

Abstract
Polycyclic aromatic hydrocarbon (PAHs) are common soil contaminants of concern due to their toxicity toward plants, animals and microorganisms. The use of indigenous or added microbes (bioaugmentation) is commonly used for bioremediation of PAHs. In this work, the biodegradation rates and changes in the bacterial community structure were evaluated. The enrichment culture was useful for unambiguously identifying members of the soil bacterial community associated with PAH degradation and yielded a low diversity community. No significant difference in the rate of PAH degradation was observed between the microcosm receiving only PAHs or PAHs and bioaugmentation. Moreover, identical matches to the bioaugmentation inoculum were only observed at the initial stages of PAH degradation on day 8. After 22 days of incubation, the substantial degradation of all PAHs had occurred in both microcosms and the PAH contaminated soil had statistically significant increases in Alphaproteobacteria. There were also increases in Betaproteobacteria. In contrast, the PAH contaminated and bioaugmented soil was not enriched in PAH degrading Proteobacteria genera and, instead, an increase from 1.6% to 8% of the population occurred in the phylum Bacteroidetes class Flavobacteria, with Flavobacterium being the only identified genus. In addition, the newly discovered genus Ohtaekwangia increased from 0% to 3.2% of the total clones. These results indicate that the same soil microbial community can give rise to different PAH degrading consortia that are equally effective in PAH degradation efficiency. Moreover, these results suggest that the lack of efficacy of bioaugmentation in soils can be attributed to a lack of persistence of the introduced microbes, yet nonetheless may alter the microbial community that arises in response to PAH contamination in unexpected ways.
Key words: polycyclic aromatic hydrocarbon, PAH, soil microbial community, biodegradation, bioaugmentation
Introduction
PAHs contamination of soil is wide spread as a result of the use of petroleum fuels, lubricants and petrochemicals and the associated spills, aerosols, disposal. In addition, there is significant release of PAH’s from natural oil reservoirs, both terrestrially and marine. A primary concern regarding PAHs is their toxicity toward plants, animals and microorganisms, with some being known or suspected carcinogens (Eisler 1987; Petry et al. 1996). Currently there are 32 PAH compounds listed as priority pollutants by the US EPA; these include naphthalene, phenanthrene, anthracene and pyrene.
PAHs of environmental concern are biodegradable, however, their rate of degradation tends to decrease with increases in ring number, ring arrangement and substitutions to the aromatic rings making many of the higher molecular weight PAHs persist in soils for extended periods (Juhasz and Naidu 2000). PAH degradation has been identified in wide a range bacteria of both Gram-positive phyla including Actinobacteria, Deinococcus-Thermus and Firmicutes, and Gram-negative phyla, including Bacteroidetes, Cyanobacteria, and all classes of Proteobacteria accept the Deltaproteobacteria (Prince et al. 2010). Nonetheless, PAH degradation in soils and sediments is complex and still poorly understood with respect to the dynamics of the response of the PAH degrading microbial community after PAH contamination. This can be attributed in part to the fact that these environments typically contain a variety of different PAH degrading microorganisms with different environmental niches and metabolic strategies, metabolic pathways and substrate ranges (Janssen 2006). In addition, there are other external factors as the presence of the fungi and surfactants, which can affect the microbial community (Wyrwas et al. 2013; Szczepaniak et al. 2016).
Due to their degradability PAHs are frequently removed from soils by natural attenuation or bioremediation by the addition of limiting nutrients, usually N, P and oxygen, and/or bioaugmentation by the addition of organisms capable of degrading these compounds (Samanta et al. 2002; Van Hamme et al. 2003). Bioremediation by addition of limiting nutrients has been shown to be quite effective under nutrient limited conditions (Lindstrom et al. 1991; Philp and Atlas 2005); however, the effectiveness of bioaugmentation is less predictable with no detectable improvement in PAH removal observed in many cases (Silva et al. 2009; Sayara et al. 2011; Tyagi et al. 2011). Moreover, in some cases, PAH removal is not benefited by the either addition of nutrients or exogenous microorganisms due to the fact that in such cases these are not limiting parameters (Bento et al. 2005).
The aim of this study was to evaluate changes in the microbial community in a pristine agricultural soil contaminated with the PAHs naphthalene, phenanthrene, anthracene and pyrene (hence forth referred to as PAH amended) and with the addition of PAH’s in combination with bioaugmentation by the addition of an exogenous PAH degrading consortia (hence forth referred to as bioaugmented).
Experimental
Materials and Methods
PAH degrading isolates used for bioaugmentation. Five PAH degrading isolates identified as Bacillus sp., Bacillus brevis, Bacillus sphaericus, Bacillus subtilis, and Chromobacterium sp., isolated from hydrocarbon contaminated soils near the REPLAN oil refinery in Southeastern Brazil and from the effluent of an oil tank steam cleaning facility, were used for bioaugmentation. To verify that the strains could degrade the PAHs used in this study, cultures were inoculated separately into 30 ml of minimal salts medium (Rambeloarisoa et al. 1984), containing 30 μl of vitamin solution (Wolin et al. 1963), 9 mg of naphthalene and phenanthrene, and 6 mg of anthracene and pyrene (1 g/l total PAHs) as the carbon source. Cultures were incubated at 30°C on a rotary shaker at 150 rpm. Efficient growth was observed for all isolates used for bioaugmentation.
For inoculum preparation the selected strains were grown separately in nutrient broth medium on a rotary shaker at 150 rpm and 30°C until the desired cell density (OD590 nm = 1). Cell density in relation to OD was determined as colony forming units (CFU) from serial dilutions on nutrient agar plates. Each culture was centrifuged and resuspended in 0.75% saline solution and the isolates were mixed together for inoculation of the microcosms.
PAH enrichment cultures. Enrichments for PAH degraders from the soil used in the microcosms was performed in the same liquid culture as describe above for verification of the PAH degrading ability of the bioaugmentation consortia. Three grams of soil were suspended in 30 ml of the same media in sterile screw capped plastic tubes without PAHs and vortexed for 5 minutes. After vortexing the sediment allowed to settle for 10 minutes. A 10 ml aliquot of the supernatant was used to inoculate 250 ml of minimal media containing 250 mg/l of naphthalene, phenanthrene, anthracene and pyrene. Enrichments were performed in triplicate. One ml samples were taken for GC analysis as described below. After 22 days of incubation, at which time the efficient growth and degradation of all PAHs were observed in all enrichment replicates, a combined sample was processed using the MoBio soil DNA extraction kit in the same fashion as for soil DNA extraction and used for 16S rRNA gene sequence analysis as described below.
Microcosm setup. The soil used for the microcosms had no history of PAH contamination; it was collected in an area located at the Experimental Faculty of Agricultural Engineering, State University of Campinas, latitude 22°48’57’’ south, longitude 47°03’33’’ west at an average altitude of 640 m. The soil is characterized as dystrophic clayey Oxisol (Typic Haplorthox), which is a soil common to various regions of Brazil.
Microcosms were performed in duplicate. After correction of soil moisture to 55% of holding capacity and homogenization, 400 g of soil was added to 1.5 l glass jars with airtight lids. The PAHs were dissolved in toluene and 6 ml of the PAH solution was added to the surface of the PAH amended and PAH amended and bioaugmented (hence forth referred to as bioaugmented) microcosm to give the following mass of each PAH, naphthalene and phenanthrene 150 mg, anthracene, and pyrene 100 mg and 50 mg hexachlorobenzene as a non-biodegradable marker for use in calculating the percent degradation of the PAHs. The microcosms were left open for 48 h in a fume hood with high air-flow to allow the toluene to evaporate. After evaporation of the toluene the soils were thoroughly mixed and inoculum was added to the bioaugmented microcosms to give a final concentration of each PAH degrader of 107 CFU/g soil, after which the soils were thoroughly mixed again. A control without PAHs or inoculum was also prepared to evaluate the changes in microbial community structure over the incubation period and to evaluate the community response to the addition of PAHs and bioaugmentation.
Microcosms were incubated at room temperature and sampled every three days. At each sampling time the soils were first thoroughly mixed with a sterile spatula to aerate and homogenize the soil. One-gram samples were then taken for GC analysis and four one-gram samples were taken from different locations in the microcosm and combined for DNA extraction for construction of the 16S rRNA gene libraries.
GC analysis of PAH degradation. Samples for PAH degradation were analyzed individually for the duplicate microcosms and the average values were used for determining percent PAH degradation. The aromatic compounds were extracted from 1 g samples of soil from each microcosm at the indicated times. The samples were placed into vials containing 1 ml of dichloromethane. The vials with soil were then sonicated 10 times for 30 seconds with 30-second intervals on ice between each sonication. One gram of anhydrous Na2SO4 was then added and mixed into the soil to remove moisture from the sample. The dichloromethane supernatants were transferred to 1.5 ml Eppendorf tubes and centrifuged at 5 000 × g for 5 minutes at 4°C. An aliquot of the supernatant without any particulate material was transferred from the Eppendorf tubes and placed into 2 ml glass vials with screw cap lids with Teflon septa and stored at –20°C for subsequent GC analysis.
The degradation of PAHs was determined using a 1 μl aliquot of the dichloromethane extract injected in duplicate in splitless mode into a gas chromatograph (Shimadzu GC 14A), equipped with a flame ionization detector (GC-FID), and separated using an ID-BPX-5 column with fused silica as the stationary phase (25 m length × 0.22 mm ID × 0.25 μm film thickness, SGE-Australia) under a helium flow rate of 0.7 ml per minute. The injector temperature was set at 240°C and the detector temperature was set at 300°C. The oven temperature ramp rate was programmed as follows, 70°C hold for 1 minute, followed by an initial temperature ramp rate of 30°C per minute to 160°C, followed by an increase of 15°C per minute until reaching 310°C, at which time all PAHs had been eluted from the column. Percentage degradation was determined from the ratio of the peak height signal for each PAH to that of hexachlorobenzene obtained at T = 0 minus the ratio on the day of sampling divided by that obtained at T = 0, multiplied by 100. Percent standard deviation between duplicate injections was 10% or less for all samples.
Cloning, sequencing and analysis of 16S rRNA genes. Samples for 16S rRNA gene analysis were combined from replicate microcosms prior to DNA extraction and analysis. Total genomic DNA was extracted from 1 g of the combined soil samples using the Soil DNA Extraction Kit (MoBio Laboratories, USA) according to the procedure described by the manufacturer. PCR amplification of bacterial 16S rRNA genes was performed in 50 μl volumes containing 5 μl of 10X buffer with MgCl2 (Eppendorf), 8 mM dNTP’s (deoxyribonucleotide triphosphates), 2.5 μl of each primer, 0.5 μl of Taq polymerase (Eppendorf) and 2 μl of DNA, using the total genomic DNA. The bacteria domain specific primers 27F - 5’AGAGTTTGATCMTGGCTCAG3’ and 1492R - 5’GGTTACCTTGTTACGACTT3’ were used and the PCR amplification was performed using the following thermal cycling conditions: initial denaturation at 95°C for 5 min, 30 cycles of denaturation at 94°C for 1 min, annealing at 50°C for 1 min, extension at 72°C for 2 min, followed by a final extension at 72°C for 10 min. The amplified products were checked by electrophoresis in 2% agarose gel. All PCR amplification reactions were performed in a BioRad model iCycler. For automated sequencing, PCR products were purified using mini-columns (GFX PCR DNA and Gel Band Purification Kit, GE Health Care) and submitted to sequencing in an automated sequencer (MegaBace, GE Health Care). The sequencing reactions were performed using the DYEnamic ET Dye Terminator Cycle Sequencing Kit and MegaBace DNA Analysis Systems (GE Health Care) using the primer 338F - 5’ACTCCTACGGGAGGCAGCAG3’ (Lane 1991), which targets the hyper-variable V3 region of the 6S rRNA gene.
Phylogenetic analysis and comparison of cloned libraries. The phylogenetic diversity of the soil microcosms was determined using the CLASSIFIER algorithm provided by the Ribosome Database Project (RDP) release 10 website (Wang et al. 2007; Cole et al. 2009). This algorithm uses a naive Bayesian method of comparative statistical analyses to classify bacterial 16S rRNA sequences. Identification of specific clone sequence matches to the RDP version 11 (rdp.cme.msu. edu) and the NCBI (www.ncbi.nlm.nih.gov) nucleotide sequence (nr/nt) database was performed using the RDP online sequence comparison tool SeqMatch (Cole et al. 2005), using NCBI taxonomy, and the NCBI BLASTN DNA sequence comparison program, respectively. Microbial community comparisons between the different microcosms were performed using the RDP LIBCOMPARE program, which uses the Classifier taxonomic identifications to estimate the probability of obtaining the observed difference between two data sets for a given taxon (Cole et al. 2009). The default confidence threshold of 80% was used for assigning each taxon group. Those taxonomic groups that were identified as being significantly different at p < 0.01 were used to identify major community differences.
Results and Discussion
Bacterial population in the liquid PAH enrichment culture. The soil used for the microcosms was used as the inoculum for liquid PAH enrichment cultures containing 250 mg/l of naphthalene, phenanthrene, anthracene and pyrene as the sole carbon sources. The purpose of the liquid enrichment was to identify members of the soil community that emerged in response to growth in the presence PAHs as the sole carbon source. However, liquid culture is clearly a very different environment than soil and those organisms identified would also represent likely PAH degraders that had a competitive advantage in liquid culture. After 22 days all of the PAHs were substantially degraded (data not shown) and total genomic DNA was isolated for analysis of the community structure by comparative 16S rRNA gene sequence analysis. A total of 245 high quality partial bacterial 16S RNA gene sequences were obtained and analyzed using the RDP database suite of programs as described in Methods. The bacterial population of the PAH enrichment was of low diversity in comparison to those of the soil microcosms and was almost entirely composed of bacteria from the phylum Proteobacteria, which represented 91.3% of the total clones.
The Proteobacteria consisted of 19% Alphaproteobacteria, 28% Betaproteobacteria, and 53% Gamaproteobacteria. The Alphaproteobacteria were composed of Brevundimonas (5%), Azospirillum (17%), Shingomonas and unclassified Sphingomonadaceae (29%), and 48% were identified as members of the order Rhizobiales consisting of Hyphomicrobium and unclassified Hyphomicrobiaceae (14%), Rhizobium and unclassified Rhizobiales (20%), Bosea (5%) and Phyllobacterium (5%). The Betaproteobacteria consisted of Bordetella (87%), Achromobacter (8%) and unclassified Alcaligenaceae. The Gamaproteobacteria consisted of Pseudoxanthomonas (50%), Steroidobacter (1%), Pseudomonas (26%) and unclassified Pseudomonadaceae (18%, although all of these were identified as either Pseudomonas or Azomonas at confidence levels below 80%), and the remaining 5% were unclassified Gammaproteobacteria. All of these genera have been associated with PAH degradation (Wilson and Jones 1993; Vinas et al. 2005; Haritash and Kaushik 2009; Prince et al. 2010; Brown et al. 2012).
Members of the phylum Bacteroidetes were the only other dominant group in the enrichment representing 7.4% of the clones. The remaining 0.8% where unclassified bacteria. The Bacteroidetes clones consisted of 39% Sediminibacterium in the family Chitinophagaceae and 61% unclassified Chitinophagaceae, of these 55% were classified as Sediminibacterium at confidence levels ranging form 60% to 76%. Sediminibacterium where also found represent up to 12% of the total population in semi-continuous slurry-phase bioreactors treating PAHs (Singleton et al. 2011). In marked contrast to the soil microcosms (see below) no clones were obtained from the phylum Firmicutes, which contains the genus Bacillus.
Rates of PAH degradation in the PAH amended versus the PAH amended and bioaugmented soil. For the abiotic control there was effectively no depletion of any of the PAH over the course of the experiment with the exception of naphthalene, which saw a loss of approximately 50% on day 22 attributed to sublimation (data not shown). The percent degradation of the PAHs over time is shown for the PAH amended soil and the PAH amended and bioaugmented soil in Fig. 1 and 2, respectively. Both the PAH amended at the bioaugmented microcosms demonstrated effectively equal rates of degradation. Samples for microbial community analysis were taken on: i) day 1 for the unamended control; ii) day 8 from the PAH amended and the bioaugmented soils, at which point degradation of all of the PAHs had begun in both sets of microcosms; iii) day 22 for all microcosms at which point 30% or more of all of the PAHs had been degraded, with exception of naphthalene which was completely removed. By day 30 virtually all of the PAHs had been removed in both microcosms. The lack of a significant difference in degradation rates between the uninoculated and inoculated microcosms demonstrates that bioaugmentation did not improve the bioremediation of these compounds.
Fig. 1.
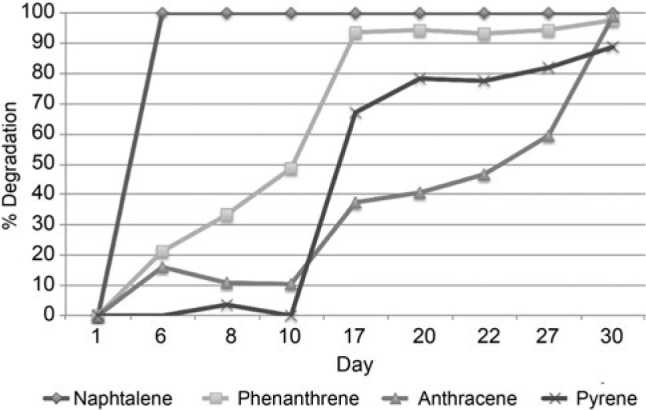
Percent PAH degradation in the PAH amended soil without bioaugmentation.
Fig. 2.
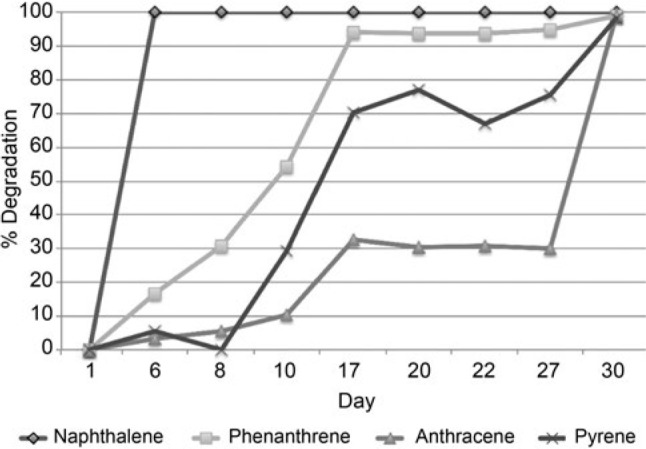
Percent PAH degradation in the PAH amended and bioaugmented soil.
Characterization of the changes in microbial populations in the soil microcosms. The microbial communities were analyzed on day 1 for the unamended control, on day 8 for the PAH treated microcosms and on day 22 for all 3 sets of microcosms to assess the effect of PAH contamination and bioaugmentation. The following number of high quality sequences over 250 bp were obtained from each microcosm (number of sequences); unamended control day 1 (145), PAH amended and bioaugmented day 8 (316 and 338, respectively), and day 22 unamended control (234), PAH amended (116) and the PAH amended and bioaugmented soil (188).
The cut off for taxons that were scored as statistically significant with respect to their relative abundance between microcosms was set at p < 0.01 and was used to identify meaningful differences between populations by the Libcompare program. There were a number of sequences that could not be classified beyond a certain taxonomic level and are designated UC (unclassified beyond that taxonomic level) in the following figures.
Comparison of the microbial communities of the unamended control and the PAH amended microcosms. The unamended control was sampled at the initiation of the microcosms and on day 22, and no statistically significant differences were observed (data not shown) indicating that exposure to microcosms conditions did not induce significant changes in the soil microbial population. The relative abundance of the identified phyla in all microcosms as well as the PAH enrichment culture on day 22 is shown in Fig. 3.
Fig. 3.

Comparison of the percentage of clones at the phylum level between the unamended control, PAH amended, bioaugmented microcosms and the soil enrichment on day 22.
* Indicates those taxonomic groups that are significantly different at p < 0.01 between the control and the PAH amended microcosms and ** indicates those taxonomic groups that are significantly different between the PAH amended and PAH amended and bioaugmented microcosms.
Comparing the bacteria from the PAH amended to the unamended control soil shows statistically significant differences. This fact was expected due to contamination of the soil by PHAs causes significant changes in the soil bacterial community (Szczepaniak et al. 2016). Microorganisms, mainly bacteria, are the prevailing organisms in soils. Bacteria as heterotrophs could mineralize contaminants, e.g. hydrocarbon (Megharaj et al. 2011; Abbasian et al. 2016b). Microbial activity depends on the hydrocarbon concentration, the length of time it contaminated the soil and the soil properties (Margesin et al. 2007; Lauber et al. 2008). Different microorganisms can be found in aged contaminated soil when compared to that of the freshly contaminated soil (Militon et al. 2010; Abbasian et al. 2016b). Changes in soil conditions may influence the composition and diversity of soil microbial community (Aleer et al. 2014; Liang et al. 2014).
At the phylum level the change occurred in the phyla Bacteroidetes, Proteobacteria and Acidobacteria as it has already been reported (Militon et al. 2010; Sutton et al. 2013; Abbasian et al. 2016a). Regarding Acidobacteria, this group of bacteria is phylogenetically diverse, and widespread in soils with many members, notably the Gp groups that are largely uncultured (Kuske et al. 1997; Naether et al. 2012). In the case of the PAH amended soil there were clones from the Gp groups 3, 4, 6 and 10, with Gp6 being the most dominant, that accounted for all of the increase in this phylum, which expanded from 0.4% to 5.1% of the total clones. However, due to the lack of cultivation of these groups their relation to PAH degradation is unclear. The obtaining of the uncultured groups is very common in the microbial diversity study. Large amounts of these bacteria were also found in the similarly contaminated soil in various geographical locations (Sutton et al. 2013; Cury et al. 2015). In contrast, the Bacteroidetes saw a large drop in numbers from 17% of the total 16S rRNA gene clones to 4% and this occurred across all classes identified. In addition, with regard to those clones that were identified as Bacteroidetes in the PAH amended soil, there was no indication of a selection for PAH degraders within this phylum.
Proteobacteria were identified as the main phylum present in hydrocarbon-contaminated soil samples and these microorganisms had an important role in the natural attenuation of hydrocarbons (Sutton et al. 2013). Among these phyla, bacteria belonging to Alpha-, Beta-, and Gammaproteobacteria degrade aliphatic and aromatic compounds into simpler forms (Kostka et al. 2011). Upon contamination with petroleum hydrocarbons a shif of the relative abundance of Proteobacteria is often noted (Sutton et al. 2013). In this work, the comparison of the microbial communities in the phylum Proteobacteria reveals a statistically significant increase in the Alphaproteobacteria, a slightly higher percentage of Gammaproteobacteria clones and an effectively equivalent number of Betaproteobacteria clones in the PAH amended soil vs. the unamended control. The Deltaproteobacteria were a tiny fraction (0.4%) of the clones from the unamended control on day 22 and were undetected in the clones form the PAH amended soil.
Within the Alphaproteobacteria the most significant difference was an almost 2-fold increase in the number of clones from the order Sphingomonadales (4% vs. 7.6%), largely in the families Erythrobacteraceae (1.6% vs. 2.5%) and Sphingomonadaceae (1.6% vs. 4.2%). In addition, there was a large increase in the unclassified Alphaproteobacteria from 1.6% to 4.2% of the total clones. The family Sphingomonadaceae contains many PAH degraders notably in the genera Sphingomonas and Sphingopyxis (Ho et al. 2000; Kertesz and Kawasaki 2010; Shokrollahzadeh et al. 2012). In this case the genus Sphingopyxis increased from 1.2% to 2.5% of the total population and the genus Sphingomonas increased from 0 to 1% of the total clones. The genus Brevundimonas in the family Caulobacteraceae remained relatively constant with only slight increase in the percentage of clones. Brevundimonas, which represented 5% of the population in the soil enrichment, have been identified frequently in PAH contaminated soils and this genus is known to contain various PAH degrading species (Seo et al. 2007; Phillips et al. 2008). There was also an increase in the order Rhizobiales (6.5% vs. 9.3%). Approximately half of the identified Rhizobiales in the PAH amended microcosm could not be identified at the genus level and the remainder were identified as members of the genera Microvirga, Bosea, and Devosia. Interestingly, Bosea represented 5% of the population in the soil enrichment culture and Bosea sp. have been isolated from PAH contaminated soil but did not demonstrate the ability to degrade PAHs themselves suggesting they may metabolize PAH degradation products produced by other microorganisms (Seo et al. 2007). Devosia been identified in PAH contaminated microcosms, however, to our knowledge, Microvirga has not associated with PAH degradation (Guazzaroni et al. 2013).
Although the total number of Betaproteobacteria clones were approximately equivalent in the unamended control versus the PAH amended soil there were differences in the distribution of identified genera; however, these differences were not scored as statistically significant at the p < 0.01 level. In particular, in the PAH amended soil, there was a 2 fold increase in the number of clones from the family Alcaligenaceae of 3.2% to 6.8% of the total clones. The increase in the Alcaligenaceae occurred within the genera Achromobacter (0.4 vs. 0.8%), Bordetella (0.4% vs. 2.5%) and Pusillimonas 1.6% vs. 3.4%), and all of these groups contain frequently identified PAH degraders (Eriksson et al. 2003; Seo et al. 2007; Hilyard et al. 2008, Prince et al. 2010). Interestingly, Achromobacter and Bordetella were the two genera identified in the Betaproteobacteria in the soil enrichment culture. The complete genome of Pusillimonas sp. T7-7, isolated from the benthal mud of a petroleum-contaminated site in Bohai Sea, China, has been sequenced (Cao et al. 2011), and a variety of aromatic degradation genes have been identified, including those involved in metabolism of methylnaphthalene, phenanthrene and anthracene that can be found in the KEGG PATHWAY Database (http://www.genome.jp/kegg/).
In the Gammaproteobacteria, a large decline occurred across all identified orders with the exception of Pseudomonadales, which remained equivalent (1.6% vs. 1.7%) with the only identified genus being Pseudomonas. In addition, although members of the Gammaproteobacteria order Xanthomonadales declined from 9 to 5.1% of the total population, the genus Pseudoxanthomonas within the order Xanthomonadales increased form 0% to 1.7% and members of this group are known to be PAH degraders (Prince et al. 2010; Patel et al. 2012).
Table I.
Putative PAH degrading genera identified in the soil enrichment and soil microcosms.
| Phylum | Soil Enrichment – Day 22 | PAH Amended – Day 8 | Augmented – Day 8 | PAH Amended – Day 22 | Bioaugmented – Day 22 |
|---|---|---|---|---|---|
| Bacteriodetes | Sediminibacterium | Flavobacterium | |||
| Arenibacter | |||||
| Alphaproteobacteria | Azospirillum | ||||
| Bosea | Bosea | ||||
| Brevundimonas | Brevundimonas | ||||
| Devosia | |||||
| Hyphomicrobium | |||||
| Phyllobacterium | |||||
| Sphingomonas | Sphingomonas | Sphingomonas | |||
| Sphingopyxis | |||||
| Betaproteobacteria | Achromobacter | Achromobacter | |||
| Bordetella | Bordetella | ||||
| Pusillimonas | Pusillimonas | ||||
| Gammaproteobacteria | Pseudomonas | Pseudomonas | |||
| Pseudoxanthomonas | Pseudoxanthomonas | Pseudoxanthomonas | |||
| Steroidobacter | Steroidobacter | ||||
| Firmicutes | Bacillus | Bacillus | Bacillus | Bacillus | |
| Paenibacillus | |||||
| Candidatus Saccharibacteria | Saccharibacteria genera incertae sedis | Saccharibacteria genera incertae sedis | Saccharibacteria genera incertae sedis | Saccharibacteria genera incertae sedis |
With regard to the Firmicutes, which contain the genus Bacillus, both the unamended control and the PAH amended soil had an effectively equivalent distribution of clones, 29% and 28%, respectively. These clones came almost entirely from the families Clostridia and Bacilliaceae, with both families representing approximately 50% of the total clones both sets of microcosms. Within the family Bacilliaceae both sets of microcosms were dominated by the genus Bacillus and unclassified members of the family Bacillaceae, which contains the genus Bacillus. Members of the genus Bacillus have frequently been identified as PAH degraders in soils.
Comparison of the microbial community structure between the PAH amended and bioaugmented microcosms. On day 8, which represents the onset of PAH degradation, the identified microbial communities were statistically equivalent between the PAH amended (316 clones) and bioaugmented (338 clones) microcosms with the exception of the genera Saccharibacteria genera incertae sedis (10% vs. 18%) within the phylum Candidatus Saccharibacteria (previously referred to as Candidate Division TM7), Paenibacillus (3% vs. 0.3%), within the phylum Firmicutes, and Pusillimonas (3% vs. 0.3%), within the Betaproteobacteria. Paenibacillus and Pusillimonas have been previously identified as PAH degraders and Saccharibacteria genera incertae sedis, the only genus of the candidate phylum Candidatus Saccharibacteria has recently been associated with benzene degradation (Xie et al. 2011), and these bacteria are ubiquitous phylum found in soils, sediments and wastewater (Ferrari et al. 2014).
Comparison of the PAH amended to the bioaugmented microcosms at the phylum level (Fig. 3) on day 22 shows a statistically significant lower abundance of Proteobacteria clones in the bioaugmented soil, 42.8% vs. 24.1% of the total clones, respectively. In contrast, a slight overall increase was observed within the Proteobacteria in the PAH amended soil in comparison to the unamended control.
Comparison of the identified Proteobacteria from the bioaugmented soil to the PAH amended soil reveals a statistically significant reduction (50%) in Alphaproteobacteria and an almost complete reduction in the Betaproteobacteria (10% vs. 0.5%). Moreover, these population reductions included virtually all of the putative aromatic degraders that increased in the soil amended with PAHs alone, with the exception of an increase in the PAH degrading Alphaproteobacteria genus Sphingomonas (0.8% to 1.6% of the total), suggesting that the added bacterial inoculum in some way suppressed this population in the later stages of PAH degradation. The Gamaproteobacteria remained roughly constant both in numbers and the distribution of genera with the exception that the genus Pseudomonas was not detected in the bioaugmented microcosms while the genus Steroidobacter was (2% vs. 0%).
In contrast to the Proteobacteria, clones identified as members of the phyla Actinobacteria were considerably more abundant comprising 9% of the total clones in the PAH amended and bioaugmented soil versus 3.4% for the PAH amended. Although the levels of Actinobacteria were significantly higher in the bioaugmented microcosms compared to the PAH amended alone, they largely mirrored those of the unamended control day 22 and there was no indication of a selection for PAH degraders.
Given the majority of the bacteria used for bioaugmentation were Bacillus species, it was of particular interest to see if these organisms persisted and may then have played a role in PAH degradation. The percentage of clones identified as members of the phylum Firmicutes are very similar between all three microcosms (Fig. 3) with all microcosms being dominated by the classes Bacilli and Clostridia, and notably in the genus Bacillus, which represented approximately 4–5% of the total in all cases. Although the genus Bacillus contains many well documented PAH degraders, the abundance of this genus in all 3 types of microcosms makes it unclear as to what extent they played a role in PAH degradation. However, 2% of the clones from bioaugmented microcosm on day 8 were identical in sequence to 2 of the Bacillus strains used for bioaugmentation suggesting these strains persisted in the microcosms up to this point. On day 22 these strains were not detected in the bioaugmented microcosm and none of the 16S sequences from the added organisms were detected at either time point in the microcosm only receiving PAHs. Nonetheless, 16S rRNA gene sequence identity does not infer secondary metabolic attributes such as PAH degradation and it is possible the matching clones identified in the bioaugmented microcosms were not from the inoculated strains.
Moreover, the fact that no matching sequences were identified on day 22 in the bioaugmented microcosm suggest that bioaugmentation did not result in the persistence of the added organisms throughout the PAH degradation period. The adaptation of the introduced microorganisms to the environmental conditions and maintenance of high metabolic activity seem to be an important aspect in the bioremediation. However, multitude of the biotic and abiotic factors may affect the success of this process (Szczepaniak et al. 2016). Among the biotic factors, the competition between indigenous microorganisms and those introduced via bioaugmentation is mentioned most frequently (Mrozik and Piotrowska-Seget 2010). Other factors like the insufficient abundance, predation and other antagonistic activities (i.e. secretion of antibiotics) also can be the causes of failure (Thompson et al. 2005; Szulc et al. 2014); hence, they may also be the most plausible causes for no persistence of bioaugmentation during this study.
On the other hand, the question then remains as to which members of the microbial community in the PAH bioaugmented are likely to be the dominant enriched PAH degraders. The most likely candidates are to be found in the clones identified as members of the phylum Bacteroidetes which had statistically significant higher percentage of the total clones (4.2% vs. 18.1) in the bioaugmented soil relative to the PAH amended soil. However, the percentage of total Bacteroidetes is effectively equivalent to that of unamended control on day 22 and as such would not suggest a significant difference between the two soils. Nonetheless, comparison of the Bacteroidetes clones at the family level reveals significant differences between the three soils as shown in Fig. 4.
Fig. 4.

Comparison of the percentage of Bacteroidetes clones at the family level (and genus level for Ohtaekwangia) between the unamended control, PAH amended and PAH amended and bioaugmented soils on day 22.
As can be seen, the distribution of the Bacteroidetes classes between the 3 soils is quite different. Both the PAH amended and PAH amended and bioaugmented soils saw relatively equivalent reduction in the percentage Sphingobacteria relative to the unamended control. The dominant groups in the Sphingobacteria in the unamended soil were classified in the genus Pedobacter in the family Sphingobacteriaceae and also in the genus Chitinophaga and uncharacterized members of the family Chitinophagaceae, and both of these groups were greatly reduced in the PAH amended and PAH amended and bioaugmented soils. However, a statistically significant difference in the PAH amended and bioaugmented soil was observed in the percentage of identified clones in the class Flavobacteria, which had a large increase from 1.6% to 8% of the total clones relative to the unamended control, and was completely undetected in the PAH amended soil. The only identified Flavobacteria genus in the PAH amended and bioaugmented and unamended control was Flavobacterium in the family Flavobacteriaceae, which are well documented to contain a diverse range of PAH degrading capabilities (Widada et al. 2002) and have also been isolated from oil contaminated soil (Haudhary and Kim 2018). The majority of the Flavobacteriaceae clones were not able to be classified at the genus level at high confidence in the PAH amended and bioaugmented soil on day 22, which is not at all uncommon in soil samples and indicates that isolates that are very closely related to the identified clones have not yet been cultivated and characterized. Nonetheless, the majority of the unclassified clones appeared to be most closely related to the genus Arenibacter (86%), an isolate of which has recently be identified as aromatic hydrocarbon-degrading bacterium and Arenibacter have also been previously isolated from oil contaminated sites (Kadali et al. 2012; Gutierrez et al. 2014). The remaining unclassified clones were identified as Kriegella and Sediminibacter (the only genus identified in the phylum Bacteroidetes in the soil enrichment culture).
In addition, Ohtaekwangia, a newly discovered genus within the phylum Bacteroidetes isolated from a sand sample collected from the west coast of the Korean peninsula using low-nutrient media (Yoon et al. 2011), increased from 0% to 3.2% of the total clones. To our knowledge it is not yet known whether this genus is associated with PAH degradation. However, given their previous association with PAH degradation it is reasonable to infer that the unclassified Flavobacteriaceae are indeed PAH degraders and it is also suggestive that Ohtaekwangia may also be PAH degraders as well. Further characterization of these and other unclassified clones is essential to a deeper understanding of their PAH degrading abilities.
Conclusions
A pristine soil was evaluated for its degradation potential and changes in the microbial community after exposure to the PAHs naphthalene, phenanthrene, anthracene and pyrene when used as an inoculum in a PAH enrichment in liquid mineral medium and in microcosms receiving either PAHs alone or the PAHs with bioaugmentation with PAH degrading isolates. The microbial community obtained in the enrichment culture degraded all the PAHs and consisted entirely of members of two phyla, Proteobacteria and Bacteroidetes. The soil enrichment culture possessed low microbial diversity with only the phyla Bacteroidetes and Proteobacteria represented. The Bacteroidetes represented a small fraction relative to the Proteobacteria and consisted only of Sediminibacter and closely related unclassified bacteria, while the Proteobacteria were represented by Alpha, Beta and Gamaproteobacteria.
The soil microcosms receiving only PAHs and receiving PAHs and bioaugmentation showed effectively equal rates of degradation of all of the PAHs with complete degradation occurring by day 30 demonstrating that microbial population of the pristine soil readily degraded these compounds and that bioaugmentation did not improve degradation rates.
At the onset of PAH degradation (day 8) the putative PAH degraders microcosms (PAH amended and bioaugmented) were similar with the exception of a significant population of the genus Pusillimonas in the PAH amended microcosms (3% of the total). Identical sequence to two of the bioaugmention inoculum strains were identified in the bioaugmented microcosms, however, these sequences were not detected at the later stages of PAH degradation (day 22) indicating the added PAH degrading isolates did not persist throughout the degradation period. On day 22 the PAH amended soil showed increases in putative PAH degraders primarily in the Alphaproteobacteria genera Sphingomonas and Sphingopyxis, and members of the order Rhizobiales and in the Betaproteobacteria in the family Alcaligenaceae within the genera Achromobacter, Bordetella, Pusillimonas, all of which contain well documented PAH degraders. In marked contrast, the increase in putative PAH degraders in the PAH amended and bioaugmented soil occurred in the Bacteroidetes family Flavobacteriaceae, and were classified as members of the genus Flavobacterium or were unclassified Flavobacteriaceae, and also the newly identified genus Ohtaekwangia. In addition, there was no evidence for a persistent increase in clones from the consortia of PAH degrading bacteria used for bioaugmentation.
These results indicate that soils can contain a diversity of microbial taxa that can degrade PAHs with equivalent efficiency, however, which groups become most dominant is flexible and dependent not only the PAH composition.
Acknowledgments
We thank the Coordinator for the Improvement of Personnel in Higher Education (CAPES, Brazil), the National Council for the Development of Science and Technology (CNPq), and the Foundation for the Support of Science in São Paulo State (FAPESP) for financial support.
Literature
- Abbasian F, Lockington R, Megharaj M, Naidu R.. 2016a. The biodiversity changes in the microbial population of soils contaminated with crude oil. Curr Microbiol. 72:663–670. [DOI] [PubMed] [Google Scholar]
- Abbasian F, Palanisami T, Megharaj M, Naidu R, Lockington R, Ramadass R.. 2016b. Microbial diversity and hydrocarbon degrading gene capacity of a crude oil field soil as determined by metagenomics analysis. Biotechnol. Prog. 32:638–648. [DOI] [PubMed] [Google Scholar]
- Aleer S, Adetutu EM, Weber J, Ball AS, Juhasz AL.. 2014. Potential impact of soil microbial heterogeneity on the persistence of hydrocarbons in contaminated subsurface soils J Environ Manag. 136:27–36. [DOI] [PubMed] [Google Scholar]
- Bento FM, Camargo FA, Okeke BC, Frankenberger WT.. 2005. Comparative bioremediation of soils contaminated with diesel oil by natural attenuation, biostimulation and bioaugmentation. Bioresour Technol. 96:1049–1055. [DOI] [PubMed] [Google Scholar]
- Brown SD, Utturkar SM, Klingeman DM, Johnson CM, Martin SL, Land ML, Lu TY, Schadt CW, Doktycz MJ, Pelletier DA.. 2012. Twenty-one genome sequences from Pseudomonas species and 19 genome sequences from diverse bacteria isolated from the rhizosphere and endosphere of Populus deltoides. J Bacteriol. 194:5991–5993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao BY, Ma T, Ren Y, Li GQ, Li P, Guo X, Ding P, Feng L.. 2011. Complete genome sequence of Pusillimonas sp. T7-7, a cold-tolerant diesel oil-degrading bacterium isolated from the Bohai Sea in China. J Bacteriol. 193:4021–4022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhary DK, Kim J.. 2018. Flavobacterium naphthae sp. nov., isolated from oil-contaminated soil. Int J Syst Evol Microbiol. 68:305–309. [DOI] [PubMed] [Google Scholar]
- Cole JR, Chai B, Farris RJ, Wang Q, Kulam SA, McGarrell DM, Garrity GM, Tiedje JM.. 2005. The Ribosomal Database Project (RDP-II), sequences and tools for high-throughput rRNA analysis. Nucleic Acids Res. 33:D294–D296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole JR, Wang Q, Cardenas E, Fish J, Chai B, Farris RJ, Kulam-Syed-Mohideen AS, McGarrell DM, Marsh T, Garrity GM, Tiedje JM.. 2009. The Ribosomal Database Project, improved alignments and new tools for rRNA analysis. Nucleic Acids Res. 37:D141–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curry JC, Jurelevicius DA, Villena HDM, Jesus HE, Peixoto RS, Schaefer CEGR, Bícego MC, Seldin L, Rosado AS.. 2015. Microbial diversity and hydrocarbon depletion in low and high diesel-polluted soil samples from Keller Peninsula, South Shetland Islands. Antarct Sci. 27:263–273. [Google Scholar]
- Eisler R. 1987. Polycyclic aromatic hydrocarbon hazards to fish, wildlife and invertebrates, a synoptic review Contaminant Hazard Reviews. Report 11: Biological Report 85(1.11). Laurel, MD (USA): U.S. Department of the Interior, Fish and Wildlife Service. [Google Scholar]
- Eriksson M, Sodersten E, Yu Z, Dalhammar G, Mohn WW.. 2003. Degradation of polycyclic aromatic hydrocarbons at low temperature under aerobic and nitrate-reducing conditions in enrichment cultures from northern soils. Appl Environ Microbiol. 69:275–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari B, Winsley T, Ji M, Neilan B.. 2014. Insights into the distribution and abundance of the ubiquitous candidatus Saccharibacteria phylum following tag pyrosequencing. Sci Rep. 4:3957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guazzaroni ME, Herbst FA, Lores I, Tamames J, Peláez AI, López-Cortés N, Alcaide M, Del Pozo MV, Vieites JM, von Bergen M, et al. 2013. Metaproteogenomic insights beyond bacterial response to naphthalene exposure and bio-stimulation. ISME J. 7:122–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutierrez T, Rhodes G, Mishamandani S, Berry D, Whitman WB, Nichols PD, Semple KT, Aitken MD.. 2014. Polycyclic aromatic hydrocarbon degradation of phytoplankton-associated Arenibacter spp. and description of Arenibacter algicola sp. nov., an aromatic hydrocarbon-degrading bacterium. Appl Environ Microbiol. 80(2): 618–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haritash AK, Kaushik CP.. 2009. Biodegradation aspects of polycyclic aromatic hydrocarbons (PAHs): A review. J Hazard Mater. 169:1–15. [DOI] [PubMed] [Google Scholar]
- Hilyard EJ, Jones-Meehan JM, Spargo BJ, Hill RT.. 2008. Enrichment, isolation and phylogenetic identification of polycyclic aromatic hydrocarbon-degrading bacteria from Elizabeth River sediments. Appl Environ Microbiol. 74:1176–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho Y, Jackson MM, Yang Y, Mueller JG, Pritchard PH.. 2000. Characterization of fluoranthene- and pyrene-degrading bacteria isolated from PAH-contaminated soils and sediments and comparison of several Sphingomonas spp. J Ind Microbiol Biotech. 24:100–112. [Google Scholar]
- Janssen PH. 2006. Identifying the dominant soil bacterial taxa in libraries of 16S rRNA and 16S rRNA genes. Appl Environ Microbiol. 72:1719–1728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juhasz AL, Naidu R.. 2000. Bioremediation of high molecular weight polycyclic aromatic hydrocarbons, a review of the microbial degradation of benzo[a]pyrene. Int Biodeterior Biodegradation. 45:57–88. [Google Scholar]
- Kadali KK, Simons KL, Skuza PP, Moore RB, Ball AS.. 2012. A complementary approach to identifying and assessing the remediation potential of hydrocarbonoclastic bacteria. J Microbiol Methods. 88:348–355. [DOI] [PubMed] [Google Scholar]
- Kertesz MA, Kawasaki A.. 2010. Hydrocarbon-degrading Sphingomonads, Sphingomonas, Sphingobium, Novosphingobium, and Sphingopyxis In: Timmis KN, McGenity T, Meer JR, Lorenzo V, editors. Handbook of hydrocarbon and lipid microbiology. Berlin Heidelberg (Germany): Springer; p. 1693–1705. [Google Scholar]
- Khan MAI, Biswas B, Smith E, Mahmud SA, Hasan NA, Khan MAW, Naidu R, Megharaj M.. 2018. Microbial diversity changes with rhizosphere and hydrocarbons in contrasting soils. Ecotoxicol Environ Saf. 156:434–442. [DOI] [PubMed] [Google Scholar]
- Kostka JE, Prakash O, Overholt WA, Green SJ. Freyer G, Canion A, Delgardio J, Norton N, Hazen TC, Huettel M.. 2011. Hydrocarbon-degrading bacteria and the bacterial community response in Gulf of Mexico beach sands impacted by the deepwater horizon oil spill. Appl Environ Microbiol. 77:7962–7974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuske CR, Barns SM, Busch JD.. 1997. Diverse uncultivated bacterial groups from soils of the arid southwestern United States those are present in many geographic regions. Appl Environ Microbiol. 63:3614–3621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane DJ. 1991. 16S/23S rRNA sequencing In: Stackebrandt E, Goodfellow M, editors. Nucleic acid techniques in bacterial systematics. New York (USA): John Wiley and Sons; p. 115–175. [Google Scholar]
- Lauber CL, Strickland MS, Bradford MA, Fierer N.. 2008. The influence of soil properties on the structure of bacterial and fungal communities across land-use types. Soil Biol Biochem. 40:2407–2415. [Google Scholar]
- Liang Y, Zhao H, Zhang X, Zhou J, Li G.. 2014. Contrasting microbial functional genes in two distinct saline-alkali and slightly acidic oil-contaminated sites. Sci Total Environ. 487:272–278. [DOI] [PubMed] [Google Scholar]
- Lindstrom JE, Prince RC, Clark JC, Grossman MJ, Yeager TR, Braddock JF, Brown, EJ.. 1991. Microbial populations and hydrocarbon biodegradation potentials in fertilized shoreline sediments affected by the T/V Exxon Valdez oil spill. Appl Environ Microbiol. 57:2514–2522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margesin R, Hämmerle M, Tscherko D.. 2007. Microbial activity and community composition during bioremediation of diesel-oil-contaminated soil: effects of hydrocarbon concentration, fertilizers, and incubation time. Microbiol Ecol. 53:259–269. [DOI] [PubMed] [Google Scholar]
- Megharaj M, Ramakrishnan B, Venkateswarlu K, Sethunathan N, Naidu R.. 2011. Bioremediation approaches for organic pollutants: a critical perspective. Environ Int. 37:1362–1375. [DOI] [PubMed] [Google Scholar]
- Militon C, Boucher D, Vachelard C, Perchet G, Barra V, Troquet J, Peyretaillade E, Peyret P.. 2010. Bacterial community changes during bioremediation of aliphatic hydrocarbon-contaminated soil. FEMS Microbiol Ecol. 74:669–681. [DOI] [PubMed] [Google Scholar]
- Mrozik A, Piotrowska-Seget Z.. 2010. Bioaugmentation as a strategy for cleaning up of soils contaminated with aromatic compounds. Microbiol Res. 165:363–375. [DOI] [PubMed] [Google Scholar]
- Naether A, Foesel BU, Naegele V, Wüst PK, Weinert J, Bonkowski M, Alt F, Oelmann Y, Polle A, Lohaus G, et al. 2012. Environmental factors affect Acidobacterial communities below the subgroup level in grassland and forest soils. Appl Environ Microbiol. 78:7398–7406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel V, Cheturvedula S, Madamwar D.. 2012. Phenanthrene degradation by Pseudoxanthomonas sp. DMVP2 isolated from hydrocarbon contaminated sediment of Amlakhadi canal, Gujarat, India. J Hazard Mater. 201–202:43–51. [DOI] [PubMed] [Google Scholar]
- Petry T, Schmid P, Schlatter C.. 1996. The use of toxic equivalency factors in assessing occupational and environmental health risk associated with exposure to airborne mixtures of polycyclic aromatic hydrocarbons (PAHs). Chemosphere. 32:639–648. [DOI] [PubMed] [Google Scholar]
- Philp JC, Atlas RM.. 2005. Bioremediation of contaminated soil and aquifers In: Atlas R M, Jim CP, editors. Bioremediation: Applied Microbial Solution for Real – World Environmental Clean Up. Washington DC (USA): ASM Press; p. 139. [Google Scholar]
- Phillips LA, Germida JJ, Farrell RE, Greer CW.. 2008. Hydrocarbon degradation potential and activity of endophytic bacteria associated with prairie plants. Soil Biol Biochem. 40:3054–3064. [Google Scholar]
- Prince R, Gramain A, McGenity T.. 2010. Prokaryotic hydrocarbon degraders In: Timmis KN, McGenity TJ, van der Meer JR, de Lorenzo V, editors. Handbook of hydrocarbon and lipid microbiology. Berlin (Germany): Springer; p. 1669–1692. [Google Scholar]
- Rambeloarisoa E, Rontani JF, Giusti G, Duvvnjak Z, Bertrand JC.. 1984. Degradation of crude oil by a mixed population of bacteria isolated from sea surface foams. Mar Biol. 83:69–81. [Google Scholar]
- Samanta SK, Singh OV, Jain RK.. 2002. Polycyclic aromatic hydrocarbons, environmental pollution and bioremediation. Trends Biotechnol. 20:243–248. [DOI] [PubMed] [Google Scholar]
- Sayara T, Borras E, Caminal G, Sarra M, Sanchez A.. 2011. Bioremediation of PAHs-contaminated soil through composting: Influence of bioaugmentation and biostimulation on contaminant biodegradation. Int Biodeterior Biodegrad. 65:859–865. [Google Scholar]
- Seo JS, Keum YS, Harada RM, Li,p QX.. 2007. Isolation and characterization of bacteria capable of degrading polycyclic aromatic hydrocarbons (PAHs) and organophosphorus pesticides from PAH-contaminated soil in Hilo, Hawaii. J Sci Food Agric. 55:5383–5389. [DOI] [PubMed] [Google Scholar]
- Seo JS, Keum YS, Li QX.. 2009. Bacterial degradation of aromatic compounds. Int J Environ Res Public Health. 6:278–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shokrollahzadeh S, Golmohammad F, Shokouhi H.. 2012. Study of Sphingopyxis isolates in degradation of polycyclic aromatic hydrocarbons. Chem Eng Trans. 27:55–60. [Google Scholar]
- Silva IS, Costa SE, Ragagnin MC, Fonseca FA, Franciscon GDE, Grossman MJ, Durrant LR.. 2009. Bioremediation of a polyaromatic hydrocarbon contaminated soil by native soil microbiota and bioaugmentation with microbial isolates and consortia. Bioresour Technol. 100:4669–4675. [DOI] [PubMed] [Google Scholar]
- Singleton DR, Richardson SD, Aitken MD.. 2011. Pyrosequence analysis of bacterial communities in aerobic bioreactors treating polycyclic aromatic hydrocarbon-contaminated soil. Biodegradation 22: 1061–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton NB, Maphosa F, Morillo JA, Al-Soud WA, Langenhoff AAM, Grotenhuis T, Smidt H.. 2013. Impact of long-term diesel contamination on soil microbial community structure. Appl Environ Microbiol. 79:619–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szczepaniak Z, Cyplik P, Juzwa W, Czarny J, Staninska J, Piotrowska-Cyplik A.. 2015. Antibacterial effect of the Trichoderma viride fungi on soil microbiome during PAH’s biodegradation. Int Biodeter Biodegr. 104:170–177. [Google Scholar]
- Szulc A, Ambrożewicz D, Sydow M, Ławniczak Ł, Piotrowska-Cyplik A, Marecik R, Chrzanowski Ł.. 2014. The influence of bioaugmentation and biosurfactant addition on bioremediation efficiency of diesel-oil contaminated soil: Feasibility during field studies. J Environ Manage. 132:121–128. [DOI] [PubMed] [Google Scholar]
- Thompson IP, Van Der Gast CJ, Ciric L, Singer AC.. 2005. Bioaugmentation for bioremediation: the challenge of strain selection. Environ Microbiol. 7(7):909–915. [DOI] [PubMed] [Google Scholar]
- Tyagi M, da Fonseca MM, de Carvalho CC.. 2011. Bioaugmentation and biostimulation strategies to improve the effectiveness of bioremediation processes. Biodegradation. 22:231–241. [DOI] [PubMed] [Google Scholar]
- Van Hamme JD, Singh A, Ward OP.. 2003. Recent advances in petroleum microbiology. Microbiol Mol Biol Rev. 67:503–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinas M, Sabate J, Espuny MJ, Solanas AM.. 2005. Bacterial community dynamics and polycyclic aromatic hydrocarbon degradation during bioremediation of heavily creosotecontaminated soil. Appl Environ Microbiol. 71:7008–7018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Garrity GM, Tiedje JM, Cole JR.. 2007. Naïve Bayesian Classifier for Rapid Assignment of rRNA Sequences into the New Bacterial Taxonomy. Appl Environ Microbiol. 73:5261–5267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widada J, Nojiri H, Kasuga K, Yoshida T, Habe H, Omori T.. 2002. Molecular detection and diversity of polycyclic aromatic hydrocarbon-degrading bacteria isolated from geographically diverse sites. Appl Microbiol Biotechnol. 58:202–209. [DOI] [PubMed] [Google Scholar]
- Wilson SC, Jones KC.. 1993. Bioremediation of soil contaminated with polynuclear aromatic hydrocarbons (PAHs): a review. Environ Pollut. 81:229–249. [DOI] [PubMed] [Google Scholar]
- Wolin EA, Wolin MJ, Wolfe RS.. 1963. Formation of methane by bacterial extracts. J Biol Chem. 238:2882–2886. [PubMed] [Google Scholar]
- Wyrwas B, Dymaczewski Z, Zgoła-Grześkowiak A, Szymański A, Frańska M, Kruszelnicka I, Ginter-Kramarczyk D, Cyplik P, Ławniczak Ł, Chrzanowski Ł.. 2013. Biodegradation of Triton X-100 and its primary metabolites by a bacterial community isolated from activated sludge. J Environ Manage. 128:292–299. [DOI] [PubMed] [Google Scholar]
- Yoon JH, Kang SJ, Lee SY, Lee JS, Park S.. 2011. Ohtaekwangia koreensis gen. nov., sp. nov. and Ohtaekwangia kribbensis sp. nov., isolated from marine sand, deep-branching members of the phylum Bacteroidetes. Int J Syst Evol Microbiol. 61:1066–1072. [DOI] [PubMed] [Google Scholar]
- Xie S, Sun W, Luo C, Cupples AM.. 2011. Novel aerobic benzene degrading microorganisms identified in three soils by stable isotope probing. Biodegradation. 22:71–81. [DOI] [PubMed] [Google Scholar]