

Abstract
Adeno-associated virus (AAV) vector gene therapy is a promising treatment for a variety of genetic diseases, including hemophilia. Systemic administration of AAV vectors is associated with a cytotoxic immune response triggered against AAV capsid proteins, which if untreated can result in loss of transgene expression. Immunosuppression (IS) with corticosteroids has limited transgene loss in some AAV gene therapy clinical trials, but was insufficient to prevent loss in other studies. We used a nonhuman primate model to evaluate intensive T cell-directed IS combined with AAV-mediated transfer of the human factor IX (FIX) gene. Early administration of rabbit anti-thymocyte globulin (ATG) concomitant with AAV administration resulted in the development of anti-FIX antibodies, whereas delayed ATG by 5 weeks administration did not. The anti-FIX immune response was associated with increases in inflammatory cytokines, as well as a skewed Th17/regulatory T cell (Treg) ratio. We conclude that the timing of T cell-directed IS is critical in determining transgene-product immunogenicity or tolerance. These data have implications for systemically administered AAV gene therapy being evaluated for hemophilia A and B, as well as other genetic diseases.
Keywords: AAV, gene therapy, gene transfer, immunogenecity, nonhuman primates, transgene immunogenecity, immunosuppression, Tregs, Th17, ATG
Graphical Abstract
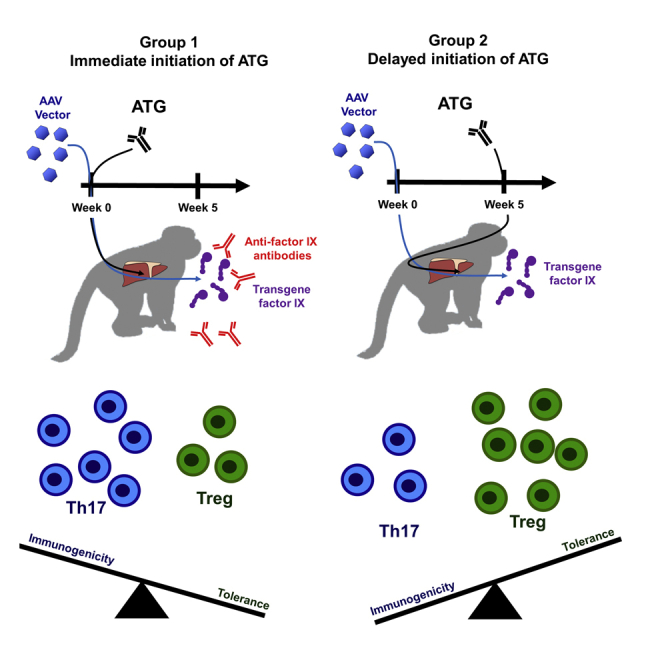
In nonhuman primates, Samelson-Jones and Finn et al. observe that early intensive immunosuppression concomitant with AAV vector administration enhances the formation of transgene-product antibodies, whereas delaying immunosuppression by 5 weeks does not. This suggests there is a critical time period when intensive immunosuppression can shift the immune system toward immunogenicity and away from tolerance.
Introduction
Adeno-associated virus (AAV) vector gene therapy has emerged as a promising treatment for a variety of genetic diseases.1,2 The recent success of newly approved AAV-based drugs was built on several decades of preclinical and clinical gene therapy studies, many focused on hemophilia B (HB).1,2 HB is an X-linked bleeding disorder due to a deficiency in coagulation factor IX (FIX).3 It affects about 1 in 30,000 male births worldwide, and without treatment is characterized by frequent spontaneous hemorrhages that can be life-threatening.3 The severity of the HB bleeding phenotype is dependent on the residual FIX activity such that modest increases in FIX activity have an important clinical impact,3 which makes HB highly amenable to gene therapy. Both the first parenterally administered in-human AAV-based gene therapy4,5 and the first in-human liver-directed AAV gene therapy were conducted in HB subjects.6
Early HB clinical liver-directed gene therapy studies helped defined key immunological complications for reliable efficacious delivery of AAV-vector mediated gene transfer.6 Successful AAV-based gene therapy must overcome at least three potential immunological obstacles: (1) impediment of vector transduction by pre-existing anti-AAV neutralizing antibodies (NAbs), (2) eradication of transduced cells by anti-AAV-capsid T cells (cellular immune response), and (3) the development of an immune response against the transgene-product.7, 8, 9, 10 The immune mechanisms responsible for each of these obstacles, and the complex relationship between them, are only partly understood. Previous preclinical studies have suggested that immunosuppression (IS) designed to address one of these obstacles may worsen others.11
The anti-AAV-capsid cellular immune response against transduced cells has remained a major limitation for liver-directed AAV-based gene therapy since its initial recognition during an AAV serotype-2 (AAV2)-based gene therapy trial for HB.6,8,12 The loss of transduced cells by this cellular immune response results in a decline in transgene expression levels, which results in diminished therapeutic efficacy. Although several vector parameters may impact the risk and intensity of the anti-AAV-capsid cellular immune response, the frequency of the anti-AAV-capsid cellular immune response increases with increasing vector dose.7, 8, 9, 10 Subsequent clinical studies demonstrated that the rapid initiation of IS with oral steroids could limit the loss of transgene expression.13, 14, 15 However, IS with oral and intravenous steroids has been insufficient to prevent the loss of transgene expression in several AAV gene therapy trials for hemophilia.7, 8, 9,16, 17, 18, 19 The incomplete efficacy of steroid IS to prevent transgene elimination has motivated studies of alternative IS regimens in preclinical models. This effort has been hampered by the lack of clinically informative in vivo models of the anti-AAV capsid cellular immune response.20, 21, 22, 23, 24 Nonhuman primate (NHP) models are advantageous given the similarity to the human immune system, which allows for the evaluation of IS with biologics, as well as comparable transgene expression levels, after liver-directed gene therapy.
Rhesus macaque FIX is 97% identical with human FIX (hFIX), differing only at 11 of 461 amino acid positions.25 Despite this similarity, about 20%–30% of NHPs that express hFIX after gene therapy develop anti-hFIX antibodies (Table 1). Anti-hFIX antibodies can also occur in NHP after administration of hFIX protein.26 About 3% of HB patients also develop neutralizing anti-hFIX antibodies, termed inhibitors, which substantially increase the morbidity of the disease.27 Thus, NHPs serve as a provocative model for assessing the immunogenicity of hFIX transgene expression.
Table 1.
Summary of Anti-hFIX Immune Response in NHPs after Liver-Directed GT
| References | Vector | Vector Dose (vg/kg) | IS | Anti-hFIX (n) | Totala (n) | Anti-hFIX (%) |
|---|---|---|---|---|---|---|
| 44 | AAV2-hFIX | 4 × 1012 | none | 1 | 5 | 20 |
| 45 | AAV5-CAGG-hFIX | 4 × 1012 | none | 1 | 4 | 25 |
| AAV8-HCR-hAAT-hFIX | 4 × 1012 | none | 0 | 1 | 0 | |
| 55 | scAAV8-LSP-hFIX | 0.4–1 × 1012 | noneb | 1 | 4 | 25 |
| 11 | AAV2-LSP-hFIX | 4 × 1012 | none | 0 | 3 | 0 |
| MMF/sirolimus | 0 | 3 | 0 | |||
| MMF/sirolimus + anti-CD25 | 3 | 3 | 100 | |||
| 46 | scAAV8-LSP-hFIX | 1 × 1012 | none | 1 | 7 | 14 |
| 43 | scAAV8-LSP-hFIX | 0.02–2 × 1012 | none | 1 | 10 | 10 |
| 47 | AAV8-hAAT-hFIX | 2 × 1013 | noneb | 2 | 2 | 100 |
| 56 | LV-FIX-Padua | 7.5 × 1013c | none | 3 | 6 | 50 |
| 57 | AAV5-hFIX-WT | 5 × 1012 | none | 1 | 3 | 33 |
| AAV5-hFIX-Padua | 0.5–9 × 1012 | none | 5 | 12 | 42 | |
| Total | 19 | 63 | 30 |
Total number of NHPs in study with detectable hFIX expression and followed for ≥12 weeks after vector administration.
Animals received rituximab and cyclosporine after inhibitor formation.
Lentivirus dosed as transduction units per kilogram.
To fulfill the promises of gene therapy for genetic disease, better approaches are required to reliably avoid or prevent anti-AAV-capsid cellular immune responses that limit transgene expression. Translatable IS approaches must promote immune tolerance induction of the transgene-product after vector administration. Herein, we evaluated the impact of the timing of intensive T cell-directed IS with rabbit anti-thymocyte globulin (ATG) in NHPs receiving therapeutically relevant doses of AAV-hFIX vectors. Animals received either early ATG concomitant with vector or delayed ATG ∼5 weeks after vector administration, which is the earliest reported onset of an anti-AAV capsid cellular immune response.10,14 Our hypothesis was that delaying intensive IS until the onset of the cellular immune response may spare early immune processes including regulatory T cells (Tregs) expansion, which would promote immune tolerance induction to transgene-expressed hFIX.
Although both ATG regimens were efficacious in producing lymphopenia, we observe that animals that received early ATG were substantially more likely to develop anti-hFIX antibodies (two out of three), whereas none of the animals that received delayed ATG developed anti-hFIX antibodies. These data for the first time indicate that the timing of IS to address immunological obstacles for gene therapy is critical for its success.
Results
Study Design
The primary study endpoints were the determination of levels of hFIX expression and the rates of immune responses to the transgene-product. As illustrated in Figure 1, six adult male rhesus macaques with low anti-AAV2 capsid NAbs (NAb titers ≤1:3) were divided into two groups to test the safety of rabbit ATG as an immune-suppressive agent either around the time of vector administration (group 1: early IS therapy) or around day 35 post-vector administration (group 2: delayed IS therapy). Day 35 post-vector administration is the approximate time of the onset of T cell cytotoxicity and progressive loss of transgene expression observed in the AAV liver-directed clinical trials.6 Likewise, the eligibility of the most current HB gene therapy trials requires a NAb titer <1:5. Although current HB gene therapy trials have shifted to alternative serotypes, the cellular immune response against AAV2 capsids remains the most extensively investigated.6,8,12 The AAV-capsid immune response is also serotype and route of administration independent.
Figure 1.

Experimental Scheme Evaluating the Safety of ATG for Immunosuppression after AAV Gene Therapy
AAV2-hAAT-hFIX at a dose of 7.5 × 1012 vg/kg was administered on day 0. Group 1 animals (early IS) received three doses of ATG around vector administration, whereas group 2 animals (delayed IS) received three doses of ATG around day 35 after vector administration, the approximate time of the onset of T cell cytotoxicity observed in the AAV2 liver-directed clinical trials.6 Both groups received MMF starting 1 week before vector administration and rapamycin starting the day of vector administration. IS was discontinued after 8 weeks from vector administration, and the animals were observed until 28 weeks after vector. Non-linear timescale is employed for clarity.
Both groups of animals were administered mycophenolate mofetil (MMF) starting on week −1 until week 8 at doses of 25 mg/kg. Both groups of animals also received rapamycin (4 mg/kg) from days 0 to 6 as loading dose to ensure therapeutic levels, followed by 2 mg/kg from day 7 until week 8 as a maintenance regimen. IS was discontinued at week 8, and the animals were followed for an additional 20 weeks. AAV2 vector expressing hFIX under control of the liver-specific promoter (AAV-hFIX) was injected at dose of 7.5 × 1012 vg/kg on day 0 via the hepatic artery under general anesthesia. Administration into the hepatic artery is standard for AAV2 vectors in both preclinical and clinical studies.6,11
ATG Is Effective in Inducing Lymphopenia and a Transient Increase in the Frequency of T Regulatory Cells
Animals from group 1 (n = 3) were administered with ATG (5 mg/kg/dose) around the time of vector administration starting at day −1 followed by additional doses on days +1 and +3. Group 2 animals (n = 3) received three doses of ATG on days +35, +37, and +39 post-AAV delivery. Peripheral total lymphocytes were monitored weekly, and we observed a marked decrease in lymphocyte numbers in each group of animals following ATG administration (Figures 2A and 2B). This rapid and efficient depletion of T cells demonstrated that the ATG regimen used was effective in these animals. We also observed a transient increase in the frequency of CD4+CD25+FoxP3+ cells in all animals shortly after ATG administration (Figures 2C and 2D); however, the magnitude of this increase was variable across animals. ATG showed no depleting effects in red cell and platelet counts, as expected (Figure S1). Plasma levels of rapamycin and MMF were monitored throughout the study. Drug levels were within the therapeutic range with the exception of one time point for monkey 1001 for rapamycin below the detection limit at week 2 and one time point for monkey 2003 for supratherapeutic MMF at week 8 (Figure S2).
Figure 2.

Peripheral Lymphocytes and Subsets before and after IS
(A and B) Total peripheral lymphocyte count for animals that received early (A) or delayed (B) ATG. (C and D) Percent CD4+CD25+FoxP3+ of total lymphocytes for animals that received early (C) or delayed (D) ATG. Each line represents an individual animal, each arrow represents an ATG dose, and the gray line indicates the timing of IS withdraw at week 8.
Early ATG at the Time of AAV Administration Is Associated with Formation of Antibodies against the hFIX Transgene-Product
Animals from group 1, early ATG, showed variable hFIX expression levels. For 1001, hFIX levels peaked at 410 ng/mL at week 2 and subsequently stabilized at 96 ng/mL (Figure 3A); notably, this animal had the highest frequency of CD4+CD25+FoxP3+ cells in the early ATG group (Figure 2C). The other two animals in the early ATG group developed antibodies to hFIX (Figure 3C; Table 2). Monkey 1002 showed undetectable hFIX expression due to low-titer neutralizing anti-FIX antibodies (∼5,000 ng/mL) and 0.5 Bethesda Unit (BU) at week 20. Monkey 1003 expressed hFIX at ∼100 ng/mL until week 10, whereas the hFIX levels dropped to undetectable levels by week 14 (Figure 3A). This drop in hFIX levels coincided with the onset of a robust anti-hFIX humoral immune response (IgG levels of ∼70,000 ng/mL) and 5.5 BU at week 20 (Figure 3C). T cell responses against hFIX were analyzed by interferon (IFN)-γ enzyme-linked immunosorbent spot (ELISpot) analysis and were negative at all time points tested (Figure S3).
Figure 3.

hFIX Expression Levels and Anti-hFIX Antibody Formation after AAV-hFIX Gene Therapy
(A and B) Plasma hFIX expression levels after AAV-hFIX gene transfer for animals that received early (A) or delayed (B) ATG. (C and D) Circulating anti-hFIX IgG levels for animals that received early (C) or delayed (D) ATG. The week 20 inhibitor titer in BU is indicated. Each line represents an individual animal.
Table 2.
Summary of Results of Early versus Delayed ATG IS with AAV Vector Administration
| NAb Titer (Pre-treatment) | Gene Copy Number/Cella | hFIX Plateau (ng/mL) | Inhibitor | |
|---|---|---|---|---|
| Group 1 (Early) | ||||
| 1001 | <1:3 | 3 | 96 | negative |
| 1002 | <1:3 | 0.48 | 0 | positive (0.5 BU) |
| 1003 | <1:3 | 0.75 | 89 (pre-inhibitor) | positive (5.5 BU) |
| Group 2 (Delayed) | ||||
| 2001 | 1:3 | 0.66 | 5 | negative |
| 2102 | <1:3 | 0.25 | 25 | negative |
| 2003 | 1:3 | 0.19 | 215 | negative |
Week 16.
Delayed Administration of ATG after AAV Administration Is Not Associated with Formation of Antibodies against the hFIX Transgene-Product
Animals from group 2, delayed ATG, also demonstrated variable hFIX expression levels. Monkey 2001 had very low expression, near the limit of detection (∼5 ng/mL), monkey 2102 peaked at 70 ng/mL at week 2 and subsequently stabilized at 10 ng/mL, and monkey 2003 peaked at 900 ng/mL at week 2 and subsequently had plateau levels of 200 ng/mL by week 28 (Figure 3B). Notably, in contrast with the animal that received ATG around vector administration (group 1), none of the animals that received delayed ATG had any detectable immune response against hFIX, either humoral (Figure 3D) or T cell mediated (IFN-γ ELIspot; Figure S3).
Inhibitor Formation to hFIX Is Associated with a Th17 Cytokine Signature and Increased Frequency of Th17 Cells
Plasma cytokine levels (interleukin-1β [IL-1β], IL-2, IL-4, IL-10, IL-17, tumor necrosis factor alpha [TNF-α], and IFN-γ) were assayed over time in all animals (Figures 4A–4G). Interestingly, the two animals that developed humoral responses against hFIX (1002 and 1003) showed transient increases in IL-2, IL-4, IL-10, IL-17, TNF-α, IL-1β, and IFN-γ levels in circulation, peaking at week 8. Given the role of TNF-α, IL-1β, and IL-17 in the production and maintenance of Th17 cells,28,29 we hypothesized that these pro-inflammatory cells might be involved in inhibitor formation. Flow cytometry was performed on peripheral blood mononuclear cells (PBMCs) using CD4-fluorescein isothiocyanate (FITC), CD196-phycoerythrin (PE)-Cy7, and IL-17a-PE after PMA stimulation. We observed a transient early (week 7) significant increase in the levels of Th17 (CD4+, CD196+, IL-17a+) cells from baseline (mean 230% ± 60% baseline) in the two animals (1002 and 1003) that developed inhibitors compared with the four animals (1001, 2001, 2102, and 2003) that did not develop an anti-hFIX immune response (mean 80% ± 30% baseline; p < 0.011) (Figure 5A). We note that one animal that did not develop an inhibitor (2001) had a late increase in Th17 levels at week 16 with no changes in hFIX expression levels. This difference in the pro-inflammatory Th17 cell frequency in the animals that developed inhibitors is even more pronounced when normalized with their Treg cell frequency (Figure 5B).
Figure 4.

Plasma Cytokine Levels after Vector Administration
(A–G) Plasma levels of IL-1β (A), IL-2 (B), IL-4 (C), IL-10 (D), IL-17 (E), TNF-α (F), and IFN-γ (G) were assayed over time by multiplex analysis. Each line represents an individual animal.
Figure 5.

Frequency of Th17 T Cells after Vector Administration
(A and B) Frequency of Th17 (CD4+, CD196+, IL-17a+) cells (A) or ratio of Th17 cells to Tregs (CD4+CD25+FoxP3+) (B) after AAV-hFIX gene therapy. The fraction CD4+ PBMCs that were also CD196+, IL-17a+ was analyzed relative to the pre-IS and pre-gene therapy (week −1) level of each animal. Each line represents an individual animal. The mean change in Th17 cell frequency and the ratio compared with Tregs from baseline at week 7 (∗), prior to the detection of the anti-hFIX immune response, was significantly higher in the two animals that developed inhibitors (1002 and 1003) compared with the four animals that did not develop inhibitors (1001, 2001, 2102, and 2003): 230% ± 60% and 880% ± 470% baseline, respectively, compared with 80% ± 30% and 84% ± 140%, respectively, baseline; p < 0.03 determined with a two-sided t test.
Local and Systemic Toxicity of ATG Coupled with MMF and Rapamycin
ATG administration was well tolerated with no effect on body weight or increased risk for opportunist infections. In all but one animal (2003, see below), liver enzyme parameters (aspartate transaminase [AST] and alanine transaminase [ALT]) were within the normal range (Figures 6A–6D). Coagulation tests (prothrombin time, activated partial thromboplastin time, and fibrinogen levels), kidney function, lipid profile, and a series of comprehensive chemical tests were within the normal range throughout the study in both groups of animals (data not shown). For 2003, we observed a transient increase ALT levels at around week 8 (Figure 6D) associated with a temporary decrease in hFIX levels (Figure 3B). No humoral immune responses to the transgene product or T cell-increased secretion of IFN-γ to hFIX or AAV2 capsid proteins by ELISpot analysis were detected (Figure S3B). Moreover, liver biopsy collected at week 10 was unremarkable (Figure S4A). The animal remained asymptomatic with no evidence of opportunistic infection. The only abnormality was a spike in the MMF levels (3.5 mg/L) at week 8. MMF was stopped at this time point, as per protocol; thus, the previous high levels of the drug could explain a transitory liver toxicity.
Figure 6.

Liver Enzymes after Vector Administration
(A–D) Plasma AST (A and B) or ALT (C and D) levels after AAV-hFIX gene transfer for animals that received early ATG (A or C, respectively) or delayed ATG (B or D, respectively). Each line represents an individual animal.
Percutaneous liver biopsies were performed in all animals at week 16, and vector genome copy number (GCN) was analyzed by quantitative real-time PCR. As seen in Table 2, all animals had detectable vector GCN, confirming effective delivery of vector to the liver. We could not correlate GCN with hFIX expression levels; however, this is not unexpected because the GCN analysis was performed on a small sample of liver and is likely not representative of the entire organ. Liver histology did not demonstrate any evidence of cellular infiltrates that would indicate an ongoing immune response or toxicity (Figure S4B).
Humoral Response to AAV2 Capsid Proteins Is Enhanced in Early ATG-Treated Animals at the Time of Vector Injection
The IS regimens did not prevent humoral responses to the vector capsid as observed by the titer of anti-AAV2 IgG antibodies (Figure 7). Early ATG administration appeared to partially suppress the onset of the humoral anti-capsid response, with antibody titers rapidly increasing after IS withdrawal in all three animals (Figure 7A). Intriguingly, only one of the three animals that received delayed ATG administration developed high anti-AAV antibody levels, with the onset occurring just prior to IS withdrawal (Figure 7B). There is a marked difference in the mean anti-AAV IgG level between the early and late cohort, although with only three animals per group, there is not a statistically significant difference.
Figure 7.

Anti-AAV Antibody Formation following Vector Administration
(A and B) Anti-AAV antibody levels after AAV-hFIX gene transfer for animals that received early (A) or delayed (B) ATG. Each line represents an individual animal, and the gray line indicates the timing of IS withdraw at week 8.
Discussion
Immune-mediated obstacles continue to impede widespread implementation of AAV-based genetic treatments.7, 8, 9, 10 Current studies generally exclude subjects with high-titer NAbs or who are at high risk for development of anti-transgene protein antibodies. However, effective strategies to consistently mitigate the anti-capsid cellular immune response remain elusive,7, 8, 9, 10 in part because several preclinical models do not recapitulate the clinical experience.20, 21, 22, 23, 24 Although IS with oral steroids has been successful in several liver-directed AAV gene therapy trials in controlling the anti-capsid cellular immune response to limit the loss of transgene expression,13, 14, 15 results from other trials reported in abstracts suggest steroids can be insufficient to prevent transgene loss.7, 8, 9,16, 17, 18, 19
In this work, we evaluated the timing of an intensive T cell-directed IS regimen as a possible strategy to better control this anti-capsid cellular immune response. MMF and rapamycin are well established IS drugs targeting T cells that are used in solid organ transplant, hematopoietic stem cell transplant (HSCT), and autoimmune diseases. ATG targets T cells, as well as a spectrum of other immune cells and molecules, and is used in similar conditions.30,31 Consistent with previous results, rabbit ATG is relatively Treg sparing compared with other lymphocytes.32 However, we observed that concomitant administration of ATG and AAV-hFIX vector breaks the tolerance to hFIX, with two out of three treated animals developing an anti-hFIX immune response, including an animal with a high-titer (>5 BU) inhibitor.
In contrast, none of the animals that received ATG around 5 weeks after AAV-hFIX delivery developed an anti-hFIX response. Because the earliest described onset of the anti-capsid cellular immune response is at ∼5 weeks after vector administration, our results suggest that the IS regimen of delayed ATG with MMF and rapamycin is a potential intensive therapy to treat the anti-capsid cellular immune response after systemic AAV vector delivery, because it does not interfere with transgene-product tolerance. However, ATG timed concomitantly with vector delivery should be avoided.
A previous NHP study evaluating intensive anti-T cell IS regimens to prevent the anti-AAV-capsid cellular immune response observed that the addition of the anti-CD25 antibody daclizumab at the time of vector delivery to MMF and rapamycin consistently resulted in the formation of inhibitory anti-hFIX antibodies, whereas dual therapy of MMF and rapamycin alone did not.11 Treatment with daclizumab was associated with a decrease in Tregs, suggesting that Tregs are critical for immune tolerance induction of the transgene-product after liver-directed gene therapy.11 Our results herein suggest that there is a critical time period surrounding vector administration when T cell- and likely Treg-dependent processes occur that initiate peripheral immune tolerance; early intensive T cell IS may disrupt these processes and result in a failure to develop immune tolerance. Indeed, the only animal in group 1 that did not develop an inhibitor (1001) had the highest frequency of Tregs of the early ATG treatment group. Supporting this model of early Treg-dependent processes initiating transgene-product immune tolerance is our previous observation that AAV-based gene therapy in HA and HB dogs with pre-existing inhibitors (against FVIII or FIX, respectively) results in an increase in Tregs within a few weeks of AAV vector administration and eradication of the inhibitors and induction of long-term immune tolerance.33, 34, 35
In the two animals in this study that developed inhibitors, we also observed a pro-inflammatory cytokine profile of increased levels of TNF-α, IL-1β, and IL-17, as well as an increase in pro-inflammatory Th17 cells several weeks before the development of anti-hFIX inhibitors. IL-1β has recently been implicated in the innate immune response to AAV.36 Th17 cells have been implicated in the initial development of anti-FVIII inhibitors in HA patients37 and after hFVIII exposure in HA mice.38 Th17 cells, and especially a skewed Th17/Treg balance, are involved in numerous autoimmune and inflammatory conditions.39, 40, 41 Consistent with this immunological mechanism, the two animals that developed inhibitors had substantial increase in their Th17/Treg ratios from baseline. This suggests that tolerance to the transgene-protein after AAV gene therapy requires sufficient Treg activity to offset the pro-inflammatory Th17 activity. Early intensive anti-T cell therapy can disrupt this balance, leading to an anti-transgene-product immune response.
Although the evaluated IS regimens were not designed to prevent anti-AAV antibody formation, it is intriguing that two out of the three NHPs that received delayed ATG did not mount an anti-AAV antibody response. The development of anti-AAV antibody in NHPs after systemic AAV vector administration is consistently observed without IS,11,42, 43, 44, 45, 46 as well as with IS regimens that have included: (1) rituximab and cyclosporine;47 (2) MMF, rapamycin, and daclizumab around vector administration;11 and (3) tacrolimus, MMF, rituximab, as well as methylprednisolone and ATG, around vector administration.48 In contrast, MMF monotherapy delayed anti-AAV antibody formation in rats,49 dual therapy with MMF and rapamycin limited anti-AAV antibody formation in NHPs,11 and the IS regimen of rituximab, rapamycin, and methylprednisolone prevented anti-AAV antibodies in a single human subject.50 More recently, concomitant administration of AAV vectors with rapamycin-containing nanoparticles that home to antigen-presenting cells have prevented anti-AAV antibody development in mice and NHPs, and allowed for vector re-administration, likely because of an increased frequency of Tregs.51 Our study suggests that the timing of anti-T cell IS may be able to modulate the likelihood of developing anti-AAV antibodies, and delaying ATG, as in our group 2, is likely preferred over administering ATG or other T cell-directed therapy around vector infusion, as in group 1 or as previously reported.11,48
Our study used rabbit ATG, which is available worldwide. However, horse ATG, which is available in the United States, is more efficacious than rabbit ATG in IS therapy for severe aplastic anemia.32 Horse ATG is less immunosuppressive than rabbit ATG, with total lymphocytes and lymphocyte subsets normalizing within a few weeks of administration. The use of horse ATG to prevent anti-AAV cellular immune response warrants further attention.
Although pre-clinical studies are promising,51 there is no current reliable procedure to re-administer AAV vectors. As such, the loss of transgene expression because of an uncontrolled anti-AAV-capsid T cell immune response represents a failure of the received AAV product, but also of the field, because individuals who have lost expression are currently prevented from ever accruing the benefits of AAV-based gene therapy. Approaches to address the anti-AAV-capsid cellular immune response include increasing the vector potency to decrease the therapeutic dose52,53 and steroid IS to prevent or treat the immune response.13, 14, 15,54 However, neither strategy is consistently effective. Intensive T cell-directed IS strategies are attractive given the lifelong consequences of a failed AAV vector administration. Previous pre-clinical studies investigating alternative IS regimens have highlighted the potential of intensifying the NAbs and the anti-transgene-product immune responses.11 Our data indicate that there is a critical time period around vector administration during which intensive IS can shift the immune system toward immunogenicity and away from immune tolerance. This phenomenon should be considered when designing IS regimens for AAV-based gene therapy.
Materials and Methods
Production of AAV2 Vectors and AAV2 Empty Capsids
Recombinant AAV2 vectors expressing hFIX under control of the human α-1 antitrypsin and apolipoprotein E promoter/enhancer were manufactured by standard triple transfection followed by double-cesium gradient centrifugation as previously described.6 Silver stain of the vector showed no evidenee of protein degradation, and bacterial endotoxin levels were 0.15 endotoxin unit /mL.
Animal Procedures
Male rhesus macaques were housed at Charles River Laboratories, Sierra Biomedical Division (Sparks, NV, USA). Study protocol was approved by the Institutional Animal Care and Use Committee of Charles River Laboratories and conducted in accordance with the US Department of Agriculture guidelines for laboratory animals and with Good Laboratory Practices. Six monkeys ranging from 2.7 to 5.9 years old (3.1–4.5 kg/body weight) were selected for the study. Animals were randomly assigned into two groups (n = 3/group). Both groups of animals were administered immune suppression via nasogastric intubation of MMF every 12 h (CellCept; Roche) and rapamycin (Rapamune; Pfizer) once a day up to week 8 post-AAV delivery (Figure 1). Rabbit ATG (Thymoglobulin; Genzyme) was injected via peripheral vein once a day. Serum chemistry and hematology parameters were assayed by Charles River Laboratories. Plasma samples collected in EDTA were monitored for the levels of MMF (by measuring mycophenolic acid levels) and rapamycin by the Dr. William Pepper’s laboratory of Clinical Medicine at the University of Pennsylvania (UPenn).
FIX Antigen and Antibody Assays
hFIX antigen levels were measured with an ELISA (Affinity Biologicals, Canada) that does not recognize endogenous rhesus macaque FIX. Pooled normal human plasma (TriniCHECK Level 1; bioMerieux) was used as standard. Undetectable hFIX levels were arbitrarily assigned 1 ng/mL (0.001% normal) to allow for logarithmic-scale plotting. Anti-hFIX and anti-AAV IgG levels were determined using an indirect ELISA as previously described.11 Inhibitory antibodies to hFIX by Bethesda assay were determined using citrate plasma samples and heat inactivation at 56°C for 1 h to inactivate rhesus macaque FIX clotting activity.6,11
NAbs to AAV2 Capsid
NAb titers to AAV were determined as previously described with a β-galactosidase colorimetric assay.4 Prior to inclusion, 20 animals were pre-screened for neutralizing anti-AAV2 antibodies, and six animals with a titer of ≤1:3 were included in the study.
ELISpot Analysis
PBMC samples were collected at 1 week before AAV delivery (week −1) and at weeks 1, 12, and 16 after vector delivery. Frozen PBMCs were thawed and used in an IFN-γ ELISpot analysis as per manufacturer’s instructions (Mabtech, Cincinnati, OH, USA). In brief, 2 × 105 PBMCs/well in AIM-V + 5% fetal bovine serum (FBS) media were incubated in filter plates (Millipore, Billerica, MA, USA) that had been pre-coated with anti-human IFN-γ antibody (cross-reacts with rhesus macaque). Cells were stimulated with 10 μg/mL hFIX protein (Mononine; CSL-Behring, King of Prussia, PA, USA) or AAV2 empty capsid particles (10 μg/mL). Phorbol myristate (50 ng/mL) and ionomycin (1 μg/mL) were used as a positive control. Cells were stimulated overnight, and IFN-γ-secreting cells were detected using a streptavidin-conjugated anti-IFN-γ antibody. Spots were counted on an ELISpot reader (Cellular Technologies, Cleveland, OH, USA) and analyzed with Immunospot software version 3 (Cellular Technologies).
Cytokine Analysis
The Radioimmunoassay and Biomarker Core at the University of Pennsylvania performed cytokine analysis using a custom MILLIPLEX-MAP NHP cytokine panel (Millipore, Billerica, MA, USA) on duplicate citrated plasma samples taken at indicated time points.
Treg and Th17 Staining
The frequency of Tregs was determined by staining fresh PBMC samples with CD4-FITC and CD25-allophycocyanin, and intracellularly staining for FoxP3-PE using rhesus macaque-specific antibodies (eBioscience, San Diego, CA, USA) according to the manufacturer’s instructions. Th17 frequencies were determined by surface staining previously frozen PBMCs with CD4-FITC and CD196-PE-Cy7, and intracellular staining with IL-17a-PE after 5-h stimulation with PMA/ionomycin + GolgiStop. Samples were run on a BD Canto flow cytometer, and data were analyzed using FlowJo software (Tree Star, Ashland, OR, USA).
AAV DNA Detection by Quantitative Real-Time PCR
Quantitative real-time PCR was performed on liver biopsy specimens collected percutaneously via ultrasound guide at week 16 that had been snap frozen in liquid nitrogen. DNA was extracted from liver sections using a commercial genomic DNA extraction kit (QIAGEN), and quantitative real-time PCR was performed as previously described.11
Data Analysis and Statistics
Data and statistical analyses were performed with OriginPro Software package. Histology images were prepared with Aperio ImageScope.
Author Contributions
B.J.S.-J. interpreted data and drafted the manuscript. J.D.F. designed and conducted experiments, interpreted data, and revised the manuscript. P.F. conducted experiments. J.F.W. provided the AAV vectors. V.R.A. designed experiments, interpreted data, provided guidance, and drafted and revised the manuscript.
Conflicts of Interest
The authors declare no competing interests.
Acknowledgments
B.J.S.-J. reports support from NIH/NHLBI (grant 1K08HL140078). V.R.A reports receiving NIH/NHLBI funding (grants P01HL64190 and R01HL084220).
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omtm.2020.05.001.
Supplemental Information
References
- 1.High K.A., Roncarolo M.G. Gene Therapy. N. Engl. J. Med. 2019;381:455–464. doi: 10.1056/NEJMra1706910. [DOI] [PubMed] [Google Scholar]
- 2.Dunbar C.E., High K.A., Joung J.K., Kohn D.B., Ozawa K., Sadelain M. Gene therapy comes of age. Science. 2018;359:eaan4672. doi: 10.1126/science.aan4672. [DOI] [PubMed] [Google Scholar]
- 3.Konkle B.A., Huston H., Nakaya Fletcher S. Hemophilia B. In: Adam M.P., Ardinger H.H., Pagon R.A., editors. GeneReviews((R)) University of Washington; 1993. [Google Scholar]
- 4.Kay M.A., Manno C.S., Ragni M.V., Larson P.J., Couto L.B., McClelland A., Glader B., Chew A.J., Tai S.J., Herzog R.W. Evidence for gene transfer and expression of factor IX in haemophilia B patients treated with an AAV vector. Nat. Genet. 2000;24:257–261. doi: 10.1038/73464. [DOI] [PubMed] [Google Scholar]
- 5.Manno C.S., Chew A.J., Hutchison S., Larson P.J., Herzog R.W., Arruda V.R., Tai S.J., Ragni M.V., Thompson A., Ozelo M. AAV-mediated factor IX gene transfer to skeletal muscle in patients with severe hemophilia B. Blood. 2003;101:2963–2972. doi: 10.1182/blood-2002-10-3296. [DOI] [PubMed] [Google Scholar]
- 6.Manno C.S., Pierce G.F., Arruda V.R., Glader B., Ragni M., Rasko J.J., Ozelo M.C., Hoots K., Blatt P., Konkle B. Successful transduction of liver in hemophilia by AAV-Factor IX and limitations imposed by the host immune response. Nat. Med. 2006;12:342–347. doi: 10.1038/nm1358. [DOI] [PubMed] [Google Scholar]
- 7.Mingozzi F., High K.A. Overcoming the Host Immune Response to Adeno-Associated Virus Gene Delivery Vectors: The Race Between Clearance, Tolerance, Neutralization, and Escape. Annu. Rev. Virol. 2017;4:511–534. doi: 10.1146/annurev-virology-101416-041936. [DOI] [PubMed] [Google Scholar]
- 8.Ertl H.C.J., High K.A. Impact of AAV Capsid-Specific T-Cell Responses on Design and Outcome of Clinical Gene Transfer Trials with Recombinant Adeno-Associated Viral Vectors: An Evolving Controversy. Hum. Gene Ther. 2017;28:328–337. doi: 10.1089/hum.2016.172. [DOI] [PubMed] [Google Scholar]
- 9.Arruda V.R., Doshi B.S., Samelson-Jones B.J. Emerging therapies for hemophilia: controversies and unanswered questions. F1000Res. 7 (F1000 Faculty Rev) 2018:489. doi: 10.12688/f1000research.12491.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Arruda V.R., Samelson-Jones B.J. Obstacles and future of gene therapy for hemophilia. Expert Opin. Orphan Drugs. 2015;3:997–1010. doi: 10.1517/21678707.2015.1069179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mingozzi F., Hasbrouck N.C., Basner-Tschakarjan E., Edmonson S.A., Hui D.J., Sabatino D.E., Zhou S., Wright J.F., Jiang H., Pierce G.F. Modulation of tolerance to the transgene product in a nonhuman primate model of AAV-mediated gene transfer to liver. Blood. 2007;110:2334–2341. doi: 10.1182/blood-2007-03-080093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mingozzi F., Maus M.V., Hui D.J., Sabatino D.E., Murphy S.L., Rasko J.E., Ragni M.V., Manno C.S., Sommer J., Jiang H. CD8(+) T-cell responses to adeno-associated virus capsid in humans. Nat. Med. 2007;13:419–422. doi: 10.1038/nm1549. [DOI] [PubMed] [Google Scholar]
- 13.Nathwani A.C., Reiss U.M., Tuddenham E.G., Rosales C., Chowdary P., McIntosh J., Della Peruta M., Lheriteau E., Patel N., Raj D. Long-term safety and efficacy of factor IX gene therapy in hemophilia B. N. Engl. J. Med. 2014;371:1994–2004. doi: 10.1056/NEJMoa1407309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nathwani A.C., Tuddenham E.G., Rangarajan S., Rosales C., McIntosh J., Linch D.C., Chowdary P., Riddell A., Pie A.J., Harrington C. Adenovirus-associated virus vector-mediated gene transfer in hemophilia B. N. Engl. J. Med. 2011;365:2357–2365. doi: 10.1056/NEJMoa1108046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.George L.A., Sullivan S.K., Giermasz A., Rasko J.E.J., Samelson-Jones B.J., Ducore J., Cuker A., Sullivan L.M., Majumdar S., Teitel J. Hemophilia B Gene Therapy with a High-Specific-Activity Factor IX Variant. N. Engl. J. Med. 2017;377:2215–2227. doi: 10.1056/NEJMoa1708538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Monahan P.E., Walsh C.E., Powell J.S., Konkle B., Josephson N., Escobar M., McPhee S., Litchev B., Cecerle M., Ewenstein B., Rottensteiner H. Congress of the International Society of Thrombosis and Haemostasis. 2015. Update on phase 1/2 open-label trial of BAX335, an adeno-associated virus 8 (AAV8) vector-based gene therapy for program for hemophilia B; p. 87. [Google Scholar]
- 17.Calcedo R., Kuri-Cervantes L., Peng H., Qin Q., Boyd S., Schneider M., Hewitt M., Betts M., Wilson J. Immune responses in 101HEMB01, a Phase 1/2 open-Label, single ascending dose-finding trial of DTX101 (AAVrh10FIX) in patients with severe hemophilia B. Blood. 2017;130(Suppl 1):3333. [Google Scholar]
- 18.Pipe S., Stine K., Rajasekhar A., Everington T., Poma A., Crombez E., Hay C. 101HEMB01 is a Phase 1/2 open-label, single ascending dose-finding trial of DTX101 (AAVrh10FIX) in patients with moderate/severe hemophilia B that demonstrated meaningful but transient expression of human factor IX (hFIX) Blood. 2017;130(Suppl 1):3331. [Google Scholar]
- 19.High K.A., George L.A., Eyster M.E., Sullivan S.K., Ragni M.V., Croteau S.E., Samelson-Jones B.J., Evans M., Joseney-Antoine M., Macdougall A. A Phase 1/2 Trial of Investigational Spk-8011 in Hemophilia a Demonstrates Durable Expression and Prevention of Bleeds. Blood. 2018;132(Suppl 1):487. [Google Scholar]
- 20.Martino A.T., Basner-Tschakarjan E., Markusic D.M., Finn J.D., Hinderer C., Zhou S., Ostrov D.A., Srivastava A., Ertl H.C., Terhorst C. Engineered AAV vector minimizes in vivo targeting of transduced hepatocytes by capsid-specific CD8+ T cells. Blood. 2013;121:2224–2233. doi: 10.1182/blood-2012-10-460733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li H., Tuyishime S., Wu T.L., Giles-Davis W., Zhou D., Xiao W., High K.A., Ertl H.C. Adeno-associated virus vectors serotype 2 induce prolonged proliferation of capsid-specific CD8+ T cells in mice. Mol. Ther. 2011;19:536–546. doi: 10.1038/mt.2010.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li C., Hirsch M., DiPrimio N., Asokan A., Goudy K., Tisch R., Samulski R.J. Cytotoxic-T-lymphocyte-mediated elimination of target cells transduced with engineered adeno-associated virus type 2 vector in vivo. J. Virol. 2009;83:6817–6824. doi: 10.1128/JVI.00278-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li C., Goudy K., Hirsch M., Asokan A., Fan Y., Alexander J., Sun J., Monahan P., Seiber D., Sidney J. Cellular immune response to cryptic epitopes during therapeutic gene transfer. Proc. Natl. Acad. Sci. USA. 2009;106:10770–10774. doi: 10.1073/pnas.0902269106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang L., Figueredo J., Calcedo R., Lin J., Wilson J.M. Cross-presentation of adeno-associated virus serotype 2 capsids activates cytotoxic T cells but does not render hepatocytes effective cytolytic targets. Hum. Gene Ther. 2007;18:185–194. doi: 10.1089/hum.2007.001. [DOI] [PubMed] [Google Scholar]
- 25.Lozier J.N., Metzger M.E., Donahue R.E., Morgan R.A. The rhesus macaque as an animal model for hemophilia B gene therapy. Blood. 1999;93:1875–1881. [PubMed] [Google Scholar]
- 26.Dumont J.A., Loveday K.S., Light D.R., Pierce G.F., Jiang H. Evaluation of the toxicology, pharmacokinetics, and local tolerance of recombinant factor IX Fc fusion protein in animals. Thromb. Res. 2015;136:371–378. doi: 10.1016/j.thromres.2015.01.020. [DOI] [PubMed] [Google Scholar]
- 27.DiMichele D. Inhibitor development in haemophilia B: an orphan disease in need of attention. Br. J. Haematol. 2007;138:305–315. doi: 10.1111/j.1365-2141.2007.06657.x. [DOI] [PubMed] [Google Scholar]
- 28.Wilson N.J., Boniface K., Chan J.R., McKenzie B.S., Blumenschein W.M., Mattson J.D., Basham B., Smith K., Chen T., Morel F. Development, cytokine profile and function of human interleukin 17-producing helper T cells. Nat. Immunol. 2007;8:950–957. doi: 10.1038/ni1497. [DOI] [PubMed] [Google Scholar]
- 29.Sutton C., Brereton C., Keogh B., Mills K.H., Lavelle E.C. A crucial role for interleukin (IL)-1 in the induction of IL-17-producing T cells that mediate autoimmune encephalomyelitis. J. Exp. Med. 2006;203:1685–1691. doi: 10.1084/jem.20060285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mohty M., Bacigalupo A., Saliba F., Zuckermann A., Morelon E., Lebranchu Y. New directions for rabbit antithymocyte globulin (Thymoglobulin(®)) in solid organ transplants, stem cell transplants and autoimmunity. Drugs. 2014;74:1605–1634. doi: 10.1007/s40265-014-0277-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Baron F., Mohty M., Blaise D., Socié G., Labopin M., Esteve J., Ciceri F., Giebel S., Gorin N.C., Savani B.N. Anti-thymocyte globulin as graft-versus-host disease prevention in the setting of allogeneic peripheral blood stem cell transplantation: a review from the Acute Leukemia Working Party of the European Society for Blood and Marrow Transplantation. Haematologica. 2017;102:224–234. doi: 10.3324/haematol.2016.148510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Scheinberg P., Nunez O., Weinstein B., Scheinberg P., Biancotto A., Wu C.O., Young N.S. Horse versus rabbit antithymocyte globulin in acquired aplastic anemia. N. Engl. J. Med. 2011;365:430–438. doi: 10.1056/NEJMoa1103975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Finn J.D., Ozelo M.C., Sabatino D.E., Franck H.W., Merricks E.P., Crudele J.M., Zhou S., Kazazian H.H., Lillicrap D., Nichols T.C., Arruda V.R. Eradication of neutralizing antibodies to factor VIII in canine hemophilia A after liver gene therapy. Blood. 2010;116:5842–5848. doi: 10.1182/blood-2010-06-288001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Crudele J.M., Finn J.D., Siner J.I., Martin N.B., Niemeyer G.P., Zhou S., Mingozzi F., Lothrop C.D., Jr., Arruda V.R. AAV liver expression of FIX-Padua prevents and eradicates FIX inhibitor without increasing thrombogenicity in hemophilia B dogs and mice. Blood. 2015;125:1553–1561. doi: 10.1182/blood-2014-07-588194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Arruda V.R., Samelson-Jones B.J. Gene therapy for immune tolerance induction in hemophilia with inhibitors. J. Thromb. Haemost. 2016;14:1121–1134. doi: 10.1111/jth.13331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kuranda K., Jean-Alphonse P., Leborgne C., Hardet R., Collaud F., Marmier S., Costa Verdera H., Ronzitti G., Veron P., Mingozzi F. Exposure to wild-type AAV drives distinct capsid immunity profiles in humans. J. Clin. Invest. 2018;128:5267–5279. doi: 10.1172/JCI122372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ettinger R.A., James E.A., Kwok W.W., Thompson A.R., Pratt K.P. Lineages of human T-cell clones, including T helper 17/T helper 1 cells, isolated at different stages of anti-factor VIII immune responses. Blood. 2009;114:1423–1428. doi: 10.1182/blood-2009-01-200725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lai J.D., Cartier D., Hartholt R.B., Swystun L.L., van Velzen A.S., den Haan J.M.M., Hough C., Voorberg J., Lillicrap D. Early cellular interactions and immune transcriptome profiles in human factor VIII-exposed hemophilia A mice. J. Thromb. Haemost. 2018;16:533–545. doi: 10.1111/jth.13936. [DOI] [PubMed] [Google Scholar]
- 39.Noack M., Miossec P. Th17 and regulatory T cell balance in autoimmune and inflammatory diseases. Autoimmun. Rev. 2014;13:668–677. doi: 10.1016/j.autrev.2013.12.004. [DOI] [PubMed] [Google Scholar]
- 40.Patel D.D., Kuchroo V.K. Th17 Cell Pathway in Human Immunity: Lessons from Genetics and Therapeutic Interventions. Immunity. 2015;43:1040–1051. doi: 10.1016/j.immuni.2015.12.003. [DOI] [PubMed] [Google Scholar]
- 41.Knochelmann H.M., Dwyer C.J., Bailey S.R., Amaya S.M., Elston D.M., Mazza-McCrann J.M., Paulos C.M. When worlds collide: Th17 and Treg cells in cancer and autoimmunity. Cell. Mol. Immunol. 2018;15:458–469. doi: 10.1038/s41423-018-0004-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Majowicz A., Salas D., Zabaleta N., Rodríguez-Garcia E., González-Aseguinolaza G., Petry H., Ferreira V. Successful Repeated Hepatic Gene Delivery in Mice and Non-human Primates Achieved by Sequential Administration of AAV5ch and AAV1. Mol. Ther. 2017;25:1831–1842. doi: 10.1016/j.ymthe.2017.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nathwani A.C., Rosales C., McIntosh J., Rastegarlari G., Nathwani D., Raj D., Nawathe S., Waddington S.N., Bronson R., Jackson S. Long-term safety and efficacy following systemic administration of a self-complementary AAV vector encoding human FIX pseudotyped with serotype 5 and 8 capsid proteins. Mol. Ther. 2011;19:876–885. doi: 10.1038/mt.2010.274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nathwani A.C., Davidoff A.M., Hanawa H., Hu Y., Hoffer F.A., Nikanorov A., Slaughter C., Ng C.Y., Zhou J., Lozier J.N. Sustained high-level expression of human factor IX (hFIX) after liver-targeted delivery of recombinant adeno-associated virus encoding the hFIX gene in rhesus macaques. Blood. 2002;100:1662–1669. doi: 10.1182/blood-2002-02-0589. [DOI] [PubMed] [Google Scholar]
- 45.Davidoff A.M., Gray J.T., Ng C.Y., Zhang Y., Zhou J., Spence Y., Bakar Y., Nathwani A.C. Comparison of the ability of adeno-associated viral vectors pseudotyped with serotype 2, 5, and 8 capsid proteins to mediate efficient transduction of the liver in murine and nonhuman primate models. Mol. Ther. 2005;11:875–888. doi: 10.1016/j.ymthe.2004.12.022. [DOI] [PubMed] [Google Scholar]
- 46.Nathwani A.C., Gray J.T., McIntosh J., Ng C.Y., Zhou J., Spence Y., Cochrane M., Gray E., Tuddenham E.G., Davidoff A.M. Safe and efficient transduction of the liver after peripheral vein infusion of self-complementary AAV vector results in stable therapeutic expression of human FIX in nonhuman primates. Blood. 2007;109:1414–1421. doi: 10.1182/blood-2006-03-010181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mingozzi F., Chen Y., Murphy S.L., Edmonson S.C., Tai A., Price S.D., Metzger M.E., Zhou S., Wright J.F., Donahue R.E. Pharmacological modulation of humoral immunity in a nonhuman primate model of AAV gene transfer for hemophilia B. Mol. Ther. 2012;20:1410–1416. doi: 10.1038/mt.2012.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Unzu C., Hervás-Stubbs S., Sampedro A., Mauleón I., Mancheño U., Alfaro C., de Salamanca R.E., Benito A., Beattie S.G., Petry H. Transient and intensive pharmacological immunosuppression fails to improve AAV-based liver gene transfer in non-human primates. J. Transl. Med. 2012;10:122. doi: 10.1186/1479-5876-10-122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Montenegro-Miranda P.S., ten Bloemendaal L., Kunne C., de Waart D.R., Bosma P.J. Mycophenolate mofetil impairs transduction of single-stranded adeno-associated viral vectors. Hum. Gene Ther. 2011;22:605–612. doi: 10.1089/hum.2010.222. [DOI] [PubMed] [Google Scholar]
- 50.Corti M., Elder M., Falk D., Lawson L., Smith B., Nayak S., Conlon T., Clément N., Erger K., Lavassani E. B-Cell Depletion is Protective Against Anti-AAV Capsid Immune Response: A Human Subject Case Study. Mol. Ther. Methods Clin. Dev. 2014;1:14033. doi: 10.1038/mtm.2014.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Meliani A., Boisgerault F., Hardet R., Marmier S., Collaud F., Ronzitti G., Leborgne C., Costa Verdera H., Simon Sola M., Charles S. Antigen-selective modulation of AAV immunogenicity with tolerogenic rapamycin nanoparticles enables successful vector re-administration. Nat. Commun. 2018;9:4098. doi: 10.1038/s41467-018-06621-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Samelson-Jones B.J., Arruda V.R. Protein-Engineered Coagulation Factors for Hemophilia Gene Therapy. Mol. Ther. Methods Clin. Dev. 2018;12:184–201. doi: 10.1016/j.omtm.2018.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Büning H., Srivastava A. Capsid Modifications for Targeting and Improving the Efficacy of AAV Vectors. Mol. Ther. Methods Clin. Dev. 2019;12:248–265. doi: 10.1016/j.omtm.2019.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mendell J.R., Al-Zaidy S., Shell R., Arnold W.D., Rodino-Klapac L.R., Prior T.W., Lowes L., Alfano L., Berry K., Church K. Single-Dose Gene-Replacement Therapy for Spinal Muscular Atrophy. N. Engl. J. Med. 2017;377:1713–1722. doi: 10.1056/NEJMoa1706198. [DOI] [PubMed] [Google Scholar]
- 55.Nathwani A.C., Gray J.T., Ng C.Y., Zhou J., Spence Y., Waddington S.N., Tuddenham E.G., Kemball-Cook G., McIntosh J., Boon-Spijker M. Self-complementary adeno-associated virus vectors containing a novel liver-specific human factor IX expression cassette enable highly efficient transduction of murine and nonhuman primate liver. Blood. 2006;107:2653–2661. doi: 10.1182/blood-2005-10-4035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Milani M., Annoni A., Moalli F., Liu T., Cesana D., Calabria A., Bartolaccini S., Biffi M., Russo F., Visigalli I. Phagocytosis-shielded lentiviral vectors improve liver gene therapy in nonhuman primates. Sci. Transl. Med. 2019;11:eaav7325. doi: 10.1126/scitranslmed.aav7325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Spronck E.A., Liu Y.P., Lubelski J., Ehlert E., Gielen S., Montenegro-Miranda P., de Haan M., Nijmeijer B., Ferreira V., Petry H., van Deventer S.J. Enhanced Factor IX Activity Following Administration of AAV5-R338L “Padua” Factor IX versus AAV5 WT Human Factor IX in NHPs. Mol. Ther. Methods Clin. Dev. 2019;15:221–231. doi: 10.1016/j.omtm.2019.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.