

Abstract
A two-compartment, magnetically responsive biomaterial system enables flexible control over the duration between pro- and anti-inflammatory cytokines deliveries. The system’s outer porous gelatin compartment can facilitate rapid infiltration and harboring of macrophages. The system’s outer compartment can be magnetically compressed, releasing payloads in earnest when stimulated with hand-held magnets. The combined 2-compartment system can release sequences of cytokines where the time between these two deliveries is controlled by the time at which a hand-held magnet is applied to the system.
Keywords: chronic wounds, drug delivery, inflammatory response, cytokine, hydrogel
It has been estimated that 1 to 2% of the population in developed countries will experience a chronic wound over their lifespan.[1] Occurrence of chronic wounds are particularly common in growing elderly populations and those who are suffering from diabetes and obesity.[2] While there are several phases in wound healing (i.e., coagulation, inflammation, proliferation, and remodeling),[3–6] chronic wounds are typically the result of prolonged and/or uncontrolled inflammation.[2,7] Despite this, inflammation is an indispensable step in the wound healing process and sets the stage for proper regeneration by staving off infection, clearing the wound site of debris, and recruiting cells to the wound that play critical roles in tissue remodeling and re-vascularization.[3,6,8,9] In fact, studies have shown that suppressing the inflammatory response actually hinders proper wound healing.[4,10] Macrophages play a key role in regulating the inflammatory response and in directing the transition to later stages of the wound healing process.[11–17] We and others believe that regulating the time at which macrophages transition from coordinating an inflammatory response (Figure 1a, Phase 1 (red)) to coordinating later pro-healing stages of the wound healing process (Phase 2 (blue)) may be key to understanding the role of inflammation in wound healing and in developing improved treatment strategies.[18–20] For example, it is apparent that proper healing requires an inflammatory phase that eventually transitions into anti-inflammatory, pro-healing phases.[21,22] However, it remains unknown how the duration of this inflammatory phase (Tih) impacts or can be used to optimize wound healing outcome. Moreover, optimal durations are likely different for different wounds and for different patients. This motivates the need for biomaterials that enable flexible control over the duration of this inflammatory period, as both an investigative and clinical tool.
Figure 1. Regulating the inflammatory period requires initial delivery of pro-inflammatory cytokines followed by delayed delivery of anti-inflammatory cytokines.
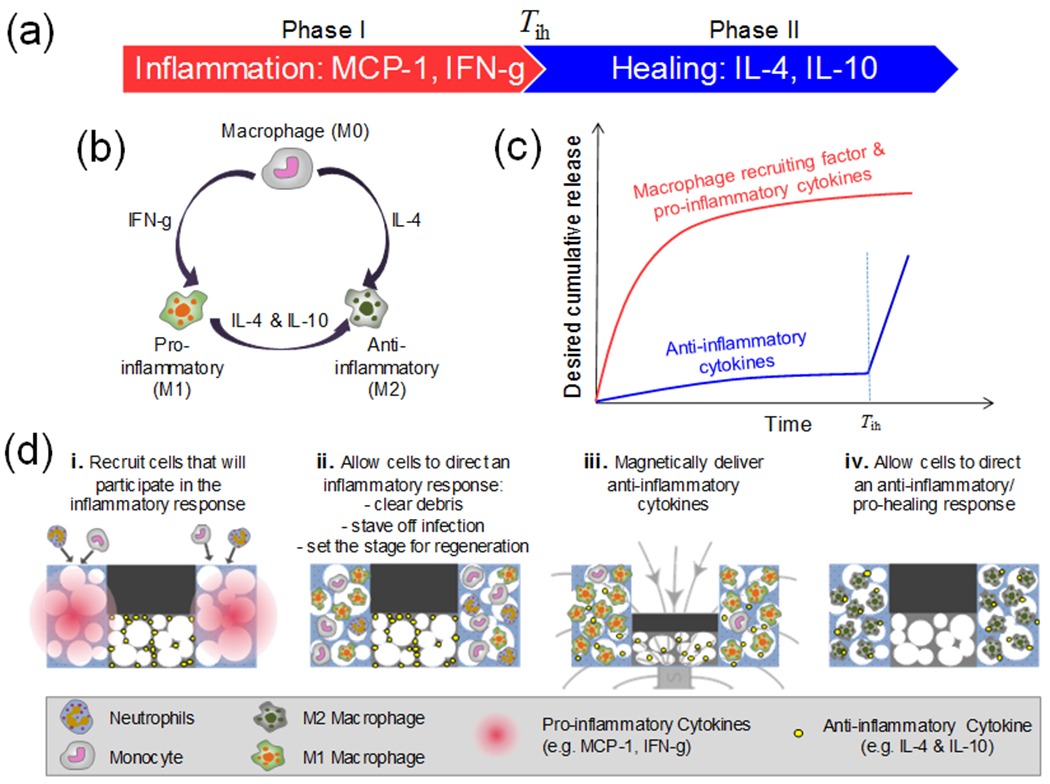
(a) Schematic describing the cytokines that regulate the inflammation phase (I, red) and healing phase (II, blue). (b) Schematic describing how M0 macrophages can be polarized into M1 (Pro-inflammatory) and/or M2 (Anti-inflammatory) phenotypes when exposed to different cytokines. (c) Illustration of the desired cumulative release profile: initial release of macrophage recruitment and pro-inflammation cytokines (red), followed by delivery of anti-inflammatory cytokines (blue). (d) Illustration of the proposed biomaterial system (top) with illustration key (bottom).
Here, we propose a biomaterial system designed to deliver immunomodulatory cytokines in a manner that can potentially regulate the inflammatory period’s duration in a flexible and on-demand manner. The inflammation phase can be initiated by establishing a population of pro-inflammatory M1 macrophages through the delivery of proteins that recruit macrophages and polarize them towards M1 phenotypes (Figure 1b, M0 to M1): for example, Monocyte Chemoattractant Protein-1 (MCP-1),[23,24] and Interferon Gamma (IFN-γ).[25] Transition from inflammatory to healing phases requires establishing a population of anti-inflammatory M2 macrophages (e.g., alternatively activated M2a, Mb, and Mc phenotypes). This can be triggered through the delivery of other proteins at the wound site: for example, Interleukin-4 (IL-4),[26,27] and Interleukin-10 (IL-10),[26,27] (Figure 1b, M0/M1 to M2). Thus, it may be possible to regulate the duration of the inflammatory response (Tih) through initial deliveries of pro-inflammatory cytokines, such as MCP-1 and IFN-γ (Figure 1c, red curve), followed by delayed deliveries of anti-inflammatory cytokines, such as IL-4 and/or IL-10 (Figure 1c, blue curve). Here, we will describe a two-compartment biomaterial system (Figure 1d) designed to: (i) initially release pro-inflammatory cytokines from an outer compartment for the recruitment and establishment of pro-inflammatory macrophage phenotypes, (ii) allow for an inflammatory period to continue until, (iii) a magnetic gradient is applied that deforms the inner compartment, releasing anti-inflammatory cytokines, which would (iv) direct macrophages to take on pro-healing phenotypes. Such a material system could enable control over the inflammatory period’s duration simply by applying a magnetic gradient (from simple hand-held magnets or electromagnets) at the time point at which one wishes inflammation to transition into an anti-inflammatory phase.
This two-compartment biomaterial system comprises an outer gelatin scaffold and an inner biphasic ferrogel (Figure 2a). The outer compartment exhibited an interconnected macroporous structure designed to permit rapid cell infiltration (Figure 2b). Also, by virtue of being made from gelatin (a hydrolyzed form of collagen), this gelatin scaffold presents binding motifs for cell binding, motility, and spreading.[28,29] For the inner compartment, we utilized a biphasic ferrogel with an Fe3O4-laden region on the top half of the cylindrical gel and an Fe3O4-free, porous, and deformable region on the bottom (Figure 2c). These biphasic ferrogels were designed to efficiently deform in the presence of a graded magnetic field (i.e., in the presence of fields emanating from simple hand-held magnets or electromagnets). When magnetically deformed, these gels would release molecular payloads stored in the Fe3O4-free region in a magnetically triggered manner. The particular ferrogel formulation adopted here (1 wt% alginate, 7 wt% Fe3O4, 2.5 mM adipic acid dihydrazide cross-linked, freeze-dried at −20ºC) was previously shown to be optimal in terms of providing magnetically triggered deliveries.[30,31]
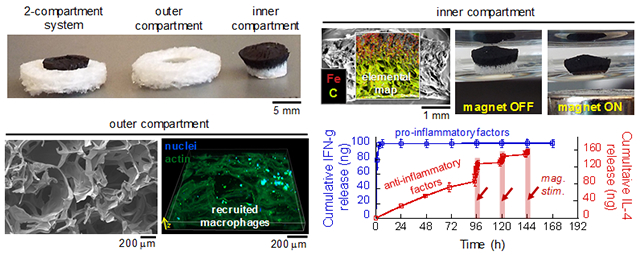
Figure 2. Two-compartment biomaterial system comprises a magnetically responsive biphasic ferrogel nested within an outer macroporous gelatin scaffold.

(a) Photographs of the 2-compartment biomaterial system at an angle (top) and from the top (bottom). (b) Cross-sectional photograph (i) and SEM micrographs (ii) of the outer porous gelatin compartment. (b) Cross-sectional photograph (i) and SEM microgarphs (ii) of the inner biphasic ferrogel compartment. Elemental map reveals the location of iron (red) and carbon (yellow-green).
The outer porous gelatin scaffold was designed to provide initial deliveries of pro-inflammatory cytokines and to recruit and permit the residence of macrophages. To test this compartment’s ability to recruit and establish macrophage populations, RAW 264.7 macrophages were seeded at 10,000 cells per well in a 12-well plate on Day −1 and allowed to establish themselves for 24 hours as a 2-dimensional (2D) colony. Then (at Day 0), gelatin scaffolds (compartment 1, Figure 2b) were placed on top of 2D macrophage colonies and left for 10 days so that macrophages could infiltrate the volume of the scaffold (Figure 3a). On Day 5, some scaffolds were removed, fixed, and stained for f-actin (FITC-Phalloidin) and nuclei (DAPI), revealing that macrophages had infiltrated and spread within the bottom volume of the gel (Figure 3b (left) and MovieS1.mov in Supporting Information). Also, by Day 5, some Macrophages had reached the top of the gel (Figure 3b (right))). DAPI-stained macrophages residing 1800 μm from the bottom of the gel (i.e., 200 μm from the top of the 2-mm gel) were quantified using fluorescence microscopy on days 5 and 10 (Figure 3c). This demonstrated that the macrophage populations could establish themselves and increase in population through the volume of these scaffolds over the course of 10 days in vitro. It must be noted that these in vitro studies utilized RAW macrophages which are more proliferative than the native macrophages that would be recruited to this material in vivo. Thus, there is no way of knowing if macrophage population increase vs. time is due to migration through the material, proliferation, or some combination thereof. Additionally, the cell densities vs. time observed here (Figure 3c) are likely higher than what would be expected in vivo. However, to enhance macrophage populations, these scaffolds could also be loaded with cytokines that could potentially expedite macrophage recruitment, as well as polarize them towards M1 phenotypes at early timepoints after implantation (e.g., MCP-1 and IFN-γ, respectively). These cytokines released rapidly at early time points (Figure 3d & 3e) due to excess cytokine being added to the outer compartment without rinsing off that excess cytokine prior to use. The total amount of cytokine delivered could be dictated simply by loading the scaffold with more or less cytokine (Figure 3d & 3e, comparing solid and dashed curves). Independent of loading, pro-inflammatory cytokine release ceased after roughly 12 hours (Figure S1), well before the times at which magnetic stimulation would be applied to trigger subsequent deliveries of anti-inflammatory cytokines.
Figure 3. The outer macroporous gelatin scaffold can recruit and harbor macrophages and can rapidly release pro-inflammatory cytokines.
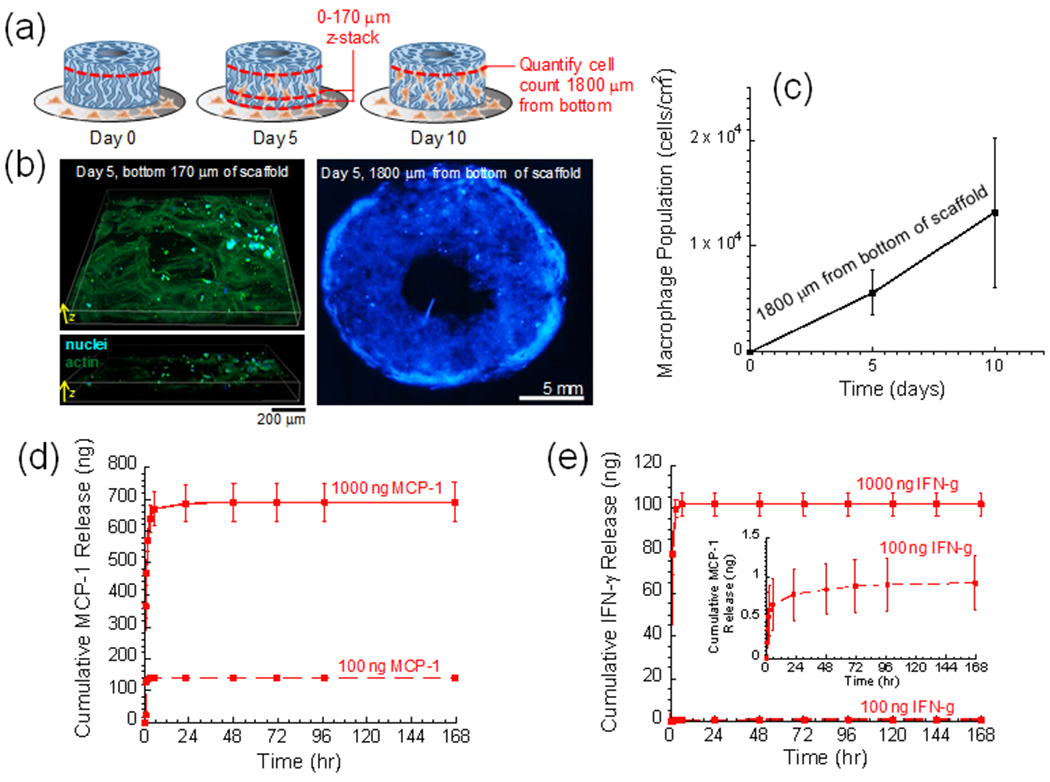
(a) Schematic detailing how macrophages were recruited to the gelatin scaffold and where in the scaffold different images and measurements were taken. (b) Left: 3D z-stack detailing DAPI- (blue) and Phalloidin- (green) stained macrophages in the bottom 170 μm of the scaffold on day 5. Right: collage image of DAPI-stained macrophages (blue) taken 1800 μm from the bottom of the gel (200 μm from the top). (c) Quantification of macrophage density vs. time recorded 1800 microns from the bottom of the scaffold. (d) Cumulative release vs. time for scaffolds loaded with 1000 ng (solid) and 100 ng (dashed) of MCP-1. (e) Cumulative release vs. time for scaffolds loaded with 1000 ng (solid) and 100 ng (dashed) of IFN-γ. Inset: zoomed-in cumulative release vs. time for scaffold loaded with 100 ng IFN-γ. Parts (c)-(e), N = 4.
The inner compartment of this biomaterial system (Figure 2c) was designed to provide delayed, on-demand, and magnetically triggered delivery of anti-inflammatory cytokines (e.g., IL-4 and IL-10). These biphasic ferrogels were designed so that cytokines could be loaded in their Fe3O4-free regions and released in earnest when magnetic gradients were used to compress the Fe3O4-free regions (Figure 4a, white region of ferrogel compresses when a hand-held magnetic is subjacently applied). See MovieS2.mov in Supporting Information for a movie of a biphasic ferrogel being magnetically compressed repeatedly at 1.4 Hz. Cytokine release rates prior to magnetic stimulation were kept at low levels by thoroughly rinsing ferrogels, as to remove excess cytokines that were not well-incorporated. Additionally, cytokine retention prior to magnetic stimulation was likely aided by the use of alginate as the polymeric constituent of these ferrogels. Alginate is heparin-mimicking, and heparin is known to bind strongly to a wide variety of cell-secreted proteins. In these studies, magnetic gradients were applied over the course of 3 hours, but with different temporal profiles (Figure 4b): (i) one where a magnetic gradient was applied at a rate of 1.4 Hz continuously over 3 hours (Stimulation Profile A, top, green) and (ii) one where a magnetic gradient was applied at a rate of 1.4 Hz intermittently, lasting for 5 minutes every hour for 3 hours (Stimulation Profile B, bottom, red). The intermittent Profile B actually yielded higher rates of cytokine delivery compared to the continuous Profile A (Figure 4c). This is possibly due to the fact that magnetic compression results in release of molecules primarily contained in the macropore space and not contained in the gel’s matrix (note that the Fe3O4-free region of these ferrogels are highly macroporous (Figure 2c)). Thus, continuous 1.4 Hz stimulation (Profile A) may initially purge these more available molecules from the pore space but may prohibit the molecules in the gel from equilibrating (i.e., molecules that were purged from the macropore space cannot be replaced by molecules contained in the matrix space due to constant 1.4 Hz gel compression). This would result in a relatively low rate of release when averaged over 3 hours. However, intermittent stimulation (i.e., Stimulation Profile B) likely permits this re-equilibrium of molecules in the 1 hour between subsequent magnetic compressions, resulting in a distribution of molecules from the matrix to the pore space. When magnetic stimulation continues, these relocated pore-space-molecules are efficiently purged. This may result in higher release rates when averaged over 3 hours. While these dynamics are outside the scope of this study, Stimulation Profile B has significant practical advantages in that it both produces higher rates of release and would be easier to implement in vivo. That is, 5 minutes of 1.4 Hz stimulation every hour can be implemented by manually brining a hand-held magnet close to the implant site whereas 3 hours of continuous 1.4 Hz stimulation could be tiring if performed manually. Nevertheless, neither magnetic stimulation profile resulted in statistically significant changes in gel mechanics (Figure S2), suggesting that magnetic stimulation does not overly damage the gels. This leaves open the possibility of magnetically stimulating at later time points for subsequent release busts.
Figure 4. The inner ferrogel compartment can produce delayed, magnetically triggered anti-inflammatory cytokine delivery profiles.
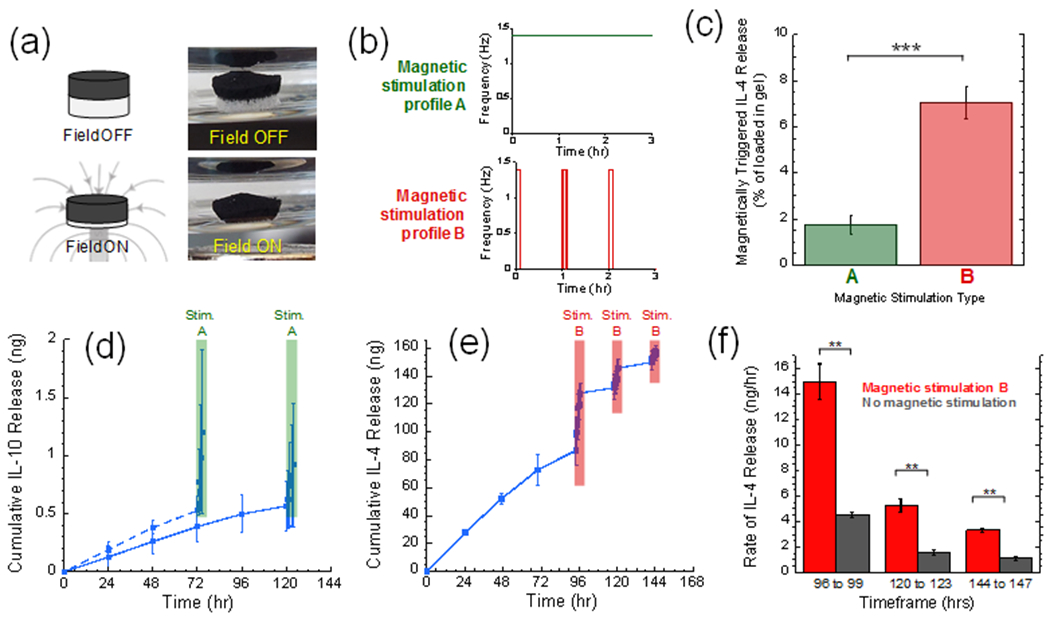
(a) Illustration (left) and photographs (right) of a biphasic ferrogel before (top) and during (bottom) magnetic compression. (b) Schematics of the two magnetic stimulation profiles used in these studies: (top, green) a cyclic magnetic field of 1.4 compressions per second continuously over 3 hours and (bottom, red) the same exposure but pulsed so that gels are cyclically compressed for 5 minutes every hour. (c) Amount released after 3 hours of stimulation profile A (green) vs. B (red). (d) Cumulative IL-4 release vs. time from ferrogels that were either magnetically stimulated on day 3 (dashed) or day 5 (solid). (e) Cumulative IL-10 release vs. time from ferrogels that were magnetically stimulated on days 4, 5, and 6. (f) Release rates over the indicated times when magnetically stimulated (red) vs. unstimulated (gray). For parts (c)-(f), ** and *** indicate statistically significant differences with p < 0.01 and 0.001, respectively (N = 4).
Despite the fact that Stimulation Profile A produced lower rates of release than Stimulation Profile B, it was nonetheless sufficient to significantly impact an anti-inflammatory cytokine’s release profile. When loaded with 500 ng of IL-10, ferrogels released baseline levels of IL-10 prior to day 3 but dramatically increased release rates on day 3 when stimulated using Magnetic Stimulation Profile A (Figure 4d, dashed curve). If this delayed IL-10 release was desired on day 5 rather than 3, magnetic stimulation could be applied on day 5 rather than day 3 (Figure 4d, solid curve). This ability to control the time at which anti-inflammatories are earnestly released could provide a powerful tool for investigating how the duration of the inflammatory response impacts wound healing outcome. Magnetic stimulation can also potentially be used to repetitively deliver anti-inflammatory cytokines on subsequent days to prevent an inflammatory response from resurging. For example, when loaded with 1000 ng of IL-4, baseline levels of IL-4 were released prior to magnetic stimulation. But, release rates were dramatically enhanced when stimulated on day 4 using Stimulation Profile B (Figure 4e, compare slope of curve before 96 hours to the slope from 96 to 99 hours). The rate of IL-4 release could be subsequently enhanced on days 5 and 6 when magnetically stimulated on those days (Figure 4e, enhanced slopes at 120 and 144 hours). These magnetically stimulated release rates on days 4, 5, and 6 were significantly higher than control gels upon which no magnetic stimulation was applied (Figure 4f). These studies demonstrate our ability to control the timing and rate of these anti-inflammatory cytokine deliveries in an on-demand, magnetically prescribed manner.
The described biomaterial system could improve control over the inflammatory response in wound healing applications by locally regulating macrophage phenotype through carefully timed immunomodulatory cytokine deliveries. There is a growing preponderance of evidence suggesting that regulating macrophage phenotype vs. time is critical to achieving desired outcomes in wound healing and regenerative therapies,[32–36] and that sequenced deliveries of immunomodulatory cytokines can provide a means for this temporal regulation.[26,37] In fact, previous studies have designed scaffolding materials to release pro- and anti-inflammatory cytokines at different rates in an attempt to temporally control macrophage phenotype.[19,38–41] While these studies yielded promising results in their ability to influence macrophage phenotype in vivo, statistically significant improvements in regeneration were not observed (e.g., larger or more well-organized vessels/tissues). This could have been due to the inability to explicitly alter and optimize the timing of different cytokine deliveries (i.e., having the delay time of anti-inflammatory cytokines be a variable parameter between conditions). The biomaterial system described here could enable explicit control over the timing of these deliveries, without having to alter the chemistry or structure of the implantable scaffold material between experiments. It should be noted, however, that with this material system’s current formulation, macrophages initially recruited to the outer compartment may be exposed to baseline levels of anti-inflammatory cytokines diffusing out of the inner ferrogel (Figure. 4d & 4e, IL-10 and IL-4 release is non-zero prior to magnetic stimulation). Even though magnetically stimulated release is significantly higher than diffusive release (Figure 4d & 4e, comparing slopes of curves with and without magnetic stimulation), if diffusive release establishes a bioactive concentration of anti-inflammatory cytokines, macrophages may begin to polarize towards pro-healing phenotypes prior to magnetic stimulation. Thus, fine-tuning of the biomaterial system will be required so that rates of release prior to magnetic stimulation result in sub-bioactive anti-inflammatory cytokine concentrations and release rates during magnetic stimulation result in bioactive concentrations. Such fine tuning can be achieved by modifying cytokine loading and ferrogel formulation (e.g., porosity, polymer concentration, polymer type, crosslinking density). Such a tuned biomaterial system will need to be tested in order to verify that this material system is capable of temporally regulating macrophage phenotype through magnetic stimulation.
In sum, we have developed a biomaterial system capable of initially delivering pro-inflammatory cytokines (MCP-1 and IFN-γ) from a macroporous gelatin structure capable of facilitating macrophage infiltration and growth. The amount of inflammatory cytokine release was dependent on the amount of cytokine loaded in the structure. This biomaterial system was also integrated with a biphasic ferrogel that was capable of delivering anti-inflammatory cytokines (IL-4 and IL-10) in a delayed and magnetically triggered manner, using common hand-held magnets. The rate of magnetically stimulated delivery could be regulated by using different magnetic stimulation profiles and the timing of delivery could be regulated simply by choosing when to apply magnetic stimulation. This biomaterial system thus has the potential to enable experimental investigations into how the rate and timing of pro- and anti-inflammatory cytokine deliveries impact biological process critical in wound healing applications. Finally, this material system could also provide the material means to therapeutically implement optimized sequential cytokine deliveries, while retaining a high degree of clinical adaptability by enabling real-time alterations in delivery profiles.
Experimental Section
Fabrication and imaging of the biomaterial system:
The outer compartment gelatin scaffolds used in these studies were purchased as 2 x 12 x 7 mm GelFoam™ sponge sheets (Pfizer, Groton, CT) and cut into hollow disks (2-mm tall, 8-mm OD, 4-mm ID) using 8-mm and 4-mm biopsy punches. Note that biopsy punches and GelFoam sponges were packaged sterile for cell experiments. Additionally, they were packaged in lyophilized form, allowing them to be sputter-coated (30 seconds in gold) and imaged under Scanning Electron Microscopy (SEM) on a Zeiss SIGMA VP Field Emission-SEM with cryogenic capability and Energy-dispersive X-ray Spectroscopy (EDS) for elemental mapping.
The inner ferrogel compartments were made similarly to those described in Cezar et al.[28] Briefly, alginate was dissolved in MES buffer (100 mM MES and 500 mM NaCl at pH = 6.0) containing HOBT and AAD crosslinker and was cast with iron oxide particles and EDC (100 mg mL−1) between two Sigmacote-treated glass plates that were separated by 2-mm spacers. During casting (~ 1 hour), a magnet was placed against one glass plate as to pull the iron oxide particles towards one side of the gel, yielding a biphasic structure. Individual biphasic ferrogels were cut into 4 x 2 mm disks using a biopsy punch and then washed in 50 mL deionized water for 3 days (with water being exchanged twice a day) so that they would fully swell and become void of residual reagents. Ferrogels were then frozen at −20 °C overnight and lyophilized. Lyophilized ferrogels were prepared for imaging by cross sectioning them using a sharp razor, sputter-coating in gold, and imaging as described above for the outer gelatin scaffolds.
Macrophage recruitment studies:
In their culture flasks, RAW 264.7 mouse macrophages were rinsed in PBS, resuspended in fresh DMEM, scraped off, collected, and plated at 10,000 cells per well on sterile 12-well plates. Macrophages were submerged in serum-containing DMEM and allowed to grow for 24 hours. A sterile gelatin scaffold (cut into a hollow disk) was then placed on top of the 2D culture in well plates (fully submerged in media) and left to recruit macrophages for 10 days. Macrophage-populated gelatin scaffolds were analyzed by fixing them in 4% PFA for 10 minutes and washed for 5 minutes in PBS, 3 times. Scaffolds were then soaked in a 0.2% Triton X-100/PBS solution for 5 minutes to permeabilize cell membranes, then washed for 5 minutes in fresh PBS, 3 times. Macrophage nuclei were DAPI-stained by soaking scaffolds in a 2 μg/mL solution of DAPI in PBS for 5 minutes and then washing for 5 minutes in fresh PBS, 3 times. Finally, macrophage actin cytoskeletons were stained by soaking scaffolds in a 0.5 μg/mL solution of FITC-phalloidin in PBS for 5 minutes and then washing for 5 minutes in fresh PBS, 3 times.
3D fluorescent image reconstructions were obtained by taking a green/blue confocal slice every 10 μm from the bottom of the scaffold to a depth of 170 μm within the scaffold using a Nikon TE2000E inverted confocal microscope and its associated NIS-Elements software package. Macrophage cell density counts were taken by inverting the gels in a fresh 12-well plate so that the top of the gels faced down against the plate. Well plates were then loaded into a BioTek Cytation 3 Cell Imaging Multi-Mode Reader which was set to capture a blue-channel image 200 μg into the scaffold (which was 1800 μm away from the side of the scaffold originally near the 2D macrophage culture). BioTek Gen5 software was used to quantify DAPI-nuclei count from these blue-channel images.
Magnetic stimulation of ferrogels:
Ferrogels were magnetically stimulated using 0.5” x 0.5” x 0.5” (1.32 x 1.32 x 1.32 cm) cylindrical neodymium magnets (K&J Magnetics, Pipersville, PA) that were integrated into a custom stimulation apparatus that enabled repetitive and prolonged magnetic field exposures. The custom stimulation apparatus consisted of an array of cylindrical neodymium magnets place on the teetering edge of a variable-speed laboratory rocker’s platform (4 magnets on one edge and 4 on the opposite edge, see MovieS2 in supporting information). This arrangement allowed 8 magnets to oscillate up and down (proximally and distally to 8 ferrogel samples) at a rate prescribed by the rocker’s speed. These studies all utilized the maximum rate of 1.4 Hz (i.e., one magnetic compression every 0.71 seconds). Ferrogels were placed in Sigmacote-treated scintillation vials and suspended above our custom stimulation apparatus with aluminum clamps. This arrangement allowed ferrogel samples to be in close proximity to the magnetics when the magnets were raised (though the magnets did not physically touch the vials) and far enough away from the magnets (~10 cm) when the magnets were lowered, allowing the ferrogels to fully compress and conform back to their original un-compressed thickness between each cycle.
Cytokine time course release studies:
Outer compartment gelatin scaffolds were unpacked and punched to shape in a lyophilized state. Thus, to load them with cytokine, concentrated solutions of protein were prepared and added dropwise directly to the dehydrated scaffolds. It was determined beforehand that when adding liquid to these scaffolds in this manner, they could fully absorb no more than 40 μL of solution. Thus, when loading the scaffolds, concentrated solutions were prepared such that the desired amount of protein to be loaded in the scaffold be contained in 40 μL volumes (e.g., 1000 ng MCP-1 loading required preparation of a concentrated solution of 1000 ng MCP-1 in 40 μL of PBS). So, scaffolds were placed in Sigmacote-treated scintillation vials (to limit protein adsorption to the surfaces of the vials) and loaded dropwise with concentrated protein solutions (MCP-1 or IFN-γ, prepared at concentrations as described above). Scintillation vials were then capped and the scaffolds were left overnight at room temperature to fully absorb the protein. Time-course release studies began after overnight protein absorption when scaffolds were submerged in 1 mL PBS with 1% BSA (t = 0). 1 mL samples were collected periodically from the vials and reserved for analysis by freezing in 1.5 mL centrifuge tubes. After sample removal, fresh 1 mL of PBS with 1% BSA was gently added back to the vial until the next sample was taken. After all samples were collected (168 hours), they were thawed and quantified for cytokine content using ELISA.
Release studies from ferrogels followed a similar procedure. As described above, ferrogels were prepared with the final step being lyophilization, thus producing macroporous and dehydrated samples. Dried ferrogels were placed in scintillation vials with the Fe3O4-free region facing up. It was determined beforehand that when adding liquid to these ferrogels that they could fully absorb no more than 20 μL of solution. They were therefore loaded using desired weights of protein dissolved in 20 μL of PBS (e.g., 1000 ng IL-4 in 20 μL PBS). Ferrogels were left to absorb the protein overnight in capped vials. Ferrogels were then rinsed in PBS with 1% BSA for 3 days to remove excess unincorporated protein, which reduced unstimulated baseline release. Ferrogels were then periodically sampled as described with the gelatin scaffolds, with sample media being fully removed and replaced with fresh media at each timepoint. Collected samples were quantitatively analyzed for IL-4 or IL-10 release using ELISA.
Statistical Analyses:
All quantitative data presented in this communication are represented as a mean ± standard deviation with 4 replicates (N = 4). Because only one-to-one statistical comparisons were made in this study (i.e., no multiple comparisons), student t-tests (two-tailed distributions, heteroscedastic) were used to calculate p-values with p < 0.05 being our benchmark for significance (Microsoft Excel).
Supplementary Material
Acknowledgements
This work was funded by a Medical Research Grant from the Rhode Island Foundation (20144262), an Early Career Development Award from the Rhode Island IDeA Network of Biomedical Research Excellence (RI-INBRE, NIH/NIGMS 2P20GM103430), a 3M Company Non-Tenure Faculty Award (32976949), a grant from the National Science Foundation (NSF-CBET 1063433), and a NSF RII Track-2 FEC grant (NSF-1539068). The authors would like to thank Everett Crissman at the RI consortium for Nanoscience & Nanotechnology for help with SEM imaging and Al Bach and Kim Andrews in the RI-INBRE Centralized Research Core Facility for help with confocal microscopy imaging.
Footnotes
Supporting Information
Additional experimental details and supplemental figures are provided in Supporting Information.
Contributor Information
Anita E. Tolouei, Department of Chemical Engineering, University of Rhode Island, Kingston, Rhode Island 02881
Nihan Dülger, Department of Chemical Engineering, University of Rhode Island, Kingston, Rhode Island 02881.
Rosa Ghatee, Department of Chemical Engineering, University of Rhode Island, Kingston, Rhode Island 02881.
Stephen Kennedy, Department of Chemical Engineering, University of Rhode Island, Kingston, Rhode Island 02881; Department of Electrical, Computer and Biomedical Engineering, University of Rhode Island, Kingston, Rhode Island 02881.
References
- 1.Sen CK, Gordillo GM, Roy S, et al. Wound Repair Regen. 2010;17(6):763–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stojadinovic A, Carlson JW, Schultz GS, Davis TA, Elster EA. Gynecol Oncol. 2008;111(2):S70–S80. [DOI] [PubMed] [Google Scholar]
- 3.SA J, CR AF. N Engl J Med. 1999;341(10):738–746. [DOI] [PubMed] [Google Scholar]
- 4.Boateng JS, Matthews KH, Stevens HNE, Eccleston GM. J Pharm Sci. 2008;97(8):2892–2923. [DOI] [PubMed] [Google Scholar]
- 5.Velnar T, Bailey T, Smrkolj V. J Int Med Res. 2009;37(5):1528–1542. [DOI] [PubMed] [Google Scholar]
- 6.Moura LIF, Dias AMA, Carvalho E, De Sousa HC. Acta Biomater. 2013;9(7):7093–7114. [DOI] [PubMed] [Google Scholar]
- 7.Frykberg RG, Banks J. Adv wound care. 2015;4(9):560–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Blakytny R, Jude E. Diabet Med. 2006;23(6):594–608. [DOI] [PubMed] [Google Scholar]
- 9.Buckley CD, Gilroy DW, Serhan CN, Stockinger B, Tak PP. Nat Rev Immunol. 2013;13(1):59–66. [DOI] [PubMed] [Google Scholar]
- 10.Eming SA, Krieg T, Davidson JM. J Invest Dermatol. 2007;127(3):514–525. [DOI] [PubMed] [Google Scholar]
- 11.Werner S, Grose R. Physiol Rev. 2003;83(3):835–870. [DOI] [PubMed] [Google Scholar]
- 12.Martinez FO, Helming L, Gordon S. Annu Rev Immunol. 2009;27(1):451–483. [DOI] [PubMed] [Google Scholar]
- 13.Park JE, Barbul A. Am J Surg. 2004;187(5 SUPPL. 1):S11–S16. [Google Scholar]
- 14.Van Amerongen MJ, Harmsen MC, Van Rooijen N, Petersen AH, Van Luyn MJA. Am J Pathol. 2007;170(3):818–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wynn TA, Chawla A, Pollard JW. Nature. 2013;496(7446):445–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Martinez FO, Sica A, Mantovani A, Locati M. 2008;13:453–461. [DOI] [PubMed] [Google Scholar]
- 17.Kim YH, Furuya H, Tabata Y. Biomaterials. 2014;35(1):214–224. [DOI] [PubMed] [Google Scholar]
- 18.Kumar VA, Taylor NL, Shi S, Wickremasinghe NC, D’Souza RN, Hartgerink JD. Biomaterials. 2015;52(1):71–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Spiller KL, Nassiri S, Witherel CE, et al. 2015;37:194–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Julier Z, Park AJ, Briquez PS, Martino MM. Acta Biomater. 2017;53:13–28. [DOI] [PubMed] [Google Scholar]
- 21.Koh TJ, DiPietro LA. Expert Rev Mol Med. 2011;13(July):1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vishwakarma A, Bhise NS, Evangelista MB, et al. Trends Biotechnol. 2016;34(6):470–482. [DOI] [PubMed] [Google Scholar]
- 23.Lin CC, Metters AT, Anseth KS. Biomaterials. 2009;30(28):4907–4914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lin CC, Boyer PD, Aimetti AA, Anseth KS. J Control Release. 2010;142(3):384–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kajahn J, Franz S, Rueckert E, et al. Biomatter. 2012;2(4):226–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pajarinen J, Tamaki Y, Antonios JK, et al. J Biomed Mater Res - Part A. 2015;103(4):1339–1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Martinez FO, Gordon S. F1000Prime Rep. 2014;6(March):1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Van Den Steen PE, Dubois B, Nelissen I, Rudd PM, Dwek RA, Opdenakker G. Crit. Rev. Biochem. Mol. Biol 2002;37(6):375–536. [DOI] [PubMed] [Google Scholar]
- 29.Gomez-Guillen MC, Gimenez B, Lopez-Caballero ME, Montero MP. Food Hydrocoll. 2011;25(8):1813–1827. [Google Scholar]
- 30.Zhao X, Kim J, Cezar C a, et al. Proc Natl Acad Sci USA. 2011;108(1):67–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cezar C a, Kennedy SM, Mehta M, et al. Adv Healthc Mater. 2014:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brown BN, Valentin JE, Stewart-Akers AM, McCabe GP, Badylak SF. Biomaterials. 2009;30(8):1482–1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Badylak SF, Valentin JE, Ravindra AK, McCabe GP, Stewart-Akers AM. Tissue Eng Part A. 2008;14(11):1835–1842. [DOI] [PubMed] [Google Scholar]
- 34.Langer R, Tirrell DA. Nature. 2004;428(6982):487–492. [DOI] [PubMed] [Google Scholar]
- 35.Roh JD, Sawh-Martinez R, Brennan MP, et al. Proc Natl Acad Sci. 2010;107(10):4669–4674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Diegelmann, Robert F; Evans MC. Front Biosci. 2004;9(1):283–289. [DOI] [PubMed] [Google Scholar]
- 37.Garash R, Bajpai A, Marcinkiewicz BM, Spiller KL. Exp Biol Med. 2016;241(10):1054–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Keeler GD, Durdik JM, Stenken JA. Acta Biomater. 2015;12:11–20. \ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Reeves Andrew RD, Spiller Kara L., Freytes Donald O., Gordana Vunjak-Novakovic and D LK. Biomaterials. 2015;344(6188):1173–1178. [Google Scholar]
- 40.Chung ES, Chauhan SK, Jin Y, et al. Am J Pathol. 2009;175(5):1984–1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Spiller KL, Anfang RR, Spiller KJ, et al. Biomaterials. 2014;35(15):4477–4488. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.