

Abstract
Therapeutic enzymes used for genetic disorders or metabolic diseases oftentimes suffer from suboptimal pharmacokinetics and stability. Nanodelivery systems have shown considerable promise for improving the performance of enzyme therapies. Here, we develop a cell membrane-camouflaged metal–organic framework (MOF) system with enhanced biocompatibility and functionality. The MOF core can efficiently encapsulate enzymes while maintaining their bioactivity. After the introduction of natural cell membrane coatings, the resulting nanoformulations can be safely administered in vivo. The surface receptors on the membrane can also provide additional functionalities that synergize with the encapsulated enzyme to target disease pathology from multiple dimensions. Employing uricase as a model enzyme, we demonstrate the utility of this approach in multiple animal disease models. The results support the use of cell membrane-coated MOFs for enzyme delivery, and this strategy could be leveraged to improve the usefulness of enzyme-based therapies for managing a wide range of important human health conditions.
Keywords: cell membrane, metal–organic framework, enzyme therapy, hyperuricemia, gout
Graphical Abstract
Enzymes are biocatalysts that help to carry out reactions essential to the normal functioning of all living organisms. The lack of critical enzymes or the loss of their functions could lead to the deleterious accumulation of biomolecule substrates, disrupting metabolic activities with potentially life-threatening consequences.1, 2 Defective enzyme production commonly occurs as a result of inherited genetic disorders. Examples include Gaucher disease and Fabry disease,3, 4 both of which are lysosomal storage disorders caused by mutations in important housekeeping hydrolase genes.5, 6 The evolutionary loss of certain genes is another cause of enzyme deficiency that can lead to some disease states.7 For instance, uricase, the enzyme responsible for uric acid conversion into allantoin, is not encoded in the human genome, which can result in gouty arthritis when urate crystals are deposited in the joints.8, 9 Enzyme replacement therapy has become the current standard of care for patients with enzyme deficiency, where exogenous proteins are intravenously infused into the bloodstream.10, 11 Despite their application in the clinic, enzymes in free form usually suffer from protease susceptibility and short-acting pharmacokinetics, both of which can severely compromise bioavailability.12, 13 In order to attain a therapeutic benefit, frequent administration is oftentimes mandatory for these therapies,14, 15 leading to high costs and negatively impacting patient compliance.16
The use of nanoparticulate systems is an emerging strategy to address some of the shortcomings associated with the administration of free enzymes.17, 18 Ideally, functional enzymes can be encased into a nanoscale network that allows their catalytic activities to be maintained, enabling substrate molecules to access the enzyme while preventing unwanted proteolytic degradation by the surrounding environment.19, 20 Among the various nanomaterials, metal–organic frameworks (MOFs) have shown great promise for enzyme delivery applications.21, 22 MOFs can be fabricated with porous structures that facilitate high-yield enzyme loading and allow size-selective exposure to the targeted substrate.23, 24 Despite these favorable characteristics, MOF-based platforms exhibit biocompatibility issues and run the risk of inducing immune reactions,25, 26 which would impede their translation into the clinic.
The use of cell membrane coatings to camouflage synthetic nanomaterials is an effective method for nanoparticle functionalization.27 The membrane-coated nanoparticles fabricated using this platform technology exhibit cell-mimicking properties that enable them to excel at in vivo applications.28 For example, red blood cell (RBC) membrane coatings can greatly prolong circulation within the bloodstream,29, 30 whereas platelet membrane coatings enable targeted delivery to bacteria, cancer, and damaged vasculature.31, 32 It was also demonstrated that nanoparticles functionalized with white blood cell membrane can be used as nanoscale decoys to absorb and neutralize inflammatory cytokines, with potential applications for autoimmune disorders and sepsis treatment.33, 34 Overall, cell membrane coatings can be derived from any type of cell, enabling researchers a wide range of options for adding functionality and creating synergies with nanoparticle-based therapeutics.27, 28, 35 Notably, it was recently demonstrated that this approach could be applied to MOF nanoparticles loaded with siRNA for anticancer applications.36
Here, we report on the fabrication of cell membrane-coated MOF nanoparticles for effective enzyme delivery (Figure 1). Taking uricase as the model enzyme, we successfully loaded the enzyme into a zeolitic imidazolate framework-8 MOF nanoparticle by a facile formulation process with precise input control.37 The uricase-loaded MOF (MOF-uricase) nanoparticles were then coated with the membrane from either RBCs or macrophages (MΦs), each of which provided application-specific benefits. In particular, RBC membrane-coated MOF-uricase (RBC-MOF-uricase) were systemically administered to catalyze the efficient degradation of serum uric acid in hyperuricemic mice. MΦ membrane-coated MOF-uricase (MΦ-MOF-uricase) were locally administered into the joints of mice with gout, where the cytokine-neutralizing property of the MΦ membrane synergized with the uricase the alleviate disease symptoms. By changing the enzyme payload and membrane coating, we envision that the reported hybrid delivery platform could be used to improve therapeutic outcomes for a wide range of conditions requiring enzyme therapy.
Figure 1.
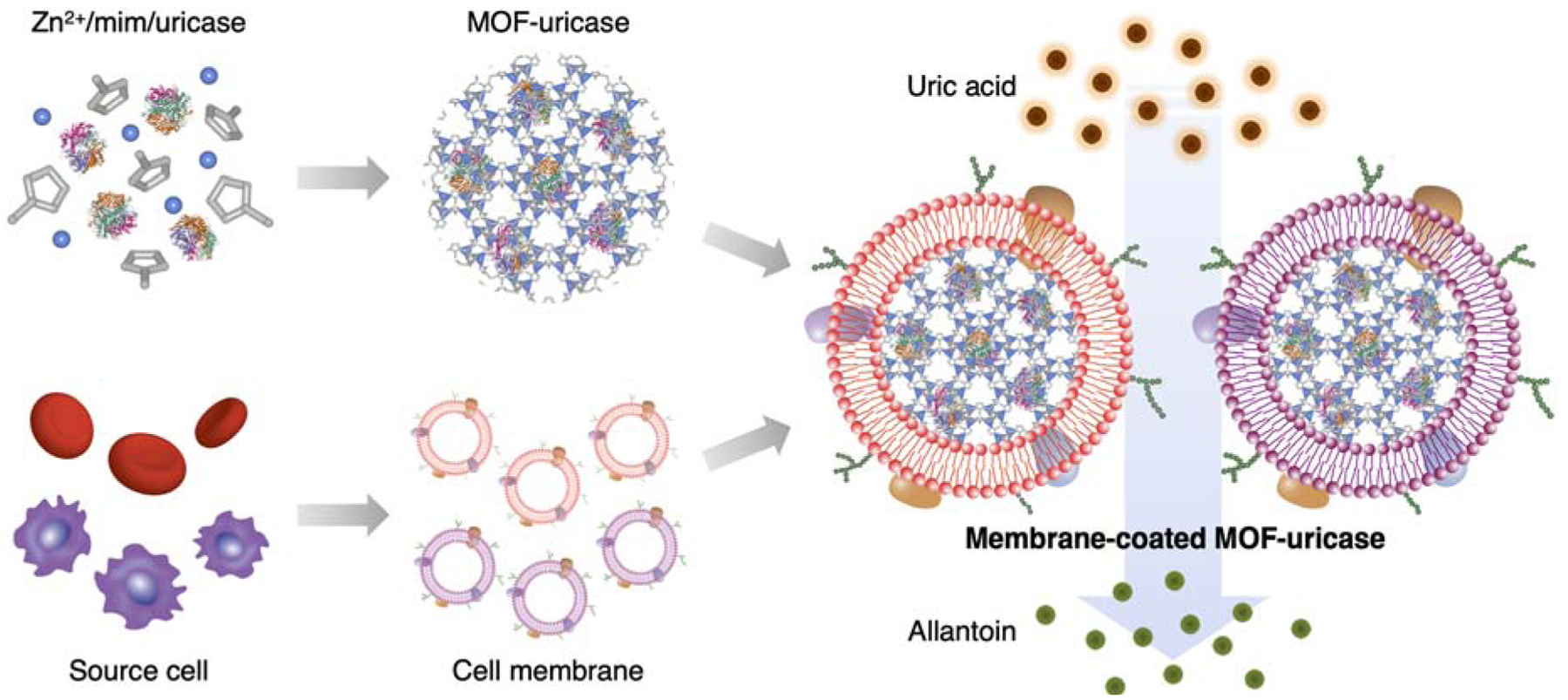
Cell membrane-coated uricase-loaded MOF nanoparticles for the enzymatic degradation of uric acid. To fabricate the formulation, MOF-uricase cores are generated by mixing the enzyme payload with Zn2+ and 2-methylimidazole (mim), followed by coating with natural cell membrane derived from source cells such as RBCs or MΦs. The resulting membrane-coated MOF-uricase nanoparticles effectively convert uric acid into allantoin, which can help to manage conditions such hyperuricemia and gout.
In the study, recombinant uricase was encapsulated within the MOF nanoparticles using a one-pot synthesis approach.36 The enzyme was first premixed with 2-methylimidazole and added to a zinc ion-containing solution to start a process of self-assembly, which yielded bare MOF-uricase nanoparticles after several hours. To coat MOF-uricase with cell membrane, the freshly formed nanoparticles were physically extruded together with purified mouse RBC membrane through porous polycarbonate membranes. The final RBC-MOF-uricase nanoparticles were isolated by centrifugation. To quantify loading efficiency, the uricase was fluorescently labeled, and then RBC-MOF-uricase were fabricated using different input amounts of the enzyme (Figure 2a). Across the inputs that were evaluated, the encapsulation efficiency was consistently around 90% up to 0.25 U of uricase. It should be noted that formulations fabricated with higher uricase inputs were unstable. The size of the RBC-MOF-uricase was highly dependent on uricase input (Figure 2b). RBC membrane-coated MOF (RBC-MOF) nanoparticles without any uricase were approximately 130 nm in size, whereas the size of RBC-MOF-uricase grew to near 270 nm at the highest input of 0.25 U. To prevent premature splenic clearance in vivo after intravenous administration,38 sub-200 nm RBC-MOF-uricase fabricated using a 0.1 U uricase input were used for further study.
Figure 2.
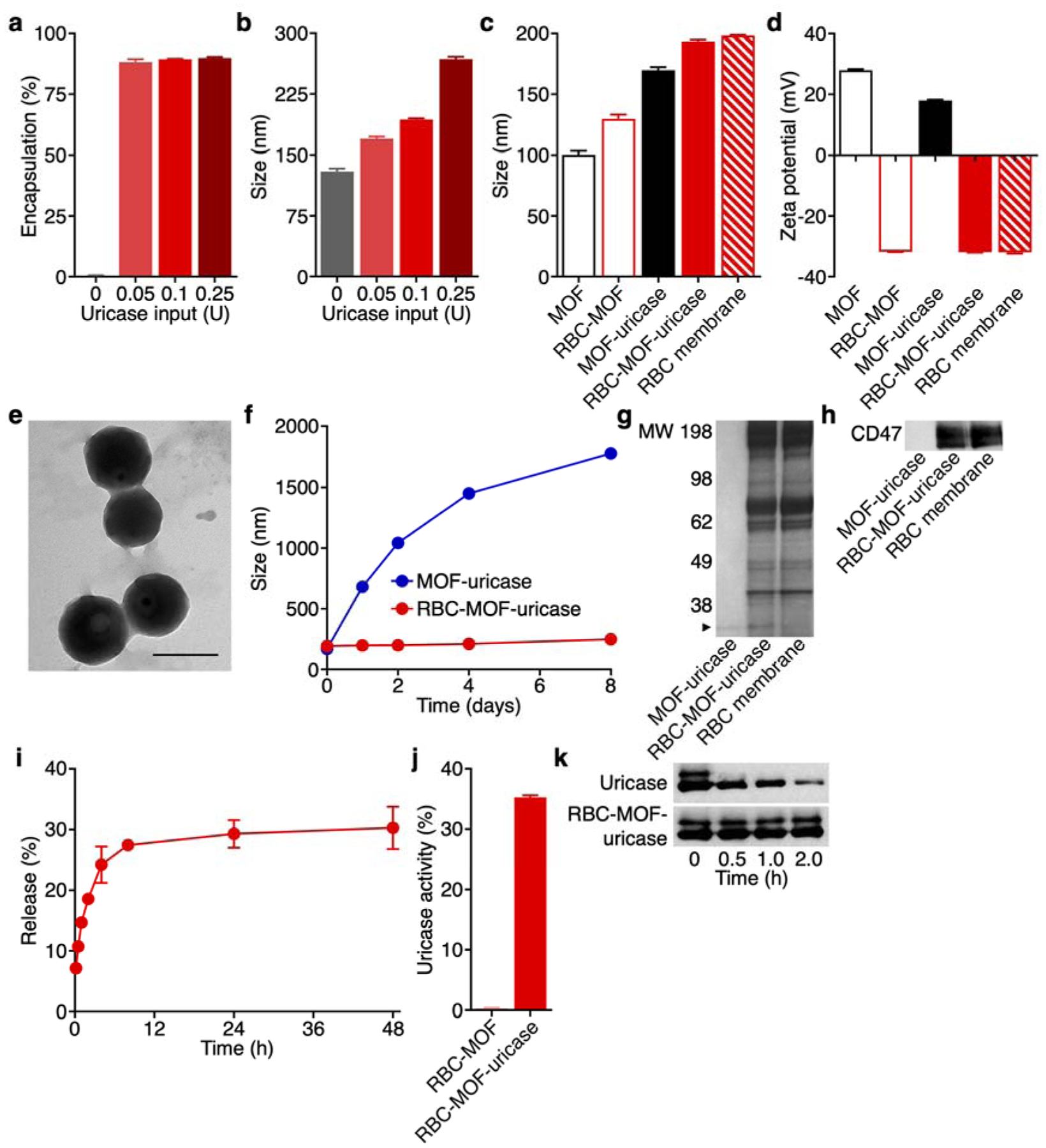
Synthesis and characterization of RBC-MOF-uricase. (a) Encapsulation efficiency of uricase in RBC-MOF-uricase at different uricase inputs (n = 3, mean + SD). (b) Diameter of RBC-MOF-uricase at different uricase inputs (n = 3, mean + SD). (c) Diameter of pristine MOF, RBC-MOF, MOF-uricase, RBC-MOF-uricase, and RBC membrane vesicles after fabrications (n = 3, mean + SD). (d) Zeta potential of pristine MOF, RBC-MOF, MOF-uricase, RBC-MOF-uricase, and RBC membrane vesicles after fabrication (n = 3, mean + SD). (e) Transmission electron microscopy image of RBC-MOF-uricase stained with uranyl acetate. Scale bar: 200 nm. (f) Stability of RBC-MOF-uricase over the course of 8 days (n = 3, mean ± SD). (g) Protein profiles of MOF-uricase, RBC-MOF-uricase, and RBC membrane. MW, molecular weight (kDa); arrow indicates uricase band. (h) Western blot for RBC surface marker CD47 (50 kDa) on MOF-uricase, RBC-MOF-uricase, and RBC membrane. (i) Uricase release from RBC-MOF-uricase at pH 7.4 in PBS over time (n = 3, mean ± SD). (j) In vitro uric acid conversion by RBC-MOF or RBC-MOF-uricase normalized to free uricase activity (n = 3, mean + SD). (k) Degradation of uricase (35 kDa), either in free form or in RBC-MOF-uricase, when exposed to trypsin for increasing amounts of time.
Compared with bare MOF, the size of MOF-uricase increased by approximately 70 nm, and coating with the RBC membrane further increased the size of the nanoparticles by about 25 nm (Figure 2c). The surface zeta potential of the MOF dropped from 30 mV to 18 mV after uricase encapsulation, while the final RBC-MOF-uricase formulation displayed a negative surface potential of −30 mV (Figure 2d). The near identical surfaces charges between the RBC-MOF-uricase and the purified RBC membrane suggested good coating coverage. To visualize the physical structure of RBC-MOF-uricase, the nanoparticles were negatively stained with uranyl acetate and examined under transmission electron microscopy (Figure 2e). The imaging confirmed a characteristic core–shell structure, which is consistent with similar cell membrane-coated nanoparticle platforms, including those employing MOF cores.36, 39, 40
The stability of the RBC-MOF-uricase formulation in physiological condition was evaluated by measuring size in phosphate buffered saline (PBS) over the course of 8 days (Figure 2f). Whereas the uncoated MOF-uricase quickly grew in size to over 1 μm, the RBC membrane-coated nanoparticles exhibited a minimal size increase. This highlighted the role of the cell membrane in enhancing the colloidal stability of MOF-based systems. Next, we sought to characterize the protein content of RBC-MOF-uricase as an additional means on confirming membrane coating. The overall protein profile of the nanoformulation was near identical to that of purified RBC membrane, indicating successful translocation of the membrane proteins (Figure 2g). The RBC-MOF-uricase sample had an additional band at approximately 35 kDa, indicating the presence of uricase; this band was present on the protein profile for the uncoated MOF-uricase sample, but not for the purified RBC membrane. Western blotting analysis was used to confirm the presence of CD47 (Figure 2h), a key membrane protein found on RBCs that helps to reduce immune clearance by acting as a ‘marker-of-self’.41
The release of the uricase payload from the RBC-MOF-uricase formulation was profiled over time in PBS (Figure 2i). A quick burst was observed in the first 4 h, after which the release plateaued near 30%. Over the course of 2 days, more than 70% of the loaded uricase was retained, suggesting that the nanocomplex was highly stable and could be used to achieve prolonged delivery of the enzyme payload. The catalytic activity of RBC-MOF-uricase against uric acid as a substrate was evaluated in vitro (Figure 2j). From the results, it was observed that unloaded RBC-MOF nanoparticles had no catalytic activity, whereas the RBC-MOF-uricase retained approximately 35% of the activity of the inputted uricase. The decrease in catalytic activity may be partly explained by the fact that not all of the uricase is immediately available to participate in reactions when loaded inside the MOF matrix. To highlight the benefit of encapsulation into RBC-MOF for protecting the enzyme payload, both free uricase and RBC-MOF-uricase were incubated with trypsin (Figure 2k). From western blot analysis, it was observed that free uricase was quickly degraded, while the integrity of the uricase inside the nanoformulation was maintained over the course of 2 h. This confirmed that the MOF matrix and membrane coating could provide a barrier to prevent unwanted contact of the encapsulated enzyme with degradative proteases, thus better preserving enzymatic activity over time.
After completing the in vitro characterizations, we next sought to characterize the performance of the RBC-MOF-uricase formulation in vivo. First, the biodistribution was studied 24 h after administration of RBC-MOF-uricase fluorescently labeled with a far-red dye (Figure 3a). The majority were found in the liver, which is the main organ mainly responsible for nanoparticle clearance.30 Notably, approximately 15% of the total fluorescent signal was found in the blood at 24 h, attesting to the long-circulating properties provided by the RBC membrane coating. To assess the in vivo activity of the uricase payload, the nanoformulation was used to treat a murine model of hyperuricemia, where elevated uric acid levels are observed in the blood (Figure 3b). Without any uricase, the serum concentration of uric acid slowly increased over time, whereas intravenous administration of RBC-MOF-uricase resulted in a rapid reduction back to basal levels. In comparison, administration of free uricase only resulted in a transient drop in uric acid levels, which reelevated after 2 h. The modest efficacy observed in mice receiving free uricase could likely be attributed to poor bioavailability, highlighting the need for nanodelivery systems capable of extending blood residence while protecting the enzyme from degradation. To evaluate the biocompatibility of the RBC-MOF-uricase formulation, major blood cell populations, including white blood cells, RBCs, and platelets, were enumerated 24 h after nanoparticle administration (Figure 3c). No significant difference was observed compared with samples obtained from mice treated with PBS. At the same timepoint, the major organs, including the heart, liver, spleen, lungs, and kidneys, were histologically sectioned and stained with hematoxylin and eosin (H&E) for analysis (Figure 3d). The overall structure, integrity, and immune infiltrate in all of these tissues were near identical to those from mice administered with PBS, demonstrating no signs of acute toxicity and further supporting the safety of RBC-MOF-uricase.
Figure 3.
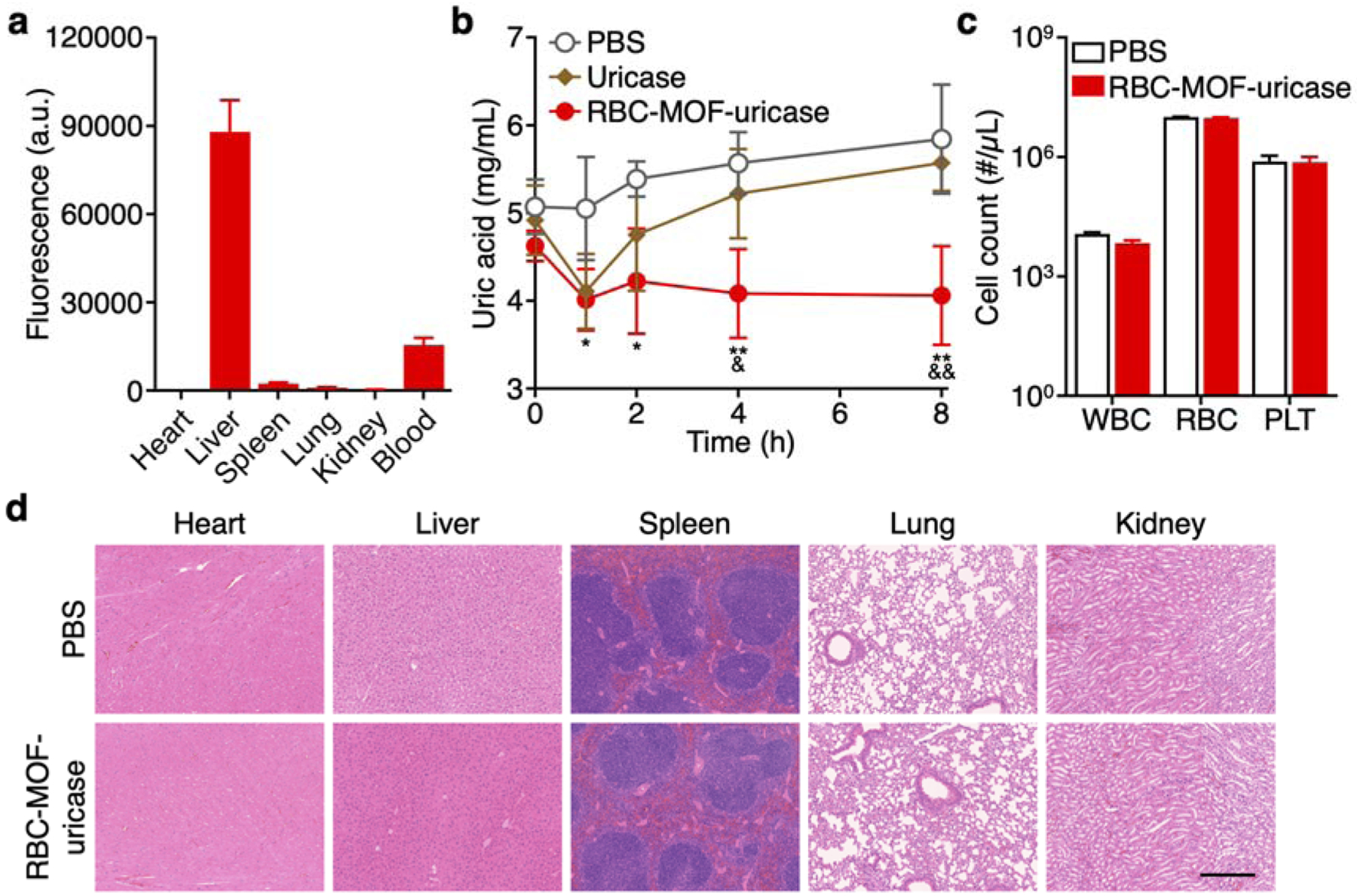
In vivo hyperuricemia management and safety. (a) Biodistribution of dye-labeled RBC-MOF-uricase in major organs, including the heart, liver, spleen, lungs, kidneys, and blood, 24 h after intravenous administration (n = 3, mean + SD). (b) Serum uric acid levels over time of hyperuricemic mice after intravenous treatment with PBS, free uricase, or RBC-MOF-uricase (n = 4, mean ± SD). *p < 0.05, **p < 0.01 (PBS compared with RBC-MOF-uricase); &p < 0.05, &&p < 0.01 (free uricase compared with RBC-MOF-uricase); one-way ANOVA. (c) Counts of various blood cells 24 h after intravenous administration of PBS or RBC-MOF-uricase (n = 3, geometric mean + SD). WBC: white blood cells, RBC: red blood cells, PLT: platelets. (d) H&E-stained histological sections from major organs 24 h after intravenous administration of PBS or RBC-MOF-uricase into healthy mice. Scale bar: 250 μm.
A major advantage of using cell membrane coatings to functionalize synthetic nanoparticle cores is the ability to custom-tailor the final formulation for different applications by changing the membrane source.27 To this end, we developed a second MOF-uricase formulation using MΦ membrane, which excels at cytokine neutralization,34 aiming to address gout. The condition is characterized by the local deposit of uric acid crystals, which can lead to excessive joint inflammation mediated by a number of proinflammatory cytokines.9 MΦ-MOF-uricase nanoparticles were fabricated similarly to the RBC-based formulation. Because nanoparticles administered locally are not subjected to the same strict size requirements as those for systemic injection, we elected to employ MOF-uricase cores inputted with 0.25 U of uricase to maximize loading, which led to a formulation approximately 270 nm in size (Figure 4a). The zeta potential of MΦ-MOF-uricase was also negative, matching closely with that of purified MΦ membrane (Figure 4b). Whereas unloaded MΦ membrane-coated MOF (MΦ-MOF) nanoparticles did not exhibit any uricase activity, MΦ-MOF-uricase retained 38% of the initial activity of the inputted uricase (Figure 4c).
Figure 4.
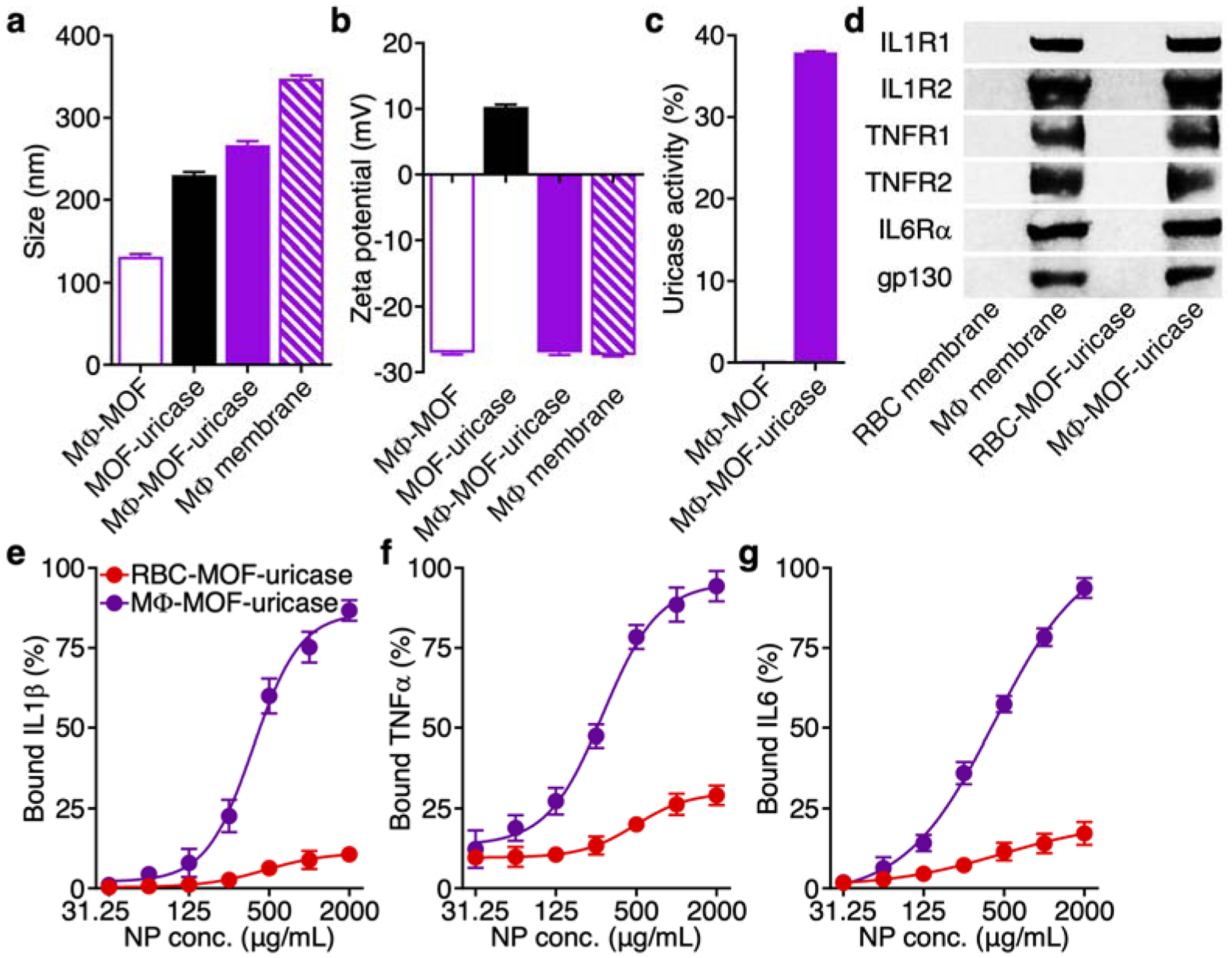
Synthesis and characterization of MΦ-MOF-uricase. (a) Diameter of MΦ-MOF, MOF-uricase, MΦ-MOF-uricase, and MΦ membrane vesicles after fabrication (n = 3, mean + SD). (b) Zeta potential of MΦ-MOF, MOF-uricase, MΦ-MOF-uricase, and MΦ membrane vesicles after fabrication (n = 3, mean + SD). (c) In vitro uric acid conversion by MΦ-MOF or MΦ-MOF-uricase normalized to free uricase activity (n = 3, mean + SD). (d) Western blot for cytokine receptors, including IL1R1 (80 kDa), IL1R2 (45 kDa), TNFR1 (55 kDa), TNFR2 (75 kDa), IL6Rα (80 kDa), and gp130 (130 kDa), on RBC membrane, MΦ membrane, RBC-MOF-uricase, and MΦ-MOF-uricase. (e-g) In vitro neutralization of cytokines, including IL1β (e), TNFα (f), and IL6 (g), by RBC-MOF-uricase and MΦ-MOF-uricase at various nanoparticle concentrations (n = 3, mean ± SD; four-parameter logistic regression).
Proinflammatory cytokines such as interleukin-1β (IL1β), tumor necrosis factor-α (TNFα), and IL6 have been confirmed to play prominent roles in the progression of gout,42 and MΦ membrane is known to possess their cognate receptors.34 Accordingly, western blot analysis was used to verify the presence of IL1 receptor type I (IL1R1), IL1R2, TNF receptor-1 (TNFR1), TNFR2, IL6 receptor α-chain (IL6Rα), and glycoprotein 130 (gp130) (Figure 4d). As expected, none of these receptors were found on RBC membrane or RBC-MOF-uricase, whereas every marker was present on both MΦ membrane and MΦ-MOF-uricase. Next, we sought to test if the presence of these receptors on the MΦ-MOF-uricase formulation could be leveraged for the neutralization of the proinflammatory cytokines often implicated in joint inflammation.33 Recombinant mouse IL1β, mouse TNFα, or mouse IL6 at a final concentration of 8 ng/mL was incubated with the nanoparticles at varying concentrations, and the percentage of bound cytokines was quantified (Figure 4e–g). Under these experimental conditions, the data indicated that MΦ-MOF-uricase exhibited half maximal inhibitory concentration values of 380 μg/mL for IL1β, 282 μg/mL for TNFα, and 425 μg/mL for IL6. In contrast, RBC-MOF-uricase did not effectively bind to any of the tested cytokines.
After confirming its activity in vitro, the therapeutic efficacy of the MΦ-MOF-uricase formulation was evaluated in vivo using a murine gout model induced by the intraarticular injection of monosodium urate crystals into the ankle joints. At 24 h after induction, when all mice had developed significant inflammation at the injection site, various treatments were administered by intraarticular injection, and the degree of ankle swelling was monitored over time (Figure 5a). Compared to the PBS control, free uricase and unloaded MΦ-MOF had a modest therapeutic effect, while MΦ-MOF-uricase showed the best efficacy and near completely alleviated the ankle swelling 48 h after treatment. With its cytokine neutralization capabilities, the MΦ membrane-coated formulation outperformed RBC-MOF-uricase, which also demonstrated considerable efficacy, likely due to its ability to enhance retention of the enzyme at the inflammation site. The uric acid remaining in the ankle tissues was quantified 48 h post-treatment, and it was shown that MΦ-MOF-uricase significantly reduced levels of the substrate molecule (Figure 5b). The effect was vastly improved compared with unloaded MΦ-MOF, where 80% of the deposited uric acid still remained within the tissues. Similarly, the proinflammatory cytokines present within the ankle tissues were evaluated at the same timepoint, and MΦ-MOF-uricase treatment was able to bring levels back down to near baseline (Figure 5c–e). This was significantly better than other treatments, including free uricase, unloaded MΦ-MOF, and RBC- MOF-uricase, all of which reduced cytokine levels to varying degrees. Lastly, histological examination revealed that the immune cell infiltrate within the periarticular ankle tissue was markedly reduced in mice treated with MΦ-MOF-uricase, indicating only a slight amount of ongoing inflammation within the region (Figure 5f). Overall, when comparing the effects of MΦ-MOF-uricase with those achieved by MΦ-MOF or RBC-MOF-uricase, the data highlights the therapeutic benefits of combining MΦ membrane-mediated cytokine neutralization with locally delivered enzyme therapy for addressing gout.
Figure 5.
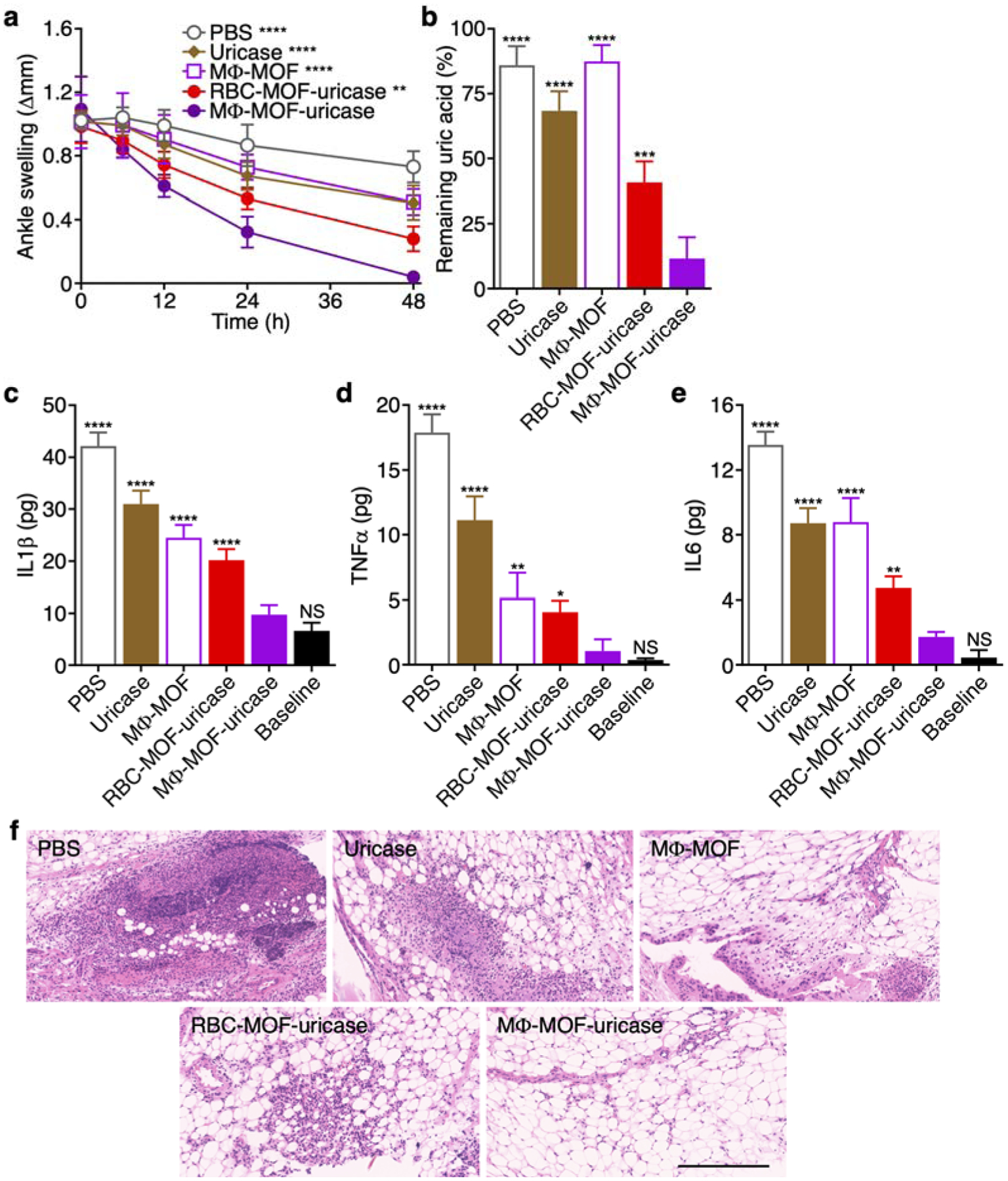
In vivo gout management and safety. (a) Change in ankle joint diameter of mice with gout after intraarticular treatment with PBS, free uricase, MΦ-MOF, RBC-MOF-uricase, or MΦ-MOF-uricase (n = 4, mean ± SD). **p < 0.01, ****p < 0.0001 (compared with MΦ-MOF-uricase at 48 h); one-way ANOVA. (b) Remaining uric acid in the ankle joints of the mice in (a) after 48 h (n = 4, mean + SD). ***p < 0.001, ****p < 0.0001 (compared with MΦ-MOF-uricase); one-way ANOVA. (c-e) Levels of cytokines, including IL1β (c), TNFα (d), and IL6 (e), in the ankle joints of the mice in (a) after 48 h (n = 4, mean + SD). NS: not significant, *p < 0.05, **p < 0.01, ****p < 0.0001 (compared with MΦ-MOF-uricase); one-way ANOVA. (f) H&E-stained histological sections of ankle joints of the mice in (a) after 48 h. Scale bar: 250 μm.
In conclusion, we have successfully fabricated a cell membrane-coated MOF platform for enhancing the activity of enzyme therapies. Using uricase as a model enzyme, it was demonstrated that high encapsulation efficiency of the enzyme could be achieved, and membrane coating further improved colloidal stability in physiological buffer. Importantly, a significant amount of enzymatic activity was retained, and incorporation of uricase within the membrane-coated MOF matrix also protected it from proteolytic degradation. To demonstrate the versatility of this approach, two separate systems were developed using membrane sourced from either RBCs or MΦs. The RBC membrane-coated MOF-uricase formulation was used for the systemic treatment of hyperuricemia, rapidly reducing serum uric levels. On the other hand, the MΦ membrane, with its ability to neutralize a wide range of proinflammatory cytokines, synergized with the uricase to effectively treat localized joint inflammation caused by insoluble uric acid deposits. In the clinic, the administration of uricase, which is not a naturally occurring human enzyme, carries major immunogenicity concerns and runs the risk of triggering anaphylaxis.43, 44 As we demonstrated here, cell membrane-coated nanodelivery systems are biocompatible and can be used to effectively shield enzyme payloads from the surrounding environment; the effect of this on long-term immune responses against the enzyme payload will need to be evaluated in future studies. It is envisioned that this strategy can be applied across a wide range of enzyme payloads, and different membrane coatings could be employed to generate synergies based on their unique biointerfacing characteristics. Membrane material can be derived from autologous sources, facilitating the fabrication of personalized therapies.45 The shape of the MOF core could also be varied to enhance nanoparticle–cell interactions, which would be particularly beneficial for targeted delivery applications.46 Overall further development along these lines could lead to novel formulations with the potential for transforming the clinical landscape of enzyme therapies.
Supplementary Material
ACKNOWLEDGMENTS
Funding: This work is supported by the National Institutes of Health under Award Number R01CA200574. Y.D. is supported by a National Institutes of Health 5T32CA153915 training grant from the National Cancer Institute.
Footnotes
The authors declare no competing financial interest.
REFERENCES
- (1).Matés JM; Pérez-Gómez C; De Castro IN, Antioxidant Enzymes and Human Diseases. Clin. Biochem 1999, 32, 595–603. [DOI] [PubMed] [Google Scholar]
- (2).López-Otín C; Bond JS, Proteases: Multifunctional Enzymes in Life and Disease. J. Biol. Chem 2008, 283, 30433–30437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Barton NW; Brady RO; Dambrosia JM; Di Bisceglie AM; Doppelt SH; Hill SC; Mankin HJ; Murray GJ; Parker RI; Argoff CE; Grewal RP; Yu K-T, Replacement Therapy for Inherited Enzyme Deficiency — Macrophage-Targeted Glucocerebrosidase for Gaucher’s Disease. N. Engl. J. Med 1991, 324, 1464–1470. [DOI] [PubMed] [Google Scholar]
- (4).Desnick RJ; Brady R; Barranger J; Collins AJ; Germain DP; Goldman M; Grabowski G; Packman S; Wilcox WR, Fabry Disease, an Under-Recognized Multisystemic Disorder: Expert Recommendations for Diagnosis, Management, and Enzyme Replacement Therapy. Ann. Intern. Med 2003, 138, 338–346. [DOI] [PubMed] [Google Scholar]
- (5).Futerman AH; van Meer G, The Cell Biology of Lysosomal Storage Disorders. Nat. Rev. Mol. Cell Biol 2004, 5, 554–565. [DOI] [PubMed] [Google Scholar]
- (6).Platt FM; Boland B; van der Spoel AC, Lysosomal Storage Disorders: The Cellular Impact of Lysosomal Dysfunction. J. Cell Biol 2012, 199, 723–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Khersonsky O; Tawfik DS, Enzyme Promiscuity: A Mechanistic and Evolutionary Perspective. Annu. Rev. Biochem 2010, 79, 471–505. [DOI] [PubMed] [Google Scholar]
- (8).Choi HK; Mount DB; Reginato AM, Pathogenesis of Gout. Ann. Intern. Med 2005, 143, 499–516. [DOI] [PubMed] [Google Scholar]
- (9).Dalbeth N; Merriman TR; Stamp LK, Gout. Lancet 2016, 388, 2039–2052. [DOI] [PubMed] [Google Scholar]
- (10).Desnick RJ; Schuchman EH, Enzyme Replacement and Enhancement Therapies: Lessons from Lysosomal Disorders. Nat. Rev. Genet 2002, 3, 954–966. [DOI] [PubMed] [Google Scholar]
- (11).Brady RO, Enzyme Replacement for Lysosomal Diseases. Annu. Rev. Med 2006, 57, 283–296. [DOI] [PubMed] [Google Scholar]
- (12).Desnick RJ; Schuchman EH, Enzyme Replacement Therapy for Lysosomal Diseases: Lessons from 20 Years of Experience and Remaining Challenges. Annu. Rev. Genomics Hum. Genet 2012, 13, 307–335. [DOI] [PubMed] [Google Scholar]
- (13).Fieker A; Philpott J; Armand M, Enzyme Replacement Therapy for Pancreatic Insufficiency: Present and Future. Clin. Exp. Gastroenterol 2011, 4, 55–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Schiffmann R; Kopp JB; Austin III HA; Sabnis S; Moore DF; Weibel T; Balow JE; Brady RO, Enzyme Replacement Therapy in Fabry Disease: A Randomized Controlled Trial. JAMA 2001, 285, 2743–2749. [DOI] [PubMed] [Google Scholar]
- (15).Kakkis ED; Muenzer J; Tiller GE; Waber L; Belmont J; Passage M; Izykowski B; Phillips J; Doroshow R; Walot I; Hoft R; Yu KT; Okazaki S; Lewis D; Lachman R; Thompson JN; Neufeld EF, Enzyme-Replacement Therapy in Mucopolysaccharidosis I. N. Engl. J. Med 2001, 344, 182–188. [DOI] [PubMed] [Google Scholar]
- (16).Rohrbach M; Clarke JTR, Treatment of Lysosomal Storage Disorders. Drugs 2007, 67, 2697–2716. [DOI] [PubMed] [Google Scholar]
- (17).Kim J; Grate JW; Wang P, Nanostructures for Enzyme Stabilization. Chem. Eng. Sci 2006, 61, 1017–1026. [Google Scholar]
- (18).Dean SN; Turner KB; Medintz IL; Walper SA, Targeting and Delivery of Therapeutic Enzymes. Ther. Deliv 2017, 8, 577–595. [DOI] [PubMed] [Google Scholar]
- (19).Zhuang J; Young AP; Tsung C-K, Integration of Biomolecules with Metal–Organic Frameworks. Small 2017, 13, 1700880. [DOI] [PubMed] [Google Scholar]
- (20).Muro S; Garnacho C; Champion JA; Leferovich J; Gajewski C; Schuchman EH; Mitragotri S; Muzykantov VR, Control of Endothelial Targeting and Intracellular Delivery of Therapeutic Enzymes by Modulating the Size and Shape of ICAM-1-Targeted Carriers. Mol. Ther 2008, 16, 1450–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Lian X; Fang Y; Joseph E; Wang Q; Li J; Banerjee S; Lollar C; Wang X; Zhou H-C, Enzyme–MOF (Metal–Organic Framework) Composites. Chem. Soc. Rev 2017, 46, 3386–3401. [DOI] [PubMed] [Google Scholar]
- (22).Mehta J; Bhardwaj N; Bhardwaj SK; Kim K-H; Deep A, Recent Advances in Enzyme Immobilization Techniques: Metal-Organic Frameworks as Novel Substrates. Coord. Chem. Rev 2016, 322, 30–40. [Google Scholar]
- (23).Shieh F-K; Wang S-C; Yen C-I; Wu C-C; Dutta S; Chou L-Y; Morabito JV; Hu P; Hsu M-H; Wu KCW; Tsung C-K, Imparting Functionality to Biocatalysts via Embedding Enzymes into Nanoporous Materials by a De Novo Approach: Size-Selective Sheltering of Catalase in Metal–Organic Framework Microcrystals. J. Am. Chem. Soc 2015, 137, 4276–4279. [DOI] [PubMed] [Google Scholar]
- (24).Liao F-S; Lo W-S; Hsu Y-S; Wu C-C; Wang S-C; Shieh F-K; Morabito JV; Chou L-Y; Wu KCW; Tsung C-K, Shielding against Unfolding by Embedding Enzymes in Metal–Organic Frameworks via a De Novo Approach. J. Am. Chem. Soc 2017, 139, 6530–6533. [DOI] [PubMed] [Google Scholar]
- (25).Giménez-Marqués M; Hidalgo T; Serre C; Horcajada P, Nanostructured Metal–Organic Frameworks and Their Bio-Related Applications. Coord. Chem. Rev 2016, 307, 342–360. [Google Scholar]
- (26).Simon-Yarza T; Mielcarek A; Couvreur P; Serre C, Nanoparticles of Metal-Organic Frameworks: On the Road to In Vivo Efficacy in Biomedicine. Adv. Mater 2018, 30, 1707365. [DOI] [PubMed] [Google Scholar]
- (27).Fang RH; Kroll AV; Gao W; Zhang L, Cell Membrane Coating Nanotechnology. Adv. Mater 2018, 30, 1706759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Kroll AV; Fang RH; Zhang L, Biointerfacing and Applications of Cell Membrane-Coated Nanoparticles. Bioconjug. Chem 2017, 28, 23–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Luk BT; Fang RH; Hu CM; Copp JA; Thamphiwatana S; Dehaini D; Gao W; Zhang K; Li S; Zhang L, Safe and Immunocompatible Nanocarriers Cloaked in RBC Membranes for Drug Delivery to Treat Solid Tumors. Theranostics 2016, 6, 1004–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Hu C-MJ; Zhang L; Aryal S; Cheung C; Fang RH; Zhang L, Erythrocyte Membrane-Camouflaged Polymeric Nanoparticles as a Biomimetic Delivery Platform. Proc. Natl. Acad. Sci. U. S. A 2011, 108, 10980–10985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Hu CM; Fang RH; Wang KC; Luk BT; Thamphiwatana S; Dehaini D; Nguyen P; Angsantikul P; Wen CH; Kroll AV; Carpenter C; Ramesh M; Qu V; Patel SH; Zhu J; Shi W; Hofman FM; Chen TC; Gao W; Zhang K; Chien S; Zhang L, Nanoparticle Biointerfacing by Platelet Membrane Cloaking. Nature 2015, 526, 118–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Hu Q; Sun W; Qian C; Wang C; Bomba HN; Gu Z, Anticancer Platelet-Mimicking Nanovehicles. Adv. Mater 2015, 27, 7043–7050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Zhang Q; Dehaini D; Zhang Y; Zhou J; Chen X; Zhang L; Fang RH; Gao W; Zhang L, Neutrophil Membrane-Coated Nanoparticles Inhibit Synovial Inflammation and Alleviate Joint Damage in Inflammatory Arthritis. Nat. Nanotechnol 2018, 13, 1182–1190. [DOI] [PubMed] [Google Scholar]
- (34).Thamphiwatana S; Angsantikul P; Escajadillo T; Zhang Q; Olson J; Luk BT; Zhang S; Fang RH; Gao W; Nizet V; Zhang L, Macrophage-Like Nanoparticles Concurrently Absorbing Endotoxins and Proinflammatory Cytokines for Sepsis Management. Proc. Natl. Acad. Sci. U. S. A 2017, 114, 11488–11493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Dehaini D; Fang RH; Zhang L, Biomimetic Strategies for Targeted Nanoparticle Delivery. Bioeng. Transl. Med 2016, 1, 30–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Zhuang J; Gong H; Zhou J; Zhang Q; Gao W; Fang RH; Zhang L, Targeted Gene Silencing In Vivo by Platelet Membrane–Coated Metal-Organic Framework Nanoparticles. Sci. Adv 2020, 6, eaaz6108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Liang K; Ricco R; Doherty CM; Styles MJ; Bell S; Kirby N; Mudie S; Haylock D; Hill AJ; Doonan CJ; Falcaro P, Biomimetic Mineralization of Metal-Organic Frameworks as Protective Coatings for Biomacromolecules. Nat. Comm 2015, 6, 7240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Blanco E; Shen H; Ferrari M, Principles of Nanoparticle Design for Overcoming Biological Barriers to Drug Delivery. Nat. Biotechnol 2015, 33, 941–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Zou MZ; Liu WL; Li CX; Zheng DW; Zeng JY; Gao F; Ye JJ; Zhang XZ, A Multifunctional Biomimetic Nanoplatform for Relieving Hypoxia to Enhance Chemotherapy and Inhibit the PD-1/PD-L1 Axis. Small 2018, 14, 1801120. [DOI] [PubMed] [Google Scholar]
- (40).Zhang L; Wang ZZ; Zhang Y; Cao FF; Dong K; Ren JS; Qu XG, Erythrocyte Membrane Cloaked Metal-Organic Framework Nanoparticle as Biomimetic Nanoreactor for Starvation-Activated Colon Cancer Therapy. ACS Nano 2018, 12, 10201–10211. [DOI] [PubMed] [Google Scholar]
- (41).Oldenborg P-A; Zheleznyak A; Fang Y-F; Lagenaur CF; Gresham HD; Lindberg FP, Role of CD47 as a Marker of Self on Red Blood Cells. Science 2000, 288, 2051–2054. [DOI] [PubMed] [Google Scholar]
- (42).So AK; Martinon F, Inflammation in Gout: Mechanisms and Therapeutic Targets. Nat. Rev. Rheumatol 2017, 13, 639–647. [DOI] [PubMed] [Google Scholar]
- (43).Sundy JS; Baraf HS; Yood RA; Edwards NL; Gutierrez-Urena SR; Treadwell EL; Vazquez-Mellado J; White WB; Lipsky PE; Horowitz Z; Huang W; Maroli AN; Waltrip RW 2nd; Hamburger SA; Becker MA, Efficacy and Tolerability of Pegloticase for the Treatment of Chronic Gout in Patients Refractory to Conventional Treatment: Two Randomized Controlled Trials. JAMA 2011, 306, 711–720. [DOI] [PubMed] [Google Scholar]
- (44).Bieber JD; Terkeltaub RA, Gout: On the Brink of Novel Therapeutic Options for an Ancient Disease. Arthritis Rheum. 2004, 50, 2400–2414. [DOI] [PubMed] [Google Scholar]
- (45).Sevencan C; McCoy RSA; Ravisankar P; Liu M; Govindarajan S; Zhu JY; Bay BH; Leong DT, Cell Membrane Nanotherapeutics: From Synthesis to Applications Emerging Tools for Personalized Cancer Therapy. Adv. Ther 2020, 3, 1900201. [Google Scholar]
- (46).Zhu J; Sevencan C; Zhang M; McCoy RSA; Ding X; Ye J; Xie J; Ariga K; Feng J; Bay BH; Leong DT, Increasing the Potential Interacting Area of Nanomedicine Enhances Its Homotypic Cancer Targeting Efficacy. ACS Nano 2020, 14, 3259–3271. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.