

Abstract
Objective—
Enhanced expression of plasminogen activator inhibitor-1 (PAI-1) has been implicated in atherosclerosis formation in humans with obesity and metabolic syndrome. However, little is known about the effects of pharmacologic targeting of PAI-1 on atherogenesis. This study examined the effects of pharmacologic PAI-1 inhibition on atherosclerosis formation in a murine model of obesity and metabolic syndrome.
Approach and Results—
LDL receptor-deficient (ldlr−/−) mice were fed a western diet (WD) high in cholesterol, fat, and sucrose to induce obesity, metabolic dysfunction, and atherosclerosis. WD triggered significant up-regulation of PAI-1 expression compared to normal diet controls. Addition of a pharmacologic PAI-1 inhibitor (either PAI-039 or MDI-2268) to WD significantly inhibited obesity and atherosclerosis formation for up to 24 weeks without attenuating food consumption. Pharmacologic PAI-1 inhibition significantly decreased macrophage accumulation and cell senescence in atherosclerotic plaques. Recombinant PAI-1 stimulated smooth muscle cell (SMC) senescence, whereas a PAI-1 mutant defective in LDL receptor-related protein 1 (LRP1) binding did not. The pro-senescent effect of PAI-1 was blocked by PAI-039 and R2629, a specific anti-LRP1 antibody. PAI-039 significantly decreased visceral adipose tissue inflammation, hyperglycemia, and hepatic triglyceride content without altering plasma lipid profiles.
Conclusions—
Pharmacologic targeting of PAI-1 inhibits atherosclerosis in mice with obesity and metabolic syndrome, while inhibiting macrophage accumulation and cell senescence in atherosclerotic plaques, as well as obesity-associated metabolic dysfunction. PAI-1 induces senescence of SMCs in an LRP1-dependent manner. These results help to define the role of PAI-1 in atherosclerosis formation and suggest a new, plasma-lipid-independent strategy for inhibiting atherogenesis.
Keywords: atherosclerosis, metabolic syndrome, plasminogen activator inhibitor-1, senescence, smooth muscle cell
Subject terms: Animal Models of Human Disease, Vascular Biology, Obesity, Metabolic Syndrome, Pharmacology, Atherosclerosis, Thrombosis
Graphical Abstract
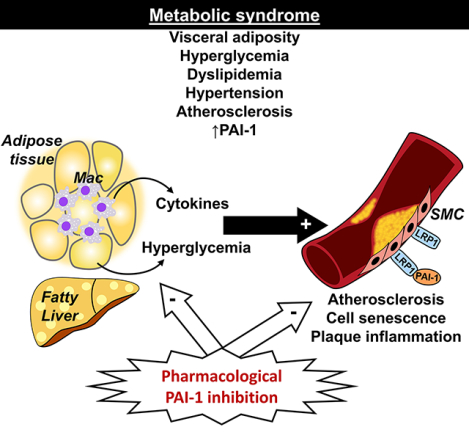
Introduction
The fibrinolytic system plays an important role in the pathogenesis of atherosclerosis by several mechanisms, including regulation of intravascular fibrin deposition, extracellular matrix turnover, and cell migration.1–3 The central reaction of the fibrinolytic system is the conversion of plasminogen to plasmin by tissue- and urinary-type plasminogen activators (tPA, uPA, respectively). Plasmin, a serine protease, degrades fibrin and several other proteins in plasma, the extracellular matrix, and on the plasma membrane of vascular cells. Plasminogen activator inhibitor-1 (PAI-1), the primary inhibitor of tPA and uPA, is a key regulator of fibrinolysis and cell migration.4 PAI-1 over-expression is associated with atherosclerosis in humans,5, 6 particularly in individuals with the metabolic syndrome,7 which is characterized by visceral obesity, insulin resistance, dyslipidemia, hypertension, and increased PAI-1 expression.8, 9
Several pharmacological inhibitors of PAI-1 have been developed and studied.10–12 These compounds have been proposed as novel treatments for multiple human diseases, including thrombosis, kidney disease, obesity, inflammatory bowel disease, and pathologic components of aging.13–23 However, no published studies have examined the effects of pharmacologic PAI-1 inhibition on atherosclerosis. It is important to study this issue for several reasons. First, there are likely to be significant differences in the biological consequences of genetic vs. pharmacologic suppression of PAI-1.24 For example, PAI-1 plays a functional role in the intracellular and extracellular compartments,25 both of which would be affected by PAI-1 gene deletion. In contrast, pharmacologic inhibitors might preferentially target the extracellular pool of PAI-1. Therefore, previous studies involving PAI-1 knockout mice26–28 cannot be presumed to predict the effects of PAI-1 inhibitors on atherosclerosis. Second, pharmacologic inhibitors of PAI-1 have begun to be studied in human clinical trials.29 If any of these drugs become approved for human therapy, it will be essential to understand their effects on atherosclerosis, which is present in many individuals. Third, several lines of evidence suggest that drug-targeting of PAI-1 has the potential to inhibit atherosclerosis,13, 16 and thus could represent a new treatment strategy to prevent disease progression and complications, including myocardial infarction and stroke.
In this study, we examined the effects of PAI-039 and MDI-2268, highly specific, small molecule inhibitors of PAI-1,11, 15 on atherosclerosis formation in a murine model of the metabolic syndrome. Specifically, we studied LDL-receptor-deficient (ldlr−/−) mice fed a western diet (WD) high in cholesterol, fat, and sucrose, which has been shown to generate the key features of human metabolic syndrome.30 We also examined the effects of pharmacologic PAI-1 inhibition on 1) atherosclerotic plaque and visceral adipose tissue (AT) inflammation, which play key roles in atherosclerosis progression and the metabolic dysfunction associated with obesity,8, 31, 32 and 2) vascular cell senescence, which is atherogenic.33, 34 Our results show that pharmacological targeting of PAI-1 inhibits atherosclerosis, AT inflammation and its downstream metabolic effects, as well as cell senescence and inflammation in atherosclerotic plaques. These results help to define the role of PAI-1 in atherogenesis and constitute the first report that drug-targeting of the fibrinolytic system inhibits atherosclerosis formation.
Materials and Methods
The authors declare that all supporting data are available within the article and its online supplementary files.
Reagents.
PAI-039, an orally available, specific PAI-1 inhibitor,15, 35 was from Pfizer, Cambridge, MA, USA. MDI-2268, an orally available, specific PAI-1 inhibitor, has been described previously.11 Recombinant human PAI-1 (Cat. #PAI-A) was from Molecular Innovations, Novi, MI. PAI-1-I91L (a recombinant human PAI-1 mutant with increased stability, normal anti-protease activity, and normal LRP1-binding affinity) and PAI-1-I91L, K80/207A (a recombinant human PAI-1 mutant with increased stability, normal anti-protease activity, normal vitronectin binding affinity, and a greater than 20-fold reduction in LRP1-binding affinity) were generated and purified as described.36 R2629, a rabbit anti-LDL receptor-related protein 1 (LRP1) antibody that does not cross-react with other LDL receptor family members,37 was a gift from Dudley Strickland, PhD, University of Maryland.
Mice.
Ldlr−/− mice, which had been back-crossed >10 generations into the C57BL/6J genetic background, were from Jackson Laboratory, Bar Harbor, ME, USA (stock number 002207). Mice were fed normal laboratory diet (ND) containing 62% of total calories from carbohydrates, 25% from proteins, <0.02% total cholesterol, and 3% sucrose by weight (#5053, LabDiet, Richmond, IN) from the time of weaning until initiation of WD, as specified for each experiment. WD contained 42% of total calories from fat, 0.2% total cholesterol, and 34% sucrose by weight (TD.88137, Envigo, South Easton, MA, USA). WD pellets lacked or contained either PAI-039 (5 mg/g of diet)38 or MDI-2268 (400 μg/g of diet). Animals consumed diet and water ad libitum. The University of Missouri Animal Care and Use Committee approved all experiments.
Plasma assays.
Plasma PAI-1 antigen was measured either by a Luminex multiplex assay39 or a standard ELISA utilizing anti-murine PAI-1 capture antibody (Molecular Innovations clone H34G6; coating concentration 1 μg/mL) and biotinylated anti-murine PAI-1 detection antibody (Molecular Innovations, ASMPAI-GF-BIO, 1 μg/mL). The Luminex PAI-1 antigen assay detects active, latent, and complexed forms of PAI-1, with the signal generated by tPA-PAI-1 complex being about 25% that of an equimolar amount of free, active PAI-1. Plasma PAI-1 activity was measured using a Luminex multiplex assay.39 Plasma total cholesterol, HDL cholesterol, LDL cholesterol, and triglycerides were measured by the University of Missouri Veterinary Diagnostic Lab. Blood samples for lipid analyses were collected after mice had fasted 4 hours. Hemoglobin A1c (HbA1c) was measured with a Siemens DCA Vantage Analyzer, per manufacturer’s instructions.
Gene expression.
Expression of PAI-1 and tissue necrosis factor-α (TNF-α) genes were assessed by quantitative real-time reverse transcriptase (RT)-PCR, as described.40 PCR primers were: PAI-1: 5’-GCTGCAGATGACCACAGCGGG-3’ and 5’-CCGCAGTACTGATCTCATTC-3’; TNF-α: ThermoFisher Scientific (Cat. #4331182); and 18S ribosomal RNA: 5’-CCTGGATACCGCAGCTAGGA-3’ and 5’-GCGGCGCAATACGAATGCCCC-3’.
Quantitative assessment of atherosclerotic plaques.
Atherosclerotic plaque formation was quantified in the aortic sinuses of Valsalva, ascending thoracic aorta, aortic arch, descending thoracic aorta, abdominal aorta, and carotid arteries, which were perfused with saline followed by 4% paraformaldehyde at physiologic pressure upon euthanasia. To quantify atherosclerosis formation in the aortic root, three cross-sections (each 10 μm thick), mounted in OCT compound and harvested at sequential intervals of 100 μm, beginning at the level of the aortic valve leaflets, were prepared for each animal and stained with Oil Red O and hematoxylin. The length of aortic root assessed in our studies (approximately 300 microns) was shorter than that advised by the American Heart Association (AHA) guidelines, which recommend assessing plaque formation throughout the entire aortic root – i.e. a distance of approximately 800 microns.41 Plaque size in aortic root microscopic images was analyzed using ImagePro software (Media Cybernetics, Rockville, MD). Atherosclerosis in the aortic arch, descending thoracic aorta, and abdominal aorta was assessed by fastidiously removing perivascular adipose tissue from the excised aorta, longitudinally incising it, staining it with Oil Red O, and performing en face quantification of total plaque area with computerized software, as described.42 This approach is consistent with AHA guidelines. Atherosclerosis in the common carotid arteries (from their origin from the aortic arch to just distal to the bifurcation into internal and external carotid artery) was measured by isolating these segments by dissection, measuring their surface area by computer-assisted planimetry of images viewed through a dissecting microscope, staining them with Oil Red O, rinsing them, incubating them in chloroform/methanol to extract Oil Red O, measuring the A520 of extracts in a spectrophotometer, and normalizing results to total surface area of the segment (i.e. A520/mm2).43
Histochemical analyses of vascular segments and AT.
Macrophage invasion into the fibrous cap of atherosclerotic plaques was assessed at the level of the aortic sinuses. Cross-sections were immunostained with anti-Mac-3 antibody (BD Pharmingen, Cat. #553322, concentration 2 μg/mL). The entire fibrous cap of the largest atherosclerotic plaque within each cross-section was selected as a region of interest. The false color segmentation technique was used to quantify macrophage content in the fibrous cap, as described.44 Smooth muscle cell (SMC)-α actin immunostaining was performed with anti-α-actin antibody (Santa Cruz biotechnology, Cat. #sc-32251, concentration 1 μg/mL).44 Picrosirius red staining for collagen was performed by incubating tissue cross-sections with Weigert’s hematoxylin (Sigma Aldrich, Cat. #HT1079–1SET), rinsing, and incubating with Direct red 80 (0.1%, Sigma Aldrich, Cat. #365548–5G) diluted in picric acid solution (1.2%, Fisher Scientific, Cat. #5860–32).45 Paraformaldehyde-fixed cross-sections of epididymal fat pad were prepared and immunostained with anti-Mac-3 antibody, as described for atherosclerotic segments of aorta. Crown-like structures (representing macrophage clusters encircling an adipocyte) per high-powered field were counted. Immunostaining of epididymal fat pads for LRP1 (Abcam, Cat. #AB92544, concentration 4.6 μg/mL) and fibrin(ogen) (Abcam, Cat. #AB34269, dilution 1:200) was performed and positive staining was quantified using the false color segmentation technique. For all immunohistochemical staining procedures, control experiments involving substitution of an isotype control primary antibody were performed to confirm assay specificity (Supplementary Fig. I). Mean adipocyte cross-sectional area was determined by counting the number of adipocytes in 4 random fields of defined area within a histologic cross-section of AT and dividing the total area by the number of adipocytes.
Cell and vascular senescence analyses.
Human coronary artery SMCs (Cascade Biologics, Portland, OR, Cat. #C0175C, lot number 1689414, from a 32 year-old female) were cultured in supplemented Medium 231 (Cascade Biologics) and passaged 6–8 times to render them more susceptible to senescence, which was defined by positive expression of senescence-associated β-galactosidase (SA-βGal). To study the effects of PAI-1 on SMC senescence, cells were grown to approximately 50% confluence, after which recombinant human PAI-1 was added to media and cells were cultured an additional 24 hours. In some experiments, PAI-039 or vehicle control was added to media along with PAI-1. Cells were fixed and scored for expression of SA-βGal using the Senescence Detection Kit (Cat. #AB65351, Abcam, Cambridge, MA, USA). Total cellular senescence in paraformaldehyde-fixed murine aortic arches was assessed by incubating them with fluorescein di‐β‐d‐galactopyranoside, as described.46 The fluorescence of the solution was measured and normalized to tissue weight. To specifically assess cell senescence in atherosclerotic plaques, paraformaldehyde-fixed, OCT mounted cross-sections of the aortic root were incubated 17 hours in citrate-phosphate buffer (37 mM citric acid, 126 mM sodium phosphate, pH 6.0) containing 150 mM NaCl, 1 mg/mL 5-bromo-4-chloro-3-indolyl β-D-galactopyranoside (X-Gal, Invitrogen Cat. #B1690), 2mM MgCl2, 5mM potassium ferrocyanide, and 5 mM potassium ferricyanide. Specimens were imaged using a Zeiss Axiovert 200M microscope and Leica DFC290 camera. Overlapping fields of view at 63x magnification were merged into a single image of the entire vascular cross-section using MetaMorph v.7.8.12 software. The region of plaque with the most intense X-gal staining (determined with examiner blinded to treatment group) was selected for comparison to aortic segments from other mice.
Hepatic analysis.
Triglyceride content of hepatic tissue was measured using the Cayman Chemical Triglyceride Colorimetric Assay Kit (Cat. #10010303).
Statistical analyses.
Results are shown as mean±standard error of the mean. Student’s t-test and one-way ANOVA were used to compare experimental groups, as appropriate, after first demonstrating that the data sets passed normality and equal variance tests. If either test failed, the Mann-Whitney U test was used for two-group comparisons. When one-way ANOVA was performed, the Holm-Sidak method was used for post hoc testing. The effect of drug treatment on mouse body weight was analyzed with a piecewise linear model with an auto-regressive error structure to accommodate the serial correlation due to repeated measurements on the same animals. Ninety-five percent confidence regions were calculated for each growth trajectory. The effect of drug treatment on diet consumption was analyzed by two-way analysis of variance with an interaction effect between diet group and time. The false discovery rate method was used to correct significance levels for multiple testing.47
Results
To determine if WD up-regulates PAI-1 expression in ldlr−/− mice, males that had been fed ND since weaning were divided into two matched groups, one fed WD (n=6, mean age 12.6 ± 0.3 weeks) the other continued on ND (n=6, mean age 12.6 ± 0.3 weeks; P>0.95 vs. WD group). After 4 weeks, plasma PAI-1 antigen was significantly increased in the WD group compared to ND controls (Supplementary Fig. IIA). Consistent with these results, PAI-1 gene expression, assessed by quantitative real-time RT-PCR analysis of RNA isolated from epididymal fat pads, was significantly higher in WD-fed ldlr−/− mice than in ND controls (Supplementary Fig. IIB). These results support the use of WD-fed ldlr−/− mice to model the metabolic syndrome, which is characterized by increased PAI-1 expression and a predisposition to atherosclerosis.7
Pharmacologic targeting of PAI-1 inhibits obesity and atherosclerosis.
To determine the effects of pharmacologic PAI-1 inhibition on atherosclerosis, we fed male adult ldlr−/− mice WD containing or lacking PAI-039 for 12–24 weeks. Mice receiving PAI-039 (n=12) were 27±3 weeks old when commencing WD vs. 27±3 weeks old for control mice receiving WD lacking PAI-039 (n=12; P>0.9). PAI-039 treatment significantly inhibited plasma PAI-1 activity, but had no significant effect on plasma PAI-1 antigen (Supplementary Fig. IIC). Body weight and diet consumption were measured weekly. Mice not treated with PAI-039 continued to gain weight throughout the 24 weeks of WD feeding. In contrast, PAI-039 treatment significantly attenuated weight gain, resulting in near plateau of body weight beyond 12 weeks of treatment (Fig. 1A), but did not decrease food consumption compared to control (Fig. 1B). At 12 weeks of WD, 7 mice from each group were euthanized for atherosclerosis studies. Atherosclerosis formation in the aortic root, aortic arch, and thoracic aorta was significantly less in mice treated with PAI-039 compared to controls (Fig. 1C–D). At 24 weeks of WD consumption, the remaining 5 mice in each group were euthanized. Atherosclerosis formation was still significantly less in the aortic arch and thoracic aorta of PAI-039-treated animals than in controls (Fig. 1D). Aortic root atherosclerosis was not assessed at the 24 week time point because a technical error in slide preparation resulted in loss of all samples. We quantified atherosclerosis in the carotid arteries of mice after 24 weeks of WD. As with the aorta, carotid atherosclerosis was significantly less in PAI-039-treated mice than in controls (Fig. 1E). Thus, pharmacologic inhibition of PAI-1 with PAI-039 was associated with a significant and sustained decrease in obesity and atherosclerosis formation.
Figure 1. PAI-039 inhibits obesity and atherosclerosis without decreasing diet consumption.
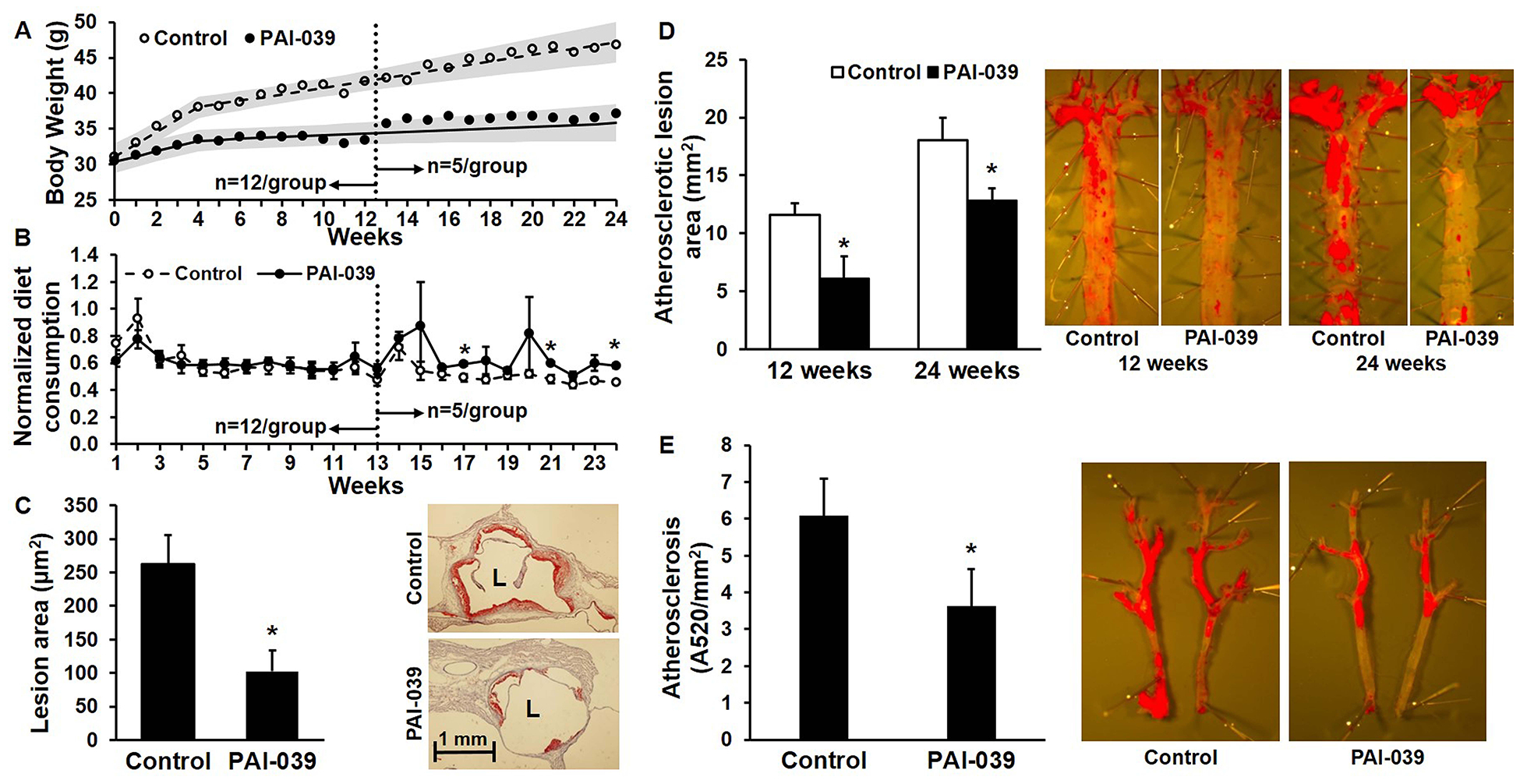
Ldlr−/− mice (n=12/group) were fed western diet (WD) containing (●) or lacking (○) PAI-039. Mean body weight and diet consumption were measured weekly. After 12 weeks, 7 mice from each group were euthanized for atherosclerosis quantification. The remaining mice (n=5/group) were continued on WD and euthanized at week 24 for atherosclerosis quantification. (A) Mean body weights. Shaded areas represent 95% confidence intervals. Differences between groups were statistically significant (P<0.05) for all data points beyond week 2. (B) Mean diet consumption/mouse/week, normalized to body weight - i.e. [diet consumed (g)/mouse/week]/[body weight (g)]; *P<0.05 vs. control. (C) Aortic root atherosclerosis, assessed after 12 weeks of WD; n=7/group; *P<0.05. Representative Oil Red O-stained images are shown. L, lumen. (D) Aortic arch and thoracic aorta atherosclerosis after 12 weeks (n=7/group) and 24 weeks (n=5/group) of WD; *P<0.05 vs. control. Representative Oil Red O-stained images are shown. (E) Carotid artery atherosclerosis after 24 weeks of WD; *P<0.05 vs. control. Representative images are shown.
To confirm the specificity of PAI-1 drug targeting, we also studied the effects of a recently described PAI-1 inhibitor on weight gain and atherosclerosis formation. For this experiment, ldlr−/− mice were fed WD containing or lacking MDI-2268 for 12 weeks. MDI-2268 is structurally unrelated and significantly more potent than PAI-039 in vivo, and was dosed more than 10-fold lower than for PAI-039 (400 μg/g of diet vs. 5 mg/g, respectively). Mice receiving MDI-2268 (n=7; 3 females, 4 males) were 19±2 weeks old when commencing WD vs 19±2 weeks old for mice receiving WD alone (n=6; 3 females, 3 males; P>0.7). Control mice not receiving MDI-2268 continued to gain weight throughout the experiment. In contrast, MDI-2268-treated mice failed to gain weight, despite diet consumption similar to that of controls (Fig. 2A–B). MDI-2268-treated mice appeared healthy throughout the experiment. At 12 weeks of WD, atherosclerosis formation in the aortic arch, thoracic and abdominal aorta was significantly less in MDI-2268-treated mice compared to controls (Fig. 2C).
Figure 2. MDI-2268 inhibits obesity and atherosclerosis, without affecting diet consumption.
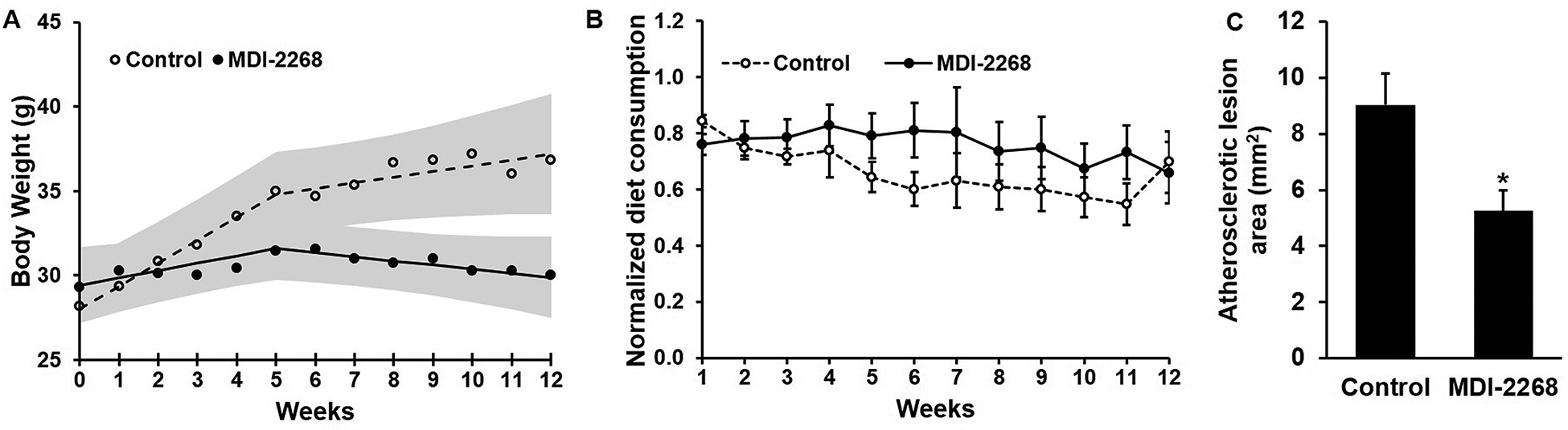
Ldlr−/− mice (n=6–7/group) were fed WD containing (●) or lacking (○) MDI-2268 for 12 weeks. Mean body weight and diet consumption were measured weekly. (A) Mean body weights. Shaded areas represent 95% confidence intervals. Differences between groups were statistically significant (P<0.05) for all data points beyond week 8. (B) Mean diet consumption, normalized to body weight - i.e. [diet consumed (g)/mouse/week]/[body weight (g)]; no statistically significant difference was achieved at any time point between groups. (C) Aortic arch, thoracic, and abdominal aorta atherosclerosis, assessed after 12 weeks of WD; *P<0.05 vs. control.
Pharmacologic targeting of PAI-1 inhibits macrophage invasion into atherosclerotic plaque without altering plaque SMC or collagen content.
We performed histochemical studies of atherosclerotic plaques to gain insights into the mechanisms by which pharmacologic PAI-1 inhibition altered atherogenesis. Macrophage accumulation in the fibrous cap was significantly decreased in mice treated with either PAI-039 or MDI-2268, as compared to controls (Fig. 3A–B). However, atherosclerotic plaque SMC and collagen content were not significantly altered by pharmacologic inhibition of PAI-1 with MDI-2268 (Fig. 3C–D).
Figure 3. Pharmacologic targeting of PAI-1 inhibits macrophage invasion into atherosclerotic plaques without altering their SMC or collagen content.
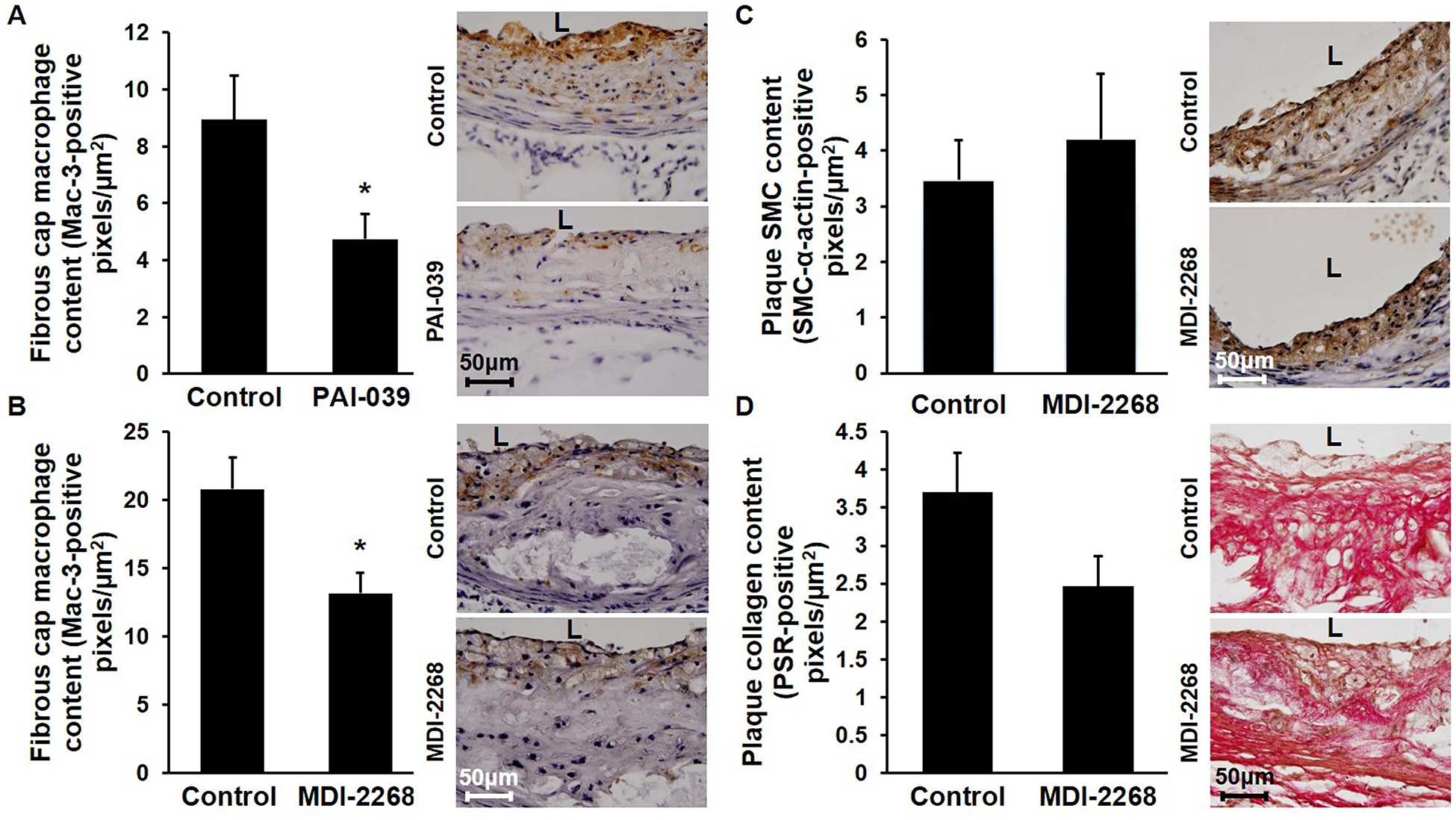
Ldlr−/− mice were fed WD containing or lacking PAI-1 inhibitor for 12 weeks, after which aortic root atherosclerotic plaque composition was assessed. (A) PAI-039 decreases fibrous cap macrophage content. Quantified data (n=5/group; *P<0.05) and representative images demonstrating macrophage invasion (brown color) are shown. (B) MDI-2268 decreases fibrous cap macrophage content. Quantified data (n=6–7/group; *P<0.05) and representative images are shown. (C) MDI-2268 does not significantly affect intimal SMC content, assessed by SMC-α actin immunostaining (brown color). Quantified data (n=4–6/group, difference between groups did not achieve statistical significance; P>0.6) and representative images are shown. (D) MDI-2268 does not significantly affect plaque collagen content, assessed by picrosirius red (PSR) staining. Quantified data (n=6–7/group, difference between groups did not achieve statistical significance; P>0.08) and representative images are shown. L, lumen.
PAI-039 attenuates visceral AT inflammation, hyperglycemia, and hepatic triglyceride content.
Visceral fat accumulation, assessed by weighing epididymal fat pads harvested 12 and 24 weeks after initiation of WD, was significantly less in PAI-039-treated mice than in controls (Fig. 4A). Formation of macrophage crown-like structures in epididymal white AT was significantly less in PAI-039-treated mice than in controls (Fig. 4B–C). Consistent with these results, quantitative RT-PCR analysis revealed a significant decrease in TNF-α gene expression in epididymal white AT of PAI-039-treated mice compared to controls (Fig. 4D). There was no significant effect of PAI-039 on mean adipocyte size (Fig. 4E). Consistent with the inhibitory effects of pharmacologic targeting of PAI-1 on obesity and AT inflammation, there was a significant reduction in blood glucose, assessed by measurement of HbA1c after 24 weeks of WD, in PAI-039-treated mice (6.5±0.8%) vs. controls (8.1±0.5%; n=5/group; P<0.05), indicating that PAI-039 reduces WD-induced hyperglycemia. PAI-039 significantly decreased liver triglyceride content, assessed after 12 weeks of WD, but had no significant effect on liver weight, assessed after 24 weeks of WD (Supplementary Fig. IIIA-B). PAI-039 had no significant effect on plasma total cholesterol, LDL cholesterol, HDL cholesterol, or triglycerides concentrations, assessed after 12 and 24 weeks of WD (Supplementary Fig. IIIC-F). Recent studies suggest that fibrin deposition in AT plays a key role in AT inflammation and metabolic dysfunction.48 However, we found no significant effect of pharmacologic PAI-1 inhibition on epididymal white AT fibrin content (Supplementary Fig. IVA-B). LRP1, which binds PAI-1, has been implicated in the pathogenesis of obesity.49 However, we found no significant effect of pharmacologic PAI-1 inhibition on visceral white AT LRP1 content, assessed by quantitative immunohistochemistry (Supplementary Fig. IVC-D). As a whole, these results suggested that the anti-atherosclerotic effects of pharmacologic PAI-1 inhibition are mediated, at least in part, by attenuation of diet-induced visceral adiposity, AT inflammation, glucose intolerance, and steatohepatosis.
Figure 4. PAI-039 decreases visceral white adipose tissue mass and inflammation.
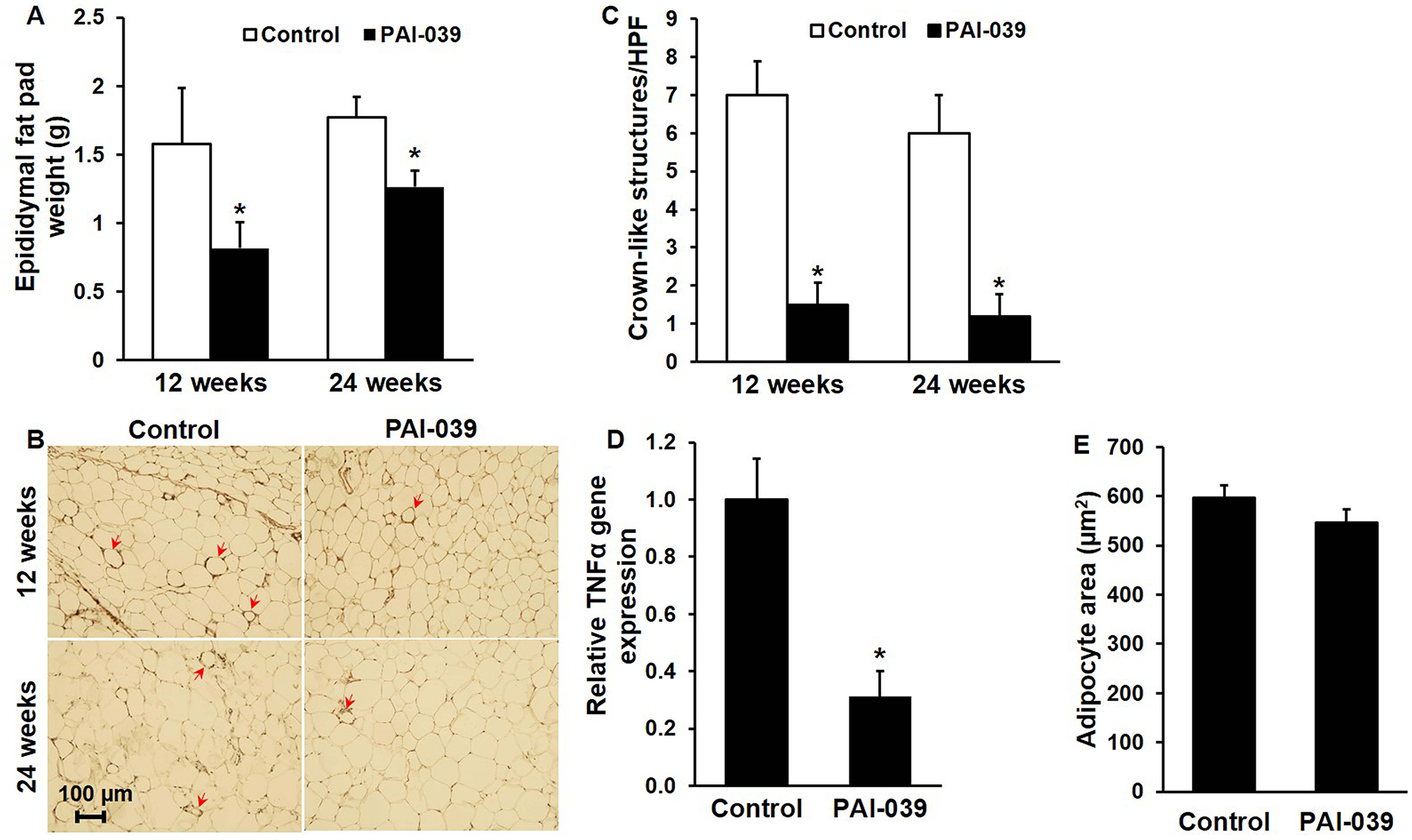
Epididymal white adipose tissue of ldlr−/− mice was harvested and analyzed after 12 and 24 weeks of WD with or without PAI-039. (A) Mean combined weights of left and right epididymal fat pads; n=5–7 mice/group; *P<0.05 vs. control. (B) Macrophage (Mac-3) immunostaining, demonstrating peri-adipocyte crown-like structures (arrows). (C) Quantification of crown-like structure formation; n=4–5/group, *P<0.01. HPF, high-power field. (D) TNF-α gene expression, assessed by real-time RT-PCR analysis after 24 weeks of WD; n=3–5 mice/group; *P<0.01 vs. control. (E) Mean adipocyte size (cross-sectional area), assessed after 12 weeks of WD; n=4–5/group; P>0.3.
PAI-1 stimulates SMC senescence, which is blocked by PAI-039 and anti-LRP1 antibody.
Senescence of vascular cells, including SMCs, plays a significant role in atherogenesis.33, 34 To determine if PAI-1 induces SMC senescence, human coronary artery SMCs were incubated with or without recombinant PAI-1 for 24 hours, after which expression of SA-βGal, a specific marker of cell senescence, was measured. PAI-1 significantly increased SMC senescence, and this effect was inhibited by PAI-039 (Fig. 5A). To elucidate potential mechanisms, SMCs were pre-treated with or without R2629, a specific anti-LRP1 antibody, then incubated with or without recombinant PAI-1. R2629 significantly inhibited the pro-senescent effect of PAI-1, while also exerting a minor pro-senescent effect when incubated with SMCs in the absence of PAI-1 (Fig. 5B). To further test the hypothesis that PAI-1 stimulates SMC senescence by binding LRP1, SMCs were incubated with vehicle control, a recombinant active, stable form of PAI-1 (I91L), or a recombinant PAI-1 mutant defective in LRP1 binding due to mutations at amino acids 80 and 207 (while also containing the activity-stabilizing I91L mutation).36 Whereas PAI-1-I91L (1 μg/mL) significantly increased SMC senescence, PAI-1-I91L, K80/207A, the PAI-1 mutant defective in LRP1 binding, had no significant effect (Fig. 5C).
Figure 5. PAI-1 stimulates smooth muscle cell (SMC) senescence in an LRP1-dependent manner.
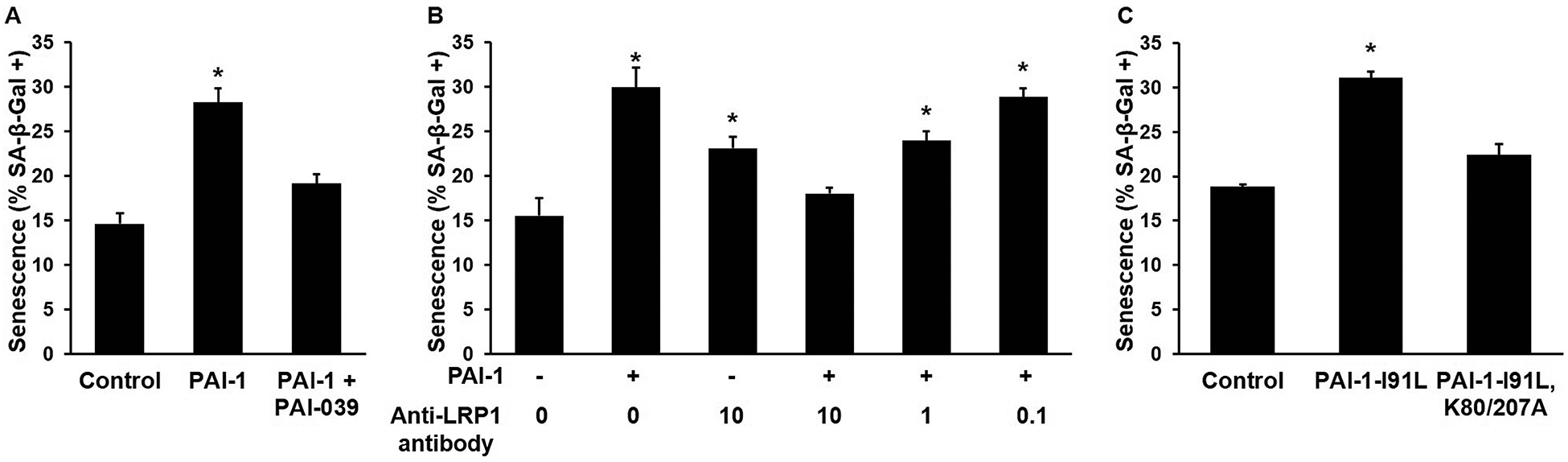
(A) SMCs were incubated for 24 hours with recombinant PAI-1 (10 μg/mL), recombinant PAI-1 and PAI-039 (25 μM), or vehicle control, after which SA-βGal expression (% positive cells) was measured; n= 5–7/group; *P<0.05 vs. other groups. (B) SMCs (passage number 6–8) were incubated 12 hours with or without anti-LRP1 antibody (at indicated concentrations [μg/mL]), after which PAI-1 (1 μg/mL, “+”) or vehicle control (“-”) was added. Cells were incubated an additional 24 hours, after which SA-βGal expression was measured; n=5/group, *P<0.05 vs. control (untreated SMCs). (C) SMCs (passage number 7) were incubated 24 hours with PAI-1-I91L (recombinant PAI-1 mutant with increased stability, normal anti-protease activity, and normal LRP1-binding affinity, concentration 1 μg/mL), PAI-1-I91L,K80/207A (recombinant PAI-1 mutant with increased stability, normal anti-protease activity, and a greater than 20-fold reduction in LRP1-binding affinity; concentration 1 μg/mL), or vehicle control, after which SA-βGal expression was measured; n=4/group; *P<0.001 vs. other groups.
PAI-039 inhibits vascular senescence in vivo.
To determine the effects of pharmacologic PAI-1 inhibition on vascular senescence in vivo, SA-βGal activity was assessed in aortic arches of ldlr−/− mice after 12 weeks of WD. Vascular cell senescence was significantly decreased in PAI-039-treated animals compared to controls fed WD alone (Fig. 6A). We used a histochemical assay to specifically assess cell senescence in atherosclerotic plaques. This analysis suggested that cell senescence was significantly reduced in atheroma from mice treated with PAI-039 compared to controls (Fig. 6B). These in vivo data, together with the in vitro SMC data, suggest that PAI-1 promotes atherosclerosis by direct effects on vascular cells, including SMCs, by pathways that are inhibited by PAI-039 and require PAI-1 binding to LRP1.
Figure 6. PAI-039 inhibits vascular senescence in vivo.
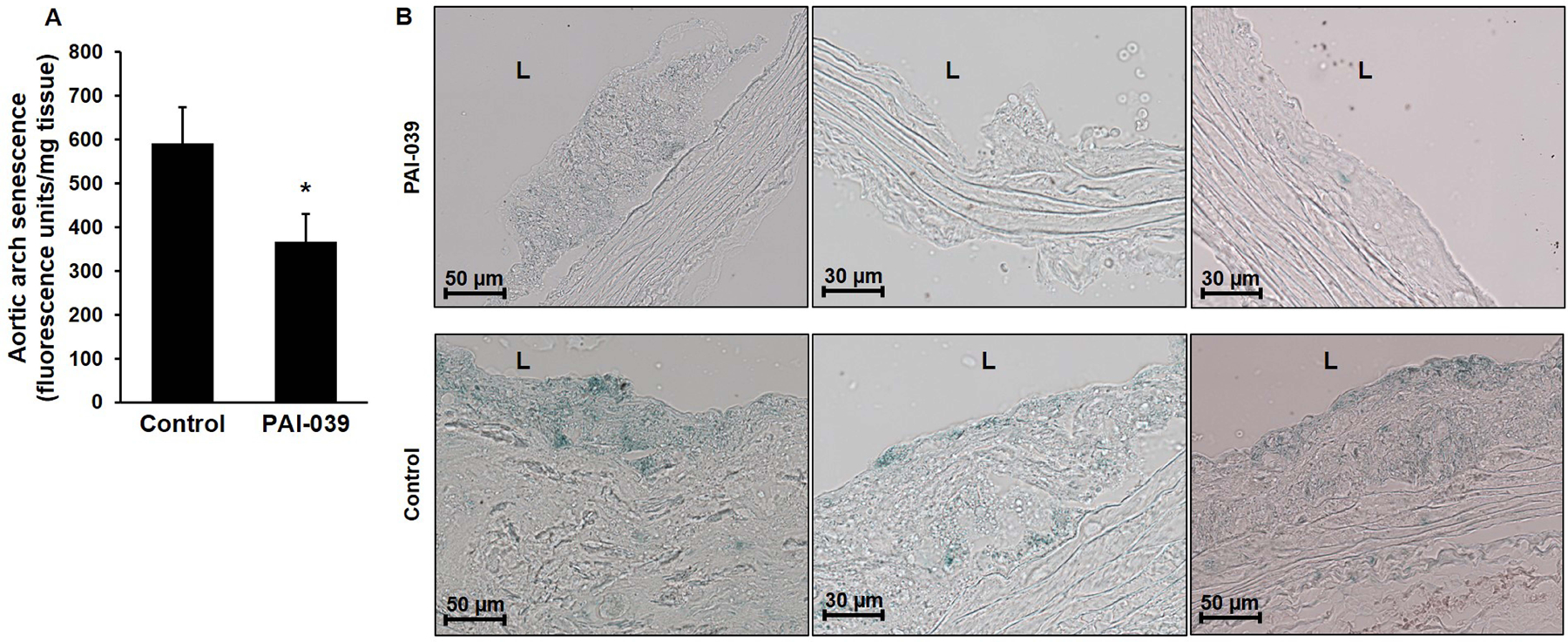
(A) Total aortic cell senescence is decreased by PAI-039. Ldlr−/− mice were fed WD containing or lacking PAI-039 for 12 weeks, after which aortas were perfusion-fixed with paraformaldehyde and excised. Periadventitial fat and other tissue were carefully excised. Cell senescence was determined by incubating aortic arches in solution containing fluorescein di-β-d-galactopyranoside for 6 hours and measuring fluorescence of the solution, with results normalized to tissue weight; n=7 mice/group, *P=0.05. (B) Ldlr−/− mice were fed WD with or without PAI-039 for 12 weeks. Paraformaldehyde-fixed aortas were excised and cross-sections from aortic roots were subjected to histochemical analysis to detect SA-β-galactosidase activity (blue color). Representative images from 3 mice in each treatment group are shown. L, lumen.
Discussion
The metabolic syndrome, which is characterized by obesity, dyslipidemia, hypertension, glucose intolerance, and atherosclerosis,50, 51 affects nearly 35% of all adults and 50% of those aged 60 years or older,52 making it a dominant driver of the total societal burden of atherosclerosis. PAI-1, the primary inhibitor of tPA and uPA, is a key regulator of fibrinolysis and cell migration that has been strongly implicated in the pathogenesis of atherosclerosis, particularly in patients with the metabolic syndrome.7, 8 In this study, we demonstrated that PAI-039 and MDI-2268 inhibit atherosclerosis formation in mice with metabolic syndrome, which is the first demonstration that drug targeting of the fibrinolytic system inhibits atherogenesis. The similar effects of two unrelated PAI-1 inhibitors suggests a drug class effect on atherogenesis, rather than a potential off-target effect of a single compound. We also showed that pharmacological targeting of PAI-1 inhibits cell senescence and macrophage accumulation in atherosclerotic plaques, while also producing beneficial metabolic effects, including attenuation of obesity, AT inflammation, hyperglycemia, and steatohepatosis. These findings have important therapeutic implications and shed new light on the complex role of PAI-1 in regulating atherosclerosis and the metabolic dysfunction associated with obesity.
In murine models, the regulatory function of PAI-1 in atherosclerosis has been examined predominantly by using PAI-1-deficient mice. Eitzman et al. found that genetic deletion of PAI-1 inhibited carotid artery atherosclerosis,27 whereas Luttun et al. reported accelerated aortic atherosclerosis in PAI-1-deficient mice.28 Sjoland et al. found no significant effect of genetic PAI-1 deficiency on atherogenesis.26 However, results of gene knockout studies do not necessarily predict the effects of pharmacologic PAI-1 inhibition on atherosclerosis and other PAI-1-dependent processes. We showed previously that genetic deletion of PAI-1 increases neointima formation in vein grafts,44 while PAI-039 significantly decreases neointima formation in the same model.24 This paradox is likely attributable to multiple factors, which we discussed previously.24 The results of the current study suggest that drug targeting of PAI-1 may be a new strategy to inhibit atherosclerotic plaque growth, which is relevant to the many patients intolerant of statin therapy.53 Pharmacologic PAI-1 inhibition could also be employed in conjunction with statin therapy, not only to provide additional anti-atherosclerotic effects, but also to inhibit thrombosis.
In the current study, we showed that PAI-1 promotes SMC senescence. This finding, which is consistent with previous studies demonstrating pro-senescent effects of PAI-1 on other cell types,13, 14, 54–58 is significant because it identifies a new trigger of SMC senescence, which itself has been identified as pro-atherosclerotic.34 We also demonstrated that PAI-039 attenuates cell senescence in vivo in atherosclerotic plaques. Therefore, our data support the hypothesis that pharmacologic targeting of PAI-1 inhibits atherosclerosis by attenuating vascular cell senescence.33 Furthermore, we demonstrated that PAI-1’s pro-senescent effect is blocked by an antibody specific for LRP1, and that a PAI-1 mutant defective in LRP1 binding has no significant effect on SMC senescence. Heretofore, little has been known about the role of LRP1 in cell senescence.59, 60 Our results suggest that binding of PAI-1 to LRP1 initiates the process. LRP1 is a recognized PAI-1 receptor that triggers intracellular signaling pathways, including JAK1/STAT1 activation.24, 61 Based on our findings and previously published data, we hypothesize that the anti-senescent effect of PAI-039 is mediated by decreasing the pool of active, pericellular PAI-1, thereby inhibiting PAI-1 binding to uPA, which is expressed by SMCs.62, 63 This, in turn, will reduce the interaction of PAI-1 with LRP1, as PAI-1-uPA complex binds LRP1 approximately 100-fold more avidly than either active or latent PAI-1.36, 64 Our studies also suggest another mechanism by which pharmacologic inhibition of PAI-1 down-regulates atherogenesis – i.e. by decreasing macrophage accumulation in developing plaques. Such an effect would be anticipated to attenuate formation of vulnerable plaques prone to rupture65 and is consistent with a study demonstrating that pharmacologic targeting of PAI-1 inhibits macrophage migration.16 Together, our cell senescence and macrophage invasion data support the hypothesis that drug-targeting of PAI-1 exerts direct effects on the vascular wall that attenuate atherogenesis. This hypothesis is also supported by our previous demonstration of inhibition of neointima formation by pharmacologic PAI-1 inhibition in mice without obesity or hyperlipidemia.24 There is yet another potential mechanisms by which pharmacologic PAI-1 inhibitors may act on the arterial wall to regulate atherosclerosis. Simone et al. demonstrated that PAI-039 fosters formation of the cleaved form of PAI-1 and increases SMC apoptosis by a pathway that appears to involve TNF-α weak inducer of apoptosis (TWEAK) and its receptor, fibroblast growth factor (FGF)-inducible 14 (FN14).20 This group also showed that PAI-039 and cleaved PAI-1 each inhibited intimal hyperplasia, a hallmark of atherogenesis, in the murine carotid artery ligation model.
In addition to direct vascular effects, pharmacologic targeting of PAI-1 has the potential to attenuate atherogenesis indirectly by dampening systemic inflammatory pathways that stimulate atherosclerosis. The metabolic syndrome is characterized by fibrin formation and leukocyte accumulation in visceral AT, which leads to secretion of inflammatory cytokines that act systemically to promote obesity, insulin resistance, and endothelial cell activation,48, 66, 67. LRP1 has also been implicated as a regulator of obesity.49, 68 We demonstrated that PAI-039 and MDI-2268 inhibit obesity. However, we found no significant effect of pharmacologic PAI-1 inhibitors on visceral AT fibrin or LRP1 content, assessed by immunostaining. These results suggest that promotion of fibrinolysis or changes in LRP1 expression in AT did not play major roles in the suppression of obesity and hyperglycemia by PAI-1 inhibition in our model. However, the methods employed in our study do not allow us to exclude the possibility that subtle changes in AT fibrin and LRP1 content, cleavage, or distribution contribute to the pro-metabolic and anti-atherosclerotic effects of PAI-1 inhibition. Crandall et al. and Lijnen et al. demonstrated that PAI-039 inhibits short-term weight gain in mice with normal lipid metabolism.19, 38 Our data significantly extend the prior work by showing that pharmacologic inhibition of PAI-1 produces a durable anti-obesity effect (i.e. still present after 24 weeks of therapy, which is 6-fold longer than the previously published studies), including in obese mice with hyperglycemia and severe dyslipidemia. Unlike the studies of Crandall and Lijnen, we found no significant effect of PAI-039 on adipocyte size, raising the possibility that the attenuation of visceral obesity by pharmacologic targeting of PAI-1 under the experimental conditions in our study might be linked to decreases in adipocyte number, possibly mediated by effects of PAI-1 targeting on preadipocyte survival or differentiation.69 Further studies are warranted to address this issue. We showed in the current study that PAI-039 significantly inhibits macrophage accumulation and TNF-α expression in visceral white AT, while also inhibiting hyperglycemia and steatohepatosis, key features of the metabolic syndrome that are associated with atherogenesis.70 These findings are consistent with a previous report demonstrating that TM5441, another PAI-1 inhibitor, significantly attenuates macrophage accumulation in white AT and insulin resistance in mice, assessed by glucose- and insulin tolerance testing.71 Henkel et al. showed that pharmacologic PAI-1 inhibition attenuates steatohepatosis.72 As a whole, our findings support the hypothesis that pharmacologic inhibition of PAI-1 inhibits atherogenesis by significantly attenuating the AT inflammation and systemic metabolic dysfunction that are characteristic of the metabolic syndrome.
In conclusion, we have demonstrated that pharmacologic targeting of PAI-1 significantly attenuates atherosclerosis in mice with obesity and features of the metabolic syndrome, a common clinical entity that is strongly associated with increased PAI-1 expression. Our results provide important insights into the role of PAI-1 in atherogenesis that complement and extend studies involving PAI-1 knockout mice. The finding that drug targeting of PAI-1 attenuates atherogenesis is highly significant from the clinical perspective. The fibrinolytic pathway is not directly involved in lipid metabolism, which is the target of currently used anti-atherosclerotic drugs, such as statins and PCSK9 inhibitors.73, 74 Thus, our results suggest a potential new direction in anti-atherosclerotic therapy. They also suggest that the anti-atherosclerotic effects of pharmacologic PAI-1 inhibition involve multiple mechanisms, including attenuation of cell senescence and macrophage accumulation in atherosclerotic plaques, as well as reductions in visceral AT inflammation and glucose intolerance. We also have shown that PAI-1 promotes SMC senescence, a key step in atherogenesis, by an LRP1-dependent mechanism. Our data suggest that drug targeting of PAI-1 provides long-term attenuation of weight gain, even in the presence of severe metabolic dysfunction, a significant and clinically relevant advance from prior reports. More studies are indicated to define the effects of PAI-1 inhibitors on specific AT depots (i.e. subcutaneous, perirenal, perivascular, and brown), including their size, degree of inflammation, and thermogenic properties. In future work, it also will be of interest to study the capacity of pharmacologic PAI-1 inhibitors to reverse established obesity and atherosclerosis.
Supplementary Material
Highlights.
PAI-1 inhibitors significantly decrease atherosclerosis formation in a murine model of obesity and metabolic syndrome.
Pharmacologic targeting of PAI-1 inhibits macrophage accumulation and cell senescence in atherosclerotic plaques.
PAI-1 stimulates vascular smooth muscle cell senescence by binding to LDL receptor-related protein 1 (LRP1).
Pharmacologic PAI-1 inhibition exerts durable anti-obesity and pro-metabolic effects in LDL-receptor-deficient (ldlr−/−) mice fed western diet for up to 24 weeks.
Overall, these results suggest that pharmacologic targeting of PAI-1 may be an effective strategy to inhibit obesity-associated atherosclerosis and metabolic dysfunction.
Acknowledgments
Greg Petroski, PhD, Department of Health Management & Informatics, University of Missouri, assisted with statistical methods. Robert Baker, PhD, Molecular Cytology Core, University of Missouri, assisted with imaging of cell senescence in atherosclerotic plaques. Histology slides were prepared by IDEXX Laboratories, Columbia, MO.
Sources of Funding
This work was supported by NIH R01 HL136746 (WPF), HL55374 (DAL), American Heart Association Grant-in-Aid 17GRNT33671082 (WPF), and Department of Veterans Affairs Merit Review Award CARA-007-12S (WPF).
Nonstandard abbreviations and acronyms
- AT
adipose tissue
- Ldlr−/−
low-density lipoprotein-receptor-deficient
- LRP1
low-density lipoprotein receptor-related protein 1
- ND
normal diet
- PAI-1
plasminogen activator inhibitor-1
- SA-βgal
senescence-associated β-galactosidase
- SMC
smooth muscle cell
- tPA
tissue-type plasminogen activator
- uPA
urinary-type plasminogen activator
- WD
western diet
- X-Gal
5-bromo-4-chloro-3-indolyl β-D-galactopyranoside
Footnotes
Disclosures
DAL and CDE have an equity interest in MDI Therapeutics, which holds an option to license MDI-2268 from the University of Michigan.
References
- 1.Juhan-Vague I and Collen D. On the role of coagulation and fibrinolysis in atherosclerosis. Ann Epidemiol. 1992;2:427–438. [DOI] [PubMed] [Google Scholar]
- 2.Xiao Q, Danton MJS, Witte DP, Kowala MC, Valentine MT, Bugge TH and Degen JL. Plasminogen deficiency accelerates vessel wall disease in mice predisposed to atherosclerosis. Proc Natl Acad Sci USA. 1997;94:10335–10340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cozen AE, Moriwaki H, Kremen M, DeYoung MB, Dichek HL, Slezicki KI, Young SG, Veniant M and Dichek DA. Macrophage-targeted overexpression of urokinase causes accelerated atherosclerosis, coronary artery occlusions, and premature death. Circulation. 2004;109:2129–2135. [DOI] [PubMed] [Google Scholar]
- 4.Declerck PJ and Gils A. Three decades of research on plasminogen activator inhibitor-1: a multifaceted serpin. Semin Thromb Hemost. 2013;39:356–364. [DOI] [PubMed] [Google Scholar]
- 5.Hamsten A, Wiman B, De Faire U and Blomback M. Increased plasma levels of a rapid inhibitor of tissue plasminogen activator in young survivors of myocardial infarction. N Eng J Med. 1985;313:1557–1563. [DOI] [PubMed] [Google Scholar]
- 6.Vaughan DE. PAI-1 and atherothrombosis. J Thromb Haemost. 2005;3:1879–1883. [DOI] [PubMed] [Google Scholar]
- 7.Alessi MC and Juhan-Vague I. PAI-1 and the Metabolic Syndrome: Links, Causes, and Consequences. Arterioscler Thromb Vasc Biol. 2006;26:2200–2207. [DOI] [PubMed] [Google Scholar]
- 8.Alexopoulos N, Katritsis D and Raggi P. Visceral adipose tissue as a source of inflammation and promoter of atherosclerosis. Atherosclerosis. 2014;233:104–112. [DOI] [PubMed] [Google Scholar]
- 9.Song C, Burgess S, Eicher JD, O’Donnell CJ and Johnson AD. Causal Effect of Plasminogen Activator Inhibitor Type 1 on Coronary Heart Disease. J Am Heart Assoc. 2017;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vaughan DE, De Taeye BM and Eren M. PAI-1 antagonists: predictable indications and unconventional applications. Curr Drug Targets. 2007;8:962–970. [DOI] [PubMed] [Google Scholar]
- 11.Reinke AA, Li S-H, Warnock M, Shaydakov ME, Guntaka NS, Su EJ, Diaz JA, Emal CD and Lawrence DA. Dual-reporter high-throughput screen for small-molecule in vivo inhibitors of plasminogen activator inhibitor type-1 yields a clinical lead candidate. J Biol Chem. 2019;294:1464–1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brown NJ. Therapeutic potential of plasminogen activator inhibitor-1 inhibitors. Ther Advances Cardiovasc Dis. 2010;4:315–324. [DOI] [PubMed] [Google Scholar]
- 13.Boe AE, Eren M, Murphy SB, Kamide CE, Ichimura A, Terry D, McAnally D, Smith LH, Miyata T and Vaughan DE. Plasminogen Activator Inhibitor-1 Antagonist TM5441 Attenuates N-Nitro-l-Arginine Methyl Ester-Induced Hypertension and Vascular Senescence. Circulation. 2013;128:2318–2324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Eren M, Boe AE, Murphy SB, Place AT, Nagpal V, Morales-Nebreda L, Urich D, Quaggin SE, Budinger GRS, Mutlu GkM, Miyata T and Vaughan DE. PAI-1−regulated extracellular proteolysis governs senescence and survival in Klotho mice. Proc Natl Acad Sci USA. 2014;111:7090–7095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hennan JK, Elokdah H, Leal M, Ji A, Friedrichs GS, Morgan GA, Swillo RE, Antrilli TM, Hreha A and Crandall DL. Evaluation of PAI-039 [{1-benzyl-5-[4-(trifluoromethoxy)phenyl]-1H-indol-3-yl}(oxo)acetic acid], a novel plasminogen activator inhibitor-1 inhibitor, in a canine model of coronary artery thrombosis. The Journal of pharmacology and experimental therapeutics. 2005;314:710–6. [DOI] [PubMed] [Google Scholar]
- 16.Ichimura A, Matsumoto S, Suzuki S, Dan T, Yamaki S, Sato Y, Kiyomoto H, Ishii N, Okada K, Matsuo O, Hou FF, Vaughan DE, van Ypersele de Strihou C and Miyata T. A Small Molecule Inhibitor to Plasminogen Activator Inhibitor 1 Inhibits Macrophage Migration. Arterioscler Thromb Vasc Biol. 2013;33:935–942. [DOI] [PubMed] [Google Scholar]
- 17.Jeong BY, Uddin MJ, Park JH, Lee JH, Lee HB, Miyata T and Ha H. Novel Plasminogen Activator Inhibitor-1 Inhibitors Prevent Diabetic Kidney Injury in a Mouse Model. PLOS ONE. 2016;11:e0157012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rebalka IA, Raleigh MJ, D’Souza DM, Coleman SK, Rebalka AN and Hawke TJ. Inhibition of PAI-1 Via PAI-039 Improves Dermal Wound Closure in Diabetes. Diabetes. 2015;64:2593–2602. [DOI] [PubMed] [Google Scholar]
- 19.Lijnen RH, Alessi MC, Frederix L, Collen D and Juhan-Vague I. Tiplaxtinin impairs nutritionally induced obesity in mice. Thromb Haemost. 2006;96:731–737. [PubMed] [Google Scholar]
- 20.Simone TM, Higgins SP, Archambeault J, Higgins CE, Ginnan RG, Singer H and Higgins PJ. A small molecule PAI-1 functional inhibitor attenuates neointimal hyperplasia and vascular smooth muscle cell survival by promoting PAI-1 cleavage. Cellular Signalling. 2015;27:923–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Suzuki J-i, Ogawa M, Muto S, Yamaguchi Y, Itai A and Isobe M. The effects of pharmacological PAI-1 inhibition on thrombus formation and neointima formation after arterial injury. Expert Opin Therapeut Targets. 2008;12:783–794. [DOI] [PubMed] [Google Scholar]
- 22.Weisberg AD, Albornoz F, Griffin JP, Crandall DL, Elokdah H, Fogo AB, Vaughan DE and Brown NJ. Pharmacological inhibition and genetic deficiency of plasminogen activator inhibitor-1 attenuates angiotensin II/salt-induced aortic remodeling. Arterioscler Thromb Vasc Biol. 2005;25:365–71. [DOI] [PubMed] [Google Scholar]
- 23.Kaiko GE, Chen F, Lai C-W, Chiang I-L, Perrigoue J, Stojmirović A, Li K, Muegge BD, Jain U, VanDussen KL, Goggins BJ, Keely S, Weaver J, Foster PS, Lawrence DA, Liu T-C and Stappenbeck TS. PAI-1 augments mucosal damage in colitis. Sci Transl Med. 2019;11:eaat0852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ji Y, Weng Z, Fish P, Goyal N, Luo M, Myears SP, Strawn TL, Chandrasekar B, Wu J and Fay WP. Pharmacological Targeting of Plasminogen Activator Inhibitor-1 Decreases Vascular Smooth Muscle Cell Migration and Neointima Formation. Arterioscler Thromb Vasc Biol. 2016;36:2167–2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yahata T, Ibrahim AA, Muguruma Y, Eren M, Shaffer AM, Watanabe N, Kaneko S, Nakabayashi T, Dan T, Hirayama N, Vaughan DE, Miyata T and Ando K. TGF-β-induced intracellular PAI-1 is responsible for retaining hematopoietic stem cells in the niche. Blood. 2017;130:2283–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sjöland H, Eitzman DT, Gordon D, Westrick R, Nabel EG and Ginsburg D. Atherosclerosis Progression in LDL Receptor–Deficient and Apolipoprotein E–Deficient Mice Is Independent of Genetic Alterations in Plasminogen Activator Inhibitor-1. Arterioscler Thromb Vasc Biol. 2000;20:846–852. [DOI] [PubMed] [Google Scholar]
- 27.Eitzman DT, Westrick RJ, Xu Z, Tyson J and Ginsburg D. Plasminogen activator inhibitor-1 deficiency protects against atherosclerosis progression in the mouse carotid artery. Blood. 2000;96:4212–5. [PubMed] [Google Scholar]
- 28.Luttun A, Lupu F, Storkebaum E, Hoylaerts MF, Moons L, Crawley J, Bono F, Poole AR, Tipping P, Herbert JM, Collen D and Carmeliet P. Lack of plasminogen activator inhibitor-1 promotes growth and abnormal matrix remodeling of advanced atherosclerotic plaques in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol. 2002;22:499–505. [DOI] [PubMed] [Google Scholar]
- 29.Khan SS, Shah SJ, Klyachko E, Baldridge AS, Eren M, Place AT, Aviv A, Puterman E, Lloyd-Jones DM, Heiman M, Miyata T, Gupta S, Shapiro AD and Vaughan DE. A null mutation in SERPINE1 protects against biological aging in humans. Science Advances. 2017;3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Neuhofer A, Wernly B, Leitner L, Sarabi A, Sommer N, Staffler G, Zeyda M and Stulnig T. An accelerated mouse model for atherosclerosis and adipose tissue inflammation. Cardiovasc Diabetol. 2014;13:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kohlgruber A and Lynch L. Adipose tissue inflammation in the pathogenesis of type 2 diabetes. Current diabetes reports. 2015;15:92. [DOI] [PubMed] [Google Scholar]
- 32.Richardson VR, Smith KA and Carter AM. Adipose tissue inflammation: Feeding the development of type 2 diabetes mellitus. Immunobiology. 2013;218:1497–1504. [DOI] [PubMed] [Google Scholar]
- 33.Childs BG, Baker DJ, Wijshake T, Conover CA, Campisi J and van Deursen JM. Senescent intimal foam cells are deleterious at all stages of atherosclerosis. Science. 2016;354:472–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang J, Uryga AK, Reinhold J, Figg N, Baker L, Finigan A, Gray K, Kumar S, Clarke M and Bennett M. Vascular Smooth Muscle Cell Senescence Promotes Atherosclerosis and Features of Plaque Vulnerability. Circulation. 2015;132:1909–1919. [DOI] [PubMed] [Google Scholar]
- 35.Elokdah H, Abou-Gharbia M, Hennan JK, McFarlane G, Mugford CP, Krishnamurthy G and Crandall DL. Tiplaxtinin, a novel, orally efficacious inhibitor of plasminogen activator inhibitor-1: design, synthesis, and preclinical characterization. J Med Chem. 2004;47:3491–4. [DOI] [PubMed] [Google Scholar]
- 36.Migliorini M, Li S-H, Zhou A, Emal CD, Lawrence DA and Strickland DK. High affinity binding of plasminogen-activator inhibitor 1-complexes to LDL receptor-related protein 1 requires lysines 80, 88 and 207. J Biol Chem. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Muratoglu SC, Mikhailenko I, Newton C, Migliorini M and Strickland DK. Low Density Lipoprotein Receptor-related Protein 1 (LRP1) Forms a Signaling Complex with Platelet-derived Growth Factor Receptor-β in Endosomes and Regulates Activation of the MAPK Pathway. J Biol Chem. 2010;285:14308–14317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Crandall DL, Quinet EM, El Ayachi S, Hreha AL, Leik C, Savio DA, Juhan-Vague I and Alessi MC. Modulation of Adipose Tissue Development by Pharmacological Inhibition of PAI-1. Arterioscler Thromb Vasc Biol. 2006;26:2209–2215. [DOI] [PubMed] [Google Scholar]
- 39.Courey AJ, Horowitz JC, Kim KK, Koh TJ, Novak ML, Subbotina N, Warnock M, Xue B, Cunningham AK, Lin Y, Goldklang MP, Simon RH, Lawrence DA and Sisson TH. The vitronectin-binding function of PAI-1 exacerbates lung fibrosis in mice. Blood. 2011;118:2313–2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ji Y, Fish PM, Strawn TL, Lohman AW, Wu J, Szalai AJ and Fay WP. C-reactive protein induces expression of tissue factor and plasminogen activator inhibitor-1 and promotes fibrin accumulation in vein grafts. J Thromb Haemost. 2014;12:1667–1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Daugherty A, Tall AR, Daemen M, Falk E, Fisher EA, Garcia-Cardena G, Lusis AJ, Owens AP 3rd, Rosenfeld ME and Virmani R. Recommendation on Design, Execution, and Reporting of Animal Atherosclerosis Studies: A Scientific Statement From the American Heart Association. Arterioscler Thromb Vasc Biol. 2017;37:e131–e157. [DOI] [PubMed] [Google Scholar]
- 42.Febbraio M, Podrez EA, Smith JD, Hajjar DP, Hazen SL, Hoff HF, Sharma K and Silverstein RL. Targeted disruption of the class B scavenger receptor CD36 protects against atherosclerotic lesion development in mice. J Clin Invest. 2000;105:1049–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Beattie JH, Duthie SJ, Kwun IS, Ha TY and Gordon MJ. Rapid Quantification of Aortic Lesions in ApoE–/– Mice. J Vasc Res. 2009;46:347–352. [DOI] [PubMed] [Google Scholar]
- 44.Ji Y, Strawn TL, Grunz EA, Stevenson MJ, Lohman AW, Lawrence DA and Fay WP. Multifaceted Role of Plasminogen Activator Inhibitor-1 in Regulating Early Remodeling of Vein Bypass Grafts. Arterioscler Thromb Vasc Biol. 2011;31:1781–1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kiernan JA. Histological and Histochemical Methods - Theory and Practice. 5th ed Banbury, UK: Scion Publishing Ltd; 2015. [Google Scholar]
- 46.Forouzandeh F, Salazar G, Patrushev N, Xiong S, Hilenski L, Fei B and Alexander RW. Metformin Beyond Diabetes: Pleiotropic Benefits of Metformin in Attenuation of Atherosclerosis. J Amer Heart Assoc. 2014;3:e001202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Benjamini Y and Hochberg Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J Royal Stat Soc Series B (Methodological). 1995;57:289–300. [Google Scholar]
- 48.Kopec AK, Abrahams SR, Thornton S, Palumbo JS, Mullins ES, Divanovic S, Weiler H, Owens AP III, Mackman N, Goss A, van Ryn J, Luyendyk JP and Flick MJ. Thrombin promotes diet-induced obesity through fibrin-driven inflammation. J Clin Invest. 2017;127:3152–3166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Masson O, Chavey C, Dray C, Meulle A, Daviaud D, Quilliot D, Muller C, Valet P and Liaudet-Coopman E. LRP1 receptor controls adipogenesis and is up-regulated in human and mouse obese adipose tissue. PLoS One. 2009;4:e7422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kaur J A comprehensive review on metabolic syndrome. Cardiol Res Pract. 2014;2014:943162. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 51.Kodaman N, Aldrich MC, Sobota R, Asselbergs FW, Brown NJ, Moore JH and Williams SM. Plasminogen Activator Inhibitor‐1 and Diagnosis of the Metabolic Syndrome in a West African Population. J Amer Heart Assoc. 2016;5:e003867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Aguilar M, Bhuket T, Torres S, Liu B and Wong RJ. Prevalence of the metabolic syndrome in the United States, 2003–2012. JAMA. 2015;313:1973–1974. [DOI] [PubMed] [Google Scholar]
- 53.Bitzur R, Cohen H, Kamari Y and Harats D. Intolerance to statins: mechanisms and management. Diabetes Care. 2013;36 Suppl 2:S325–S330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ghosh AK, Rai R, Park KE, Eren M, Miyata T, Wilsbacher LD and Vaughan DE. A small molecule inhibitor of PAI-1 protects against doxorubicin-induced cellular senescence. Oncotarget 2016;7:72443–72457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kortlever RM, Higgins PJ and Bernards R. Plasminogen activator inhibitor-1 is a critical downstream target of p53 in the induction of replicative senescence. Nat Cell Biol. 2006;8:877–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Elzi DJ, Lai Y, Song M, Hakala K, Weintraub ST and Shiio Y. Plasminogen activator inhibitor 1 - insulin-like growth factor binding protein 3 cascade regulates stress-induced senescence. Proc Natl Acad Sci USA. 2012;109:12052–12057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Eren M, Boe AE, Klyachko EA and Vaughan DE. Role of Plasminogen Activator Inhibitor-1 in Senescence and Aging. Semin Thromb Hemost. 2014;40:645–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Vaughan DE, Rai R, Khan SS, Eren M and Ghosh AK. Plasminogen Activator Inhibitor-1 Is a Marker and a Mediator of Senescence. Arterioscler Thromb Vasc Biol. 2017;37:1446–1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Huang SS, Ling TY, Tseng WF, Huang YH, Tang FM, Leal SM and Huang JS. Cellular growth inhibition by IGFBP-3 and TGF-β1 requires LRP-1. FASEB Journal. 2003;17:2068–2081. [DOI] [PubMed] [Google Scholar]
- 60.Kuilman T and Peeper DS. Senescence-messaging secretome: SMS-ing cellular stress. Nature Reviews Cancer. 2009;9:81. [DOI] [PubMed] [Google Scholar]
- 61.Degryse B, Neels JG, Czekay RP, Aertgeerts K, Kamikubo Y and Loskutoff DJ. The low density lipoprotein receptor-related protein is a motogenic receptor for plasminogen activator inhibitor-1. J Biol Chem. 2004;279:22595–604. [DOI] [PubMed] [Google Scholar]
- 62.Booyse FM, Scheinbuks J, Radek J, Osikowicz G, Feder S and Quarfoot AJ. Immunological identification and comparison of plasminogen activator forms in cultured normal human endothelial cells and smooth muscle cells. Thromb Res. 1981;24:495–504. [DOI] [PubMed] [Google Scholar]
- 63.Clowes AW, Clowes MM, Au YP, Reidy MA and Belin D. Smooth muscle cells express urokinase during mitogenesis and tissue-type plasminogen activator during migration in injured rat carotid artery. Circ Res. 1990;67:61–67. [DOI] [PubMed] [Google Scholar]
- 64.Stefansson S, Muhammad S, Cheng X-F, Battey FD, Strickland DK and Lawrence DA. Plasminogen Activator Inhibitor-1 Contains a Cryptic High Affinity Binding Site for the Low Density Lipoprotein Receptor-related Protein. J Biol Chem. 1998;273:6358–6366. [DOI] [PubMed] [Google Scholar]
- 65.Weber C, Zernecke A and Libby P. The multifaceted contributions of leukocyte subsets to atherosclerosis: lessons from mouse models. Nat Rev Immunol. 2008;8:802–815. [DOI] [PubMed] [Google Scholar]
- 66.Jansson PA. Endothelial dysfunction in insulin resistance and type 2 diabetes. J Intern Med. 2007;262:173–183. [DOI] [PubMed] [Google Scholar]
- 67.An X, Yu D, Zhang R, Zhu J, Du R, Shi Y and Xiong X. Insulin resistance predicts progression of de novo atherosclerotic plaques in patients with coronary heart disease: a one-year follow-up study. Cardiovasc Diabetol. 2012;11:71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Konaniah ES, Kuhel DG, Basford JE, Weintraub NL and Hui DY. Deficiency of LRP1 in Mature Adipocytes Promotes Diet-Induced Inflammation and Atherosclerosis. Arterioscler Thromb Vasc Biol. 2017;37:1046–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Correia MLG and Haynes WG. A Role for Plasminogen Activator Inhibitor-1 in Obesity: From Pie to PAI? Arterioscler Thromb Vasc Biol. 2006;26:2183–2185. [DOI] [PubMed] [Google Scholar]
- 70.Williams T Metabolic Syndrome: Nonalcoholic Fatty Liver Disease. FP Essent. 2015;435:24–9. [PubMed] [Google Scholar]
- 71.Piao L, Jung I, Huh JY, Miyata T and Ha H. A novel plasminogen activator inhibitor‐1 inhibitor, TM5441, protects against high‐fat diet‐induced obesity and adipocyte injury in mice. Br J Pharmacol. 2016;173:2622–2632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Henkel AS, Khan SS, Olivares S, Miyata T and Vaughan DE. Inhibition of Plasminogen Activator Inhibitor 1 Attenuates Hepatic Steatosis but Does Not Prevent Progressive Nonalcoholic Steatohepatitis in Mice. Hepatology Communications. 2018;2:1479–1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wang C-Y, Liu P-Y and Liao JK. Pleiotropic effects of statin therapy: molecular mechanisms and clinical results. Trends Mol Med. 2008;14:37–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kuhnast S, van der Hoorn J, Pieterman EJ, van den Hoek AM, Sasiela WJ, Gusarova V, Peyman A, Schafer HL, Schwahn U, Jukema JW and Princen HMG. Alirocumab inhibits atherosclerosis, improves the plaque morphology, and enhances the effects of a statin. J Lipid Res. 2014;55:2103–2112. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.