

Introduction
Juvenile xanthogranuloma (JXG) is an uncommon benign histiocytic tumor that classically presents as a 0.2- to 2-cm, well demarcated, dome-shaped, red-orange to brown nodule or papule that becomes increasingly yellow and flat over time.1,2 JXG is the most common of the cutaneous and mucocutaneous histiocytoses, but the exact incidence and prevalence are unknown.2 Lesions are often present at birth (up to 34.5% of cases)3 or develop during the first years of life; however, JXG can also present in adulthood. JXG may increase in size or number but typically become stable or begin to spontaneously regress and disappear by the age of 3 to 6 years.1 The condition is overall self-limiting, carrying an excellent prognosis; however, some cases of extracutaneous JXG, such as those involving the eye or visceral organs, can have significant morbidity and mortality.4 An association between JXG, neurofibromatosis type 1 (NF1), and juvenile myelomonocytic leukemia (JMML) has been well-described in the literature. The incidence of JMML in patients with both NF1 and JXG is 20 to 32 times higher than the incidence of JMML with NF1 alone.5
Case presentation
A 5-week-old infant was referred to the dermatology clinic for evaluation of subcutaneous nodules on his scalp, trunk, groin, and extremities. Many lesions were present since birth, but the mother reported the appearance of new nodules appearing throughout the body over the last 2 weeks. The infant had an uncomplicated vaginal delivery at 38 weeks, at which time he was noted to have petechiae and purpura covering much of his body and several nodules on his trunk and extremities (Fig 1). Laboratory values at birth revealed a platelet count of 25,000, and the baby spent 2 weeks in the neonatal intensive care unit where alloimmune thrombocytopenia, unrelated to his skin nodules, was eventually diagnosed. He received multiple platelet transfusions and intravenous immunoglobulins infusions with eventual resolution of his thrombocytopenia.
Fig 1.
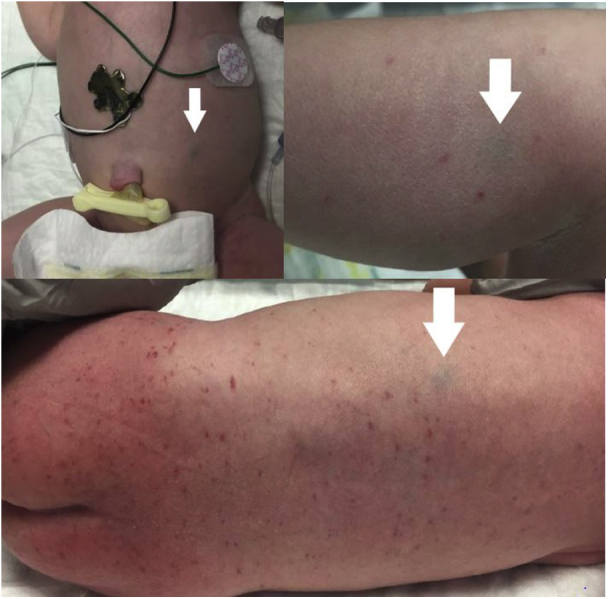
White arrows point to blue-gray patches and nodules on the abdomen, leg, and back present at birth representing JXG.
The patient was referred to the dermatology clinic by his pediatrician for further evaluation. The skin examination in office found at least 28 diffuse mobile subcutaneous nodules on the trunk, head, groin, and extremities, many of which had an overlying bluish hue (Fig 2). The initial clinical differential diagnosis included multifocal lymphangioendotheliomatosis with thrombocytopenia, leukemia cutis, and widespread lymphovascular malformations. A punch biopsy of a nodule on the thigh was performed. Biopsy results showed a dermal proliferation of histiocytes (Fig 3). Immunohistochemistry stained positive for CD68 and negative for S100 and CD1a. These results were consistent with the diagnosis of JXG, and the patient was referred to the ophthalmology department to rule out ocular involvement, although the family declined to follow through with the appointment. Ultrasound scans of the abdomen, head, and neck were unremarkable and did not show evidence of visceral JXG. Because of the association between JXG and JMML, a workup was also performed to rule out concomitant JMML. This patient lacked the hallmark findings of hepatomegaly and splenomegaly (seen in 97% of cases) and did not show any of the characteristic hematologic abnormalities such as monocytosis, anemia, and thrombocytopenia on repeat complete blood count over time.6 Follow-up at 4 months found the same distribution of subcutaneous nodules but with the orange-to-tan coloration typical of JXG (Fig 4). At 10 months, follow-up examination found only residual hyperpigmented macules, and at 12 months there was no evidence of JXG present on examination.
Fig 2.

At 5 weeks of age, white arrows point to subcutaneous nodules with overlying blue hue on the leg.
Fig 3.

A, Low-power view of punch biopsy shows dermal proliferation of histiocytes. B, High-power view shows mononuclear and touton giant cells admixed with lymphocytes and occasional eosinophils. (A and B, Hematoxylin-eosin stain; original magnifications: A, ×20; B, ×400.)
Fig 4.

By 4 months of age, the subcutaneous nodules have become orange to tan in color as seen on the leg, back, and abdomen.
Discussion
The etiology of JXG is unknown; however, there may be potential differences in pathogenesis between solitary and systemic JXG and the role that genetic mutations play. When looking at solitary cutaneous JXG, 89% (17 of 19) had no identifiable genetic changes.7 In comparison, some cases of systemic JXG have been shown to have mutations in the MAPK pathway, 58% (7 of 12) with mutations in the MAP2K1 (3 of 12), KRAS (2 of 12), and NRAS + ARAF (2 of 12) genes.8
This case of JXG was atypical because of the number of lesions, many of which could be palpated but not visualized, and the color of the visible nodules. JXG overwhelmingly presents as a single lesion. Two of the largest series to date found that 88% (114 of 129 and 153 of 174)3,9 of all JXG presented as a solitary lesion. Additionally, when looking solely at cutaneous JXG, 90% of lesions were solitary with only 2% of cases having between 5 and 9 lesions and an additional 2% having between 10 and 20.3 For JXG presenting in the subcutaneous fat, deep soft tissue, and muscle, 100% were solitary.3 JXG also has a strong preference for cutaneous presentation, 74%9 to 81%,3 compared with extracutaneous sites such as the soft tissue, internal organs, or the eye. Finally, the common presenting colors of JXG are described as a range that includes red, orange, yellow, brown, and tan. This case of JXG with 28 subcutaneous nodules with overlying blue hue represents a highly atypical variant of JXG with a unique clinical presentation.
Footnotes
Funding sources: None.
Conflicts of interest: None disclosed.
References
- 1.Gianotti F., Caputo R. Histiocytic syndromes: a review. J Am Acad Dermatol. 1985;13(3):383–404. doi: 10.1016/s0190-9622(85)70181-8. [DOI] [PubMed] [Google Scholar]
- 2.Hernandez-Martin A., Baselga E., Drolet B.A., Esterly N.B. Juvenile xanthogranuloma. J Am Acad Dermatol. 1997;36(3):355–367. doi: 10.1016/s0190-9622(97)80207-1. [DOI] [PubMed] [Google Scholar]
- 3.Janssen D., Harms D. Juvenile xanthogranuloma in childhood and adolescence: a clinicopathologic study of 129 patients from the kiel pediatric tumor registry. Am J Surg Pathol. 2005;29(1):21–28. doi: 10.1097/01.pas.0000147395.01229.06. [DOI] [PubMed] [Google Scholar]
- 4.Freyer D.R., Kennedy R., Bostrom B.C., Kohut G., Dehner L.P. Juvenile xanthogranuloma: forms of systemic disease and their clinical implications. J Pediatr. 1996;129(2):227–237. doi: 10.1016/s0022-3476(96)70247-0. [DOI] [PubMed] [Google Scholar]
- 5.Zvulunov A., Barak Y., Metzker A. Juvenile xanthogranuloma, neurofibromatosis, and juvenile chronic myelogenous leukemia: world statistical analysis. Arch Derm. 1995;131(8):904–908. [PubMed] [Google Scholar]
- 6.Niemeyer C.M., Arico M., Basso G. Chronic myelomonocytic leukemia in childhood: a retrospective analysis of 110 cases. Blood. 1997;89(10):3534–3543. [PubMed] [Google Scholar]
- 7.Paxton C.N., O'Malley D.P., Bellizzi A.M. Genetic evaluation of juvenile xanthogranuloma: genomic abnormalities are uncommon in solitary lesions, advanced cases may show more complexity. Mod Pathol. 2017;30(9):1234–1240. doi: 10.1038/modpathol.2017.50. [DOI] [PubMed] [Google Scholar]
- 8.Diamond E.L., Durham B.H., Haroche J. Diverse and targetable kinase alterations drive histiocytic neoplasms. Cancer Discov. 2016;6(2):154–165. doi: 10.1158/2159-8290.CD-15-0913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dehner L.P. Juvenile xanthogranulomas in the first two decades of life: a clinicopathologic study of 174 cases with cutaneous and extracutaneous manifestations. Am J Surg Pathol. 2003;27(5):579–593. doi: 10.1097/00000478-200305000-00003. [DOI] [PubMed] [Google Scholar]