

Abstract

Inland sources of particulate chloride for atmospheric nitryl chloride (ClNO2) formation remain unknown and unquantified, hindering air quality assessments. Globally each winter, tens of millions of tons of road salt are spread on roadways for deicing. Here, we identify road salt aerosol as the primary chloride aerosol source, accounting for 80–100% of ClNO2 formation, at an inland urban area in the wintertime. This study provides experimental evidence of the connection between road salt and air quality through the production of this important reservoir for nitrogen oxides and chlorine radicals, which significantly impact atmospheric composition and pollutant fates. A numerical model was employed to quantify the contributions of chloride sources to ClNO2 production. The traditional method for simulating ClNO2 considers chloride to be homogeneously distributed across the atmospheric particle population; yet, we show that only a fraction of the particulate surface area contains chloride. Our new single-particle parametrization considers this heterogeneity, dramatically lowering overestimations of ClNO2 levels that have been routinely reported using the prevailing methods. The identification of road salt as a ClNO2 source links this common deicing practice to atmospheric composition and air quality in the urban wintertime environment.
Short abstract
Road salt was identified as the primary source of chloride-containing aerosol in the wintertime urban atmosphere. This links deicing practices to air quality via the production of the trace gas ClNO2.
1. Introduction
In coastal urban areas, dinitrogen pentoxide (N2O5), formed by NO2 reaction with the photolabile nitrate radical (NO3, reaction R1), reacts on the surface of chloride (Cl–)-containing sea spray aerosol particles to produce nitryl chloride (ClNO2, R2).1−3 Upon sunrise, ClNO2 photolysis releases chlorine atoms (Cl) and nitrogen dioxide (NO2) (R3), altering pollutant fate, ozone levels, and particulate nitrate formation.1,4
| R1 |
| R2 |
| R3 |
While this chlorine chemistry was originally thought to only occur in marine and coastal locations, Thornton et al.5 first showed observations of ClNO2 far inland (near Boulder, Colorado, 1400 km inland). Model predictions, constrained to measurements of particulate chloride across the United States, indicate that the majority of ClNO2 formation occurs over land, particularly during winter.5,6 However, the source(s) of inland particulate chloride are highly uncertain.2,7
The wintertime application of road salt for deicing purposes is ubiquitous,8,9 with tens of millions of tons of salt applied globally each year.8−12 In the United States, over 20 million tons of salt were applied to roadways in 2014 alone.8 This road salt contributes to chloride-containing atmospheric particles,11 saline snowpacks,13 and urban grime.14,15 Road salt is mechanically aerosolized by vehicular traffic,16 with deposition observed hundreds of meters away from roadways.17 Observed increases in wintertime PM2.5 (particulate matter <2.5 μm) chloride concentrations are often correlated with snowfall, due to the application of deicing salts before, during, and after snowfall events.11 These wintertime inland PM2.5 chloride concentrations can even rival those of coastal areas influenced by sea spray.11 For flights downwind from New York City, NY, after a snow storm, Haskins et al.18 reported observations of gas phase Cly (≡ HCl + ClNO2 + HOCl + 2Cl2) that could not be not explained by sea salt displacement, suggesting road salt influence. In Calgary, Alberta, where road salts are used, increasing ClNO2 was observed following the first snowfall in November 2010,19 suggesting that road salt could be an important chloride source for ClNO2 production. Further, the slower thermal dissociation of N2O5 (R1, reverse) under the lower winter temperatures,20 when road salts are used, leads to greater importance of this chemistry in wintertime environments. However, the contribution of road salt chloride to ClNO2 formation is unknown,2 despite being hypothesized for several decades.1
To model ClNO2 production in the atmosphere, laboratory-based measurements of N2O5 uptake (γN2O5) and ClNO2 yields (φClNO2) have informed the current parametrization based on aerosol mass concentrations of chloride, nitrate, and liquid water content.21 This “bulk” parametrization21 assumes that all aerosol chemical components are equally distributed across the entire aerosol size distribution and that all particles contain chloride.22 Yet, even in the polluted marine atmosphere, chloride is not homogeneously mixed across the entire aerosol population.23,24 The γN2O5 and φClNO2 values are often overestimated when compared to field-derived values,5,25−29 including recent comprehensive assessments by McDuffie et al.30,31 Further, observed variability and trends in γN2O5 and φClNO2 are often not reproduced, pointing to an inaccurate parametrization and reducing the accuracy of associated air quality simulations.26,27,30,31
2. Results and Discussion
2.1. Winter Measurements of ClNO2 and Identification of Road Salt Aerosol
From February 1 to March 10, 2016 in Ann Arbor, Michigan, ClNO2 production was investigated using a comprehensive suite of atmospheric trace gas and particle measurements. Using chemical ionization mass spectrometry (CIMS),32 N2O5 and ClNO2 were continuously measured at 12 m above ground level, with average nighttime levels of 141 ± 7 ppt (pmol mol–1, 95% confidence interval) and 23 ± 1 ppt, respectively (Figure S1 and Section S2.1 of the Supporting Information). Nocturnal ClNO2 reached a maximum of 220 ppt. Lower temperatures in the winter favor N2O5 (reducing thermal dissociation),20 and the application of road salt for deicing provides a unique chloride source. Previously, in Calgary, Alberta, Mielke et al.19,33 observed elevated ClNO2 in March and April (up to 338 and 250 ppt, respectively), compared to September (max. 30 ppt), prior to road salt application. Similarly, in Ann Arbor, N2O5 and ClNO2 levels were lower in October 2016, with average nighttime levels of 71 ± 5 ppt and 3.5 ± 0.5 ppt, respectively, and maximum ClNO2 of 29 ppt (see Section S2.2).
To test the hypothesis that road salt aerosol chloride contributes to ClNO2 production, we present comprehensive observations of individual particle chemical composition with source attribution, complemented by modeling of two representative case periods (February 17–18 and March 7–8, 2016). These case periods encompass a range of observed ClNO2 levels, and no precipitation or rapid changes in wind speed and direction occurred, making them well-suited for numerical modeling. Although surface-level observations can vary from those made aloft,34,35 ClNO2 and N2O5 mole ratios during the case periods were not impacted by N2O5 depletion from NO3 titration,36 or morning ClNO2 enhancement due to entrainment from the residual layer37 (Figure S1). The model results, using our new parametrization, show that the ClNO2 observed in Ann Arbor was predominantly produced from N2O5 reaction with particulate chloride from widespread road salt application, thereby quantifying the link between wintertime deicing practices and chlorine activation (Figure 1).
Figure 1.

The role of road salt aerosol in wintertime ClNO2 production in inland areas. Vehicular traffic causes lofting of road salt, providing a chloride-containing particle surface for N2O5 uptake, producing ClNO2 at night. Upon sunrise, ClNO2 photolysis leads to chlorine radical formation and NOx recycling, linking the common deicing practice to wintertime inland air quality.
Individual particle measurements of chemical composition and morphology were conducted using computer-controlled scanning electron microscopy with energy dispersive X-ray spectroscopy (CCSEM-EDX)38 and aerosol time-of-flight mass spectrometry (ATOFMS),39 with 8,052 and 83,597 individual particles analyzed by each method, respectively. These methods identified and quantified the chloride-containing particles. This analysis complemented bulk PM2.5 inorganic ion measurements using ambient ion monitor-ion chromatography (AIM-IC).40 Concurrent measurements of atmospheric PM2.5 chloride ranged from 0.01 to 0.35 μg m–3 (Section S2.1), similar to concentrations during elevated ClNO2 levels in Boulder, CO.5
Five individual particle types—road salt, aged road salt, biomass burning, soot, and road dust (Figures 2 and S2, and discussed in Section 4.3)—were observed by ATOFMS and CCSEM-EDX. Only road salt particles contained significant chloride. Road salt particles were primarily composed of sodium and chloride (average Cl/Na atomic ratio of 0.87 ± 0.03, 95% confidence interval, determined by CCSEM-EDX), consistent with road salt used by the city of Ann Arbor and the University of Michigan for winter maintenance (Section S2.3). Sulfate, known to suppress aerosol ClNO2 production,41 was not detected in the individual nascent road salt particles. Aged road salt particles were depleted in chloride (average Cl/Na atomic ratio of 0.10 ± 0.01) and enriched in nitrate and sulfate, compared to the nascent road salt particles. Biomass burning particles have been suggested as a possible ClNO2 source, depending on their fuel chloride content.42 However, these particles were identified as a mixture of mainly organic carbon, nitrate, sulfate, ammonium, and potassium (Figure S2),43 and contained less than 2% chlorine (atomic percentage), representing a minor chloride source. Therefore, the observed ammonium was primarily in the form of ammonium nitrate and sulfate, rather than ammonium chloride (see Section S2.4). Less than 3% of road dust particles, by number, and no soot particles contained chloride (Section 4.5.2). The frequent nightly observations of elevated N2O5 and ClNO2, concurrent with nascent and aged road salt particles, suggest that ClNO2 is mainly formed from the multiphase reaction of N2O5 on road salt aerosols, as confirmed by the model simulations described below.
Figure 2.

Identification and quantitation of nascent and aged road salt aerosol. Representative EDX spectra and SEM images and average ATOFMS mass spectra of individual (a, b) nascent and (c, d) aged road salt particles. *Al and Si peaks in the EDX spectra are from substrate and detector backgrounds. Aged road salt is characterized by chloride depletion and nitrate and/or sulfate enrichment. (e) Average aerosol surface area fractions (0.015–20 μm) attributed to the five particle types identified by CCSEM-EDX and ATOFMS for the February 17–18 and March 7–8 cases.
2.2. Considering Aerosol Heterogeneity in Model Simulations of ClNO2 Production
A one-dimensional atmospheric numerical model simulated ClNO2 production constrained by measurements of N2O5, HCl, and aerosol surface area and composition for the February 17–18 and March 7–8 cases. The influence of aerosol chemical composition on the uptake of N2O5 (γN2O5) and yield of ClNO2 (φClNO2) was accounted for through two different model scenarios: (1) using the “traditional” parametrization by Bertram & Thornton,21 which is based on bulk particulate inorganic ion mass concentrations, here constrained by hourly PM2.5 measurements using AIM-IC, and (2) a new parametrization developed based on measured single-particle chemical composition. Our single-particle measurements showed that chloride-containing particles, dominated by nascent and aged road salt, only contributed 20–26% of the aerosol surface area concentration (Figure 2), suggesting that the bulk parametrization should overestimate ClNO2 by assuming that all particles contain chloride. Indeed, for southeast Michigan, previously simulated mean wintertime ClNO2 mole ratios6 (using the bulk parametrization) were up to an order of magnitude higher than our observations.
With knowledge of the distribution of chloride among individual particles, the new single particle parametrization, described in Section 4.5, considers that N2O5 reacts with a heterogeneous aerosol population, producing ClNO2 only from particles that contain chloride. We assigned chemically specific γN2O5 and φClNO2 values to the five observed particle types using the most relevant laboratory model systems20,21,44−46 (Table S1). Since N2O5 reacts on the surface of particles,3 these γN2O5 and φClNO2 values were then weighted by the fraction of the measured temporally varying aerosol surface area concentration corresponding to each particle type. This method results in significantly lower estimates of γN2O5 and φClNO2 compared to the bulk parametrization, by directly accounting for additional chemical components that alter γN2O5 and φClNO2, such as sulfate, carboxylate, and nitrate,21,41 within the heterogeneous individual particle population. A primary advantage of this single-particle approach is that only chloride-containing particles can produce ClNO2, and that aerosol components that suppress production are accounted for within the individual particle types. The main uncertainty in the single-particle method comes from the choice of laboratory proxy γN2O5 and φClNO2 values for the observed particle types, and this uncertainty can be reduced with further laboratory studies of varying aerosol composition.
The average γN2O5 values calculated for the single-particle method (γN2O5 = 0.018 and 0.013 for February 17–18 and March 7–8) were approximately half of those predicted by the bulk method (γN2O5,bulk = 0.030 and 0.031, respectively). Likewise, the average φClNO2 values for the single-particle method (φClNO2 = 0.28 and 0.20 for the two cases, respectively) were about one-third of the bulk chemical composition method (φClNO2,bulk = 0.8 ± 0.1 and 0.7 ± 0.2, respectively). Notably, the single-particle-weighted γN2O5 and φClNO2 are both closer to previous wintertime field-based estimates (γN2O5 = 0.02; φClNO2 = 0.005–0.15) in Calgary, Alberta,19,33 where road salting is also commonplace.
By constraining to measurements of N2O5, two model simulations of ClNO2 production were conducted: using the (1) traditional bulk parametrization and (2) new single-particle parametrization, which employs the chemically dependent γN2O5 and φClNO2 values. The predicted ClNO2 mole ratios using the single-particle parametrization generally agree within the measurement uncertainty of the observations, while the bulk parametrization overpredicts ClNO2 by an average of 120 ± 20 ppt (340%) and 12 ± 3 ppt (150%), respectively, for the February and March cases (Figure 3). The results of the single-particle method identify aerosolized road salt as the dominant ClNO2 source, accounting for 80–100%, on average, of the total simulated ClNO2 (Figure 3). This study demonstrates road salt as the dominant source of photolyzable chlorine in the wintertime inland environment, despite only representing a fraction (20–26%) of the aerosol surface area available for N2O5 uptake and ClNO2 production (Figure 2). Application of this new parametrization to modeling in both marine and inland environments will improve predictions of ClNO2 production because chloride is typically not distributed equally among all particles, even in the coastal marine atmosphere.23
Figure 3.

Single-particle parametrization of N2O5 uptake (γN2O5) and ClNO2 yield (φClNO2) improves model agreement with measured ClNO2 in Ann Arbor, Michigan. (a) Schematic comparing the bulk and single-particle methods to parametrize N2O5 uptake and ClNO2 yield. For both (b) February 17–18 and (c) March 7–8 cases, the modeled ClNO2, using γN2O5 and φClNO2 from the single-particle chemical composition and surface area parametrization (solid green lines), agrees well with the magnitude and shape of the ClNO2 measurements (black lines), while the traditional bulk aerosol composition parametrization (gray lines) significantly overpredicts ClNO2. The dashed blue lines represent modeled ClNO2 from road salt aerosol (nascent + aged) only, demonstrating that road salt aerosol is the dominant ClNO2 source (80–100%).
3. Atmospheric Implications
For decades, road salt use for wintertime deicing has been increasing across North America8,9 and has led to increased salinity of surface and ground waters with severe ecosystem impacts.47 Here, this common deicing practice is linked to wintertime air quality due to the role of road salt chloride as a large and potent source of photolyzable chlorine. Single-particle measurements identified aerosolized road salt as the dominant source of particulate chloride. This road salt aerosol was calculated to be responsible for 80–100% of atmospheric ClNO2 in this inland wintertime environment. The key to the improved modeling of ClNO2 is for only the chloride-containing particle surface area to produce ClNO2, as was achieved with our single-particle parametrization. Laboratory studies of additional model aerosols, as well as authentic road salt aerosol, are needed to measure γN2O5 and φClNO2 to improve the accuracy of the application of the single-particle parametrization. Although the chloride-containing particles were primarily fresh road salt in this study, environments influenced by other particle types may need to consider additional single-particle properties. For example, different classes of organic compounds can result in particle phase separation,48 impact N2O5 uptake,49,50 and/or limit chloride availability,51 thereby altering ClNO2 production. If additional properties are known or can be modeled, then appropriate laboratory γN2O5 and φClNO2 can be applied through the single-particle, surface area-weighted approach, which can also be applied to other multiphase reactions. While additional measurements of single-particle chemical composition are needed for other environments influenced by chlorine chemistry, modeling approaches that describe the distribution of chemical components across the aerosol population, from zero-dimensional to regional/global model levels, are now available22 and should be used to further evaluate the single-particle, surface area-weighted approach presented here.
The chlorine radicals produced from ClNO2 photolysis, which contributed 44–99% of the daytime Cl production rate (Figure S3), impact the fate of atmospheric hydrocarbons, particularly in the winter when hydroxyl radicals are less abundant due to reduced sunlight and water vapor.52 In addition, ClNO2 photolysis also significantly alters the levels of NOx, O3, and particulate matter,5,37,53,54 impacting downwind areas struggling with wintertime air quality.55 Other road salt contaminated surfaces, including buildings, road surfaces, and snowpacks, which were not explored in this study, likely also support ClNO2 production15 and should be examined in future work. Future efforts are also needed to quantify road salt emissions and generate inventories for models, as this substantial chloride source is currently not included in emissions inventories for atmospheric modeling of nitryl chloride,6,56 thereby limiting the quantitation of the broad implications of this chlorine chemistry.
4. Methods
Atmospheric sampling of trace gases and particles was conducted at 12 m above ground level (agl) in Ann Arbor, MI, at the University of Michigan (UM, 42.2786°N, 83.7369°W) from February 1 to March 10, 2016. ClNO2, N2O5, HNO3, and Cl2 were monitored using CIMS (Section 4.1 and Section S1.1), and soluble inorganic trace gases, including HCl and SO2, and PM2.5 Cl– and NO3– were measured using the AIM-IC system (Section S1.2). Measurements of individual particle chemical composition were conducted online using ATOFMS (Section 4.2.1) and offline using CCSEM-EDX (Section 4.2.2). Sections S1.3–S1.6 of the Supporting Information contain additional methods details, including measurements of NO, volatile organic compounds, and aerosol size distributions. Single-particle identifications are described in Section 4.3. Measurements and calculations pertaining to the single-particle parametrization for ClNO2 formation are described in Sections 4.4 and 4.5. Two 24 h case study periods (February 17–18 and March 7–8, 2016), each starting and ending at 12:00 EST, were investigated using a one-dimensional atmospheric model described in Section 4.6.
Roadway and sidewalk deicing maintenance using road salt and brine was regularly conducted adjacent to the sampling site by the city of Ann Arbor and UM. Meteorological data (temperature, relative humidity, wind speed, and wind direction) were obtained from a weather station (42.2769°N 83.7655°W) located 2.3 km west of the measurement site. Wind speed and direction were measured at ∼12 m agl. Temperature and relative humidity were measured at ∼5 m agl. No unexpected or unusually high safety hazards were encountered.
4.1. Chemical Ionization Mass Spectrometry
A chemical ionization mass spectrometer (CIMS, THS Instruments)32 quantified ambient ClNO2, N2O5, and HNO3 from February 1 to March 10, 2016, and again from October 23–28, 2016 for comparison to autumn conditions prior to road salting. The CIMS used I(H2O–)n reagent ions to react with analytes, forming iodide ion adducts that were subsequently detected by a quadrupole mass analyzer. Ambient air was pulled through a heated (25–30 °C), 0.95 cm I.D., 2.5 m long FEP tube at 7.1 L min–1, at atmospheric pressure, into a custom three-way valve used for calibration and background measurements, as described by Liao et al.32 The CIMS ion molecule reaction region (IMR, maintained at 19 Torr) sampled 0.9 L min–1, and an ozone monitor (model 205, 2B Technologies) sampled 1.7 L min–1, with the remaining flow sent to exhaust. The I–(H2O)n reagent ions were formed by passing 5 L min–1 of CH3I (5 ppm in N2, Scott-Marrin, Inc.) through a 210Po source (20 mCi), forming I–, and combining with humidified N2 (from a room temperature 1 L bubbler) in the CIMS IMR. Water vapor was added, as in previous work,57 to prevent fluctuations in CIMS sensitivity due to ambient humidity. The following ions were monitored (with dwell times noted): ClNO2 as I35ClNO2– at m/z 208 (1 s) and I37ClNO2– at m/z 210 (0.5 s), N2O5 as IN2O5– at m/z 235 (0.5 s), HNO3 as IHNO3– at m/z 190 (0.2 s), and Cl2 as I–(35Cl35Cl) at m/z 197 (1 s) and I–(37Cl35Cl) at m/z 199 (0.5 s). The measured isotopic ratio for 37ClNO2 to 35ClNO2 (0.31) confirmed the identity of the observed ClNO2 (theoretical ratio = 0.32). Ambient Cl2 was not observed above the detection limit during the study and was therefore not quantified. Reagent ions were monitored at m/z 147 (IH218O–). Considering all monitored masses, the total measurement time for the CIMS was 14.6 s. Background measurements were conducted for 2 min every 15 min by passing the airflow through a glass wool and stainless steel wool scrubber heated to 120 °C, removing ClNO2 and N2O5 with >95% efficiency, and HNO3 with 88% efficiency.
Online Cl2 calibration was completed every 2 h by adding 200 mL min–1 of 190 ± 10 ppb Cl2 (in N2) from a permeation source (VICI Metronics) to the ambient airflow. The Cl2 permeation rate (110 ng min–1) was confirmed every 1–2 days using the optical absorption method described by Liao et al.32 The Cl2 sensitivity at m/z 197 was 3.0 ± 0.4 Hz ppt–1. Calibrations of HNO3, N2O5, and ClNO2 were completed offline using calibration factors relative to Cl2 (Section S1.1). Limits of detection (LODs, 3σ), corresponding to 2 min background periods, were 0.9, 4, 34, and 2 ppt for ClNO2, N2O5, HNO3, and Cl2, respectively. Taking into account counting statistics,32 we report 10 min averaged LODs of 0.4, 2, 15, and 1 ppt for ClNO2 N2O5, HNO3, and Cl2, respectively. Measurement uncertainties, which include the propagated calibration uncertainties and fluctuations in CIMS background signals, for 10 min averaged ClNO2, N2O5, and HNO3 were 17% + 0.4 ppt, 17% + 2 ppt, and 27% + 15 ppt, respectively. Following the study, the CIMS sampling line was characterized for wall losses of ClNO2, N2O5, and HNO3 by flowing each trace gas through the sampling line and comparing to its direct injection into the CIMS inlet. Measured line losses for each species (ClNO2 = 20 ± 7% (±standard deviation), N2O5 = 14 ± 15%, and HNO3 = 3 ± 9%) were used to adjust the CIMS measured values to ambient mole ratios. The CIMS sampling line was tested postcampaign for potential inlet artifacts, and little to no ClNO2 generation was observed following the addition of N2, N2O5, and Cl2 into the sampling line, as discussed in Section S1.1.
4.2. Measurements of Individual Particle Chemical Composition and Morphology
4.2.1. Aerosol Time-of-Flight Mass Spectrometry (ATOFMS)
An aerosol time-of-flight mass spectrometer (ATOFMS), based on the design of Pratt et al.,39 measured individual particle size and chemical composition for particles 0.1–1.5 μm (vacuum aerodynamic diameter) from February 3 to March 9, 2016. Air was sampled from a manifold (shared with additional aerosol instruments, Section S1.4) through a 0.17 cm ID copper sampling line at 0.1 L min–1. The operation of the ATOFMS is described in detail elsewhere.39 Positive and negative ion mass spectra were collected for 83,597 individual particles during the study. Mass spectral peak lists were generated using custom LabVIEW software and imported into Matlab for analysis with YAADA, a custom toolkit. Individual particle mass spectra were clustered using an ART-2a algorithm with a vigilance factor of 0.8 and a learning rate of 0.05 for 20 iterations.58 Particle types were identified based on the most likely m/z from previous field and laboratory studies (Section 4.3).
4.2.2. Computer-Controlled Scanning Electron Microscopy with Energy Dispersive X-ray Spectroscopy (CCSEM-EDX)
Atmospheric particles were impacted onto transmission electron microscopy (TEM) grids (Formvar Type-B Copper grids, Ted Pella, Inc.) and aluminum foil substrates (MSP Corp.) using a rotating 10-stage micro-orifice uniform deposit impactor (MOUDI, model 110R, MSP Corp.). Ambient air flowed at 20 L min–1 through a 3.7 m long, 1.1 cm ID unheated, insulated copper sampling inlet with 10 L min–1 of particle-free dilution air sampled through a HEPA capsule (Pall Laboratory), resulting in a 30 L min–1 flow into the MOUDI. Particles analyzed herein were collected on stages with 50% efficiency upper-limit size cuts of 3.2, 1.0, 0.32, and 0.10 μm (aerodynamic diameter, da). Individual particles were analyzed by computer-controlled scanning electron microscopy with energy dispersive X-ray spectroscopy (CCSEM-EDX) using FEI Quanta SEM and FEI Helios SEM instruments. Both instruments were equipped with a field emission gun operating at 20 keV and high angle annular dark field scanning TEM detectors and Everhart-Thornley secondary electron detectors, for analysis of morphology, including projected area diameter, of particles on TEM grids and aluminum foil substrates, respectively. An EDX detector (EDAX, Inc.) collected X-ray spectra for individual particles to determine relative atomic abundances of C, N, O, Na, Mg, P, S, Cl, K, Ca, Ti, Fe, and Ni. Samples from the two case days were analyzed by CCSEM-EDX: February 17–18, 2016 19:00–07:45 EST (2,915 particles analyzed) and March 7–8, 2016 19:30–07:15 EST (5,137 particles analyzed). K-means cluster analysis of the individual particle EDX spectra38 resulted in 50 clusters, which were grouped into particle classes based on elemental composition. Particle classes were identified based on similarity to individual particle EDX spectral signatures in published literature.38 The CCSEM-EDX relative atomic percentages for Cl and Na were used to calculate individual road salt particle Cl/Na atomic ratios.
4.3. Identification of Atmospheric Particle Types from Single-Particle Measurements
The clustering analyses of ATOFMS and CCSEM-EDX individual particle spectra (Section 4.2) resulted in the identification of five major particle types: road salt, aged road salt, biomass burning, soot, and road dust (Figure 2 and Figure S2). Road salt was characterized by intense Na and Cl peaks in the EDX spectra;38,59 by ATOFMS, road salt was identified by an intense m/z 23 (Na+) peak, as well as less intense ion peaks at m/z 39 (K+), 40 (Ca+), 81 (Na2Cl+), −35/–37 (Cl–), and −93/95 (NaCl2–) (Figure 2). The road salt mass spectra were consistent with previous ATOFMS ambient observations,60 as well as our laboratory measurements of local road salt and brine samples, as discussed in Section S2.3. Aged road salt also contained N and/or S, in addition to Na, and was depleted in Cl, as determined by EDX. Through ATOFMS analysis, aged road salt was also characterized by positive ions at m/z 23 (Na+), 39 (K+), and 40 (Ca+), with negative ion peaks at m/z −46 (NO2–), −62 (NO3–), and −97 (HSO4–), indicative of atmospheric processing.61 Biomass burning particles, attributed to residential wood burning,43 were identified by ATOFMS by an intense peak at m/z 37 (C3H+) and 39 (K+), with less intense organic carbon peaks at m/z 27 (C2H3+), 43 (C2H3O+), and 50 (C4H2+), as well as ammonium (m/z 18 (NH4+)) (Figure S2). The EDX spectra of these particles were characterized by intense C and O peaks, in addition to S, N, and/or K.62,63 Road dust particles were identified by intense Fe and Al and/or Si peaks, in addition to O, in the EDX spectra;38 corresponding ATOFMS spectra were characterized by an intense peak at m/z 56 (Fe+), with less intense peaks at m/z 72 (FeO+) and 88 (FeO2+), with iron likely from vehicular brake wear consistent with local road dust (Figure S2).64 Using CCSEM-EDX, soot particles were primarily composed of carbon and characterized by chain-like agglomerate morphology (Figure S2).62,65 Corresponding soot particle mass spectra contained elemental carbon peaks at m/z 12, 24, 36, 48, and 60 (C+, C2+, C3+, C4+, C5+); these particles are consistent with local vehicle combustion.66,67
4.4. Calculation of Chemically Resolved Surface Area Concentrations
Combined aerosol size distributions were obtained, using the method of Khlystov et al.,68 assuming a shape factor of 1 and a density of 1.5 g cm–3, using measurements from a scanning mobility particle sizer spectrometer (SMPS) for 15–600 nm particles (mobility diameter, dm) and an aerodynamic particle sizer (APS) for 0.60–20 μm (da) particles (Section S1.4). This results in continuous particle size distributions from 0.015 to 20 μm (da). Total particle surface area concentrations were then calculated based on these size-resolved number concentrations for the February 17–18 and March 7–8 cases. These surface area concentrations were then scaled by the size-resolved number fractions of the five particle types (identified in Section 4.3) to calculate time-resolved fractions of the aerosol surface area corresponding to each particle type using the following method.
First, particle diameters measured by CCSEM-EDX were converted from projected area diameter to da using the method of Wagner & Leith69 (Section S1.5). Then, the number fractions of the five particle types, determined by CCSEM-EDX for 0.13–2.29 μm, were applied to each size bin of the full aerosol size distribution, measured by SMPS and APS for 0.015–20 μm, to calculate size-resolved number fractions for each particle type. Since the measurements of aerosol size distributions extended down to 0.015 μm, the number fractions of each particle type below 0.13 μm were assumed to be the same as for the smallest CCSEM-EDX size bin (0.13–0.17 μm), which consisted of 20% road salt, 25% biomass burning, 2% aged road salt, 29% soot, and 24% road dust. Likewise, the aerosol size distribution measurements extended up to 20 μm; therefore, particles larger than 2.29 μm were assumed to be the same as for the number fractions of particle types from the largest CCSEM-EDX size bin (1.84–2.29 μm), which consisted of 70% road salt, 3% biomass burning, 10% aged road salt, 0% soot, and 18% road dust. Particles smaller than 0.13 μm composed 46 ± 11% and 36 ± 3%, on average, of the total particle surface area concentrations during the February 17–18 and March 7–8 case days, respectively. The percentage of total surface area corresponding to 2.29–20 μm particles was an average of 3 ± 2% during February 17–18 and 2 ± 1% during March 7–8.
For each case period, the size-resolved particle type number fractions were scaled by the time-varying particle number distributions (0.015–20 μm, da, Section S1.4) using the method described by Reinard et al.70 This generated number concentrations for each of the five particle types as a function of time. These chemically resolved, time-dependent number fractions were then used to determine the surface area concentrations for each particle type, with particles assumed to be spherical. The resulting chemically resolved, time-dependent surface area concentrations were used to calculate the single-particle weighted N2O5 uptake and ClNO2 yield (Section 4.5).
4.5. Determination of the Single-Particle Weighted γN2O5 and φClNO2
Reactions between particles and gas-phase N2O5 (γN2O5), and the yield of ClNO2 (φClNO2), are dependent on particle chemical composition. To generate time-resolved γN2O5 and φClNO2 values used for the new single-particle parametrization, previously determined laboratory values of γN2O5 and φClNO2 for laboratory proxies were weighted by the time-resolved fractions of aerosol surface area for each ambient particle type (Section 4.4). These laboratory-based γN2O5 and φClNO2 are provided in Table S1, and the selection criteria are discussed below.
4.5.1. Road Salt Particles (Nascent and Aged)
There are no reported studies of N2O5 uptake onto road salt. Given the observed RH range for the case days (Section S2.5) and the mechanism of road salt aerosolization (spray generated from vehicular traffic on wet roads),16 the road salt particles were likely deliquesced at the relative humidities observed for February 17–18 and March 7–8: 54–87% and 58–78%, with minimum temperatures of −12 and 10 °C, respectively (Section S2.5). For reference, the deliquescence and efflorescence RH points for NaCl particles were previously determined to be 75–80% and 40%, respectively, over a wide range of temperatures (253–298 K).71 Since the road salt aerosol was primarily NaCl (Cl/Na atomic ratio of 0.87 ± 0.03; Figure 2), the corresponding γN2O5 and φClNO2 values for road salt aerosol were assumed to be that of NaCl.20,21,44 However, the aged road salt particles were largely depleted in chloride and enriched in nitrate, as observed by ATOFMS; CCSEM-EDX showed that 64% of these particles contained little chlorine (2–5% atomic percentage), with an overall average Cl/Na atomic ratio of 0.10 ± 0.01 (Figure 2). The particulate nitrate suppresses N2O5 uptake, and the lack of available chloride reduces the ClNO2 yield.21 Therefore, the γN2O5 and φClNO2 values for aged road salt were chosen based on studies of mixed nitrate and NaCl particles.21,44
4.5.2. Biomass Burning, Road Dust, and Soot Particles
Recently, Ahern et al.42 confirmed the production of ClNO2 from reaction of N2O5 on biomass burning particles, showing that it is a function of the chloride mass fraction, but γN2O5 and φClNO2 values were not quantified in that study. However, only 8–18%, by number, of the observed biomass burning particles contained chloride (determined by ATOFMS), with measured atomic percentages of less than 2% chlorine (determined by CCSEM-EDX). It is likely that these particles corresponded to both fresh and aged residential wood burning emissions.72 Therefore, the uptake of N2O5 onto malonic acid (RH = 50–70%)20 was used as a proxy for the primarily organic biomass burning particles. The φClNO2 of the biomass burning particles was assumed to be the same as the aged road salt particles, approximately five times lower than that for nearly pure NaCl (nascent road salt), likely representing an upper limit of ClNO2 production. For the road dust particles, γN2O5 values are based on literature review recommendations,45 and since <3% of these particles, by number, contained Cl, φClNO2 values were set to zero. For the soot particles, γN2O5 values are based on chamber studies by Saathoff et al.,46 and φClNO2 values were set to zero, since no chloride was observed by CCSEM-EDX or ATOFMS. Surface area fractions of the single-particle types (Figure 2) were then used to calculate the single-particle surface area-weighted γN2O5 and φClNO2 values.
4.6. One-Dimensional (1-D) Atmospheric Model Description
A one-dimensional (1-D) numerical model simulated ClNO2 mole ratios for the February 17–18 and March 7–8 case study periods. The 1-D model incorporates turbulent transport, dry deposition, and multiphase aerosol chemistry in the urban atmospheric boundary layer, building upon the 0-D model described by Wang & Pratt.73 The model consists of 22 vertical atmospheric layers (heights: 0.02, 0.3, 1, 2, 3, 4, 6, 8, 12, 20, 30, 60, 100, 200, 300, 400, 500, 600, 700, 800, 900, and 1,000 m agl). The differential equations describing the temporal evolution of chemical species are solved using IGOR Pro software (https://www.wavemetrics.com/).
Temporal evolution of each chemical species is described by
| E1 |
The term 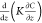 describes the vertical transport, where z is the layer height (m agl), and K is
the turbulent eddy diffusivity (m2 s–1), described in Section S1.6. P, L, and D are chemical
production, loss, and deposition terms, respectively (all in molecules
cm–3 s–1). Chemical reactions
are based on those in the 0-D model described by Wang & Pratt73 and from the Master Chemical Mechanism (C1–C4
hydrocarbon precursors from the MCM version 3.2, http://mcm.leeds.ac.uk/MCM/), except that bromine chemistry is not included. Time-dependent
photolysis frequencies were calculated using the Tropospheric Ultraviolet
and Visible Radiation Model (https://www2.acom.ucar.edu/modeling/tropospheric-ultraviolet-and-visible-tuv-radiation-model). Trace gas dry deposition was calculated using resistance-analog
methods, with the exception of N2O5 and NO2, for which deposition velocities previously determined for
snow-covered ground were applied.74,75 The term
describes the vertical transport, where z is the layer height (m agl), and K is
the turbulent eddy diffusivity (m2 s–1), described in Section S1.6. P, L, and D are chemical
production, loss, and deposition terms, respectively (all in molecules
cm–3 s–1). Chemical reactions
are based on those in the 0-D model described by Wang & Pratt73 and from the Master Chemical Mechanism (C1–C4
hydrocarbon precursors from the MCM version 3.2, http://mcm.leeds.ac.uk/MCM/), except that bromine chemistry is not included. Time-dependent
photolysis frequencies were calculated using the Tropospheric Ultraviolet
and Visible Radiation Model (https://www2.acom.ucar.edu/modeling/tropospheric-ultraviolet-and-visible-tuv-radiation-model). Trace gas dry deposition was calculated using resistance-analog
methods, with the exception of N2O5 and NO2, for which deposition velocities previously determined for
snow-covered ground were applied.74,75 The term 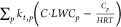 describes
the mass transport between the
gas and aqueous particle phases. C is the concentration
of a given species (molecules cm–3) in the gas-phase. Cp is the corresponding particle-phase
concentration of liquid water, and kt,p (s–1) is the phase-transfer coefficient for particles.
describes
the mass transport between the
gas and aqueous particle phases. C is the concentration
of a given species (molecules cm–3) in the gas-phase. Cp is the corresponding particle-phase
concentration of liquid water, and kt,p (s–1) is the phase-transfer coefficient for particles.
A number of measurements were used as model inputs: (1) Daily sounding data and ground-based meteorological data (Section S2.5) were used to calculate altitude- and time-varying eddy diffusivities. (2) Ambient measurements of N2O5, O3, HCl, and PM2.5 chloride and nitrate were constrained in the corresponding model layer (12 m agl). Measurements of NO (Figure S4) and select volatile organic compounds (VOCs, Table S2) were also used as constraints. (3) Measured aerosol total surface area (Figure S5) was constrained and assumed to be the same throughout all model atmospheric layers. (4) PM2.5 inorganic composition was used as inputs for the thermodynamic model (E-AIM Model IV, http://www.aim.env.uea.ac.uk/aim/model4/model4a.php) to calculate aerosol liquid water content. When ambient temperatures were below 265 K, the input temperature was set to 265 K due to the specified limits of the E-AIM model. The traditional parametrization of γN2O5 and φClNO2, based on Bertram and Thornton21 and constrained by hourly measurements of PM2.5 Cl– and NO3−, and our new parametrization, constrained to measured single-particle chemical composition (Section 4.5), were employed in the model simulations.
Acknowledgments
Funding was provided by the University of Michigan (UM), an Alfred P. Sloan Foundation Research Fellowship in Chemistry (FG-2017-9431), and the National Science Foundation (NSF) Atmospheric Chemistry program (AGS-1738588). K.R.K. was partially funded by a Dow Postdoctoral Fellowship in Sustainability from UM. S.M.M. and N.W.M. were partially funded by U.S. Department of Education Graduate Assistance in Areas of National Need fellowships. CCSEM-EDX analyses were performed at the Environmental Molecular Sciences Laboratory, a national scientific user facility located at the Pacific Northwest National Laboratory and sponsored by the Office of Biological and Environmental Research of the U.S. Department of Energy. Additional CCSEM-EDX analyses were conducted at the Michigan Center for Materials Characterization, which is thanked for use of the instruments and staff assistance. The National Center for Atmospheric Research is sponsored by the NSF; any opinions, findings, and conclusions or recommendations expressed in this publication are those of the authors and do not necessarily reflect the views of the NSF. The authors thank D. J. Tanner and L. G. Huey (Georgia Institute of Technology) for providing the NO analyzer, T. B. Ryerson (National Oceanic and Atmospheric Administration) for providing the photolytic NO2 converter, C. Mattson, A. Barget, A. Kevelin, M. Morales, M. Parks, G. Verwer, and A. Leemon (UM) for experimental assistance, C. R. Thompson (NOAA) for assistance with NOx measurements and helpful discussions, and A. P. Ault, P. K. Peterson (UM), and H. Osthoff (University of Calgary) for helpful discussions.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscentsci.9b00994.
Supplementary tables and figures, and additional methods details, including the PM2.5 sampling information and other measurements used to constrain the 1-D model. Additional ambient observations, analysis of the road salt and brine standards, and meteorology information (PDF)
Author Present Address
# (K.R.K.) Air Sciences Inc., Portland, OR, USA.
Author Present Address
∇ (S.W.) Atmospheric Chemistry Observations & Modeling, National Center for Atmospheric Research, Boulder, CO, USA.
Author Present Address
□ (A.L.) Department of Chemistry, Purdue University, West Lafayette, IN, USA.
The authors declare no competing financial interest.
Notes
The CIMS dataset is archived through PANGAEA: McNamara, S. M., Pratt, K. A. 10-min averages of ambient ClNO2, N2O5, HNO3, and O3 measured at 12 m above ground level in Ann Arbor, Michigan, USA, from February 1 to March 10, 2016. PANGAEA, 2020, https://doi.org/10.1594/PANGAEA.912746.
Supplementary Material
References
- Finlayson-Pitts B. J.; Ezell M. J.; Pitts J. N. Formation of Chemically Active Chlorine Compounds by Reactions of Atmospheric NaCl Particles with Gaseous N2O5 and ClONO2. Nature 1989, 337 (6204), 241–244. 10.1038/337241a0. [DOI] [Google Scholar]
- Simpson W. R.; Brown S. S.; Saiz-Lopez A.; et al. Tropospheric Halogen Chemistry: Sources, Cycling, and Impacts. Chem. Rev. 2015, 115, 4035–4062. 10.1021/cr5006638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaston C. J.; Thornton J. A. Reacto-Diffusive Length of N2O5 in Aqueous Sulfate- and Chloride-Containing Aerosol Particles. J. Phys. Chem. A 2016, 120, 1039. 10.1021/acs.jpca.5b11914. [DOI] [PubMed] [Google Scholar]
- Osthoff H. D.; Roberts J. M.; Ravishankara A. R.; et al. High Levels of Nitryl Chloride in the Polluted Subtropical Marine Boundary Layer. Nat. Geosci. 2008, 1 (5), 324–328. 10.1038/ngeo177. [DOI] [Google Scholar]
- Thornton J. A.; Kercher J. P.; Riedel T. P.; et al. A Large Atomic Chlorine Source Inferred from Mid-Continental Reactive Nitrogen Chemistry. Nature 2010, 464 (7286), 271–274. 10.1038/nature08905. [DOI] [PubMed] [Google Scholar]
- Sarwar G.; Simon H.; Bhave P.; et al. Examining the Impact of Heterogeneous Nitryl Chloride Production on Air Quality across the United States. Atmos. Chem. Phys. 2012, 12 (14), 6455–6473. 10.5194/acp-12-6455-2012. [DOI] [Google Scholar]
- Sherwen T.; Schmidt J. A.; Evans M. J.; Carpenter L. J.; Großmann K.; Eastham S. D.; Jacob D. J.; Dix B.; Koenig T. K.; Sinreich R.; Ortega I.; Volkamer R.; Saiz-Lopez A.; Prados-Roman C.; Mahajan A. S.; Ordonez C. Global Impacts of Tropospheric Halogens (Cl, Br, I) on Oxidants and Composition in GEOS-Chem. Atmos. Chem. Phys. 2016, 16, 12239–12271. 10.5194/acp-2016-424. [DOI] [Google Scholar]
- Lilek J.Roadway deicing in the United States, Factsheet. https://www.americangeosciences.org/critical-issues/factsheet/roadway-deicing-united-states.
- Ramakrishna D. M.; Viraraghavan T. Environmental Impact of Chemical Deicers - A Review. Water, Air, Soil Pollut. 2005, 166 (1–4), 49–63. 10.1007/s11270-005-8265-9. [DOI] [Google Scholar]
- Breining G.We’re pouring millions of tons of salt on roads each winter. Here’s why that’s a problem. https://ensia.com/features/road-salt/ (accessed September 18, 2019).
- Kolesar K. R.; Mattson C. N.; Peterson P. K.; et al. Increases in Wintertime PM2.5 Sodium and Chloride Linked to Snowfall and Road Salt Application. Atmos. Environ. 2018, 177, 195–202. 10.1016/j.atmosenv.2018.01.008. [DOI] [Google Scholar]
- Denby B. R.; Ketzel M.; Ellermann T.; et al. Road Salt Emissions: A Comparison of Measurements and Modelling Using the NORTRIP Road Dust Emission Model. Atmos. Environ. 2016, 141, 508–522. 10.1016/j.atmosenv.2016.07.027. [DOI] [Google Scholar]
- Sansalone J. J.; Glenn D. W. Accretion and Partitioning of Heavy Metals Associated with Snow Exposed to Urban Traffic and Winter Storm Maintenance Activities. I. J. Environ. Eng. 2002, 128 (2), 151–166. 10.1061/(ASCE)0733-9372(2002)128:2(151). [DOI] [Google Scholar]
- Baergen A. M.; Styler S. A.; Van Pinxteren D.; et al. Chemistry of Urban Grime: Inorganic Ion Composition of Grime vs Particles in Leipzig, Germany. Environ. Sci. Technol. 2015, 49 (21), 12688–12696. 10.1021/acs.est.5b03054. [DOI] [PubMed] [Google Scholar]
- Baergen A. M.; Donaldson D. J. Seasonality of the Water-Soluble Inorganic Ion Composition and Water Uptake Behavior of Urban Grime. Environ. Sci. Technol. 2019, 53 (10), 5671–5677. 10.1021/acs.est.9b00532. [DOI] [PubMed] [Google Scholar]
- Denby B. R.; Kupiainen K. J.; Gustafsson M.. Review of Road Dust Emissions. In Non-Exhaust Emissions: An Urban Air Quality Problem for Public Health; Impact and Mitigation Measures; Elsevier Inc., 2018; pp 183–203. 10.1016/b978-0-12-811770-5.00009-1. [DOI] [Google Scholar]
- Lazarcik J.; Dibb J. E. Evidence of Road Salt in New Hampshire’s Snowpack Hundreds of Meters from Roadways. Geosciences 2017, 7 (3), 54. 10.3390/geosciences7030054. [DOI] [Google Scholar]
- Haskins J. D.; Jaeglé L.; Shah V.; et al. Wintertime Gas-Particle Partitioning and Speciation of Inorganic Chlorine in the Lower Troposphere over the Northeast United States and Coastal Ocean. J. Geophys. Res.: Atmos. 2018, 123, 1–20. 10.1029/2018JD028786. [DOI] [Google Scholar]
- Mielke L. H.; Furgeson A.; Odame-Ankrah C. A.; Osthoff H. D.; et al. Ubiquity of ClNO2 in the Urban Boundary Layer of Calgary, Alberta, Canada. Can. J. Chem. 2016, 94, 414. 10.1139/cjc-2015-0426. [DOI] [Google Scholar]
- Chang W. L.; Bhave P. V.; Brown S. S.; et al. Heterogeneous Atmospheric Chemistry, Ambient Measurements, and Model Calculations of N2O5: A Review. Aerosol Sci. Technol. 2011, 45 (6), 665–695. 10.1080/02786826.2010.551672. [DOI] [Google Scholar]
- Bertram T. H.; Thornton J. A. Toward a General Parameterization of N2O5 Reactivity on Aqueous Particles: The Competing Effects of Particle Liquid Water, Nitrate and Chloride. Atmos. Chem. Phys. 2009, 9, 8351–8363. 10.5194/acp-9-8351-2009. [DOI] [Google Scholar]
- Riemer N.; Ault A. P.; West M.; Craig R. L.; Curtis J. H. Aerosol Mixing State: Measurements, Modeling, and Impacts. Rev. Geophys. 2019, 57, 187–6. 10.1029/2018RG000615. [DOI] [Google Scholar]
- Gaston C. J.; Quinn P. K.; Bates T. S.; et al. The Impact of Shipping, Agricultural, and Urban Emissions on Single Particle Chemistry Observed Aboard the R/V Atlantis during CalNex. J. Geophys. Res. Atmos. 2013, 118 (10), 5003–5017. 10.1002/jgrd.50427. [DOI] [Google Scholar]
- Healy R. M.; Hellebust S.; Kourtchev I.; et al. Source Apportionment of PM2.5 in Cork Harbour, Ireland Using a Combination of Single Particle Mass Spectrometry and Quantitative Semi-Continuous Measurements. Atmos. Chem. Phys. 2010, 10 (19), 9593–9613. 10.5194/acp-10-9593-2010. [DOI] [Google Scholar]
- Tham Y. J.; Wang Z.; Li Q.; Wang W.; Wang X.; Lu K.; Ma N.; Yan C.; Kecorius S.; Wiedensohler A.; Zhang Y.; Wang T. Heterogeneous N2O5 Uptake Coefficient and Production Yield of ClNO2 in Polluted Northern China: Roles of Aerosol Water Content and Chemical Composition. Atmos. Chem. Phys. 2018, 18, 13155–13171. 10.5194/acp-18-13155-2018. [DOI] [Google Scholar]
- Phillips G. J.; Thieser J.; Tang M. J.; et al. Estimating N2O5 Uptake Coefficients Using Ambient Measurements of NO3, N2O5, ClNO2 and Particle-Phase Nitrate. Atmos. Chem. Phys. 2016, 3 (August), 1–34. 10.5194/acp-2016-693. [DOI] [Google Scholar]
- Mielke L. H.; Stutz J.; Tsai C.; et al. Heterogeneous Formation of Nitryl Chloride and Its Role as a Nocturnal NOx Reservoir Species during CalNex-LA 2010. J. Geophys. Res. Atmos. 2013, 118 (18), 10638–10652. 10.1002/jgrd.50783. [DOI] [Google Scholar]
- Riedel T. P.; Bertram T. H.; Ryder O. S.; et al. Direct N2O5 Reactivity Measurements at a Polluted Coastal Site. Atmos. Chem. Phys. 2012, 12 (6), 2959–2968. 10.5194/acp-12-2959-2012. [DOI] [Google Scholar]
- Bertram T. H.; Thornton J. A.; Riedel T. P.; Middlebrook A. M.; Bahreini R.; Bates T. S.; Quinn P. K.; Coffman D. J. Direct Observations of N2O5 Reactivity on Ambient Aerosol Particles. Geophys. Res. Lett. 2009, 36 (19), 1–5. 10.1029/2009GL040248. [DOI] [Google Scholar]
- McDuffie E. E.; Fibiger D. L.; Dubé W. P.; et al. ClNO2 Yields from Aircraft Measurements during the 2015 WINTER Campaign and Critical Evaluation of the Current Parameterization. J. Geophys. Res.: Atmos. 2018, 123, 1–22. 10.1029/2018JD029358. [DOI] [Google Scholar]
- McDuffie E. E.; Fibiger D. L.; Dubé W. P.; et al. Heterogeneous N2O5 Uptake During Winter: Aircraft Measurements During the 2015 WINTER Campaign and Critical Evaluation of Current Parameterizations. J. Geophys. Res. Atmos. 2018, 123 (8), 4345–4372. 10.1002/2018JD028336. [DOI] [Google Scholar]
- Liao J.; Sihler H.; Huey L. G.; et al. A Comparison of Arctic BrO Measurements by Chemical Ionization Mass Spectrometry and Long Path-Differential Optical Absorption Spectroscopy. J. Geophys. Res. 2011, 116, D00R02. 10.1029/2010JD014788. [DOI] [Google Scholar]
- Mielke L. H.; Furgeson A.; Osthoff H. D. Observation of ClNO2 in a Mid-Continental Urban Environment. Environ. Sci. Technol. 2011, 45 (20), 8889–8896. 10.1021/es201955u. [DOI] [PubMed] [Google Scholar]
- Riedel T. P.; Wagner N. L.; Dubé W. P.; et al. Chlorine Activation within Urban or Power Plant Plumes: Vertically Resolved ClNO2 and Cl2 Measurements from a Tall Tower in a Polluted Continental Setting. J. Geophys. Res. Atmos. 2013, 118 (15), 8702–8715. 10.1002/jgrd.50637. [DOI] [Google Scholar]
- Wagner N. L.; Riedel T. P.; Young C. J.; et al. N2O5 Uptake Coefficients and Nocturnal NO2 Removal Rates Determined from Ambient Wintertime Measurements. J. Geophys. Res. Atmos. 2013, 118 (16), 9331–9350. 10.1002/jgrd.50653. [DOI] [Google Scholar]
- Stutz J.; Alicke B.; Ackermann R.; et al. Vertical Profiles of NO3, N2O5, O3, and NOx in the Nocturnal Boundary Layer: 1. Observations during the Texas Air Quality Study 2000. J. Geophys. Res. 2004, 109 (12), 1–15. 10.1029/2003JD004209. [DOI] [Google Scholar]
- Tham Y. J.; Wang Z.; Li Q.; et al. Significant Concentrations of Nitryl Chloride Sustained in the Morning: Investigations of the Causes and Impacts on Ozone Production in a Polluted Region of Northern China. Atmos. Chem. Phys. 2016, 16, 14959–14977. 10.5194/acp-16-14959-2016. [DOI] [Google Scholar]
- Ault A. P.; Peters T. M.; Sawvel E. J.; et al. Single-Particle SEM-EDX Analysis of Iron-Containing Coarse Particulate Matter in an Urban Environment: Sources and Distribution of Iron within Cleveland, Ohio. Environ. Sci. Technol. 2012, 46 (8), 4331–4339. 10.1021/es204006k. [DOI] [PubMed] [Google Scholar]
- Pratt K. A.; Mayer J. E.; Holecek J. C.; et al. Development and Characterization of an Aircraft Aerosol Time-of-Flight Mass Spectrometer. Anal. Chem. 2009, 81 (5), 1792–1800. 10.1021/ac801942r. [DOI] [PubMed] [Google Scholar]
- Markovic M. Z.; VandenBoer T. C.; Murphy J. G. Characterization and Optimization of an Online System for the Simultaneous Measurement of Atmospheric Water-Soluble Constituents in the Gas and Particle Phases. J. Environ. Monit. 2012, 14 (7), 1872. 10.1039/c2em00004k. [DOI] [PubMed] [Google Scholar]
- Staudt S.; Gord J. R.; Karimova N. V.; et al. Sulfate and Carboxylate Suppress the Formation of ClNO2 at Atmospheric Interfaces. ACS Earth Sp. Chem. 2019, 3 (9), 1987–1997. 10.1021/acsearthspacechem.9b00177. [DOI] [Google Scholar]
- Ahern A. T.; Goldberger L.; Jahl L.; et al. Production of N2O5 and ClNO2 through Nocturnal Processing of Biomass-Burning Aerosol. Environ. Sci. Technol. 2018, 52 (2), 550–559. 10.1021/acs.est.7b04386. [DOI] [PubMed] [Google Scholar]
- Pratt K. A.; Murphy S. M.; Subramanian R.; et al. Flight-Based Chemical Characterization of Biomass Burning Aerosols within Two Prescribed Burn Smoke Plumes. Atmos. Chem. Phys. 2011, 11 (24), 12549–12565. 10.5194/acp-11-12549-2011. [DOI] [Google Scholar]
- Behnke W.; George C.; Scheer V.; et al. Production and Decay of ClNO2 from the Reaction of Gaseous N2O5 with NaCl Solution : Bulk and Aerosol Experiments. J. Geophys. Res. 1997, 102 (D3), 3795–3804. 10.1029/96JD03057. [DOI] [Google Scholar]
- Crowley J. N.; Ammann M.; Cox R. A.; et al. Evaluated Kinetic and Photochemical Data for Atmospheric Chemistry: Volume v -Heterogeneous Reactions on Solid Substrates. Atmos. Chem. Phys. 2010, 10 (18), 9059–9223. 10.5194/acp-10-9059-2010. [DOI] [Google Scholar]
- Saathoff H.; Naumann K.-H.; Riemer N.; et al. The Loss of NO2, HNO3, NO3/N2O5, and HO2 /HOONO2 on Soot Aerosol: A Chamber and Modeling Study. Geophys. Res. Lett. 2001, 28 (10), 1957–1960. 10.1029/2000GL012619. [DOI] [Google Scholar]
- Dugan H. A.; Bartlett S. L.; Burke S. M.; et al. Salting Our Freshwater Lakes. Proc. Natl. Acad. Sci. U. S. A. 2017, 114 (17), 4453–4458. 10.1073/pnas.1620211114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaston C. J.; Thornton J. A.; Ng N. L. Reactive Uptake of N2O5 to Internally Mixed Inorganic and Organic Particles: The Role of Organic Carbon Oxidation State and Inferred Organic Phase Separations. Atmos. Chem. Phys. 2014, 14 (11), 5693–5707. 10.5194/acp-14-5693-2014. [DOI] [Google Scholar]
- Anttila T.; Kiendler-Scharr A.; Tillmann R.; et al. On the Reactive Uptake of Gaseous Compounds by Organic-Coated Aqueous Aerosols: Theoretical Analysis and Application to the Heterogeneous Hydrolysis of N2O5. J. Phys. Chem. A 2006, 110 (35), 10435–10443. 10.1021/jp062403c. [DOI] [PubMed] [Google Scholar]
- Ryder O. S.; Campbell N. R.; Morris H.; et al. Role of Organic Coatings in Regulating N2O5 Reactive Uptake to Sea Spray Aerosol. J. Phys. Chem. A 2015, 119 (48), 11683–11692. 10.1021/acs.jpca.5b08892. [DOI] [PubMed] [Google Scholar]
- Ryder O. S.; Ault A. P.; Cahill J. F.; et al. On the Role of Particle Inorganic Mixing State in the Reactive Uptake of N2O5 to Ambient Aerosol Particles. Environ. Sci. Technol. 2014, 48 (3), 1618–1627. 10.1021/es4042622. [DOI] [PubMed] [Google Scholar]
- Lelieveld J.; Gromov S.; Pozzer A.; et al. Global Tropospheric Hydroxyl Distribution, Budget and Reactivity. Atmos. Chem. Phys. 2016, 16 (19), 12477–12493. 10.5194/acp-16-12477-2016. [DOI] [Google Scholar]
- Wang T.; Tham Y. J.; Xue L.; Li Q.; Zha Q.; Wang Z.; Poon S. C. N.; Dube W. P.; Blake D. R.; Louie P. K. K.; Luk C. W. Y.; Tsui W.; Brown S. S. Observations of Nitryl Chloride and Modeling Its Source and Effect on Ozone in the Planetary Boundary Layer of Southern China. J. Geophys. Res. Atmos. 2016, 121, 2476–2489. 10.1002/2015JD024556. [DOI] [Google Scholar]
- Sarwar G.; Simon H.; Xing J.; et al. Importance of Tropospheric ClNO2 Chemistry across the Northern Hemisphere. Geophys. Res. Lett. 2014, 41 (11), 4050–4058. 10.1002/2014GL059962. [DOI] [Google Scholar]
- Stanier C.; Singh A.; Adamski W.; et al. Overview of the LADCO Winter Nitrate Study: Hourly Ammonia, Nitric Acid and PM2.5 Composition at an Urban and Rural Site Pair during PM2.5 Episodes in the US Great Lakes Region. Atmos. Chem. Phys. 2012, 12 (22), 11037–11056. 10.5194/acp-12-11037-2012. [DOI] [Google Scholar]
- Wang X.; Jacob D. J.; Eastham S. D.; Sulprizio M. P.; Zhu L.; Chen Q.; Alexander B.; Sherwen T.; Evans M. J.; Lee B. H.; Haskins J. D.; Lopez-Hilfiker F. D.; Thornton J. A.; Huey G. L.; Liao H. The Role of Chlorine in Tropospheric Chemistry. Atmos. Chem. Phys. 2019, 19, 3981–4003. 10.5194/acp-19-3981-2019. [DOI] [Google Scholar]
- McNamara S. M.; Raso A. R. W.; Wang S.; et al. Springtime Nitrogen Oxide-Influenced Chlorine Chemistry in the Coastal Arctic. Environ. Sci. Technol. 2019, 53 (14), 8057–8067. 10.1021/acs.est.9b01797. [DOI] [PubMed] [Google Scholar]
- Song X. H.; Hopke P. K.; Fergenson D. P.; et al. Classification of Single Particles Analyzed by ATOFMS Using an Artificial Neural Network, ART-2A. Anal. Chem. 1999, 71 (4), 860–865. 10.1021/ac9809682. [DOI] [Google Scholar]
- Kumar P.; Hopke P. K.; Raja S.; et al. Characterization and Heterogeneity of Coarse Particles across an Urban Area. Atmos. Environ. 2012, 46, 449–459. 10.1016/j.atmosenv.2011.09.018. [DOI] [Google Scholar]
- Corbin J. C.; Rehbein P. J. G.; Evans G. J.; et al. Combustion Particles as Ice Nuclei in an Urban Environment: Evidence from Single-Particle Mass Spectrometry. Atmos. Environ. 2012, 51, 286–292. 10.1016/j.atmosenv.2012.01.007. [DOI] [Google Scholar]
- Sullivan R. C.; Guazzotti S. A.; Sodeman D. A.; et al. Direct Observations of the Atmospheric Processing of Asian Mineral Dust. Atmos. Chem. Phys. 2007, 7 (5), 1213–1236. 10.5194/acp-7-1213-2007. [DOI] [Google Scholar]
- Pósfai M.; Simonics R.; Li J.; Hobbs P. V.; Buseck P. R.; Individual Aerosol Particles from Biomass Burning in Southern Africa: 1. Compositions and Size Distributions of Carbonaceous Particles. J. Geophys. Res. Atmos. 2003, 108 ( (D13), ). 10.1029/2002JD002291. [DOI] [Google Scholar]
- Moffet R. C.; Henn T. R.; Tivanski A. V.; et al. Microscopic Characterization of Carbonaceous Aerosol Particle Aging in the Outflow from Mexico City. Atmos. Chem. Phys. 2010, 10 (3), 961–976. 10.5194/acp-10-961-2010. [DOI] [Google Scholar]
- Dall’Osto M.; Beddows D. C. S.; Gietl J. K.; et al. Characteristics of Tyre Dust in Polluted Air: Studies by Single Particle Mass Spectrometry (ATOFMS). Atmos. Environ. 2014, 94, 224–230. 10.1016/j.atmosenv.2014.05.026. [DOI] [Google Scholar]
- Németh Z.; Pósfai M.; Nyiro-Kósa I.; et al. Images and Properties of Individual Nucleated Particles. Atmos. Environ. 2015, 123, 166–170. 10.1016/j.atmosenv.2015.10.051. [DOI] [Google Scholar]
- Toner S. M.; Sodeman D. A.; Prather K. A. Single Particle Characterization of Ultrafine and Accumulation Mode Particles from Heavy Duty Diesel Vehicles Using Aerosol Time-of-Flight Mass Spectrometry. Environ. Sci. Technol. 2006, 40 (12), 3912–3921. 10.1021/es051455x. [DOI] [PubMed] [Google Scholar]
- Toner S. M.; Shields L. G.; Sodeman D. A.; et al. Using Mass Spectral Source Signatures to Apportion Exhaust Particles from Gasoline and Diesel Powered Vehicles in a Freeway Study Using UF-ATOFMS. Atmos. Environ. 2008, 42 (3), 568–581. 10.1016/j.atmosenv.2007.08.005. [DOI] [Google Scholar]
- Khlystov A.; Stanier C.; Pandis S. N. An Algorithm for Combining Electrical Mobility and Aerodynamic Size Distributions Data When Measuring Ambient Aerosol Special Issue of Aerosol Science and Technology on Findings from the Fine Particulate Matter Supersites Program. Aerosol Sci. Technol. 2004, 38 (sup1), 229–238. 10.1080/02786820390229543. [DOI] [Google Scholar]
- Wagner J.; Leith D. Passive Aerosol Sampler. Part I: Principle of Operation. Aerosol Sci. Technol. 2001, 34 (2), 186–192. 10.1080/027868201300034808. [DOI] [Google Scholar]
- Reinard M. S.; Adou K.; Martini J. M.; et al. Source Characterization and Identification by Real-Time Single Particle Mass Spectrometry. Atmos. Environ. 2007, 41 (40), 9397–9409. 10.1016/j.atmosenv.2007.09.001. [DOI] [Google Scholar]
- Cziczo D. J.; Abbatt J. P. D. Infrared Observations of the Response of NaCl, MgCl2, NH4HSO4, and NH4NO3 Aerosols to Changes in Relative Humidity from 298 to 238 K. J. Phys. Chem. A 2000, 104 (10), 2038–2047. 10.1021/jp9931408. [DOI] [Google Scholar]
- Tan P. V.; Evans G. J.; Tsai J.; et al. On-Line Analysis of Urban Particulate Matter Focusing on Elevated Wintertime Aerosol Concentrations. Environ. Sci. Technol. 2002, 36 (16), 3512–3518. 10.1021/es011448i. [DOI] [PubMed] [Google Scholar]
- Wang S.; Pratt K. A. Molecular Halogens Above the Arctic Snowpack: Emissions, Diurnal Variations, and Recycling Mechanisms. J. Geophys. Res. Atmos. 2017, 122 (21), 11991–12007. 10.1002/2017JD027175. [DOI] [Google Scholar]
- Valdez M. P.; Bales R. C.; Stanley D. A.; et al. Gaseous Deposition to Snow: 1. Experimental Study of SO2 and NO2 Deposition. J. Geophys. Res. 1987, 92 (D8), 9779. 10.1029/JD092iD08p09779. [DOI] [Google Scholar]
- Huff D. M.; Joyce P. L.; Fochesatto G. J.; et al. Deposition of Dinitrogen Pentoxide, N2O5, to the Snowpack at High Latitudes. Atmos. Chem. Phys. 2011, 11 (10), 4929–4938. 10.5194/acp-11-4929-2011. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.