

Abstract
Aims
This first‐in‐human clinical trial of P218, a novel dihydrofolate reductase inhibitor antimalarial candidate, assessed safety, tolerability, pharmacokinetics and food effects in healthy subjects.
Methods
The study consisted of two parts. Part A was a double‐blind, randomized, placebo‐controlled, parallel group, ascending dose study comprising seven fasted cohorts. Eight subjects/cohort were randomized (3:1) to receive either a single oral dose of P218 (10, 30, 100, 250, 500, 750 and 1000 mg) or placebo. Part B was an open‐label, cross‐over, fed/fasted cohort (eight subjects) that received a 250 mg single dose of P218 in two treatment periods.
Results
P218 was generally well tolerated across all doses; 21 treatment‐emergent adverse events occurred in 15/64 subjects. Nine adverse events in five subjects, all of mild intensity, were judged drug related. No clinically relevant abnormalities in ECG, vital signs or laboratory tests changes were observed. P218 was rapidly absorbed, with C max achieved between 0.5 and 2 hours post dose. Plasma concentrations declined bi‐exponentially with half‐life values ranging from 3.1 to 6.7 hours (10 and 30 mg), increasing up to 8.9 to 19.6 hours (doses up to 1000 mg). Exposure values increased dose‐proportionally between 100 and 1000 mg for P218 (parent) and three primary metabolites (P218 β‐acyl glucuronide, P218‐OH and P218‐OH β‐acyl glucuronide). Co‐administration of P218 with food reduced C max by 35% and delayed absorption by 1 hour, with no significant impact on AUC.
Conclusion
P218 displayed favourable safety, tolerability and pharmacokinetics. In view of its short half‐life, a long‐acting formulation will be needed for malaria chemoprotection.
Keywords: chemoprotection, clinical trial, dihydrofolate reductase inhibitor, malaria, P218, pharmacodynamics, pharmacokinetics
What is already known about this subject
Dihydrofolate reductase is a known target for antimalarial drugs, though resistance to existing agents, for example pyrimethamine, has curtailed their utility for malaria treatment and prevention.
P218 is a novel antimalarial candidate with in vitro inhibitory activity against Plasmodium falciparum dihydrofolate reductase comparable to or greater than pyrimethamine.
P218 has potent in vitro and in vivo activity against pyrimethamine‐resistant P. falciparum.
What this study adds
In this first‐in‐human study of P218, there were no concerning safety signal signals observed for adverse events, laboratory parameters and QTcF prolongation.
P218 metabolites observed pre‐clinically were confirmed in humans and pharmacokinetics for P218 and metabolites were dose‐proportional with no relevant effect of food on drug exposure, safety or tolerability.
P218 appears to possess the required safety profile for further development as an antimalarial drug and pharmacokinetic properties that will require development of an extended release formulation. Further clinical studies should be considered.
1. INTRODUCTION
The Plasmodium parasites Plasmodium falciparum and P. vivax caused an estimated 219 million malaria cases in 2017 and around half of the world's population remains at risk of this serious infection.1
Preventive therapies targeting P. falciparum are recommended by the World Health Organization for the most vulnerable groups in malaria endemic areas of sub‐Saharan Africa. These include intermittent preventive treatment of pregnant women (IPTp),2 intermittent preventive treatment of infants (IPTi)3 and seasonal malaria chemoprevention in children (SMC).4 Migrant and forest workers in Africa and Asia are another group that could benefit from chemoprotection.5
The artemisinin‐based combination therapy (ACT) dihydroartemisinin‐piperaquine has been investigated for chemoprotection.6, 7 However, in South‐East Asia, P. falciparum resistance to dihydroartemisinin‐piperaquine is already widespread, undermining treatment efficacy.8 As ACTs are the only agents available for malaria treatment, there is concern that wider use of these drugs for malaria prevention in Africa could accelerate the development of drug resistance, with devastating consequences. Consequently, preventive strategies in Africa are currently dependent upon the antimalarial efficacy of sulphadoxine‐pyrimethamine, though widespread resistance has developed to both components of this regimen.9, 10
The candidate antimalarial agent P218 (3‐[2‐[3‐[[2,4‐diamino‐6‐ethylpyrimidin‐5‐yl]oxy]propoxy]phenyl]propanoic acid hydrochloride) is a selective inhibitor of P. falciparum bifunctional dihydrofolate reductase‐thymidylate synthase (PfDHFR‐TS), an essential enzyme in the formation of parasite DNA, recycling folate to its reduced form.11, 12 PfDHF‐TS is a validated target for malaria treatment and is shared with several approved antimalarial drugs, including pyrimethamine.9, 10 Previous studies indicated that P218 is highly specific for PfDHFR‐TS, with binding properties that cause it to be active against drug‐resistant mutants and to have a low potential for resistance induction.11, 12 In vitro, P218 was highly potent against P. falciparum blood stages (TM4 strain; IC50 4.6 ± 1.9 nM) and the pyrimethamine‐resistant quadruple mutant strain V1/S (IC50 56 ± 20 nM).11 P218 also had high in vivo antimalarial efficacy in the humanised mouse model.11, 12 Similar to other dihydrofolate inhibitors, P218 has activity against P. falciparum liver stages and is envisaged as a malaria chemoprotective agent.11, 12 Preclinical safety findings with P218 were favourable and the molecule was found to have high selectivity for the PfDHFR‐TS target versus human DHFR.11
The primary objective of the first‐in‐human study was to investigate the safety and tolerability of single escalating oral doses of P218 in fasted healthy human volunteers. Additionally, the study aimed to describe the pharmacokinetics (PK) of P218 and its major metabolites observed pre‐clinically (P218‐OH, P218 β‐acyl glucuronide and P218‐OH β‐acyl glucuronide) and the effect of a high‐fat meal on P218 PK and safety outcomes.
2. METHODS
2.1. Ethical statement
The study was conducted at Richmond Pharmacology, UK, according to the Declaration of Helsinki, International Conference on Harmonization Good Clinical Practice guidelines, European Union Directive 21CRF50, and complied with all local and international laws pertaining to the use of human subjects. The trial is registered at ClinicalTrials.gov with the identifier NCT02885506. The protocol and informed consent form were approved by an independent ethics committee (Health Research Authority Ethics Committee, South Central—Berkshire B, UK) and the Medicines and Healthcare Products Regulatory Authority. All subjects voluntarily provided signed informed consent before participation in the study. All drug/molecular target nomenclature conforms to the IUPHAR/BPS Guide to PHARMACOLOGY nomenclature classification 2017.13
2.2. Study design
This first‐time‐in‐human study of the antifolate inhibitor P218 consisted of two parts. Study plans for parts A and B are shown in Figure 1. Part A was a double‐blind, randomized, placebo‐controlled, parallel group ascending dose study conducted in seven fasted cohorts, each including eight healthy human volunteers (two randomized to placebo and six to P218). Each subject received a single oral dose of study drug on day 1. Standard meals were given: lunch 4 hours after dosing and dinner 9‐10 hours post dose, plus an evening snack. P218 doses started at 10 mg and, following a formal evaluation by the safety review committee, were escalated to 30, 100, 250, 500, 750 and 1000 mg (cohorts 1‐7).
Figure 1.
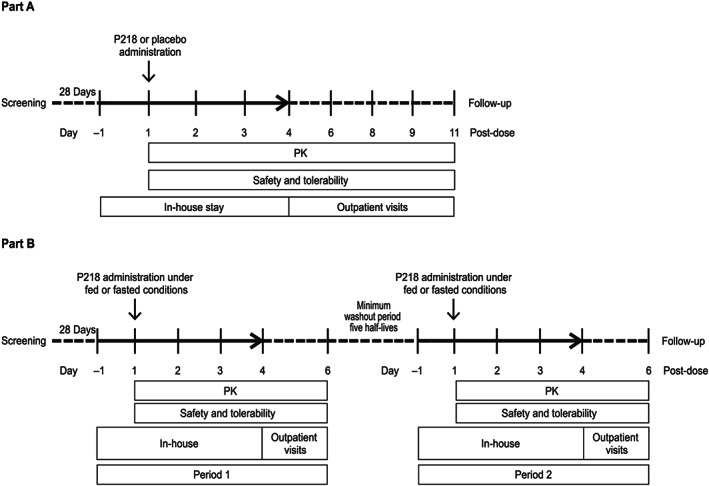
Study flow for Part A: Safety and PK assessment of P218 and Part B: Food effect study
The maximum starting dose (10 mg) was selected as per recommendations from the US Food and Drug Administration14 based on preclinical good laboratory practice‐compliant toxicology data obtained in the rat and dog (data on file available from MF Chughlay [chughlayf@mmv.org]). The 10 mg starting dose was predicted to generate mean maximum drug plasma concentrations (C max) and area under the concentration‐time curve (AUC) values of 0.220 μg/mL and 1.27 μg·h/mL, respectively. In the most conservative scenario, these values provided a predicted safety margin of 4.2‐fold when compared to the C max (0.921 μg/mL) and 34.7‐fold for the cumulative AUC0‐336 (44.1 μg·h/mL) in the most sensitive species (dog) at 10 mg/kg/day. The cumulative AUC was used as the only effects of P218 could be ascribed to folate depletion that required multiple doses to manifest. When compared to unbound C max (0.198 μg/mL) and unbound cumulative AUC0‐336 (9.48 μg·h/mL), the margins increased to 15.2 and 124.7, respectively. A maximum dose of 1000 mg is considered the highest feasible dose for use in malaria‐endemic countries owing to cost limitations. The predicted P218 exposure at this dose (16.5 μg·h/mL) and the resulting margin to P218 AUC cap was 4.4‐fold. No toxicity that was not related to DHFR inhibition (ie weight loss, gastrointestinal symptoms, decreased reticulocyte counts and increased neutrophil counts) was observed in either rats or dogs, supporting the conclusion that none of the P218 metabolites posed an additional toxicological risk. Thus, doses were chosen based on predicted P218 exposure.
Part B evaluated the effect of food on P218 PK and safety in an open‐label, randomized fed/fasted crossover design in eight additional subjects who received two single oral P218 administrations, the second after a washout period of at least five times the P218 half‐life. The investigational P218 dose was determined based on pharmacokinetic and safety findings in Part A to achieve a predicted P218 exposure at least 3‐fold below the maximum observed exposure achieved that was safe and well tolerated, in order to account for a possible increase in exposure with food. A standard high‐fat breakfast (800‐1000 kilocalories, with 50% fat15) was given 0.5 hours pre dose to be completed within 20 minutes.
For Parts A and B, subjects were screened within 28 days of study start, with eligible subjects enrolled on day −1 and randomly assigned to treatment on day 1, according to a randomization schedule provided by the study statistician. P218 was formulated in 10, 50 or 250 mg capsules without excipients with matching 50 mg placebo capsules. Study drugs were administered with 240 mL of water.
2.3. Subjects
Eligible subjects were healthy males or females with no childbearing potential aged 18‐45 years with a body mass index between 18 and 25 kg/m2 and a total body weight >50 kg. Main exclusion criteria were evidence or history of clinically significant medical conditions or infections, including any history of gallbladder disease, cholecystitis and/or cholelithiasis/gallbladder medical conditions, megaloblastic anaemia or folate deficiency; any clinically significant abnormal laboratory, vital signs or other safety findings. Subjects should also not have taken any drug or substances that might affect the safety or pharmacokinetic assessment. Full inclusion and exclusion details are provided in Supporting Information Methods S1.
2.4. Safety assessments
At screening and enrolment, a medical history was taken and vital signs and physical assessments performed. The full schedule of safety assessments is shown in Supporting Information Table S1 for Parts A and B. Adverse events were evaluated throughout the study and categorized using the Medical Dictionary for Regulatory Activities (MedDRA version 20.0) and their severity noted according to the WHO Toxicity Grading Scale.16 Biochemistry, haematology, coagulation tests and urinalysis were conducted at screening, enrolment and during follow‐up. Additionally, owing to the mechanism of action of P218, serum folate levels were determined at baseline and throughout the study.
2.5. Pharmacokinetic assessments and bioanalytical assay
Frequent pharmacokinetic blood sampling was conducted in Parts A and B (Supporting Information Table S2), and pharmacokinetic urine samples were collected in Part A only (Supporting Information Table S2). Plasma samples for determination of P218 and metabolite concentrations were analysed by Swiss BioQuant (Reinach, Switzerland) using high performance liquid chromatography‐tandem mass spectrometry (LC‐MS/MS). The analytical range for P218 and all P218 metabolites was 0.2–200 ng/mL; samples with values greater than >200 ng/mL were diluted with blank human plasma and corrected for the dilution factor. The assay was validated as the relative standard deviation (percentage coefficient of variation [%CV]) of the intra‐ and inter‐batch precision of the quality control samples did not exceed 15% (range 4.2‐6.9%) and the accuracy of the mean determined concentrations to the nominal concentration of analytes did not exceed 15% (range 98.0‐100.7%).
2.6. Electrocardiograms
Triplicate 12‐lead ECGs were performed at baseline, pre dose and post dose, paired with a PK sample. Continuous ECG telemetry from pre dose to 12 hours post dose was done in Part A using a Marquette MAC1200®/MAC1200ST® recorder connected to the MUSE® Cardiology Information System (both from GE Medical Systems, Bloomington, IL, USA) (Supporting Information Table S1). Static 12‐lead ECG recordings were done after the subjects had been resting in a supine position for at least 10 minutes. Repeat ECGs were performed until at least three 10‐second ECG records per scheduled time point met the quality criteria to enable analysis of at least five complexes per derivation. ECGs were read automatically by the Marquette® 12SL™ ECG Analysis Program (MEAP) and manually adjudicated by a cardiologist blinded to treatment, time and date.
2.7. Ex vivo malaria assay
In Part A only, and as typically performed during first‐in‐human trials with new antimalarial compounds, blood samples for the ex vivo malaria assay were collected from subjects receiving 250 or 500 mg P218 at pre dose on day 1, and post dose at 0.5, 1, 2, 6, 12, 24, 48, 120 and 240 hours. Serum was extracted and stored at −20 to −70°C. The assay was performed by the Swiss Tropical and Public Health Institute (Basel, Switzerland) using the 3H‐hypoxanthine incorporation assay as previously described,17, 18 with the P. falciparum strain NF54 cultured in O+ erythrocytes (Blood Bank, Basel, Switzerland) and parasite inhibition expressed as IC50 values. To determine whether antimalarial activity originated with P218 or metabolites, the P218 concentration‐time profiles determined by LC‐MS/MS analysis were compared to concentration‐time profiles calculated from the antimalarial activity of P218 in the collected serum samples, normalized to a known IC50 value, obtained in the same experimental run using a reference serum sample spiked with a known amount of P218 (see Supporting Information Methods S2 for details).
2.8. Outcomes
The primary outcome was the incidence, severity and relationship to the investigational product of adverse events. Secondary endpoints were the estimation of PK parameters for P218 and primary metabolites (P218‐OH, P218 β‐acyl glucuronide and P218‐OH β‐acyl glucuronide): AUClast, AUCinf C max, time of C max (T max), elimination half‐life (t 1/2), mean residence time (MRT), apparent total body clearance of the drug from plasma (CL/F; for P218 only), apparent volume of distribution during terminal elimination (Vz/F; for P218 only) and metabolites ratio. Exploratory endpoints were the efficacy of P218 against parasites using an ex vivo malaria assay, the absolute change from baseline P218 on serum folate levels over time, the paired PK and QTc interval parameters pre dose compared to post administration of P218, clinically significant ECG morphology and interval changes from baseline. A concentration‐response analysis was also conducted for the effect of P218 on Fridericia‐corrected QT interval (QTcF), to be published separately.
2.9. Statistical analysis
All enrolled subjects who received a dose of the study drug were included in the safety population. The PK population included all subjects in the safety population with at least one drug concentration measurement. All outcomes were presented using descriptive statistics. Because the primary objective was an initial assessment of safety, each treatment group was limited to six subjects receiving P218. This provided a 47%, 62%, 74% or 82% probability of observing at least one occurrence of any adverse event with a true incidence rate for a given dose group of 10%, 15%, 20% or 25%, respectively.
Pharmacokinetic outcomes were estimated using non‐compartmental methods with Phoenix WINNONLIN version 8.0 (Certara, Princeton, NJ, USA). In Part A, dose proportionality for C max and AUC was assessed using a power model with log (PK parameter) as response variable and log (dose) as predictor fitted to the data for plasma C max, AUClast and AUCinf. In Part B, AUCinf and C max were compared among the fasting dose groups with dose as a fixed effect. A 90% confidence interval (CI) was constructed on the ratio of C max fed to C max fasting and a similar one constructed for AUC to obtain a preliminary estimate of the food effect of P218 on transformed parameters; fasting/fed treatment, period and sequence were fixed effects and subject to a random effect. Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to Pharmacology.
3. RESULTS
3.1. Study participants and exposure
In Part A, 42 subjects received P218 at single doses of 10‐1000 mg (n = 6 per group) and 14 subjects received placebo (Table 1). In Part B, four subjects received a single dose of 250 mg P218 when fasted in Period 1 and fed in Period 2, four other subjects received 250 mg P218 when fed in Period 1 and fasted in Period 2. Baseline characteristics of the subjects were similar across all groups (Table 1). All subjects were included in the safety and pharmacokinetic analyses.
Table 1.
Subject disposition and demographic characteristics
| Characteristic | Part A: P218 dose group | Part B | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 10 mg (n = 6) | 30 mg (n = 6) | 100 mg (n = 6) | 250 mg (n = 6) | 500 mg (n = 6) | 750 mg (n = 6) | 1000 mg (n = 6) | Placebo (n = 14) | 250 mg fed‐fasted (n = 4) | 250 mg fasted‐fed (n = 4) | |
| Enrolled | 6 (100) | 6 (100) | 6 (100) | 6 (100) | 6 (100) | 6 (100) | 6 (100) | 14 (100) | 4 (100) | 4 (100) |
| Safety population, n (%) | 6 (100) | 6 (100) | 6 (100) | 6 (100) | 6 (100) | 6 (100) | 6 (100) | 14 (100) | 4 (100) | 4 (100) |
| PK population, n (%) | 6 (100) | 6 (100) | 6 (100) | 6 (100) | 6 (100) | 6 (100) | 6 (100) | 0 | 4 (100) | 4 (100) |
| PD population, n (%) | 6 (100) | 6 (100) | 6 (100) | 6 (100) | 6 (100) | 6 (100) | 6 (100) | 14 (100) | 3 (75) | 4 (100) |
| Mean (SD) age (years) | 32.2 (8.8) | 26.8 (6.9) | 29.7 (8.0) | 27.2 (7.7) | 26.2 (6.2) | 27.5 (4.1) | 28.7 (7.6) | 25.4 (6.2) | 23.0 (5.0) | 32.0 (6.2) |
| Male gender, n (%) | 6 (100) | 6 (100) | 6 (100) | 6 (100) | 6 (100) | 6 (100) | 6 (100) | 14 (100) | 4 (100) | 4 (100) |
| Mean (SD) weight (kg) | 73.4 (8.1) | 70.8 (8.7) | 66.1 (5.9) | 76.6 (5.4) | 77.0 (7.0) | 74.2 (6.4) | 70.9 (3.6) | 70.2 (7.6) | 73.9 (12.4) | 80.0 (6.9) |
| Mean (SD) height (cm) | 177.7 (6.6) | 177.3 (10.2) | 169.5 (5.9) | 182.0 (3.3) | 183.2 (4.7) | 182.2 (4.4) | 177.8 (8.6) | 179.0 (7.5) | 184.3 (6.1) | 180.0 (3.6) |
| Body mass index (kg/m2) | 23.2 (1.4) | 22.5 (2.0) | 23.0 (1.9) | 23.2 (2.0) | 23.0 (2.4) | 22.4 (1.7) | 22.6 (2.4) | 21.9 (1.9) | 21.7 (2.5) | 24.7 (1.3) |
| Race, n (%) | ||||||||||
| Caucasian | 5 (83.3) | 3 (50.0) | 4 (66.7) | 3 (50.0) | 3 (50.0) | 5 (83.3) | 5 (83.3) | 8 (57.1) | 3 (75.0) | 3 (75.0) |
| Black African | 1 (16.7) | 1 (16.7) | 0 | 1 (16.7) | 1 (16.7) | 1 (16.7) | 0 | 3 (21.4) | 1 (25.0) | 1 (25.0) |
| Asian | 0 | 1 (16.7) | 1 (16.7) | 1 (16.7) | 1 (16.7) | 0 | 0 | 2 (14.3) | 0 | 0 |
| Other | 0 | 1 (16.7) | 1 (16.7) | 1 (16.7) | 1 (16.7) | 0 | 1 (16.7) | 1 (7.1) | 0 | 0 |
Abbreviations: PD, pharmacodynamic; PK, pharmacokinetics.
3.2. Adverse events
Overall, 21 treatment‐emergent adverse events occurred in 15 of the 64 subjects enrolled (Table 2). Except for one moderate case of back pain reported after 750 mg P218, all other adverse events were mild in severity. There were no serious adverse events, no deaths and no study withdrawals owing to adverse events. All adverse events resolved spontaneously. An unblinded review of the adverse events occurring in Part A found that four subjects had five adverse events that were possibly related to P218 administration based upon their temporality (constipation, abdominal pain plus soft faeces, headache and abdominal pain). In Part B, after receiving the 250 mg dose in a fed state, one subject of the eight enrolled experienced four mild adverse events, all of which were considered possibly treatment related (Table 2). The symptoms (dizziness, nausea, hot flush, mild headache) appeared to be related to postural hypotension that occurred when standing up for a scheduled blood pressure measurement at 2 hours post dose and resolved within 30 minutes. Overall, nine adverse events in five subjects were regarded as related to P218 administration.
Table 2.
Treatment‐emergent adverse events occurring with P218 at doses of 10‐1000 mg or placebo in Part A and following 250 mg P218 in fed or fasted state in Part B
| Adverse event, n subjects (%) | Part A | Part B (250 mg P218) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 10 mg (n = 6) | 30 mg (n = 6) | 100 mg (n = 6) | 250 mg (n = 6) | 500 mg (n = 6) | 750 mg (n = 6) | 1000 mg (n = 6) | Placebo (n = 14) | Fasted (n = 8) | Fed (n = 8) | |
| Any adverse events | 0 | 0 | 2 (33.3) | 1 (16.7) | 2 (33.3) | 3 (50.0) | 3 (50.0) | 3 (21.4) | 0 | 1 (12.5) |
| Presyncope | 0 | 0 | 0 | 0 | 1 (16.7) | 0 | 0 | 0 | 0 | 0 |
| Dizziness | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (12.5) |
| Abdominal pain | 0 | 0 | 0 | 0 | 0 | 0 | 2 (33.3) | 0 | 0 | 0 |
| Constipation | 0 | 0 | 1 (16.7) | 1 (16.7) | 0 | 0 | 0 | 0 | 0 | 0 |
| Faeces soft | 0 | 0 | 0 | 0 | 0 | 0 | 1 (16.7) | 0 | 0 | 0 |
| Nausea | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (12.5) |
| Nasopharyngitis | 0 | 0 | 0 | 0 | 0 | 2 (33.3) | 0 | 1 (7.1) | 0 | 0 |
| Sinusitis | 0 | 0 | 1 (16.7) | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Back pain | 0 | 0 | 0 | 0 | 0 | 2 (33.3) | 0 | 0 | 0 | 0 |
| Headache | 0 | 0 | 0 | 0 | 1 (16.7) | 0 | 1 (16.7) | 1 (7.1) | 0 | 1 (12.5) |
| Paraesthesia | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (12.5) |
| Oropharyngeal pain | 0 | 0 | 0 | 0 | 0 | 0 | 1 (16.7) | 1 (7.1) | 0 | 0 |
All events were mild, except one case of moderate back pain (Part A: 750 mg).
3.3. Laboratory parameters
In Part A, there were no major deviations from normal values at screening for any haematology parameter and no abnormal changes of clinical significance in any haematology parameter (Supporting Information Table S3). In Part B, there were no notable changes in the fed or fasted state for haematology parameters (Supporting Information Table S3). For clinical biochemistry, in Part A, although some isolated pre‐ to post‐dose biochemistry changes were noted (direct bilirubin, potassium, creatine phosphokinase and C‐reactive protein), these were without clinical findings and not considered clinically relevant (Supporting Information Table S4). In Part B, there were no clinically relevant abnormalities or deviations from normal biochemistry values (Supporting Information Table S4). For coagulation parameters, there were no clinically significant abnormal values in any subject at any time point in either study part, nor were any clinically significant changes from baseline observed (Supporting Information Table S5). Most subjects showed a general trend of reducing serum folate levels after single‐dose administration during the study, however, the same trend was observed in placebo subjects and there was no apparent relationship to P218 dose (Supporting Information Table S6).
3.4. Pharmacokinetics and food effect
Plasma concentration‐time data were available from all completed cohorts for P218 as well as the three primary metabolites P218 β‐acyl glucuronide, P218‐OH and P218‐OH β‐acyl glucuronide (Figure 2 and Supporting Information Fig. S1). P218 was rapidly absorbed with C max values achieved between 30 minutes and 2 hours post dose (Table 3). Plasma concentrations of P218 declined in a bi‐exponential manner with P218 t 1/2 values ranging from 3.1 hours at 10 mg to 19.6 hours at 1000 mg (Table 3). P218 β‐acyl glucuronide and P218‐OH β‐acyl glucuronide were confirmed as the major metabolites with P218‐OH exposure at lower levels (Table 4). P218 parent and metabolite exposure values as assessed by C max and AUCinf increased in a dose‐proportional manner between 100 and 1000 mg (Supporting Information Table S7). Co‐administration of P218 with food reduced C max and delayed absorption, but with no significant impact on AUC (Table 5 Supporting Information Fig. S2). Excretion of unchanged P218 in the urine accounted for 6.73–11.94% of the administered dose with higher values observed at higher doses (Table 6).
Figure 2.
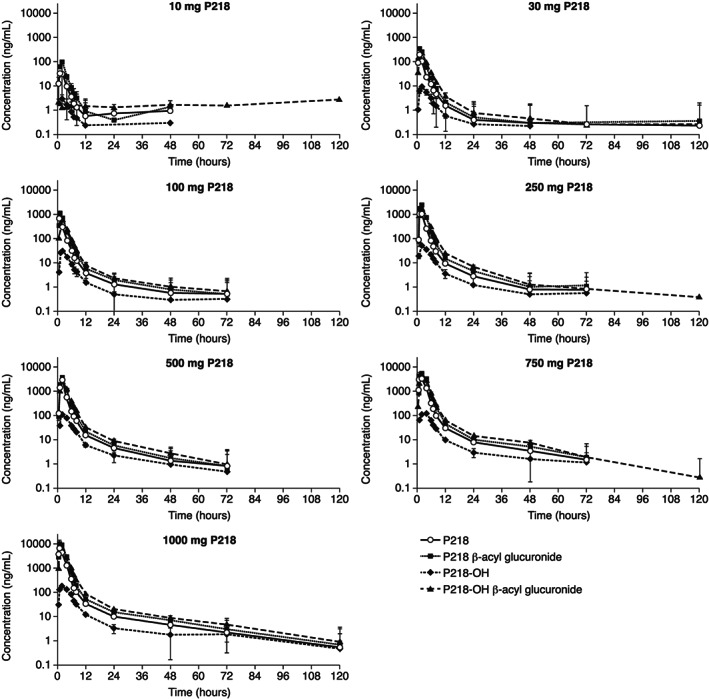
Geometric mean (±SD) plasma concentration‐time profiles for P218 compared to metabolites following single P218 doses of 10‐1000 mg. Note: Data plotted for P218 and metabolites shown individually for all doses are given in Supporting Information Fig. S1
Table 3.
Pharmacokinetic parameters for P218 in healthy subjects following single oral doses
| Dose (mg) | C max (ng/mL) | T max (h) | AUClast (h.ng/mL) | AUCinf (h.ng/mL) | t 1/2 (h) | CL/F (L/h) | Vz/F (L) |
|---|---|---|---|---|---|---|---|
| 10 | 64.33 (58.4) | 1.0 (0.5‐2.0) | 125.97 (27.5) | 129.20 (28.8) | 3.14 (50.9) | 77.40 (28.8) | 350.35 (38.5) |
| 30 | 203.19 (24.2) | 1.0 (1.0‐2.0) | 435.84 (21.2) | 413.35 (16.4) | 6.87 (82.7) | 72.58 (16.4) | 718.98 (74.0) |
| 100 | 929.13 (45.8) | 0.75 (0.5‐2.0) | 1574.05 (24.8) | 1578.35 (27.3) | 17.17 (25.8) | 63.36 (27.3) | 1569.59 (44.2) |
| 250 | 2093.29 (17.5) | 1.0 (0.5‐2.0) | 3904.39 (25.3) | 3924.78 (25.1) | 8.91 (104.7) | 63.70 (25.1) | 818.99 (108.1) |
| 500 | 4158.80 (41.2) | 1.5 (0.5‐2.0) | 7731.40 (33.3) | 7757.98 (33.1) | 12.71 (58.9) | 64.45 (33.1) | 1181.52 (61.1) |
| 750 | 5979.78 (35.2) | 1.0 (0.5‐4.0) | 13403.09 (18.8) | 13568.48 (20.4) | 16.02 (60.3) | 55.28 (20.4) | 1277.11 (81.3) |
| 1000 | 8641.96 (31.1) | 1.0 (1.0‐2.0) | 17608.63 (29.7) | 17814.77 (28.3) | 19.61 (79.1) | 56.13 (28.3) | 1587.73 (89.5) |
C max, AUClast, AUCinf, t 1/2, CL/F and Vz/F are described as geometric mean (CV%); T max is described as median (range).
Abbreviations: AUClast, area under the concentration‐time curve for the sampling period; AUCinf, area under the concentration‐time curve extrapolated to infinity; CL/F, clearance; C max, maximum plasma concentration; CV%, percentage coefficient of variation; t 1/2, terminal half‐life; T max, time to C max; Vz/F, volume of distribution during the terminal phase.
Table 4.
Pharmacokinetic parameters for P218 metabolites in healthy subjects
| P218 β‐acyl glucuronide | ||||||
|---|---|---|---|---|---|---|
| Dose (mg) | C max (ng/mL) | T max (h) | AUClast (h.ng/mL) | AUCinf (h.ng/mL) | t 1/2 (h) | Mean ratio of AUClast metabolite/P218 (%) |
| 10 | 140.42 (48.0) | 1.5 (0.5‐2.0) | 313.80 (33.8) | 316.57 (34.0) | 3.74 (26.8) | 252.68 |
| 30 | 384.45 (20.4) | 1.0 (1.0‐2.0) | 858.69 (20.1) | 863.40 (20.3) | 8.12 (82.9) | 202.22 |
| 100 | 1372.49 (46.9) | 1.0 (0.5‐2.0) | 2857.73 (26.2) | 3111.35 (19.1) | 16.50 (16.6) | 188.45 |
| 250 | 3807.83 (26.1) | 1.5 (1.0‐2.1) | 8226.07 (27.7) | 8251.06 (27.6) | 9.24 (90.3) | 211.14 |
| 500 | 4853.60 (48.6) | 1.5 (1.0‐2.0) | 11459.20 (42.9) | 11494.98 (42.7) | 14.15 (61.7) | 151.94 |
| 750 | 8325.86 (15.9) | 1.5 (1.0‐4.0) | 22895.45 (16.2) | 22996.04 (16.4) | 15.71 (59.8) | 176.74 |
| 1000 | 12567.43 (44.9) | 1.0 (1.0‐2.0) | 31835.68 (49.5) | 32077.15 (49.0) | 21.45 (66.3) | 187.86 |
| P218‐OH | ||||||
|---|---|---|---|---|---|---|
| Dose (mg) | C max (ng/mL) | T max (h) | AUClast (h.ng/mL) | AUCinf (h.ng/mL) | t 1/2 (h) | Mean ratio of AUClast metabolite/P218 (%) |
| 10 | 3.16 (27.3) | 2.0 (1.0‐2.0) | 11.89 (23.7) | 13.37 (26.2) | 2.69 (55.9) | 9.75 |
| 30 | 9.81 (44.9) | 2.0 (1.0‐2.0) | 46.87 (41.0) | 49.48 (40.0) | 4.53 (65.8) | 11.16 |
| 100 | 35.98 (18.8) | 1.0 (1.0‐2.0) | 160.62 (32.1) | 163.76 (35.5) | 16.44 (90.6) | 10.41 |
| 250 | 57.74 (33.8) | 2.0 (1.0‐4.0) | 312.94 (24.9) | 322.63 (25.1) | 8.66 (61.2) | 8.36 |
| 500 | 110.94 (27.0) | 2.0 (1.0‐2.0) | 619.88 (28.6) | 650.81 (24.4) | 20.91 (50.9) | 8.23 |
| 750 | 143.65 (17.7) | 2.0 (1.0‐4.0) | 863.77 (7.2) | 890.45 (8.0) | 15.20 (36.5) | 6.52 |
| 1000 | 192.70 (21.3) | 2.0 (1.0‐2.0) | 1168.07 (32.5) | 1187.56 (27.7) | 15.41 (44.8) | 6.75 |
| P218‐OH β‐acyl glucuronide | ||||||
|---|---|---|---|---|---|---|
| Dose (mg) | C max (ng/mL) | T max (h) | AUClast (h.ng/mL) | AUCinf (h.ng/mL) | t 1/2 (h) | Mean ratio of AUClast metabolite/P218 (%) |
| 10 | 86.89 (34.4) | 2.0 (1.0‐2.0) | 258.37 (24.4) | 260.63 (24.2) | 3.56 (41.3) | 212.89 |
| 30 | 270.40 (36.0) | 2.0 (1.0‐2.0) | 887.21 (23.6) | 853.04 (22.7) | 7.50 (118.9) | 209.09 |
| 100 | 888.30 (33.0) | 1.0 (1.0‐2.0) | 2479.41 (19.8) | 2595.61 (19.5) | 16.86 (28.9) | 159.44 |
| 250 | 2288.02 (29.0) | 1.5 (1.0‐2.1) | 6963.97 (20.8) | 6984.84 (20.6) | 7.62 (57.1) | 181.96 |
| 500 | 2888.72 (20.3) | 2.0 (1.0‐2.0) | 9672.66 (22.4) | 9719.67 (22.5) | 14.85 (43.8) | 128.83 |
| 750 | 4539.26 (20.0) | 2.0 (1.0‐4.0) | 17427.14 (16.6) | 16489.84 (10.1) | 12.60 (28.8) | 137.09 |
| 1000 | 6569.84 (31.7) | 2.0 (1.0‐2.0) | 23715.16 (41.5) | 22295.25 (25.2) | 11.98 (49.1) | 136.00 |
C max, AUClast, AUCinf and t 1/2, are described as geometric mean (CV%); T max is described as median (range).
Metabolite ratios were calculated as AUCinf of analyte/(sum of all AUCinf analytes).
Abbreviations: AUClast, area under the concentration‐time curve for the sampling period; AUCinf, area under the concentration‐time curve extrapolated to infinity; C max, maximum plasma concentration; CV%, percentage coefficient of variation; t 1/2, terminal half‐life; T max, time to C max.
Table 5.
Effect of food on P218 plasma PK parameters
| Dose | Fasted, 250 mg (n = 8) | Fed, 250 mg (n = 8) | Food effect, ratio (95%CI) |
|---|---|---|---|
| C max (ng/mL) | 1581.03 (57.6) | 1026.12 (87.2) | 0.65 (0.38, 1.11) |
| T max (h) | 1.0 (0.5‐2.0) | 2.0 (1.0‐4.0) | … |
| AUClast (h.ng/mL) | 3267.11 (32.2) | 3165.14 (42.4) | 0.97 (0.84, 1.12) |
| AUCinf (h.ng/mL) | 3301.99 (31.5) | 3215.84 (42.2) | 0.97 (0.85, 1.12) |
| t 1/2 (h) | 19.50 (71.5) | 23.04 (58.9) | … |
| CL/F (L/h) | 75.71 (31.5) | 77.74 (42.2) | … |
| Vz/F (L) | 2129.58 (81.7) | 2583.99 (72.8) | … |
C max, AUClast, AUCinf, t 1/2, CL/F and Vz/F are described as geometric mean (CV%); T max is described as median (range).
Abbreviations: AUClast, area under the concentration‐time curve for the sampling period; AUCinf, area under the concentration‐time curve extrapolated to infinity; CL/F, clearance; C max, maximum plasma concentration; CV%, percentage coefficient of variation; t 1/2, terminal half‐life; T max, time to Cmax; Vz/F, volume of distribution during the terminal phase.
Table 6.
P218 urine PK parameters
| P218 dose (mg) | Amount of P218 excreted in urine (ng) at time | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0‐6 h | 6‐12 h | 12‐24 h | 24‐48 h | In 48 h | Dose recovered unchanged (%) | Clearance | ||||||||
| Geometric mean | CV | Geometric mean | CV | Geometric mean | CV | Geometric mean | CV | Geometric mean | CV | Geometric mean | CV | Geometric mean | CV | |
| 10 | 568525.1 | 55.9 | 65452.7 | 31.3 | 21132.3 | 20.7 | 18833.8 | 24.4 | 685987.1 | 44.4 | 6.88 | 44.0 | 5445.9 | 56.9 |
| 30 | 1757637.5 | 55.1 | 142613.1 | 67.2 | 50970.6 | 32.6 | 33948.2 | 30.0 | 2025482.6 | 50.1 | 6.73 | 50.6 | 4647.3 | 43.8 |
| 100 | 6432925.0 | 35.1 | 266978.0 | 36.0 | 115412.4 | 56.4 | 93966.9 | 24.7 | 6924034.7 | 34.8 | 6.92 | 34.5 | 4398.0 | 21.8 |
| 250 | 15823908.7 | 24.1 | 614413.3 | 76.2 | 204287.6 | 123.3 | 92206.9 | 482.5 | 17059380.1 | 22.0 | 6.83 | 22.0 | 4369.3 | 27.2 |
| 500 | 35188232.0 | 28.2 | 2016567.8 | 63.0 | 529863.2 | 79.2 | 340479.6 | 92.3 | 38470150.7 | 28.5 | 7.70 | 28.6 | 4975.8 | 28.2 |
| 750 | 79236662.7 | 40.8 | 2415818.1 | 116.1 | 735012.7 | 46.6 | 723125.7 | 52.3 | 84699011.0 | 36.6 | 11.31 | 36.7 | 6319.4 | 29.9 |
| 1000 | 1.2E+08 | 54.3 | 2371949.5 | 78.1 | 712410.0 | 34.1 | 809023.4 | 46.2 | 1.2E+08 | 52.3 | 11.94 | 52.6 | 6787.1 | 53.4 |
Abbreviation: CV, geometric coefficient of variation; PK, pharmacokinetic.
3.5. Electrocardiograms
There were no clinically significant abnormalities in ECG intervals; no prolongations of QTcF >30 milliseconds and no QTcF values >480 milliseconds were observed (Supporting Information Tables S8 and S9, Supporting Information Fig. S3). Aggregate QTcF values exceeding 450 milliseconds were observed in one subject in Part A (maximum 474 milliseconds 8 hours post dose), including pre dose. There was no deviation from baseline and no discernible trend in QTcF readings for this subject throughout the study. Further PK/pharmacodynamic (PD) modelling for QTcF was performed, to be reported separately.
3.6. Ex vivo antimalarial activity
For both the 250 and 500 mg P218 doses, within the first 12‐24 hours the concentration‐time profiles calculated from antimalarial activity of all six subjects were increased compared to the matching profiles provided by LC‐MS/MS (Figure 3 and Supporting Information Fig. S4). P218 T max was the same whether derived from the concentration‐time profiles obtained from LC‐MS/MS or calculated from the antimalarial activity of serum samples (Supporting Information Table S10), but C max was around 2‐3‐fold higher (Figure 3). The ratio of LC‐MS/MS versus calculated P218 concentration tended to increase over time (Supporting Information Table S10 and Fig. S4). These data suggest that the metabolites contribute to the antimalarial activity and the persistence of metabolites as P218 concentrations decline could explain the augmentation of antimalarial activity. As an additional investigation, using the same 3H‐hypoxanthine incorporation assay, the in vitro activity of P218‐OH against P. falciparum was determined, with a mean IC50 of 4.6 ± 0.35 nM, similar to that reported for the P218 parent (IC50 4.6 ± 1.9 nM11).
Figure 3.
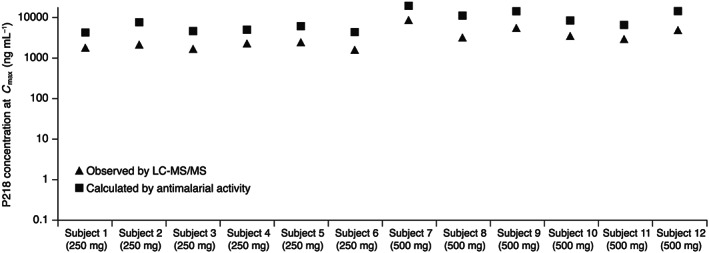
Observed P218 C max using LC‐MS/MS compared to the P218 C max calculated from the ex vivo antimalarial activity of serum samples collected at the same time point. Note: See Supporting Information Fig. S4 for concentration‐time curves
4. DISCUSSION
PfDHFR‐TS inhibitors are an important class of antimalarial, seeing widespread use for malaria treatment until parasite resistance curtailed their utility. Despite resistance, and because of a lack of feasible alternatives, pyrimethamine is still used in SMC in children and for the intermittent preventive treatment of malaria in pregnancy and infants in combination with sulphadoxine. These indications are key components of malaria programmes in Africa, substantially reducing morbidity and mortality.4 However, resistance to the combination is now widespread; mutations in dihydrofolate reductase (Pfdhfr gene) confer resistance to pyrimethamine while mutations in the dihydropteroate synthetase (Pfdhps gene) confer resistance to sulphadoxine.19 Malaria treatment was compromised with the emergence in East Africa of the quintuple mutant comprising a triple mutant dhfr allele (N51I, C59R, S108N) and a double mutant in dhps (A437G, K540E).20, 21, 22, 23 Although outcomes in chemoprevention may still be maintained in the presence of the quintuple mutant, the acquisition of additional mutations in dhps (540E or 581G) and/or in dhfr undermines the preventive efficacy of the sulphadoxine‐pyrimethamine regimen.24, 25
P218 has in vitro inhibitory activity on PfDHFR comparable to or greater than pyrimethamine and has potent in vitro and in vivo activity against Plasmodium strains resistant to pyrimethamine.11 This first‐in‐human study examined P218 safety, tolerability and PK following single oral dosing, as well as investigating the effect of food on PK and the ex vivo antimalarial activity of P218. P218 was well tolerated up to doses of 1000 mg, with infrequent and mild adverse events that resolved spontaneously. Although the sample size was small and adverse events infrequent, a potential for P218 to increase the frequency of gastrointestinal adverse events cannot be excluded at the highest dose level. However, overall the highest dose tested in the study (1000 mg) was well tolerated. Antifolate drugs are known to be associated with gastrointestinal adverse effects.26, 27, 28 Although there was a trend for reductions in serum folate with P218 when samples were taken at study end (day 11), these changes were smaller than those observed in the placebo group, not clinically relevant and there was no relationship to dose.
Four adverse events were reported by a single subject after dosing in the fed state, all occurring within the approximate same time frame and thus likely representing a single clinical event. Clinical presentations such as these are often seen in healthy volunteer trials and commonly represent vasovagal episodes or pre‐syncopal episodes caused by the trial procedures. Notably, this subject had higher P218 exposures in the fasted state without adverse events. Thus, overall, there appears to be no effect of food on P218 safety.
Preclinical findings of P218 receptor binding against thromboxane A2/PGH2 did not translate into any clinically relevant effect on blood clotting. No in vivo cardiac safety signal was identified for P218.
P218 was rapidly absorbed with exposures dose‐proportional at doses between 100 and 1000 mg, and although food delayed absorption by 1 hour and reduced C max by 35%, there was no effect on AUC. These findings are consistent with the potential for oral dosing identified preclinically.11
Based on an in vitro activity for P218 against P. falciparum of around 4 ng/mg,11 C max concentrations for all dose levels were in the pharmacologically relevant range. However, the t 1/2 for P218 and metabolites was less than 24 hours, even at the highest doses, which is considerably shorter than pyrimethamine (t 1/2 96 hours). This represents a challenge in developing P218 as a drug for chemoprotection.5 However, there is ongoing work to develop a P218 modified‐release formulation. The objective is to target at least once weekly, ideally monthly dosing, and a number of forms and formulations are being investigated.
P218 renal clearance appeared to decrease with increasing dose. The low level of P218 observed in urine indicates that, while renal clearance contributes to the elimination, it is not the primary route and the majority of the clearance takes place through drug metabolism.
PK characteristics were also reported for two major P218 metabolites in human plasma, P218 β‐acyl glucuronide and P218‐OH β‐acyl‐glucuronide, plus a minority metabolite, P218‐OH. Both acyl‐glucuronides appeared rapidly after dosing and were more abundant than unchanged parent. This is in contrast to the rat, in which P218 β‐acyl‐glucuronide was the most abundant drug‐related molecule, and the dog, in which unchanged P218 was most abundant. However, the current study was designed using a ‘total body burden’ approach which summed exposure to all acyl‐glucuronides in the animal toxicology studies.29 Thus, all metabolites had been adequately assessed in the toxicology studies and their total cumulative exposure was higher in animals at the no effect dose than the predicted exposure in humans.
The IC50 values derived from human serum following P218 dosing showed potent activity against P. falciparum in vitro. The ex vivo antimalarial bioassay indicated that at least one P218 metabolite must be active against P. falciparum because the antimalarial activity in serum samples was greater than that predicted by the P218 measured concentrations. Similar findings using the same ex vivo assay have been reported for the investigational antimalarial ACT‐451840, and an active metabolite was subsequently confirmed in human serum.30 The in vivo antimalarial activity of P218 metabolites against P. falciparum will require further evaluation.
In conclusion, P218 has a favourable safety and tolerability profile with no significant effect of food on safety, tolerability or PK. These findings support further clinical investigation of P218 in a controlled human malaria study for chemoprotection.31, 32
COMPETING INTERESTS
M.F.C., C.D., M.E.G., J.M. and S.Ch. are current employees of the study sponsor MMV. E.R. was previously employed at MMV when the study was conducted. G.L. and T.H. are paid consultants of MMV, U.L. and S.Co. are employees of Richmond Pharmacology, UK where the study was performed.
CONTRIBUTORS
M.F.C., E.R., C.D., M.E.G., U.L., G.L., T.H., J.M. and S.Ch. contributed to study design, protocol development and data analysis and interpretation. S.Co. contributed to data analysis and interpretation and development of the clinical study report. U.L. was the principal investigator and contributed to data acquisition. S.Ch., M.F.C. and U.L. provided medical oversight. M.F.C. developed the first draft of this article. All authors contributed to the development of this article, agreed the final version for submission and take full responsibility for the results reported.
Supporting information
Methods S1 Full subject inclusion and exclusion criteria
Methods S2 Calculation of P218 concentrations based on an ex vivo antimalarial assay
Table S1 Study safety assessments
Table S2 Study pharmacokinetic assessments
Table S3 Mean (SD) values for haematological measures following single doses of P218 or placebo
Table S4 Mean (SD) values for laboratory measures following single doses of P218 or placebo
Table S5 Coagulation measures at baseline and change from baseline following single‐dose P128 or placebo
Table S6 Baseline serum folate levels (µg/mL) and change in serum folate levels from baseline following single‐dose P218 or placebo
Table S7 Dose proportionality analysis of P218 single doses for P218 and metabolites
Table S8 Categorical QTcF interval data following a single dose of P218 or placebo
Table S9 Categorical QTcF interval data versus baseline following a single dose of P218 or placebo
Table S10 Concentration‐time data determined by LC‐MS/MS analysis (limit of quantification 0.200 ng/mL) versus concentration‐time data calculated from the P .falciparum antimalarial activity of P218 in serum samples
Figure S1 Geometric mean(±SD) plasma concentration‐time profiles for P218 and metabolites following single P218 doses of 10–1000 mg
Figure S2 Concentration‐time profiles for P218 and metabolites in subjects administered P218 250 mg in the fasted or fed state
Figure S3 Change in QTcF interval from baseline following a single dose of P218 or placebo
Figure S4 Comparison of concentration‐time profiles determined by LC‐MS/MS analysis (limit of quantification 0.200 ng/mL)versus concentration‐time data calculated from the P. falciparum antimalarial activity of P218 in serum samples following a 250 mg or 500 mg single dose of P218
ACKNOWLEDGEMENTS
The authors acknowledge Janssen Pharmaceuticals, Belgium and BIOTEC, Thailand for their ongoing collaboration with Medicines for Malaria Venture (MMV) in the development of P218. Funding for this study was provided by MMV and Janssen Pharmaceuticals. The authors acknowledge the support of Sumalee Kamchonwongpaisan, Darin Kongkasuriyachai, Yongyuth Yuthavong all of BIOTEC, Thailand. The Swiss Tropical and Public Health Institute (Basel, Switzerland) performed the ex vivo malaria assay and the authors acknowledge the contribution of Sergio Wittlin. The authors acknowledge the medical writing support provided by Naomi Richardson of Magenta Communications Ltd, Abingdon, UK, funded by MMV.
This study was funded by Medicines for Malaria Venture, Geneva Switzerland and Janssen Pharmaceuticals, Beerse, Belgium.
Chughlay MF, Rossignol E, Donini C, et al. First‐in‐human clinical trial to assess the safety, tolerability and pharmacokinetics of P218, a novel candidate for malaria chemoprotection. Br J Clin Pharmacol. 2020;86 1113–1124. 10.1111/bcp.14219
The authors confirm that the PI for this paper is Ulrike Lorch and that she had direct clinical responsibility for patients.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1. World Health Organisation . World malaria report. Geneva: WHO; [Cited 2019 19 July]. Available from: https://www.who.int/malaria/publications/world-malaria-report-2018/en/. [Google Scholar]
- 2. World Health Organization . Intermittent preventive treatment in pregnancy (IPTp). Geneva: WHO; [Cited 2018 30 June]. Available from: http://www.who.int/malaria/areas/preventive_therapies/pregnancy/en/. [Google Scholar]
- 3. Meremikwu MM, Donegan S, Sinclair D, Esu E, Oringanje C. Intermittent preventive treatment for malaria in children living in areas with seasonal transmission. Cochrane Database Syst Rev. 2012;2:CD003756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. World Health Organisation . Malaria prevention works: let's close the gap. Geneva: WHO; [Cited 2018 30 June]. Available from: http://www.who.int/malaria/publications/atoz/malaria-prevention-works/en/. [Google Scholar]
- 5. Burrows JN, Duparc S, Gutteridge WE, et al. New developments in anti‐malarial target candidate and product profiles. Malar J. 2017;16:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Olaleye A, Okusanya BO, Oduwole O, Esu E, Meremikwu M. A systematic review and meta‐analysis of dihydroartemisinin‐piperaquine versus sulphadoxine‐pyrimethamine for malaria prevention in pregnancy. Int J Gynaecol Obstet. 2019;146:43‐55. [DOI] [PubMed] [Google Scholar]
- 7. Chotsiri P, Zongo I, Milligan P, et al. Optimal dosing of dihydroartemisinin‐piperaquine for seasonal malaria chemoprevention in young children. Nat Commun. 2019;10:480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. World Health Organization . Emergency response to artemisinin resistance in the Greater Mekong subregion: Regional framework for action 2013‐2015. Geneva: WHO; [Cited 2018 26 March]. Available from: http://www.who.int/malaria/publications/atoz/9789241505321/en/. [Google Scholar]
- 9. Sirawaraporn W, Prapunwattana P, Sirawaraporn R, Yuthavong Y, Santi DV. The dihydrofolate reductase domain of Plasmodium falciparum thymidylate synthase‐dihydrofolate reductase. Gene synthesis, expression, and anti‐folate‐resistant mutants. J Biol Chem. 1993;268:21637‐21644. [PubMed] [Google Scholar]
- 10. Sirawaraporn W, Sathitkul T, Sirawaraporn R, Yuthavong Y, Santi DV. Antifolate‐resistant mutants of Plasmodium falciparum dihydrofolate reductase. Proc Natl Acad Sci USA. 1997;94:1124‐1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yuthavong Y, Tarnchompoo B, Vilaivan T, et al. Malarial dihydrofolate reductase as a paradigm for drug development against a resistance‐compromised target. Proc Natl Acad Sci USA. 2012;109:16823‐16828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Abbat S, Jain V, Bharatam PV. Origins of the specificity of inhibitor P218 toward wild‐type and mutant PfDHFR: a molecular dynamics analysis. J Biomol Struct Dyn. 2015;33(9):1913‐1928. [DOI] [PubMed] [Google Scholar]
- 13. Alexander SP, Kelly E, Marrion NV, et al. The concise GUIDE to PHARMACOLOGY 2017/18. Br J Pharmacol. 2017;174(Suppl 1):S1‐S446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. U.S. Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research (CDER) . Guidance for Industry ‐ Estimating the maximum safe starting dose in initial clinical trials for therapeutics in adult healthy subjects. Rockville: FDA; [Cited 2018 24 October]. Available from: https://www.fda.gov/downloads/Drugs/Guidances/UCM078932.pdf%23search=%27guidekines+for+industry+sfe+starting%27. [Google Scholar]
- 15. U.S. Department of Health and Human Services Food and Drug Administration Center for Drug Evaluation and Research (CDER) . Guidance for Industry: Food‐Effect Bioavailability and Fed Bioequivalence Studies. Rockville: FDA; [Cited 2019 21 September]. Available from:. [Google Scholar]
- 16. Miller AB, Hoogstraten B, Staquet M, Winkler A. Reporting results of cancer treatment. Cancer. 1981;47:207‐214. [DOI] [PubMed] [Google Scholar]
- 17. Le Manach C, Scheurer C, Sax S, et al. Fast in vitro methods to determine the speed of action and the stage‐specificity of anti‐malarials in Plasmodium falciparum . Malar J. 2013;12:424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Snyder C, Chollet J, Santo‐Tomas J, Scheurer C, Wittlin S. In vitro and in vivo interaction of synthetic peroxide RBx11160 (OZ277) with piperaquine in Plasmodium models. Exp Parasitol. 2007;115(3):296‐300. [DOI] [PubMed] [Google Scholar]
- 19. Okell LC, Griffin JT, Roper C. Mapping sulphadoxine‐pyrimethamine‐resistant Plasmodium falciparum malaria in infected humans and in parasite populations in Africa. Sci Rep. 2017;7:7389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Omar SA, Adagu IS, Warhurst DC. Can pretreatment screening for dhps and dhfr point mutations in Plasmodium falciparum infections be used to predict sulfadoxine‐pyrimethamine treatment failure? Trans R Soc Trop Med Hyg. 2001;95:315‐319. [DOI] [PubMed] [Google Scholar]
- 21. Staedke SG, Sendagire H, Lamola S, Kamya MR, Dorsey G, Rosenthal PJ. Relationship between age, molecular markers, and response to sulphadoxine‐pyrimethamine treatment in Kampala, Uganda. Trop Med Int Health. 2004;9(5):624‐629. [DOI] [PubMed] [Google Scholar]
- 22. Kublin JG, Dzinjalamala FK, Kamwendo DD, et al. Molecular markers for failure of sulfadoxine‐pyrimethamine and chlorproguanil‐dapsone treatment of Plasmodium falciparum malaria. J Infect Dis. 2002;185(3):380‐388. [DOI] [PubMed] [Google Scholar]
- 23. Desai M, Gutman J, Taylor SM, et al. Impact of sulfadoxine‐pyrimethamine resistance on effectiveness of intermittent preventive therapy for malaria in pregnancy at clearing infections and preventing low birth weight. Clin Infect Dis. 2016;62:323‐333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gesase S, Gosling RD, Hashim R, et al. High resistance of Plasmodium falciparum to sulphadoxine/pyrimethamine in northern Tanzania and the emergence of dhps resistance mutation at codon 581. PLoS One. 2009;4:e4569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Braun V, Rempis E, Schnack A, et al. Lack of effect of intermittent preventive treatment for malaria in pregnancy and intense drug resistance in western Uganda. Malar J. 2015;14:372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Shiroky JB, Neville C, Esdaile JM, et al. Low‐dose methotrexate with leucovorin (folinic acid) in the management of rheumatoid arthritis. Results of a multicenter randomized, double‐blind, placebo‐controlled trial. Arthritis Rheum. 1993;36(6):795‐803. [DOI] [PubMed] [Google Scholar]
- 27. Shea B, Swinden MV, Ghogomu ET, et al. Folic acid and folinic acid for reducing side effects in patients receiving methotrexate for rheumatoid arthritis. J Rheumatol. 2014;41:1049‐1060. [DOI] [PubMed] [Google Scholar]
- 28. Nzila A, Okombo J, Molloy AM. Impact of folate supplementation on the efficacy of sulfadoxine/pyrimethamine in preventing malaria in pregnancy: the potential of 5‐methyl‐tetrahydrofolate. J Antimicrob Chemother. 2014;69(2):323‐330. [DOI] [PubMed] [Google Scholar]
- 29. Smith DA, Hammond T, Baillie TA. Safety assessment of acyl glucuronides ‐ a simplified paradigm. Drug Metab Dispos. 2018;46(6):908‐912. [DOI] [PubMed] [Google Scholar]
- 30. Bruderer S, Hurst N, de Kanter R, et al. First‐in‐humans study of the safety, tolerability, and pharmacokinetics of ACT‐451840, a new chemical entity with antimalarial activity. Antimicrob Agents Chemother. 2015;59:935‐942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Roestenberg M, Hoogerwerf MA, Ferreira DM, Mordmuller B, Yazdanbakhsh M. Experimental infection of human volunteers. Lancet Infect Dis. 2018;18(10):e312‐e322. [DOI] [PubMed] [Google Scholar]
- 32. Stanisic DI, McCarthy JS, Good MF. Controlled human malaria infection: applications, advances, and challenges. Infect Immun. 2018;86:e00479‐17. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Methods S1 Full subject inclusion and exclusion criteria
Methods S2 Calculation of P218 concentrations based on an ex vivo antimalarial assay
Table S1 Study safety assessments
Table S2 Study pharmacokinetic assessments
Table S3 Mean (SD) values for haematological measures following single doses of P218 or placebo
Table S4 Mean (SD) values for laboratory measures following single doses of P218 or placebo
Table S5 Coagulation measures at baseline and change from baseline following single‐dose P128 or placebo
Table S6 Baseline serum folate levels (µg/mL) and change in serum folate levels from baseline following single‐dose P218 or placebo
Table S7 Dose proportionality analysis of P218 single doses for P218 and metabolites
Table S8 Categorical QTcF interval data following a single dose of P218 or placebo
Table S9 Categorical QTcF interval data versus baseline following a single dose of P218 or placebo
Table S10 Concentration‐time data determined by LC‐MS/MS analysis (limit of quantification 0.200 ng/mL) versus concentration‐time data calculated from the P .falciparum antimalarial activity of P218 in serum samples
Figure S1 Geometric mean(±SD) plasma concentration‐time profiles for P218 and metabolites following single P218 doses of 10–1000 mg
Figure S2 Concentration‐time profiles for P218 and metabolites in subjects administered P218 250 mg in the fasted or fed state
Figure S3 Change in QTcF interval from baseline following a single dose of P218 or placebo
Figure S4 Comparison of concentration‐time profiles determined by LC‐MS/MS analysis (limit of quantification 0.200 ng/mL)versus concentration‐time data calculated from the P. falciparum antimalarial activity of P218 in serum samples following a 250 mg or 500 mg single dose of P218
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.