

Abstract
The SARS-CoV-2 is the causative agent of COVID-19 pandemic that is causing a global
health emergency. The lack of targeted therapeutics and limited treatment options have
triggered the scientific community to develop new vaccines or small molecule therapeutics
against various targets of SARS-CoV-2. The main protease (Mpro) is a well characterized
and attractive drug target because of its crucial role in processing of the polyproteins
which are required for viral replication. In order to provide potential lead molecules
against the Mpro for clinical use, we docked a set of 65 bioactive molecules of Tea plant
followed by exploration of the vast conformational space of protein-ligand complexes by
long term molecular dynamics (MD) simulations (1.50 µs). Top three bioactive molecules
(Oolonghomobisflavan-A, Theasinensin-D, and Theaflavin-3-O-gallate) were selected by
comparing their docking scores with repurposed drugs (Atazanavir, Darunavir, and
Lopinavir) against SARS-CoV-2. Oolonghomobisflavan-A molecule showed a good number of
hydrogen bonds with Mpro and higher MM-PBSA binding energy when compared to all three
repurposed drug molecules. during the time of simulation. This study showed
Oolonghomobisflavan-A as a potential bioactive molecule to act as an inhibitor for the
Mpro of SARS-CoV-2. 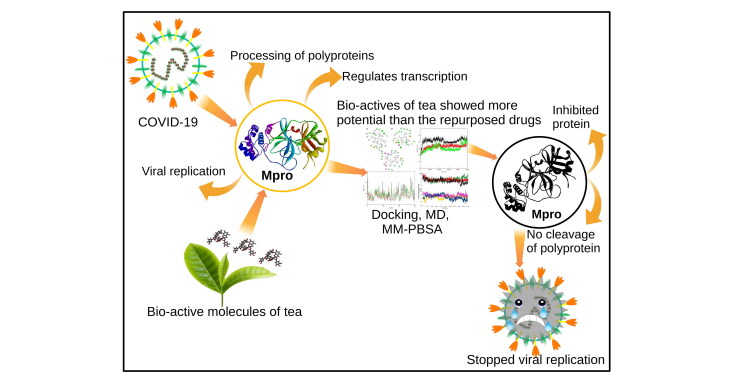
Communicated by Ramaswamy H. Sarma
Keywords: COVID-19, SARS-CoV-2, main protease, bioactive molecules
Introduction
Coronaviruses (CoVs) contain single positive-stranded RNA, enveloped inside a capsid with projections of peplomers. CoVs give rise to respiratory, neurological, and gastrointestinal diseases in humans (Zumla et al., 2016). CoVs were the causative agents of the 2002 severe acute respiratory syndrome (SARS) and the middle east respiratory syndrome (MERS) of 2012 (De Wit et al., 2016; Song et al., 2019). A new CoV designated as SARS-CoV-2 (Gorbalenya et al., 2020) caused an outbreak of viral pneumonia, named COVID-19, in the Wuhan city of China (F. Wu et al., 2020; Zhou et al., 2020). The worldwide spread of the disease at an extraordinary pace triggered the scientific community to rapidly develop efficient testing kits and a cure for infected patients. The COVID-19 outbreak was subsequently declared as a pandemic by the World Health Organization on March 11, 2020. The short term and non-specific approach for the treatment of COVID-19 patients is drug repurposing (Wang et al., 2020). However, efficient and potent drugs for specific targets are highly desirable.
Through extensive research, scientists around the world have suggested suitable drug targets against CoVs, which includes the Angiotensin-converting enzyme II (ACE2) entry receptor, the main protease (Mpro), and the RNA-dependent RNA polymerase (RdRp) (Li & De Clercq, 2020; Borgio et al., 2020). The drugs targeted to the ACE2 entry receptor, and the RdRp showed significant side effects and lower potency (Cameron & Castro, 2001; Han et al., 2006). The Mpro is one of the best-characterized and most promising drug targets in CoVs (Anand et al., 2003; Blanchard et al., 2004). The Mpro is responsible for the processing of the polyproteins that are products of the viral RNA transcription (Hilgenfeld, 2014). The Mpro targets and cleaves upto 11 sites on a large replicase protein (1ab, ∼790kDa). The inhibition of Mpro would essentially block viral replication. There are no known homologs of Mpro in humans with identical cleavage specificity. Hence, its inhibition is unlikely to show adverse toxic outcomes.
The amino acid sequences of the Mpro of all the SARS-like CoVs are highly conserved from humans to other animals (Ortega et al., 2020). The Mpro tends to form a dimer (protomer A and B). The protomers can be divided into three domains. Domain I (residues 8–101) and domain II (residues 102–184) forms an antiparallel β-barrel structure. The substrate-binding pocket is present in a cleft between these two domains. The domain III (residues 201-303) forms an antiparallel globular cluster consisting of five α-helices (Jin et al., 2020). Domain III is primarily involved in the regulation of dimerization of two protomers by a salt-bridge interaction between Glu290 of one protomer and Arg4 of the other. The dimerization of Mpro is essential for the catalytic activity of the enzyme because the S1 pocket of the catalytic site is shaped by the interaction of N-terminal residues (N-finger) of each of the two protomers with Glu166 of the other protomer (Anand et al., 2003). In SARS-CoV-2 Mpro, the dimer has a contact interface, mainly between the domain II of protomer A and the N-finger residues of protomer B. All these structural features of the Mpro of SARS-CoV-2 are similar to SARS-CoV (Báez-Santos et al., 2015; Lee et al., 2005). The T(SARS-CoV-2)285A(SARS-CoV) Mpro mutation allowed the two domains (Domain III) of different protomers to come closer, which resulted in a slightly increased catalytic efficiency of SARS-CoV-2 Mpro than the SARS-CoV. This was a result of a change in the enzyme dynamics that transmitted the effect of mutation to the catalytic center (Zhang et al., 2020).
High-throughput screening and drug repurposing have suggested some potential hit compounds against SARS-CoV-2 (Jin et al., 2020). However, no therapeutic medication has been established for the remedy of human CoVs. Many natural molecules, their products, and molecules inspired by natural lead compounds have entered in different stages of drug design, including clinical trials. Among these, Tea polyphenols are an attractive source of molecules showing anti-HIV effect (Hashimoto et al., 1996), anti-cancerous (Fujiki et al., 1998), anti-oxidative (Tomita et al., 1994), anti-mutagenic (Yen & Chen, 1995), and anti-diabetic (Murase et al., 2002), and hypocholesterolemic activities (Ikeda et al., 1992). The beneficial effects of green Tea, oolong Tea, and black Tea are well-known for many years. Although, all types of Tea are prepared from Camellia sinensis L., the difference lies in the process of preparation (C. D. Wu & Wei, 2002). The three main objectives of this study were to screen a set of 65 potential bioactive molecules of Tea against the Mpro of SARS-CoV-2. Secondly, to perform and compare the molecular docking and molecular dynamics simulations results of Tea bioactive molecules with three potential repurposed drugs (Atazanavir, Darunavir, and Lopinavir) against the Mpro of SARS-CoV-2. Lastly, to provide a lead molecule that could be developed as an inhibitor against the Mpro of SARS-Cov-2.
Materials and methods
Data sets
Three-dimensional structure of SARS-CoV-2 Mpro (PDB ID: 6Y2F) with resolution 1.95 Å was collected from Protein Data Bank (Zhang et al., 2020) and an assemblage of FDA approved drugs and bioactive molecules from Tea were constituted for the study. The preparation of the protein structure was conducted by the Discovery studio package protocols “prepare protein” (Studio, 2015). A total number of 65 bioactive molecules (Green Tee, 2000; Nakai et al., 2005; Sai et al., 2011) of Tea plant were drawn and saved in .SDF format. The repurposed FDA drug molecules (Atazanavir, Darunavir, and Lopinavir) were retrieved from PubChem (Atazanavir | C38H52N6O7 - PubChem; Darunavir | C27H37N3O7S - PubChem; Lopinavir | C37H48N4O5 - PubChem.). Ligand geometry of every molecule was optimized by the Gaussian16 with DFT (minimization protocols) (Zheng & Frisch, 2017).
Molecular docking
The CDOCKER utility of Discovery Studio (Studio, 2015) was adopted for the study of molecular docking. CDOCKER is an application of a CHARMM (Chemistry at Harvard Macromolecular Mechanics energy) (Brooks et al., 1983) based semi-flexible docking tool. The flexible conformation region grabbed by ligand molecules explored using High-temperature kinetics. The optimization at the binding site of each conformation is completed by employing the simulated annealing process to achieve accurate results of docking. The default values of CDOCKER parameters were applied. During docking, the receptor is set as rigid while the ligands are flexible. The ligand strain with interaction energy (CHARMM energy) and alone interaction energy, which defines the ligand-binding affinity is calculated for every complex. The water molecules are usually expelled out in semi-flexible and rigid docking because the formation of the receptor-ligand complex might be affected by the fixed water molecules. After removing water, hydrogen atoms were added to the protein. The binding site was assigned with an 8.0 Å radius throughout the initial inhibitor, which included all the binding site amino acids of the SARS-CoV-2 Mpro protein. The structures of recognized hits were fixed and docked into the binding pocket of SARS-CoV-2 Mpro. Different poses for each molecule were created and interpreted based on -CDOCKER interaction energy.
Molecular dynamics simulations
The molecular dynamics (MD) simulations were performed on the Mpro of SARS-CoV-2 protein with the selected inhibitors. The MD simulations were executed by the GROningen MAchine for Chemical Simulations (GROMACS) version 5.1 (Abraham et al., 2015; Hess et al., 2008; Van Der Spoel et al., 2005). The protein topology was prepared by the ‘pdb2gmx’ script, while the ligand topologies were obtained front the GlycoBioChem PRODRG1 server. The generated ligand topologies were rejoined to the processed protein structures. The energy minimized conformations of all the processed complexes were obtained by the GROMOS96 43a1 force field. The energy minimized structures were then solvated with a single point charge (SPC) water model. A total of 30076 water molecules were added to a cubic simulation box containing Mpro and Atazanavir. Similarly, 30083, 30075, 30065, 30075, and 30077 water molecules were added to the complexes having Darunavir, Lopinavir, Oolonghomobisflavan-A, Theasinensin-D, and Theaflavin-3-O-gallate respectively. To neutralize the net charges in the system a total of 2 Na+ counter-ions were added to the system by using the ‘gmx genion’ script. The energy minimization of the complexes was achieved by employing the steepest descent minimization algorithm with a maximum of 50,000 steps and < 10.0 kJ/mol force. The equilibration was of the system was obtained in two steps. In the first step, a NVT ensemble having constant number of particles, volume and temperature was maintained for 2 ns, while in the second step, a NPT ensemble containing a constant number of particles, pressure and temperature was equilibrated for 10 ns. Both the ensembles (NVT and NPT) were subjected to positions restraints MD for 100ps. In this step, the backbone C- α atoms were restrained, while all the solvent molecules were allowed free movement to ensure the solvent equilibrium in the system is maintained. The covalent bonds the system were constrained by the linear constraint solver algorithm (Hess et al., 1997). The long range electrostatic interactions were obtained by the particle mesh Eshwald method with a 1.2 nm cut-off and 1.2 nm Fourier spacing (Essmann et al., 1995). The well equilibrated and solvated structures in terms of geometry and solvent orientation were subjected to further steps of simulation. The temperature of the system was regulated by the V-rescale weak coupling method (Berendsen et al., 1984) at 310 K. To equilibrate and set the pressure (1 atm), density, and total energy of the system, the Parrinello − Rahman method (Parrinello & Rahman, 1981) was used. Further, the well-equilibrated complexes (six protein-ligand complex) were then subjected to the production phase without any restrains for a period of 250 ns with a time step of 2 fs, and after every 2 ps the structural coordinates were saved. The produced trajectories of MD simulations were then used to generate the root mean square deviation (RMSD) and hydrogen bond graphs by various in-built scripts of GROMACS. MD simulations were used in several integrative studies to tackle various conditions such as cancers (John, Sivashanmugam, et al., 2020; John et al., 2020), identification of novel inhibitors (Sadhasivam et al., 2019; Sadhasivam & Vetrivel, 2019; Sharma et al., 2020; Singh et al., 2019), role of various mutations (Nagarajan et al., 2020; Rajendran et al., 2018), and exploration of drug resistance mechanisms (Rajendran & Sethumadhavan, 2014).
MM-PBSA calculation
The binding free energy of protein and ligand complexes can be calculated by combining the molecular Mechanic/Poisson-Boltzmann Surface Area (MM-PBSA) with MD. The MD scripts were extracted to perform MM-PBSA calculations. The binding free energy provides an overview of the biomolecular interactions between protein and ligand. The binding energy constitutes of potential energy, polar and non-polar solvation energies. The MM-PBSA binding free energies were calculated by utilizing the ‘g_mmpbsa’ (Kumari et al., 2014) script of GROMACS. The binding energy calculations in this method were calculated by using the following equation:
The ΔGbinding represents the total binding energy of the complex, while the binding energy of free receptor is Greceptor, and that of unbounded ligand is represented by Gligand.
Results and discussion
Molecular docking
A complete set of 65 bioactive molecules and FDA approved repurposed drugs against Mpro of SARS-CoV-2 was docked to obtain the molecules manifesting the best interaction energy (Table S1), which could act as promising inhibitors. The computational strategy is based on the best fitting molecules in the active site of a target protein structure, accompanied by the ranking of these molecules based on their interaction profile. The crystal structure of SARS-CoV-2 Mpro was adopted as an origin point for computer-aided drug design due to its functional relevance in the survival of CoV. A set of 68 molecules was docked and six molecules were prioritized with notable -CDOCKER interaction energy scores as depicted in Table 1.
Table 1.
Selected bioactive molecules and FDA approved drugs based on -CDOCKER interaction energy.
| S. No. | Molecules | -CDOCKER Interaction Energy |
|---|---|---|
| 1. | Oolonghomobisflavan-A | 75.54 |
| 2. | Theasinensin D | 71.58 |
| 3. | Theaflavin-3′-O-Gallate | 71.56 |
| 4. | Atazanavir | 64.85 |
| 5. | Lopinavir | 56.17 |
| 6. | Darunavir | 46.02 |
The interacting residues between the active site and the selected molecules were thoroughly examined by employing the discovery studio visualizer. Validation of docking protocol is confirmed by re-docking of the initial inhibitor of SARS-CoV-2 Mpro protein and found 0.00 Å Root Mean Square Deviation (RMSD) between the docked pose and the crystal structure (Figure S1,supplementary material). We found a similar interaction pattern in the experimental co-crystal structure. Furthermore, to validate the robustness for the adapted docking protocol, we compared the selected structure with another newly submitted SARS-CoV-2 Mpro crystal structure with PDB ID: 5R7Z (http://www.rcsb.org/structure/5R7Z). In both the complexes, molecules formed interactions with the same residue (Glu166, His41) and some other critical residues essential for inhibition, as shown in Figure S2 (supplementary material). The establishment of a similar bonding pattern confirmed that the CDOCKER module was extremely reliable for reproducing the experimentally noticed binding mode of SARS-CoV-2 Mpro. After exploring the molecular interactions, it was perceived that the bioactive molecule Oolonghomobisflavan-A manifests hydrogen bond with residues Thr25, Thr25, Asn142, His163, Glu166, Arg188, and His164. One carbon-hydrogen bond (C-H bond) with residues Gly143, two Pi-alkyl (Met165, His41), and one Pi-Pi T-shaped interaction (His41). It also formed Van der Waal (VdW) interaction with residues Phe140, Leu141, His172, Ser144, Thr24, Cys145, Leu27, Met49, Asp187, Gln189, Gln192, Val186, Thr190, Pro168, and Leu167. In molecule Theasinensin-D, residues Gln166, His163, Gly143 formed double, and residues Gln189, His164, Ser144, Cys145, Thr45, Cys44 formed single hydrogen bond. Several residues formed other interactions like Met165, Met49 (Pi-alkyl), His41, Asn142 (C-H bond), His41 (Pi-sigma), Cys145 (Pi-sulfur), and residues Gln192, Leu167, Arg188, Leu27, Thr26, Ser46 showed VdW interactions. In the case of Theaflavin-3-O-gallate, residue Glu166 formed triple and Asn142, Phe140, His163, Thr26, His41, Arg188, Met165 single hydrogen bond. Residues Cys145, His41, Met165 interacted via alkyl and Pi-alkyl interactions. Residues Pro168, Val186, Asp187, Gln192, Thr190, Gln189, Met49, Val42, Thr25, Lue27, Gly143, Ser144, His172, and Leu141, formed VdW interactions (Figure 1).
Figure 1.

2-D interactions of SARS-CoV-2 Mpro protein with bio-actives (a) Oolonghomobisflavan-A (b) Theasinensin-D, and (c) Theaflavin-3-O-gallate.
Figure 2.

2-D Interactions of SARS-CoV-2 Mpro protein with FDA approved drugs (a) Atazanavir (b) Darunavir, and (c) Lopinavir.
The binding of FDA approved drugs to target protein was also significantly mediated by the critical residues and conferred the best interactions. In Atazanavir, hydrogen bonds were formed by residues Glu166, Asn142, and C-H bond by Asn142, Asp187, Arg188, Leu141. Residues also formed other interactions, including one Sulfur-X (Met49), Pi-alkyl (His41, Pro168, Cys145, His163), alkyl (Met165, Met49, Leu41). Residues His172, Ser144, GLly43, His164, Thr25, Thr26, Leu27, Gln189, Thr190, Tyr154, Pro52, Cys44, and GLN192 showed VdW interactions. In the case of Darunavir, hydrogen bond formed by residues Glu166, Gly143, Phe140, and C-H bond by Pro168, Asn142, Leu167. Residues also formed other interactions, including one Pi-sulfur (Cys145), Pi-stacked (Leu41), alkyl (His41, Leu27, Cys145). Residues His164, Thr26, Ser144, Gly170, Thr25, Gln189, Thr190, Arg188, Val186, Gln192, and Met49 displayed VdW interactions. Lopinavir exhibits a triple C-H bond with residue ASN142 and one with Glu166, alkyl (Met49, His163, Leu167, Met165), and one Pi-Pi T-shaped interaction (His41), Pi-sulfur (Cys145), one stacked with residue Leu167. It also formed VdW interaction with residues Ala191, Thr190, Gln192, Arg188, Asp187, Tyr54, His164, Thr25, Thr26, Phe140, His172, Leu141, Ser144, and Leu27 (Figure 2).
The binding style of molecules is biologically appealing as it contributes a plausible rationale for the significance of interacting residues in viral protein inhibition. The docking results unveiled that among picked molecules, Oolonghomobisflavan-A showed the highest interaction energy compared to other molecules. All six chosen molecules exhibited notable interactions. The selected molecules were subjected to dynamic simulation examinations accompanied by binding free energy computation to determine the stability of these molecules.
Stability of docked complexes
Molecular docking provides static poses of the most favored conformations of molecules in the binding pocket of a protein to present a stable complex. The static images are not able to present the other crucial features involved in providing stability to a protein. These features include the flexibility of residues and secondary structural elements (Purohit, 2014). The conformational changes arising from the dynamic behavior of a protein might affect its actual biological functioning (Bhardwaj & Purohit, 2020). The actual movement and structural perturbations of a protein in its biological environment could be visualized by MD simulations. The RMSD is a well-known estimator of equilibration and protein stability. To validate our docking poses and analyze the average behavior of complete protein during MD simulations, we calculated the RMSD of backbone C-α atoms of all the selected complexes (Figure 3).
Figure 3.

RMSD of backbone C-α atoms of complexes with (a) bioactives: Oolonghomobisflavan-A (black), Theasinensin-D (Red), and Theaflavin-3-O-gallate (green). (b) FDA approved drugs, Atazanavir (black), Darunavir (red), and Lopinavir (green).
All the complexes having selected molecules from Tea showed deviations lower than 0.45 nm (Figure 3(a)). The complex with Theaflavin-3-O-gallate deviated at a higher trajectory than the other two complexes throughout the simulation period. The average RMSD values for complexes with Oolonghomobisflavan-A, Theasinensin-D, and Theaflavin-3-O-gallate were ∼0.43 nm, ∼0.36 nm, and ∼0.51 nm respectively. The complex with Theaflavin-3-O-gallate displayed higher simulation trajectory after ∼120ns than the complexes with Oolonghomobisflavan-A and Theasinensin-D. For complexes having proposed repurposed drugs (Figure 3(b)) Atazanavir, Darunavir, and Lopinavir, the average RMSD values were ∼0.41 nm, ∼0.35 nm, and ∼0.37 nm respectively. For the first ∼25ns, the complex with Lopinavir showed average RMSD around ∼0.26 nm, while for the rest simulation, it deviated at a higher trajectory with a mean value around ∼0.37 nm. The complex with Atazanavir showed the highest average RMSD value of ∼0.44 ns, while the complex with Darunavir showed the lowest average value of 0.32 ns till 200 ns. After 200 ns, the RMSD trajectory of complex with Darunavir showed similar deviations as shown by other two complexes. The minimal fluctuations in the RMSD trajectories and low difference in average RMSD values showed that the protein complexes were stable and comparable to experimental structures.
To check the stability of the selected molecules in the binding pocket of Mpro, we extracted MD simulation conformations at different intervals and visualized the interactions between protein and ligands (Figure S3, supplementary material). The selected molecules from Tea (Theaflavin-3-O-gallate, Oolonghomobisflavan-A, and Theasinensin-D) formed more number of hydrogen bonds and hydrophobic interactions than repurposed drugs (Atazanavir, Darunavir, and Lopinavir). All the bioactive molecules remained in the binding pocket throughout the simulation period.
Hydrogen bond analysis
Hydrogen bonds are one of the crucial elements responsible for the molecular interactions in biological systems. Hydrogen bonds provide the basis for molecular recognition and selectivity by imparting directionality and explicitness to molecular interactions. The protein-ligand interactions were guided by the changes in the secondary structures, which in turn were regulated by the hydrogen bonds. MD simulations provided different conformations in which a protein could be found in actual biological conditions. Each conformation of a protein is supposed to have its own interaction pattern with the ligand. We calculated the number of hydrogen bonds formed during the complete run of MD simulations for selected complexes, as presented in Figure 4.
Figure 4.

Hydrogen bond profiles of the Mpro complexes having with (a) bioactives: Oolonghomobisflavan-A (black), Theasinensin-D (Red), and Theaflavin-3-O-gallate (green). (b) FDA approved drugs, Atazanavir (black), Darunavir (red), and Lopinavir (green).
In complexes with Oolonghomobisflavan-A and Theaflavin-3-O-gallate, the most number of conformations formed up to 10 hydrogen bonds during the simulation. A very few conformations showed less than 5 and greater than 13 hydrogen bonds. The complex with Theasinensin-D formed an average of 12 hydrogen bonds. However, in complexes with repurposed drugs against Mpro of SARS-CoV-2, the average number of hydrogen bonds formed was 6. In complex having Darunavir, the average numbers of hydrogen bonds for the first 20 ns were 4 with few conformations showing up to 8 hydrogen bonds. In all the three complexes, a very few conformations were able to form more than 5 hydrogen bonds except the complex with Lopinavir. These results showed that the selected bioactive molecules formed a greater number of hydrogen bonds with Mpro during the simulation than repurposed drugs. The bioactive molecules were able to maintain strong interaction with the binding pocket of Mpro throughout the simulation period. The simulation trajectories were further exploited to study the interaction between the ligand and protein.
MM-PBSA binding free energy
We utilized a python script MmPbSaStat.py to calculate average free binding energy of the selected complexes (Table 2), is provided in the g_mmpbsa package. This script calculates the average free binding energy and its standard deviation/error from the output files, which were obtained from g_mmpbsa.
Table 2.
MM-PBSA calculations of binding free energy for six selected complexes.
| Complexes | ΔEbinding (kj/mol) | SASA (kJ/mol) | ΔEpolar solvation (kj/mol) | ΔEElectrostatic (kj/mol) | ΔEVan der Waal (kj/mol) |
|---|---|---|---|---|---|
| Oolonghomobisflavan -A | −256.875+/−33.239 | −28.582+/−2.030 | 283.652+/−40.481 | −127.505+/−31.872 | −384.439+/−26.921 |
| Thiasinensin-D | −217.823+/−25.637 | −26.989+/−1.763 | 335.817+/−26.114 | −190.397+/−25.384 | −336.254+/−25.716 |
| Theaflavin-3-O-gallate | −187.134+/−28.808 | −24.782+/−1.562 | 246.131+/−25.512 | −96.231+/−26.248 | −312.252+/−30.289 |
| Atazanavir | −229.499+/−40.040 | −24.212+/−3.588 | 124.302+/−30.707 | −37.184+/−13.389 | −292.405+/−48.889 |
| Darunavir | −220.260+/−20.520 | −20.565+/−1.407 | 102.674+/−17.016 | −31.549+/−10.590 | −270.820+/−22.563 |
| Lopinavir | −250.285+/−25.615 | −23.230+/−2.051 | 102.931+/−17.001 | −35.608+/−9.809 | −294.378+/−24.776 |
The energy liberated during the process of bond formation, or alternatively, the interaction between a ligand and protein is shown in the form of binding energy. Lesser the binding energy, the better is the binding of the ligand and protein. The final binding energy is a cumulative sum of van der Wall, electrostatic, polar solvation, and SASA energy. Except for the polar solvation energy, all other forms of energy contributed favorably to the interaction between different molecules and Mpro. The bioactive molecule Oolonghomobisflavan-A showed the least binding free energy (-256.875 kJ/mol) among all the selected molecules. The repurposed drug Lopinavir showed the second least binding free energy of -250.585 kJ/mol. A comparison of the binding free energies of all the complexes were made by plotting the binding energy versus time graphs (Figure 5). These results showed that Oolonghomobisflavan-A could outperform the FDA approved repurposed drugs in inhibiting the Mpro of SARS-CoV-2.
Figure 5.

Graphical representation of the Delta_E_Binding free energy kJ/mol showing (a) Bio-actives, Oolonghomobisflavan-A (black), Theasinensin-D (Red), and Theaflavin-3-O-gallate (green). (b) FDA approved drugs, Atazanavir (blue), Darunavir (pink), and Lopinavir (yellow).
Further, we examined the contribution of each residue of Mpro in terms of binding free energy to the interaction with the selected molecules. The contribution of each residue was calculated by decomposing the total binding free energy of the system into per residue contribution energy (Figure 6).
Figure 6.

Graphical representation of per residue contribution plot for (a) Bio-actives, Oolonghomobisflavan-A (black), Theasinensin-D (Red), and Theaflavin-3-O-gallate (green). (b) FDA approved drugs, Atazanavir (Cyan), Darunavir (yellow), and Lopinavir (pink).
Many residues (His41, Thr45, Met49, Phe140, Met165, and Glu166) showed favourable contribution energy. On comparing the complexes having Oolonghomobisflavan-A and Atazanavir, we found that the key residue involved in the interaction showed a significant difference in its contribution energy. The residue Glu166 showed -18.58 kJ/mol contribution energy in complex with Oolonghomobisflavan-A, while with Atazanavir the contribution energy was -4.29 kJ/mol. The contribution energy of other key residues are shown in Table S2. The residue Glu166 was also involved in the formation of a biologically functional dimeric form of Mpro (Anand et al., 2003). This suggests that the binding of bioactive molecule Oolonghomobisflavan-A to the catalytic site of one protomer could also interfere with the dimerization of Mpro as the residue Glu166 would not be available to interact with N-finger residues of the other protomer. This would increase the efficacy of the molecule against SARS-CoV-2.
Conclusions
SARS-CoV-2 has caused a state of a global health emergency. This situation demands to develop effective and targeted strategies to counter this disease. In this study, we explored the conformational space of 65 bioactive molecules of Tea plant by targeting the Mpro of SARS-CoV-2. These molecules were compared to three proposed repurposed drugs (Atazanavir, Darunavir, and Lopinavir) against COIVID-19. Our molecular docking results revealed that the molecules Oolonghomobisflavan-A, Theasinensin-D, and Theaflavin-3-O-gallate had higher docking scores than the repurposed drugs. These molecules were selected for MD simulation studies. The RMSD trajectories showed that the selected complexes were stable and comparable to experimental structures. Further, we calculated the total number of hydrogen bonds formed during the simulation time in all the selected complexes. The complexes with bioactive molecules with Tea formed a greater number of hydrogen bonds than the complexes with repurposed drugs suggesting stronger interaction and greater stability of the bioactive molecules in the binding pocket of the Mpro of SARS-CoV-2. These results were further evaluated and validated by MM-PBSA binding free energy calculations. Oolonghomobisflavan-A showed the least binding free energy among all the simulated complexes. Hence, this study reports Oolonghomobisflavan-A as a more potent inhibitor of the Mpro of SARS-CoV-2 than previously suggested repurposed anti-HIV drugs. Oolonghomobisflavan-A is one of the most abundant polymerized polyphenol present in Tea. The tea extract containing Oolonghomobisflavan-A, or the purified compound could be tested for its inhibitiory potential against Mpro of SARS-CoV-2 using various in-vitro and in-vivo studies. Moreover, the backbone structure of Oolonghomobisflavan-A could be further exploited to develop more potent inhibitors of SARS-CoV-2 Mpro.
Author contribution
RP conceived of and designed the study. VKB, RS, JS and VR analyzed and interpreted the data. RS, VKB, VR, RP and SK critically revised it for important intellectual content. All authors gave final approval of the version to be published.
Supplementary Material
Glossary
Abbreviations
- ACE2
Angiotensin-Converting Enzyme II
- CoV
Coronavirus
- GROMACS
GROningen MAchine for Chemical Simulations
- Mpro
Main Protease
- MD
Molecular Dynamics
- MERS
Middle East Respiratory Syndrome
- MM-PBSA
Molecular Mechanics Poisson-Boltzmann Surface Area
- RMSD
Root Mean Square Deviation
- SARS
Severe Acute Respiratory Syndrome
Acknowledgements
We gratefully acknowledge to the Director, CSIR-Institute of Himalayan Bioresource Technology, Palampur for providing the facilities to carry out this work. RP gratefully acknowledges the Board of Research in Nuclear Sciences, Department of Atomic Energy, Mumbai, India for financial support vide letter No: 37(1)/14/26/2015/BRNS. The CSIR support in the form of projects MLP0201 for bioinformatics studies and HCP0007 for providing project fellowship is highly acknowledged. VB acknowledges the Department of Science and Technology, New Delhi, India for providing junior research fellowship SERB File No: ECR/2016/000031. This manuscript represents CSIR-IHBT communication no. 4608 and Academy of Scientific and Innovative Research.
Disclosure statement
No potential conflict of interest was reported by the author(s).
References
- Abraham M. J., Murtola T., Schulz R., Páll S., Smith J. C., Hess B., & Lindahl E. (2015). GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX, 1-2, 19–25. 10.1016/j.softx.2015.06.001 [DOI] [Google Scholar]
- Anand K., Ziebuhr J., Wadhwani P., Mesters J. R., & Hilgenfeld R. (2003). Coronavirus main proteinase (3CLpro) structure: Basis for design of anti-SARS drugs. Science (New York, N.Y.).), 300(5626), 1763–1767. 10.1126/science.1085658 [DOI] [PubMed] [Google Scholar]
- Atazanavir | C38H52N6O7 - PubChem (n.d.). Retrieved April 18, 2020, from https://pubchem.ncbi.nlm.nih.gov/compound/Atazanavir
- Báez-Santos Y. M., St. John S. E., & Mesecar A. D. (2015). The SARS-coronavirus papain-like protease: Structure, function and inhibition by designed antiviral compounds. Antiviral Research, 115, 21–38. 10.1016/j.antiviral.2014.12.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berendsen H. J. C., Postma J. P. M., van Gunsteren W. F., DiNola A., & Haak J. R. (1984). Molecular dynamics with coupling to an external bath. The Journal of Chemical Physics, 81(8), 3684–3690. 10.1063/1.448118 [DOI] [Google Scholar]
- Bhardwaj V. K., & Purohit R. (2020). A new insight into protein-protein interactions and the effect of conformational alterations in PCNA. International Journal of Biological Macromolecules, 148, 999–1009. 10.1016/j.ijbiomac.2020.01.212 [DOI] [PubMed] [Google Scholar]
- Blanchard J. E., Elowe N. H., Huitema C., Fortin P. D., Cechetto J. D., Eltis L. D., & Brown E. D. (2004). High-throughput screening identifies inhibitors of the SARS coronavirus main proteinase. Chemistry & Biology, 11(10), 1445–1453. 10.1016/j.chembiol.2004.08.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borgio J. F., Alsuwat H. S., Al Otaibi W. M., Ibrahim A. M., Almandil N., Al Asoom L. I., Salahuddin M., Kamaraj B., & AbdulAzeez S. (2020). State-of-the-art tools unveil potent drug targets amongst clinically approved drugs to inhibit helicase in SARS-CoV-2. Archives of Medical Science, 16(3), 508–518. 10.5114/aoms.2020.94567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks B. R., Bruccoleri R. E., Olafson B. D., States D. J., Swaminathan S., & Karplus M. (1983). CHARMM: A program for macromolecular energy, minimization, and dynamics calculations. Journal of Computational Chemistry, 4(2), 187–217. 10.1002/jcc.540040211 [DOI] [Google Scholar]
- Cameron C. E., & Castro C. (2001). The mechanism of action of ribavirin: Lethal mutagenesis of RNA virus genomes mediated by the viral RNA-dependent RNA polymerase. Current Opinion in Infectious Diseases, 14(6), 757–764. 10.1097/00001432-200112000-00015 [DOI] [PubMed] [Google Scholar]
- Darunavir | C27H37N3O7S - PubChem. (n.d.). Retrieved April 18, 2020, from https://pubchem.ncbi.nlm.nih.gov/compound/213039
- De Wit E., Van Doremalen N., Falzarano D., & Munster V. J. (2016). SARS and MERS: Recent insights into emerging coronaviruses. Nature Reviews. Microbiology, 14(8), 523–534. 10.1038/nrmicro.2016.81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Essmann U., Perera L., Berkowitz M. L., Darden T., Lee H., & Pedersen L. G. (1995). A smooth particle mesh Ewald method. The Journal of Chemical Physics, 103(19), 8577–8593. 10.1063/1.470117 [DOI] [Google Scholar]
- Fujiki H., Suganuma M., Okabe S., Sueoka N., Komori A., Sueoka E., Kozu T., Tada Y., Suga K., Imai K., & Nakachi K. (1998). Cancer inhibition by green tea. Mutation Research, 402(1/2), 307–310. 10.1016/S0027-5107(97)00310-2 [DOI] [PubMed] [Google Scholar]
- Gorbalenya A. E., Baker S. C., Baric R. S., de Groot R. J., Drosten C., Gulyaeva A. A., Haagmans B. L., Lauber C., Leontovich A. M., Neuman B. W., Penzar D., Perlman S., Poon L. L. M., Samborskiy D. V., Sidorov I. A., Sola I., & Ziebuhr J. (2020). The species Severe acute respiratory syndrome-related coronavirus: Classifying 2019-nCoV and naming it SARS-CoV-2. Nature Microbiology, 10.1038/s41564-020-0695-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green Tee J. (2000). Chemical composition of various kinds of Japanese green tea chemical constituents of Japanese green tea at different stages of growth [Google Scholar]
- Han D. P., Penn-Nicholson A., & Cho M. W. (2006). Identification of critical determinants on ACE2 for SARS-CoV entry and development of a potent entry inhibitor. Virology, 350(1), 15–25. 10.1016/j.virol.2006.01.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto F., Kashiwada Y., Nonaka G. I., Nishioka I., Nohara T., Cosentino L. M., & Lee K. H. (1996). Evaluation of tea polyphenols as anti-HIV agents. Bioorganic & Medicinal Chemistry Letters, 6(6), 695–700. 10.1016/0960-894X(96)00095-9 [DOI] [Google Scholar]
- Hess B., Hess B., Bekker H., Berendsen H. J. C., & Fraaije J. G. E. M. (1997). LINCS: A linear constraint solver for molecular simulations. Journal of Computational Chemistry, 18(12), 1463–1463. http://citeseerx.ist.psu.edu/viewdoc/summary?doi=10.1.1.48.2727 [Google Scholar]
- Hess B., Kutzner C., van der Spoel D., & Lindahl E. (2008). GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. Journal of Chemical Theory and Computation, 4(3), 435–447. 10.1021/ct700301q [DOI] [PubMed] [Google Scholar]
- Hilgenfeld R. (2014). From SARS to MERS: Crystallographic studies on coronaviral proteases enable antiviral drug design. The FEBS Journal, 281(18), 4085–4096. 10.1111/febs.12936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda I., Imasato Y., Sasaki E., Nakayama M., Nagao H., Takeo T., Yayabe F., & Sugano M. (1992). Tea catechins decrease micellar solubility and intestinal absorption of cholesterol in rats. Biochimica et Biophysica Acta (Bba) – Lipids and Lipid Metabolism, 1127(2), 141–146. 10.1016/0005-2760(92)90269-2 [DOI] [PubMed] [Google Scholar]
- Jin Z., Du X., Xu Y., Deng Y., Liu M., Zhao Y., Zhang B., Li X., Zhang L., Peng C., Duan Y., Yu J., Wang L., Yang K., Liu F., Jiang R., Yang X., You T., Liu X., … Yang H. (2020). Structure of Mpro from COVID-19 virus and discovery of its inhibitors. BioRxiv, 10.1101/2020.02.26.964882 [DOI] [PubMed] [Google Scholar]
- John A., Sivashanmugam M., Natarajan S. K., & Umashankar V. (2020). Computational modeling of novel inhibitory peptides targeting proteoglycan like region of carbonic anhydrase IX and in vitro validation in HeLa cells. Journal of Biomolecular Structure & Dynamics, 38(7), 1995–2006. 10.1080/07391102.2019.1623075 [DOI] [PubMed] [Google Scholar]
- John A., Vetrivel U., Sivashanmugam M., & Natarajan S. K. (2020). Microsecond simulation of the proteoglycan-like region of carbonic anhydrase IX and design of chemical inhibitors targeting pH homeostasis in cancer cells. ACS Omega, 5(8), 4270–4281. 10.1021/acsomega.9b04203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumari R., Kumar R., & Lynn A., & Open Source Drug Discovery Consortium (2014). g_mmpbsa-a GROMACS tool for high-throughput MM-PBSA calculations . Journal of Chemical Information and Modeling, 54(7), 1951–1962. 10.1021/ci500020m [DOI] [PubMed] [Google Scholar]
- Lee T. W., Cherney M. M., Huitema C., Liu J., James K. E., Powers J. C., Eltis L. D., & James M. N. G. (2005). Crystal structures of the main peptidase from the SARS coronavirus inhibited by a substrate-like aza-peptide epoxide. Journal of Molecular Biology, 353(5), 1137–1151. 10.1016/j.jmb.2005.09.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G., & De Clercq E. (2020). Therapeutic options for the 2019 novel coronavirus (2019-nCoV). In. Nature Reviews. Drug Discovery, 19(3), 149–150. NLM (Medline). 10.1038/d41573-020-00016-0 [DOI] [PubMed] [Google Scholar]
- Lopinavir | C37H48N4O5 - PubChem (n.d.). Retrieved April 18, 2020, from https://pubchem.ncbi.nlm.nih.gov/compound/92727
- Murase T., Nagasawa A., Suzuki J., Hase T., & Tokimitsu I. (2002). Beneficial effects of tea catechins on diet-induced obesity: Stimulation of lipid catabolism in the liver. International Journal of Obesity and Related Metabolic Disorders: Journal of the International Association for the Study of Obesity, 26(11), 1459–1464. 10.1038/sj.ijo.0802141 [DOI] [PubMed] [Google Scholar]
- Nagarajan H., Narayanaswamy S., & Vetrivel U. (2020). Mutational landscape screening of methylene tetrahydrofolate reductase to predict homocystinuria associated variants: An integrative computational approach. Mutation Research, 819-820, 111687 10.1016/j.mrfmmm.2020.111687 [DOI] [PubMed] [Google Scholar]
- Nakai M., Fukui Y., Asami S., Toyoda-Ono Y., Iwashita T., Shibata H., Mitsunaga T., Hashimoto F., & Kiso Y. (2005). Inhibitory effects of oolong tea polyphenols on pancreatic lipase in vitro . Journal of Agricultural and Food Chemistry, 53(11), 4593–4598. 10.1021/jf047814+ [DOI] [PubMed] [Google Scholar]
- Ortega J. T., Serrano M. L., Pujol F. H., & Rangel H. R. (2020). Unrevealing sequence and structural features of novel coronavirus using in silico approaches: The main protease as molecular target. EXCLI Journal, 19, 400–409. 10.17179/excli2020-1189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrinello M., & Rahman A. (1981). Polymorphic transitions in single crystals: A new molecular dynamics method. Journal of Applied Physics, 52(12), 7182–7190. 10.1063/1.328693 [DOI] [Google Scholar]
- Purohit R. (2014). Role of ELA region in auto-activation of mutant KIT receptor: A molecular dynamics simulation insight. Journal of Biomolecular Structure & Dynamics, 32(7), 1033–1046. 10.1080/07391102.2013.803264 [DOI] [PubMed] [Google Scholar]
- Singh R., Bhardwaj V., Das P., & Purohit R. (2019). Natural analogues inhibiting selective cyclin-dependent kinase protein isoforms: A computational perspective. Journal of Biomolecular Structure and Dynamics, 10.1080/07391102.2019.1696709 [DOI] [PubMed] [Google Scholar]
- Rajendran V., Gopalakrishnan C., & Sethumadhavan R. (2018). Pathological role of a point mutation (T315I) in BCR-ABL1 protein-A computational insight. Journal of Cellular Biochemistry, 119(1), 918–925. 10.1002/jcb.26257 [DOI] [PubMed] [Google Scholar]
- Rajendran V., & Sethumadhavan R. (2014). Drug resistance mechanism of PncA in Mycobacterium tuberculosis. Journal of Biomolecular Structure & Dynamics, 32(2), 209–221. 10.1080/07391102.2012.759885 [DOI] [PubMed] [Google Scholar]
- Sadhasivam A., Nagarajan H., & Umashankar V. (2019). Structure-based drug target prioritisation and rational drug design for targeting Chlamydia trachomatis eye infections. Journal of Biomolecular Structure and Dynamics, 1–13. 10.1080/07391102.2019.1652691 [DOI] [PubMed] [Google Scholar]
- Sadhasivam A., & Vetrivel U. (2019). Identification of potential drugs targeting L,L‐diaminopimelate aminotransferase of Chlamydia trachomatis : An integrative pharmacoinformatics approach. Journal of Cellular Biochemistry, 120(2), 2271–2288. 10.1002/jcb.27553 [DOI] [PubMed] [Google Scholar]
- Sharma J., Bhardwaj V. K., Das P., & Purohit R. (2020). Identification of naturally originated molecules as γ-aminobutyric acid receptor antagonist. Journal of Biomolecular Structure and Dynamics, 1–12. 10.1080/07391102.2020.1720818 [DOI] [PubMed] [Google Scholar]
- Sai V., Chaturvedula P., & Prakash I. (2011). The aroma, taste, color and bioactive constituents of tea. Journal of Medicinal Plants Research, 5(11), 2110–2124. http://www.academicjournals.org/JMPR [Google Scholar]
- Song Z., Xu Y., Bao L., Zhang L., Yu P., Qu Y., Zhu H., Zhao W., Han Y., & Qin C. (2019). From SARS to MERS, thrusting coronaviruses into the spotlight. Viruses, 11(1), 59 MDPI AG. 10.3390/v11010059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Studio D. (2015). Dassault Systemes BIOVIA, Discovery Studio Modelling Environment, Release 4.5. Accelrys Software, Inc. [Google Scholar]
- Van Der Spoel D., Lindahl E., Hess B., Groenhof G., Mark A. E., & Berendsen H. J. C. (2005). GROMACS: Fast, flexible, and free. Journal of Computational Chemistry, 26(16), 1701–1718. 10.1002/jcc.20291 [DOI] [PubMed] [Google Scholar]
- Wang M., Cao R., Zhang L., Yang X., Liu J., Xu M., Shi Z., Hu Z., Zhong W., & Xiao G. (2020). Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Research, ( 30Issue (3), 269–271. Springer Nature. 10.1038/s41422-020-0282-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C. D., & Wei G. X. (2002). Tea as a functional food for oral health. Nutrition (Burbank, Los Angeles County, Calif.), 18(5), 443–444. 10.1016/S0899-9007(02)00763-3 [DOI] [PubMed] [Google Scholar]
- Wu F., Zhao S., Yu B., Chen Y. M., Wang W., Song Z. G., Hu Y., Tao Z. W., Tian J. H., Pei Y. Y., Yuan M. L., Zhang Y. L., Dai F. H., Liu Y., Wang Q. M., Zheng J. J., Xu L., Holmes E. C., & Zhang Y. Z. (2020). A new coronavirus associated with human respiratory disease in China. Nature, 579(7798), 265–269. 10.1038/s41586-020-2008-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yen G. C., & Chen H. Y. (1995). Antioxidant activity of various tea extracts in relation to their antimutagenicity. Journal of Agricultural and Food Chemistry, 43(1), 27–32. 10.1021/jf00049a007 [DOI] [Google Scholar]
- Zhang L., Lin D., Sun X., Curth U., Drosten C., Sauerhering L., Becker S., Rox K., & Hilgenfeld R. (2020). Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science, eabb3405, 10.1126/science.abb3405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng J., & Frisch M. J. (2017). Efficient geometry minimization and transition structure optimization using interpolated potential energy surfaces and iteratively updated Hessians. Journal of Chemical Theory and Computation, 13(12), 6424–6432. 10.1021/acs.jctc.7b00719 [DOI] [PubMed] [Google Scholar]
- Zhou P., Yang X.-L., Wang X.-G., Hu B., Zhang L., Zhang W., Si H.-R., Zhu Y., Li B., Huang C.-L., Chen H.-D., Chen J., Luo Y., Guo H., Jiang R.-D., Liu M.-Q., Chen Y., Shen X.-R., Wang X., … Shi Z.-L. (2020). A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature, 579(7798), 270–273. 10.1038/s41586-020-2012-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zumla A., Chan J. F. W., Azhar E. I., Hui D. S. C., & Yuen K. Y. (2016). Coronaviruses - drug discovery and therapeutic options. Nature Reviews. Drug Discovery, 15(5), 327–347. 10.1038/nrd.2015.37 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.