

Abstract
PURPOSE
Recently developed clinical guidelines suggest that men in families with specific cancer syndromes, such as hereditary breast and ovarian cancer (HBOC), consider genetic testing, especially in the setting of aggressive disease. However, although a family history (FH) of the same disease among close relatives is an established risk factor for prostate cancer (PC), a direct comparison of PC risk for men with each syndrome in a single population is needed.
METHODS
The Utah Population Database was used to identify 619,630 men, age ≥ 40 years, who were members of a pedigree that included at least 3 consecutive generations. Each man was evaluated for FH of hereditary PC (HPC), HBOC, and Lynch syndrome (LS) and for his own PC status. PC occurrences (N = 36,360) were classified into one or more subtypes: early onset (EO), lethal, and/or clinically significant. Relative risks (RRs) associated with each subtype, adjusted for important covariables, were calculated in STATA using a modified Poisson regression with robust error variances to obtain corresponding RR CIs for each FH definition.
RESULTS
An FH of HPC conveyed the greatest relative risk for all PC subtypes combined (RR, 2.30; 95% CI, 2.22 to 2.40), followed by HBOC and LS (both with 1 < RR < 2 and statistically significant). The strongest risks associated with FH were observed for EO disease in all pedigree types, consistent with the contribution of genetic factors to disease occurrence.
CONCLUSION
In this large, population-based, family database, the risk of PC varied by cancer FH and was most strongly associated with EO disease. These results are critically valuable in understanding and targeting high-risk populations that would benefit from genetic screening and enhanced surveillance.
INTRODUCTION
Prostate cancer (PC) is the most common invasive cancer diagnosed among men in the United States, with an estimated 175,000 new occurrences projected to be diagnosed in 2019.1 It currently is the second most common cause of cancer-related death among men, with a projected 31,600 deaths in 2019.1 Although 5-year survival is essentially 100% in men diagnosed with localized or regional PC, it declines significantly (to approximately 30%) among men first diagnosed with metastatic disease,2 emphasizing the importance of earlier detection and treatment. Established PC risk factors include age, ethnicity, and family history (FH) of PC.3 The relative risk (RR) of PC increases approximately 2- to 3-fold for those with an FH of the disease in ≥ 1 first-degree relatives. Risk increases with an increasing number of affected relatives and is inversely related to the age at time of diagnosis among those relatives.4-6 Inherited susceptibility is suspected to account for approximately 40% of all diagnosed PCs; however, genetic variants and mutations discovered to date account for only a subset of these cancers.7,8
Research and clinical studies have used different definitions to identify men who may be at genetic risk for PC according to their FH of PC. Familial PC (FPC) has been defined as families with either 2 first-degree relatives diagnosed with PC at any age or 1 first-degree relative and ≥ 2 second-degree relatives diagnosed at any age.6 A more stringent definition of hereditary PC (HPC) has been used to characterize families with a particularly strong history of PC and includes those families with ≥ 3 affected first-degree relatives, or PC diagnosed in 3 successive generations of the same lineage (paternal or maternal), or 2 first-degree relatives both diagnosed with early-onset disease (≤ 55 years).9
PC risk also has been implicated in other familial cancer syndromes, including hereditary breast and ovarian cancer (HBOC) syndrome and Lynch syndrome (LS). Associated with a germline mutation in the tumor suppressor DNA repair genes BRCA1 and BRCA2, HBOC syndrome typically is found in families with multiple breast, ovarian, and pancreatic cancer diagnoses, particularly with early age of cancer onset. Studies suggest that the RR of PC for men < 65 years with a BRCA1 mutation is nearly 2-fold and more than 7-fold for mutations in BRCA2.10-12 Deleterious BRCA2 mutations also are associated with clinically aggressive disease, progression, and higher rates of cancer-specific mortality.13,14 As a result, the National Comprehensive Cancer Network (NCCN) recommends that BRCA2 mutation carriers begin PC screening with prostate-specific antigen (PSA) testing and a digital rectal exam by age 40 and that BRCA1 mutation carriers consider testing at this age.15
LS, or hereditary non-polyposis colorectal cancer, is associated with germline DNA mismatch repair defects and is clinically defined by the Amsterdam II criteria, which requires at least 3 family members to have a related cancer (eg, colorectal, endometrial, small intestinal, renal pelvic, or ureteral).16 Individuals with LS are 2-5 times more likely to develop PC during their lifetimes.16-19 However, in contrast to PC in the setting of HBOC syndrome, no current NCCN recommendations exist for PC screening in this cohort.
Given the importance of family cancer history in risk assessment for earlier detection and treatment recommendations, the objective of this population-based study was to quantify the RRs of PC associated with different family cancer histories (ie, HPC and FPC, HBOC, and LS). The RR for PC in general as well as the risks for three PC subgroups—early-onset, lethal, and clinically significant PCs—was evaluated.
METHODS
This study used data from the Utah Population Database (UPDB). The UPDB contains data on more than 11 million individuals from the late 18th century to the present. The UPDB links pedigree information from the Genealogical Society of Utah and state vital records to a number of data sources, including but not limited to statewide birth and death certificates, medical records, and the Utah Cancer Registry (UCR). The UCR is a statewide cancer registry established in 1966 and has been part of the SEER registry since 1973. This study was approved by the University of Utah institutional review board and by the Utah Resource for Genetic and Epidemiologic Research, the regulatory oversight board of the UPDB.
Men were included in the study if they met the following inclusion criteria: were members of a pedigree with at least 3 consecutive generations in the record, were age ≥ 40 years by January 1, 2017, or at time of death, were residents of Utah during the year 1966 or after (when a diagnosis of PC could be captured in the UCR), and had known birth years and birth states.
Qualifying patients with PC were identified using the UCR and death certificates according to the following criteria: histopathologically confirmed PC reported in the UCR and/or PC as primary cause of death recorded either in the UCR or on the death certificate. All eligible PC occurrences were then classified as ≥ 1 of the following PC subtypes: early onset, lethal, and/or clinically significant. Early-onset PC was defined as a PC diagnosed at age ≤ 55 years. Lethal PC was identified if PC was listed as the primary cause of death on a death certificate or if a UCR record indicated that the patient died of PC. A PC occurrence was considered clinically significant if any one of 3 conditions were met: Gleason score ≥ 7; missing Gleason score but UCR stage 2 (regional, direct extension only), 3 (regional, regional lymph nodes only), 4 (regional, direct extension, and regional lymph nodes), 5 (regional), or 7 (distant metastases/systemic disease); and lethal PC.
Each included man was evaluated for a positive or negative FH of HPC, FPC, HBOC, and LS. The man’s own cancer disease status was not used in any of the FH definitions. HPC was defined by Carter et al9 in 1993. Although the established criteria can delineate HPC families, it cannot be applied immediately to determine if an individual has an FH of HPC. As such, the criteria were adapted for use in this study, as follows: ≥ 3 first-degree relatives with PC; ≥ 3 affected relatives spanning 3 generations out to third-degree relatives and all on the same ancestral lineage; and ≥ 2 first- or second-degree relatives diagnosed with PC at age ≤ 55 years (Fig 1). A positive history of FPC was defined as having ≥ 2 first- or second-degree relatives with PC on the same side of the pedigree.
FIG 1.

Example pedigrees for hereditary prostate cancer (HPC), with probands meeting the following family history criteria: (A) ≥ 3 first-degree relatives with prostate cancer (PC), (B) ≥ 3 affected relatives spanning 3 generations (out to third-degree relative and all on same side), and (C) ≥ 2 first- or second-degree relatives diagnosed with PC at age ≤ 55 years.
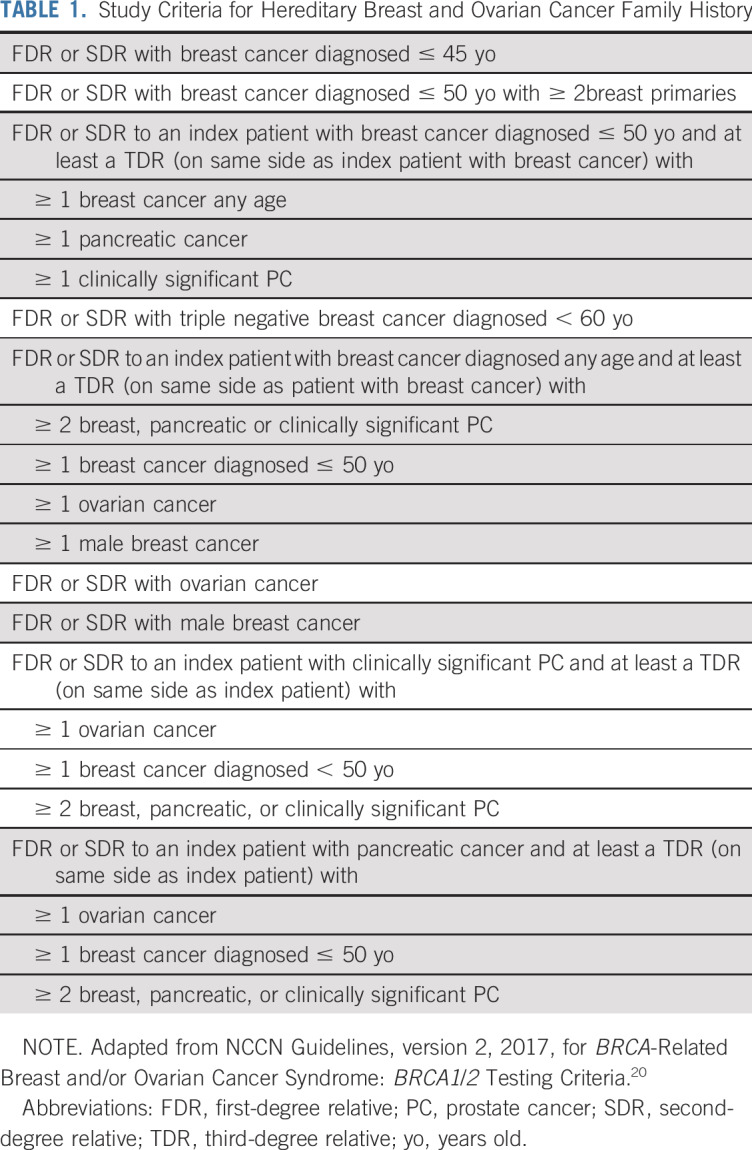
For an FH of HBOC, the NCCN Guidelines, version 2.2017, for BRCA-Related Breast and/or Ovarian Cancer Syndrome were adapted to determine if an individual would meet the BRCA1/2 testing criteria on the basis of FH alone (Table 1).20 Some criteria refer to relatives who had PC with Gleason scores ≥ 7. Because Gleason scores were not available for all PC occurrences, the clinically significant PC definition described earlier in the Methods was used to identify these relatives.
TABLE 1.
Study Criteria for Hereditary Breast and Ovarian Cancer Family History
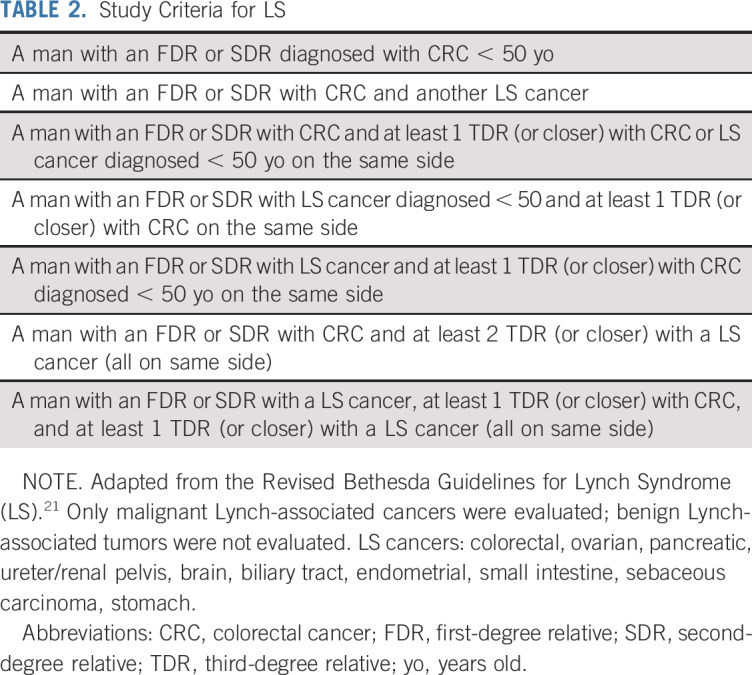
Finally, the Revised Bethesda Guidelines for LS were adapted to determine if an individual had a positive FH of LS (Table 2).21 The LS-associated cancers included in the criteria were colorectal, ovarian, pancreatic, ureter/renal pelvis, brain, biliary tract, endometrial, small intestine, sebaceous carcinoma, and stomach cancers. Only malignant LS-associated cancers were evaluated; benign LS-associated tumors were not evaluated.
TABLE 2.
Study Criteria for LS
Statistical Methods
RRs of PC were calculated for each FH definition. This calculation was completed in STATA (STATA Corp, College Station, TX) using a modified Poisson regression with robust error variances to obtain the RR CIs. Covariables representing 5-year birth year groups, birth state (Utah or non-Utah), and number of male relatives were included in the model to control for possible confounding. The 5-year birth year group was included to control for differences over time (PSA-based diagnosis, older individuals with more FH). Birth state (Utah or non-Utah), was included to control for the fact that the men born in Utah families likely have more data in UPDB than those migrating to Utah. Number of male relatives was included to control for the possible increased likelihood of a positive FH of PC with more men in the family available to evaluate. All calculations were repeated with early-onset, lethal, and clinically significant PC each as the dependent variable.
RESULTS
A total of 619,630 men met the inclusion criteria for the study, and 421,827 men (68.1%) were still alive at the time of study query (August 2018). The median age at time of query among those still alive was 57.5 years (interquartile range [IQR], 48.1-66.9 years). The median age at time of death for the remaining 31.9% of the cohort was 75.5 years (IQR, 64.6-83.9 years). The prevalence of PC was 5.9% for the cohort (n = 36,360); 2,562 patients met the criteria for early-onset disease (7.0%), 4,044 patients (11.1%) met the criteria for lethal disease, and 15,201 patients (41.8%) met the criteria for clinically significant disease (Table 3). The median age at time of diagnosis was 69.0 years (IQR, 63.0-76.0 years; Data Supplement). Just more than 70% of men were diagnosed with organ-confined disease, and approximately 6% were first diagnosed with metastatic disease. Approximately 71% of patients were missing information on Gleason score. However, among those patients with information on Gleason score (n = 10,523 men), 46.7% were diagnosed with Gleason score of ≤ 6; 39.0%, with a Gleason score of 7; and 14.3%, with a Gleason score of ≥ 8. When cancers were categorized into early-onset PC, lethal PC, and clinically significant PC, temporal trends were observed: in recent decades, more patients had early-onset PC and fewer had lethal PC. No consistent linear trends were observed for clinically significant PC within the timeframe of the study (Data Supplement).
TABLE 3.
Prevalence of PC in Study Population, Overall and by PC Subtype
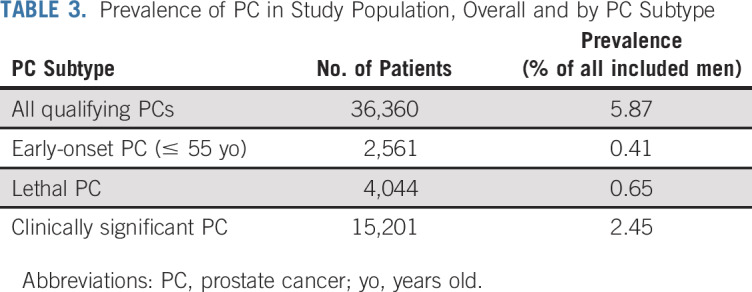
Table 4 lists the RRs for PC according to FH. The risks were highest among men with HPC, who experienced a 2.3-fold increase in risk for PC overall (RR, 2.30; 95% CI, 2.22 to 2.40). An FH of PC (FPC), HBOC, and LS all were associated with an increase in risk, albeit more modestly (RRs, 1.81 [95% CI, 1.76 to 1.86], 1.47 [95% CI, 1.43 to 1.50], and 1.16 [95% CI, 1.12 to 1.19], respectively). Generally, the greatest risk associated with any FH was for early-onset disease. HPC was associated with a near 4-fold increase in risk for early-onset PC (RR, 3.93; 95% CI, 3.33 to 4.61). HPC also was associated with higher risks for both lethal PC (RR, 2.21; 95% CI, 1.95 to 2.50) and clinically significant disease (RR, 2.32; 95% CI, 2.17 to 2.48). Overall, modest elevations in risk were associated with LS, with a 34% increase in risk for early-onset disease (RR, 1.34; 95% CI, 1.18 to 1.52) and a small increase in risk for clinically significant disease (RR, 1.15; 95% CI, 1.10 to 1.21).
TABLE 4.
Relative Risks for PC for Men with Different Family Cancer Histories, by PC Subtypes

Given the notable elevation in PC risk observed in HPC families, we evaluated how each of the 3 definitions of HPC (≥ 3 first-degree relatives with PC; ≥ 3 affected relatives spanning 3 generations out to third-degree relatives and all on same side [eg, a paternal grandfather, father, and brother]); and ≥ 2 first- or second-degree relatives diagnosed with PC at age ≤ 55 years) affected an individual’s risk for PC (Table 5). The most common HPC criteria was ≥ 3 affected relatives spanning 3 generations (n = 11,104), whereas the other 2 criteria were uncommon. Men with ≥ 3 affected first-degree relatives (n = 2,618) or men with > 1 first-degree and/or second-degree relative with PC (n = 893) had increased risks for being diagnosed with early-onset PC: 8.72 (95% CI, 6.60 to 11.5) and 8.92 (95% CI, 6.07 to 13.1), respectively.
TABLE 5.
Relative Risks for PC for Men Meeting the Different Criteria for Hereditary PC by PC Subtypes

DISCUSSION
To our knowledge, this is largest study to date about RR of PC associated with pedigree characteristics indicative of hereditary cancer syndromes. We observed that characteristics consistent with HPC in the family were associated with the greatest risk of PC, irrespective of clinical subtype, followed by FPC, HBOC, and LS. Furthermore, risk estimates generally were highest for early-onset disease, especially for men in families with HPC or FPC. The highest risks for lethal and clinically significant PC also were observed in HPC families (RRs, 2.21 and 2.32, respectively). This result was unexpected, given reports of DNA repair gene mutations (eg, BRCA1/2 and mismatch repair genes) in men with metastatic PC.22,23 Prior research using families with HPC for susceptibility gene identification have been partially unsuccessful, because clinical heterogeneity of disease often was ignored. This observation also suggests a renewed focus on families with HPC by specifically segregating the lethal phenotype for gene discovery research.
Our results are consistent with the larger body of literature estimating PC risk as a function of family cancer history. Genetic variants have been notoriously difficult to replicate across studies, with only a few exceptions. The G84E mutation in HOXB13 was first discovered in a linkage study of PC families, with near complete cosegregation of the mutation with disease in these families.24 The strongest estimates of risk were observed among men with early-onset disease.24 The mutation is considered moderately penetrant, with a 4- to 5-fold increase in risk for PC.25-27 However, the G84E mutation is observed almost exclusively in populations of European descent, occurs in less than 2% of men, and has not been linked consistently to aggressive disease; thus, its use for genetic screening, at least in the general population, is limited.24,28,29
The breast cancer gene BRCA2 also has been implicated in PC, particularly in HBOC families. The reported frequency of BRCA2 mutations ranges from 1.3%-3.2% depending on the study design and population.30 Unlike HOXB13, the function of BRCA2 and its importance in DNA repair are well known. Mutations in BRCA2 are associated with more aggressive PC clinical features, mortality, and response to poly (ADP-ribose) polymerase inhibitors, so it is an actionable mutation.11,13,31 In fact, recommendations about genetic testing for BRCA2 mutations in the context of PC are emerging from specific national consensus panels.32
In addition to these rare moderately to highly penetrant mutations, genome-wide association studies have identified approximately 100 common loci that associate with PC and have validation in independent cohorts that explain an estimated 30% of familial PC risk.7 Although these loci have associations with PC at a high level of statistical significance, the magnitude of risk remains modest. In many cases, the functional significance of identified genome-wide association study variants remains unclear, with ongoing efforts at functional annotation.33,34 Furthermore, no common variants have been consistently associated with PC-specific survival,35 which is an important factor for the clinical use of germline variants in informing management. Nevertheless, polygenic genetic risk scores using single-nucleotide polymorphisms consistently associated with PC increasingly are being used to help stratify populations and identify intermediate to high risks according to FH.36
This study adds to the existing body of literature by using data from the UPDB and UCR. These unique resources have been used before to evaluate the RRs of PC among men with different FH patterns of PC. In a study by Albright et al,37 the overall PC rate was 2.85% in men with sufficient information in 3 vertical generations (n = 635,443). The RR for men with 1 affected first-degree relative was approximately 2.5-fold, with increases to nearly 4-fold for 4 affected first-degree relatives. A subsequent study led by the same team observed similar risk trends in examining lethal PC.38 These investigations did not consider the impact on early-onset disease or other familial cancer syndromes that include PC. The observation in our study that men with either 3 first-degree relatives with PC or 2 first- or second-degree relatives diagnosed with early-onset PC had a more than 8-fold increase in the diagnosis of early-onset PC is important and merits replication in other studies, because this confirmation would affect the age at which PC screening should be initiated.
The study has notable strengths, including its large sample size, which allowed for relatively precise estimates of risk in the evaluation of rare pedigree phenotypes (HBOC, LS). The linkage of UPDB and UCR is a unique resource to study well-characterized family pedigrees with lengthy and complete follow-up for postdiagnostic outcomes. We chose to focus our investigation on men age ≥ 40 years, recognizing that PC diagnoses at age < 40 years are exceedingly rare in the general population, even among families demonstrating a predisposition to disease.
The study also has several limitations. First, PSA data were not available for UPDB participants. In the Selenium and Vitamin E Cancer Prevention Trial (SELECT), men with a FH of PC were more likely undergo a prostate biopsy even after adjusting for PSA levels and DRE results which increased the likelihood of being diagnosed with prostate cancer.39 Although analysis of PSA screening could not be assessed, it is important to note that 23% of patients with PC in our study were diagnosed before 1990, thus reflecting clinical diagnoses. Furthermore, Bratt et al40 showed that men with an FH of PC have an increased risk for all forms of PC, including high-risk disease. Our analysis showed an elevation in risk for diagnosis of clinically significant disease that is similar to the risk for diagnosis of any PC (Table 4), which mitigates the concern that excess PSA screening in men with an FH of PC resulted in inflated PC risks according to the identification of men with low-risk disease. Also, genetic data are not currently available on UPDB participants, so the impact of known susceptibility genes cannot be assessed. The Utah population with deep pedigree information is relatively homogenous with respect to race/ethnicity, so these findings may not be generalizable to nonwhite populations. The Gleason score was missing for more than 70% of patients, so classification of clinically significant disease relied on stage at diagnosis and/or cause of death, introducing the potential for misclassification. Finally, as in all studies of FH, the exposures of interest are not static, so changes in FH during the study are not considered.
In conclusion, results from this investigation indicate that pedigrees exhibiting HPC and other hereditary cancer syndromes predict risk of PC, particularly early-onset disease. Future studies using 3 pedigree phenotypes will be critical for the discovery of new PC susceptibility genes that can be used in genetic screening and risk assessment.
ACKNOWLEDGMENT
We thank the Pedigree and Population Resource of the Huntsman Cancer Institute, University of Utah (funded in part by the Huntsman Cancer Foundation) for its role in the ongoing collection, maintenance, and support of the Utah Population Database (UPDB). We also acknowledge partial support for the UPDB through Grant No. P30 CA2014 from the National Cancer Institute, University of Utah, and the University of Utah’s Program in Personalized Health and Center for Clinical and Translational Science. Support for the Utah Cancer Registry is provided by National Cancer Institute's SEER Program, Contract No. HHSN261201800016I, the US Center for Disease Control and Prevention's National Program of Cancer Registries, Cooperative Agreement No. NU58DP0063200, with additional support from the University of Utah and Huntsman Cancer Foundation.
SUPPORT
Supported by the Huntsman Cancer Foundation, National Institutes of Health Grant No. P30 CA2014, the University of Utah, the University of Utah’s Program in Personalized Health and Center for Clinical and Translational Science, the National Cancer Institute's SEER Program (Contract No. HHSN261201800016I), and the US Center for Disease Control and Prevention's National Program of Cancer Registries (Cooperative Agreement No. NU58DP0063200).
AUTHOR CONTRIBUTIONS
Conception and design: Jennifer L. Beebe-Dimmer, Ashley Kapron, Ken R. Smith, Kathleen A. Cooney
Collection and assembly of data: Ashley L. Kapron, Alison M. Fraser, Ken R. Smith, Kathleen A. Cooney
Data analysis and interpretation: Jennifer L. Beebe-Dimmer, Ashley L. Kapron, Alison M. Fraser, Ken R. Smith, Kathleen A. Cooney
Provision of study material or patients: Alison M. Fraser, Ken R. Smith, Kathleen A. Cooney
Administrative support: Ashley L. Kapron, Alison M. Fraser, Ken R. Smith, Kathleen A. Cooney
Financial support: Kathleen A. Cooney
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Risk of Prostate Cancer Associated With Familial and Hereditary Cancer Syndromes
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated unless otherwise noted. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/jco/authors/author-center.
Open Payments is a public database containing information reported by companies about payments made to US-licensed physicians (Open Payments).
Kathleen A. Cooney
Patents, Royalties, Other Intellectual Property: patent awarded for discovery of HOXB13 as prostate cancer susceptibility gene (Inst)
Travel, Accommodations, Expenses: Boston Scientific (I)
No other potential conflicts of interest were reported.
REFERENCES
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69:7–34. doi: 10.3322/caac.21551. [DOI] [PubMed] [Google Scholar]
- 2.Howlader N NA, Krapcho M, Miller D, et al. http://seer.cancer.gov/csr/1975_2013/ SEER Cancer Statistics Review, 1975-2013.
- 3.Attard G, Parker C, Eeles RA, et al. Prostate cancer. Lancet. 2016;387:70–82. doi: 10.1016/S0140-6736(14)61947-4. [DOI] [PubMed] [Google Scholar]
- 4.Kiciński M, Vangronsveld J, Nawrot TS. An epidemiological reappraisal of the familial aggregation of prostate cancer: A meta-analysis. PLoS One. 2011;6:e27130. doi: 10.1371/journal.pone.0027130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Johns LE, Houlston RS. A systematic review and meta-analysis of familial prostate cancer risk. BJU Int. 2003;91:789–794. doi: 10.1046/j.1464-410x.2003.04232.x. [DOI] [PubMed] [Google Scholar]
- 6.Stanford JL, Ostrander EA. Familial prostate cancer. Epidemiol Rev. 2001;23:19–23. doi: 10.1093/oxfordjournals.epirev.a000789. [DOI] [PubMed] [Google Scholar]
- 7.Eeles RA, Olama AA, Benlloch S, et al. : Identification of 23 new prostate cancer susceptibility loci using the iCOGS custom genotyping array. Nat Genet 45:385-391,2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mucci LA, Hjelmborg JB, Harris JR, et al. Familial risk and heritability of cancer among twins in Nordic countries. JAMA. 2016;315:68–76. doi: 10.1001/jama.2015.17703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Carter BS, Bova GS, Beaty TH, et al. Hereditary prostate cancer: Epidemiologic and clinical features. J Urol. 1993;150:797–802. doi: 10.1016/s0022-5347(17)35617-3. [DOI] [PubMed] [Google Scholar]
- 10.Na R, Zheng SL, Han M, et al. Germline Mutations in ATM and BRCA1/2 distinguish risk for lethal and indolent prostate cancer and are associated with early age at death. Eur Urol. 2017;71:740–747. doi: 10.1016/j.eururo.2016.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mateo J, Boysen G, Barbieri CE, et al. DNA repair in prostate cancer: Biology and clinical implications. Eur Urol. 2017;71:417–425. doi: 10.1016/j.eururo.2016.08.037. [DOI] [PubMed] [Google Scholar]
- 12.Akbari MR, Wallis CJ, Toi A, et al. The impact of a BRCA2 mutation on mortality from screen-detected prostate cancer. Br J Cancer. 2014;111:1238–1240. doi: 10.1038/bjc.2014.428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Castro E, Goh C, Olmos D, et al. Germline BRCA mutations are associated with higher risk of nodal involvement, distant metastasis, and poor survival outcomes in prostate cancer. J Clin Oncol. 2013;31:1748–1757. doi: 10.1200/JCO.2012.43.1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Narod SA, Neuhausen S, Vichodez G, et al. Rapid progression of prostate cancer in men with a BRCA2 mutation. Br J Cancer. 2008;99:371–374. doi: 10.1038/sj.bjc.6604453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Carroll PR, Parsons JK, Andriole G, et al. NCCN Clinical Practice Guidelines: Prostate cancer early detection, version 2.2015. J Natl Compr Canc Netw. 2015;13:1534–1561. doi: 10.6004/jnccn.2015.0181. [DOI] [PubMed] [Google Scholar]
- 16.Samadder NJ, Smith KR, Wong J, et al. Cancer risk in families fulfilling the Amsterdam Criteria for Lynch syndrome. JAMA Oncol. 2017;3:1697–1701. doi: 10.1001/jamaoncol.2017.0769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Raymond VM, Mukherjee B, Wang F, et al. Elevated risk of prostate cancer among men with Lynch syndrome. J Clin Oncol. 2013;31:1713–1718. doi: 10.1200/JCO.2012.44.1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Haraldsdottir S, Hampel H, Wei L, et al. Prostate cancer incidence in males with Lynch syndrome. Genet Med. 2014;16:553–557. doi: 10.1038/gim.2013.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bauer CM, Ray AM, Halstead-Nussloch BA, et al. Hereditary prostate cancer as a feature of Lynch syndrome. Fam Cancer. 2011;10:37–42. doi: 10.1007/s10689-010-9388-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Daly MB, Pilarski R, Barry R, et al. NCCN guidelines insights: Genetic/familial high-risk assessment: Breast and ovarian, version 2. JNCCN. 2017;15:9–20. doi: 10.6004/jnccn.2017.0003. [DOI] [PubMed] [Google Scholar]
- 21.Umar A, Boland CR, Terdiman JP, et al. Revised Bethesda Guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. J Natl Cancer Inst. 2004;96:261–268. doi: 10.1093/jnci/djh034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Robinson D, Van Allen EM, Wu YM, et al. Integrative clinical genomics of advanced prostate cancer. Cell. 2015;162:454. doi: 10.1016/j.cell.2015.06.053. [DOI] [PubMed] [Google Scholar]
- 23.Pritchard CC, Offit K, Nelson PS. DNA-repair gene mutations in metastatic prostate cancer. N Engl J Med. 2016;375:1804–1805. doi: 10.1056/NEJMc1611137. [DOI] [PubMed] [Google Scholar]
- 24.Ewing CM, Ray AM, Lange EM, et al. Germline mutations in HOXB13 and prostate-cancer risk. N Engl J Med. 2012;366:141–149. doi: 10.1056/NEJMoa1110000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Beebe-Dimmer JL, Hathcock M, Yee C, et al. The HOXB13 G84E mutation is associated with an increased risk for prostate cancer and other malignancies. Cancer Epidemiol Biomarkers Prev. 2015;24:1366–1372. doi: 10.1158/1055-9965.EPI-15-0247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huang H, Cai B. G84E mutation in HOXB13 is firmly associated with prostate cancer risk: A meta-analysis. Tumour Biol. 2014;35:1177–1182. doi: 10.1007/s13277-013-1157-5. [DOI] [PubMed] [Google Scholar]
- 27.Xu J, Lange EM, Lu L, et al. HOXB13 is a susceptibility gene for prostate cancer: Results from the International Consortium for Prostate Cancer Genetics (ICPCG) Hum Genet. 2013;132:5–14. doi: 10.1007/s00439-012-1229-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Beebe-Dimmer JL, Isaacs WB, Zuhlke KA, et al. Prevalence of the HOXB13 G84E prostate cancer risk allele in men treated with radical prostatectomy. BJU Int. 2014;113:830–835. doi: 10.1111/bju.12522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Giri VN, Beebe-Dimmer JL. Familial prostate cancer. Semin Oncol. 2016;43:560–565. doi: 10.1053/j.seminoncol.2016.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Castro E, Eeles R. The role of BRCA1 and BRCA2 in prostate cancer. Asian J Androl. 2012;14:409–414. doi: 10.1038/aja.2011.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mateo J, Carreira S, Sandhu S, et al. DNA-repair defects and olaparib in metastatic prostate cancer. N Engl J Med. 2015;373:1697–1708. doi: 10.1056/NEJMoa1506859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Giri VN, Knudsen KE, Kelly WK, et al. Role of genetic testing for inherited prostate cancer risk: Philadelphia Prostate Cancer Consensus Conference 2017. J Clin Oncol. 2018;36:414–424. doi: 10.1200/JCO.2017.74.1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Larson NB, McDonnell S, French AJ, et al. Comprehensively evaluating cis-regulatory variation in the human prostate transcriptome by using gene-level allele-specific expression. Am J Hum Genet. 2015;96:869–882. doi: 10.1016/j.ajhg.2015.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hazelett DJ, Rhie SK, Gaddis M, et al. Comprehensive functional annotation of 77 prostate cancer risk loci. PLoS Genet. 2014;10:e1004102. doi: 10.1371/journal.pgen.1004102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Szulkin R, Karlsson R, Whitington T, et al. Genome-wide association study of prostate cancer–specific survival. . Cancer Epidemiol Biomarkers Prev. 2015;24:1796–1800. doi: 10.1158/1055-9965.EPI-15-0543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Helfand BT, Chen H, Fantus RJ, et al. Differences in inherited risk among relatives of hereditary prostate cancer patients using genetic risk score. Prostate. 2018;78:1063–1068. doi: 10.1002/pros.23664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Albright F, Stephenson RA, Agarwal N, et al. Prostate cancer risk prediction based on complete prostate cancer family history. Prostate. 2015;75:390–398. doi: 10.1002/pros.22925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Albright FS, Stephenson RA, Agarwal N, et al. Relative risks for lethal prostate cancer based on complete family history of prostate cancer death. Prostate. 2017;77:41–48. doi: 10.1002/pros.23247. [DOI] [PubMed] [Google Scholar]
- 39.Tangen CM, Goodman PJ, Till C, et al. Biases in recommendations for and acceptance of prostate biopsy significantly affect assessment of prostate cancer risk factors: Results from two large randomized clinical trials. J Clin Oncol. 2016;34:4338–4344. doi: 10.1200/JCO.2016.68.1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bratt O, Drevin L, Akre O, et al. Family history and probability of prostate cancer, differentiated by risk category: A nationwide population-based study. J Natl Cancer Inst. 2016;108:djw110. doi: 10.1093/jnci/djw110. [DOI] [PubMed] [Google Scholar]