

Abstract
Objective:
Pulmonary hypertension due to left heart disease (PH-LHD; Group 2), especially in the setting of heart failure with preserved ejection fraction (HFpEF), is the most common cause of PH worldwide, however, at present, there is no proven effective therapy available for its treatment. PH-HFpEF is associated with insulin resistance and features of metabolic syndrome. The stable prostacyclin analog, treprostinil, is an effective and widely used FDA-approved drug for the treatment of pulmonary arterial hypertension. While the effect of treprostinil on metabolic syndrome is unknown, a recent study suggests that the prostacyclin analog beraprost can improve glucose intolerance and insulin sensitivity. We sought to evaluate the effectiveness of treprostinil in the treatment of metabolic syndrome-associated PH-HFpEF.
Approach and Results:
Treprostinil treatment was given to mice with mild metabolic syndrome-associated PH-HFpEF induced by high-fat diet (HFD) and to SU5416/Obese ZSF1 rats, a model created by the treatment of rats with a more profound metabolic syndrome due to double leptin receptor defect (obese ZSF1) with a vascular endothelial growth factor receptor blocker SU5416. In HFD-exposed mice, chronic treatment with treprostinil reduced hyperglycemia and pulmonary hypertension. In SU5416/Obese ZSF1 rats, treprostinil improved hyperglycemia with similar efficacy to that of metformin (a first-line drug for type 2 diabetes); the glucose-lowering effect of treprostinil was further potentiated by the combined treatment with metformin. Early treatment with treprostinil in SU5416/Obese ZSF1 rats lowered pulmonary pressures, and a late treatment with treprostinil together with metformin improved pulmonary artery acceleration time to ejection time ratio (PAAT/ET) and tricuspid annular plane systolic excursion (TAPSE) with AMP-activated protein kinase (AMPK) activation in skeletal muscle and the right ventricle.
Conclusions:
Our data suggest a potential use of treprostinil as an early treatment for mild metabolic syndrome-associated PH-HFpEF and that combined treatment with treprostinil and metformin may improve hyperglycemia and cardiac function in a more severe disease.
Keywords: Pulmonary Hypertension, Metabolic syndrome, HFpEF, Treprostinil, Metformin
Graphical Abstract

Introduction
Pulmonary hypertension due to left heart disease (PH-LHD; Group 2), especially in the setting of heart failure with preserved ejection fraction (HFpEF), is among the most frequent causes of pulmonary hypertension worldwide. Features of metabolic syndrome, including obesity, diabetes, hyperlipidemia, and hypertension, are recognized as risk factors for developing PH-HFpEF.1–3 In fact, two or more of these features are commonly seen in patients with PH-HFpEF.4 Although the exact prevalence varies based on definitions and diagnostic methods, the range in the reported occurrence of pulmonary hypertension (simply defined as having a mean pulmonary artery pressure greater than or equal to 25 mm Hg) is 23%−83% in patients with HFpEF.5–7 A large recent retrospective analysis of patients with right heart catheterization suggested that using more stringent values for high intrinsic pulmonary vascular resistance, such as a transpulmonary gradient > 12 mm Hg, a pulmonary vascular resistance > 3 woods units, or a diastolic pressure gradient > 7 mm Hg, was present in 48.9%, 34.2%, and 13.7% of patients with HFpEF, respectively.7–9 Patients with PH-HFpEF develop more severe symptoms than those with HFpEF and suffer significant exercise intolerance, frequent hospitalization and reduced survival.1 At present, there are no approved specific therapies for PH-HFpEF. Most drugs which target only the pulmonary vasculature tested thus far have failed to demonstrate any significant benefit in the treatment of PH-HFpEF.10–14
Our group has recently developed a two-hit model of PH-HFpEF, which combines endothelial injury using the VEGF receptor blocker SU5416 in rats with multiple features of metabolic syndrome due to double leptin receptor defects (obese ZSF1).15 Clinical features seen in PH-HFpEF patients, including elevated left ventricular (LV) end-diastolic pressure, right ventricular (RV) systolic pressure and right atrial pressure, preserved LV ejection fraction, as well as biventricular hypertrophy, are consistently observed in this model.15 In addition, SU5416/obese ZSF1 rats develop impaired insulin sensitivity characterized by high fasting glucose levels, elevated glycated hemoglobin (HbA1c) and defective glucose tolerance. Using this model, we have reported that drugs which target both cardiopulmonary pathophysiological defects and metabolic syndrome, such as metformin and nitrite, a drug that is metabolized to nitric oxide, reduce pulmonary pressures and vascular remodeling, and improve glucose intolerance, glucose uptake, and insulin resistance.15 Moreover, metformin has also been recently shown to improve metabolic syndrome-associated PH and/or HFpEF in preclinical models induced by high-fat diet (HFD) alone or in combination with supra-coronary aortic banding and the antidepressant olanzapine.16, 17
The prostacyclin analog, treprostinil, is one of the US Food and Drug Administration-approved drugs that consistently leads to hemodynamic improvement of pulmonary arterial hypertension (PAH, Group 1).18 While the effect of treprostinil on metabolic syndrome is unknown, a prostacyclin analog, beraprost sodium, has been recently shown to reverse features of metabolic syndrome in obese Zucker rats.19 In addition, prostacyclin precursor L-carnitine has been shown to prevent the development of HFpEF in rats fed with a high-salt diet.20 Still, the mechanism behind these observations remains elusive. Due to a potential ability of treprostinil to regulate both systemic metabolic defects and pulmonary vascular disease, in the present work we sought to evaluate the effectiveness and mechanism of treprostinil in the treatment of metabolic syndrome-associated PH-HFpEF.
Methods
All experimental procedures were approved by the Institutional Animal Care and Use Committee of the University of Pittsburgh and the Indiana University School of Medicine. All the experiments were blindly performed and analyzed. Detailed methods and Major Resources Table are available in the Supplemental Materials. The data that support the findings of this study are available from the corresponding author upon reasonable request.
Animal studies
The PH-HFpEF models were developed as described before.15, 21, 22 For the rat model, we used eight-week old male obese ZSF1 rats, which present with multiple features of metabolic syndrome, diabetic nephropathy and diastolic dysfunction,23, 24 as well as their lean littermates (Charles River, Wilmington, MA). VEGFR2 antagonist SU5416 (Sigma-Aldrich, St. Louis, MO), which induces lung endothelial injury and/or apoptosis,25 was dissolved in CMC buffer (0.5 % sodium carboxymethyl cellulose, 0.9 % sodium chloride, 0.4 % polysorbate 80, and 0.9 % benzyl alcohol) and given as a single 100 mg/kg subcutaneous injection to obese and/or lean ZSF1 rats at day 0. As female obese ZSF1 rats are resistant to the development of diabetes (hyperglycemia) and renal disease (proteinuria),26 only male rats were used in this study. For the mouse model, eight-week old male C57BL/6J mice (The Jackson Laboratory, Bar Harbor, ME) were fed a regular diet (RD; 15% lipids/kcals) or HFD (60% lipids/kcals; Research Diets, New Brunswick, NJ) for 16 weeks. Treprostinil (40, 300 and 900 ng/kg/min; United Therapeutics Corporation, Silver Spring, MD) was delivered through osmotic minipumps (model 2ML4 for rats; model 2006 for mice; duration of 4 or 6 weeks, respectively; Alzet, Cupertino, CA). After 28 or 42 days, used minipumps were replaced with fresh minipumps. Maximum of 3 implantations were performed in each animal. Metformin (300 mg/kg/day; Spectrum Chemical, New Brunswick, NJ) was given in drinking water. While this dosage of metformin is higher than the oral dose of metformin (~30 mg/kg/day) given to patients with type 2 diabetes, it is considered to be clinically relevant as therapeutic effects of 300 mg/kg/day metformin in rats are similar to the effects of 30 mg/kg/day of metformin in humans.27, 28 All animals were maintained in a normoxic environment.
Hemodynamic and ventricular measurements
In brief, mice and rats were anesthetized with isoflurane (1–2 % v/v). The trachea was cannulated, and rats were ventilated at the rate of 75 to 80 bpm with a tidal volume of 2.5 mL. RV systolic pressure, LV end-diastolic pressure and LV ejection fraction were measured using an admittance pressure-volume catheter. Weights of RV and LV+septum were normalized to tibial length as indexes of ventricular mass.
Transthoracic echocardiography
Transthoracic echocardiography was performed and analyzed using the Vevo 770 System (VisualSonics, ON, Canada). Rats were induced into anesthesia for handling with ~3 % v/v isoflurane and maintained with ~1.5 % v/v. The end-diastolic thickness of left ventricular posterior wall and intraventricular septum, early diastolic mitral annular velocity, mitral peak velocity of late filling, pulmonary artery acceleration time, ejection time, tricuspid annular plane systolic excursion and right ventricular dimension at end-diastole were measured.
Statistical Analysis
Statistical analyses were performed using Prism 8.2.0 software (La Jolla, CA). Statistical comparison between two groups for in vitro studies were performed using the unpaired Student’s t-test after testing for normality (Shapiro-Wilk test; assuming equal variance). Comparison among ≥ 3 groups was performed using one-way analysis of variance (ANOVA) followed by Tukey’s post hoc test if the data followed a normal distribution, otherwise nonparametric Kruskal-Wallis test with Dunn’s post hoc analysis was used. For differences in blood glucose levels during the glucose tolerance test, two-way ANOVA followed by Bonferroni’s post hoc test was performed. A limitation of the study is a small sample size in some experiments, in which only 3 animals were used. Values of P < 0.05 were considered statistically significant.
Results
Chronic treprostinil administration improves hyperglycemia and pulmonary hypertension in HFD-exposed mice
To evaluate the preventative effect of treprostinil on metabolic syndrome-associated PH-HFpEF, treprostinil (40 ng/kg/min, the effective dose for PAH therapy with similar average infusion rate achieved in clinical trials, on a ng/m2 basis) was given through osmotic minipumps for 16 weeks in mice fed with a HFD (Figure 1A), which has been shown to induce PH and/or HFpEF phenotype in mice.16, 21, 22, 29, 30 Consistent with previous studies, HFD-exposed mice had significantly higher body weights, hyperglycemia and glucose intolerance when compared to RD-treated mice (Figure 1B through 1D). Additionally, HFD-exposed mice developed mild PH and/or HFpEF phenotype as evidenced by elevated right ventricular systolic pressure (RVSP), higher left ventricular end diastolic pressure (LVEDP), preserved left ventricular ejection fraction (LVEF) and both LV and RV hypertrophy (Figure 1E through 1I). In contrast, chronic treprostinil treatment significantly lowered HbA1c levels and improved glucose intolerance independent of changes in body weight (Figure 1B through 1D). Furthermore, a tendency for treprostinil to lower pulmonary pressures was observed in HFD-exposed mice (P = 0.058; Figure 1E), although treprostinil caused no significant differences in LVEDP and biventricular hypertrophy (Figure 1F, 1H and 1I). Collectively, these data demonstrate that chronic treprostinil treatment improves glucose metabolism and lowers pulmonary hypertension in a mild mouse model of PH-HFpEF induced by HFD.
Figure 1. Chronic treprostinil treatment reduces hyperglycemia, glucose intolerance and pulmonary hypertension in mice with high-fat diet-induced PH-HFpEF.
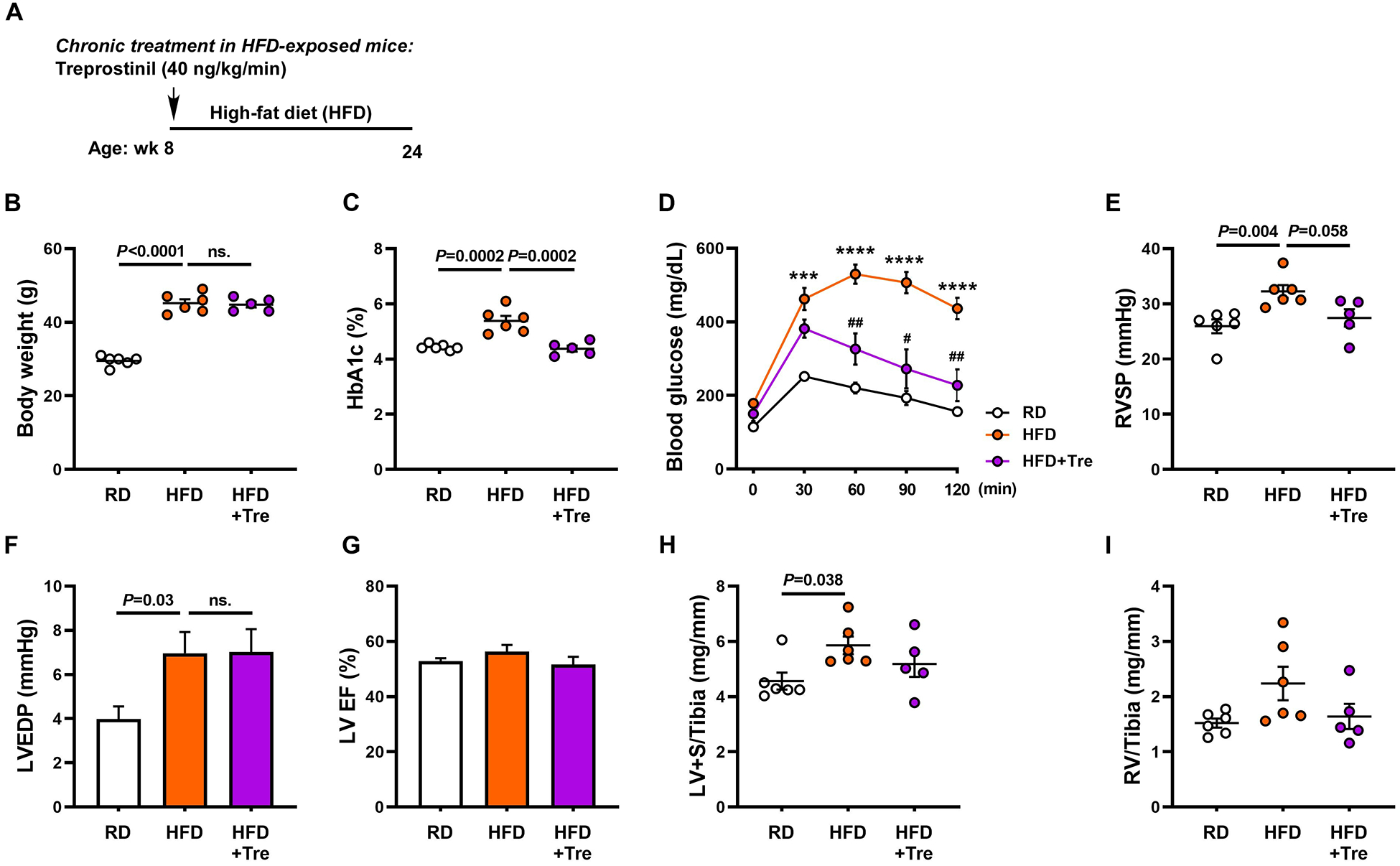
(A) Treprostinil (40 ng/kg/min) was given through osmotic minipumps for 16 weeks in mice fed with a HFD (60% lipids/kcal). Body weights (B), HbA1c levels (C), glucose tolerant abilities (D), right ventricular systolic pressures (RVSP, E), left ventricular end diastolic pressure (LVEDP, F), left ventricular ejection fraction (LVEF, G) were measured. H and I, LV and RV mass normalized to tibial length. Data are mean ± SEM; n = 5–6 mice/group; For D, ***P < 0.001 and ****P < 0.0001 vs. RD; #P < 0.05 and ##P < 0.01 vs. HFD.
Early treatment with treprostinil reduces hyperglycemia, improves lipemia and lowers pulmonary pressures in SU5416/obese ZSF1 rats
We next evaluated the early treatment effect of treprostinil in rats with a more profound metabolic syndrome-associated with PH-HFpEF. Treprostinil (40, 300 and 900 ng/kg/min) was given through osmotic minipumps concurrently with SU5416 exposure to 8-week old obese ZSF1 rats (day 0, Figure 2A and Supplemental Figure IA) for 14 weeks. Note that these rats already had insulin resistance and glucose intolerance at this time point, but have not yet developed HFpEF and/or PH phenotypes.24 The effect of treprostinil on PH-HFpEF was compared to that of metformin (300 mg/kg), the first-line drug for type 2 diabetes, as well as a newly identified early intervention in this PH-HFpEF model.15 While the low dose of treprostinil (40 ng/kg/min) was found to be effective in reducing hyperglycemia and pulmonary pressures in the mild mouse model of metabolic syndrome-associated PH-HFpEF induced by HFD (Fig 1), no significant effect of 40 ng/kg/min of treprostinil on hyperglycemia and pulmonary pressures was observed in Ob-Su rats (Supplemental Figure I). On the other hand, moderate dose of treprostinil (300 ng/kg/min) significantly reduced fasting blood glucose levels with similar efficacy to that of metformin, independent of changes in body weight (Figure 2B and 2C). A dose of 300 ng/kg/min of treprostinil treatment also significantly lowered HbA1c levels and improved glucose intolerance compared to the untreated Ob-Su animals (Figure 2D and 2E). Plasma obtained from Ob-Su rats was noted to be lipemic and milky (lactescent), and this phenomenon was improved by treprostinil treatment (Figure 2F). Additionally, treprostinil (300 ng/kg/min) treatment resulted in lower right ventricular systolic pressure compared to the untreated Ob-Su rats (Figure 2G), although no effect on medial wall thickness was observed (Supplemental Figure II). Similar effects of treprostinil on improving hyperglycemia and lipemia was observed with the higher dose (900 ng/kg/min), but not the lower dose (40 ng/kg/min) of treprostinil in Ob-Su rats (Supplemental Figure IB through IE). Collectively, our results demonstrate that a treatment with moderate dose of treprostinil can lead to a beneficial effect on glucose metabolism with efficacy similar to that of metformin. While an infusion rate of 300 ng/kg/min of treprostinil in Ob-Su rats is 8 times the average rate used in PAH clinical trials, this dose significantly improves hyperglycemia and pulmonary hypertension in Ob-Su rat model of metabolic syndrome-associated PH-HFpEF. As patients with PH-HFpEF usually present with a higher body mass index (42.1 ± 10.1) compared to that of patients with PAH (30.7 ± 5.7),46 it is possible that a higher dose (infusion rate) of treprostinil may be needed for the treatment of PH-HFpEF. Since metformin shows a better efficacy with respect to lowering pulmonary pressures than does treprostinil and has been shown to reverse PH-HFpEF via inhibiting IL6/STAT3-mediated pulmonary vascular remodeling,17 these data suggest that a combination therapy of treprostinil and metformin may be beneficial for the late treatment of metabolic syndrome-associated PH-HFpEF.
Figure 2. Early treatment with treprostinil improves hyperglycemia and lowers pulmonary pressures in SU5416/obese ZSF1 rats.
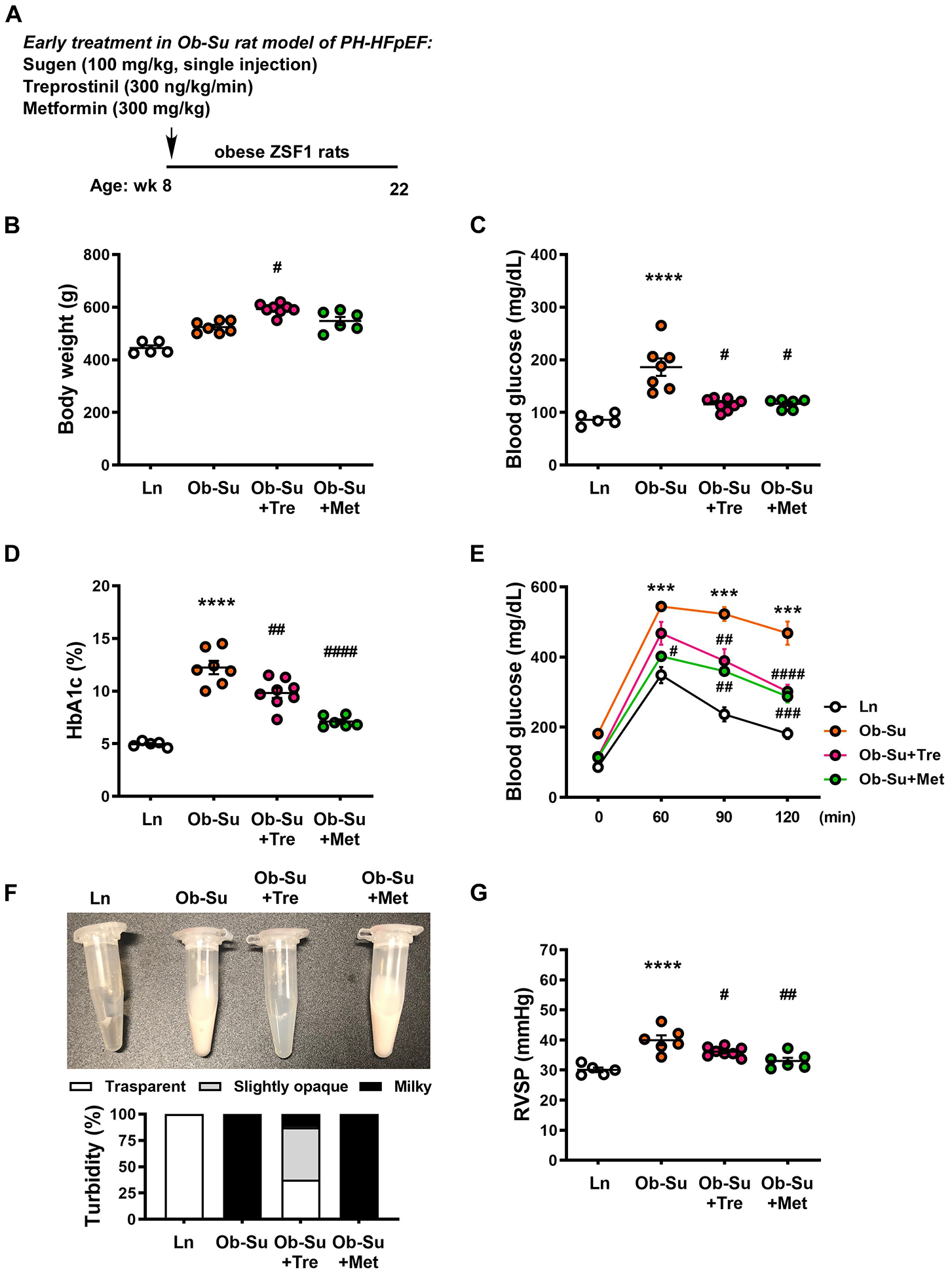
A, Treprostinil (300 ng/kg/min) and metformin (300 mg/kg) were given through osmotic minipumps and drinking water, respectively, for 14 weeks to 8-week old SU5416/obese ZSF1 rats (Ob-Su). Body weights (B), fasting blood glucose levels (C), HbA1c levels (D), glucose tolerant abilities (E) and right ventricular systolic pressures (RVSP, G) were measured. F, Representative images and percentage of milky, slightly opaque and transparent plasma samples in different groups. Data are mean ± SEM; n = 5–8 rats/group; *P < 0.05, ***P < 0.001 and ****P < 0.0001 vs. ln; #P < 0.05, ##P < 0.01, ###P < 0.001 and ####P < 0.0001 vs. Ob-Su.
SU5416/obese ZSF1 rats progressively develop metabolic syndrome-associated PH-HFpEF
In order to assess disease progression and to provide baseline features for the late treatment study in SU5416/obese ZSF1 rats, we monitored echocardiographic, metabolic and hemodynamic changes after 7 and 14 weeks of SU5416 administration in obese ZSF1 rats. Echocardiographic findings in SU5416-exposed obese ZSF1 rats showed a time-independent increase in left ventricular posterior wall (LVPW) and interventricular septum (IVS) thickness and decrease in the ratio of early diastolic mitral annular velocity to mitral peak velocity of late filling (E/A), which reflects grade I diastolic dysfunction, compared to lean rats throughout the entire study (Figure 3A through 3D). In line with progressive decrease in pulmonary artery acceleration time to ejection time ratio (PAAT/ET) and tricuspid annular plane systolic excursion (TAPSE), the latter of which reflects reduced RV systolic function, together with increased RV diastolic dimension (RVDd), SU5416/obese ZSF1 rats also exhibited elevated RV systolic pressure, fasting blood glucose and HbA1c levels in a time-dependent manner (Figure 3E through 3G; Supplemental Figure III). Note that SU5416 administration alone in lean rats had no effect on the echocardiographic, metabolic and hemodynamic features of metabolic syndrome-associated PH-HFpEF compared to untreated lean animals (Fig 3 and Supplemental Figure III). We also note that obese ZSF1 rats do not develop resting PH (~ 27.5 mm Hg vs. 25 mmHg in lean rats; P = NS; data not shown). Together, these results demonstrate that SU5416/obese ZSF1 rats progressively develop metabolic syndrome-associated PH-HFpEF.
Figure 3. SU5416/obese ZSF1 rats progressively develop metabolic syndrome-associated PH-HFpEF.

(A) Representative echocardiographic images assessed after 14 weeks of SU5416 administration to obese ZSF1 rats (SU5416/obese ZSF1, labeled as Ob-Su). Seven and 14 weeks after SU5416 administration in obese ZSF1 rats, the end-diastolic thickness of left ventricular posterior wall (LVPW, B) and interventricular septum (IVS, C), the ratio of early diastolic mitral annular velocity to mitral peak velocity of late filling (E/A, D), the ratio of pulmonary artery acceleration time to ejection time (PAAT/ET, E), tricuspid annular plane systolic excursion (TAPSE, F) and right ventricular dimension at end-diastole (RVDd, G) were measured. Data are mean ± SEM; n = 3–8 rats/group.
Late treatment with treprostinil and metformin normalizes hyperglycemia and improves cardiac function in SU5416/obese ZSF1 rats
To evaluate the late treatment effect of treprostinil, alone or in combination with metformin, treprostinil (300 ng/kg/min) and metformin (300 mg/kg) were given 7 weeks after a single injection of SU5416 to obese ZSF1 rats (Figure 4A). Note that these rats already exhibited mild LV diastolic dysfunction accompanied by more profound hyperglycemia and elevated right ventricular systolic pressure at this time point (Figure 3; Supplemental Figure III). Late treatment with treprostinil significantly lowered HbA1c levels and improved glucose intolerance independent of changes in body weight. This was further potentiated by the combined treatment with metformin in more severely affected SU5416/obese ZSF1 rats (Figure 4B through 4D). Additionally, relatively mild lipemia was observed in Ob-Su rats treated with treprostinil alone or in combination with metformin (Supplemental Figure IV). While the late treatment with treprostinil alone or in combination with metformin failed to reverse the increased right ventricular systolic pressure and had no effect on E/A, biventricular hypertrophy, LVPW, LV EF and cardiac output (Figure 4E and 4F; Supplemental Figure V), our data showed that the combination of treprostinil and metformin significantly increased PAAT/ET and TAPSE relative to the untreated, but more severely affected Ob-Su animals (Figure 4G and 4H). Together, these data demonstrate the benefits of a late treatment with treprostinil specifically related to improving hyperglycemia and glucose intolerance. These data also suggest that combination treatment with treprostinil and metformin may improve cardiac function in metabolic syndrome-associated PH-HFpEF.
Figure 4. Treatment with treprostinil and metformin normalizes hyperglycemia and improves cardiac function in SU5416/obese ZSF1 rats.
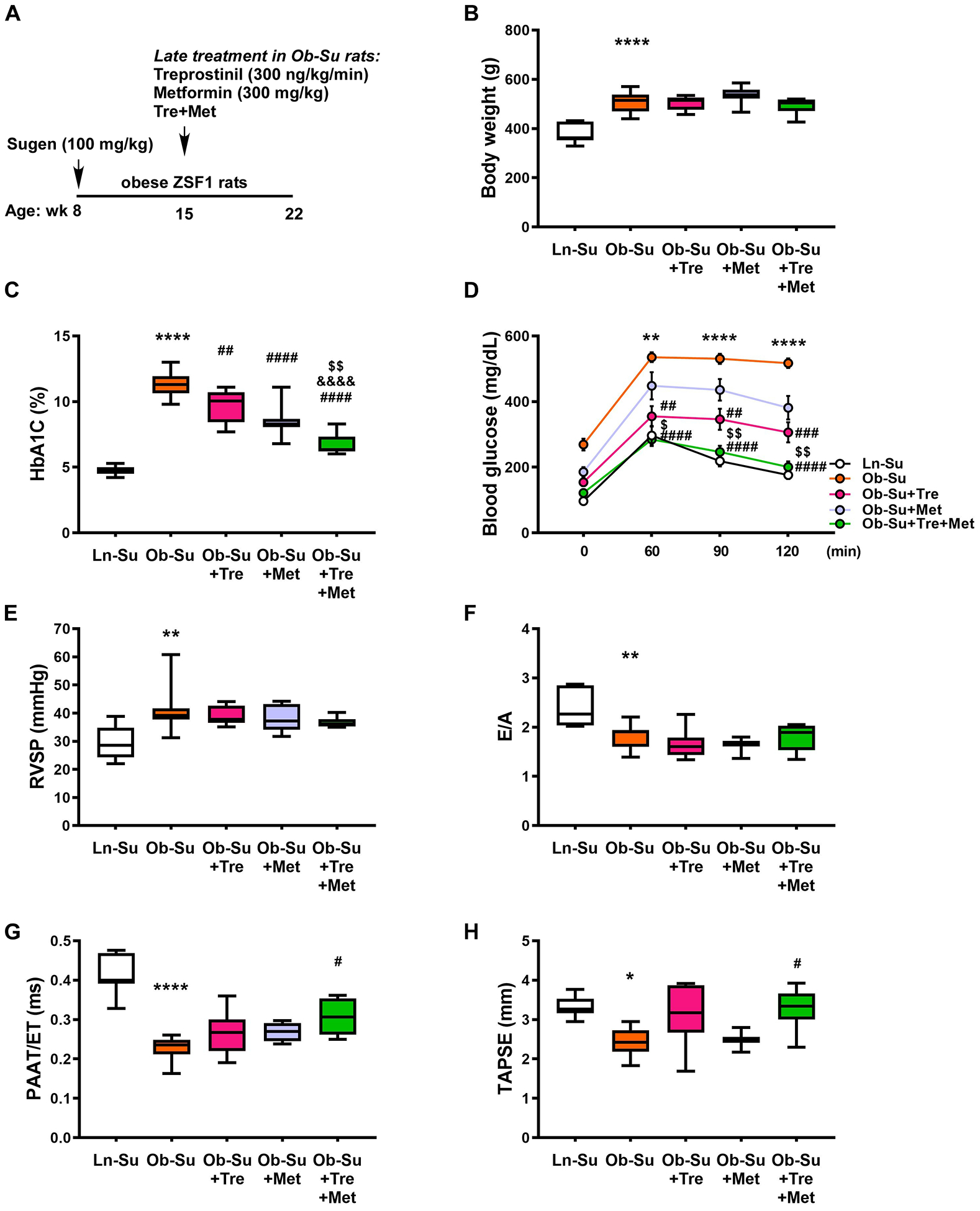
A, Treprostinil (300 ng/kg/min, osmotic minipumps), alone or in combination with metformin (300 mg/kg, drinking water), was given 7 weeks after a single injection of SU5416 (100 mg/kg) to obese ZSF1 rats. Body weights (B), HbA1c levels (C), glucose tolerant abilities (D), right ventricular systolic pressures (RVSP, E) and the ratio of early diastolic mitral annular velocity to mitral peak velocity of late filling (E/A, F) were measured. The ratio of pulmonary artery acceleration time to ejection time (PAAT/ET, G) and tricuspid annular plane systolic excursion (TAPSE, H) were measured. Data are mean ± SEM; n = 4–13 rats/group; *P < 0.05, **P < 0.01 and ****P < 0.0001 vs. Ln-Su; #P < 0.05, ##P < 0.01, ###P < 0.001 and ####P < 0.0001 vs. Ob-Su; &&&&P < 0.0001 vs. Ob-Su+Tre; $P < 0.05 and $ $P < 0.01 vs. Ob-Su+Met.
Treatment with treprostinil and metformin improves metabolic syndrome-associated PH-HFpEF via skeletal muscle activation of AMPK-GLUT4 and systemic improvement of insulin resistance
To determine whether the beneficial effect of treprostinil on improving hyperglycemia and lipemia in Ob-Su rats is related to changes in insulin secretion and lipid contents, plasma insulin, cholesterol and triglyceride levels were measured. Our data showed that late treatment with treprostinil alone in our severe model had no effect on plasma insulin, cholesterol and triglyceride levels in Ob-Su rats compared to the untreated animals (Figure 5A through 5C), however, combination treatment with treprostinil and metformin significantly lowered plasma insulin in Ob-Su rats, suggesting improved insulin resistance (Figure 5A).
Figure 5. Treatment with treprostinil and metformin ameliorates hyperinsulinemia and hypercreatininemia.

Treprostinil (300 ng/kg/min, osmotic minipumps), alone or in combination with metformin (300 mg/kg, drinking water), was given 7 weeks after a single injection of SU5416 (100 mg/kg) to obese ZSF1 rats. Seven weeks after the treatment, plasma samples were collected and circulating levels of insulin (A), cholesterol (B) and triglyceride (C) were measured. Data are mean ± SEM; n = 4–8 rats/group; *P < 0.05 and **P < 0.01 vs. Ln-Su; #P < 0.05 vs. Ob-Su.
To further evaluate the mechanism by which treprostinil improves glucose metabolism, we measured AMPK levels in liver and skeletal muscle, the main organs for glucose metabolism through inhibition of gluconeogenesis and stimulation of glucose uptake, respectively. As shown in Supplememtal Figure VIA, phosphorylation of AMPK was not altered in liver of treprostinil-treated Ob-Su rats compared to the untreated animals, suggesting the glucose-lowering effect of treprostinil is independent of suppression of hepatic glucose production. On the other hand, our data showed a significant increase in AMPK phosphorylation in skeletal muscle of treprostinil-treated Ob-Su rats, accompanied by increased membrane translocation of glucose transporter 4 (GLUT4) compared to the untreated animals (Figure 6A and 6B). Together, our data suggest that treprostinil treatment improves hyperglycemia and glucose intolerance via AMPK-GLUT4-mediated glucose uptake in skeletal muscle. Our data also suggest that the combination of treprostinil and metformin improves PH-HFpEF-associated metabolic syndrome via both local (skeletal muscle) and systemic improvement of glucose uptake and insulin resistance.
Figure 6. Treprostinil and metformin improves metabolic syndrome-associated PH-HFpEF via activation of LKB1/SIRT3-AMPK-GLUT4 signaling pathway.
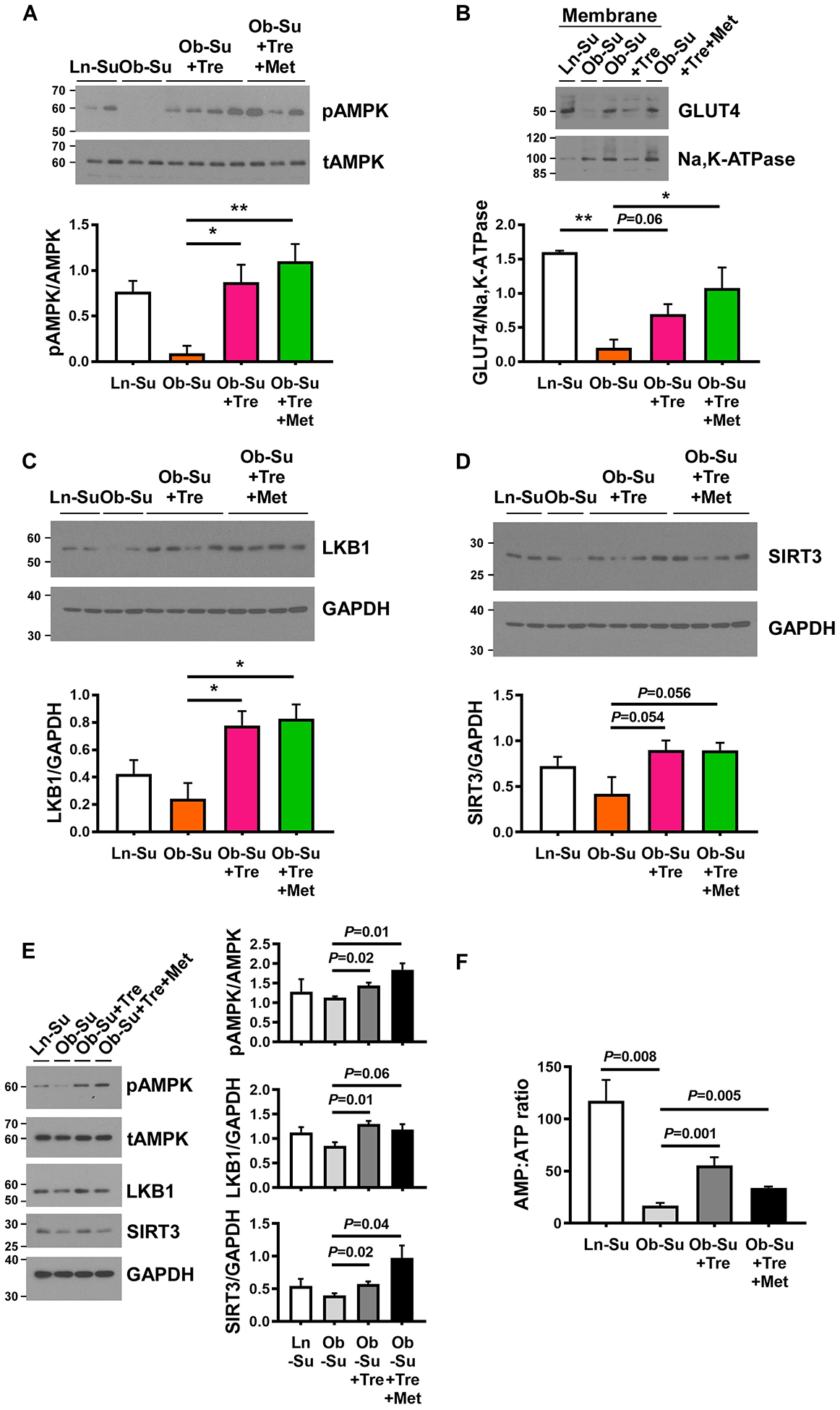
A through D, Skeletal muscle samples were collected from SU5416 (100 mg/kg) treated lean and obese ZSF1 rats (labeled as Ln-Su and Ob-Su, respectively), in the presence or absence of treprostinil (300 ng/kg/min, osmotic minipumps) alone or in combination with metformin (300 mg/kg, drinking water). Effect of treprostinil on AMPK-stimulated glucose uptake was detected by Western blot analyses. B, Representative Western blots for GLUT4 expression in membrane protein extracts from skeletal muscle. Representative Western blots for LKB1 expression (C) and SIRT3 activation (D) levels in skeletal muscle. E and F, C2C12 skeletal muscle myoblasts were treated with plasma obtained from Ln-Su rats or Ob-Su rats, treated with or without treprostinil, alone or in combination with metformin, for 48 hours at the end of the differentiation period. AMPK phosphorylation, LKB1 expression and SIRT3 activation levels (E), as well as the AMP:ATP ratio (F) were measured. Data are mean ± SEM; n = 3–6 (in-vivo) or 3 (in-vitro); *P < 0.05 and **P < 0.01.
Treprostinil activates skeletal muscle AMPK-GLUT4 signaling pathway via upstream activators, LKB1 and SIRT3
Given that liver kinase B1 (LKB1), transforming growth factor-β-activated kinase-1 (TAK1), calcium/calmodulin-dependent protein kinase kinase 2 (CaMKK2) and sirtuin-3 (SIRT3) are known upstream activators of AMPK,31–34 we next investigated their expression/activation levels in skeletal muscle of treprostinil-treated Ob-Su rats. As shown in Figure 6C, 6D and Supplememtal Figure VIB and VIC, expression levels of CaMKK2 and TAK1 are not altered in the skeletal muscle of treprostinil-treated Ob-Su rats compared to the untreated animals, while the increased LKB1 expression and elevated SIRT3 activation levels (the antibody we used specifically recognizes the short active form of SIRT3 at ≈ 28 kDa, which contains the catalytic domain and regulates deacetylation of downstream substrates) was observed. The same AMPK/LKB1/SIRT3-activating effect was observed when C2C12 skeletal muscle myoblasts were treated with plasma obtained from treprostinil-treated Ob-Su rats (Figure 6E), whereas direct stimulation of treprostinil at relevant concentrations did not result in significant activation of AMPK, LKB1 and SIRT3 (Supplemental Figure VID). A higher ratio of intercellular AMP to ATP levels was also observed in C2C12 skeletal muscle myoblasts stimulated with plasma obtained from treprostinil-treated Ob-Su rats (Figure 6F). Together, these data suggest that treprostinil activates AMPK through upstream regulation of LKB1 and SIRT3, along with a substantial increase in the AMP:ATP ratio. These data also suggest that the metabolite of treprostinil may be required for the activation of LKB1/SIRT3-AMPK signaling pathway.
To probe for further evidence, we examined the effect of siRNA-mediated knockdown of LKB1 and SIRT3 on treprostinil-mediated AMPK activation. C2C12 cells were transiently transfected with siRNA targeting LKB1, SIRT3 or scrambled control for 24 hours before stimulation with plasma obtained from Ob-Su rats, in the presence or absence of treprostinil. As shown in Figure 7, the ability of treprostinil-contained plasma to activate AMPK was reduced in LKB1 and SIRT3 knockdown cells, further suggesting that LKB1 and SIRT3 are required for treprostinil-mediated AMPK activation.
Figure 7. LKB1 and SIRT3 are required for treprostinil-mediated AMPK activation in C2C12 skeletal muscle cells.

C2C12 skeletal muscle cells were transiently transfected with siRNA targeting LKB1, SIRT3 or scrambled control 24 hours before stimulation with plasma obtained from treprostinil-treated Ob-Su rats for an additional 48 hours at the end of the differentiation period. Effect of LKB1 or SIRT3 knockdown (KD) on treprostinil-mediated AMPK activation was measured by Western blots. Dot plots show the fold change of AMPK phosphorylation relative to stimulation with plasma obtained from Ob-Su rats. Data are mean ± SEM; n = 3; *P < 0.05 and **P < 0.01 compared to correlated scrambled control groups.
Treatment with treprostinil and metformin activates AMPK in the RV
To understand how late treatment with treprostinil and metformin might improve PAAT/ET and TAPSE (Figure 4), we investigated pulmonary vascular changes, along with AMPK activation in the RV, since AMPK is thought to be a central player in regulating cardiac function, cardiomyocyte contractility and lipid accumulation in the RV.16, 35 While the late treatment with treprostinil alone or in combination with metformin failed to reverse the elevated medial wall thickness in the pulmonary arteries (Figure 8A) and had no effect on PDGF-BB-induced proliferation of pulmonary arterial smooth muscle cells (PASMCs; Supplemental Figure VIIB), an increase in AMPK activation levels was observed in the RV of Ob-Su rats treated with treprostinil and metformin (Figure 8B). AMPK has been shown to modulate cardiac contractile function through upstream LKB1 and downstream target Ser150 of cardiac Troponin I.36–38 However, our data showed that increased AMPK activation by treprostinil and metformin was not accompanied by changes in LKB1 nor was associated with phosphorylation levels of cardiac Troponin I (Supplemental Figure VIIC and VIID). Additionally, SIRT3 activation levels were not altered by the combination treatment with treprostinil and metformin in the RV (Supplemental Figure VIIC). Together, these data suggest that LKB1/SIRT3-indipendent activation of AMPK in the RV by treprostinil and metformin may be one of the mechanisms associated with improved cardiac function in metabolic syndrome-associated PH-HFpEF.
Figure 8. Treatment with treprostinil and metformin activates AMPK in the RV.

Treprostinil (300 ng/kg/min, osmotic minipumps), alone or in combination with metformin (300 mg/kg, drinking water), was given 7 weeks after a single injection of SU5416 (100 mg/kg) to obese ZSF1 rats. A, Seven weeks after the treatment, lung tissue sections were subjected to pulmonary artery (PA) medial wall thickness (MT) analyses. Scale bar = 50 μm. 40X. B, Effects of treprostinil and/or metformin on AMPK activation in the RV were detected by Western blot analyses. Data are mean ± SEM; n = 3–5 rats/group; *P < 0.05 vs. Ln-Su and #P < 0.05 vs. Ob-Su.
Discussion
Using high-fat feeding, which induces mild metabolic syndrome-associated PH-HFpEF, we demonstrate that chronic treprostinil treatment reduces hyperglycemia, glucose intolerance and pulmonary hypertension. We also demonstrate that treprostinil has a beneficial effect on glucose homeostasis, with comparable efficacy to metformin alone in rats with more profound metabolic syndrome-associated PH-HFpEF. Additionally, our data show that early chronic supplementation of treprostinil lowers pulmonary pressures and that a late treatment with both treprostinil and metformin normalizes hyperglycemia and improves cardiac function by a mechanism involving, at least in part, AMPK activation in skeletal muscle and the RV, along with improved insulin resistance. These findings are important as both cardiopulmonary and metabolic defects in metabolic syndrome-associated PH-HFpEF were highly modifiable in our study by treprostinil, a US Food and Drug Administration-approved drug for PAH, and metformin, which is the most commonly prescribed drug for the treatment of type 2 diabetes, demonstrating the potential for repurposing these drugs for the early treatment of PH-HFpEF.
To date, no approved specific medication or consensus therapeutic strategy for PH-HFpEF is available. Targeting left-sided filling pressure with PA pressure monitoring device to adjust doses of diuretic therapy has been shown to reduce HF hospitalization, but no changes with respect to PA pressure was observed in HFpEF patients.39 Additionally, the use of the current approved treatments for PAH, such as sildenafil, riociguat, bosentan and macitentan, which act on nitric oxide or endothelin-1 pathways, is controversial and has been shown to be ineffective or even harmful in patients with PH-HFpEF.10–14, 40 While the search for effective therapies for PH-HFpEF remains challenging, a significant breakthrough in cardiology represented by a recent finding that some antidiabetic drugs, such as metformin and sodium glucose co-transporter 2 inhibitors, are associated with reduction in HF hospitalizations and lower mortality.41–43 These exciting findings have made metabolic syndrome-targeting therapies a potential strategy for treating heart failure, with several ongoing clinical trials evaluating the effects of SGLT2 inhibitors in HFpEF patients taking place at present (NCT03057951, NCT03619213 and NCT03030235).
In addition to HFpEF, several new discoveries over the past decade have revealed a ~35% prevalence of metabolic syndrome in PAH patients4. It has also been shown that the prevalence of insulin resistance is higher among female PAH patients and is associated with reduced survival compared to the insulin sensitive counterparts.44 Moreover, coexistence of diabetes is associated with reduced 10-year survival in PAH patients compared to those without diabetes.45 Targeting metabolic syndrome with rosiglitazone, an antidiabetic drug, as well as a peroxisome proliferator-activated receptor-gamma activator, has been shown to increase plasma adiponectin, reduce hyperglycemia and reverse increased RVSP and RV hypertrophy in HFD-treated apolipoprotein E mice.29 Additionally, preventative treatment with metformin in HFD-treated C57/B16 mice has been shown to improve insulin resistance, reduce lipid accumulation and decrease right ventricular systolic pressure.16 These data further support the idea that targeting metabolic syndrome may be a viable strategy for the treatment of metabolic syndrome-associated PAH and have led to initiation of a clinical trial designed to evaluate the effect of metformin in PAH patients (NCT03617458).
In contrast to patients with HFpEF or PAH, whose prevalence of metabolic syndrome is approximately 35%, clinical observations have shown that patients with PH-HFpEF have higher prevalence of hypertension, obesity, diabetes and hyperlipidemia. According to Robbins et al, more than 90% of PH-HFpEF patients have been shown to have two or more features of metabolic syndrome.4 Consistent with these findings, more than 90% comorbidities of hypertension, diabetes and hyperlipidemia with a high body mass index value of 42 were observed in a patient cohort of PH-HFpEF recruited by Simon et al for evaluating the effect of nitrite,46 a drug which has been shown to improve metabolic syndrome and cardiopulmonary hemodynamics in the Ob-Su rat model of PH-HFpEF with a similar effect to metformin.15 Most recently, Ranchoux et al developed a new pre-clinical model of metabolic syndrome-associated PH-HFpEF using SAB with a combination treatment of olanzapine, an antidepressant known to induce the adverse effect of metabolic syndrome, and HFD.17 This model confirms the idea that metabolic syndrome exacerbates Group 2 PH. Using this model, metformin treatment significantly reversed plasma leptin, visceral fat and elevated pulmonary pressure, further supporting the therapeutic potential of metabolic syndrome-targeting strategy for the treatment of PH-HFpEF.17 While the effects of the first-line and second-line drugs for metabolic syndrome, metformin and SGLT2 inhibitor empagliflozin, respectively, are currently under investigation in clinical trials which include PH-HFpEF or HF patients with diabetes (NCT03629340 and NCT03030222), our new findings provide additional evidence that improved metabolic syndrome and insulin resistance may be a strategy for the early treatment of metabolic syndrome-associated PH-HFpEF. Our observations may open a new avenue for an early combination therapy of treprostinil and metformin in the management of PH-HFpEF in future studies.
Several studies have suggested that microvascular endothelial dysfunction caused by systemic inflammation and oxidative stress is the likely driving force that links insulin resistance to the development of metabolic syndrome-associated PH-HFpEF.47–49 This may be related to nitric oxide deficiency and the subsequent impairment of cyclin guanosine monophosphate/protein kinase G signaling in cardiomyocytes, which results in LV stiffness and abnormal relaxation, left atrial impairment and ultimately an increase in pulmonary artery pressures.50, 51 As endothelial dysfunction contributes to insulin resistance, impaired insulin action has been shown to reverberate a negative feedback loop to worsen endothelial dysfunction and further disturb glucose and lipid metabolism, which in turn trigger inflammation, induce oxidative stress and exacerbate endothelial dysfunction and insulin resistance. In fact, lipid accumulation in skeletal muscle has been shown to create a pro-inflammatory state and promote insulin resistance in mice with bone morphogenetic protein receptor 2 mutation, the most commonly identified genetic mutation in PAH, as well as in patients with HFpEF.52–54 Lipid accumulation in the RV has also been linked to the development of PH.16 Besides, hyperglycemia-induced overproduction of mitochondrial reactive oxygen species, elevated oxidative stress and mitochondrial dysfunction have been suggested as major factors in the pathogenesis of diabetic complications.55 Impaired mitochondrial function and elevated oxidative stress by persistent hyperglycemia in cardiomyocytes has been associated with LV diastolic dysfunction and disease worsening in HFpEF.56 While the impact of hyperglycemia on the stabilization of hypoxia-inducible factor 1α is still unclear, hyperglycemia-induced cellular hypoxia has been shown to promote hyperglycemic complications via induction of endothelin-1 and fibronection in endothelial cells.57 Hyperglycemia also regulates PASMCs proliferation and migration through increased expression of Smad ubiquitin ligase (Smurf-1) to down-regulate BMP signaling in PAH.58 Moreover, skeletal muscle glucose intolerance, mitochondrial dysfunction and metabolism defects have also been closely associated with exercise intolerance and worsened functional capacity in patients with either PAH or HFpEF.59–61
We are intrigued by the chronically sustainable effect of treprostinil in improving glucose homeostasis, including hyperglycemia and glucose intolerance, with a comparable efficacy to that of metformin alone in rats with more profound diabetes associated with dyslipidemia and hypertension. While a prostacyclin derivative, beraprost sodium, has been recently shown to suppress the development of diabetes by improving glucose intolerance and insulin resistance in obese Zucker fatty rats, the mechanism behind this observation has not been established.19 Here, we show that the glucose-lowering effect of treprostinil seems to be mediated predominantly by insulin-independent skeletal muscle glucose uptake through increased muscle LKB1/SIR3-AMPK-GLUT4 activation. Consistent with recent findings that activation of skeletal muscle AMPK by pan-AMPK activators is capable of promoting insulin-independent glucose lowering and uptake in diabetic mice and monkeys without affecting hepatic glucose production,62, 63 our data suggest that skeletal muscle AMPK activation by treprostinil may be crucial in the regulation of antihyperglycemic action. Yet, whether this skeletal muscle AMPK activation-mediated glucose lowering effect can be extended to all prostanoids and/or to various routes of drug administration (e.g. inhalation and oral), needs to be further investigated. Based on the US Food and Drug Administration report, among 33,568 people who have adverse effects when taking treprostinil, 0.1% have hypoglycemia, particularly to those who are female, age 60 or older, and also take bosentan. While the mechanism related to this observation is unknown, one of the metabolites of treprostinil, the glucuronoconjugate of treprostinil, has been shown to increase glucose consumption during metabolism.64, 65 In the present study, our results show that plasma from treprosinil-treated animals, rather than direct addition of treprostinil, induces LKB1/SIRT3-AMPK activation in vitro. Whether or not the glucose lowering effect of treprostinil is dependent on the glucuronidated metabolites needs to be further evaluated.
Our data also unexpectedly reveal that treprostinil synergizes with metformin in improving abnormal glucose regulation to normal glucose tolerance via skeletal muscle LKB1/SIR3-AMPK-mediated glucose uptake and improved insulin resistance. These phenomena are associated with improved TAPSE and PAAT/ET in more severely affected SU5416/obese ZSF1 rats. Similar to SIRT3, the mitochondrial deacetylase that has been shown to modulate diabetes through the maintenance of skeletal muscle insulin action, glucose disposal and mitochondrial function,66 the tumor suppressor LKB1 is involved in the regulation of insulin sensitivity and glucose homeostasis.67 LKB1 has also been shown to regulate lipid oxidation independently of AMPK during exercise.68 Given that treprostinil improves lipemia without affecting triglycerides and total cholesterol, future experiments are needed to determine whether LKB1-mediated lipid oxidation is involved in this context. Along with improved hyperglycemia and pulmonary pressures by nitrite and metformin alone via skeletal muscle SIRT3-AMPK activation,15 our data suggest a common mechanism contributed, at least in part, by skeletal muscle LKB1/SIRT3-AMPK signaling to the early treatment of metabolic syndrome-associated PH-HFpEF. However, the relative contributions to improved cardiac function and/or pulmonary pressure observed with these drugs by skeletal muscle-mediated improvement of glucose metabolism and lung signaling remain uncertain.
In addition to skeletal muscle AMPK activation, our data show a connection between RV AMPK activation and improved RV function in Ob-Su rats treated with treprostinil and metformin. AMPK activation has been shown to inhibit cardiomyocyte hypertrophy via suppression of protein synthesis and gene transcription,69, 70 however, RV mass/hypertrophy was not affected by the combination treatment with treprostinil and metformin in this study. AMPK has also been shown to play a role in regulating cardiac function and cardiomyocyte contractility through phosphorylating cardiac Troponin I at Ser150.35–37 We found that treprostinil and metformin-mediated RV AMPK activation is not associated with phosphorylation levels of cardiac Troponin I, suggesting the beneficial effect of treprostinil and metformin on the RV is independent of improved contractile function. Our data indicate that late treatment with treprostinil alone does not alter RV hypertrophy, TAPSE and phosphorylation levels of AMPK and Troponin I in the RV of Ob-Su rats. This observation is consistent with previous findings that treprostinil treatment has no significant effect on cardiomyocyte contractility, RV hypertrophy and RV function in rat cardiomyocytes or in pulmonary trunk banding model of pressure overload-induced right ventricular hypertrophy and failure.71, 72 Recently, metformin has been shown to prevent RV dysfunction and myocardial steatosis via improved insulin resistance and reduced RV lipid accumulation in HFD-treated C57/B16 mice.16 Infusion of epoprostenol has also been shown to reduce RV size, decrease PA resistance and improve RV function via its vasodilating effect in patients with PAH and in dogs with load-induced acute RV failure.73, 74 Further studies are needed to determine whether the reduction of RV lipid content together with pulmonary and/or systemic vasodilation are the mechanisms through which treprostinil and metformin may exert their beneficial effects.
Although not conclusive, our data suggest a potential role of treprostinil as an early treatment for mild metabolic syndrome-associated PH-HFpEF. Our data also show that treprostinil has similar effects to metformin, improving insulin-independent glucose lowering and disposal by activating LKB1/SIRT3-AMPK-GLUT4 in skeletal muscle. In addition, we show that early chronic supplementation of treprostinil lowers pulmonary pressures and that a late combination therapy of treprostinil and metformin normalizes hyperglycemia and improves cardiac function via skeletal muscle and RV AMPK activation in rats with more severe PH-HFpEF. As a whole, our study provides insights into the development of skeletal muscle LKB1/SIRT3-AMPK- and/or RV AMPK-targeted therapies and presents an early treatment strategy for a combination therapy of treprostinil and metformin for PH-HFpEF in the future.
Supplementary Material
Highlights.
Chronic treprostinil treatment lowers hyperglycemia, glucose intolerance and pulmonary pressures.
Treprostinil has a beneficial effect on lowering glucose via activation of LKB1/SIRT3-AMPK-GLUT4 pathway in skeletal muscle with efficacy comparable to metformin.
Late treatment with treprostinil and metformin normalizes hyperglycemia and improves cardiac function by AMPK activation in skeletal muscle and the RV.
This study provides insights into the development of skeletal muscle LKB1/SIRT3-AMPK- and/or RV AMPK-targeted therapies for metabolic syndrome-associated PH-HFpEF.
Acknowledgments
The authors thank Dr. Sergei Snovida for helpful comments on the manuscript. The authors also thank Qinzi Xu for technical assistance. Dr. Gladwin receives research support from NIH grants 5R01HL098032-11, 2R01HL125886-05, and 5P01HL103455-09, 5T32HL110849-08, the Burroughs Wellcome Fund, the Institute for Transfusion Medicine and the Hemophilia Center of Western Pennsylvania.
Sources of Funding
The research was supported by a United Therapeutics Corp grant to Y-C.L. and M.T.G. The research was also supported by NIH/NHLBI R01HL142638 to Y-C. L., NIH/NHLBI P01HL103455 to M.T.G., China Council Scholarship CSC201706370266 to L.W. and American Cancer Society 132013-RSG-18-010-01-CCG to A.B.
Abbreviations
- AMPK
AMP-activated protein kinase
- CaMKK2
calcium/calmodulin-dependent protein kinase kinase 2
- GLUT4
glucose transporter 4
- HbA1c
hemoglobin A1c
- HFD
high-fat diet
- HFpEF
heart failure with preserved ejection fraction
- LKB1
liver kinase B1
- LV
left ventricle and or left ventricular
- PAAT/ET
pulmonary artery acceleration time to ejection time ratio
- PAH
pulmonary arterial hypertension
- PH
pulmonary hypertension
- PH-HFpEF
pulmonary hypertension associated with heart failure with preserved ejection fraction
- RV
right ventricle and/or right ventricular
- SIRT3
sirtuin-3
- TAK1
transforming growth factor-β-activated kinase-1
- TAPSE
tricuspid annular plane systolic excursion
Footnotes
Disclosures
Dr. Gladwin is a co-inventor of pending patent applications and planned patents directed to the use of recombinant neuroglobin and heme-based molecules as antidotes for CO poisoning, which have been licensed by Globin Solutions, Inc. Globin Solutions, Inc. also has an option to a potential therapeutic for CO poisoning from VCU, hydroxycobalamin. Dr. Gladwin is a shareholder, advisor, and director in Globin Solutions, Inc. Dr. Gladwin is also co-inventor on patents directed to the use of nitrite salts in cardiovascular diseases, which were previously licensed to United Therapeutics and Hope Pharmaceuticals, and is now licensed to Globin Solutions. Additionally, Dr. Gladwin is a co-investigator in a research collaboration with Bayer Pharmaceuticals to evaluate riociguat as a treatment for patients with sickle cell disease, and is a consultant for Bayer and Complexa pharmaceuticals. The other authors report no conflicts.
References
- 1.Guazzi M Pulmonary hypertension in heart failure preserved ejection fraction: prevalence, pathophysiology, and clinical perspectives. Circulation Heart failure. 2014;7:367–77. [DOI] [PubMed] [Google Scholar]
- 2.Thenappan T, Shah SJ, Gomberg-Maitland M, Collander B, Vallakati A, Shroff P and Rich S. Clinical characteristics of pulmonary hypertension in patients with heart failure and preserved ejection fraction. Circulation Heart failure. 2011;4:257–65. [DOI] [PubMed] [Google Scholar]
- 3.Shapiro BP, McGoon MD and Redfield MM. Unexplained pulmonary hypertension in elderly patients. Chest. 2007;131:94–100. [DOI] [PubMed] [Google Scholar]
- 4.Robbins IM, Newman JH, Johnson RF, Hemnes AR, Fremont RD, Piana RN, Zhao DX and Byrne DW. Association of the metabolic syndrome with pulmonary venous hypertension. Chest. 2009;136:31–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vachiéry J-L, Adir Y, Barberà JA, Champion H, Coghlan JG, Cottin V, De Marco T, Galiè N, Ghio S, Gibbs JSR, Martinez F, Semigran M, Simonneau G, Wells A and Seeger W. Pulmonary Hypertension Due to Left Heart Diseases. Journal of the American College of Cardiology. 2013;62:D100. [DOI] [PubMed] [Google Scholar]
- 6.Guha A, Amione-Guerra J and Park MH. Epidemiology of Pulmonary Hypertension in Left Heart Disease. Progress in cardiovascular diseases. 2016;59:3–10. [DOI] [PubMed] [Google Scholar]
- 7.Lai YC, Wang L and Gladwin MT. Insights into the pulmonary vascular complications of heart failure with preserved ejection fraction. J Physiol. 2019;597:1143–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vanderpool RR, Saul M, Nouraie M, Gladwin MT and Simon MA. Association Between Hemodynamic Markers of Pulmonary Hypertension and Outcomes in Heart Failure With Preserved Ejection Fraction. JAMA Cardiol. 2018;3:298–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Levine AR, Simon MA and Gladwin MT. Pulmonary vascular disease in the setting of heart failure with preserved ejection fraction. Trends Cardiovasc Med. 2019;29:207–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hoendermis ES, Liu LC, Hummel YM, van der Meer P, de Boer RA, Berger RM, van Veldhuisen DJ and Voors AA. Effects of sildenafil on invasive haemodynamics and exercise capacity in heart failure patients with preserved ejection fraction and pulmonary hypertension: a randomized controlled trial. European heart journal. 2015;36:2565–73. [DOI] [PubMed] [Google Scholar]
- 11.Redfield MM, Chen HH, Borlaug BA, Semigran MJ, Lee KL, Lewis G, LeWinter MM, Rouleau JL, Bull DA, Mann DL, Deswal A, Stevenson LW, Givertz MM, Ofili EO, O’Connor CM, Felker GM, Goldsmith SR, Bart BA, McNulty SE, Ibarra JC, Lin G, Oh JK, Patel MR, Kim RJ, Tracy RP, Velazquez EJ, Anstrom KJ, Hernandez AF, Mascette AM, Braunwald E and Trial R. Effect of phosphodiesterase-5 inhibition on exercise capacity and clinical status in heart failure with preserved ejection fraction: a randomized clinical trial. Jama. 2013;309:1268–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vachiery JL, Delcroix M, Al-Hiti H, Efficace M, Hutyra M, Lack G, Papadakis K and Rubin LJ. Macitentan in pulmonary hypertension due to left ventricular dysfunction. The European respiratory journal. 2018;51. [DOI] [PubMed] [Google Scholar]
- 13.Bonderman D, Pretsch I, Steringer-Mascherbauer R, Jansa P, Rosenkranz S, Tufaro C, Bojic A, Lam CSP, Frey R, Ochan Kilama M, Unger S, Roessig L and Lang IM. Acute hemodynamic effects of riociguat in patients with pulmonary hypertension associated with diastolic heart failure (DILATE-1): a randomized, double-blind, placebo-controlled, single-dose study. Chest. 2014;146:1274–1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Koller B, Steringer-Mascherbauer R, Ebner CH, Weber T, Ammer M, Eichinger J, Pretsch I, Herold M, Schwaiger J, Ulmer H and Grander W. Pilot Study of Endothelin Receptor Blockade in Heart Failure with Diastolic Dysfunction and Pulmonary Hypertension (BADDHY-Trial). Heart, lung & circulation. 2017;26:433–441. [DOI] [PubMed] [Google Scholar]
- 15.Lai YC, Tabima DM, Dube JJ, Hughan KS, Vanderpool RR, Goncharov DA, St Croix CM, Garcia-Ocana A, Goncharova EA, Tofovic SP, Mora AL and Gladwin MT. SIRT3-AMP-Activated Protein Kinase Activation by Nitrite and Metformin Improves Hyperglycemia and Normalizes Pulmonary Hypertension Associated With Heart Failure With Preserved Ejection Fraction. Circulation. 2016;133:717–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brittain EL, Talati M, Fortune N, Agrawal V, Meoli DF, West J and Hemnes AR. Adverse physiologic effects of Western diet on right ventricular structure and function: role of lipid accumulation and metabolic therapy. Pulmonary circulation. 2019;9:2045894018817741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ranchoux B, Nadeau V, Bourgeois A, Provencher S, Tremblay E, Omura J, Cote N, Abu-Alhayja’a R, Dumais V, Nachbar RT, Tastet L, Dahou A, Breuils-Bonnet S, Marette A, Pibarot P, Dupuis J, Boucherat O, Paulin R, Archer SL, Bonnet S and Potus F. Metabolic Syndrome Exacerbates Pulmonary Hypertension due to Left Heart Disease. Circulation research. 2019. [DOI] [PubMed] [Google Scholar]
- 18.Simonneau G, Barst RJ, Galie N, Naeije R, Rich S, Bourge RC, Keogh A, Oudiz R, Frost A, Blackburn SD, Crow JW and Rubin LJ. Continuous subcutaneous infusion of treprostinil, a prostacyclin analogue, in patients with pulmonary arterial hypertension: a double-blind, randomized, placebo-controlled trial. American journal of respiratory and critical care medicine. 2002;165:800–4. [DOI] [PubMed] [Google Scholar]
- 19.Sato N, Kaneko M, Tamura M and Kurumatani H. The prostacyclin analog beraprost sodium ameliorates characteristics of metabolic syndrome in obese Zucker (fatty) rats. Diabetes. 2010;59:1092–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Omori Y, Ohtani T, Sakata Y, Mano T, Takeda Y, Tamaki S, Tsukamoto Y, Kamimura D, Aizawa Y, Miwa T, Komuro I, Soga T and Yamamoto K. L-Carnitine prevents the development of ventricular fibrosis and heart failure with preserved ejection fraction in hypertensive heart disease. Journal of hypertension. 2012;30:1834–44. [DOI] [PubMed] [Google Scholar]
- 21.Meng Q, Lai YC, Kelly NJ, Bueno M, Baust JJ, Bachman TN, Goncharov D, Vanderpool RR, Radder JE, Hu J, Goncharova E, Morris AM, Mora AL, Shapiro SD and Gladwin MT. Development of a Mouse Model of Metabolic Syndrome, Pulmonary Hypertension, and Heart Failure with Preserved Ejection Fraction. American journal of respiratory cell and molecular biology. 2017;56:497–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kelly NJ, Radder JE, Baust JJ, Burton CL, Lai YC, Potoka KC, Agostini BA, Wood JP, Bachman TN, Vanderpool RR, Dandachi N, Leme AS, Gregory AD, Morris A, Mora AL, Gladwin MT and Shapiro SD. Mouse Genome-Wide Association Study of Preclinical Group II Pulmonary Hypertension Identifies Epidermal Growth Factor Receptor. American journal of respiratory cell and molecular biology. 2017;56:488–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tofovic SP, Kusaka H, Kost CK Jr. and Bastacky S. Renal function and structure in diabetic, hypertensive, obese ZDFxSHHF-hybrid rats. Renal failure. 2000;22:387–406. [DOI] [PubMed] [Google Scholar]
- 24.Hamdani N, Franssen C, Lourenco A, Falcao-Pires I, Fontoura D, Leite S, Plettig L, Lopez B, Ottenheijm CA, Becher PM, Gonzalez A, Tschope C, Diez J, Linke WA, Leite-Moreira AF and Paulus WJ. Myocardial titin hypophosphorylation importantly contributes to heart failure with preserved ejection fraction in a rat metabolic risk model. Circulation Heart failure. 2013;6:1239–49. [DOI] [PubMed] [Google Scholar]
- 25.Taraseviciene-Stewart L, Kasahara Y, Alger L, Hirth P, Mc Mahon G, Waltenberger J, Voelkel NF and Tuder RM. Inhibition of the VEGF receptor 2 combined with chronic hypoxia causes cell death-dependent pulmonary endothelial cell proliferation and severe pulmonary hypertension. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2001;15:427–38. [DOI] [PubMed] [Google Scholar]
- 26.Su Z, Widomski D, Ma J, Namovic M, Nikkel A, Leys L, Olson L, Salte K, Donnelly-Roberts D, Esbenshade T and McGaraughty S. Longitudinal Changes in Measured Glomerular Filtration Rate, Renal Fibrosis and Biomarkers in a Rat Model of Type 2 Diabetic Nephropathy. Am J Nephrol 2016;44:339–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wessels B, Ciapaite J, van den Broek NM, Nicolay K and Prompers JJ. Metformin impairs mitochondrial function in skeletal muscle of both lean and diabetic rats in a dose-dependent manner. PloS one. 2014;9:e100525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Penicaud L, Hitier Y, Ferre P and Girard J. Hypoglycaemic effect of metformin in genetically obese (fa/fa) rats results from an increased utilization of blood glucose by intestine. Biochem J. 1989;262:881–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hansmann G, Wagner RA, Schellong S, Perez VA, Urashima T, Wang L, Sheikh AY, Suen RS, Stewart DJ and Rabinovitch M. Pulmonary arterial hypertension is linked to insulin resistance and reversed by peroxisome proliferator-activated receptor-gamma activation. Circulation. 2007;115:1275–84. [DOI] [PubMed] [Google Scholar]
- 30.Kelley EE, Baust J, Bonacci G, Golin-Bisello F, Devlin JE, St Croix CM, Watkins SC, Gor S, Cantu-Medellin N, Weidert ER, Frisbee JC, Gladwin MT, Champion HC, Freeman BA and Khoo NK. Fatty acid nitroalkenes ameliorate glucose intolerance and pulmonary hypertension in high-fat diet-induced obesity. Cardiovascular research. 2014;101:352–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hawley SA, Pan DA, Mustard KJ, Ross L, Bain J, Edelman AM, Frenguelli BG and Hardie DG. Calmodulin-dependent protein kinase kinase-beta is an alternative upstream kinase for AMP-activated protein kinase. Cell Metab. 2005;2:9–19. [DOI] [PubMed] [Google Scholar]
- 32.Momcilovic M, Hong SP and Carlson M. Mammalian TAK1 activates Snf1 protein kinase in yeast and phosphorylates AMP-activated protein kinase in vitro. J Biol Chem. 2006;281:25336–43. [DOI] [PubMed] [Google Scholar]
- 33.Woods A, Johnstone SR, Dickerson K, Leiper FC, Fryer LG, Neumann D, Schlattner U, Wallimann T, Carlson M and Carling D. LKB1 is the upstream kinase in the AMP-activated protein kinase cascade. Curr Biol. 2003;13:2004–8. [DOI] [PubMed] [Google Scholar]
- 34.Shaw RJ, Kosmatka M, Bardeesy N, Hurley RL, Witters LA, DePinho RA and Cantley LC. The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:3329–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sung MM, Zordoky BN, Bujak AL, Lally JS, Fung D, Young ME, Horman S, Miller EJ, Light PE, Kemp BE, Steinberg GR and Dyck JR. AMPK deficiency in cardiac muscle results in dilated cardiomyopathy in the absence of changes in energy metabolism. Cardiovascular research. 2015;107:235–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nixon BR, Thawornkaiwong A, Jin J, Brundage EA, Little SC, Davis JP, Solaro RJ and Biesiadecki BJ. AMP-activated protein kinase phosphorylates cardiac troponin I at Ser-150 to increase myofilament calcium sensitivity and blunt PKA-dependent function. J Biol Chem. 2012;287:19136–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Oliveira SM, Zhang YH, Solis RS, Isackson H, Bellahcene M, Yavari A, Pinter K, Davies JK, Ge Y, Ashrafian H, Walker JW, Carling D, Watkins H, Casadei B and Redwood C. AMP-activated protein kinase phosphorylates cardiac troponin I and alters contractility of murine ventricular myocytes. Circulation research. 2012;110:1192–201. [DOI] [PubMed] [Google Scholar]
- 38.Behunin SM, Lopez-Pier MA, Birch CL, McKee LAK, Danilo C, Khalpey Z and Konhilas JP. LKB1/Mo25/STRAD uniquely impacts sarcomeric contractile function and posttranslational modification. Biophys J. 2015;108:1484–1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Adamson PB, Abraham WT, Bourge RC, Costanzo MR, Hasan A, Yadav C, Henderson J, Cowart P and Stevenson LW. Wireless pulmonary artery pressure monitoring guides management to reduce decompensation in heart failure with preserved ejection fraction. Circulation Heart failure. 2014;7:935–44. [DOI] [PubMed] [Google Scholar]
- 40.Guazzi M, Vicenzi M, Arena R and Guazzi MD. Pulmonary hypertension in heart failure with preserved ejection fraction: a target of phosphodiesterase-5 inhibition in a 1-year study. Circulation. 2011;124:164–74. [DOI] [PubMed] [Google Scholar]
- 41.MacDonald MR, Eurich DT, Majumdar SR, Lewsey JD, Bhagra S, Jhund PS, Petrie MC, McMurray JJ, Petrie JR and McAlister FA. Treatment of type 2 diabetes and outcomes in patients with heart failure: a nested case-control study from the U.K. General Practice Research Database. Diabetes Care. 2010;33:1213–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zinman B, Wanner C, Lachin JM, Fitchett D, Bluhmki E, Hantel S, Mattheus M, Devins T, Johansen OE, Woerle HJ, Broedl UC, Inzucchi SE and Investigators E-RO. Empagliflozin, Cardiovascular Outcomes, and Mortality in Type 2 Diabetes. N Engl J Med. 2015;373:2117–28. [DOI] [PubMed] [Google Scholar]
- 43.Mahaffey KW, Neal B, Perkovic V, de Zeeuw D, Fulcher G, Erondu N, Shaw W, Fabbrini E, Sun T, Li Q, Desai M, Matthews DR and Group CPC. Canagliflozin for Primary and Secondary Prevention of Cardiovascular Events: Results From the CANVAS Program (Canagliflozin Cardiovascular Assessment Study). Circulation. 2018;137:323–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zamanian RT, Hansmann G, Snook S, Lilienfeld D, Rappaport KM, Reaven GM, Rabinovitch M and Doyle RL. Insulin resistance in pulmonary arterial hypertension. The European respiratory journal. 2009;33:318–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Benson L, Brittain EL, Pugh ME, Austin ED, Fox K, Wheeler L, Robbins IM and Hemnes AR. Impact of diabetes on survival and right ventricular compensation in pulmonary arterial hypertension. Pulmonary circulation. 2014;4:311–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Simon MA, Vanderpool RR, Nouraie M, Bachman TN, White PM, Sugahara M, Gorcsan J 3rd, Parsley EL and Gladwin MT. Acute hemodynamic effects of inhaled sodium nitrite in pulmonary hypertension associated with heart failure with preserved ejection fraction. JCI insight. 2016;1:e89620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Olver TD, Edwards JC, Jurrissen TJ, Veteto AB, Jones JL, Gao C, Rau C, Warren CM, Klutho PJ, Alex L, Ferreira-Nichols SC, Ivey JR, Thorne PK, McDonald KS, Krenz M, Baines CP, Solaro RJ, Wang Y, Ford DA, Domeier TL, Padilla J, Rector RS and Emter CA. Western Diet-Fed, Aortic-Banded Ossabaw Swine: A Preclinical Model of Cardio-Metabolic Heart Failure. JACC Basic Transl Sci. 2019;4:404–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Duplain H, Burcelin R, Sartori C, Cook S, Egli M, Lepori M, Vollenweider P, Pedrazzini T, Nicod P, Thorens B and Scherrer U. Insulin resistance, hyperlipidemia, and hypertension in mice lacking endothelial nitric oxide synthase. Circulation. 2001;104:342–5. [DOI] [PubMed] [Google Scholar]
- 49.Paulus WJ and Tschope C. A novel paradigm for heart failure with preserved ejection fraction: comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. Journal of the American College of Cardiology. 2013;62:263–71. [DOI] [PubMed] [Google Scholar]
- 50.Greene SJ, Gheorghiade M, Borlaug BA, Pieske B, Vaduganathan M, Burnett JC, Jr., Roessig L, Stasch JP, Solomon SD, Paulus WJ and Butler J. The cGMP signaling pathway as a therapeutic target in heart failure with preserved ejection fraction. J Am Heart Assoc. 2013;2:e000536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Thenappan T, Prins KW, Cogswell R and Shah SJ. Pulmonary hypertension secondary to heart failure with preserved ejection fraction. The Canadian journal of cardiology. 2015;31:430–9. [DOI] [PubMed] [Google Scholar]
- 52.West J, Niswender KD, Johnson JA, Pugh ME, Gleaves L, Fessel JP and Hemnes AR. A potential role for insulin resistance in experimental pulmonary hypertension. The European respiratory journal. 2013;41:861–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Upadhya B, Haykowsky MJ, Eggebeen J and Kitzman DW. Exercise intolerance in heart failure with preserved ejection fraction: more than a heart problem. J Geriatr Cardiol. 2015;12:294–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Weiss K, Schar M, Panjrath GS, Zhang Y, Sharma K, Bottomley PA, Golozar A, Steinberg A, Gerstenblith G, Russell SD and Weiss RG. Fatigability, Exercise Intolerance, and Abnormal Skeletal Muscle Energetics in Heart Failure. Circulation Heart failure. 2017;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nishikawa T, Edelstein D, Du XL, Yamagishi S, Matsumura T, Kaneda Y, Yorek MA, Beebe D, Oates PJ, Hammes HP, Giardino I and Brownlee M. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature. 2000;404:787–90. [DOI] [PubMed] [Google Scholar]
- 56.Brown DA, Perry JB, Allen ME, Sabbah HN, Stauffer BL, Shaikh SR, Cleland JG, Colucci WS, Butler J, Voors AA, Anker SD, Pitt B, Pieske B, Filippatos G, Greene SJ and Gheorghiade M. Expert consensus document: Mitochondrial function as a therapeutic target in heart failure. Nat Rev Cardiol. 2017;14:238–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sada K, Nishikawa T, Kukidome D, Yoshinaga T, Kajihara N, Sonoda K, Senokuchi T, Motoshima H, Matsumura T and Araki E. Hyperglycemia Induces Cellular Hypoxia through Production of Mitochondrial ROS Followed by Suppression of Aquaporin-1. PloS one. 2016;11:e0158619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Barnes JW, Kucera ET, Tian L, Mellor NE, Dvorina N, Baldwin WW 3rd, Aldred MA, Farver CF, Comhair SA, Aytekin M and Dweik RA. Bone Morphogenic Protein Type 2 Receptor Mutation-Independent Mechanisms of Disrupted Bone Morphogenetic Protein Signaling in Idiopathic Pulmonary Arterial Hypertension. American journal of respiratory cell and molecular biology. 2016;55:564–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Heresi GA, Malin SK, Barnes JW, Tian L, Kirwan JP and Dweik RA. Abnormal Glucose Metabolism and High-Energy Expenditure in Idiopathic Pulmonary Arterial Hypertension. Ann Am Thorac Soc. 2017;14:190–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kitzman DW, Nicklas B, Kraus WE, Lyles MF, Eggebeen J, Morgan TM and Haykowsky M. Skeletal muscle abnormalities and exercise intolerance in older patients with heart failure and preserved ejection fraction. American journal of physiology Heart and circulatory physiology. 2014;306:H1364–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Batt J, Ahmed SS, Correa J, Bain A and Granton J. Skeletal muscle dysfunction in idiopathic pulmonary arterial hypertension. American journal of respiratory cell and molecular biology. 2014;50:74–86. [DOI] [PubMed] [Google Scholar]
- 62.Myers RW, Guan HP, Ehrhart J, Petrov A, Prahalada S, Tozzo E, Yang X, Kurtz MM, Trujillo M, Gonzalez Trotter D, Feng D, Xu S, Eiermann G, Holahan MA, Rubins D, Conarello S, Niu X, Souza SC, Miller C, Liu J, Lu K, Feng W, Li Y, Painter RE, Milligan JA, He H, Liu F, Ogawa A, Wisniewski D, Rohm RJ, Wang L, Bunzel M, Qian Y, Zhu W, Wang H, Bennet B, LaFranco Scheuch L, Fernandez GE, Li C, Klimas M, Zhou G, van Heek M, Biftu T, Weber A, Kelley DE, Thornberry N, Erion MD, Kemp DM and Sebhat IK. Systemic pan-AMPK activator MK-8722 improves glucose homeostasis but induces cardiac hypertrophy. Science. 2017;357:507–511. [DOI] [PubMed] [Google Scholar]
- 63.Cokorinos EC, Delmore J, Reyes AR, Albuquerque B, Kjobsted R, Jorgensen NO, Tran JL, Jatkar A, Cialdea K, Esquejo RM, Meissen J, Calabrese MF, Cordes J, Moccia R, Tess D, Salatto CT, Coskran TM, Opsahl AC, Flynn D, Blatnik M, Li W, Kindt E, Foretz M, Viollet B, Ward J, Kurumbail RG, Kalgutkar AS, Wojtaszewski JFP, Cameron KO and Miller RA. Activation of Skeletal Muscle AMPK Promotes Glucose Disposal and Glucose Lowering in Non-human Primates and Mice. Cell Metab. 2017;25:1147–1159 e10. [DOI] [PubMed] [Google Scholar]
- 64.Kelly NJ, Dandachi N, Goncharov DA, Pena AZ, Radder JE, Gregory AD, Lai YC, Leme AS, Gladwin MT, Goncharova EA, St Croix CM and Shapiro SD. Automated Measurement of Blood Vessels in Tissues from Microscopy Images. Curr Protoc Cytom. 2016;78:12 44 1–12 44 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.McNulty MJ, Sailstad JM and Steffen RP. The pharmacokinetics and pharmacodynamics of the prostacyclin analog 15AU81 in the anesthetized beagle dog. Prostaglandins Leukot Essent Fatty Acids. 1993;48:159–66. [DOI] [PubMed] [Google Scholar]
- 66.Lantier L, Williams AS, Williams IM, Yang KK, Bracy DP, Goelzer M, James FD, Gius D and Wasserman DH. SIRT3 Is Crucial for Maintaining Skeletal Muscle Insulin Action and Protects Against Severe Insulin Resistance in High-Fat-Fed Mice. Diabetes. 2015;64:3081–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Koh HJ, Arnolds DE, Fujii N, Tran TT, Rogers MJ, Jessen N, Li Y, Liew CW, Ho RC, Hirshman MF, Kulkarni RN, Kahn CR and Goodyear LJ. Skeletal muscle-selective knockout of LKB1 increases insulin sensitivity, improves glucose homeostasis, and decreases TRB3. Mol Cell Biol. 2006;26:8217–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Jeppesen J, Maarbjerg SJ, Jordy AB, Fritzen AM, Pehmoller C, Sylow L, Serup AK, Jessen N, Thorsen K, Prats C, Qvortrup K, Dyck JR, Hunter RW, Sakamoto K, Thomson DM, Schjerling P, Wojtaszewski JF, Richter EA and Kiens B. LKB1 regulates lipid oxidation during exercise independently of AMPK. Diabetes. 2013;62:1490–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chan AY, Soltys CL, Young ME, Proud CG and Dyck JR. Activation of AMP-activated protein kinase inhibits protein synthesis associated with hypertrophy in the cardiac myocyte. J Biol Chem. 2004;279:32771–9. [DOI] [PubMed] [Google Scholar]
- 70.Chan AY, Dolinsky VW, Soltys CL, Viollet B, Baksh S, Light PE and Dyck JR. Resveratrol inhibits cardiac hypertrophy via AMP-activated protein kinase and Akt. J Biol Chem. 2008;283:24194–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Fontana M, Olschewski H, Olschewski A and Schluter KD. Treprostinil potentiates the positive inotropic effect of catecholamines in adult rat ventricular cardiomyocytes. Br J Pharmacol. 2007;151:779–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Axelgaard S, Holmboe S, Ringgaard S, Hillgaard TK, Andersen S, Hansen MS, Andersen A and Nielsen-Kudsk JE. Effects of chronic treprostinil treatment on experimental right heart hypertrophy and failure. Cardiol Young. 2017;27:90–100. [DOI] [PubMed] [Google Scholar]
- 73.Hinderliter AL, Willis PWt, Barst RJ, Rich S, Rubin LJ, Badesch DB, Groves BM, McGoon MD, Tapson VF, Bourge RC, Brundage BH, Koerner SK, Langleben D, Keller CA, Murali S, Uretsky BF, Koch G, Li S, Clayton LM, Jobsis MM, Blackburn SD Jr., Crow JW and Long WA. Effects of long-term infusion of prostacyclin (epoprostenol) on echocardiographic measures of right ventricular structure and function in primary pulmonary hypertension. Primary Pulmonary Hypertension Study Group. Circulation. 1997;95:1479–86. [DOI] [PubMed] [Google Scholar]
- 74.Kerbaul F, Brimioulle S, Rondelet B, Dewachter C, Hubloue I and Naeije R. How prostacyclin improves cardiac output in right heart failure in conjunction with pulmonary hypertension. American journal of respiratory and critical care medicine. 2007;175:846–50. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.