

Abstract
Antimicrobial peptides (AMPs) are a valuable source of antimicrobial agents and a potential solution to the multi-drug resistance problem. In particular, short-length AMPs have been shown to have enhanced antimicrobial activities, higher stability, and lower toxicity to human cells. We present a short-length (≤30 aa) AMP prediction method, Deep-AmPEP30, developed based on an optimal feature set of PseKRAAC reduced amino acids composition and convolutional neural network. On a balanced benchmark dataset of 188 samples, Deep-AmPEP30 yields an improved performance of 77% in accuracy, 85% in the area under the receiver operating characteristic curve (AUC-ROC), and 85% in area under the precision-recall curve (AUC-PR) over existing machine learning-based methods. To demonstrate its power, we screened the genome sequence of Candida glabrata—a gut commensal fungus expected to interact with and/or inhibit other microbes in the gut—for potential AMPs and identified a peptide of 20 aa (P3, FWELWKFLKSLWSIFPRRRP) with strong anti-bacteria activity against Bacillus subtilis and Vibrio parahaemolyticus. The potency of the peptide is remarkably comparable to that of ampicillin. Therefore, Deep-AmPEP30 is a promising prediction tool to identify short-length AMPs from genomic sequences for drug discovery. Our method is available at https://cbbio.cis.um.edu.mo/AxPEP for both individual sequence prediction and genome screening for AMPs.
Keywords: antimicrobial peptide, drug discovery, convolutional neural network, reduced amino acid composition, machine learning, Candida glabrata, ampicillin, AmPEP, AxPEP
Graphical Abstract
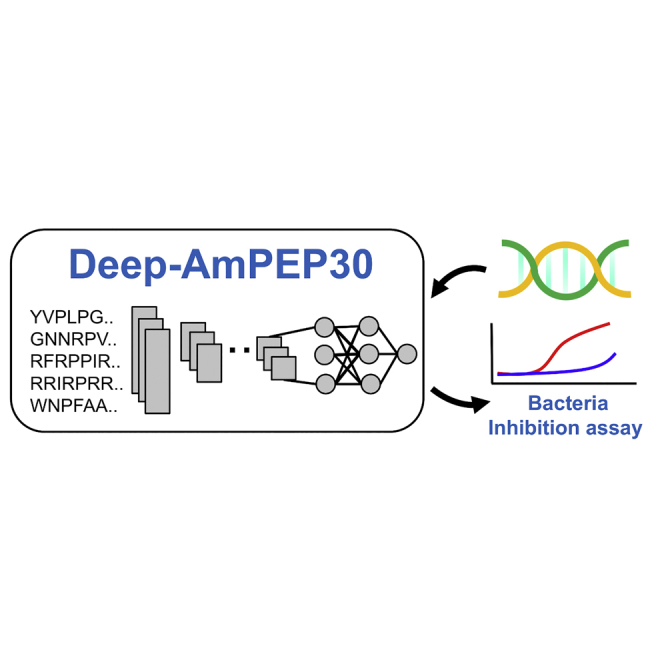
Siu and colleagues report the first short antimicrobial peptide classifier based on convolutional neural network. The results show that the prediction accuracy of Deep-AmPEP30 reaches 77%, outperforming state-of-the-art models. By screening the genome of Candida glabrata, Deep-AmPEP30 successfully identified a novel AMP with strong experimental anti-bacterial activity comparable to ampicillin.
Introduction
Antimicrobial peptides (AMPs; also called host defense peptides) are produced by most organisms as an innate immune response against microbes. They represent a large repertoire of antimicrobial agents and hence a valuable resource for drug discovery. In particular, cationic AMPs have received intense interest in recent years for their potential in the development of novel antibacterial drugs.1,2 These peptides often fold into amphiphilic α helices displaying both hydrophobic and hydrophilic surfaces. They accumulate by several fold at the negatively charged surface of the Gram-negative (outer membrane) or Gram-positive (cell wall) bacteria, leading to surface destabilization and permeation.3 Once having entered the bacteria cell, AMPs are able to interact with the cytoplasmic membrane, forming membrane-spanning channels or carpeting the bilayer with peptides.4 Some AMPs can target intracellular molecules and interfere with their activities.5 For example, AMPs were reported in different studies of their ability to inhibit protein synthesis, DNA binding and transcription, protein folding, and enzymatic activities. AMPs were also suggested as the next generation of anticancer drug candidates.6 By virtue of a strong electrostatic force, AMPs bind to the negatively charged surface of cancer cells, leading to membrane disruption.7 Despite their potentialities, there are limitations in using AMPs as pharmaceutical agents, such as some toxicity to human cells, lack of stability, and high manufacturing cost.8,9 While the former two limitations may be overcome through careful optimization and peptide engineering, the high cost of production inevitably makes screening a large number of peptides expensive. This, together with the possible need and costs for improving stability and toxicity, renders the peptide screening approach cost-prohibitive and hence non-appealing for pharmaceutical companies and research laboratories to undertake.
Cost constraints have been partially overcome through the development of several sequence-based computational methods for AMP prediction. These methods generally exploit machine learning algorithms to learn the sequential, compositional, and physicochemical properties of known AMP sequences. For example, Xiao et al.10 proposed iAMP-2L to predict AMPs using pseudo amino acid composition (PseAAC) and fuzzy k-nearest neighbor. Thomas et al.11 built the Collection of Anti-Microbial Peptides (CAMP) database and then constructed prediction tools using random forest (RF), support vector machines (SVMs), and discriminant analysis (DA). Meher et al.12 attempted to improve the prediction accuracy by combining structural features with compositional and physicochemical properties, and they used SVMs for the classification algorithm. In our previous work, we proposed the use of distribution patterns of amino acid properties as the sole feature type and RF classifier to generate a highly accurate prediction model, AmPEP.13 Vishnepolsky et al.14 used a semi-supervised machine learning approach based on physicochemical descriptors and the DBSCAN clustering algorithm. More recently, Veltri et al.15 introduced the first deep learning method for AMP prediction, in which they used a neural network model with convolutional and recurrent layers and the primary sequence composition as predictive features. In recent years, many promising targeted peptides prediction methods have been proposed based on sequence information. Wei et al.16 developed an anti-cancer peptides (ACPs) predictor called ACPred-FL. To build an effective predictive model, they proposed a feature representation learning scheme to integrate the class information of data into features. The class information was obtained from a pool of SVM classifiers based on sequence-based feature descriptors. They further used a two-step feature selection technique and represented the most informative five-dimensional feature vector for the final peptide representation. Similarly, Manavalan et al.17 used the predicted probability of a baseline predictor as an input feature into the final predictor to identify antihypertensive peptides (AHTPs). They extracted informative features using extremely randomized tree (ERT) algorithms. The predicted probability of AHTPs using ERT algorithms was used as an input feature with other features to four different machine learning algorithms (ERT, gradient boosting, RF, SVM). Finally, they integrated the four meta-predictors into an ensemble model for final prediction. Wei et al.18 also developed a prediction model, CPPred-RF, to identify cell-penetrating peptides (CPPs) and their uptake efficiency. They integrated multiple sequence-based feature descriptors to sufficiently explore distinct information embedded in CPPs and used the maximal relevance-maximal distance (MRMD) method to select important features. Alternatively, Wu et al.19 dealt with biologically important features to identify neoantigens. They proposed a recurrent neural network (RNN)-based method, DeepHL-Apan, for high-confidence neoantigen prediction considering both the possibility of mutant peptide presentation and the potential immunogenicity of the peptide-human leukocyte antigen complex (pHLA). Recently, Wang et al.20 proposed a feature representation named general dipeptide composition (G-DipC) based on short CPP sequences and they used the XGBoost algorithm as a classifier.
Recent discovery of several short AMPs possessing potent inhibitory activity against bacteria with high specificity and low toxicity to human cells have led to an exciting turning point in AMP research.9,21, 22, 23 This new class of AMPs (known as short-length AMPs), in general, has enhanced antimicrobial activities, lower toxicity, and higher stability.9,21 More importantly, the nature of its shorter length also means that the synthesis, modification, and optimization of these AMPs are relatively easier and cheaper than with regular AMPs. The significant improvements introduced with the use of short-length AMPs have collectively made them an attractive and affordable class of molecules for drug screening purpose. To date, several AMPs are already being tested as antibacterial agents in clinical trials.6
Efforts to identify AMPs from the vast amount of freely available genome sequences (especially the genome of those organisms constantly living and interacting with microbes) are expected to yield many novel AMPs with diverse pharmaceutical properties, including anti-cancer activity, which is commonly found in AMPs. However, there is currently no prediction program for identifying AMPs from assembled genome sequences. Moreover, it was unclear whether existing AMP prediction methods, which were originally developed based on collection of AMPs without length consideration, would be suitable for predicting short-length AMPs. To answer this question, we first tested four state-of-the-art AMP prediction methods10,12,13,15 using peptides 5–33 residues in length. Unexpectedly, the prediction accuracy achieved with the models was between 65% and 73%, much worse than the previously reported accuracy of 90%–95% for peptides of any length. We hypothesized that many of the long sequences included in the datasets for model construction may not be at their optimal composition for antimicrobial function. This means that they may include sequence segments that do not contribute to antimicrobial activities of the peptides. However, deducing the optimal subsequence for antimicrobial function from a given sequence is not a trivial task. For the purpose of developing a prediction model targeting short sequences, we proposed to lower the maximum sequence cutoff length used in the training dataset to 30, instead of the 80–255 range used in previous studies.10,12,13,15 In this way, a large proportion of suboptimal sequences would be removed, which should raise the accuracy and specificity in identification of short-length AMPs.
Herein, we present a method, Deep-AmPEP30, for short-length AMP prediction based on the reduced dataset using a deep convolutional neural network (CNN)24 and reduced AAC (RAAC). In RAAC, amino acids are clustered on the basis of evolutionary information, substitution score, hydrophobicity, and contact potential energy. Clustering is a simple but effective approach to reduce the dimensionality in protein sequence encoding, to reduce over-fitting, and to improve model performance. For example, it enhances the sensitivity and selectivity in fold recognition25 and helps to identify families of heat shock proteins more accurately.26 Compared with the 20 natural amino acids composition, the RAACs exhibited superior predictive capability with reduced protein complexity and the ability to withdraw conservative features hidden in noise signals.27,28 In 2017, Zuo et al.29 established the first online web server, which performed high-potential roles in dealing with protein sequence analysis, such as protein folding,30 protein defensins,31 animal toxins,32 heat shock protein,26 type VI secreted effectors,33 DNA-binding proteins,34 and so on. With the wide applications of PseKRAAC (pseudo K-tuple RAAC), more and more online services were proposed for protein sequence-dependent inference during the last decade. In this study, we combine the power of CNN and different types of RAAC as features in order to increase the prediction accuracy for short functional AMPs.
To further validate our method, we applied Deep-AmPEP30 to identify potential short-length AMPs from the genome sequence of Candida glabrata, a commensal fungus living with trillions of other microbes in the gut. We hypothesized that C. glabrata may have evolved the means (e.g., via AMPs) to interact with and/or inhibit other microorganisms in the competitive gut environment and that if AMPs were involved our program would be able to identify them. Indeed, a short peptide of 20 aa was selected by Deep-AmPEP from sequences extracted from the C. glabrata genome and experimentally validated to have anti-bacterial activities as potent as that of ampicillin. Encouraged by the positive prediction outcome, we have constructed a web service to process not only individual peptide sequences, but also fully assembled genome DNA sequences for AMP prediction using Deep-AmPEP. To the best of our knowledge, our web service is the first and so far the only one designed for processing and predicting AMPs from genome DNA sequences, and it is expected to facilitate screening the colossal amount of genome sequences of diverse organisms for novel and active short-length AMPs.
Results and Discussion
Feature Selection and Model Performance
In an attempt to select the best features among four commonly used sequence-encoding methods for AMP prediction—namely, AAC, composition-transition-distribution (CTD), general PseAAC (PseAAC-General), and PseKRAAC—and the best feature modes from the two latter methods, we performed extensive 10-fold cross-validation (CV) experiments using the training set on the proposed CNN model. Based on the wrapper-based sequential forward selection (SFS) approach, the best performing PseAAC-General feature mode was series-correlation PseAAC (SC-PseAAC), with an accuracy of 75.1%. Next, we tested SC-PseAAC features generated using combinations of parameters with λ = {1, 2, 3, 4} and w = {0.1, 0.2, …, 1.0}. The optimal parameters were identified as λ = 4 and w = 0.2, giving an accuracy of 76.2%. The relatively small value of w suggests that the residue-pair correlation only plays a minor role in AMP identification as compared with the AAC.
For PseKRAAC, the feature selection procedure yielded a set of five feature modes that gave the highest prediction accuracy at 76.5%. These “five-best” feature modes were type 3A-cluster 19, type 7-cluster 15, type 8-cluster 17, type 12-cluster 17, and type 12-cluster 18. A brief description of these feature modes and the respective reduced amino acids alphabets are listed in Table 1.
Table 1.
Five-Best Reduced Amino Acid Types and Clusters Selected from PseKRAAC for AMP Prediction
| Type | Description | No. of Clusters | Reduced Amino Acid Alphabets |
|---|---|---|---|
| 3A | based on PAM (point accepted mutation) matrix | 19 | {(FA),(P),(G),(S),(T),(D),(E),(Q),(N),(K),(R),(H),(W),(Y),(M),(L),(I),(V),(C)} |
| 7 | based on inter-residue contact energies using the Miyazawa-Jernigan matrix | 15 | {(C),(K),(R),(W),(Y),(A),(FILV),(M),(D),(E),(Q),(H),(TP),(GS),(N)} |
| 8 | based on properties of JTT (Jones-Taylor-Thornton) rate matrices | 17 | {(AT),(C),(DE),(F),(G),(H),(IV),(K),(L),(M),(N),(P),(Q),(R),(S),(V),(W)} |
| 12a | based on the substitution scores using database of aligned protein structures | 17 | {(TVLI),(M),(F),(W),(Y),(C),(A),(H),(G),(N),(Q),(P),(R),(K),(S),(T),(DE)} |
| 12a | based on the substitution scores using database of aligned protein structures | 18 | {(TVLI),(M),(F),(W),(Y),(C),(A),(H),(G),(N),(Q),(P),(R),(K),(S),(T),(D),(E)} |
Type 12 in the PseKRAAC web server corresponds to type 11 in Zuo et al.29
A comparison of the best CNN models from all four feature types is shown in Table 2. Experiments of 10 times 10-fold CV were performed to obtain sufficient statistics about these models. The results confirm that the five-best PseKRAAC with 86 features achieves the highest auto-cross covariance (ACC) of 77%, area under the receiver operating characteristic curve (AUC-ROC) of 82%, area under the precision-recall curve (AUC-PR) of 80%, kappa of 53%, and MCC of 54%. The second- and third-best models are SC-PseAAC with 32 features and AAC with 20 features, respectively. It is noteworthy that the sequence-encoding methods used by the top two models are PseAAC-based variants, suggesting that grouping amino acids is crucial for detecting the correlation between amino acid properties and peptide functionality. Interestingly, in the top three models either all or most dominant features are compositional. While it is well recognized that AMPs often adopt helical conformation with a hydrophobic surface on one side and a hydrophilic surface on the other side, one may assume that not only the composition but also the inter-residue correlation should be strong to uphold the AMP function. However, as shown in Table S1, accuracies of CNN models using SC-PseAAC features generated by different λ values with the same w differ by only 1%–3%. In PseAAC, λ defines the maximum positional distance between two residues from which sequence order correlation is calculated, while w is the weight factor of the calculated sequence order effect. The larger the value of λ the farther the two residues are in terms of sequential distance to be considered. It is observed that λ of the best model is 4, coincident with the number of residues per turn in α helices, which is 3.6. Nevertheless, the best model used only a small weight of w = 0.2, and an increase in w did not show improvement in the prediction accuracy. A possible explanation for the minor role of inter-residue correlation in AMP identification is that many AMPs in the dataset do not adopt helical conformation or only segments of peptides are helical. This agrees with the structural statistics published in the Antimicrobial Peptide Database (APD) server that only 14% of AMPs are helices and around 4% of AMPs are in combined helix and β structures; meanwhile, the structures of as many as 60% of AMPs are unknown.
Table 2.
Comparison of CNN Classifiers of Different Feature Sets By 10 Times 10-Fold Cross-Validation
| Feature Set {#} | ACC | AUC-ROC | AUC-PR | Kappa | Sn | Sp | MCC |
|---|---|---|---|---|---|---|---|
| T {21} | 71.22 ± 0.51 | 77.41 ± 0.22 | 73.97 ± 0.53 | 42.43 ± 1.01 | 78.22 ± 1.1 | 64.21 ± 0.91 | 42.86 ± 1.06 |
| C {21} | 72.65 ± 0.35 | 78.33 ± 0.12 | 75.54 ± 0.32 | 45.30 ± 0.69 | 77.85 ± 1.66 | 67.45 ± 1.29 | 45.57 ± 0.78 |
| CTD {147} | 73.71 ± 0.34 | 79.96 ± 0.21 | 76.61 ± 0.48 | 47.41 ± 0.67 | 79.05 ± 1.93 | 68.36 ± 1.51 | 47.71 ± 0.79 |
| D {105} | 73.74 ± 0.23 | 79.92 ± 0.17 | 76.73 ± 0.30 | 47.48 ± 0.46 | 79.33 ± 1.2 | 68.14 ± 1.01 | 47.79 ± 0.52 |
| AAC {20} | 74.27 ± 0.26 | 80.48 ± 0.19 | 77.52 ± 0.31 | 48.55 ± 0.51 | 80.92 ± 0.85 | 67.63 ± 1.05 | 48.99 ± 0.48 |
| SC-PseAAC {32}a | 75.62 ± 0.27 | 82.07 ± 0.19 | 79.04 ± 0.37 | 51.24 ± 0.54 | 82.33 ± 0.78 | 68.91 ± 1.18 | 51.72 ± 0.45 |
| Five-best PseKRAAC {86} | 76.50 ± 0.37 | 82.48 ± 0.20 | 79.55 ± 0.5 | 53.00 ± 0.74 | 83.35 ± 0.86 | 69.65 ± 0.67 | 53.51 ± 0.78 |
Values shown are mean ± SD (values were multiplied by 100).
Parameters used for SC-PseAAC (series-correlation pseudo amino acid composition, commonly known as type-2 PseAAC) are λ = 4 and w = 0.2.
Deep learning has been mainly applied in learning problems with large datasets. In this short AMP prediction problem, our dataset is relatively small, with only 1,529 positive samples. To examine whether our model architecture and the training procedure were appropriate, we tested the CNN model on progressively increased size of the training set. As a reference, we took the larger AMP dataset, which is a dataset containing >3,000 samples used in our previous study,13 to understand the size effect of a dataset on model performance. Figure 1 shows that all performance measures of our CNN model reached stability when the size of the dataset was between 2,000 and 3,000. The result of the same test using the larger AMP dataset shows a similar trend for datasets of 3,000 and 6,000 (see Figure S1). Hence, we can conclude that our model is suitable for learning small datasets of AMP and short AMPs. We anticipate that when a larger dataset will be available in the future, the model may need to be revised for learning more complex data.
Figure 1.

Size Effect of the Train Dataset on Model Performance
CNN versus Other Learning Algorithms
Using the CNN-selected best features from PseKRAAC, we compared the performances of two popular machine learning algorithms, RF and SVM, to the CNN model using 10 times 10-fold CV against the training dataset. The best RF classifier was selected by running different combinations of hyperparameter values (mtry for the number of variables at the split of the tree, and ntree for the number of trees). A two-stage approach of grid search was executed. First, a coarse search was performed to find out promising ranges of the parameter values, after which a finer search was conducted to decide the optimal parameters. Experiments were performed using the training dataset. As shown in Figure S2, the model with mtry = 1 and ntree = 1,200 performs the best, giving an average accuracy of 75.42% in repeated 10-fold CV experiment.
Performances of four models using different learning algorithms are compared in Table 3. The RF classifier with tuned parameters performed close to the CNN model but with a 1%–6% reduction in performance across different aspects. Both SVM models—one using the linear kernel and the other radial kernel—performed inferiorly. Henceforth, the two best models are further compared with other methods; we name the five-best PseKRAAC CNN classifier as Deep-AmPEP30 and the RF classifier as RF-AmPEP30.
Table 3.
Comparison of CNN, RF, and SVM Using Five-Best PseKRAAC Features by 10 Times 10-Fold Cross-Validation
| Algorithm | ACC | AUC-ROC | AUC-PR | Kappa | Sn | Sp | MCC |
|---|---|---|---|---|---|---|---|
| CNN (from Table 2 five-best PseKRAAC) | 76.50 ± 0.37 | 82.48 ± 0.20 | 79.55 ± 0.5 | 53.00 ± 0.74 | 83.35 ± 0.86 | 69.65 ± 0.67 | 53.51 ± 0.78 |
| RF | 75.42 ± 0.23 | 80.58 ± 0.09 | 74.85 ± 0.21 | 50.84 ± 0.46 | 81.55 ± 0.42 | 69.28 ± 0.22 | 51.23 ± 0.48 |
| SVMlinear | 72.37 ± 0.24 | 77.90 ± 0.07 | 76.08 ± 0.14 | 44.75 ± 0.49 | 68.36 ± 0.29 | 76.39 ± 0.42 | 44.95 ± 0.50 |
| SVMradiala | 56.90 | 38.95 | 47.80 | 13.75 | 74.39 | 39.36 | 23.56 |
Parameters used were as follows: RF, mtry = 1, ntree = 1,200; SVMlinear, cost = 1; SVMradial, gamma= 0.008569952, cost = 0.25. Values shown are mean ± SD (values were multiplied by 100). In the text, the model of CNN is referred to as Deep-AmPEP30 and RF as RF-AmPEP30.
One 10-CV was performed.
Comparison with State-of-the-Art Methods
Using the benchmark dataset, we compared our short-length AMP classifiers, Deep-AmPEP30 and RF-AmPEP30, to three state-of-the-art general AMP prediction methods and our previous AmPEP method. As shown in Table 4, our classifiers Deep-AmPEP30 and RF-AmPEP30 outperformed general AMP methods in most performance metrics. In particular, they achieved an accuracy of 77%, AUC-ROC of 85%, and MCC of 54%. The overall measures ACC, MCC, and kappa suggest that Deep-AmPEP30 is marginally better than RF-AmPEP30. The AUC-ROC curves shown in Figure 2A confirm that both models have high accuracy at different classification thresholds.
Table 4.
Comparison of Our Prediction Models with Existing Methods Using the Benchmark Dataset
| Method | ACC | AUC-ROC | AUC-PR | Kappa | Sn | Sp | MCC | Reference |
|---|---|---|---|---|---|---|---|---|
| iAMP-2L | 65.43 | – | – | 31.85 | 82.98 | 47.87 | 32.95 | Xiao et al.10 |
| iAMPpred | 70.74 | – | – | 41.49 | 80.85 | 60.64 | 42.36 | Meher et al.12 |
| AmPEP | 68.09 | 75.14 | 68.63 | 36.17 | 93.62 | 42.55 | 42.07 | Bhadra et al.13 |
| AMP Scanner DNN | 73.40 | 80.66 | 77.78 | 46.81 | 80.85 | 65.96 | 47.34 | Veltri et al.15 |
| RF-AmPEP30 | 77.12 | 85.46 | 86.83 | 54.25 | 77.65 | 76.59 | 54.25 | This study |
| Deep-AmPEP30 | 77.13 | 85.31 | 85.36 | 54.26 | 76.60 | 77.66 | 54.26 | This study |
All values were multiplied by 100.
Figure 2.

Performance of AMP Classifiers
(A) Receiver operator characteristic curves of different AMP classifiers and (B) their run time performances on the benchmark dataset.
Run Time Performance
The run time performance of the two methods, RF-AmPEP30 and Deep-AmPEP30, were compared to our previously developed general prediction method AmPEP. We used all data from the two datasets for training the final models of RF-AmPEP30 and Deep-AmPEP30, i.e., 1,651 positive sequences and 1,646 negative sequences. For testing the run time performance, we randomly selected sequences of 5–30 residues from the open reading frames of C. glabrata genome to form a test dataset that was subjected to prediction subsequently by three methods. The experiment was repeated five times and the run time of each method was recorded. The computation was done on a HP workstation with 32-core Intel Xeon E5-2650 processors at 2.6 GHz and 16 GB memory. Run time performances of the three methods are also shown in Figure 2B. It was observed that both short-length AMP classifiers run efficiently and are able to complete the prediction within 70 s for 20,000 sequences. Although Deep-AmPEP30 is slightly better in accuracy than RF-AmPEP30 (see Tables 3 and 4), it requires also increased run time when the prediction involves 9,000 or more sequences (Figure 2B, inset). Nonetheless, results of this run time performance test suggest that short-length classifiers are efficient tools for running massive prediction of AMPs, such as for virtual screening of genome sequences.
Screening of the C. glabrata Genome for Novel AMPs
To showcase an application of the short-length classifiers developed in this study, we performed screening of the C. glabrata genome using the Deep-AmPEP30 method. To this end, we first converted the entire genomic DNA sequences into peptide sequences from all six translation frames. Among 456,723 peptide sequences extracted, 243,072 sequences were of length from 5 to 30 residues and subjected to AMP prediction. To minimize false-positive results, we applied a strong classification threshold of 0.998 to the Deep-AmPEP30 classified peptides; with the classification filter, 24 sequences with lengths from 15 to 29 residues were obtained (see Table S2). To select the most promising AMPs for experimental validation, we investigated the peptide-membrane interactions of all 24 sequences using the CPPpred method of the CellPM server.35 CPPpred predicts the peptide ability to cross the lipid bilayer by calculating its optimal spatial position in the membrane, the energy of membrane binding, and the lowest energy translocation pathway across the lipid bilayer. The predictions were performed using the following parameters: T = 310 K, pH 7.4, and dioleoylphosphatidylcholine (DOPC) bilayer for the membrane type (the only membrane type available). For calculation of the lowest transfer energy pathway, ΔG(z), we tried both dragging optimization and global energy optimization methods. The predicted values of ΔG(z) of the 24 sequences range from −14.3 to −2.7 kcal/mol, with a log of partition coefficient (log Pcalc) ranging from −5.5 to −34.8 using the dragging optimization method and from 3.1 to −35.3 using the global energy optimization method. Two sequences, P3 and P10, which gave the lowest ΔG were selected for further biological experiments. In addition, the highest scoring sequence of Deep-AmPEP30, P26, was also selected for subsequent experimental testing. The three selected sequences and their respective scores are summarized in Table 5. All sequences in the list were checked to confirm that none of them was a previously known AMP.
Table 5.
The Three Selected C. glabrata Genome Sequences for Experimental Validation and Their Predicted Ability to Cross Lipid Bilayer by CPPpred
| ID | Sequence | Net Charge | Length | Deep-AmPEP30 | RF-AmPEP30 | ΔGa (kcal/mol) | Log Pcalc1 | Log Pcalc2 |
|---|---|---|---|---|---|---|---|---|
| P3 | FWELWKFLKSLWSIFPRRRP | +4 | 20 | 0.999090 | 0.785833 | −14.3 | −19.3 | −3.9 |
| P10 | ICTTLNWMVKLTCLTHVTLTTRWC | +2 | 24 | 0.998451 | 0.627500 | −12.8 | −5.5 | 3.1 |
| P26 | RWPPTTTLCYLSRPRRCSWTSSVCRCTLT | +7 | 29 | 0.999972 | 0.709167 | −9.2 | −31.7 | −13.4 |
Log Pcalc of membrane permeability coefficient predicted using the dragging optimization method (log Pcalc1) and the global optimization method (log Pcalc2).
Water-to-membrane transfer free energy by CPPpred..
To validate the prediction, we commercially synthesized the three predicted AMPs and tested them in an anti-bacterial assay against three Gram-negative (Vibrio parahaemolyticus, Pseudomonas aeruginosa, and Escherichia coli) and one Gram-positive (Bacillus subtilis) bacteria. Interestingly, there are bacteria-specific effects for the three peptides (Figure 3). For example, P3 showed strong inhibitory effect against B. subtilis (Gram positive) and V. parahaemolyticus (Gram negative), and the effect was as strong as, if not even better than, that by ampicillin (see V. parahaemolyticus at a later time, around 9–12 h). In contrast, P3 did not inhibit growth of P. aeruginosa and E. coli under the same culturing conditions. Alternatively, P10 and P26 had no noticeable effects against B. subtilis, V. parahaemolyticus, and P. aeruginosa; interestingly, however, they seemed to slow down the growth of E. coli between 5 and 18 h after inoculation. This subtle growth defect is consistently observed across all three biological repeats, and it is therefore biologically significant and meaningful, although we do not understand the cause at the moment. Nevertheless, the bacteria assay results nicely validated the prediction of our Deep-AmPEP30 method. More importantly, when applied on the vast amount of freely available genome sequences, the program can offer an easy but powerful way to realize the potentials of short-length AMPs for drug discovery.
Figure 3.

Anti-Bacterial Effect of Three Top-Ranked Predicted AMPs against Four Different Bacteria Species
Growth assay of Bacillus subtilis, Vibrio parahaemolyticus, Pseudomonas aeruginosa, and Escherichia coli in the absence (H2O) or presence of P3, P10, P26, and a control peptide (Pcontrol) that is known to have no anti-bacterial effect. Ampicillin was used as a positive control. Growth of bacteria was measured by absorbance at OD600 over time. The average of three independent experiments is presented. Treatment showing an inhibitory effect against the assayed bacteria is highlighted by a red box. A pink box indicates a subtle but significant (e.g., consistent in all three biological repeats) effect.
Web Server
To facilitate the use of our methods, we implemented a web server, called AxPEP (https://cbbio.cis.um.edu.mo/AxPEP), to allow submission and prediction of AMPs from peptide sequences as well as large DNA sequence files such as assembled genome DNA sequences. The web system consists of a front-end HTTP server to accept user inputs and to manage job queues, and a back-end application server to execute prediction tasks. The application server is equipped with 2.6-GHz Intel Xeon processors that consist of a total of 32 cores and 16-GB memory running on the CentOS 7.0 platform.
Users can either directly enter amino acid sequences in FASTA format or upload them in a file. Only the one-letter codes of the 20 standard amino acids are accepted in a sequence. Three officially-released prediction methods are currently available for selection: AmPEP, Deep-AmPEP30, and RF-AmPEP30. The former is our first AMP prediction method constructed for general AMP prediction using RF13 (general here means the sequence length is not considered as a factor in the prediction model) and the latter two are the methods introduced in the present work. All input sequences should have at least 5 residues and no more than 250 residues. To predict using Deep-AmPEP30 or RF-AmPEP30, only sequences of up to 30 residues will be considered. Users can enter a job description and/or an email address (both optional) for each submitted job; this email address can be used later to retrieve results of all past jobs associated with the address.
Once a job is submitted, a unique ID is assigned to the job and the status page is displayed. This page is refreshed every 3 s to check the queue and execution status. As soon as all prediction tasks are finished, a result page is displayed. This page shows the tables of classification and prediction scores in two separate tabs. Sequences are displayed in rows and methods are shown in columns; cells in the table are color-coded to aid visualization, namely, green for positive sequences (class = 1 or score ≥0.5) and pale yellow for negative sequences (class = 0 or score <0.5). Sequences that are considered invalid for a method (e.g., sequence length >30 for Deep-AmPEP) will be displayed as −1 (error). All result data are available for download in a comma-separated values (CSV) file.
Beside individual peptide sequences, our server also accepts genome DNA sequences in FASTA format for screening AMPs from the entire genome sequence. The uploaded genome DNA sequence file will be processed and converted to peptide sequences from all six translation frames using the user-selected codon table. Peptide sequences with lengths of 5–30 aa will be sent for AMP prediction using Deep-AmPEP30.
Conclusions
Antimicrobial peptides are known to exhibit a broad spectrum of activity, including antibacterial, anticancer, antifungal, and antiviral, among others. Increasing reports of bacterial resistance to conventional antibiotics urgently demand effective methods to discover new AMPs. Short AMPs are better drug options because of their low production cost, higher stability, and minimal damage to host cells. To aid the discovery of short AMPs, we have developed a sequence-based short AMP classification model, named Deep-AmPEP30, using RAAC and a convolutional neural network. With a small dataset, the architecture of the deep learning model, the parameters, and the training process have to be carefully designed to ensure fast convergence and sufficient training, but not overfitting. To this end, we have designed a CNN model with a small number of layers, i.e., two convolutional layers, two maximum pooling layers, and only one hidden layer with 10 nodes in the dense network. We selected the rectified linear unit (ReLU) function as the activation function to facilitate fast convergence in learning and the batch normalization and dropout strategy to prevent overfitting. This model was obtained from an extensive experiment of different combinations of architectures and parameters. For comparison, we tested some traditional machine learning methods. Based on the RF classifier and tuning using grid search, we obtained a RF model that performed very close to the CNN model, with slightly inferior performance in the train and test datasets experiments. This model was named RF-AmPEP30.
Although there are a few existing AMP prediction methods available, the development of the short AMP models is different from those methods in several ways. To the best of our knowledge, this is the first time AMPs with short chain lengths (5–30 aa) are targeted computationally. Deep-AmPEP30 and RF-AmPEP30 have higher accuracy for the recognition of short-length AMPs compared to other existing AMP prediction models. We have shown that a reduced amino acid alphabet is enough to accurately recognize short AMPs. To investigate how a class or property of amino acids affects recognition accuracy and to determine the minimum amount of information needed for recognition, a large number of reduced amino acid sets were studied. We found that a combination of five reduced sets of amino acids based on evolutionary information, substitution score, hydrophobicity, and contact potential energy similarity preserve high recognition accuracy. Deep-AmPEP30 and RF-AmPEP30 outperform existing AMP prediction methods in terms of ACC, AUC-ROC, AUC-PR, kappa, and MCC. With a prediction speed of 70 s per 20,000 sequences, Deep-AmPEP30 and RF-AmPEP30 are the methods of choice for large-scale prediction tasks requiring high efficiency, such as virtual screening of sequences from a genome or computational mutagenesis studies of peptides.
In addition to the methodology development, here we have also demonstrated the use of Deep-AmPEP30 to discover novel AMPs from the C. glabrate genome, a gut commensal fungus expected to interact with and/or inhibit other microbes of the gut microbiome. Thirty short AMPs were identified with high prediction scores above 0.998. Three sequences, P3, P10, and P26, of length 20, 24, and 29, respectively, were chosen for antibacterial assay. Remarkably, all three peptides exhibited varying levels of potency against different bacteria, with P3 having the strongest inhibitory effect that is comparable to ampicillin. Taken together, our results suggest that Deep-AmPEP30 is a promising tool in identifying short-length AMPs, and its application on the vast amount of publicly available genomic sequences would leverage short-length AMPs to their full potential in novel drug discovery and development.
Feature selection is important to find out the most compact and informative feature subsets for machine learning. This has been a topic of intense research for the past two decades.36 Besides the popular wrapper-based feature selection approach that was adopted in this study, more recently developed methods are worthy of further exploration. For example, the minimum-Redundancy-Maximum-Relevance (mRMR) selects features using mutual information for computing relevance and redundancy among features;37 the Max-Relevance-Max-Distance (MRMD) selects features with strong correlation with the dependent variable and a subset with lowest redundancy features.38 Both of these methods are filter-based and were demonstrated to be effective in bioinformatics predictive problems. Strategies that combine both filter-based and wrapper-based methods might achieve higher accuracy in faster speed than the wrapper-based method alone.37
Materials and Methods
Datasets
Training Dataset
The training dataset used for model construction and CV is a subset of the training data from our previous work for AmPEP.13 Only sequences with a length of 5- to 30-aa residues were included, yielding a set of 1,529 positive non-duplicated samples. To generate a balanced training dataset, we selected an equal number of negative samples randomly from the AmPEP’s non-AMP set while maintaining the same sequence-length distribution as the positive set.
Benchmark Dataset
For comparing our proposed method to existing AMP prediction methods, an independent dataset was constructed from the benchmark dataset of a recent publication.39 Sequences that are 5–30 aa in length were taken as positive samples while the negatives were selected in the same way as in our training dataset. We checked to ensure that this benchmark dataset does not contain highly similar sequences (>90%) to the training dataset of either our method or existing AMP methods with which we made comparisons in this study. Moreover, to get rid of redundancy and avoid bias, we used the CD-HIT40 software with a cutoff of 80% to remove highly similar sequences within the set. Finally, we obtained 94 positive and 94 negative samples in the benchmark dataset.
Sequence-Encoding Methods
One of the most challenging problems in the development of sequence-based prediction methods is to effectively encode a primary sequence into feature vectors. Here, we explored four different commonly used features and their combinations for short AMP prediction, including AAC, CTD,41 all modes of PseAAC for protein sequencing,42 and all types of PseKRAAC.29 Below, we introduce them briefly.
AAC features are normalized counts of single amino acids or pairs of amino acids. Such features are simple and powerful and have been shown to perform on par with features based on protein domains or sequence information for protein interactions prediction.43
CTD41 describes the composition, transition, and distribution of a particular physicochemical property of amino acids in the sequence. It clusters all amino acids into three groups depending on the property being examined. There are seven properties recommended in the CTD method, which include charge, hydrophobicity, normalized van der Waals volume, polarity, polarizability, secondary structure, and solvent accessibility.
PseAAC-General from the Pse-in-One server42 contains eight different modes. They are the basic kmer (Kmer), auto covariance (AC), cross covariance (CC), ACC, parallel-correlation PseAAC (PC-PseAAC), SC-PseAAC, general PC-PseAAC (PC-PseAAC-General), and general SC-PseAAC (SC-PseAAC-General). The Kmer method uses the occurrence frequencies of k neighboring amino acids as features to account for the local short-range compositional effect in sequences. AC, CC, and ACC use the correlation of the same property (AC), different properties (CC), or both (AAC) between 2 aa separated by lag positions. A larger lag value takes longer range sequential effects into account. The remaining PseAAC-related features combine the AAC and global sequence-order effects via parallel correlation or series correlation. The former computes correlation factors as mean square differences of the selected physicochemical properties between residue pairs, while the latter computes correlation factors by the multiplication of property values. All general methods accept new user-defined physicochemical properties in addition to the 547 properties pre-populated in the Pse-in-One server. Two important parameters in all PseAAC methods are λ and the weight factor w. In the case of λ, the maximum positional distance of the two residues is specified for which correlations are calculated; meanwhile, w ∈ (0, 1) scales the correlation factors by a user-defined value to control their impact relative to the AAC features.
PseKRAAC employs reduced amino acid alphabets to encode a protein sequence. These are a set of alphabets, each of which contains one or more residues clustered together by their evolutionary, physiochemical, structural, or functional similarity. The PseKRAAC server supports 17 types of reduced amino acids, and each of them allows from 2 to 19 clusters. Following the concept of PseAAC, users can specify three characteristic parameters: K is the tuple size, gap is the positional separation between the tuple, and λ-correlation is the positional separation within the tuple. The PseKRAAC features with gap = 0, λ-correlation = 0, and K = 1 are simply AAC using the reduced amino acid alphabets. We illustrate in Figure S3 the computation of converting a sequence into the corresponding feature vector using an example sequence “FVKKRAAT” and type 7-cluster 15 reduced amino acid alphabets. In the type 7-cluster 15 alphabet set, amino acids are grouped into 15 clusters based on their inter-residue contact energies using the Miyazawa-Jernigan matrix {(C),(K),(R),(W),(Y),(A),(FILV),(M),(D),(E),(Q),(H),(TP),(GS),(N)}. Each cluster is labeled as Ri, where i is the cluster ID. To encode the example sequence into RAAC using the type 7-cluster 15 alphabet set, the sequence is first converted into “R7R7R2R2R3R6R6R13” and then its composition is encoded in a feature vector of {0,2,1,0,0,2,2,0,0,0,0,0,1,0,0}, where the first element indicates that there are no R1s in the transformed sequence, two R2s, one R3, and so forth.
Besides these four aforementioned feature encoding methods, we also tested the co-variance methods (auto, cross, and auto-cross), autocorrelation methods (Moran, Geary, and normalized Moreau-Broto), and dimer and quasi-sequence-order encoding methods. The performances of RF and SVM models from these features were far from satisfactory and hence they were not included for further study. Also note that as RAAC methods generate clusters of amino acids based on the evolutionary data, substitution scores, hydrophobicity, and contact potential energy, as well as other factors, RAAC encodings represent evolutionary, physicochemical, and structure properties of protein sequences in some ways. Hence, we did not explore specific evolutionary-based, structure-based, and physicochemical properties-based encoding methods.
Classifiers
We proposed a CNN classifier comprised of an eight-layer architecture of the convolutional layers and fully connected neural network. The final optimal model was the result of extensive 10-fold CV experiments based on model accuracies. All hyper-parameters (e.g., number of convolutional layers, number of kernels, activation function, dropout rate, batch size, number of hidden layers and hidden nodes, optimizer, number of epochs) were determined by systematically testing ranges of different values.
Our final optimal model is depicted in Figure 4. The first layer of our network was the input layer, followed by the first convolutional layer, a maximum pooling layer, the second convolutional layer, the second maximum pooling layer, the flattened layer, the fully connected layer, and the output layer. For each data sample, it was transformed into a feature matrix of size N × 1 (where N is the number of features) to be fed into the input layer. Batch normalization was applied to the input layer with a batch size of 64. The first convolutional layer had 128 kernels with a kernel size of 3 × 1. Each kernel was slid through the input vector with a stride value of 1 and no padding. Thus, each kernel generated N × 1 convoluted features via the convolution operation. The final convoluted feature size was 128 × N × 1. After convolution, the rectified linear function (ReLU)44 was applied as the activation function to generate outputs that were greater than 0. Then, the maximum pooling layer reduced the dimensionality of the convoluted features to 128 × floor(N/2) × 1. The second convolutional and maximum pooling layers performed convolution operation and maximum pooling on the output of the first maximum pooling layer using the same parameters. Outputs of the second maximum pooling layer were reshaped into a vector of 128 × floor(N/4) × 1 convoluted features to be fed into the fully connected neural network with 10 nodes in the dense layer. While ReLU was used again as the activation function in the dense layer nodes, the sigmoid function was used at the output layer; the output layer, in turn, computed the probability of a sample to be an AMP or not with a classification threshold of 0.5. To prevent overfitting, a dropout rate of 20%45 was used in each maximum pooling layer.
Figure 4.

The Architecture of Our CNN-Based Classifier for Short AMP Prediction
The model accepts a feature vector of N elements as input. First, the data values are normalized using a batch size of 64; then, the input is transferred into convoluted features by two convolutional layers and two maximum pooling layers. Each convolutional layer applies 128 kernels using a kernel size of 3 × 1 with stride 1, while each maximum pooling layer pools together data using a kernel size of 2 × 1 with stride 2. A dropout rate of 20% is applied in the maximum pooling step to prevent overfitting. Finally, all convoluted features are flattened and fed into a fully connected neural network with 10 hidden nodes and 1 output node. The rectified linear function (ReLU) is used as the activation function in the convolutional layer and by the hidden nodes, but the sigmoid function is used by the output node.
The training process of our CNN consisted of two phases: feed forward and back propagation. In the feed-forward phase, samples were passed through the input layer to the output layer and errors were computed using the cross-entropy loss function. Based on the obtained error, bias and weights were updated by the RMSprop optimizer in the back-propagation phase. The training was executed with 100 epochs.
Besides the CNN model, we tested two other traditional machine learning algorithms, RF and SVM, and compared their performances to those of the CNN model.
Feature Selection
Both PseAAC-General and PseKRAAC offer many feature modes; to select the best feature modes from the available sequence-encoding methods, we implemented the wrapper-based SFS method.46 Starting with an empty feature set, we added a feature mode, one at a time, to the set and evaluated the CNN classifier with this feature set using 10-fold CV. Importantly, unlike general feature selection methods where each feature is selected and included in the set independently, in our implementation, all features of a feature mode were evaluated together. This means that once a feature mode is selected based on its better performance, all of its features are included into the subset. The procedure continued until the classifier accuracy was not further improved.
For PseAAC-General, the following feature modes (with default parameters) were subjected to feature selection: Kmer, AC, CC, ACC, PC-PseAAC, SC-PseAAC, PC-PseAAC-General, and SC-PseAAC-General. Once the best feature mode was determined, combinations of parameter values were systematically tested to find the optimal parameters with the highest prediction accuracy in CV. In total, 74 CNN classifiers were evaluated.
For PseKRAAC, the same feature selection procedure as for PseAAC-General was performed. All 17 reduced amino acid types and up to 19 clusters for each type were evaluated for performances in the first feature selection iteration; this corresponded to 323 CNN classifiers. Subsequently, only the best 10 feature modes were subjected to further feature selection iterations due to the intractable large number of feature modes of PseKRAAC.
Evaluation Metrics
The performance of a prediction method was systematically assessed by seven metrics: sensitivity (Sn), specificity (Sp), accuracy (ACC), Matthew’s correlation coefficient (MCC),47 kappa statistic,48 AUC-ROC,49,50 and AUC-PR.51 The first four metrics are defined as follows:
| (1) |
| (2) |
| (3) |
| (4) |
where TP is the number of true positives, TN is the number of true negatives, FP is the number of false positives, and FN is the number of false negatives. MCC considers all aspects of the prediction method, making it one of the most comprehensive metrics.33 Values of MCC are in the range of −1 (worst) to 1 (best). The value of −1 implies total disagreement between prediction and observation, and 0 implies that the prediction method is the same as any random prediction.
To measure the reliability of the result, the kappa statistic can be used. It is defined as follows:
| (5) |
| (6) |
| (7) |
The kappa value indicates the level of agreement between the prediction and the actual values. Its value is between −1 (perfect disagreement) and 1 (perfect agreement), where 0 indicates a chance (random) agreement. Classifiers with kappa greater than 0.75 are considered high reliability, between 0.4 and 0.75 are considered moderate reliability, and less than 0.4 are deemed low reliability.48
For methods that can return a numerical value such as the probability of a sample belonging to a class, their overall predictive performance can be analyzed using all possible classification thresholds. A curve of Sn versus 1 − Sp is the ROC curve, and the area under this curve (AUC-ROC) gives the probability of the method to rank a randomly chosen positive sample higher than a randomly chosen negative sample.35,36 Likewise, the precision-recall curve focuses on the positive samples by analyzing the correctly predicted positives across all predicted positives (the precision) versus the sensitivity (also called recall) at a range of classification thresholds. The area under this curve (AUC-PR) gives the probability of a predicted positive sample to be a real positive.51
In Silico Whole-Genome Screening of C. glabrata for Novel AMP Discovery
The genome sequences of C. glabrata (version s02-m07-r07) were downloaded from the Candida Genome Database.52 Using our in-house-developed script based on the Biopython package (version 1.68, Python version 2.7.5), all six reading frames of each chromosome sequence were processed to extract the open reading frames (ORFs) using the standard genetic codon table (NCBI). An ORF is a sequence of nucleotide triplets that begins with the start codon, ends with the stop codon, and with no other stop codons in between. As shown in Table S3, a total of 12,338,305 nt of the C. glabrata genome were processed to obtain 456,723 ORFs; among them, 243,072 are in the range of 5- to 30-aa sequence length. These short sequences were subsequently subjected to AMP prediction.
Software Implementation
CNN classifiers were implemented in R (version 3.4.2) using the Keras deep learning library kerasR (version 0.6.1). Sequence features were generated using the propy package (version 1.0), Pse-in-One server at http://bioinformatics.hitsz.edu.cn/Pse-in-One/home/, and the PseKRAAC server (http://bigdata.imu.edu.cn/psekraac.ashx).
Peptide Synthesis and Preparation
Selected AMPs were commercially synthesized by ChinaPeptides (Shanghai, China). Lyophilized peptides were dissolved in water to a final concentration of 1 mg/mL. Dissolved peptides were kept at −80°C in small aliquots until use.
Bioactivity Assay on Bacteria
Bacterial inhibition assay was carried out at 37°C in the 96-well plate format using the BioTek Cytation 3 plate reader. Peptides were added to bacteria cells at a concentration of 100 μg/uL. Bacterial growth was measured by optical density (OD)600 every 15 min during 24 h and plotted as absorbance at OD600 over time.
Author Contributions
Conceptualization, Supervision, and Funding Acquisition, K.H.W. and S.W.I.S.; Methodology, Software, Investigation, and Web System, J.Y., P.B., and H.K.T.; Biological Investigation, A.L., L.Q., P.S., and K.H.W.; Writing – Original Draft, J.Y. and P.B.; Writing – Review & Editing, K.H.W. and S.W.I.S.
Conflicts of Interest
The authors declare no competing interests.
Acknowledgments
This work was supported by the University of Macau, Macao (grants MYRG2019-00098-FST and MYRG2017-00146-FST to H.K.T. and J.Y., respectively) and the Science and Technology Development Fund from Macao S.A.R., Macao (grant FDCT-066/2016/A to P.B. and S.W.I.S.; grant FDCT-085/2014/A2 and Faculty of Health Science internal grant 2018 to L.Q., P.S., and K.H.W.). Part of the computational analysis was performed at the High Performance Computing Cluster (HPCC), which is supported by the Information and Communication Technology Office (ICTO) of the University of Macau.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omtn.2020.05.006.
Supplemental Information
References
- 1.Hancock R.E.W., Sahl H.G. Antimicrobial and host-defense peptides as new anti-infective therapeutic strategies. Nat. Biotechnol. 2006;24:1551–1557. doi: 10.1038/nbt1267. [DOI] [PubMed] [Google Scholar]
- 2.Afacan N.J., Yeung A.T.Y., Pena O.M., Hancock R.E.W. Therapeutic potential of host defense peptides in antibiotic-resistant infections. Curr. Pharm. Des. 2012;18:807–819. doi: 10.2174/138161212799277617. [DOI] [PubMed] [Google Scholar]
- 3.Bechinger B., Gorr S.-U. Antimicrobial peptides: mechanisms of action and resistance. J. Dent. Res. 2017;96:254–260. doi: 10.1177/0022034516679973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ciumac D., Gong H., Hu X., Lu J.R. Membrane targeting cationic antimicrobial peptides. J. Colloid Interface Sci. 2019;537:163–185. doi: 10.1016/j.jcis.2018.10.103. [DOI] [PubMed] [Google Scholar]
- 5.Scocchi M., Mardirossian M., Runti G., Benincasa M. Non-membrane permeabilizing modes of action of antimicrobial peptides on bacteria. Curr. Top. Med. Chem. 2016;16:76–88. doi: 10.2174/1568026615666150703121009. [DOI] [PubMed] [Google Scholar]
- 6.Felício M.R., Silva O.N., Gonçalves S., Santos N.C., Franco O.L. Peptides with dual antimicrobial and anticancer activities. Front Chem. 2017;5:5. doi: 10.3389/fchem.2017.00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ma R., Wong S.W., Ge L., Shaw C., Siu S.W.I., Kwok H.F. In vitro and MD simulation study to explore physicochemical parameters for antibacterial peptide to become potent anticancer peptide. Mol. Ther. Oncolytics. 2019;16:7–19. doi: 10.1016/j.omto.2019.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Matsuzaki K. Control of cell selectivity of antimicrobial peptides. Biochim. Biophys. Acta. 2009;1788:1687–1692. doi: 10.1016/j.bbamem.2008.09.013. [DOI] [PubMed] [Google Scholar]
- 9.Kim H., Jang J.H., Kim S.C., Cho J.H. De novo generation of short antimicrobial peptides with enhanced stability and cell specificity. J. Antimicrob. Chemother. 2014;69:121–132. doi: 10.1093/jac/dkt322. [DOI] [PubMed] [Google Scholar]
- 10.Xiao X., Wang P., Lin W.Z., Jia J.H., Chou K.C. iAMP-2L: a two-level multi-label classifier for identifying antimicrobial peptides and their functional types. Anal. Biochem. 2013;436:168–177. doi: 10.1016/j.ab.2013.01.019. [DOI] [PubMed] [Google Scholar]
- 11.Thomas S., Karnik S., Barai R.S., Jayaraman V.K., Idicula-Thomas S. CAMP: a useful resource for research on antimicrobial peptides. Nucleic Acids Res. 2010;38:D774–D780. doi: 10.1093/nar/gkp1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Meher P.K., Sahu T.K., Saini V., Rao A.R. Predicting antimicrobial peptides with improved accuracy by incorporating the compositional, physico-chemical and structural features into Chou’s general PseAAC. Sci. Rep. 2017;7:42362. doi: 10.1038/srep42362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bhadra P., Yan J., Li J., Fong S., Siu S.W.I. AmPEP: sequence-based prediction of antimicrobial peptides using distribution patterns of amino acid properties and random forest. Sci. Rep. 2018;8:1697. doi: 10.1038/s41598-018-19752-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vishnepolsky B., Gabrielian A., Rosenthal A., Hurt D.E., Tartakovsky M., Managadze G., Grigolava M., Makhatadze G.I., Pirtskhalava M. Predictive model of linear antimicrobial peptides active against gram-negative bacteria. J. Chem. Inf. Model. 2018;58:1141–1151. doi: 10.1021/acs.jcim.8b00118. [DOI] [PubMed] [Google Scholar]
- 15.Veltri D., Kamath U., Shehu A. Deep learning improves antimicrobial peptide recognition. Bioinformatics. 2018;34:2740–2747. doi: 10.1093/bioinformatics/bty179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wei L., Zhou C., Chen H., Song J., Su R. ACPred-FL: a sequence-based predictor using effective feature representation to improve the prediction of anti-cancer peptides. Bioinformatics. 2018;34:4007–4016. doi: 10.1093/bioinformatics/bty451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Manavalan B., Basith S., Shin T.H., Wei L., Lee G. mAHTPred: a sequence-based meta-predictor for improving the prediction of anti-hypertensive peptides using effective feature representation. Bioinformatics. 2019;35:2757–2765. doi: 10.1093/bioinformatics/bty1047. [DOI] [PubMed] [Google Scholar]
- 18.Wei L., Xing P., Su R., Shi G., Ma Z.S., Zou Q. CPPred-RF: a sequence-based predictor for identifying cell-penetrating peptides and their uptake efficiency. J. Proteome Res. 2017;16:2044–2053. doi: 10.1021/acs.jproteome.7b00019. [DOI] [PubMed] [Google Scholar]
- 19.Wu J., Wang W., Zhang J., Zhou B., Zhao W., Su Z., Gu X., Wu J., Zhou Z., Chen S. DeepHLApan: a deep learning approach for neoantigen prediction considering both HLA-peptide binding and immunogenicity. Front. Immunol. 2019;10:2559. doi: 10.3389/fimmu.2019.02559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang S., Cao Z., Li M., Yue Y. G-DipC: an improved feature representation method for short sequences to predict the type of cargo in cell-penetrating peptides. IEEE/ACM Trans. Comput. Biol. Bioinform. 2019 doi: 10.1109/TCBB.2019.2930993. Published online July 25, 2019. [DOI] [PubMed] [Google Scholar]
- 21.Ramesh S., Govender T., Kruger H.G., de la Torre B.G., Albericio F. Short antimicrobial peptides (SAMPs) as a class of extraordinary promising therapeutic agents. J. Pept. Sci. 2016;22:438–451. doi: 10.1002/psc.2894. [DOI] [PubMed] [Google Scholar]
- 22.Thennarasu S., Tan A., Penumatchu R., Shelburne C.E., Heyl D.L., Ramamoorthy A. Antimicrobial and membrane disrupting activities of a peptide derived from the human cathelicidin antimicrobial peptide LL37. Biophys. J. 2010;98:248–257. doi: 10.1016/j.bpj.2009.09.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li X., Li Y., Han H., Miller D.W., Wang G. Solution structures of human LL-37 fragments and NMR-based identification of a minimal membrane-targeting antimicrobial and anticancer region. J. Am. Chem. Soc. 2006;128:5776–5785. doi: 10.1021/ja0584875. [DOI] [PubMed] [Google Scholar]
- 24.LeCun Y. Proceedings of the 2015 IEEE Hot Chips 27 Symposium. (HCS) (IEEE); 2016. Deep learning & convolutional networks; pp. 1–95. [DOI] [Google Scholar]
- 25.Peterson E.L., Kondev J., Theriot J.A., Phillips R. Reduced amino acid alphabets exhibit an improved sensitivity and selectivity in fold assignment. Bioinformatics. 2009;25:1356–1362. doi: 10.1093/bioinformatics/btp164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Feng P.M., Chen W., Lin H., Chou K.C. iHSP-PseRAAAC: identifying the heat shock protein families using pseudo reduced amino acid alphabet composition. Anal. Biochem. 2013;442:118–125. doi: 10.1016/j.ab.2013.05.024. [DOI] [PubMed] [Google Scholar]
- 27.Wang J., Wang W. A computational approach to simplifying the protein folding alphabet. Nat. Struct. Biol. 1999;6:1033–1038. doi: 10.1038/14918. [DOI] [PubMed] [Google Scholar]
- 28.Liu D., Li G., Zuo Y. Function determinants of TET proteins: the arrangements of sequence motifs with specific codes. Brief. Bioinform. 2019;20:1826–1835. doi: 10.1093/bib/bby053. [DOI] [PubMed] [Google Scholar]
- 29.Zuo Y., Li Y., Chen Y., Li G., Yan Z., Yang L. PseKRAAC: a flexible web server for generating pseudo K-tuple reduced amino acids composition. Bioinformatics. 2017;33:122–124. doi: 10.1093/bioinformatics/btw564. [DOI] [PubMed] [Google Scholar]
- 30.Huang J.T., Wang T., Huang S.R., Li X. Reduced alphabet for protein folding prediction. Proteins. 2015;83:631–639. doi: 10.1002/prot.24762. [DOI] [PubMed] [Google Scholar]
- 31.Zuo Y., Lv Y., Wei Z., Yang L., Li G., Fan G. iDPF-PseRAAAC: a web-server for identifying the defensin peptide family and subfamily using pseudo reduced amino acid alphabet composition. PLoS ONE. 2015;10:e0145541. doi: 10.1371/journal.pone.0145541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pan Y., Wang S., Zhang Q., Lu Q., Su D., Zuo Y., Yang L. Analysis and prediction of animal toxins by various Chou’s pseudo components and reduced amino acid compositions. J. Theor. Biol. 2019;462:221–229. doi: 10.1016/j.jtbi.2018.11.010. [DOI] [PubMed] [Google Scholar]
- 33.Wang J., Yang B., Leier A., Marquez-Lago T.T., Hayashida M., Rocker A., Zhang Y., Akutsu T., Chou K.C., Strugnell R.A. Bastion6: a bioinformatics approach for accurate prediction of type VI secreted effectors. Bioinformatics. 2018;34:2546–2555. doi: 10.1093/bioinformatics/bty155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu B., Xu J., Lan X., Xu R., Zhou J., Wang X., Chou K.C. iDNA-Prot|dis: identifying DNA-binding proteins by incorporating amino acid distance-pairs and reduced alphabet profile into the general pseudo amino acid composition. PLoS ONE. 2014;9:e106691. doi: 10.1371/journal.pone.0106691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lomize M.A., Pogozheva I.D., Joo H., Mosberg H.I., Lomize A.L. OPM database and PPM web server: resources for positioning of proteins in membranes. Nucleic Acids Res. 2012;40:D370–D376. doi: 10.1093/nar/gkr703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Basith S., Manavalan B., Hwan Shin T., Lee G. Machine intelligence in peptide therapeutics: a next-generation tool for rapid disease screening. Med. Res. Rev. 2020 doi: 10.1002/med.21658. Published online January 10, 2020. [DOI] [PubMed] [Google Scholar]
- 37.Peng H., Long F., Ding C. Feature selection based on mutual information: criteria of max-dependency, max-relevance, and min-redundancy. IEEE Trans. Pattern Anal. Mach. Intell. 2005;27:1226–1238. doi: 10.1109/TPAMI.2005.159. [DOI] [PubMed] [Google Scholar]
- 38.Zou Q., Zeng J., Cao L., Ji R. A novel features ranking metric with application to scalable visual and bioinformatics data classification. Neurocomputing. 2016;173:346–354. [Google Scholar]
- 39.Manavalan B., Basith S., Shin T.H., Choi S., Kim M.O., Lee G. MLACP: machine-learning-based prediction of anticancer peptides. Oncotarget. 2017;8:77121–77136. doi: 10.18632/oncotarget.20365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fu L., Niu B., Zhu Z., Wu S., Li W. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics. 2012;28:3150–3152. doi: 10.1093/bioinformatics/bts565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Govindan G., Nair A.S. Proceedings of the 2011 Annual IEEE India Conference. IEEE; 2011. Composition, Transition and Distribution (CTD)—a dynamic feature for predictions based on hierarchical structure of cellular sorting; pp. 1–6. [DOI] [Google Scholar]
- 42.Liu B., Liu F., Wang X., Chen J., Fang L., Chou K.C. Pse-in-One: a web server for generating various modes of pseudo components of DNA, RNA, and protein sequences. Nucleic Acids Res. 2015;43(W1):W65–W71. doi: 10.1093/nar/gkv458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Roy S., Martinez D., Platero H., Lane T., Werner-Washburne M. Exploiting amino acid composition for predicting protein-protein interactions. PLoS ONE. 2009;4:e7813. doi: 10.1371/journal.pone.0007813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nair V., Hinton G.E. Proceedings of the 27th International Conference on Machine Learning. (ICML) (Omnipress); 2010. Rectified linear units improve restricted Boltzmann machines; pp. 807–814.https://www.cs.toronto.edu/∼fritz/absps/reluICML.pdf [Google Scholar]
- 45.Hinton G.E., Srivastava N., Krizhevsky A., Sutskever I., Salakhutdinov R.R. Improving neural networks by preventing co-adaptation of feature detectors. arXiv. 2012 https://arxiv.org/pdf/1207.0580.pdf arXiv:1207.0580. [Google Scholar]
- 46.Guyon I., Gunn S., Nikravesh M., Zadeh L.A. Springer-Verlag; 2006. Feature Extraction: Foundations and Applications. [Google Scholar]
- 47.Matthews B.W. Comparison of the predicted and observed secondary structure of T4 phage lysozyme. Biochim. Biophys. Acta. 1975;405:442–451. doi: 10.1016/0005-2795(75)90109-9. [DOI] [PubMed] [Google Scholar]
- 48.Li J.Y., Fong S., Mohammed S., Fiaidhi J. Improving the classification performance of biological imbalanced datasets by swarm optimization algorithms. J. Supercomput. 2016;72:3708–3728. [Google Scholar]
- 49.Hanley J.A., McNeil B.J. The meaning and use of the area under a receiver operating characteristic (ROC) curve. Radiology. 1982;143:29–36. doi: 10.1148/radiology.143.1.7063747. [DOI] [PubMed] [Google Scholar]
- 50.Davis J., Goadrich M. Proceedings of the 23rd International Conference on Machine Learning. 2006. The relationship between precision-recall and ROC curves; pp. 233–240.https://www.biostat.wisc.edu/∼page/rocpr.pdf [Google Scholar]
- 51.Raghavan V., Bollmann P., Jung G.S. A critical investigation of recall and precision as measures of retrieval system performance. ACM Trans. Inf. Syst. 1989;7:205–229. [Google Scholar]
- 52.Skrzypek M.S., Binkley J., Binkley G., Miyasato S.R., Simison M., Sherlock G. The Candida Genome Database (CGD): incorporation of Assembly 22, systematic identifiers and visualization of high throughput sequencing data. Nucleic Acids Res. 2017;45(D1):D592–D596. doi: 10.1093/nar/gkw924. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.