

Abstract

AlkB and its human homologue AlkBH2 are Fe(II)- and 2-oxoglutarate (2OG)-dependent oxygenases that repair alkylated DNA bases occurring as a consequence of reactions with mutagenic agents. We used molecular dynamics (MD) and combined quantum mechanics/molecular mechanics (QM/MM) methods to investigate how structural dynamics influences the selectivity and mechanisms of the AlkB- and AlkBH2-catalyzed demethylation of 3-methylcytosine (m3C) in single (ssDNA) and double (dsDNA) stranded DNA. Dynamics studies reveal the importance of the flexibility in both the protein and DNA components in determining the preferences of AlkB for ssDNA and of AlkBH2 for dsDNA. Correlated motions, including of a hydrophobic β-hairpin, are involved in substrate binding in AlkBH2–dsDNA. The calculations reveal that 2OG rearrangement prior to binding of dioxygen to the active site Fe is preferred over a ferryl rearrangement to form a catalytically productive Fe(IV)=O intermediate. Hydrogen atom transfer proceeds via a σ-channel in AlkBH2–dsDNA and AlkB–dsDNA; in AlkB–ssDNA, there is a competition between σ- and π-channels, implying that the nature of the complexed DNA has potential to alter molecular orbital interactions during the substrate oxidation. Our results reveal the importance of the overall protein–DNA complex in determining selectivity and how the nature of the substrate impacts the mechanism.
Short abstract
Our study explores the effects of structural variations and the nature of the protein complexed DNA on the dynamics and mechanisms of 2-oxoglutarate- and iron-dependent DNA damage repair enzymes.
1. Introduction
Alkylation of DNA by endogenous and exogenous sources can cause cytotoxicity and/or cancer-linked mutations.1−4 Direct repair of damaged DNA bases occurs via processes involving DNA glycosylases, O6-methylguanine DNA methyltransferases, and AlkB type oxygenases.5,6 AlkB family oxygenases utilize 2-oxoglutarate (2OG) and Fe(II) to catalyze demethylation of alkylated DNA bases. AlkB homologues exist in most bacteria and many eukaryotes. In humans, nine AlkB homologues have been identified (AlkBH1 to AlkBH8 and FTO), most of which are reported to act on DNA and/or RNA.6−9 The AlkB oxygenases that are involved in repair have distinct substrate selectivities for different types of alkylated DNA. For example, AlkB and AlkBH3 prefer to repair methylation damage in single-stranded nucleic acids (ssDNA) rather than double-stranded DNA (dsDNA), whereas AlkBH2 more efficiently acts on duplex dsDNA compared to ssDNA.6,10,11,12a The effectiveness of the AlkB-related enzymes also depends on the identity of the nucleobase and the nature and position of the alkylated group. AlkB-related enzymes are reported to act on multiple monoalkylated DNA nucleobases, i.e., 1-methyladenine (m1A), 3-methylcytosine (m3C), 1-methylguanine (m1G), 3-methylthymine (m3T), 6-methyladenine (m6A), 4-methylcytosine (m4C), and exocyclic bridge-containing lesions, e.g., ethenoadenine (εA) and ethenocytosine (εC).6,13a,14 In the cases of AlkB, AlkBH2, and AlkBH3, alkylated adenines and cytosines are more efficiently repaired than their guanine and thymine analogues, and alkyl groups on the endo nitrogen nucleobase atoms are more efficiently repaired than alkyl groups on exocyclic amines.6,10,11,12a AlkB and AlkBH2 are proposed to efficiently repair m1A and m3C owing to their cationic nature which is believed to enhance the rate of dealkylation.6,10,11,12a
Crystal structures of AlkB (Escherichia coli) and its homologues reveal that they have a modified double-stranded β-helix (DSBH) core fold,1,10,11 which is made up of eight β-strands that support the active site, and which is conserved in 2OG oxygenases.6,10,11 AlkB interacts almost exclusively with the DNA strand that contains the damaged base and uses a base-flipping mechanism to access the damaged base, resulting in significant distortions in the DNA; AlkB compresses the two bases that flank the flipped-out bases to maintain base stacking.10,11,12a By contrast, AlkBH2 interacts extensively with both strands of its dsDNA substrate. AlkBH2 contains a catalytically important hydrophobic hairpin motif, located between β3 and β4, which bears an aromatic “finger” Phe102 that intercalates into the duplex stack, filling the gap resulting from the flipping of the damaged base and thus helping maintain the normal length and stacking of dsDNA in the enzyme–substrate complex.10,11 Compared to AlkB, AlkBH2 has extra DNA-binding motifs that grasp the complementary strand of the dsDNA; AlkBH2 uses a positively charged RKK loop (Arg241, Lys242, and Lys243) and an additional long, flexible loop containing DNA-binding residues Arg198, Gly204, and Lys205 to bind the complementary DNA strand.10,11,12a
The available evidence implies that the AlkB homologues have three Fe coordinating residues (His1, Asp, and His2) and employ a typical 2OG–oxygenase catalytic cycle (Scheme 1), which has two main stages: dioxygen activation and substrate oxidation. The first stage involves oxidative decarboxylation of 2OG, to give succinate and CO2, leading to the formation of an Fe(IV)=O intermediate, which for some 2OG oxygenases has been shown to have a high-spin quintet ground state (S = 2, M = 5).15−17 Crystallographic analyses show that the 2OG ligates the metal in an “off-line” mode where the C2 carbonyl oxygen is positioned trans to a carboxylate oxygen of the Fe-coordinating aspartate.10,11 One of the C1 carboxylate oxygens of 2OG occupies an axial position (trans to His2); the sixth coordination site of the octahedral geometry is occupied by a water. Subsequent formation of the reactive Fe(IV)=O intermediate via such a binding mode (“off-line” geometry) of O2 would require rearrangement to position the ferryl adjacent to the oxidized substrate C—H bond (“in-line” geometry). This could occur via a 2OG C1 carboxylate rearrangement at the five-coordination state or a “ferryl flip” to position the activated oxygen near the substrate.18a Studies on PHF8, another 2OG-dependent demethylase acting on N-methyl lysine residues in histones, imply that the former is more likely.18b
Scheme 1. Proposed Catalytic Cycle for Demethylation of a Monoalkyl Substrate (Exemplified with m3C) by AlkB Family Members.

In the second stage of the catalytic cycle, the methyl group of the substrate is hydroxylated by the Fe(IV)=O species. The hydroxylated hemiaminal product can then undergo a spontaneous, nonenzymatic reaction to give the final demethylated product with the release of formaldehyde, the rate of which is dependent on the product.19
Biochemical and structural studies have led to a consensus mechanism for 2OG oxygenases4,6,12a,12b,13a (Scheme 1). Prior to O2 binding, the resting state (C) contains an Fe(II) complex which is then ligated by 2OG (in a bidentate manner), two histidines (His1 and His2), and one aspartate residue (Scheme 1). The sixth coordination site is normally occupied by a water molecule.10,11,20a Binding of the substrate (e.g., 3-methylcytosine (m3C) DNA) induces the loss of the ligating water, leading to the opening of the site to give a five-coordinate complex (D) with (at least in some cases, as observed by crystallography) an “off-line” 2OG binding mode. From the five-coordinate complex (D), (at least) two reactions paths are possible from the “off-line” mode, i.e., paths A and B (Scheme 1). In path A, dioxygen binds to the five-coordinate complex (D) in an “end on” manner to give an Fe(III)–superoxo complex (E) (Figure 1, Figure S1). Oxidative decarboxylation of 2OG, which in some cases proceeds via an Fe(II)-peroxysuccinate intermediate (F),20b would result in the formation of an Fe(IV)–oxo complex (G), which is incorrectly oriented to react with substrate. The ferryl intermediate (G) could then reorient (“ferryl flip”) toward the methyl group of the substrate to orientate the reactive Fe(IV)=O species (H) in a catalytically productive manner. In path B, the “off-line” 2OG binding mode of the five-coordinate complex (D) first rearranges to an “in-line” 2OG binding mode of the five-coordinate complex (D′) with an open site adjacent to the substrate C—H bond (trans to His2). Dioxygen binds to the open site to give the “in-line” Fe(III)–superoxo complex (E′) which undergoes decarboxylation of 2OG to give the reactive Fe(IV)=O species (H). The active Fe(IV)=O complex enables hydrogen atom abstraction followed by rebound hydroxylation to give the hydroxylated product (J).
Figure 1.

Average structure of AlkBH2–dsDNA (A) and the view of the active site (B) derived from the Fe(III)–superoxo intermediate MD simulations. Coloring: double-stranded beta helix (DSBH) core fold (green) and the hydrophobic β-hairpin (pink). NTD and CTD are N-terminal domain and C-terminal domain, respectively.
Owing to their pathological and biological roles, 2OG oxygenases have been the subject of computational analyses.21−33 Studies have been reported on the mechanism of bacterial AlkB in the repair of 1-methyl adenine (m1A) and exocyclic bridge-containing ethenoadenine (εA) single-stranded DNA (ssDNA) substrates using DFT and quantum mechanics/molecular mechanics (QM/MM) methods.22,24,27−29 Several dynamics studies have shown the importance of conformational dynamics for catalysis in AlkB and its human homologues.34a−34c,35a−35d For example, dynamics studies on human AlkB homologue 5 (AlkBH5) imply that its 2OG binding pocket undergoes conformational changes that expand the active site to permit catalytically productive substrate binding.34a Dynamics studies with the homology modeled AlkB human homologue 1 (AlkBH1) show the importance of two predicted disulfide bridges and a zinc finger domain for DNA recognition and binding.34b Recent molecular dynamics studies on the enzyme–substrate complex of bacterial AlkB using m1A and m6A ssDNA and dsDNA substrates have shown how conformational flexibility can influence the structure–function relationships and substrate selectivity.34c Bleijlevens and co-workers have reported that AlkB is a dynamic protein that exhibits different folding states in its apo and holo forms, and that a fully folded and catalytically competent complex can occur only when both 2OG and Fe2+ are bound to the enzyme.35a It has also been shown that AlkB exhibits significantly different dynamics properties when bound with 2OG or succinate; this is substantially because 2OG makes interactions with both active site ion and the larger β-sheet of the DSBH, helping to maintain a well-folded conformation.35b Dynamics studies on AlkB by Pang et al. reveal that the DNA undergoes substantial structural relaxation upon binding to AlkB which changes the protein–DNA interaction, highlighting that crystal packing may have an important role for the structural features of protein–DNA complexes.35c Similarly, studies on AlkB dynamics by Ergel et al. show that an important conformational transition during the catalysis by AlkB involves the movement of the nucleotide recognition lid away from the active site into a more open position where it participates in fewer constraining interactions with the Fe(II)/2OG core.35d However, the studies on nucleic acid oxygenases to date24,27−29 have not investigated the 2OG rearrangement or “ferryl flip” processes that may be required to produce a catalytically productive Fe(IV)=O species. They also did not consider the effect of the nature of the substrate (ss/dsDNA) and conformational dynamics of the entire complex on determining substrate selectivity and mechanism. Although some studies have explored the substrate oxidation step by modeling the ferryl complex,24,27,29,32 none have analyzed the dynamics and collective motions of the reactive oxidizing intermediates.
To investigate the roles of structural dynamics in the bacterial and eukaryotic AlkB homologue selectivity and mechanism, we performed molecular dynamic simulations to explore variations in the conformational behavior of Fe(III)–superoxo and ferryl complexes in AlkB–ssDNA, AlkB–dsDNA, and AlkBH2–dsDNA. The human homologue of AlkB 2 (AlkBH2) was chosen to explore the generality of the results for AlkB and because of its role in human DNA repair.10,11 The m3C monoalkylated substrates were studied because of their biological importance and lack of previous computational work on them. Combined QM/MM methods were used to explore the mechanisms of the three enzyme–substrate complexes during demethylation of 3-methylcytosine (m3C) substrates, including dioxygen activation, “ferryl flip”/2OG rearrangement, and substrate hydroxylation steps.
2. Results and Discussion
2.1. The Nature of the Substrate Influences the Conformational Dynamics of the Enzyme–Substrate Complexes for O2 Activation in AlkB–ssDNA, AlkB–dsDNA, and AlkBH2–dsDNA Systems
2.1.1. Overall Dynamics of the Enzyme–Substrate Complexes
To obtain insight into the structural dynamics of the three enzyme–substrate complexes of interest (AlkB–ssDNA, AlkB–dsDNA, and AlkBH2–dsDNA), we first carried out the MD simulations on their Fe(III)–superoxo complexes with coordinated 2OG. The results reveal that the active sites, proteins, and the protein–DNA complexes of the three enzymes are stable with average root mean square deviations (RMSDs) of 0.38, 1.03, and 1.13 Å, respectively, for AlkB–ssDNA; 0.51, 1.52, and 3.05 Å, respectively, for AlkBH2–dsDNA; and 0.42, 1.59, and 2.95 Å, respectively, for AlkB–dsDNA (Figure S2). The dsDNA in both AlkBH2–dsDNA and AlkB–dsDNA exhibited large fluctuations during the simulations, which affects the overall RMSD of the protein–DNA complexes. Hence, the DNA contributes substantially to the overall flexibility of the complexes, in agreement with conclusions from previous studies on the dynamics of the resting state of AlkB.34c,35c By contrast, AlkB–ssDNA shows a more compact structure than both AlkB–dsDNA and AlkBH2–dsDNA, due to the smaller number of nucleotides. This observation is supported by analyses on the distance between the center of mass of the protein and the DNA (Figures S3–S6). Center of mass analyses reveal average values of 15.8, 19.7, and 25.2 Å between the protein and DNA for AlkB–ssDNA, AlkBH2–dsDNA, and AlkB–dsDNA, respectively. This trend implies that the AlkBH2–dsDNA complex structure is more compact than that of AlkB–dsDNA, in agreement with crystallographic observations; i.e., AlkBH2–dsDNA forms a stabilizing interaction with both DNA chains, whereas AlkB–dsDNA only forms direct interactions with one of the DNA chains.10,11
2.1.2. Flexibility of the 2OG Binding Sites
In the 2OG binding site of AlkBH2–dsDNA, the hydrogen bonding interactions between the C5 carboxylate oxygens of 2OG and Tyr161 (98% of the MD snapshots) and Arg248 (93%) are stable. O4 of the 2OG C5 carboxylate makes an electrostatic interaction with Arg248 (93%). Similarly, with AlkB–ssDNA, the 2OG makes analogous strong hydrogen bonding interactions with Tyr122 (>99%) and Arg204 (99%) and an electrostatic interaction with Arg204 (99%). In AlkB–dsDNA, 2OG (O3, O4) make interactions with Ser145 (61%, 54%) and Trp178 (41%, 30%). These interactions are less stable along the MD time course when compared with AlkBH2–dsDNA and AlkB–ssDNA, implying that 2OG is more effectively stabilized in both of these enzyme–substrate complexes compared to AlkB–dsDNA.
2.1.3. Interactions of the Iron-Coordinating Residues
Hydrogen bonding interactions involving the Fe-coordinating histidines (His1 and His2) (100% of the MD snapshots in all AlkBH2–dsDNA, AlkB–ssDNA, and AlkB–dsDNA) may enhance active site stability. The noncoordinating oxygen of the Fe-coordinating aspartate is also apparently stabilized via hydrogen bonding interactions, with Arg254 (>99%), Arg210 (99%), and Arg210 (47%) in AlkBH2–dsDNA, AlkB–ssDNA, and AlkB–dsDNA, respectively (Figures S7–S9). The Fe-coordinating axial histidine (His2) residues are stabilized via π–π stacking with nearby Phe and Trp residues in AlkBH2–dsDNA (Phe195 and Phe197) and AlkB–ssDNA (Phe154 and Trp178); these stacking interactions are apparently weaker in AlkB–dsDNA (Phe154 and Trp178) (Figures S10–S12), possibly reflecting weaker binding of the DNA to AlkB in this complex, as the protein only interacts with one of the duplex DNA chains.10,11
2.1.4. Dynamics of the Substrate Binding Sites
In the three enzyme–substrate complexes, the heteroaromatic ring of the substrate base (m3C) is stabilized via π-stacking interactions with aromatic residues, which are important in substrate recognition.10,11 Phe124 and Tyr122 (AlkBH2–dsDNA), Trp69 and Tyr76 (AlkB–ssDNA), and Trp69 and Tyr76 (AlkB–dsDNA), as well as the imidazole groups of the coordinating equatorial histidine (His1) residues, participate in π-stacking interactions, which likely help promote a catalytically productive orientation of the substrate. The exocyclic amine (N4) of the substrate is stabilized by interactions with Glu175 (AlkBH2–dsDNA), Asp135 (AlkB–ssDNA), and Asp135 (AlkB–dsDNA), but the interaction is weaker in the latter case. The intercalating residue, Phe102, that helps in flipping of the damaged base into the active site in AlkBH2–dsDNA,10,11 forms a stable and continuous π-stacking interaction with the nearby bases; this enhances the stability of the duplex DNA in AlkBH2–dsDNA.
2.1.5. Long-Range Correlated Motions
Collective dynamics provides insight into correlated motions between remote regions of the enzyme–substrate complex. In the AlkBH2–dsDNA and AlkB–dsDNA complexes, dynamic cross correlation analysis (DCCA) (Figure 2A, Figures S13–S16) shows that the β-sheets of the DSBH core have positive correlations with one another. These are more intense in AlkBH2–dsDNA and AlkB–dsDNA than in AlkB–ssDNA, implying that the nature of DNA substrate is capable of influencing the overall correlation motion of the Fe center region via a long-range interaction.
Figure 2.

Dynamic cross correlation (A) and principal component analysis (B) for the AlkBH2–dsDNA Fe(III)–superoxo complex. In part A, residue numbers are as follows: 1–206 (protein), 207 (Fe), 208 (O2), 209 (2OG), 210–235 (DNA), and 216 (m3C substrate). NTD and CTD are the N-terminal and C-terminal domains, respectively. Yellow to blue represents the direction of motion of residues in part B.
In AlkBH2–dsDNA, residues Tyr161 and Arg248, that bind to 2OG, have a positive correlation with β6 and β7 that form the substrate recognition lid, as well as one of the β-strands (β9) of the DSBH, indicating that binding of 2OG might influence the substrate binding site and the orientation of the Fe center. The DNA binding residues (198–214) manifest a positive correlation with Fe, the metal ion coordinating His236 and nearby residues (230–238), implying that such correlated motions might also be of importance for productive substrate orientation. With AlkB–ssDNA, the 2OG binding residues (Tyr122 and Arg204) manifest a positive correlation with Fe, the metal coordinating aspartate, and the loop bearing the metal coordinating HXD motif. The 2OG binding residues (Ser145 and Trp178) in AlkB–dsDNA only show positive correlation with residues on β7 (147–150). These observations imply that the correlated motions of the 2OG binding residues in AlkB–ssDNA likely influence substrate binding more than in AlkB–dsDNA. Further, the DNA binding residues (155–166) in AlkB–ssDNA show positive correlation with the Fe coordinating 2OG, His131, Asp133, and His187, and the nearby residues (126–135 and 183–192). The DNA binding residues (155–166) in AlkB–dsDNA only show positive correlation with the two metal ion coordinating histidines (His131 and His187), implying that there are stronger correlated motions in AlkB–ssDNA compared to AlkB–dsDNA. Differences in correlation motions could reflect the stronger binding of AlkB to ssDNA compared to dsDNA (where the protein only interacts with the strand that contains the damaged base). Overall, the analyses imply that complex correlated motions of the molecules are important during catalysis.
Biophysical analyses reveal that AlkBH2–dsDNA contains a hydrophobic β-hairpin (β3- β4) (aa 89–108) that is close to the active site and which is important in enabling the preference of AlkBH2 for dsDNA substrates.10,11 DCCA reveals that this hydrophobic β-hairpin has a strong positive correlation with residues 123–128 and the substrate. Residues 123–128 belong to β6 and β7 and are proposed to form a substrate recognition lid.10,11 Such correlated motions might contribute to the binding of the substrate as proposed on the basis of experimental studies.10,11
Studies on AlkBH2 have revealed that the D173A, H236A, and R203H substitutions cause loss of activity.36 DCCA shows that D173 and H236 have a strong positive correlation with the Fe center, 2OG, and the coordinating histidines and aspartate. R203 which is located in α4, shows a strong correlation with the DNA-binding flexible long loop (residues 200–206). Such correlated motions might potentially contribute to the loss of activity with the D173A and H236A variants, while the R203H substitution might affect DNA binding. Additional studies are needed to validate these proposals.
PCA shows that AlkB–ssDNA (Figure S17) has limited motion for the loop connecting β6 to β7 and its N- and C-terminal regions. AlkBH2–dsDNA (Figure 2B) has limited motions at its N- and C-terminal regions, whereas AlkB–dsDNA (Figure S18) shows major motion in its complexed DNA and limited motions at its N- and C-terminal regions. These observations support the preferences of AlkB and AlkBH2 for ssDNA and dsDNA, respectively.
2.1.6. Dynamics of AlkB–ssDNA, AlkB–dsDNA, and AlkBH2–dsDNA with Undamaged DNA
MD simulations with the nonmethylated DNA substrate reveal that it can bind to the enzymes (Figures S19–S21). The studies show that the protein–DNA complexes in both AlkBH2–dsDNA and AlkB–ssDNA are more rigid than the AlkB–dsDNA complex (Figures S22–S24). The measured distance between the Fe center and N3 of the substrate in comparison with the N-methylated DNA substrates in all the three systems implies that the undamaged DNA systems are not catalytically productive (Figures S25 and S26), even though the complexes are stable. Binding free energy calculations using the molecular mechanics/generalized born surface area (MM/GBSA)13b method reveal weaker binding of undamaged DNA to the respective enzyme compared with the results obtained for the damaged DNA substrates. In the undamaged DNA complexes, the relative free energies of binding of DNA to protein in AlkBH2–dsDNA, AlkB–ssDNA, and AlkB–dsDNA are −125.26, −26.55, and −32.87 kcal/mol, respectively. Values of −143.03, −37.10, and −45.99 kcal/mol for the damaged DNA complexes of AlkBH2–dsDNA, AlkB–ssDNA, and AlkB–dsDNA complexes, respectively, are consistent with the catalytically productive nature of the latter complexes.
2.2. Reaction Mechanism of the Dioxygen Activation
2.2.1. O2 Activation for the “Off-Line” Fe(III)–Superoxo Complex
QM/MM calculations were carried out using a snapshot from the productive MD trajectories of the “off-line” Fe(III)–superoxo complex, taking the distance between the distal oxygen (Od) of the superoxide and the C2 of the 2OG as a reaction coordinate (Figures S2D, S28, and S29). The QM region (Figure 3) contains the iron–dioxygen (Fe–O2) unit, 2OG, the methylimidazole groups of His171 (His1) (His131 for AlkB) and His236 (His2) (His187 for AlkB), the acetate group of Asp173 (Asp133 for AlkB), the methylguanidium group of Arg254 (Arg210 for AlkB), and the 3-methylcytosine part of the DNA substrate.
Figure 3.
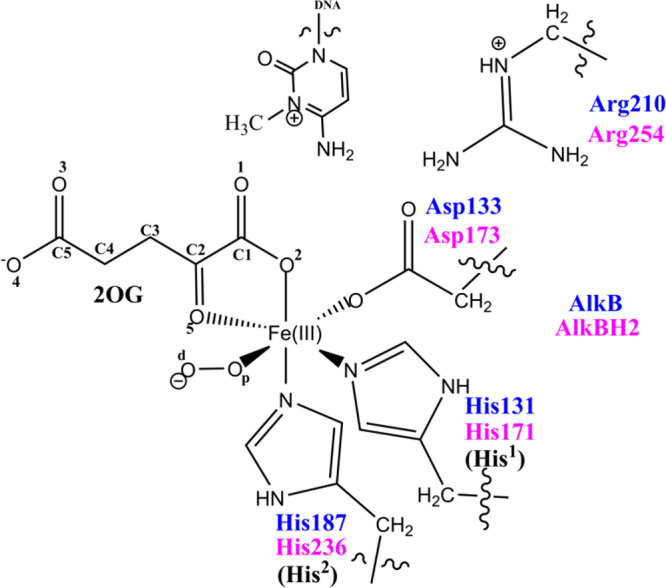
QM region of AlkB/AlkBH2 used in the QM/MM calculations.
The potential energy profile and the optimized geometries of the stationary points obtained for the dioxygen activation process (Scheme 2) are presented in Figures 4 and 5, respectively. In the optimized QM/MM model of the reactant (RC1), the Fe–Op and Op–Od distances are 2.11 and 1.27 Å, respectively. The Fe center oxidation state was determined via a spin density analysis. The spin densities of Fe and dioxygen are 4.19 and −0.68, respectively, supporting the Fe(III) oxidation state of this complex; the values compare favorably with previous studies on 2OG oxygenases.23−26 The relatively long Op–Od bond and the spin density value of O2 support the superoxide character of RC1.23,24 In the first transition state (TS1) the Op–Od bond distance increases to 1.37 Å, while the Fe–Op and Od–C2 bond lengths shorten to 2.00 and 1.42 Å, respectively, in agreement with the results of previous studies.24,25a The C1–C2 distance increases from 1.55 to1.83 Å, pointing to the partial cleavage of the bond in readiness to eliminate CO2.
Scheme 2. Mechanism of Dioxygen Activation.

Figure 4.
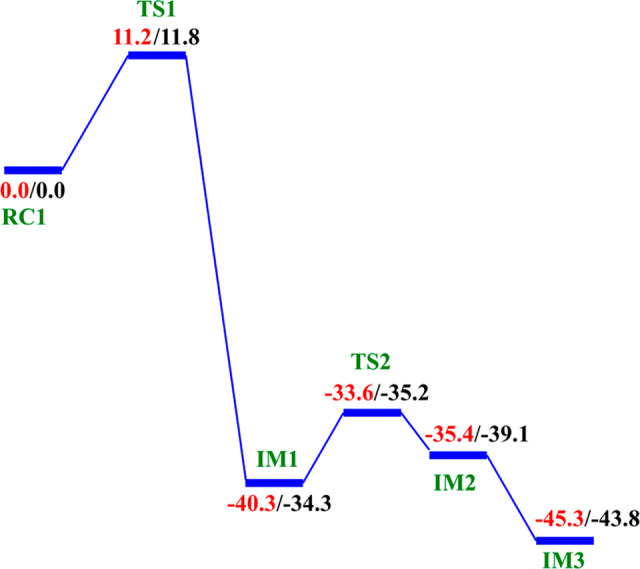
QM/MM reaction profile for the dioxygen activation step by AlkBH2–dsDNA. Relative energies are in kcal/mol at UB3LYP/def2-TZVP (BS2) (in red) and BS2 with ZPE (in black).
Figure 5.

Geometries of the stationary points along the dioxygen activation step in AlkBH2–dsDNA. Distances (Å) and the spin densities are in black and pink, respectively.
Decarboxylation proceeds via an Fe(II)-peroxysuccinate intermediate20b (IM1), involving the cleavage of C1–C2 of 2OG and formation of a bond between Od and C2 of 2OG, with an activation barrier of 11.8 kcal/mol, including the zero-point energy contribution, calculated at the UB3LYP/def2-TZVP level. Formation of this intermediate (IM1) is highly exergonic with an energy of −34.3 kcal/mol. The highly exergonic nature of this step is likely due to release of CO2 and formation of succinate. At this stage, the C1–C2 bond is completely cleaved with a distance of 3.48 Å while the peroxy bridge Op–Od bond is elongated to 1.45 Å. In AlkBH2–dsDNA, hydrophobic interactions of Met226 with Phe197 and Ile184 including π-stacking interaction of Phe195 and Phe197 enhance stabilization of TS1. TS1 is further stabilized by networks of hydrogen bonding interactions of Arg254 with Thr252 and the noncoordinating oxygen of the iron ligating Asp173. TS1 derived from AlkB–ssDNA is stabilized by hydrophobic interaction of Ile143 with Trp178, hydrogen bonding interaction of Arg210 with the noncoordinating oxygen of the iron ligating Asp133, and hydrogen bonding interaction of Thr208 with the noncoordinating oxygen of C1-carboxylate of the 2OG. In addition to the hydrophobic interaction of Ile143 with Trp178, TS1 in AlkB–dsDNA is stabilized by hydrogen bonding networks of Arg183 and Arg210 with Glu136 and the noncoordinating oxygen of the iron ligating Asp133, as well as by a salt bridge interaction of Arg210 with the 2OG C1-carboxylate. The DCCA shows that the residues involved in the stabilization of TS1 in AlkBH2–dsDNA have positive correlation with the Fe-center and DSBH core residues. In AlkB–dsDNA, the TS1 stabilizing residues manifest positive correlation with nucleotide recognition lid residues, while in AlkB–ssDNA the residues have positive correlation with residues that make up the DSBH core. This indicates that long-range interactions with DSBH residues might be more important for O2 activation in AlkBH2–dsDNA and AlkB–ssDNA than in AlkB–dsDNA. The results suggest that modification of residues in the DSBH might selectively influence the dioxygen activation process in AlkBH2–dsDNA and AlkB–ssDNA, whereas with AlkB–dsDNA targeting residues in the nucleotide recognition lid may be more productive in this regard.
The next step, i.e., reaction of IM1 to IM2, involves homolytic cleavage of the Op—Od bond of IM1; this is faster than the decarboxylation step with a barrier of 6.7 kcal/mol and a barrierless process with ZPE contribution. This result implies that the decarboxylation step is rate-limiting in the oxygen activation phase of catalysis. IM2 has a partial bond of length 2.10 Å between the two oxygen atoms, and the Fe—Op bond length is 1.77 Å. The partial Op—Od bond then breaks, and IM2 rearranges to form IM3 with a new Fe—Op bond length of 1.62 Å. The spin density of 3.16 for Fe reveals the formation of the ferryl (Fe(IV)=O) species. IM3 is thermodynamically stable with an overall reaction energy of −43.8 kcal/mol at the BS2+ZPE theory level, inferring that dioxygen activation is exergonic. The Fe(IV)–oxo group in IM3 is incorrectly positioned (“off-line” geometry) to react with the substrate, and thus, if an intermediate, it must undergo rearrangement, i.e., the Fe(IV)–oxo migrates toward the methyl group of the nucleobase substrate (“in-line” geometry).
The dioxygen activation step in AlkB–ssDNA and AlkB–dsDNA with the same substrate (m3C) manifests similar behavior as observed in AlkBH2–dsDNA with the rate-determining step having barriers of 11.3 and 13.2 kcal/mol with ZPE correction, respectively. The detailed geometries of the stationary points are presented in the SI (Figures S30 and S31).
2.2.2. Formation of the “In-Line” Fe(IV)=O Intermediate via a Potential “Ferryl Flip” in AlkBH2
We then investigated the two proposed mechanistic possibilities for obtaining a productive ferryl intermediate using QM/MM calculations. In one mechanism, formation of the “in-line” Fe(IV)=O intermediate from IM3 (wherein the oxo group is not productively oriented to react with the substrate) has been proposed to occur via oxygen atom exchange with the use of a water molecule.13a,18a,23 Such a process has been termed a “ferryl flip”; i.e., the oxo group in IM3 flips toward the target methyl group of the m3C substrate to give a productive Fe(IV)=O intermediate (Scheme 3). IM3′ comprises IM3 with two water molecules added to the QM region. The ferryl flip process begins with the binding of one water molecule to the open coordination site of the iron center, leading to the formation of a six-coordinate ferryl complex (IM4) with bond lengths of 2.12 and 1.62 Å for Fe—O1 and Fe—Op, respectively. One of the protons (H1) from the coordinated water molecule is then transferred to the noncoordinating carboxylate oxygen of the succinate to give IM5. This step passes through TS3 with a barrier 6.6 kcal/mol at the BS2+ZPE level of theory. The so-transferred proton (H1) to succinate is then transferred to the oxo (Op) group of the “off-line” Fe(IV)=O to give a “dihydroxylated” Fe(IV) complex, IM6, with Fe—O1 and Fe—Op bond lengths of 1.82 and 1.74 Å, respectively. The formation of this “dihydroxylated” complex is slightly endergonic with an energy of 5.2 kcal/mol at the BS2+ZPE level of theory. The equatorial hydroxyl group is then converted to H2O using the proton (Ha) from the Arg254 guanidino group, concomitant with the spontaneous transfer of a proton (H2) from the axial hydroxyl group to the noncoordinating carboxylate oxygen (O2) of the coordinating aspartate, to give IM7. This passes thorough TS5 with a barrier of 18.3 kcal/mol; the so-formed IM7 is slightly endergonic with an energy of 6.7 kcal/mol at the BS2+ZPE level of theory. Subsequently, the proton (H2) from the noncoordinating carboxylate of the coordinating aspartate in IM7 is transferred to the deprotonated NH (NaH) group of Arg254, resulting in the “flipped” Fe(IV)=O complex, IM8. IM8 can then release the bound water molecule to give the “in-line” 5-coordinate complex, IM9. The optimized “ferryl flip” reaction states geometries and energy profile are presented in Figure 6 and Figure S32, and Figure 7, respectively. The overall “ferryl flip” process proceeds with a very high barrier of 21.7 kcal/mol, at the BS2+ZPE level of theory, which is higher than the previously reported barrier of 10.9 and 18.1 kcal/mol for oxygen atom exchange for Asqj23 and PHF8,18b respectively. We also explored the possibility of direct transfer of the proton (H2) from the axial hydroxyl (O1H2) group to the equatorial hydroxyl group (OpH1) of the “dihydroxylated” Fe(IV) complex, IM6 (Figure S33). This transfer results in a barrier of 24.3 kcal/mol at the BS2+ZPE level of theory, which is 2.6 kcal/mol higher than the one observed via proton transfer through Arg254; thus, this process is not energetically viable in agreement with the studies on the histone demethylase, PHF8.18b These high barriers could in part be due to the compact nature of the active site and steric effects arising from the nucleobase ring (cytosine) of the substrate, as well as the second sphere residue Arg254.
Scheme 3. Proposed Ferryl Flip Mechanism by AlkBH2–dsDNA.

Figure 6.

Geometries of the stationary points along the proposed ferryl flip mechanism in AlkBH2–dsDNA. Distances are in Å.
Figure 7.

QM/MM reaction profile for the proposed ferryl flip mechanism by AlkBH2–dsDNA. Relative energies are in kcal/mol at UB3LYP/def2-TZVP (BS2) (in red) and BS2 with ZPE (in black).
2.2.3. Rotation of the C1 Carboxylate of 2OG from the “Off-Line” to an “In-Line” Binding Mode in AlkBH2
The change in the 2OG binding mode from the “off-line” to the “in-line” geometry to enable a productive ferryl orientation was then explored by performing a potential energy scan via the changing of the dihedral angle Nε (His1)–Fe–O5 (2OG)–O2 (2OG) of the five-coordinate enzyme–substrate (ES1) complex of AlkBH2–dsDNA. The five-coordinate ES1 complex contains an Fe(II) center (high spin state S = 2, M = 5) ligated by two histidinyl residues (His1 and His2), one aspartyl residue, and the 2OG cosubstrate which is bound in an “off-line” bidentate manner. First, we performed a 1 μs MD of the five-coordinate enzyme–substrate complex (Figures S34 and S35). We then used an MD snapshot to perform QM/MM calculations for the proposed 2OG reorientation. QM/MM optimizations were first carried out on the ES1 snapshots of the system. The potential energy scans were then performed on the optimized ES1 complex with 2° increment of the Nε (His1)–Fe–O5–O2 dihedral angle.
The optimized stationary point geometries of the 2OG rotation are presented in Figure 8. In the optimized QM/MM model of the MD simulated ES1 complex snapshot, the bond lengths of Fe–O2 and Fe–O5 are 2.05 and 2.34 Å, respectively. The bonds are marginally elongated compared to the Fe(III)–superoxo complexes (RC1), where the Fe–O2 and Fe–O5 bond lengths are 2.03 and 2.18 Å, respectively. The shorter bond lengths observed in the Fe(III)–superoxo complex are likely due to the oxidation of Fe(II) to Fe(III), which strengthens metal–ligand bonds. The dihedral angle potential energy scan resulted in the rotation of the 2OG C1 carboxylate from the “off-line” binding mode (ES1) to the “in-line” mode (ES2). The transition state has a dihedral angle of 151.6° with Fe–O2 and Fe–O5 bond lengths 2.07 and 2.36 Å, respectively. The rotation proceeds rapidly with barriers of 2.94 kcal/mol at the BS2+ZPE level. This value is about 1.5 kcal/mol higher than the value obtained for another 2OG oxygenase, PHF8,18b possibly due to increased steric effects in the active site as well as the strong hydrogen bonding interaction of the second-sphere arginine residue (Arg254) with the C1 carboxylate of 2OG in the enzyme, which is absent in PHF8.
Figure 8.

Geometries of the stationary points along the 2OG rotation in AlkBH2–dsDNA. The distances and angles are in Å and degrees, respectively.
Formation of the “in-line” five-coordinate AlkBH2–dsDNA enzyme substrate complex (ES2) is slightly exergonic with relative energies of −1.98 kcal/mol at the BS2+ZPE level. The dihedral angle of the formed “in-line” complex (ES2) is 153.6° while the Fe–O2 and Fe–O5 bond lengths are 2.04 and 2.35 Å, respectively. Overall, these calculations imply that 2OG rotation is energetically favorable in agreement with the results for PHF8.18b
2.2.4. O2 Activation for “In-Line” Fe(III)–Superoxo Complex
After the formation of the “in-line” five-coordinate enzyme substrate complex (ES2), dioxygen binds to the vacant Fe coordination site to give the “in-line” Fe(III)superoxo complex, which was then subjected to 1 μs MD simulations (Figures S36 and S37). An MD snapshot was then used for the QM/MM study. The obtained results are similar to those obtained for the “off-line” Fe(III)superoxo complex. The rate-determining decarboxylation step has a barrier of 9.6 kcal/mol, at the BS2+ZPE level of theory, in comparison to 11.8 kcal/mol obtained for the “off-line” Fe(III)superoxo complex. Therefore, for the “in-line” Fe(III)superoxo complex, the rate-determining decarboxylation barrier is 2.2 kcal/mol lower than for the “off-line” Fe(III)–superoxo complex, implying that the dioxygen activation is faster in “in-line” than “off-line” geometry. Formation of the “in-line” Fe(IV)=O complex (IM3) is also thermodynamically favorable with a reaction energy of −63.4 kcal/mol at the BS2+ZPE level of theory, in comparison to −43.8 kcal/mol obtained for the “off-line” Fe(IV)=O via “off-line” Fe(III)–superoxo complex, indicating the exergonic nature of the dioxygen activation reaction. The geometries of the stationary points are presented in Figure S38.
2.3. Conformational Flexibility Modulates the Structure of the Reactive Complex (Fe(IV)=O) for the Substrate Hydroxylation in AlkB–ssDNA, AlkB–dsDNA, and AlkBH2–dsDNA Systems
RMSD analyses of the active site, protein, protein–DNA complex, and DNA for all the three Fe(IV)=O complexes of the three systems show stable structures with average RMSD values of 2.05, 1.67, and 1.75 Å for the protein–DNA complex in AlkBH2–dsDNA, AlkB–ssDNA, and AlkB–dsDNA, respectively (Figure S39). These analyses reveal the stability of the active site, protein, protein–DNA complex, and the DNA in both AlkB–ssDNA and AlkBH2–dsDNA while they show some flexibility in AlkB–dsDNA. DNA contributes significantly to the overall RMSD of the protein–DNA complexes in both AlkB–substrate complexes, while minimal contribution is observed in AlkBH2–dsDNA. Center of mass analyses shows a similar trend to that observed for the Fe(III)–superoxo complexes with average values of 17.5, 26.7, and 19.3 Å for AlkB–ssDNA, AlkB–dsDNA, and AlkBH2–dsDNA, respectively (Figure S40). This observation indicates that AlkBH2–dsDNA is more structurally compact than AlkB–dsDNA in the ferryl complex as observed for the superoxo complex, a difference arising from the observed stronger interactions between the protein and dsDNA in AlkBH2–dsDNA.10,11 Hydrogen bonding analysis shows that the nonmetal-coordinating C4 carboxylate oxygens (O3, O4) of the succinate interact with Arg 248 (76%, 84%) and Tyr 161 (84%, 18%) in AlkBH2–dsDNA. Similar hydrogen bonding interactions are observed in the AlkBs where the succinate C4 carboxylate oxygens (O3, O4) interact with Ser 145 and Trp 178 with (55%, 61%) and (18%, 17%), respectively, in AlkB–ssDNA and (38%, 37%), (37%, 39%), respectively, in AlkB–dsDNA. These results imply more flexibility in the binding of succinate compared with that of 2OG, likely reflecting stronger binding of the 2OG cosubstrate compared to the succinate coproduct. These observations could be of relevance to inhibitor design in terms of optimizing the chain length of bonding in the 2OG/succinate pocket. In AlkBH2–dsDNA, the residues involved in the stabilization of the succinate have positive correlation with residues 159–166 (β8), 188–192 (β10), and 238–250 (β14, which contains coordinating His236 and β15). In the AlkBs, the residues that stabilize the succinate manifest positive correlation with 174–178 (loop connecting β9 and β10) and 206–209 (β12). All these residues are in the vicinity of the noncoordinating C4-carboxylate of the succinate, indicating that the correlated motions likely aid in the overall binding of the succinate. The results reveal stable hydrogen bonding interactions between the iron coordinating His1 and His2 residues (>99% of the structures in the MD simulations) in the AlkBH2–dsDNA, AlkB–ssDNA, and AlkB–dsDNA complexes. Analysis of the distances between the oxo group of the Fe(IV)=O intermediate and the methyl carbon of the substrate (m3C) reveals that the substrate is closer to the metal center in the ferryl complex than in the superoxo complex (Figures S39D, S41, and S42). This observation is supported by molecular mechanics/generalized born surface area (MM/GBSA)13b calculations, which predict how strongly the substrates bind to the proteins. In the Fe(III)–superoxo complex, the relative free energies of binding of m3C DNA substrates to AlkBH2–dsDNA, AlkB–ssDNA, and AlkB–dsDNA are −143.03, −37.10, and −45.99 kcal/mol, respectively, while values of −168.89, −56.01, and −67.91 kcal/mol are obtained for the ferryl complex of AlkBH2–dsDNA, AlkB–ssDNA, and AlkB–dsDNA, respectively. These results show that the m3C substrate binds better to the proteins in the ferryl complex than in the superoxo complex in all three enzymes, reflecting formation of more productive complexes necessary for hydroxylation. The calculations also show that the substrate in AlkBH2–dsDNA binds better than in AlkB-dsDNA complexes, possibly due to stronger interactions between the two strands of the dsDNA and the protein in AlkBH2–dsDNA. The MM/GBSA method provides binding free energies of a set of ligands with similar size and structure.13c,13d We compared the relative free energies of the AlkBH2–dsDNA and AlkB–dsDNA complexes, and the Fe(III)–superoxo and Fe(IV)–oxo intermediates in the same system. The effect of the size of the substrate is reduced by calculating the relative free energies for AlkB and the methylated nucleotide only (excluding the rest of the DNA fragments) in both AlkB–ssDNA and AlkB–dsDNA. The results show stronger binding between AlkB and methylated ssDNA (−11.26 and −18.19 kcal/mol for the Fe(III)–superoxo and Fe(IV)–oxo complexes, respectively) compared to dsDNA (−6.46 and −9.11 kcal/mol for the Fe(III)–superoxo and Fe(IV)–oxo complexes, respectively), in accord with experimental observations.10,11
DCCA (Figure 9A, Figures S15, S16, S43, and S44) for the ferryl proteins reveals correlated motions that are similar but differ in details to that observed in the Fe(III)–superoxo systems, indicating that complex correlated motions are involved in the binding of DNA and the succinate coproduct and contribute to the proper positioning of the substrate with respect to Fe(IV)=O for the hydrogen abstraction step.
Figure 9.

Dynamic cross correlation (A) and principal component analysis (B) for AlkBH2–dsDNA at the ferryl complex stage. In part A, residue numbers are as follows: 1–206 (protein), 207 (Fe), 208 (O), 209 (succinate), 210–235 (DNA), and 216 (m3C substrate). NTD and CTD are the N-terminal domain and C-terminal domain, respectively. Yellow to blue represents the direction of motion of protein residues in part B.
PCA provides insight into the essential dynamics and the direction of motion in flexible regions of proteins.37a,37b PCA of AlkBH2–dsDNA (Figure 9B) has limited motions in the N- and C-terminal regions of the protein and major motions at the hydrophobic β-hairpin region. The observed motion of the hydrophobic β-hairpin, which is not observed in the Fe(III)–superoxo complex, is toward the Fe center, resulting in compaction of the complex, and hence likely a complex that favors catalysis. AlkB–ssDNA (Figure S45) has motions in its N- and C-terminal regions, while AlkB–dsDNA (Figure S46) shows major motions at the loops connecting β6 to β7 and β4 to α2, in the DNA substrate (reduced motion when compared with the superoxo complex) and limited motions in the N- and C-terminal regions. The PCA results also support experimental observations that the protein in the AlkBH2–dsDNA complex makes strong interactions with both DNA chains while AlkB–dsDNA protein makes interactions with one of the DNA chains only.10,11 PCA indicates that the duplex DNA in AlkBH2 is more rigid than in AlkB, arising from the above stated variation in the proteins’ interaction with dsDNA.
2.4. Mechanism of DNA Substrate Hydroxylation in AlkB–ssDNA, AlkB–dsDNA, and AlkBH2–dsDNA Complexes
Steps involved in the hydroxylation of the m3C DNA substrate (Scheme 4) comprise (i) hydrogen atom abstraction (HAT) followed by (ii) rebound hydroxylation. HAT involves the cleavage of the C—H bond in the substrate, which subsequently attacks the Fe(IV)–oxo group from the equatorial or axial position.25b,38 An electron is transferred from the substrate into the 3d-orbitals of the Fe(IV) center. HAT has been reported to occur via two possible channels for 2OG oxygenases: (i) a σ-channel and (ii) a π-channel25b,25c,38 (Figure 10). In the σ-channel, an α-spin electron shifts from σCH of the substrate to σ*z2 of the metal; this results in a TS that assumes a trajectory with linear Fe—O—H arrangement (approximately 180°). In the π-channel, the hydrogen atom approaches the Fe(IV)–oxo metal center in a sideways manner, leading to the transfer of a β-spin electron into the antibonding π* orbitals of the Fe(IV) center. This overlap results in a TS with an optimal Fe—O—H angle of roughly 120°. As the substrate approaches the Fe(IV)=O group, the Fe—O bond gradually elongates and polarizes to form the electron acceptor, a ferric-oxyl species.25b,25c,38
Scheme 4. Mechanism of Hydrogen Atom Abstraction and Rebound Hydroxylation Steps.

Figure 10.

Reaction channels for HAT by the Fe(IV)=O complexes.
2.4.1. Hydrogen Atom Abstraction (HAT)
The reactive Fe(IV)=O species (RC2) generated from the dioxygen activation phase abstracts a hydrogen atom from the methyl group of lesioned DNA substrate (m3C), leading to formation of the ferric-hydroxo (Fe(III)—OH) group and a methylene (R—•CH2) radical (IM10). The formation of IM10 passes through TS7.
2.4.1.1. Effects of the Conformational Flexibility on HAT
To explore the effects of conformational flexibility, QM/MM calculations were performed using five (5) well-equilibrated snapshots from the ferryl complex production MD trajectories. The snapshots were used to explore the effect of conformational variations on the potential energy barrier of the rate-determining hydrogen atom abstraction step. Calculations were carried out with the quintet spin state of the Fe(IV)–oxo intermediate because previous studies have shown the preference of non-heme iron enzymes for this spin state.15,23−26 Our calculations reveal the HAT to be the rate-determining step in substrate hydroxylation in agreement with other studies on non-heme iron enzymes.24,27−30 The calculated barriers at the B3LYP/def2-TZVP level (BS2) for the five snapshots vary between 24.9 and 27.5 kcal/mol without zero-point energy (ZPE) correction, and with ZPE, they vary between 21.6 and 24.7 kcal/mol (Table 1). The average barriers calculated using the Boltzmann weighted average39 were found to be 22.2 and 25.3 kcal/mol with and without ZPE, respectively. The Boltzmann weighted average has been employed in various QM/MM studies of enzyme reactions, where multiple snapshots have been used in the mechanistic studies31,39−43 in order to eliminate the contributions of unreasonably high barriers and stronger weighs the lower and more representative barriers.43 The calculated barrier is consistent with the experimentally derived value of 20.1 kcal/mol, calculated from the kcat value of 2.6 min–1 at 37 °C.44 In AlkB–ssDNA, the Boltzmann weighted average barriers are 25.6 and 21.9 kcal/mol, without and with ZPE, respectively, while 25.1 and 22.3 kcal/mol were obtained for AlkB–dsDNA without and with ZPE, respectively (Tables S1 and S3). Our calculated energy barriers match with previously reported HAT values for other similar non-heme iron enzyme models using QM/MM or DFT methods.24,27−30 Comparisons of key distances and angles in both RC2 and TS7 for all the five snapshots used for AlkBH2–dsDNA are presented in Table 1 and are included in the SI for both AlkB complexes (Tables S2 and S4). Subsequent calculations and molecular orbital analysis were performed using the snapshots giving the lowest barrier for the HAT.
Table 1. Selected Distances, Angles, And Barriers for the Different Snapshots of RC2 and TS7 for the HAT Step in AlkBH2–dsDNA, Calculated at the B3LYP/def2-TZVP Level.
| d(Fe–Op) (Å) | d(Op–H) (Å) | d(C–H) (Å) | <(Fe–Op–H) (deg) | <(C–Hp–O) (deg) | Csubstrate spin density in TS7 | barrier without ZPE (kcal/mol) | barrier with ZPE (kcal/mol) | |
|---|---|---|---|---|---|---|---|---|
| Snapshot 1 | ||||||||
| RC2 | 1.610 | 2.689 | 1.102 | 125.46 | 158.54 | –0.379 | ||
| TS7 | 1.744 | 1.233 | 1.313 | 145.15 | 169.88 | –0.379 | 24.9 | 21.6 |
| Snapshot 2 | ||||||||
| RC2 | 1.610 | 3.203 | 1.099 | 114.00 | 146.87 | –0.337 | ||
| TS7 | 1.778 | 1.285 | 1.281 | 141.89 | 166.29 | –0.337 | 27.5 | 24.7 |
| Snapshot 3 | ||||||||
| RC2 | 1.617 | 3.337 | 1.101 | 130.74 | 154.64 | –0.392 | ||
| TS7 | 1.750 | 1.239 | 1.321 | 151.36 | 173.07 | –0.392 | 25.2 | 22.6 |
| Snapshot 4 | ||||||||
| RC2 | 1.613 | 2.737 | 1.102 | 130.18 | 172.29 | –0.377 | ||
| TS7 | 1.743 | 1.242 | 1.307 | 148.76 | 176.37 | –0.377 | 26.3 | 23.3 |
| Snapshot 5 | ||||||||
| RC2 | 1.615 | 2.522 | 1.102 | 127.94 | 155.59 | –0.369 | ||
| TS7 | 1.739 | 1.260 | 1.305 | 148.99 | 170.17 | –0.369 | 24.9 | 22.0 |
| energy barrier Boltzmann weighted average | 25.3 | 22.2 | ||||||
2.4.1.2. Molecular Orbital Interactions Driving σ- and π-Pathways
During hydrogen atom abstraction, the substrate C–H bond cleaves and reacts with the Fe(IV)–oxo group, leading to the elongation of the Fe–Op bond and its polarization to form an Fe(III)-oxyl radical (Fe(III)–O•–) at TS7. The obtained spin density of 4.00 for Fe at the TS7 confirms the 3+ oxidation state for the Fe center. Figure 11 shows the electron shift with orbital occupations in RC2 and TS7 in HAT. In RC2, the σ*x2–y2 orbital is half filled, and σ*z2 is a virtual orbital. An α-electron is transferred from the substrate (σCH) into the antibonding σ*z2 orbital located along the Fe–O axis to give the radical carbon IM10 intermediate. HAT proceeds via a σ-channel, and the calculated spin density for the carbon atom of the substrate at TS7 varies between −0.392 and −0.337 in all the snapshots. These observations support transfer of an α-electron to the 3d orbital of the Fe metal (Table 1). The Fe–Op–H angle in all the snapshots varies between 141.89° and 151.36° at the transition state (TS7) (Table 1); i.e., it deviates from 180°, likely because of constraints in geometry as the m3C DNA substrate cannot move freely in the protein environment. Previous studies on other non-heme iron enzymes have also reported an analogous Fe–Op–H angle that deviates from 180°, but which still proceeds via the σ-channel for hydrogen atom transfer.28,29,32
Figure 11.

Orbital occupancy diagram during hydrogen atom abstraction.
With both AlkB–ssDNA and AlkB–dsDNA, the HAT transition state (TS7) Fe–Op–H angle in all the snapshots varies between 118.37° and 145.55° (AlkB–ssDNA) and 129.88° and 139.31° (AlkB–dsDNA) (Tables S2 and S4). These results and the calculated spin densities (Table S4) at the carbon of the m3C substrate, which vary between −0.342 and −0.314, imply that AlkB–dsDNA hydrogen atom transfer can also proceed via a σ-channel, with the transfer of an α-electron from the substrate to the 3d orbitals of the Fe center. However, with AlkB–ssDNA, the calculated spin density for the carbon varies between −0.366 and 0.469 (Table S2). This implies that there is a competition between σ- and π-channels, as proposed in previous studies on AlkB.28 The observed σ and π competition in the hydrogen atom transfer channel in AlkB–ssDNA could be due to reduced steric constraints experienced by m3C substrate in AlkB–ssDNA when compared to the substrate in duplex DNA, so that it can relatively more easily adopt a conformation which can undergo hydrogen abstraction via both σ- and π-channels. With AlkBH2–dsDNA and AlkB–dsDNA, m3C is more restrained due to the duplex nature of the dsDNA as the second strand of the DNA enhances the stability of the strand that contains the substrate, making the HAT proceed only through the σ-channel.
2.4.1.3. Effects of Residues from the Second Sphere and beyond on HAT
In AlkBH2–dsDNA, the transition state is stabilized by second sphere residues. Thr252, Arg110, and Tyr122 stabilize the noncoordinating oxygen of succinate, O2 of the cytosine ring of the m3C substrate (hydrogen bonding interaction), and the exocyclic amine (N4) of the substrate (hydrogen bonding interaction), respectively. Further, a strong π-stacking interaction of Phe124 with the cytosine ring of the substrate enhances the stability and proper orientation of the substrate. Networks of hydrogen bonding interactions of second sphere residues, Glu175, Arg254, and Tyr122 enhance the stability of the TS. These residues are in the vicinity of the Fe center and the substrate. However, in both AlkB–ssDNA and AlkB–dsDNA the transition states are stabilized by T-shaped π-stacking interactions between Trp69 and Tyr76 and the cytosine ring of the substrate; the Fe center is stabilized via the hydrogen bonding interactions of Arg210 with the noncoordinating oxygen of the coordinating aspartate. The residues that stabilize the TS for HAT in the AlkBH2–dsDNA show positive correlated motions with the Fe, metal coordinating residues (His171, Asp173, and His236), substrate recognition lid residues, and the double-stranded β-helix (DSBH) core residues, while in bacterial AlkB (both with ss- and dsDNA), the residues show positive correlation with Fe, metal ligating residues, and DSBH residues, indicating that more second sphere residues and stronger correlation motions participate in the overall stabilization of TS7 in human homologue than in bacterial AlkB and thus confirm their importance in catalysis. The finding suggests that while the three systems follow the same overall mechanism, modification of the orientation of second sphere residues in the substrate binding lid could influence HAT in AlkBH2, whereas such modifications are less likely to affect AlkB. Thus, although the details are complex, targeting second sphere residues could be a way to obtain selective inhibitors for specific 2OG–oxygenases.
At the IM10 stage, the Fe(IV)=O species is completely reduced to the Fe(III)—OH intermediate with the generation of a substrate carbon radical. The formation of IM10 is slightly endergonic with the energy of 6.5 kcal/mol, including ZPE. The Fe—Op and Op—H distances are 1.85 and 0.95 Å, respectively, confirming the formation of the Fe(III)—OH complex. The calculated spin density of 4.24 for the Fe center supports the 3+ oxidation state assignment for iron at this stage. The optimized reaction state geometries for the HAT and the spin natural orbitals (SNOs) of the HAT transition states for the snapshots with the lowest barrier in AlkBH2–dsDNA and AlkBs are presented in Figures 12 and 13, and Figures S47 and S48, respectively.
Figure 12.

Geometries of the stationary points along the hydrogen atom abstraction step in AlkBH2–dsDNA. Distances (Å) and spin densities are in black and pink, respectively.
Figure 13.

Spin natural orbitals (SNOs) with their respective populations (in parentheses) for the hydrogen atom abstraction transition state in the AlkBH2–dsDNA complex.
2.4.2. Mechanism of Rebound Hydroxylation
The radical (IM10) formed by HAT undergoes a radical rebound process via TS8, in which a hydroxyl (OH) group is transferred from the Fe(III)–OH complex to the radical, leading to reduction of Fe(III) to Fe(II) and formation of the hydroxylated product (PD). The rebound reaction occurs rapidly in AlkBH2–dsDNA with a barrier of 12.6 kcal/mol, and the overall reaction is highly exothermic (−31.6 kcal/mol), indicating that the formation of the product (PD) is both thermodynamically and kinetically favored. The rebound barriers in AlkB–ssDNA and AlkB–dsDNA are 17.4 and 15.8 kcal/mol, respectively. These barriers imply that the rebound process in both AlkB enzymes occurs at a slower rate than with AlkBH2–dsDNA. The slightly higher rebound barrier found in AlkB–ssDNA compared to AlkB–dsDNA could be due to the fact that the Fe(III)-linked OH group in IM10 makes a hydrogen bonding interaction with the iron-coordinating Asp133 in agreement with previous studies.28,29 The hydroxylated products (PD) in both AlkB complexes are stable with energies of −23.1 and −33.9 kcal/mol in AlkB–ssDNA and AlkB–dsDNA, respectively. In the PD, the hydroxyl group of the hydroxylated product forms a hydrogen bonding interaction with the iron-ligating aspartate in AlkBH2–dsDNA and AlkB–ssDNA. With AlkBH2–dsDNA, PD is further stabilized via a strong π-stacking interaction of the substrate base with Phe124. The exocyclic amine (N4) of the substrate forms a strong hydrogen bonding interaction with Glu175 and Tyr122, and the O2 of the nucleobase ring (cytosine) forms a stable interaction with Arg110; in both AlkB–ssDNA and AlkB–dsDNA, the stability of the product is enhanced by interaction with Tyr76. The obtained distances and spin densities are in good agreement with reported values.24,28,29,31−33 The reaction state geometries for TS8 and PD as well as the substrate hydroxylation step energy profile for AlkBH2–dsDNA are presented in Figure S49 and Figure 14, respectively.
Figure 14.

QM/MM reaction profile for the substrate hydroxylation step by AlkBH2–dsDNA as calculated using UB3LYP/def2-TZVP (BS2) (in red) and BS2 with ZPE (in black). Relative energies are in kcal/mol.
3. Computational Methods
3.1. Model Preparation
X-ray crystal structures of AlkBH2–dsDNA, AlkB–dsDNA, and AlkB–ssDNA (PDB codes 3RZJ,103O1M,45 and 3I49,46 respectively, in complex with 3-methylcytosine (m3C) substrates) were used. Crystallographically unobserved residues from the loop region (residues 204 to 206) of AlkBH2–dsDNA were added using Modeller.47 This process was followed by replacement of the Mn(II) used for crystallization with Fe(II) in the AlkBH2–dsDNA and AlkB–dsDNA structures using GaussView 6.0. The protonation states of the ionizable side chains were assessed with the H++ server48a and with PROPKA software (Table S5);48b the histidine residues that are coordinated to the Fe(II) center were assigned protonation states based on visual inspection of their local environments.
The Fe(II) center is in the high spin state (S = 2, M = 5);15−17 the octahedrally coordinated Fe binds 2OG (in a bidentate fashion), two histidines (His1, His2), and one aspartate residue. The sixth coordination site in AlkB enzymes is occupied by a water in the crystal structures;10,11 this was substituted with a dioxygen (O2), bound in an end on manner, to give an Fe(III)–superoxo complex. The parameters for the active site were prepared using the Metal Center Parameter Builder (MCPB.py)49 as implemented in Amber. The metal center parameters were derived based on the bonded and electrostatic model approach in which the coordinating ligands are connected to the metal through covalent bonds. The bond and the angle force constants were derived using the Seminario method;50 point charge parameters for the electrostatic potential were obtained using the ChgModB method. MCPB tools have been successfully used for description of the mononuclear non-heme iron center and iron–sulfur Rieske cluster.34c,51,52 Molecular dynamics simulations using parameters generated via the MCPB tool have successfully reproduced the crystallographically observed geometry of metal–ligand complexes for other 2OG oxygenases.34c,51,52 The parameters for 2OG and the substrate (m3C) were generated using the Antechamber module of Amber 16.53 The Leap module in Amber was used to add missing hydrogen atoms, and the protein systems were neutralized using counterions: Na+ for AlkBH2–dsDNA and AlkB–dsDNA and Cl– for AlkB–ssDNA, to compensate for the negative charge of the AlkBH2–dsDNA and AlkB–dsDNA systems, and the positive charge of the AlkB–ssDNA system. The systems were surrounded by a rectangular box solvated with Transferable Intermolecular Potential 3-Point (TIP3P) water molecules54 within a distance of at least 10 Å from the surface of the proteins.
A two-stage minimization of the geometries using MM was performed to eliminate clashes and bad contacts, prior to the MD simulations. In the first stage of minimization, only water molecules and ions were minimized, while the solute molecules were restrained with a 500 kcal/(mol Å2) harmonic potential. In the final stage of minimization, all atoms were optimized with no restraints. The systems were subjected to 5000 steps of steepest descent, followed by 5000 steps of conjugate gradient energy minimization. The minimization steps were done with the CPU version of SANDER in Amber16.
Similar procedures were used to prepare the parameters for the ferryl complex of the three enzyme–DNA complexes, where the 2OG cosubstrate was substituted with succinate, which was modeled with monodentate carboxylate binding.
3.2. Molecular Dynamic Simulations
Molecular dynamics simulations were performed using the GPU version55 of the PMEMD engine integrated with Amber16.56 The FF14SB57 force field was used in all the simulations, and periodic boundary conditions were employed in all simulations. Long-range electrostatic interactions were calculated using the Particle Mesh Ewald (PME) method58 with a direct space and vdW cutoff of 10 Å. The minimized systems were first heated for 50 ps by linearly increasing the temperature from 0 to 300 K in a canonical ensemble (NVT) using the Langevin thermostat.59 The heated systems were further subjected to constant temperature heating (at 300 K) for 1 ns in an NPT ensemble. The solute molecules were restrained with the harmonic potential of 10 kcal/(mol Å2) during the heating processes. Thereafter, systems were equilibrated within an NPT ensemble at a fixed temperature and pressure of 300 K and 1 bar, respectively, for 3 ns without any restraints on solute molecules. The MD productive runs were performed for 1 μs in an NPT ensemble with a target pressure set at 1 bar and constant pressure coupling of 2 ps. The pressure was held constant using the Berendsen barostat,60 and the SHAKE algorithm61 was used to constrain the bond lengths of those bonds involving hydrogen atoms.
Trajectories were analyzed using CPPTRAJ,62 VMD,63 UCSF Chimera,64 and R (Bio3D).65 The analyses of the root mean square deviation (RMSD) of the enzymes with respect to the minimized crystal structure, root mean square fluctuations (RMSFs), electrostatic interactions, and hydrogen bonding were performed. The Bio3D package in R was used to produce principal component analysis (PCA) and dynamic cross correlation analysis (DCCA) as used in the previous studies.34c,51
3.3. QM/MM Calculations
All QM/MM calculations were performed using the ChemShell package,66 that combines Turbomole67 and DL_POLY.68 The former was used for the QM region while the MM region was treated with the latter. The electronic embedding scheme, which includes the polarization of the QM region by the MM charge distribution in the QM calculation, was used to describe the interaction between the QM and MM regions. Hydrogen atoms were used as the linked atoms to complete valences of bonds spanning between the two regions. The MM region was described with the Amber force field, and the QM part was accounted for with density functional theory (DFT) using the unrestricted B3LYP (UB3LYP) functional. The QM region used for the Fe(III)–superoxo complex involves the non-heme iron center, its coordinating residues (two His (His1 and His2), Asp, 2OG, and O2), the methylguanidinium group of Arg (Arg254 in AlkBH2–dsDNA and Arg210 in AlkBs), and the 3-methyl-cytosine part of the substrate (Figure 3). In the ferryl complex, 2OG and O2 were replaced with succinate and an oxygen atom, respectively. All geometry optimizations were performed with the def2-SVP basis set (labeled as BS1) for all the atoms. After the full geometry optimizations, linear transit scans along the reaction coordinate were performed with 0.1 Å increments to locate the transition states (the geometry that corresponds to the highest energy in the PES) using DL-find optimizer.69 Transition states were reoptimized using the partitioned rational function optimization (P-RFO) algorithm implemented in the HDLC code.70 The fully optimized geometries of the minima and the transition states were characterized via frequency calculations. The energies of the optimized stationary points were recalculated via single-point energy calculation using a larger basis set, def2-TZVP (labeled as BS2), for all the atoms.
4. Conclusions
Our MD and QM/MM computations inform on the roles of dynamics in influencing the selectivity and mechanisms of methylated DNA repair by AlkB and its human homologue AlkBH2. Based on the general mechanism of 2OG oxygenases, reaction path calculations of dioxygen activation and substrate oxidation steps were performed. Dioxygen activation involves oxidative decarboxylation of the 2OG cosubstrate to succinate, leading to formation of the Fe(IV)=O intermediate. Following dioxygen activation, decarboxylation is the rate-limiting step, with barriers of 11.3, 11.8, and 13.2 kcal/mol for AlkB–ssDNA, AlkBH2–dsDNA, and AlkB–dsDNA, respectively. The DCCA implies that the correlated motions of the residues that stabilize the decarboxylation process transition state are more important for catalysis in AlkBH2–dsDNA and AlkB–ssDNA than in AlkB–dsDNA. We also explored the 2OG rearrangement and “ferryl flip” mechanistic paths in the formation of the reactive Fe(IV)=O intermediate from the “off-line” binding mode of the 2OG cosubstrate in the enzymes. The calculations reveal that 2OG rearrangement is more energetically viable than the “ferryl flip”.
Prior to the QM/MM calculations, we performed a series of 1 μs MD simulations on both the Fe(III)–superoxo and the ferryl complexes. The dynamics studies reveal the importance of conformational flexibility of the DNA component in the overall motion of the protein–DNA complexes. They also reveal flexibility in the binding of succinate compared to 2OG, likely reflecting preferred binding of the 2OG cosubstrate compared to the succinate coproduct. Residues Tyr161 and Arg248 (AlkBH2–dsDNA), Tyr122 and Arg204 (AlkB–ssDNA), and Ser145 and Trp178 (AlkB–dsDNA) are involved in binding 2OG and the succinate product, while His171, Tyr122, and Phe124 (AlkBH2–dsDNA) and Trp69, Tyr76, and His131 (AlkBs) enhance DNA substrate binding. MD studies also reveal the importance of the hydrophobic β-hairpin in AlkBH2–dsDNA as these residues have a strong positive correlation with the substrate recognition lid residues, and such correlated motion might contribute significantly to the binding of the substrate. The dynamics studies reveal that this hydrophobic β-hairpin is more flexible in the ferryl complex than in the Fe(III)–superoxo complex. In the ferryl complex, it moves toward the Fe center, leading to the compaction of the complex and, hence, a mode that favors catalysis. The results also reveal that AlkB–ssDNA is more structurally compact than AlkB–dsDNA with the protein in the latter only interacting with one of the duplex DNA strands. The m3C DNA substrate binds better to the active site of the protein in the ferryl compared to the Fe(III)–superoxo complex, resulting in a more productive complex for the substrate oxidation step.
After dioxygen activation, the reactive Fe(IV)=O species enables hydrogen atom abstraction (HAT) of the substrate methyl group to give a methylene radical intermediate which subsequently undergoes rebound hydroxylation to give the hydroxylated product. The QM/MM calculations imply that HAT is rate-limiting for substrate oxidation in agreement with studies on some 2OG oxygenases.27−30 However, if one considers the entire catalytic cycle in 2OG–oxygenases, including substrate binding and the product release, the rate-determining step can vary and, at least in some cases, is dissociation of the enzyme product complex.19
To explore the effect of conformational variations of the protein on the energy barrier, we used multiple snapshots (5 snapshots for each of the systems studied) to investigate the HAT for the three systems. The results reveal that the hydrogen abstraction occurs via the σ-channel in both AlkBH2–dsDNA and AlkB–dsDNA. With AlkB–ssDNA, the σ- and π-channels compete, due to the reduced steric constraints experienced by the m3C DNA substrate in AlkB–ssDNA when compared to AlkBH2–dsDNA and AlkB–dsDNA. The rebound hydroxylation occurs at a faster rate than HAT, resulting in hydroxylated products that are both thermodynamically and kinetically stable.
The studies reported here provide in-depth insight into the relationship between dynamic behaviors and the catalytic mechanisms of AlkB–ssDNA, AlkB–dsDNA, and AlkBH2–dsDNA. Various 2OG oxygenases are current targets for medicinal chemistry, with the vast majority of inhibitors reported being active site Fe chelators/2OG competitors, resulting in blockage of catalysis.71 Our results on the roles of dynamics in catalysis suggest that analogous studies aimed at understanding the modes of action of active site binding inhibitors, including those in clinical/agrochemical use and trials, may be productive.71 In addition, inhibitors that bind to other regions of the protein (for example, substrate binding lid region) might provide better selectivity for a desired AlkB target over the other analogous enzymes. Our studies have suggested potential targets in this regard.
At least in some cases there is a desire to identify allosteric inhibitors or even compounds enhancing 2OG oxygenase activity. Indeed, in some cases structure–activity relationships for 2OG oxygenase inhibitors are difficult to rationalize on the basis of active site interactions alone, especially those observed by “static” crystallography.72 The dynamic and selective roles of the different regions of the overall enzyme–substrate complexes in substrate recognition and catalysis by the 2OG oxygenase–substrate complexes imply that there is scope for identifying modulators that act on specific steps during catalysis.
Acknowledgments
This research was supported by a Michigan Technological University Graduate Teaching Assistantship to S.O.W. and start-up grants to C.Z.C. and T.G.K.-C. C.J.S. thanks Cancer Research UK (C8717/A18245), the EPRC, and the Wellcome Trust (106244/Z/14/Z) for support. C.Z.C. acknowledges NSF grant 1904215 (testing the methodology).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscentsci.0c00312.
Analysis of molecular dynamics and QM/MM optimized QM geometries with spin densities and Cartesian coordinates of the QM region in QM/MM calculations (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Clifton I. J.; McDonough M. A.; Ehrismann D.; Kershaw N. J.; Granatino N.; Schofield C. J. Structural studies on 2-oxoglutarate oxygenases and related double-stranded β-helix fold proteins. J. Inorg. Biochem. 2006, 100 (4), 644–669. 10.1016/j.jinorgbio.2006.01.024. [DOI] [PubMed] [Google Scholar]
- Aik W.; Demetriades M.; Hamdan M. K. K.; et al. Structural Basis for Inhibition of the Fat Mass and Obesity Associated Protein (FTO). J. Med. Chem. 2013, 56 (9), 3680–3688. 10.1021/jm400193d. [DOI] [PubMed] [Google Scholar]
- Loenarz C.; Ge W.; Coleman M. L.; Rose N. R.; Cooper C. D.; Klose R. J.; Ratcliffe P. J.; Schofield C. J. PHF8, a gene associated with cleft lip/palate and mental retardation, encodes for an N 1-dimethyl lysine demethylase. Hum. Mol. Genet. 2010, 19 (2), 217–222. 10.1093/hmg/ddp480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schofield C. J.; Zhang Z. Structural and mechanistic studies on 2-oxoglutarate-dependent oxygenases and related enzymes. Curr. Opin. Struct. Biol. 1999, 9 (6), 722–731. 10.1016/S0959-440X(99)00036-6. [DOI] [PubMed] [Google Scholar]
- Samson L.; Cairns J. A new pathway for DNA repair in Escherichia coli. Nature 1977, 267 (5608), 281–283. 10.1038/267281a0. [DOI] [PubMed] [Google Scholar]
- Fedeles B. I.; Singh V.; Delaney J. C.; Li D.; Essigmann J. M. The AlkB Family of Fe(II)/α-Ketoglutarate-dependent Dioxygenases: Repairing Nucleic Acid Alkylation Damage and Beyond. J. Biol. Chem. 2015, 290 (34), 20734–20742. 10.1074/jbc.R115.656462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi C.; He C. DNA Repair by Reversal of DNA Damage. Cold Spring Harbor Perspect. Biol. 2013, 5 (1), a012575–a012575. 10.1101/cshperspect.a012575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aravind L.; Koonin E. V. The DNA-repair protein AlkB, EGL-9, and leprecan define new families of 2-oxoglutarate- and iron-dependent dioxygenases. Genome Biol. 2001, 2 (3), research0007.1. 10.1186/gb-2001-2-3-research0007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedgwick B.; Bates P.; Paik J.; Jacobs S.; Lindahl T. Repair of alkylated DNA: Recent advances. DNA Repair 2007, 6 (4), 429–442. 10.1016/j.dnarep.2006.10.005. [DOI] [PubMed] [Google Scholar]
- Yi C.; Chen B.; Qi B.; Zhang W.; Jia G.; Zhang L.; Li C. J.; Dinner A. R.; Yang C. G.; He C. Duplex interrogation by a direct DNA repair protein in search of base damage. Nat. Struct. Mol. Biol. 2012, 19 (7), 671–676. 10.1038/nsmb.2320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang C.-G.; Yi C.; Duguid E. M.; Sullivan C. T.; Jian X.; Rice P. A.; He C. Crystal structures of DNA/RNA repair enzymes AlkB and ABH2 bound to dsDNA. Nature 2008, 452 (7190), 961–965. 10.1038/nature06889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Zheng G.; Fu Y.; He C. Nucleic Acid Oxidation in DNA Damage Repair and Epigenetics. Chem. Rev. 2014, 114 (8), 4602–4620. 10.1021/cr400432d. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Chaturvedi S. S.; Ramanan R.; Waheed S. O.; Karabencheva-Christova T. G.; Christov C. Z. Structure-function relationships in KDM7 histone demethylases. Adv. Protein Chem. Struct. Biol. 2019, 117, 113–125. 10.1016/bs.apcsb.2019.08.005. [DOI] [PubMed] [Google Scholar]
- a Hausinger R. P. Fe(II)/α-Ketoglutarate-Dependent Hydroxylases and Related Enzymes. Crit. Rev. Biochem. Mol. Biol. 2004, 39 (1), 21–68. 10.1080/10409230490440541. [DOI] [PubMed] [Google Scholar]; b Miller III B. R.; McGee T. D. Jr.; Swails J. M.; Homeyer N.; Gohlke H.; Roitberg A. E. MMPBSA.py: An efficient program for end-state energy calculations. J. Chem. Theory Comput. 2012, 8 (9), 3314–3321. 10.1021/ct300418h. [DOI] [PubMed] [Google Scholar]; c Genheden S.; Ryde U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discovery 2015, 10 (5), 449–461. 10.1517/17460441.2015.1032936. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Hou T.; Wang J.; Li Y.; Wang W. Assessing the performance of the MM/PBSA and MM/GBSA methods. 1. The accuracy of binding free energy calculations based on molecular dynamics simulations. J. Chem. Inf. Model. 2011, 51 (1), 69–82. 10.1021/ci100275a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu C.; Yi C. Switching demethylation activities between AlkB family RNA/DNA demethylases through exchange of active-site residues. Angew. Chem., Int. Ed. 2014, 53 (14), 3659–3662. 10.1002/anie.201310050. [DOI] [PubMed] [Google Scholar]
- Krebs C.; Fujimori D. G.; Walsh C. T.; Bollinger J. M. Non-heme Fe(IV)-oxo intermediates. Acc. Chem. Res. 2007, 40 (7), 484–492. 10.1021/ar700066p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomon E. I. Geometric and Electronic Structure Contributions to Function in Bioinorganic Chemistry: Active Sites in Non-Heme Iron Enzymes. Inorg. Chem. 2001, 40 (15), 3656–3669. 10.1021/ic010348a. [DOI] [PubMed] [Google Scholar]
- Solomon E. I.; Light K. M.; Liu L. V.; Srnec M.; Wong S. D. Geometric and Electronic Structure Contributions to Function in Non-heme Iron Enzymes. Acc. Chem. Res. 2013, 46 (11), 2725–2739. 10.1021/ar400149m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Zhang Z.; Ren J.; Harlos K.; McKinnon C. H.; Clifton I. J.; Schofield C. J. Crystal structure of a clavaminate synthase-Fe(II)-2-oxoglutarate-substrate-NO complex: evidence for metal centred rearrangements. FEBS Lett. 2002, 517, 7–12. 10.1016/S0014-5793(02)02520-6. [DOI] [PubMed] [Google Scholar]; b Chaturvedi S. S.; Ramanan R.; Lehnert N.; Schofield C. J.; Karabencheva-Christova T. G.; Christov C. Z. Catalysis by the Non-heme Fe(II) Histone Demethylase PHF8 Involves Iron Center Rearrangement and Conformational Modulation of Substrate Orientation. ACS Catal. 2020, 10 (2), 1195–1209. 10.1021/acscatal.9b04907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shishodia S.; Zhang D.; El-Sagheer A. H.; Brown T.; Claridge T. D. W.; Schofield C. J.; Hopkinson R. J. NMR analyses on N-hydroxymethylated nucleobases - implications for formaldehyde toxicity and nucleic acid demethylases. Org. Biomol. Chem. 2018, 16, 4021–4032. 10.1039/C8OB00734A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Solomon E. I.; Brunold T. C.; Davis M. I.; Kemsley J. N.; Lee S. K.; Lehnert N.; Neese F.; Skulan A. J.; Yang Y. S.; Zhou J. Geometric and Electronic Structure/Function Correlations in Non-Heme Iron Enzymes. Chem. Rev. 2000, 100 (1), 235–350. 10.1021/cr9900275. [DOI] [PubMed] [Google Scholar]; b Mitchell A. J.; Dunham N. P.; Martinie R. J.; Bergman J. A.; Pollock C. J.; Hu K.; Allen B. D.; Chang W.; Silakov A.; Bollinger J. M.; Krebs C.; Boal A. K. Visualizing the reaction cycle in an Iron(II)- and 2-(oxo)-glutarate dependent hydroxylase. J. Am. Chem. Soc. 2017, 139, 13830–13836. 10.1021/jacs.7b07374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B.; Cao Z.; Sharon D. A.; Shaik S. Computations reveal a rich mechanistic variation of demethylation of N-methylated DNA/RNA nucleotides by FTO. ACS Catal. 2015, 5, 7077–7090. 10.1021/acscatal.5b01867. [DOI] [Google Scholar]
- a Wang B.; Usharani D.; Li C.; Shaik S. Theory uncovers an unusual mechanism of DNA repair of a lesioned adenine by AlkB enzymes. J. Am. Chem. Soc. 2014, 136 (39), 13895–13901. 10.1021/ja507934g. [DOI] [PubMed] [Google Scholar]; b Torabifard H.; Cisneros G. A. Computational investigation of O2 diffusion through an intra-molecular tunnel in AlkB; Influence of polarization on O2 transport. Chem. Sci. 2017, 8 (9), 6230–6238. 10.1039/C7SC00997F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song X.; Lu J.; Lai W. Mechanistic insights into dioxygen activation, oxygen atom exchange and substrate epoxidation by AsqJ dioxygenase from quantum mechanical/molecular mechanical calculations. Phys. Chem. Chem. Phys. 2017, 19 (30), 20188–20197. 10.1039/C7CP02687K. [DOI] [PubMed] [Google Scholar]
- Liu H.; Llano J.; Gauld J. W. A DFT study of nucleobase dealkylation by the DNA repair enzyme AlkB. J. Phys. Chem. B 2009, 113 (14), 4887–4898. 10.1021/jp810715t. [DOI] [PubMed] [Google Scholar]
- a Wójcik A.; Radoń M.; Borowski T. Mechanism of O2 Activation by α-Ketoglutarate Dependent Oxygenases Revisited. A Quantum Chemical Study. J. Phys. Chem. A 2016, 120 (8), 1261–1274. 10.1021/acs.jpca.5b12311. [DOI] [PubMed] [Google Scholar]; b Geng C.; Ye S.; Neese F. Analysis of reaction channels for alkane hydroxylation by Nonheme Iron(IV)-oxo complexes. Angew. Chem., Int. Ed. 2010, 49, 5717–5720. 10.1002/anie.201001850. [DOI] [PubMed] [Google Scholar]; c Neidig M. L.; Decker A.; Choroba O. W.; Huang F.; Kavana M.; Moran G. R.; Spencer J. B.; Solomon E. I. Spectroscopic and electronic structure studies of aromatic electrophilic attack and hydrogen atom abstraction by non-heme iron enzymes. Proc. Natl. Acad. Sci. U. S. A. 2006, 103 (35), 12966–12973. 10.1073/pnas.0605067103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortopassi W. A.; Simion R.; Honsby C. E.; França T. C. C.; Paton R. S. Dioxygen Binding in the Active Site of Histone Demethylase JMJD2A and the Role of the Protein Environment. Chem. - Eur. J. 2015, 21 (52), 18983–18992. 10.1002/chem.201502983. [DOI] [PubMed] [Google Scholar]
- Cisneros G. A. DFT study of a model system for the dealkylation step catalyzed by AlkB. Interdiscip. Sci.: Comput. Life Sci. 2010, 2 (1), 70–77. 10.1007/s12539-010-0092-z. [DOI] [PubMed] [Google Scholar]
- Quesne M. G.; Latifi R.; Gonzalez-Ovalle L. E.; Kumar D.; De Visser S. P. Quantum mechanics/molecular mechanics study on the oxygen binding and substrate hydroxylation step in AlkB repair enzymes. Chem. - Eur. J. 2014, 20 (2), 435–446. 10.1002/chem.201303282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang D.; Lord R. L.; Cisneros G. A. Ab initio QM/MM calculations show an intersystem crossing in the hydrogen abstraction step in dealkylation catalyzed by AlkB. J. Phys. Chem. B 2013, 117 (21), 6410–6420. 10.1021/jp403116e. [DOI] [PubMed] [Google Scholar]
- Torabifard H.; Cisneros G. A. Insight into wild-type and T1372E TET2-mediated 5hmC oxidation using ab initio QM/MM calculations. Chem. Sci. 2018, 9 (44), 8433–8445. 10.1039/C8SC02961J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Álvarez-Barcia S.; Kästner J. Atom Tunneling in the Hydroxylation Process of Taurine/α-Ketoglutarate Dioxygenase Identified by Quantum Mechanics/Molecular Mechanics Simulations. J. Phys. Chem. B 2017, 121 (21), 5347–5354. 10.1021/acs.jpcb.7b03477. [DOI] [PubMed] [Google Scholar]
- Fang D.; Cisneros G. A. Alternative pathway for the reaction catalyzed by DNA dealkylase AlkB from Ab initio QM/MM calculations. J. Chem. Theory Comput. 2014, 10 (11), 5136–5148. 10.1021/ct500572t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godfrey E.; Porro C. S.; de Visser S. P. Comparative Quantum Mechanics/Molecular Mechanics (QM/MM) and density functional theory calculations on the oxo-iron species of taurine/α- ketoglutarate dioxygenase. J. Phys. Chem. A 2008, 112 (11), 2464–2468. 10.1021/jp710999v. [DOI] [PubMed] [Google Scholar]
- a Purslow J. A.; Nguyen T. T.; Egner T. K.; Dotas R. R.; Khatiwada B.; Venditti V. Active site breathing of human Alkbh5 revealed by solution NMR and accelerated molecular dynamics. Biophys. J. 2018, 115 (10), 1895–1905. 10.1016/j.bpj.2018.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Silvestrov P.; Muller T. A.; Clark K. N.; Hausinger R. P.; Cisneros G. A. Homology modeling molecular dynamics, and site-directed mutagensis study of AlkB human homolog 1 (ALKBH1). J. Mol. Graphics Modell. 2014, 54, 123–130. 10.1016/j.jmgm.2014.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Waheed S. O.; Ramanan R.; Chaturvedi S. S.; Ainsley J.; Evison M.; Ames J. M.; Schofield C. J.; Christov C. Z.; Karabencheva-Christova T. G. Conformational flexibility influences structure-function relationships in nucleic acid N-methyl demethylases. Org. Biomol. Chem. 2019, 17 (8), 2223–2231. 10.1039/C9OB00162J. [DOI] [PubMed] [Google Scholar]
- a Bleijlevens B.; Shivarattan T.; van de Boom K. S.; de Haan A.; van der Zwan G.; Simpson P. J.; Mathews S. J. Changes in protein dynamics of the DNA repair dioxygenase AlkB upon binding of Fe(2+) and 2-oxoglutarate. Biochemistry 2012, 51, 3334–3341. 10.1021/bi201699e. [DOI] [PubMed] [Google Scholar]; b Bleijlevens B.; Shivarattan T.; Flashman E.; Yang Y.; Simpson P. J.; Koivisto P.; Sedgwick B.; Schofield C. J.; Mathews S. J. Dynamic states of the DNA repair enzyme AlkB regulate product release. EMBO Rep. 2008, 9, 872–877. 10.1038/embor.2008.120. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Pang X.; Han K.; Cui Q. A simple but effective modeling strategy for structural properties of non-heme Fe(II) sites in proteins: Test of force field models and application to proteins in the AlkB family. J. Comput. Chem. 2013, 34 (19), 1620–1635. 10.1002/jcc.23305. [DOI] [PubMed] [Google Scholar]; d Ergel B.; Gill M. L.; Brown L.; Yu B.; Palmer A. G. 3rd; Hunt J. F. Protein dynamics control the progression and efficiency of the catalytic reaction cycle of the Escherichia coli DNA-repair enzyme AlkB. J. Biol. Chem. 2014, 289, 29584–29601. 10.1074/jbc.M114.575647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee D. H.; Jin S. G.; Cai S.; Chen Y.; Pfeifer G. P.; O’Connor T. R. Repair of methylation damage in DNA and RNA by mammalian AlkB homologues. J. Biol. Chem. 2005, 280 (47), 39448–39459. 10.1074/jbc.M509881200. [DOI] [PubMed] [Google Scholar]
- a Hünenberger P. H.; Mark A. E.; van Gunsteren W. F. Fluctuation and Cross-Correlation Analysis of Protein Motions Observed in Nanosecond Molecular Dynamics Simulations. J. Mol. Biol. 1995, 252, 492–503. 10.1006/jmbi.1995.0514. [DOI] [PubMed] [Google Scholar]; b Balsera M. A.; Wriggers W.; Oono Y.; Schulten K. Principal Component Analysis and Long Time Protein Dynamics. J. Phys. Chem. 1996, 100, 2567–2572. 10.1021/jp9536920. [DOI] [Google Scholar]
- Shaik S.; Chen H.; Janardanan D. Exchange-enhanced reactivity in bond activation by metal-oxo enzymes and synthetic reagents. Nat. Chem. 2011, 3, 19–26. 10.1038/nchem.943. [DOI] [PubMed] [Google Scholar]
- Logunov I.; Schulten K. Quantum chemistry: Molecular dynamics study of the dark-adaptation process in bacteriorhodopsin. J. Am. Chem. Soc. 1996, 118 (40), 9727–9735. 10.1021/ja953091m. [DOI] [Google Scholar]
- Christov C. Z.; Lodola A.; Karabencheva-Christova T. G.; Wan S.; Coveney P. V.; Mulholland A. J. Conformational effects on the pro-s hydrogen abstraction reaction in Cyclooxygenase-1: An integrated QM/MM and MD study. Biophys. J. 2013, 104 (5), L5–L7. 10.1016/j.bpj.2013.01.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonsdale R.; Hoyle S.; Grey D. T.; Ridder L.; Mulholland A. J. Determinants of reactivity and selectivity in soluble epoxide hydrolase from quantum mechanics/molecular mechanics modeling. Biochemistry 2012, 51 (8), 1774–1786. 10.1021/bi201722j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonsdale R.; Houghton K. T.; Zurek J.; Bathelt C. M.; Foloppe N.; de Groot M. J.; Harvey J. N.; Mulholland A. J. Quantum mechanics/molecular mechanics modeling of regioselectivity of drug metabolism in cytochrome P450 2C9. J. Am. Chem. Soc. 2013, 135 (21), 8001–8015. 10.1021/ja402016p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonsdale R.; Harvey J. N.; Mulholland A. J. Compound I reactivity defines alkene oxidation selectivity in cytochrome p450cam. J. Phys. Chem. B 2010, 114 (2), 1156–1162. 10.1021/jp910127j. [DOI] [PubMed] [Google Scholar]
- Chen F.; Bian K.; Tang Q.; Fedeles B. I.; Singh V.; Humulock Z. T.; Essigmann J. M.; Li D. Oncometabolites d- and l-2-Hydroxyglutarate Inhibit the AlkB Family DNA Repair Enzymes under Physiological Conditions. Chem. Res. Toxicol. 2017, 30 (4), 1102–1110. 10.1021/acs.chemrestox.7b00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi C.; Jia G.; Hou G.; Dai Q.; Zhang W.; Zheng G.; Jian X.; Yang C. G.; Cui Q.; He C. Iron-catalysed oxidation intermediates captured in a DNA repair dioxygenase. Nature 2010, 468 (7321), 330–333. 10.1038/nature09497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu B.; Hunt J. F. Enzymological and structural studies of the mechanism of promiscuous substrate recognition by the oxidative DNA repair enzyme AlkB. Proc. Natl. Acad. Sci. U. S. A. 2009, 106 (34), 14315–14320. 10.1073/pnas.0812938106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiser A.; Šali A. Modeller: Generation and Refinement of Homology-Based Protein Structure Models. Methods Enzymol. 2003, 374, 461–491. 10.1016/S0076-6879(03)74020-8. [DOI] [PubMed] [Google Scholar]
- a Gordon J. C.; Myers J. B.; Folta T.; Shoja V.; Heath L. S.; Onufriev A. H++: A server for estimating pKas and adding missing hydrogens to macromolecules. Nucleic Acids Res. 2005, 33, W368–371. 10.1093/nar/gki464. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Olsson M. H.; Sondergaard C. R.; Rostkowski M.; Jensen J. H. PROPKA3: Consistent treatment of internal and surface residues in empirical pKa predictions. J. Chem. Theory Comput. 2011, 7 (2), 525–537. 10.1021/ct100578z. [DOI] [PubMed] [Google Scholar]
- Li P.; Merz K. M. MCPB.py: A Python Based Metal Center Parameter Builder. J. Chem. Inf. Model. 2016, 56 (4), 599–604. 10.1021/acs.jcim.5b00674. [DOI] [PubMed] [Google Scholar]
- Seminario J. M. Calculation of intramolecular force fields from second-derivative tensors. Int. J. Quantum Chem. 1996, 60 (7), 1271–1277. . [DOI] [Google Scholar]
- Chaturvedi S. S.; Ramanan R.; Waheed S. O.; Ainsley J.; Evison M.; Ames J. M.; Schofield C. J.; Karabencheva-Christova T. G.; Christov C. Z. Conformational Dynamics Underlies Different Functions of Human KDM7 Histone Demethylases. Chem. - Eur. J. 2019, 25 (21), 5422–5426. 10.1002/chem.201900492. [DOI] [PubMed] [Google Scholar]
- Pabis A.; Geronimo I.; York D. M.; Paneth P. Molecular dynamics simulation of nitrobenzene dioxygenase using AMBER force field. J. Chem. Theory Comput. 2014, 10 (6), 2246–2254. 10.1021/ct500205z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.; Wang W.; Kollman P. A.; Case D. A. Automatic atom type and bond type perception in molecular mechanical calculations. J. Mol. Graphics Modell. 2006, 25 (2), 247–260. 10.1016/j.jmgm.2005.12.005. [DOI] [PubMed] [Google Scholar]
- Jorgensen W. L.; Chandrasekhar J.; Madura J. D.; Impey R. W.; Klein M. L. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79, 926–935. 10.1063/1.445869. [DOI] [Google Scholar]
- Götz A. W.; Williamson M. J.; Xu D.; Poole D.; LeGrand S.; Walker R. C. Routine microsecond molecular dynamics simulations with AMBER on GPUs. 1. generalized born. J. Chem. Theory Comput. 2012, 8 (5), 1542–1555. 10.1021/ct200909j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case D. A.; Betz R. M.; Cerutti D. S.; Cheatham T. E.; Daeden T. A.; Duke R. E.; Giese T. J.; Gohlke H.; Goetz A. W.; Homeyer N.; et al. AMBER 2016; University of California: San Francisco, 2016.
- Maier J. A.; Martinez C.; Kasavajhala K.; Wickstrom L.; Hauser K. E.; Simmerling C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11 (8), 3696–3713. 10.1021/acs.jctc.5b00255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deserno M.; Holm C. How to mesh up Ewald sums. I. A theoretical and numerical comparison of various particle mesh routines. J. Chem. Phys. 1998, 109 (18), 7678–7693. 10.1063/1.477414. [DOI] [Google Scholar]
- Davidchack R. L.; Ouldridge T. E.; Tretyakov M. V. New Langevin and gradient thermostats for rigid body dynamics. J. Chem. Phys. 2015, 142, 144114. 10.1063/1.4916312. [DOI] [PubMed] [Google Scholar]
- Bresme F. Equilibrium and nonequilibrium molecular-dynamics simulations of the central force model of water. J. Chem. Phys. 2001, 115 (16), 7564–7574. 10.1063/1.1407288. [DOI] [Google Scholar]
- Ryckaert J.-P.; Ciccotti G.; Berendsen H. J. Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23 (3), 327–341. 10.1016/0021-9991(77)90098-5. [DOI] [Google Scholar]
- Roe D. R.; Cheatham T. E. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013, 9 (7), 3084–3095. 10.1021/ct400341p. [DOI] [PubMed] [Google Scholar]
- Humphrey W.; Dalke A.; Schulten K. VMD: Visual Molecular Dynamics. J. Mol. Graphics 1996, 14, 33–38. 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- Pettersen E. F.; Goddard T. D.; Huang C. C.; Couch G. S.; Greenblatt D. M.; Meng E. C.; Ferrin T. E. UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25 (13), 1605–1612. 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Grant B. J.; Rodrigues A. P. C.; ElSawy K. M.; McCammon J. A.; Caves L. S. D. Bio3d: An R package for the comparative analysis of protein structures. Bioinformatics 2006, 22 (21), 2695–2696. 10.1093/bioinformatics/btl461. [DOI] [PubMed] [Google Scholar]
- Sherwood P.; De Vries A. H.; Guest M. F.; et al. QUASI: A general purpose implementation of the QM/MM approach and its application to problems in catalysis. J. Mol. Struct.: THEOCHEM 2003, 632, 1–28. 10.1016/S0166-1280(03)00285-9. [DOI] [Google Scholar]
- Ahlrichs R.; Bär M.; Häser M.; Horn H.; Kölmel C. Electronic structure calculations on workstation computers: The program system turbomole. Chem. Phys. Lett. 1989, 162 (3), 165–169. 10.1016/0009-2614(89)85118-8. [DOI] [Google Scholar]
- Smith W.; Forester T. R. DL-POLY-2.0: A general-purpose parallel molecular dynamics simulation package. J. Mol. Graphics 1996, 14 (3), 136–141. 10.1016/S0263-7855(96)00043-4. [DOI] [PubMed] [Google Scholar]
- Kästner J.; Carr J. M.; Keal T. W.; Thiel W.; Wander A.; Sherwood P. DL-FIND: An open-source geometry optimizer for atomistic simulations. J. Phys. Chem. A 2009, 113 (43), 11856–11865. 10.1021/jp9028968. [DOI] [PubMed] [Google Scholar]
- Billeter S. R.; Turner A. J.; Thiel W. Linear scaling geometry optimization and transition state search in hybrid delocalised internal coordinates. Phys. Chem. Chem. Phys. 2000, 2 (10), 2177–2186. 10.1039/a909486e. [DOI] [Google Scholar]
- Rose N. R.; McDonough M. A.; King O. N. F.; Kawamura A.; Schofield C. J. Inhibition of 2-oxoglutarate dependent oxygenase. Chem. Soc. Rev. 2011, 40, 4364–4397. 10.1039/c0cs00203h. [DOI] [PubMed] [Google Scholar]
- Yeh T.-L.; Leissing T. M.; Abboud M. I.; Thinnes C. C.; Atasoylu O.; Holt-Martyn J. P.; Zhang D.; Tumber A.; Lippl K.; Lohans C. T.; Leung I. K. H.; Morcrette H.; Clifton I. J.; Claridge T. D. W.; Kawamura A.; Flashman E.; Lu X.; Ratcliffe P. J.; Chowdhury R.; Pugh C. W.; Schofield C. J. Inhibition of 2-oxoglutarate dependent oxygenase. Chem. Sci. 2017, 8, 7651–7668. 10.1039/C7SC02103H. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.