcription factors[20,21], GATA transcription factors[22], NRF2 and the oxidative stress response[23,24], and insulin signaling[25,26], among others, play important roles under different infection scenarios (reviewed in[27]). Despite all this important knowledge, which helped identify new pathways in mammalian systems[18,28], the question remained: how does C. elegans detect infection to trigger host response pathways?
Neural regulation of host defense gene expression
Recent evidence points to the nervous system in general, and the chemosensory system in particular, as key for pathogen sensing in C. elegans[29]. C. elegans is one of the most popular model organisms to study the nervous system, because several features make the C. elegans nervous system an appealing model for higher organisms. Compared to the human brain, which contains about 100 billion (1011) neurons[30], the C. elegans nervous system comprises just 302 neurons[31•]. These neurons are connected through about 5,000 synapses and a few hundred gap junctions, which have been comprehensively mapped[31•].
At the molecular level, the C. elegans nervous system shares many features with its mammalian counterparts because its genome encodes many pathways that are conserved in mammals[32]. For example, C. elegans produce most neurotransmitters that are present in mammals (with the exception of glycine and histamine)[32,33]. Moreover, the C. elegans nervous system expresses neurotransmitter receptors, ion-channels, and GPCRs with mammalian counterparts[34]. Thus, C. elegans is poised to facilitate fundamental understanding of neural regulation of innate immunity.
Insulin and insulin receptor
The first study to demonstrate a key role of the nervous system in C. elegans host defense found that mutants defective in neurotransmitter secretion were resistant to P. aeruginosa[35••]. Concordantly, compared to wild type such mutants exhibited increased expression of genes encoding antimicrobial factors (e.g. saposin spp-1, antimicrobial peptide abf-2, and lysozyme lys-7). On the other hand, mutants with enhanced neuronal secretion were more susceptible to P. aeruginosa than wild type, which correlated with decreased expression of antimicrobial peptides and other host defense genes in the intestinal epithelium[35••]. Thus, the nervous system appeared to function in communication with the intestinal epithelium to repress host defense.
The molecular mechanism was shown to involve the insulin pathway. DAF-16, homologous to mammalian transcription factor FOXO3, functions downstream of insulin receptor DAF-2. DAF-2 represses DAF-16 to prevent the expression of a broad range of longevity and stress defense genes[26]. Under stress conditions, DAF-2 activity diminishes, causing DAF-16 activation by nuclear translocation and expression of stress resistance genes. In the mentioned study, the pathogen resistance phenotype of neurotransmitter defective mutants was suppressed by mutation of daf-16, implicating the insulin pathway. Moreover, such neurotransmitter defective mutants exhibited constitutive nuclear translocation of DAF-16 (FOXO3) in the intestinal cells in the absence of infection. Thus, the nervous system functions to repress DAF-16, presumably by activating DAF-2 via an agonistic ligand.
Consistent with this idea, the insulin gene ins-7, which encodes a putative DAF-2 ligand[36], was upregulated in uninfected neurotransmission-defective mutants and in infected wild type animals. Moreover, ins-7 mutants exhibited increased nuclear translocation of DAF-16 (FOXO3) and enhanced survival during infection, similar to neurotransmitter defective mutants. Furthermore, these phenotypes were reversed by neuronal rescue with wild type ins-7. These results implied that neurotransmission was a key mechanism of control of ins-7 expression and downstream DAF-16 (FOXO3) activity via DAF-2[35••]. While INS-7 induction during infection appears to be counterproductive for the nematode, whether this is an example of the pathogen “hacking” the host response, or if INS-7 confers an undescribed advantage to the host in a more ecologically relevant scenario, is not clear.
In mammals, insulin signaling plays multiple roles in immunity, including both pro- and anti-inflammatory functions[37]. For example, hyperglycemia, a metabolic disorder presenting excessive blood glucose, causes increased production of pro-inflammatory mediators and cytokines[38]. Insulin, which acts to reduce blood glucose, opposes this effect[38]. Furthermore, insulin directly modulates immune cells such as human monocytes to enhance immunocompetence[38]. Thus, the repression of host defense genes by INS-7 and the anti-inflammatory function of insulin in mammals may be shared evolutionarily ancient functions of insulin.
Neuropeptides and NPR-1
The G-protein coupled receptor (GPCR) family, which includes olfactory and bacterial molecule receptors, is expanded in the C. elegans genome compared with vertebrates[34]. Because GPCRs are known to function as receptors for molecules of bacterial origin (e.g. mammalian formylated peptide receptors FPR1 and FPR2), mutants lacking individual GPCR genes were screened for defective survival of P. aeruginosa infection. This screen identified npr-1 {homologous to mammalian neuropeptide Y (NPY) receptor (NPYR)} to be required for defense[39].
npr-1 had been previously implicated in avoidance of oxygen and attraction to food[40,41]. Using multiple approaches, the study demonstrated that npr-1 mutants exhibited defective induction of host defense genes, such as C-type lectin clec-85 and lysozymes lys-2 and lys-8, which are controlled by the p38 MAPK pathway[13,26]. Moreover, the promotion of defense gene expression by npr-1 required genes encoding known signaling components downstream of NPR-1 (NPYR), including guanylate cyclase gcy-35 and cGMP-gated ion channel subunits tax-2 and tax-4[42]. Although the mechanism of NPR-1 activation during infection was not identified, this study found that npr-1 function in AQR, PQR, and URX sensory neurons was sufficient for protection against infection[39]. The proposed model was that NPR-1 (NPYR) inhibits the activity of sensory neurons AQR, PQR, and URX, while GCY-35, TAX-2, and TAX-4 are required for their activation (Figure 1). Whether infection reduced the secretion of a peptide agonist for NPR-1 (NPYR) was not known, nor how this pathway controlled p38 MAPK activity to enact gene induction during infection. In addition, subsequent work showed that NPR-1 is involved in aversive learning to avoid infection with P. aeruginosa after initial exposure[43]. Thus, the emerging consensus is that NPR-1 (NPYR) signaling contributes to host defense via control of both gene expression and behavioral immunity.
Figure 1. Multiple neuronal pathways regulate intestinal host defense in C. elegans.

NPR-1 (NPYR) positively controls the expression of host defense genes during P. aeruginosa infection. In parallel, GCY-35, TAX-2, and TAX-4 negatively regulate their induction. FLP-18 and FLP-21 neuropeptides are upregulated during infection, and act through NPR-1 (NPYR) for behavioral avoidance of infection, but they have not been shown to affect defense gene expression[79]. Octopamine, released from RIC sensory neurons, acts through receptor OCTR-1 in ASH neurons to inhibit expression, presumably in the intestinal epithelium, of non-canonical UPR genes that promote host defense. This may be a mechanism of preventing inappropriate activation in noninfected animals. Dopamine, released during infection, acts on DOP-3 receptor on neurons to drive “conditioning” to EPEC. This was dependent on both insulin signaling and p38 MAPK, presumably via induction of host defense genes in the intestinal epithelium. Acetylcholine, released during S. aureus infection, drives muscarinic signaling in the intestinal epithelium and induces the expression of Wnt homolog CWN-2 in the same tissue. CWN-2 activates canonical Wnt signaling through MIG-1 (Frizzled), resulting in the intestinal epithelial expression of host defense genes. Created with BioRender.
Mammalian NPY and NPYRs have been shown to play a role in the immune response and inflammation[44]. Sympathetic neurons that innervate lymphoid organs produce NPY[45,46]. NPY1 and NPY2 receptors are expressed in immune cells, such as macrophages, neutrophils, granulocytes, and lymphocytes[44], and NPY has been shown to promote both pro- and anti-inflammatory activities in these cells[47,48]. NPY may also function directly in gastrointestinal immunity, since NPY-positive nerve fibers in the mouse ileal lamina propria are found in close proximity with gut immune cells, including IgA+ lymphocytes[49]. Further, there is evidence that NPY exerts a proinflammatory function in the mouse intestine[47,50]. Thus, NPR-1 (NPYR) may regulate an important, evolutionarily ancient pathway for host defense in nematodes and mammals.
Octopamine and OCTR-1
The same genetic screen that identified npr-1 revealed that mutations in octr-1 resulted in enhanced resistance of P. aeruginosa infection[51]. octr-1 encodes a GPCR specific for octopamine, which is an invertebrate neurotransmitter that is closely related to mammalian noradrenaline[52]. Consistent with their resistance phenotype, octr-1 mutants exhibited increased expression of a specific set of host defense genes, which are thought to participate in a noncanonical unfolded protein response (UPR)[51]. Using a similar neuron-specific rescue approach as for NPR-1, OCTR-1 was found to function in ASH and ASI sensory neurons. These findings led to the proposal that OCTR-1 functions in ASH and ASI neurons to suppress noncanonical UPR genes in uninfected animals, which are required for host defense during bacterial infection (Figure 1). However, the cellular source of octopamine in uninfected animals and the mechanism of control of the UPR genes remained unclear.
More recently, tbh-1 mutants unable to synthesize octopamine were found to be resistant to P. aeruginosa and to express higher levels of host defense genes compared with wild type, consistent with the previous findings using octr-1 mutants[53••]. Furthermore, nematodes lacking RIC octopaminergic neurons phenocopied tbh-1 mutants in infection assays. Conversely, exogenous octopamine supplementation was sufficient to repress host defense genes and cause hypersusceptibility to infection, indicating that octopamine is necessary and sufficient to repress host defense against P. aeruginosa[53••]. In vivo neuronal activity, recorded using genetically-encoded Ca2+− sensitive fluorophore GCAMP6, revealed that RIC activity diminished during P. aeruginosa infection in comparison with uninfected animals[53••]. Thus, octopamine released from RIC neurons may function as a repressor of unwanted host defense gene expression in uninfected animals, acting via OCTR-1 expressed in postsynaptic ASH neurons. During infection, an unknown mechanism may reduce RIC activity, enhancing ASH neuron activity and increasing expression of specific host defense genes that enhance survival (Figure 1).
C. elegans OCTR-1 is considered to be an ortholog of human α2A adrenergic receptor for norepinephrine[53••]. Norepinephrine regulates innate immune responses in mammals and other vertebrates, suggesting that the role of octopamine and/or norepinephrine in immunomodulation is evolutionarily conserved[54]. A recent study showed that elimination of noradrenergic neurons using 6-hydroxydopamine improved survival of mice during Klebsiella pneumoniae peritonitis[55]. Two complementary mechanisms of action were revealed: sympathetic-derived norepinephrine directly reduced production of monocyte chemoattractant protein-1 (MCP-1) by peritoneal macrophages, and splenic-nerve-derived norepinephrine prevented egress of splenic monocytes[55]. Elimination of noradrenergic neurons thus abrogated these two mechanisms, enhancing the ability of the innate immune system to clear the infection.
Dopamine and DOP receptors
Enteropathogenic E. coli (EPEC) kills C. elegans via secreted toxins, which were found to be indole and other derivatives of tryptophan[56,57]. Brief exposure to virulent or avirulent EPEC significantly improved the survival of C. elegans to subsequent infection with the pathogen, a phenomenon then termed “conditioning”[57]. Conditioned animals expressed higher levels of several host defense genes in the intestine, which candidate analysis showed to depend on the insulin and p38 MAPK pathways. Tissue-specific rescue experiments revealed that the insulin receptor DAF-2 and FOXO3 homolog DAF-16 functioned in the nervous system for conditioning[57].
Because serotonin and dopamine had been previously implicated in food sensing[58,59], these two neurotransmitters were examined as potential upstream signals in the neural control of conditioning. Mutants defective in synthesis of dopamine, but not serotonin, exhibited defective conditioning, implicating dopamine in the conditioning of C. elegans for EPEC infection[57]. Furthermore, mutants defective in dopamine synthesis, reuptake, or signaling through D2-like dopamine receptor DOP-3 were also defective in conditioning. The proposed model posited that exposure to EPEC triggered dopamine release, which drove DAF-16 (FOXO3) and p38 MAPK activation to increase the expression of antimicrobial peptides in conditioned animals[57]. However, the mechanism of EPEC detection to induce dopamine release, and the mechanism connecting dopaminergic signaling to the insulin and p38 pathways were not elucidated.
In contrast, a recent study showed that dopamine signaling through D1-like DOP-4 receptor resulted in the inhibition of p38 MAPK, downregulation of host defense genes, and increased susceptibility during P. aeruginosa infection[60••]. In this study, the cellular source of dopamine was the CEP mechanosensory neurons, and required active DOP-4 in downstream chemosensory ASG neurons. Thus, the neural circuits and biological role of dopaminergic signaling may depend on the specific pathogenic insult.
Additionally, dopamine signaling has been implicated in the modulation of behavioral plasticity, such as during food sensing, in C. elegans[61,62]. C. elegans possess eight pairs of dopaminergic neurons, with sensory projections that are directly exposed to the external environment[63]. Their anatomical position and exposure to the environment makes dopaminergic sensory neurons prime candidates for pathogen sensing by the nervous system. However, whether the sensory endings of dopaminergic neurons directly detect pathogenic bacteria remains unknown.
Multiple immune cells in mammals express dopamine receptors, and dopamine is known to modulate their functions. For example, human monocytes and macrophages express all five subtypes of dopamine receptors [64] and dopamine alters their cytokine production upon stimulation[65]. In resting macrophages, dopamine caused increased production of IL-6 and CCL2[65]. After LPS stimulation, dopamine caused increased production of IL-6, CCL2, CXCL8, and IL-10, and decreased production of TNF-α[65]. Several studies have shown that neurological disorders that arise from dysregulation of dopaminergic system in humans, such as Parkinson’s disease and schizophrenia, correlate with abnormal immune function[66]. Thus, dopamine appears to be another shared and evolutionarily ancient signaling molecule in host defense.
Acetylcholine and muscarinic receptors
A reverse genetics approach was used to identify GPCRs that are required for the induction of C-type lectin gene clec-60 (used as a reporter of the broader host response) during S. aureus infection[67••]. Simultaneous silencing of genes gar-2 and gar-3, which encode muscarinic acetylcholine receptors, prevented the induction of clec-60 and other S. aureus-induced genes. Consistent with the notion that muscarinic signaling was important for the host response, the same study defined a potential signaling pathway downstream of GAR-2 and GAR-2, comprised of EGL-30 (Gαq), PLC-1 (phospholipase Cε), diacylglycerol, and EGL-8 (protein kinase Cβ) that was necessary and sufficient for induction of clec-60 and other host defense genes(Figure 1)[67••]. Moreover, infection with S. aureus increased endogenous acetylcholine release, and acetylcholine analog arecoline was sufficient to trigger this pathway in the absence of infection. Because gar-2 and gar-3 were found to function in the intestine, and because acetylcholine is produced only in the nervous system in C. elegans[68,69•], these results suggested the existence of a novel neuro-intestinal axis.
Downstream of muscarinic signaling, the same study defined a canonical Wingless-integration site (Wnt) signaling pathway, which was activated by acetylcholine via the transcriptional induction of Wnt gene cwn-2 and its putative receptor mig-1 (Frizzled) (Figure 1)[67••]. Activation of this Wnt pathway was necessary and sufficient to drive the expression of host defense genes like clec-60[67••]. However, the mechanism of S. aureus detection by the nervous system to trigger acetylcholine release remained unknown.
Nonetheless, a similar muscarinic-Wnt signaling axis was recently implicated in the development of gastric cancer in mice and humans[70•], suggesting that this mechanism is evolutionarily conserved. Moreover, murine nociceptive sensory neurons can directly sense S. aureus infection. For example, two recent studies showed that S. aureus α-hemolysin and N-formylated peptides, released during infection, cause Ca2+ influx in nociceptor neurons, thereby activating them[71••,72]. Activation of nociceptive neurons by S. aureus α-hemolysin led to the release of neuropeptide CGRP in a dose-dependent manner. Furthermore, microarray analysis showed high expression levels of CGRP receptor in innate immune cells, such as neutrophils and macrophages, in addition to nociceptive neurons themselves, and deletion of CGRP receptor prevented the inhibition of pro-inflammatory signaling caused by α-hemolysin[72]. These studies demonstrated how a pore-forming toxin can trigger release of neuropeptides from neurons, which can act on innate immune cells, thus modulating the inflammatory response[72].
Although nociceptor neurons can be cholinergic, there is no known direct connection of intestinal epithelial cells with cholinergic nerve fibers in mammals. However, acetylcholine has been shown to induce secretion of antimicrobials in the mammalian intestinal epithelium. For example, activation of muscarinic acetylcholine receptors in small intestine isolated from mice increased secretion of antimicrobial defensins from Paneth cells and enhanced crypt antimicrobial activity ex vivo[73–75]. Conversely, muscarinic inhibitor atropine abolished antimicrobial secretion by Paneth cells[74]. Moreover, acetylcholine induced the expression of antimicrobial peptides in mouse intestinal epithelial cells[76•]. Recently, a study showed that muscarinic signaling is essential for clearing Salmonella infection in mice[77•]. Thus, muscarinic signaling to the gut epithelium appears to be an evolutionary ancient form of regulation of intestinal host defense.
Conclusion
From the studies described above and others, it is increasingly clear that different neuronal circuits and neurotransmitters have distinct relevance to C. elegans host defense under different infection scenarios. What is lacking is an integrated understanding of how these distinct circuits and neurotransmitter systems are collectively regulated during any particular pathogenic encounter. It is important to understand how they become activated or inhibited at the whole nervous system level by any particular pathogen, for a clear view of the various pathogen-sensing functions of the nervous system in the context of the whole organism.
Similarly, it is not clear exactly what signals are being sensed to detect the presence of pathogen. Most likely some of these involve soluble chemical compounds or macromolecules that sensory neurons can detect in the context of the thin watery films in which C. elegans live. Recent studies suggest that secondary metabolites produced by P. aeruginosa modulate behavior through one such mechanism of detection[78]. However, other mechanisms of pathogen detection may also operate, such as detection of translational repression by bacterial toxins[21] or morphological alterations to the intestine caused by bacterial colonization of the lumen[79••,80••]. In many cases, potential detected signals have been identified, but the mechanisms of detection remain unclear.
Finally, the exact mechanisms of neuron-epithelium communication to regulate epithelial gene expression are not known. In mammals, recent work has demonstrated direct synapsing of neurons unto epithelial enteroendocrine cells of the intestine[81••] and extensive neural innervation of the mucosa to the immediate vicinity of the intestinal epithelium[81••]. Unlike mammals, direct synapsing of neurons on epithelial cells has not been observed in C. elegans; however, in the tiny nematode body several neurons are in anatomical positions that would be consistent with distal chemical communication, as has been demonstrated in mammals. Most mediators of such communication in nematodes have not been identified.
Given these considerations, much further work is required to completely understand neuronal control of intestinal epithelial gene expression even in a simple organism like C. elegans. The next few years will likely witness great progress in addressing these outstanding questions, with great potential implications for the mammalian microbiota-gut-brain axis[82].
Highlights.
C. elegans responds to pathogens by transcriptional induction of evolutionarily-conserved host defense genes
Neuron-intestine communication is essential for host defense gene induction in several infection paradigms
Conserved neurotransmitter pathways can function as negative or positive regulators of host defense gene expression
Further dissection of the mechanisms of neuro-epithelial communication is needed for full understanding of organismal responses to infection
Acknowledgements
The authors apologize for not being able to include much important work in this short review. Funding was provided by the National Science Foundation of the USA (grant number 1656925 to JEI) and the National Institute of General Medical Sciences of the National Institutes of Health of the USA (grant number R01GM101056 to JEI).
Footnotes
Conflict of interest statement
The authors declare no conflicts of interest.
References and recommended reading
Papers of particular interest, published within the period of review, have been highlighted as:
• of special interest
•• of outstanding interest
- 1.Schulenburg H, Felix MA: The Natural Biotic Environment of Caenorhabditis elegans. Genetics 2017, 206:55–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kim DH, Ausubel FM: Evolutionary perspectives on innate immunity from the study of Caenorhabditis elegans. Curr Opin Immunol 2005, 17:4–10. [DOI] [PubMed] [Google Scholar]
- 3.Irazoqui JE, Urbach JM, Ausubel FM: Evolution of host innate defence: insights from Caenorhabditis elegans and primitive invertebrates. Nat Rev Immunol 2010, 10:47–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nyholm SV, Graf J: Knowing your friends: invertebrate innate immunity fosters beneficial bacterial symbioses. Nat Rev Microbiol 2012, 10:815–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ewbank JJ: Tackling both sides of the host-pathogen equation with Caenorhabditis elegans. Microbes Infect 2002, 4:247–256. [DOI] [PubMed] [Google Scholar]
- 6.Tan MW, Mahajan-Miklos S, Ausubel FM: Killing of Caenorhabditis elegans by Pseudomonas aeruginosa used to model mammalian bacterial pathogenesis. Proc Natl Acad Sci U S A 1999, 96:715–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Labrousse A, Chauvet S, Couillault C, Kurz CL, Ewbank JJ: Caenorhabditis elegans is a model host for Salmonella typhimurium. Curr Biol 2000, 10:1543–1545. [DOI] [PubMed] [Google Scholar]
- 8.Ermolaeva MA, Schumacher B: Insights from the worm: the C. elegans model for innate immunity. Semin Immunol 2014, 26:303–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Irazoqui JE, Troemel ER, Feinbaum RL, Luhachack LG, Cezairliyan BO, Ausubel FM: Distinct pathogenesis and host responses during infection of C. elegans by P. aeruginosa and S. aureus. PLoS Pathog 2010, 6:e1000982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.George DT, Behm CA, Hall DH, Mathesius U, Rug M, Nguyen KC, Verma NK: Shigella flexneri infection in Caenorhabditis elegans: cytopathological examination and identification of host responses. PLoS One 2014, 9:e106085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lievin-Le Moal V, Servin AL: The front line of enteric host defense against unwelcome intrusion of harmful microorganisms: mucins, antimicrobial peptides, and microbiota. Clin Microbiol Rev 2006, 19:315–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cash HL, Whitham CV, Behrendt CL, Hooper LV: Symbiotic bacteria direct expression of an intestinal bactericidal lectin. Science 2006, 313:1126–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim DH, Feinbaum R, Alloing G, Emerson FE, Garsin DA, Inoue H, Tanaka-Hino M, Hisamoto N, Matsumoto K, Tan MW, et al. : A conserved p38 MAP kinase pathway in Caenorhabditis elegans innate immunity. Science 2002, 297:623–626. [DOI] [PubMed] [Google Scholar]
- 14.Kim DH, Liberati NT, Mizuno T, Inoue H, Hisamoto N, Matsumoto K, Ausubel FM: Integration of Caenorhabditis elegans MAPK pathways mediating immunity and stress resistance by MEK-1 MAPK kinase and VHP-1 MAPK phosphatase. Proc Natl Acad Sci U S A 2004, 101:10990–10994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Irazoqui JE, Ng A, Xavier RJ, Ausubel FM: Role for beta-catenin and HOX transcription factors in Caenorhabditis elegans and mammalian host epithelial-pathogen interactions. Proc Natl Acad Sci U S A 2008, 105:17469–17474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Luhachack LG, Visvikis O, Wollenberg AC, Lacy-Hulbert A, Stuart LM, Irazoqui JE: EGL-9 controls C. elegans host defense specificity through prolyl hydroxylation-dependent and -independent HIF-1 pathways. PLoS Pathog 2012, 8:e1002798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Singh V, Aballay A: Heat-shock transcription factor (HSF)-1 pathway required for Caenorhabditis elegans immunity. Proc Natl Acad Sci U S A 2006, 103:13092–13097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Visvikis O, Ihuegbu N, Labed SA, Luhachack LG, Alves AF, Wollenberg AC, Stuart LM, Stormo GD, Irazoqui JE: Innate host defense requires TFEB-mediated transcription of cytoprotective and antimicrobial genes. Immunity 2014, 40:896–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Haskins KA, Russell JF, Gaddis N, Dressman HK, Aballay A: Unfolded protein response genes regulated by CED-1 are required for Caenorhabditis elegans innate immunity. Dev Cell 2008, 15:87–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dunbar TL, Yan Z, Balla KM, Smelkinson MG, Troemel ER: C. elegans detects pathogen-induced translational inhibition to activate immune signaling. Cell Host Microbe 2012, 11:375–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McEwan DL, Kirienko NV, Ausubel FM: Host translational inhibition by Pseudomonas aeruginosa Exotoxin A Triggers an immune response in Caenorhabditis elegans. Cell Host Microbe 2012, 11:364–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shapira M, Hamlin BJ, Rong J, Chen K, Ronen M, Tan MW: A conserved role for a GATA transcription factor in regulating epithelial innate immune responses. Proc Natl Acad Sci U S A 2006, 103:14086–14091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chavez V, Mohri-Shiomi A, Maadani A, Vega LA, Garsin DA: Oxidative stress enzymes are required for DAF-16-mediated immunity due to generation of reactive oxygen species by Caenorhabditis elegans. Genetics 2007, 176:1567–1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hoeven R, McCallum KC, Cruz MR, Garsin DA: Ce-Duox1/BLI-3 generated reactive oxygen species trigger protective SKN-1 activity via p38 MAPK signaling during infection in C. elegans. PLoS Pathog 2011, 7:e1002453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Garsin DA, Villanueva JM, Begun J, Kim DH, Sifri CD, Calderwood SB, Ruvkun G, Ausubel FM: Long-lived C. elegans daf-2 mutants are resistant to bacterial pathogens. Science 2003, 300:1921. [DOI] [PubMed] [Google Scholar]
- 26.Troemel ER, Chu SW, Reinke V, Lee SS, Ausubel FM, Kim DH: p38 MAPK regulates expression of immune response genes and contributes to longevity in C. elegans. PLoS Genet 2006, 2:e183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim DH, Ewbank JJ: Signaling in the innate immune response. WormBook 2018, 2018:1–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Najibi M, Labed SA, Visvikis O, Irazoqui JE: An Evolutionarily Conserved PLC-PKD-TFEB Pathway for Host Defense. Cell Rep 2016, 15:1728–1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hoffman C, Aballay A: Role of neurons in the control of immune defense. Curr Opin Immunol 2019, 60:30–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Herculano-Houzel S: The human brain in numbers: a linearly scaled-up primate brain. Front Hum Neurosci 2009, 3:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.•.Cook SJ, Jarrell TA, Brittin CA, Wang Y, Bloniarz AE, Yakovlev MA, Nguyen KCQ, Tang LT, Bayer EA, Duerr JS, et al. : Whole-animal connectomes of both Caenorhabditis elegans sexes. Nature 2019, 571:63–71. [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper described the first complete wiring diagram of the nervous system of C. elegans. The study included both males and hermaphrodites and revealed substantial differences between them.
- 32.Bargmann CI: Neurobiology of the Caenorhabditis elegans genome. Science 1998, 282:2028–2033. [DOI] [PubMed] [Google Scholar]
- 33.Chase DL, Koelle MR: Biogenic amine neurotransmitters in C. elegans. WormBook 2007:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hobert O: The neuronal genome of Caenorhabditis elegans. WormBook 2013:1–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.••.Kawli T, Tan MW: Neuroendocrine signals modulate the innate immunity of Caenorhabditis elegans through insulin signaling. Nat Immunol 2008, 9:1415–1424. [DOI] [PubMed] [Google Scholar]; The first paper that demonstrated that C. elegans nervous system plays a major role in the regulation of host defense against pathons. The paper showed that neuronal signals inhibit host defense via insulin signaling pathway.
- 36.Murphy CT, McCarroll SA, Bargmann CI, Fraser A, Kamath RS, Ahringer J, Li H, Kenyon C: Genes that act downstream of DAF-16 to influence the lifespan of Caenorhabditis elegans. Nature 2003, 424:277–283. [DOI] [PubMed] [Google Scholar]
- 37.Boucher J, Kleinridders A, Kahn CR: Insulin receptor signaling in normal and insulin-resistant states. Cold Spring Harb Perspect Biol 2014, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sun Q, Li J, Gao F: New insights into insulin: The anti-inflammatory effect and its clinical relevance. World J Diabetes 2014, 5:89–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Styer KL, Singh V, Macosko E, Steele SE, Bargmann CI, Aballay A: Innate immunity in Caenorhabditis elegans is regulated by neurons expressing NPR-1/GPCR. Science 2008, 322:460–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.de Bono M, Bargmann CI: Natural variation in a neuropeptide Y receptor homolog modifies social behavior and food response in C. elegans. Cell 1998, 94:679–689. [DOI] [PubMed] [Google Scholar]
- 41.Chang AJ, Chronis N, Karow DS, Marletta MA, Bargmann CI: A distributed chemosensory circuit for oxygen preference in C. elegans. PLoS Biol 2006, 4:e274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Morton DB, Hudson ML, Waters E, O’Shea M: Soluble guanylyl cyclases in Caenorhabditis elegans: NO is not the answer. Curr Biol 1999, 9:R546–547. [DOI] [PubMed] [Google Scholar]
- 43.Reddy KC, Andersen EC, Kruglyak L, Kim DH: A polymorphism in npr-1 is a behavioral determinant of pathogen susceptibility in C. elegans. Science 2009, 323:382–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.El-Salhy M, Hausken T: The role of the neuropeptide Y (NPY) family in the pathophysiology of inflammatory bowel disease (IBD). Neuropeptides 2016, 55:137–144. [DOI] [PubMed] [Google Scholar]
- 45.Lundberg JM, Rudehill A, Sollevi A: Pharmacological characterization of neuropeptide Y and noradrenaline mechanisms in sympathetic control of pig spleen. Eur J Pharmacol 1989, 163:103–113. [DOI] [PubMed] [Google Scholar]
- 46.Romano TA, Felten SY, Felten DL, Olschowka JA: Neuropeptide-Y innervation of the rat spleen: another potential immunomodulatory neuropeptide. Brain Behav Immun 1991, 5:116–131. [DOI] [PubMed] [Google Scholar]
- 47.Farzi A, Reichmann F, Holzer P: The homeostatic role of neuropeptide Y in immune function and its impact on mood and behaviour. Acta Physiol (Oxf) 2015, 213:603–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wheway J, Mackay CR, Newton RA, Sainsbury A, Boey D, Herzog H, Mackay F: A fundamental bimodal role for neuropeptide Y1 receptor in the immune system. J Exp Med 2005, 202:1527–1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shibata M, Hisajima T, Nakano M, Goris RC, Funakoshi K: Morphological relationships between peptidergic nerve fibers and immunoglobulin A-producing lymphocytes in the mouse intestine. Brain Behav Immun 2008, 22:158–166. [DOI] [PubMed] [Google Scholar]
- 50.Chandrasekharan B, Bala V, Kolachala VL, Vijay-Kumar M, Jones D, Gewirtz AT, Sitaraman SV, Srinivasan S: Targeted deletion of neuropeptide Y (NPY) modulates experimental colitis. PLoS One 2008, 3:e3304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sun J, Singh V, Kajino-Sakamoto R, Aballay A: Neuronal GPCR controls innate immunity by regulating noncanonical unfolded protein response genes. Science 2011, 332:729–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Roeder T, Seifert M, Kahler C, Gewecke M: Tyramine and octopamine: antagonistic modulators of behavior and metabolism. Arch Insect Biochem Physiol 2003, 54:1–13. [DOI] [PubMed] [Google Scholar]
- 53.••.Sellegounder D, Yuan CH, Wibisono P, Liu Y, Sun J: Octopaminergic Signaling Mediates Neural Regulation of Innate Immunity in Caenorhabditis elegans. MBio 2018, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper showed that octopamine functions as a ligand for the OCTR-1 pathway to suppress host defense against pathogens.
- 54.Kohm AP, Sanders VM: Norepinephrine: a messenger from the brain to the immune system. Immunol Today 2000, 21:539–542. [DOI] [PubMed] [Google Scholar]
- 55.Seeley EJ, Barry SS, Narala S, Matthay MA, Wolters PJ: Noradrenergic neurons regulate monocyte trafficking and mortality during gram-negative peritonitis in mice. J Immunol 2013, 190:4717–4724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Anyanful A, Dolan-Livengood JM, Lewis T, Sheth S, Dezalia MN, Sherman MA, Kalman LV, Benian GM, Kalman D: Paralysis and killing of Caenorhabditis elegans by enteropathogenic Escherichia coli requires the bacterial tryptophanase gene. Mol Microbiol 2005, 57:988–1007. [DOI] [PubMed] [Google Scholar]
- 57.Anyanful A, Easley KA, Benian GM, Kalman D: Conditioning protects C. elegans from lethal effects of enteropathogenic E. coli by activating genes that regulate lifespan and innate immunity. Cell Host Microbe 2009, 5:450–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhang Y, Lu H, Bargmann CI: Pathogenic bacteria induce aversive olfactory learning in Caenorhabditis elegans. Nature 2005, 438:179–184. [DOI] [PubMed] [Google Scholar]
- 59.Chase DL, Pepper JS, Koelle MR: Mechanism of extrasynaptic dopamine signaling in Caenorhabditis elegans. Nat Neurosci 2004, 7:1096–1103. [DOI] [PubMed] [Google Scholar]
- 60.••.Cao X, Aballay A: Neural Inhibition of Dopaminergic Signaling Enhances Immunity in a Cell-Non-autonomous Manner. Curr Biol 2016, 26:2329–2334. [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper showed that dopamine by acting through D1-like DOP-4 receptor, negatively regulates host defense by the downregulation of p38 MAPK pathway.
- 61.Sawin ER, Ranganathan R, Horvitz HR: C. elegans locomotory rate is modulated by the environment through a dopaminergic pathway and by experience through a serotonergic pathway. Neuron 2000, 26:619–631. [DOI] [PubMed] [Google Scholar]
- 62.Kindt KS, Quast KB, Giles AC, De S, Hendrey D, Nicastro I, Rankin CH, Schafer WR: Dopamine mediates context-dependent modulation of sensory plasticity in C. elegans. Neuron 2007, 55:662–676. [DOI] [PubMed] [Google Scholar]
- 63.Sulston J, Dew M, Brenner S: Dopaminergic neurons in the nematode Caenorhabditis elegans. J Comp Neurol 1975, 163:215–226. [DOI] [PubMed] [Google Scholar]
- 64.McKenna F, McLaughlin PJ, Lewis BJ, Sibbring GC, Cummerson JA, Bowen-Jones D, Moots RJ: Dopamine receptor expression on human T- and B-lymphocytes, monocytes, neutrophils, eosinophils and NK cells: a flow cytometric study. J Neuroimmunol 2002, 132:34–40. [DOI] [PubMed] [Google Scholar]
- 65.Gaskill PJ, Carvallo L, Eugenin EA, Berman JW: Characterization and function of the human macrophage dopaminergic system: implications for CNS disease and drug abuse. J Neuroinflammation 2012, 9:203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Basu S, Dasgupta PS: Dopamine, a neurotransmitter, influences the immune system. J Neuroimmunol 2000, 102:113–124. [DOI] [PubMed] [Google Scholar]
- 67.••.Labed SA, Wani KA, Jagadeesan S, Hakkim A, Najibi M, Irazoqui JE: Intestinal Epithelial Wnt Signaling Mediates Acetylcholine-Triggered Host Defense against Infection. Immunity 2018, 48:963–978 e963. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study showed that infection-triggered neuronal acetylcholine release drives intestinal host defense gene expression via conserved muscarinic and Wnt signaling pathways.
- 68.Duerr JS, Han HP, Fields SD, Rand JB: Identification of major classes of cholinergic neurons in the nematode Caenorhabditis elegans. J Comp Neurol 2008, 506:398–408. [DOI] [PubMed] [Google Scholar]
- 69.•.Pereira L, Kratsios P, Serrano-Saiz E, Sheftel H, Mayo AE, Hall DH, White JG, LeBoeuf B, Garcia LR, Alon U, et al. : A cellular and regulatory map of the cholinergic nervous system of C. elegans. Elife 2015, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper thoroughly mapped the cholinergic nervous system of C. elegans, and showed that acetylcholine is the most broadly used neurotransmitter in this nematode.
- 70.•.Zhao CM, Hayakawa Y, Kodama Y, Muthupalani S, Westphalen CB, Andersen GT, Flatberg A, Johannessen H, Friedman RA, Renz BW, et al. : Denervation suppresses gastric tumorigenesis. Sci Transl Med 2014, 6:250ra115. [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper demonstrated that the cholinergic nervous system can drive gastric cancer via Wnt signaling. Moreover, the paper implied that inhibition of muscarinic signaling can potentially be used to decelerate tumor progression.
- 71.••.Blake KJ, Baral P, Voisin T, Lubkin A, Pinho-Ribeiro FA, Adams KL, Roberson DP, Ma YC, Otto M, Woolf CJ, et al. : Staphylococcus aureus produces pain through pore-forming toxins and neuronal TRPV1 that is silenced by QX-314. Nat Commun 2018, 9:37. [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper demonstrated that pain-sensing neurons are directly activated by bacterial toxins.
- 72.Chiu IM, Heesters BA, Ghasemlou N, Von Hehn CA, Zhao F, Tran J, Wainger B, Strominger A, Muralidharan S, Horswill AR, et al. : Bacteria activate sensory neurons that modulate pain and inflammation. Nature 2013, 501:52–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Satoh Y, Ishikawa K, Oomori Y, Takeda S, Ono K: Bethanechol and a G-protein activator, NaF/AlCl3, induce secretory response in Paneth cells of mouse intestine. Cell Tissue Res 1992, 269:213–220. [DOI] [PubMed] [Google Scholar]
- 74.Stockinger S, Albers T, Duerr CU, Menard S, Putsep K, Andersson M, Hornef MW: Interleukin-13-mediated paneth cell degranulation and antimicrobial peptide release. J Innate Immun 2014, 6:530–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ayabe T, Satchell DP, Wilson CL, Parks WC, Selsted ME, Ouellette AJ: Secretion of microbicidal alpha-defensins by intestinal Paneth cells in response to bacteria. Nat Immunol 2000, 1:113–118. [DOI] [PubMed] [Google Scholar]
- 76.•.Dhawan S, Hiemstra IH, Verseijden C, Hilbers FW, Te Velde AA, Willemsen LE, Stap J, den Haan JM, de Jonge WJ: Cholinergic receptor activation on epithelia protects against cytokine-induced barrier dysfunction. Acta Physiol (Oxf) 2015, 213:846–859. [DOI] [PubMed] [Google Scholar]; This paper showed that acetylcholine acts on muscarinic receptors in intestinal epithelial cells to maintain barrier function under inflammatory conditions in mice.
- 77.•.Al-Barazie RM, Bashir GH, Qureshi MM, Mohamed YA, Al-Sbiei A, Tariq S, Lammers WJ, Al-Ramadi BK, Fernandez-Cabezudo MJ: Cholinergic Activation Enhances Resistance to Oral Salmonella Infection by Modulating Innate Immune Defense Mechanisms at the Intestinal Barrier. Front Immunol 2018, 9:551. [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper demonstrated that acetycholine positively regulates host defense at intestinal barrier surfaces against Salmonella infection in mice.
- 78.Meisel JD, Panda O, Mahanti P, Schroeder FC, Kim DH: Chemosensation of bacterial secondary metabolites modulates neuroendocrine signaling and behavior of C. elegans. Cell 2014, 159:267–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.••.Singh J, Aballay A: Microbial Colonization Activates an Immune Fight-and Flight Response via Neuroendocrine Signaling. Dev Cell 2019, 49:89–99 e84. [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper showed that microbial colonization and distention of the C. elegans intestine function as danger signals that activate a fight-and-flight response.The paper proposes that inputs from the intestine could help in the recognition of pathogenic microbes and thus, modulate host behavior via neuroendocrine signaling.
- 80.Kumar S, Egan BM, Kocsisova Z, Schneider DL, Murphy JT, Diwan A, Kornfeld K:Lifespan Extension in C. elegans Caused by Bacterial Colonization of the Intestine and Subsequent Activation of an Innate Immune Response. Dev Cell 2019, 49:100–117 e106. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study described connections between C. elegans longevity, bacterial colonization, innate immune response, and behavioral avoidance to pathogens.
- 81.Kaelberer MM, Buchanan KL, Klein ME, Barth BB, Montoya MM, Shen X, Bohorquez DV: A gut-brain neural circuit for nutrient sensory transduction. Science 2018, 361. [DOI] [PMC free article] [PubMed] [Google Scholar]; This elegant paper showed for the first time that enteroendocrine cells of the intestine directly synapse with vagal neurons to transduce gut luminal signals by using glutamate as a neurotransmitter.
- 82.Long-Smith C, O’Riordan KJ, Clarke G, Stanton C, Dinan TG, Cryan JF: Microbiota-Gut-Brain Axis: New Therapeutic Opportunities. Annu Rev Pharmacol Toxicol 2019. [DOI] [PubMed] [Google Scholar]