

Abstract
The field of preimplantation genetic testing (PGT) is evolving fast and best practice advice is essential for regulation and standardisation of diagnostic testing. The previous ESHRE guidelines on best practice for PGD, published in 2005 and 2011, are considered outdated, and the development of new papers outlining recommendations for good practice in PGT was necessary.
The current paper provides recommendations on the technical aspects of PGT for monogenic/single-gene defects (PGT-M) and covers recommendations on basic methods for PGT-M and testing strategies. Furthermore, some specific recommendations are formulated for special cases, including de novo pathogenic variants, consanguineous couples, HLA typing, exclusion testing and disorders caused by pathogenic variants in the mitochondrial DNA. This paper is one of a series of four papers on good practice recommendations on PGT. The other papers cover the organisation of a PGT centre, embryo biopsy and tubing and the technical aspects of PGT for chromosomal structural rearrangements/aneuploidies.
Together, these papers should assist scientists interested in PGT in developing the best laboratory and clinical practice possible.
Keywords: ESHRE, preimplantation genetic testing, monogenic disorders, HLA, pathogenic variants, exclusion testing, good practice, mitochondrial DNA
WHAT DOES THIS MEAN FOR PATIENTS?
The paper describes good practice recommendations for preimplantation genetic testing (or PGT). Similar documents have been published in 2011, but these needed updating to the new techniques used in IVF and genetics labs.
The recommendations should help laboratory personnel and geneticist to perform PGT according to the best laboratory and clinical practice possible. The current paper provides recommendations on the genetic testing for monogenic disorders, which are diseases caused by a change in one of the genes (and where in the gene this change occurs is known). The aim of the genetic testing is to select an embryo that does not have the change in the gene causing the disorder, which can then be transferred back to the mother.
These technical recommendations are not directly relevant for patients, but they should ensure that PGT patients receive the best care possible.
Disclaimer
This Good Practice Recommendations (GPR) document represents the views of ESHRE, which are the result of consensus between the relevant ESHRE stakeholders and are based on the scientific evidence available at the time of preparation.
ESHRE GPRs should be used for information and educational purposes. They should not be interpreted as setting a standard of care or be deemed inclusive of all proper methods of care, nor exclusive of other methods of care reasonably directed to obtaining the same results. They do not replace the need for application of clinical judgment to each individual presentation, nor variations based on locality and facility type.
Furthermore, ESHRE GPRs do not constitute or imply the endorsement, or favouring of any of the included technologies by ESHRE.
Introduction
The previous terms of preimplantation genetic diagnosis (PGD) and preimplantation genetic screening (PGS) have been replaced by the term preimplantation genetic testing (PGT), following a revision of terminology used in infertility care (Zegers-Hochschild et al., 2017). PGT is defined as a test performed to analyse the DNA from oocytes (polar bodies) or embryos (cleavage stage or blastocyst) for HLA typing or for determining genetic abnormalities. This includes PGT for aneuploidy (PGT-A), PGT for monogenic/single gene defects (PGT-M) and PGT for chromosomal structural rearrangements (PGT-SR) (Zegers-Hochschild et al., 2017). PGT for chromosomal numerical aberrations of high genetic risk are included within PGT-SR in the data collections of the ESHRE PGT consortium.
PGT began as an experimental procedure in the 1990s with polymerase chain reaction (PCR)-based methods used for sex selection and the detection of monogenic diseases. Interphase fluorescence in situ hybridisation (FISH) was introduced a few years later and became the standard method for sexing embryos and for detecting numerical and structural chromosomal aberrations. Genome-wide technologies began to replace the gold standard methods of FISH and PCR over the last decade and this trend was most apparent for PGT-A. PGT-A has been carried out mainly for in vitro fertilisation (IVF) patients with original aims of increasing pregnancy rates and decreasing miscarriage rates. Other outcome measures such as increasing elective single embryo transfer and reduced time to pregnancy have been added more recently. Cited indications for PGT-A include advanced maternal age (AMA), recurrent implantation failure (RIF), severe male factor (SMF) and couples with normal karyotypes who have experienced recurrent miscarriage (RM). The value of the procedure for all IVF patients and/or appropriate patient selection remains an ongoing discussion, but this is outside the scope of this manuscript (Harper et al., 2018).
The goal of this series of papers is to bring forward best practices to be followed in all types of PGT services, offering PGT-A as well as PGT-M and PGT-SR.
In order to take PGT to the same high-quality level as routine genetic testing, guidelines for best practice have been designed by several societies. The PGD International Society has drafted guidelines (The Preimplantation Genetic Diagnosis International Society (PGDIS) (2004), Preimplantation Genetic Diagnosis International Society (2008)) while the American Society for Reproductive Medicine reviewed PGT practice in the USA (Practice Committee of the Society for Assisted Reproductive Technology and Practice Committee of the American Society for Reproductive Medicine, 2008) and published several opinion papers (on blastocyst culture, on embryo transfer and on PGT-A). The first guidelines of the ESHRE PGT Consortium were published in 2005, as one of the missions of the Consortium was to bring overall standardisation and improve quality standards (Thornhill et al., 2005). In collaboration with the Cytogenetic European Quality Assessment (CEQA) and the UK National External Quality Assessment Service (UKNEQAS), now together in Genomics Quality Assessment (GenQA), the ESHRE PGT Consortium also initiated External Quality Assessment (EQA) schemes to provide an independent evaluation of laboratories and help them improving their techniques and reports. A review of the original guidelines yielded four sets of recommendations on different aspects of PGT: one on the organisation of PGT and three relating to the methods used: embryo biopsy, amplification-based testing and FISH-based testing (Harton et al., 2011a, Harton et al., 2011b, Harton et al., 2011c, Harton et al., 2011d). These four guidelines are now being updated and extended, taking into account the fast changes in the provision of PGT services. In these updated guidelines, the laboratory performing the diagnosis will be referred to as the PGT centre and the centre performing the IVF as the IVF centre.
General aspects of PGT, including patient selection, counselling, pregnancy and children follow-up and transport PGT, will be covered in the paper on organisation of PGT. Technical recommendations for embryo biopsy and tubing will be covered in the paper on embryo biopsy. Recommendations for genetic testing will be covered in the papers on detection of numerical and structural chromosomal aberrations and on detection of monogenic disorders. The content of the different papers is aligned with the IVF/PGT clinical procedure in Fig. 1.
Figure 1.
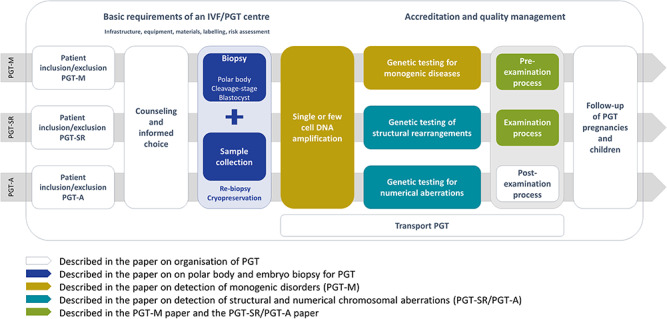
Overview of the IVF/PGT process, and how all aspects are covered by one of the four recommendations papers. IVF: in vitro fertilisation, PGT: preimplantation genetic testing.
The ESHRE PGT Consortium recognises that owing to variations in local or national regulations and specific laboratory practices, there will remain differences in the ways in which PGT is practiced (from initial referral through IVF treatment, genetic testing to follow-up of pregnancies, births and children). This does not preclude a series of consensus recommendations for best practice based on experience and available evidence. These recommendations are not intended as the only approved standard of practice nor are they legally binding. The unique needs of individual patients may justify deviation, and the recommendations must be applied according to individual patient’s needs using professional judgement. However, recommendations and opinions may be used to frame laws and regulations, and practitioners should ensure that they comply with statutory requirements or clinical practice guidelines in their own countries. To keep the papers concise, repetitions have been excluded as much as possible and many cross-references were included. Therefore, it is recommended to not consult the papers independently but always as a set when one is seeking guidance on a PGT issue.
Materials and Methods
The current paper was developed according to the published methodology for ESHRE Recommendations for good practice papers (Vermeulen et al., 2019). A working group was composed of geneticists with hands-on expertise in the described techniques, aiming at a representation of different settings and nationalities. The working group members assessed the previous guidelines (Harton et al., 2011b) and created an outline for the current paper. As the aim was to provide technical guidance and support, it was not considered relevant to perform a formal literature search and as a result, no references were added, except for references to other guidance documents. All group members, according to their expertise, wrote a section that was later discussed in depth with the entire group until consensus. Eleven online meetings were organised for discussion. The final draft of the paper was checked for consistency with the other papers of the series. The draft was then submitted for stakeholder review; it was published on the ESHRE website between 10 June and 11 July 2019, and ESHRE members were invited to send in comments. All comments were checked by the working group, discussed in an online meeting and incorporated in the final version where relevant. A review report is published on the ESHRE website.
For easier use of the recommendations, specific terms are explained in a glossary (Supplementary Table SI) and abbreviations are listed (Supplementary Table SII).
Results/Recommendations
Introduction to PGT-M
This paper provides detailed technical recommendations for the most applied methods for PGT-M.
PGT-M refers to testing for nuclear DNA pathogenic variant(s) causing monogenic disorders, with an autosomal dominant, autosomal recessive or X-linked transmission pattern, but also mitochondrial DNA (mtDNA) pathogenic variant(s). It also refers to exclusion testing and to HLA typing with or without concurrent testing for a monogenic disorder.
One of the greatest challenges for PGT-M is the low amount of input DNA, for which sensitive DNA amplification techniques are needed. Biopsied single (after polar body (PB) or single blastomere biopsy) or few cells [i.e. 5–10 trophectoderm (TE) cells] undergo either a targeted amplification reaction via multiplex PCR or a whole-genome amplification (WGA) step followed by downstream applications (targeted or genome-wide) such as PCR, single nucleotide polymorphism (SNP) arrays or next-generation sequencing (NGS) (Fig. 2). Each method has its advantages and its limitations. The principle of most of these methods is based on haplotyping (i.e. determination of the group of alleles within a genetic segment on a single chromosome being inherited together). Therefore, genetic markers located close to the gene of interest are genotyped in DNA samples from the couple and relevant family members with known genetic status during the preclinical work-up. Genetic markers that are informative, flank the locus of interest and allow discrimination of the parental haplotypes are selected for use in the clinical test. The haplotype which is common in the family members with the familial pathogenic variant is referred to as the high-risk haplotype (or mutant), whereas the haplotype without the familial pathogenic variant is referred to as the wild-type or low-risk haplotype. The clinical test can be either direct, when the pathogenic variant plus linked genetic markers are assessed, or indirect, when testing is based on haplotyping only.
Figure 2.
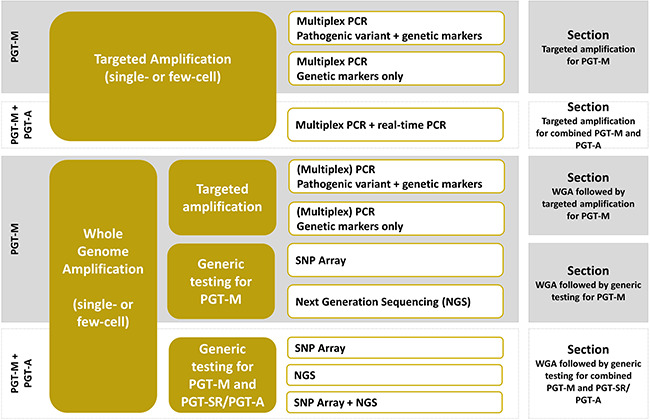
Overview of the testing strategies that can be applied for PGT-M. PGT-M: PGT for monogenic/single-gene defects, PGT-A: PGT for aneuploidy, PGT-SR: PGT for chromosomal structural rearrangements, SNP: single nucleotide polymorphism, NGS: next-generation sequencing.
The limitations of low DNA quantity are related to the increased risk of either DNA amplification failure (AF), DNA contamination or the phenomenon of allele drop-out (ADO), in which one of two alleles in a heterozygous sample is amplified while the other remains undetected. This is often more challenging for single-cell analysis compared with analysis of a few cells. The occurrence of any of these events may have a severe impact on the reliability of the diagnostic result, and precautions must be taken to minimise their occurrence or improve their detection during the test set-up and its clinical implementation.
The recommendations formulated in this section are independent of the testing method applied.
Training and personnel
Genetic testing procedures should be performed under the supervision of a specialised geneticist, competent and authorised to perform clinical diagnostics according to local or national regulations.
All personnel undertaking genetic testing should be trained adequately as required in a genetic laboratory and should follow written standard operating procedures (SOPs).
-
Training for each technique should be documented. Prior to working on clinical specimens, the following recommendations apply for each trainee;
-
∘
For tubing, training is discussed in the paper on PB and embryo biopsy for PGT (ESHRE PGT Consortium and SIG-Embryology Biopsy Working Group et al., 2020).
For targeted PCR, it is recommended that 30 to 50 single- or few-cell samples are subjected to multiplex PCR, in two or three separate testing rounds and successfully processed. Negative controls should be included to monitor contamination in each round.
For WGA, it is recommended that 30 to 50 single- or few-cell samples are subjected to WGA and that the WGA products are successfully processed in downstream application(s), in multiple separate testing rounds. Negative controls should be included to monitor contamination in each round.
-
∘
Laboratory infrastructure, equipment and materials
General aspects of infrastructure, equipment and materials are covered in the paper on organisation of PGT (ESHRE PGT Consortium Steering committee et al., 2020).
Labelling and witnessing
General guidance on labelling and witnessing is covered in the paper on organisation of PGT (ESHRE PGT Consortium Steering committee et al., 2020).
Single- or few-cell methods
PGT-M can be subdivided into the pre-examination process and the clinical cycle (examination process). The pre-examination process includes preclinical work-up with informativity/segregation analysis and eventually test development followed by validation. For informativity/segregation analysis, short tandem repeat markers (STR) or SNP marker genotyping is performed on DNA samples from the couple and related family member(s) to identify informative markers and to establish which combination of marker alleles (haplotype) segregates with the pathogenic variant. If the high-risk haplotype is determined during work-up, an indirect testing method can be applied. Alternatively, a direct method is chosen where the detection of the pathogenic variant is combined with the genetic markers for haplotype confirmation. For some cases, it will not be possible to determine the high-risk haplotype during work-up, for instance when a de novo pathogenic variant is present or when no relevant family DNA samples can be obtained (see also section De novo pathogenic variant(s)). In these cases, it may be determined during the clinical cycle based on the results from the biopsied embryo cells.
The following section describes pathogenic variant and genetic marker loci and the most applied methods for their detection.
Pathogenic variant and genetic marker loci
Pathogenic variant loci can be nuclear or mitochondrial and involve germline genetic variant(s) proven to be disease causing (previously termed mutation). Whether the pathogenic variants themselves are incorporated in the clinical test depends on multiple factors, including the nature of the pathogenic variant (familial or de novo), the availability of relevant family DNA samples, the variant type and the preclinical work-up results. For mitochondrial diseases the variant is always included, because the test is based on the determination of the percentage of the genetic variant present in the embryo.
STR markers are short tandemly repeated DNA sequences (dinucleotides are the most common), which are highly polymorphic and quite abundant in the human genome (one STR per 2000–10 000 bp). Useful STR markers are taken from published papers or in silico selected from public databases and usually involve many alleles (high heterozygosity). STR loci are targeted with fluorescently labelled primers and coamplified in a multiplex PCR reaction.
A fully informative STR marker will have different amplicon sizes for each of the four parental alleles, allowing discrimination of all possible embryo genotypes and detection of problems of contamination, ADO, recombination and copy number aberrations. A partially informative (semi- or limited informative) STR marker indicates that not all embryo genotypes can be distinguished and is less powerful in detecting additional problems. A non-informative STR marker is a marker that cannot distinguish between an affected and an unaffected embryo. This is illustrated in an example for an autosomal dominant disorder (Table I).
Table I.
Example of short tandem repeat informativity results for an autosomal dominant disorder.
| Affected male partner | Unaffected female partner | Informativity | ADO detection in the embryo | Detection of maternal contamination | Additional info on monosomy/trisomy | Comments | Recommendation for PGT-M (ranking1) |
|---|---|---|---|---|---|---|---|
| 124–126* | 120–122 | Fully informative | Yes | Yes | Yes | 4 distinctive parental alleles | Preferred marker (1) |
| 124–126* | 120–120 | Informative | Yes | No | Partially | 3 distinctive parental alleles, the affected partner is heterozygous, the unaffected partner is homozygous for a third allele. The wild-type allele is a unique allele. | Good marker (2) |
| 124–126* | 120–126 | Partially informative | Partially | Partially | Partially | 3 distinctive parental alleles, both partners are heterozygous, but the mutant allele of the affected partner is shared with an allele of the unaffected partner. The wild-type allele is a unique allele. Unaffected embryos (124–120 or 124–126) can be distinguished, as well as one genotype of affected embryos (126*-120). The second genotype of affected embryos is homozygous (126*-126), therefore it is uncertain if both paternal and maternal alleles are present. | Usable marker (3) |
| 124–126* | 126–126 | Partially informative | Partially | No | Partially | 2 distinctive parental alleles, the wild-type allele is a unique allele; the marker yields only information about the wild-type allele and is therefore limited in use | Usable marker (4) |
| 124–126* | 120–124 | Partially informative | Partially | Partially | Partially | 3 distinctive parental alleles, both partners are heterozygous, but the wild-type allele of the affected partner is shared by the unaffected partner. One genotype of unaffected embryos (124–120) can be distinguished from affected embryos (126*-120 or 126*-124); the second genotype of unaffected embryos is homozygous (124–124), therefore it is uncertain if both paternal and maternal alleles are present. | Usable marker (5) |
| 124–126* | 124–124 | Partially informative | Partially | No | Partially | 2 distinctive parental alleles, the mutant allele is a unique allele; the marker yields limited information about the mutant allele. | Usable marker (6) |
| 124–126* | 124–126 | Partially informative | Partially | No | No | 2 distinctive parental alleles, no unique alleles; the marker yields very limited information (to be used in combination with other markers) | Usable marker (7) |
| 126–126* | Any genotype | Non-informative | No information about the monogenic disorder but may yield information on parental contribution. | Not recommended |
1The utility of the markers is ranked from very good (1) to very low (7)
The pathogenic allele is indicated with * after validation of segregation analysis with a suitable reference.
ADO: allele drop-out, PGT-M: preimplantation genetic testing for monogenic/single-gene defects
The ranking of the marker according to its informativity takes into consideration the presence of unique alleles on the low-risk haplotype, confirming the presence of this haplotype. The ranking can be used when developing a new test, but any (partially) informative marker included in an existing protocol can be helpful, independent of its ranking.
SNPs are mostly biallelic and have a lower information content per marker compared to STRs. Three informative SNPs provide equivalent information to a single informative STR, but SNPs are much more abundant (one SNP per 300–1000 bp), easier to interpret and amenable to high-throughput analysis.
A SNP combination in a couple is informative when a clear distinction between the high-risk and low-risk allele(s) can be made. An informative SNP marker in which the wild-type allele is unique is the most powerful, as unaffected embryos are then distinguished by heterozygous SNPs, limiting the misdiagnosis risk due to ADO. This is illustrated in an example for an autosomal dominant disorder (Table II).
Table II.
Example of single nucleotide polymorphism informativity results for an autosomal dominant disorder.
| Affected male partner | Unaffected female partner | Informativity | ADO detection in the embryo | Detection of maternal contamination | Additional info on monosomy/trisomy | Comments | Recommendation for PGT-M (ranking1) |
|---|---|---|---|---|---|---|---|
| A*B | AA | Informative | Partially | No | Partially | The wild-type allele is a unique allele, therefore unaffected embryos are heterozygous and can be distinguished | Preferred marker (1) |
| AB* | BB | ||||||
| AB* | AA | Informative | Partially | No | Partially | The mutant allele is a unique allele therefore unaffected embryos are homozygous; therefore, it is uncertain whether both paternal and maternal alleles are present | Usable marker (2) |
| A*B | BB | ||||||
| A*B or AB* | AB | Non-informative | No | No | No | The marker yields very limited information (to be used in combination with other markers) | Usable marker (3) |
| AA or BB | Any genotype | Non-informative | - | - | - | No information | Not recommended |
1The utility of the markers is ranked from very good (1) to very low (3)
The pathogenic allele is indicated with *after validation of segregation with a suitable reference.
Informativity results are first evaluated for each marker separately; afterwards, the overall effectiveness of the selected set of markers to be used in the clinical test is assessed for its ability to evaluate the status of the embryo relative to the monogenic disorder, as well as other parameters such as occurrence of ADO, monosomy, trisomy and parental (mostly maternal) contamination.
Basic methods for allele discrimination
Pathogenic variant and marker loci are amplified with PCR primer pairs in which one primer is fluorescently labelled, allowing sensitive detection of the amplification products afterwards. The method is designed so that wild-type and pathogenic or high-risk allele discrimination is part of the amplification itself [e.g. double amplification refractory mutation system (D-ARMS)], or allele discrimination is carried out in a post-amplification step (e.g. mini-sequencing). In some cases, a DNA purification step may be required to remove primers and buffer components of the amplification reaction, before starting the post-amplification reaction.
Fragment length analysis
Principle of the test
This approach is based on different migration patterns of fluorescently labelled DNA molecules according to their molecular weight or size. Fragment length analysis is usually carried out via capillary electrophoresis on an automated sequencer. Allele discrimination for STR markers and insertion/deletion pathogenic variant loci is performed via fragment length analysis directly following PCR.
Limitations of the test
For pathogenic variant(s), direct allele discrimination following PCR via fragment length analysis is limited to variants that generate DNA fragments of different size. Although it is technically feasible to distinguish fragments differing by 1 bp, this may require another strategy for more accurate discrimination. For other loci, such as SNPs and single nucleotide variant(s) which do not generate PCR products of different size, direct amplification methods exist (e.g. D-ARMS), but often amplification is followed by post-PCR reactions for allele discrimination. The direct detection of complex and/or larger gene rearrangements may not be feasible, as the exact break points are often unknown, or their amplification is not possible as single- or few-cell targeted PCR fragments usually remain below 500 bp.
For STRs, especially with dinucleotide repeats, stutter patterns (one repeat unit less in size) may complicate allele discrimination and make data interpretation more difficult.
Restriction enzyme digestion
Principle of the test
A common form of DNA sequence variation detection is based on the ability of restriction enzymes to recognise specific DNA sequences and cleave the strands very close to, or at the site of, the variant. As a variant can create or destroy a restriction site, fragment length analysis will reveal the presence or absence of the variant. This method is a post-PCR reaction, which may require a prior DNA purification step. The restriction enzyme digestion is followed by fragment length analysis. It is recommended to always check for complete restriction digestion.
Limitations of the test
This approach can be used if the pathogenic variant creates or destroys a restriction site. If not, primer design may be adapted in order to generate an artificial restriction site.
It is preferable to apply this method in cases where the pathogenic variant destroys rather than when it creates a restriction site. When the pathogenic variant destroys a restriction site, the normal allele will be digested whereas the mutant allele will remain undigested. When the pathogenic variant creates a restriction site, failed or incomplete digestion could lead to misdiagnosis.
Double amplification refractory mutation system
Principle of the test
Double amplification refractory mutation system (D-ARMS) allows the amplification of both the wild-type and the pathogenic or high-risk allele for single nucleotide pathogenic variant(s). The test relies on a set of three PCR primers: a common fluorescently labelled primer, and two primers located at the target site with the last 3′ nucleotide overlapping the single nucleotide pathogenic variant, one primer being specific for the normal allele and one specific for the mutant allele. A tail is added at the 5′ end of one primer to enable sizing discrimination between wild-type and pathogenic or high-risk alleles following single-round PCR and fragment length analysis. For ARMS primers, it is recommended to introduce an additional mismatch between three and five nucleotides upstream of the 3′ end of each specific primer to increase the discrimination potential between pathogenic or high-risk and wild-type alleles.
Limitations of the test
D-ARMS is not recommended when the pathogenic variant is part of a nucleotide stretch, since the difference in amplification specificity between pathogenic or high-risk and wild-type alleles may be insufficient.
Real-time PCR for pathogenic variant detection and genotyping
Principle of the test
Real-time PCR is a closed-tube system where amplification is monitored in real time and post-PCR processing steps are not required. A first-round multiplex PCR precedes the nested real-time PCR to enable multiplexing for concurrent amplification of the variant locus (or loci) and informative markers. Probe design is flexible, and the most commonly used are hybridisation and hydrolysis probes. There are a number of real-time PCR platforms and chemistries suitable for PGT-M genotyping.
Limitations of the test
This approach requires dedicated instruments, and the possibility of multiplexing is limited depending on the real-time PCR platform (limited number of detector channels).
Mini-sequencing
Principle of the test
Mini-sequencing is based on Sanger sequencing but without sequencing the entire PCR product. The mini-sequencing reaction requires purified PCR products as template, together with a specific unlabelled mini-sequencing primer (forward and/or reverse), designed to anneal adjacent to the target site. The mini-sequencing primer is extended with a single dideoxy nucleotide, complementary to the target site. Each dideoxy nucleotide is labelled with a different fluorochrome, allowing alleles to be distinguished on an automated sequencer. This detection method is mainly used in cases of base substitutions, but it can also be applied for small insertions or deletions.
Limitations of the test
When applying this detection method in cases of small insertions or deletions, the nucleotide may be the same in the presence or absence of the pathogenic variant, and mini-sequencing primer design should be adapted.
Single- or few-cell targeted amplification
Following embryo biopsy, biopsied cell samples are washed, transferred to reaction tubes and lysed. Amplification reaction components are then added directly to the lysed cell(s) without prior DNA purification. Samples undergo either targeted amplification by means of multiplex PCR or WGA (see ‘Single- or few-cell whole-genome amplification’). The prevention of external DNA contamination is mandatory, together with accurate and strict sample processing. This requires a specialised laboratory environment and working practice.
When performing targeted amplification on single or few cells, the following recommendations apply.
Laboratory infrastructure, equipment and materials
General aspects on infrastructure, equipment and materials are covered in the paper on organisation of PGT (ESHRE PGT Consortium Steering committee et al., 2020). For targeted amplification-based PGT specifically, the following recommendations are made:
Infrastructure
There should be a physical separation between the genetic laboratories and the biopsy laboratory.
There should be a physical separation of the preamplification (preferentially a positive pressure room with a dedicated laminar flow hood) and the post-amplification (preferentially a negative pressure room) areas. It is recommended to have the thermal cyclers in a dedicated room (amplification area). If this is not possible, it is acceptable to have them in the post-amplification area.
When positive and negative pressure rooms are present, they are preferably enclosed by a locked chamber.
Secondary amplification reactions can be performed in the post-amplification area in a simple cabinet such as a PCR workstation or a dedicated area in which one has a restricted area to process the samples.
A dedicated set of equipment (including thermal cyclers), consumables and laboratory coats should be used for each designated area and not be exchanged between these areas.
An appropriate unidirectional workflow should be in place, avoiding any backflow of amplified products to the preamplification area.
Preferably, the pre- and post-amplification rooms/areas should be equipped with UV-C light for DNA decontamination.
Equipment
Equipment required for amplification-based analysis of samples includes the following:
-
-
class II safety cabinets, preferably equipped with UV-C light, to prevent contamination of samples at the preamplification stage;
simple cabinets;
thermal cyclers with heated lids;
micro-centrifuges, vortex and pipettes;
capillary gel electrophoresis equipment for fragment analysis following amplification.
Materials
Specific materials required for targeted amplification of samples include the following:
lysis buffers, (pre)amplification enzymes and primers/probes specific to each amplification method used;
capillary gel electrophoresis materials.
Tubing of samples
General recommendations about biopsy and transfer of samples to tubes (referred to as tubing) are provided in the paper on PB and embryo biopsy for PGT (ESHRE PGT Consortium and SIG-Embryology Biopsy Working Group et al., 2020).
Work practice controls
It is recommended to use positive and negative (no DNA) controls.
-
As a positive control sample, diluted and/or undiluted genomic DNA from the couple is recommended. DNA samples from other family members may also be included. In addition, single- or few-cell samples can be used. Positive control cell samples can be lymphocytes, buccal cells or cultured cells. If the test includes the pathogenic variant detection, it is recommended to use:
-
∘
For dominant diseases: DNA samples with high-risk and low-risk genotypes;
For X-linked diseases: DNA samples with high-risk, low-risk, male and female genotypes;
For recessive diseases: DNA samples with heterozygous pathogenic variant carrier, homozygous normal, and (if available) homozygous or compound heterozygous genotypes.
-
∘
-
Negative controls should be included to confirm that there was no contamination introduced from the procedure of sample collection or from the amplification reactions.
-
∘
A minimum of one negative control per buffer (sample collection buffer or washing media, depending on the protocols of the PGT centre) is recommended to control for contamination during each step of cell sample collection (i.e. the IVF laboratory negative control); for example, collection at two different time points for a specific cohort of embryos should yield a minimum of two negative controls of this type. As the contamination risk is substantially higher when working with single cells in comparison with few cells, the number of negative controls should preferably be increased, preferably one negative control per biopsied embryo;
A minimum of one negative control with amplification mixture only is recommended to control for contamination during setting up of amplification reactions (i.e. the genetic laboratory negative control).
-
∘
Single- or few-cell WGA
Following embryo biopsy, cell samples are washed and transferred to reaction tubes. After cell lysis, WGA reaction components are added without prior DNA purification. WGA allows the provision of sufficient DNA template from minute DNA samples to carry out subsequent DNA amplifications or to be used with other downstream techniques such as multiple standard PCR testing, array-based comparative genomic hybridisation (aCGH), SNP array or high-throughput assays such as NGS. Moreover, WGA products should be stored, according to the local quality system or legislation, for years (at −20°C) to facilitate their use later in time to confirm results/diagnosis or carry out new tests.
Several methods for WGA have been developed over time and are available as commercial kits. Any WGA technique should be evaluated with regards to genomic coverage, high fidelity of the sequence, reliable quantification of copy number variation and technical errors of ADO and allele drop-in (ADI). A WGA method should be selected in function of the downstream application, taking into account advantages and disadvantages. Currently, multiple displacement amplification (MDA) is recommended for PGT-M (e.g. SNP haplotyping), whereas displacement degenerate oligonucleotide-primed PCR (DOP-PCR) (marketed as Picoplex/Sureplex) is the method of choice for the detection of chromosomal copy number variation.
When applying WGA on single or few cells, the recommendations for laboratory infrastructure, equipment and materials, tubing and controls are described below.
Laboratory infrastructure, equipment and materials
In general, follow the recommendations as stated in section ‘Single- or few-cell targetedamplification’.
The following additional recommendations are made for infrastructure, equipment and materials, specifically for WGA.
Infrastructure
As WGA is a first round (primary) amplification step, it should be performed in the preamplification room/area. Reactions starting from WGA products are considered secondary reactions and should be performed in a separated area. Successful amplification should be confirmed before proceeding to downstream applications.
Equipment
Additional equipment includes the following:
-
–
gel electrophoresis equipment to check for successful amplification;
fluorometer for DNA quantification; the use of a DNA quantification system (to determine the amount of amplified DNA after WGA) is optional;
specific equipment, depending on the downstream application.
Materials
Specific materials required for WGA of samples include the following:
reagents specific to each WGA method used;
reagents for DNA quantification following WGA;
specific reagents, depending on the downstream application.
Tubing of samples
General recommendations about biopsy and tubing are provided in the paper on PB and embryo biopsy for PGT (ESHRE PGT Consortium and SIG-Embryology Biopsy Working Group et al., 2020).
Work practice controls
Positive and negative controls should be included to monitor the WGA reaction, as described in section ‘Single- or few-cell targeted amplification’.
It is acceptable to include these controls only at the level of the WGA reaction and omit them from downstream reactions.
Pre-examination process
The pre-examination process includes preclinical work-up with informativity/segregation analysis and test development followed by validation.
Informativity/segregation analysis.
It is recommended that the original molecular genetic reports including the description of identified variants together with the appropriate gene reference sequence are obtained from an accredited laboratory. It is advisable to confirm the pathogenic variant(s) whenever possible.
It is recommended to perform a preclinical work-up to assess PGT-M feasibility, identify informative genetic markers, establish parental haplotypes (when possible) and work on a clinical testing strategy.
It is recommended to perform the informativity/segregation analysis for STR markers as well as for SNP markers. The results allow evaluation of the expected genotypes in the embryos.
A geneticist experienced in pedigree and linkage analysis should determine which familial DNA samples are needed for a reliable and accurate diagnosis.
-
For all diseases, samples should be collected from the prospective parents and close relatives with known disease status (proven via genetic reports) to establish the high-risk and low-risk haplotypes.
-
∘
For dominant diseases, it is recommended that these samples include DNA from at least one affected (ideally two) and/or one unaffected individual as a reference.
For recessive diseases, these would include at least a homozygous or compound heterozygous affected individual, one carrier and one non-carrier individual as a reference.
For X-linked diseases, an affected individual must be used as a reference and/or one unaffected individual. A proven carrier would also be recommended.
-
∘
When no suitable family members are available, the informativity analysis should be performed in the couple and the segregation established during the clinical cycle (or on single sperm).
Testing strategies and test development
A test strategy is determined based on informativity/segregation analysis results. Different strategies for amplification and allele discrimination have been clinically applied.
The three main testing strategies for PGT-M are:
-
(i)
Targeted amplification of informative markers with or without the pathogenic variant(s) in a single-/few-cell multiplex PCR (sections ‘Targeted amplification for PGT-M’ and ‘Targeted amplification forcombined PGT-M and PGT-A’).
-
(ii)
WGA followed by targeted amplification of informative markers with or without the pathogenic variant(s) (section ‘WGA followed bytargeted amplification for PGT-M’).
-
(iii)
WGA followed by a generic method such as SNP array or NGS-based sequencing (sections ‘WGA followed by generic testing for PGT-M’ and ‘WGA followed by generic testing for combinedPGT-M and PGT-SR/PGT-A’).
The first strategy (i.e. targeted amplification of informative markers in a single-/few-cell multiplex PCR), including the development/validation of a new test, is more time consuming and labour intensive than the WGA-based strategies, and the turnaround time between referral and clinical cycle is significantly increased. The major disadvantage of this approach is that development and validation of the multiplex PCR to the single-/few-cell level has to be repeated with every new gene/locus/variant of interest. The second strategy (WGA followed by targeted amplification) is a step towards a more generic method, because the adaptation/validation of PCR reactions at the single cell level can be omitted from the preclinical work-up. Locus-specific information is available in both cases in the form of either genotypes (pathogenic variant detection, SNP) or allele length (STR). Nevertheless, due to their targeted nature, the majority of these tests do not provide a comprehensive view of the genome. The third approach, the development of genome-wide generic methods, tackles this issue. SNP arrays, as well as sequencing-based approaches, allow genome-wide haplotyping as well as copy number typing. The extent to which the whole genome is analysed depends on the platform and/or approach. SNP array-based methods are restricted by the fixed number of probes included on the platform of choice. Sequencing-based approaches can be more or less comprehensive, depending on the genome coverage, SNP density, and the depth of sequencing. Additionally, sequencing-based approaches are high-throughput and allow automation, reducing hands-on time and minimising the possibility of human errors. The WGA-based strategies are mostly coupled with TE biopsy, which often leaves insufficient time for fresh embryo transfer. This is overcome by cryopreservation and embryo transfer in a deferred cycle.
Further recommendations for test development are given in the following sections.
Targeted amplification for PGT-M
For many years, the co-amplification of genetic markers alone or in combination with the pathogenic variant at the level of single/few cells has been the ‘gold standard’ procedure for PGT-M. The inclusion of genetic markers in the clinical test improves the accuracy, as it not only allows for indirect pathogenic variant analysis but also allows for detection of ADO, contamination and recombination.
Recommendations for single- or few-cell targeted amplification concerning infrastructure, equipment, materials, tubing and work practice controls are described in section ‘Single- or few-cell targetedamplification’. At the preclinical work-up, informativity/segregation analysis is required, together with the development of a locus-specific test at the level of single or few cells. Based on the results of informativity/segregation analysis, suitable STR markers close to the locus of interest are selected for co-amplification in a multiplex PCR, alone or in combination with the pathogenic variant.
The adaptation of PCR reaction conditions is usually carried out in several steps. The selected—most suitable—amplicons are preferably first multiplexed on genomic DNA samples. Further fine tuning is then carried out with single- or few-cell samples. For test development, processing of at least one negative control with amplification mixture only for each amplification reaction is recommended. When working with single or few cells, negative controls with sample collection buffer only should be added as well, to control for contamination during sample collection. The optimised single-/few-cell PCR protocol is then validated on a series of single or few cells along with positive and negative controls (see also section ‘Pre-examination validation’).
Familial pathogenic variant + genetic markers (STRs and/or SNPs)
When developing pathogenic variant and STR and/or SNPs analysis for single or few cells, the following recommendations are made:
Amplicons should be designed ideally to be sized between 100 and 500 bp, using combinations of fluorochromes allowing loci discrimination.
Single-round multiplex PCR is preferred compared with nested or semi-nested PCR as it is less error-prone. When available, the use of STRs with tri-, tetra- or penta-nucleotide repeats is preferable, to reduce any confounding ambivalence due to the phenomenon of stutter patterns and to improve allele discrimination.
It is recommended to avoid STRs with a very wide range of allele size since the ADO risk of the large alleles is increased even at the genomic DNA level, leading to false homozygous genotypes during preclinical work-up and PGT-M.
-
Before moving on to single-cell validation, it is recommended to establish a correct discrimination of pathogenic variant/wild-type or marker alleles of the test at hand. It is recommended to test various genotypes concerning the pathogenic variant or marker of interest using the following DNA samples:
affected (autosomal dominant) DNA samples;
carrier (autosomal recessive, X-linked diseases) DNA samples;
unaffected DNA samples for the pathogenic variant to be tested;
DNA samples with heterozygous markers for indirect tests.
When a protocol is employed for PGT-M, it is recommended to apply the specific test to DNA or single cells from each particular couple, to discover any unexpected test results, which could render future blastomere results questionable (for example, a polymorphism which may exist under a primer used in the single-cell assay but not in the routine laboratory assay).
Polymorphic markers should have a high degree of heterozygosity and produce a clearly interpretable peak pattern and preferably be intragenic.
When using extragenic markers, it is recommended to stay within a 1-Mb (~1 cM) distance from the pathogenic variant of interest, to reduce the misdiagnosis risk due to recombination events (on average, loci 1 cM apart are expected to show 1% recombination). If no suitable markers are available within 1 Mb, markers within 2 Mb are acceptable but not advisable. This may be adapted in case of large genes or duplications.
The risk of misdiagnosis due to recombination should be considered for every marker and is especially important in case of large genes and genes with known recombination hot spots. Careful selection of markers flanking the pathogenic variant of interest will reduce the risk of misdiagnosis due to recombination.
Defining the minimum number of informative markers required in the single-/few-cell test: assuming validation data of AF and ADO rates per locus remain below 5%, it is recommended to include at least one STR or three SNPs proximal and one STR or three SNPs distal to the region of interest, together with the pathogenic variant locus (choose markers with rank 1 or 2 in Table I and rank 1 in Table II). In case of AF and ADO rates between 5 and 10%, either the test should be further optimised or a higher number of markers should be included. In case of insufficient markers of the highest rank, markers of lower rank can be selected for test development, but the number of markers should then be increased.
More markers will make the test more robust; analysis of at least two loci closely linked to and flanking the gene will reduce the risk of unacceptable misdiagnosis owing to ADO. Also, the risk for no diagnosis due to AF of a single amplicon in the multiplex will decrease.
Genetic markers only (STRs and/or SNPs)
Targeted indirect haplotype-only analysis of single or few cell(s) is applied for (i) exclusion testing, (ii) HLA typing, (iii) in case of an unknown pathogenic variant but the locus/genomic region of interest is proven causative, (iv) triplet repeat expansion (e.g. the fragile X mental retardation 1 (FMR1) CGG repeat expansion that is resistant to single cell amplification), (v) large deletions/insertions with unknown breakpoints, (vi) in case direct pathogenic variant testing is not feasible [presence of pseudogene(s), GC-rich sequences refractory to single-cell amplification] or (vii) linkage analysis in general (to avoid developing a test including the pathogenic variant). An indirect testing strategy is only applicable when high-risk and low-risk haplotypes have been established during preclinical work-up (exception, see section ‘De novo pathogenic variant(s)’).
In general, when developing an indirect test with STR and/or SNPs for single or few cells, follow the recommendations as stated in the previous section (familial pathogenic variant + genetic markers), except for the minimum number of markers required.
Assuming validation data of AF and ADO rates per locus remain below 5%, it is recommended to include at least two STRs or six SNPs proximal and two STRs or six SNPs distal to the locus of interest (choose markers according to Tables I and II). Here too, more markers are required in case higher AF and ADO rates are obtained and more markers will make the test more robust.
In cases where the region of interest is located close to a centromere or telomere, flanking markers may not be possible. It is then recommended to include the pathogenic variant in the test strategy and to combine the test with TE biopsy to limit the risk of ADO at the pathogenic variant locus. The risk of misdiagnosis due to recombination should be reconsidered. In exceptional cases where flanking markers are not possible and the pathogenic variant locus cannot be included [e.g. facioscapulohumeral muscular dystrophy (FSHD)], the test strategy will be linked with a higher risk of misdiagnosis. Such exceptional cases should be counselled in-depth, and the need for prenatal testing (of which the availability should be ascertained before the start of the PGT procedure) should be explained.
Targeted amplification for combined PGT-M and PGT-A.
PGT-M and PGT-A can be analysed simultaneously using the same biopsy sample in a testing strategy based on real-time PCR. The workflow involves four steps: cell sample lysis, multiplex preamplification, real-time PCR and analysis. After sample collection and cell lysis, samples are subjected to multiplex PCR preamplification for both PGT-A and PGT-M. For PGT-A, a pool of 96 loci is preamplified, representative of four independent regions for each chromosome. For PGT-M, a custom set of amplicons is added, based on preclinical work-up results. Aliquots of the preamplified samples are subsequently interrogated in triplicates or quadruplicates by real-time PCR and relative quantification. Only whole chromosome copy number changes can be detected for PGT-A by this strategy. Automation can be applied to streamline the procedure, which can be completed in 3–4 h and is compatible with fresh transfer, following biopsy and genetic analysis.
WGA followed by targeted amplification for PGT-M
The implementation of WGA for PGT-M has increased concomitantly with the development of TE biopsy and vitrification. The approach of prior single or few-cell WGA followed by standard PCR reactions for a set of STRs flanking the region of interest with or without the pathogenic variant is widely applied. The use of SNPs instead of STRs has been described but the clinical application has been very limited. It is being replaced by SNP array-based or NGS-based haplotyping, as these approaches allow assessment of a multitude of SNPs in a standardised way.
Recommendations for single- or few-cell WGA concerning infrastructure, equipment, materials, tubing and work practices are described in section ‘Single- or few-cell whole-genome amplification’. The following recommendations are made:
It is recommended to use an MDA-based WGA protocol for haplotyping applications because of better genome coverage and low genotyping error rates.
At the preclinical work-up, informativity/segregation analysis is required, together with the development of a locus-specific test, using WGA products as template DNA.
It is recommended to carry out a validation assay for the WGA protocol and the specific downstream test(s) with respect to the number of biopsied cells, to determine the rate of AF, ADO and preferential amplification.
Familial pathogenic variant + genetic markers (STRs and/or SNPs) after WGA
In general, when developing a test with WGA of single or few cells followed by familial pathogenic variant + STR and/or SNPs analysis, follow the recommendations as stated in section ‘Targeted amplification for PGT-M’.
ADO rates for WGA plus multiplex PCR at the single cell level are higher (20–30%) than for single-cell multiplex PCR. Biopsy of few cells is recommended for WGA application, as ADO rates will be lower than for single cells.
Single-cell biopsy is acceptable, but the higher ADO risk should be taken into account when defining the number of markers required in the downstream test.
Defining the minimum number of fully informative markers required in the few-cell test: assuming validation data of AF and ADO rates per locus remain below 5%, it is recommended to include at least one STRs or three SNPs proximal and one STRs or three SNPs distal to the locus of interest together with the pathogenic variant locus (choose markers according to Tables I and II). Again, more markers are required in case higher AF and ADO rates are obtained.
Genetic markers only (STRs and/or SNPs) after WGA
In general, when developing a test with WGA of single or few cells followed by indirect STR and/or SNP analysis, follow the recommendations as stated in section ‘Targeted amplification for PGT-M’ and in the previous paragraph [familial pathogenic variant + genetic markers (STRs and/or SNPs) after WGA].
ADO rates for WGA plus multiplex PCR at the single-cell level may be higher than for single-cell multiplex PCR, and this should be taken into account when defining the number of markers required in the downstream test.
Defining the minimum number of fully informative markers required in the few-cell test: assuming validation data of AF and ADO rates remain below 5%, it is recommended to include at least two STRs or six SNPs proximal and two STRs or six SNPs distal to the locus of interest (choose markers according to Tables I and II).
Here too, more markers are required in case higher ADO rates are obtained.
WGA followed by generic testing for PGT-M
SNP arrays for PGT-M only
SNP arrays are high-density oligo-arrays containing up to several million probes, which allow genotyping of hundreds of thousands of selected SNPs across all chromosomes in a single reaction. The commercially available SNP arrays use various methods for SNP genotyping of sample DNA: hybridisation to SNP allele-specific probes or single-base extension reactions is often applied. A given platform has a preset number of SNPs, and therefore, the position and number of SNPs within the region of interest will be fixed. The arrays are scanned, and SNP genotypes are called based on the total fluorescence and the ratio of hybridisation intensities for the two SNP alleles.
The following recommendations are made:
A relatively high DNA input is necessary for SNP arrays, so that a prior WGA step is required.
It is recommended to use an MDA-based WGA protocol for haplotyping applications because of better genome coverage and low genotyping error rates compared to other WGA methods.
As SNP arrays are generic platforms, preclinical work-up only requires informativity/segregation analysis for the locus of interest; the locus-specific development can be omitted.
It is recommended to carry out a validation assay for the WGA protocol and the SNP arrays in respect to the number of biopsied cells. No-call rates and ADO rates for WGA plus SNP arrays at the single-cell level will be higher than for few cells and this should be taken into account when defining the minimum number of informative SNPs required in the region of interest.
When using commercially available SNP array protocols, which already have been validated by the manufacturer, it is still recommended to carry out an implementation validation of the complete wet- and dry-laboratory workflow prior to clinical use. For specific recommendation regarding the implementation validation, see also section ‘Pre-examination validation’.
The turnaround time from sample processing to data analysis can vary from 24 h to several days, depending on the setting and the platform of choice. It is recommended that each laboratory validates in-house whether the implementation of shortened protocols has an effect on hybridisation efficiency and data quality.
Limitations of the test
SNP array haplotyping requires at least one first degree relative of the partner carrying the mutation for phase determination, as an indirect testing strategy is only applicable when high-risk and low-risk haplotypes have been established during preclinical work-up (exception, see section ‘De novo pathogenic variant(s)’).
NGS for PGT-M only
In NGS, a DNA polymerase catalyses the incorporation of deoxyribonucleotide triphosphates (dNTPs) into a DNA template during sequential cycles of DNA synthesis. Depending on the sequencing platform, each cycle of nucleotide incorporation is followed by the release of fluorophores or hydrogen ions. This procedure can take place across millions of fragments/molecules in a massively parallel manner.
Several approaches have been developed in the context of PGT-M, including both targeted locus-specific and generic genome-wide haplotyping-based methods. Some of these are commercially available.
The following recommendations are made:
A relatively high DNA input is necessary for NGS, so that a prior WGA step is required.
If long-read sequencing is applied, it is recommended to use a suitable WGA to ensure amplification of high molecular weight DNA.
Given that sequencing-based analysis is a generic approach, preclinical work-up only requires informativity/segregation testing; the locus-specific development can be omitted.
It is recommended to carry out a validation assay for the WGA protocol and the NGS protocol in respect to the number of biopsied cells.
When using commercially available NGS-based protocols, which already have been validated by the manufacturer, it is still recommended to carry out an implementation validation of the complete wet and dry-laboratory workflow prior to clinical use. Specific recommendations regarding the implementation validation are provided in section ‘Pre-examination validation’.
Each step in the NGS protocol will contribute to the overall quality of the data set. Quality control (QC) metrics should be established throughout the procedure, among others including analysis of the fragment length before and after adapter incorporation as well as quantification of the prepared library before and after possible size selection, to ensure optimal sample quality and DNA fragment representation in the multiplexed library samples. QC metrics should be established regarding the quality of the final sequencing data.
Optimal indexing of the samples should be used to ensure that different samples can be efficiently distinguished from each other during demultiplexing of the sequencing data.
The turnaround time from sample processing to data analysis can vary from 24 h to several days, depending on the setting and the platform of choice. Consequently, an embryo transfer can be planned in the current or a subsequent cycle.
Further general recommendations on NGS are covered in the paper on detection of structural and numerical chromosomal aberrations (ESHRE PGT-SR/PGT-A Working Group et al., 2020).
Limitations of the test
A major limitation of NGS methods is the length of reads they produce, a challenge tackled by long read sequencing technologies that allow the sequencing of single DNA molecules.
Generic haplotyping-based approaches require at least one first degree relative for phase determination. As an indirect test, it is not applicable in case of de novo pathogenic variant(s) for couples without previous pregnancies (see also section ‘De novo pathogenic variant(s)’).
Analysis software is only available for some of the developed approaches. In the absence of appropriate software, support of skilled bioinformaticians needs to be guaranteed and the software will require further validation.
WGA followed by generic testing for combined PGT-M and PGT-SR/PGT-A
Comprehensive PGT refers to the combination of PGT-M and PGT-A. Several methods have been developed towards that direction. These can be based on the parallel processing of the same WGA product with two different approaches, one aimed at PGT-M and the second at PGT-A. Alternatively, using genome-wide approaches enabling concurrent haplotyping and detection of copy number changes allow PGT-M and PGT-A to be simultaneously performed in the same test. These generic approaches can be SNP array-based, sequencing-based or a combination of the two.
The following recommendations are made:
When combined PGT-M and PGT-A are offered, it is recommended that the couple receives comprehensive counselling regarding the possible findings and the consequences on the transfer policy, according to the method used.
Regardless of the centre-specific transfer policy, it is recommended that if following analysis unaffected embryos free of aneuploidies are available, they are given priority for transfer.
The preclinical work-up for PGT-M should be performed, as described in section ‘WGA followed by generic testing for PGT-M’.
These approaches can also be used for inherited chromosomal structural variants in PGT-SR. Depending on the size of the involved segments, aberrant intensity ratios may or may not be detectable for the region(s) of interest. If detectable, it is recommended that the diagnosis is supported by log R ratio and B allele frequency values.
Additionally, even if a commercially available platform is used, an implementation validation to determine or confirm the lower size limit for the detection of segmental aneuploidies is recommended. These values may differ between platforms. It is recommended to perform the validation assay with WGA products from single or few cell samples of known karyotype and/or WGA products from embryonic cell(s) diagnosed with a formerly validated method.
Further recommendations specific to PGT-A are covered in the paper on detection of structural and numerical chromosomal aberrations (ESHRE PGT-SR/PGT-A Working Group et al., 2020).
Limitations of the test (for combined PGT-M and PGT-A)
As these tests (for combined PGT-M and PGT-A) require the presence of phasing reference(s), they are not applicable to all PGT-A indications.
Ploidy changes cannot be detected by all approaches; methods based on aCGH or NGS cannot reliably detect all types of polyploidy and haploidy (see also Table I in the paper on detection of structural and numerical chromosomal aberrations (ESHRE PGT-SR/PGT-A Working Group et al., 2020); SNP arrays and NGS-based haplotyping can identify polyploidy and haploidy.
Meiotic errors cannot be distinguished from mitotic in all cases and by all approaches.
Defining the threshold of mosaicism detection is recommended.
Pre-examination validation
For PGT-M
Validation criteria are dependent on the number of cells biopsied (single cell at cleavage stage, or few cells at blastocyst stage) and on the type of strategy used for PGT-M. It is acceptable to perform the validation on cell(s) from embryos donated to research or on other cell types such as peripheral blood lymphocytes.
Misdiagnosis risk needs to be established.
-
The following criteria apply for targeted STR-based testing and variant analysis, with or without prior WGA:
-
∘
Validation assays will determine amplification efficiency, accuracy and ADO rate. Accuracy should be >99% for single- or few-cell samples of known genotype.
The amplification efficiency per locus should be >95%. An amplification efficiency of >90% is acceptable, but more markers need to be included.
The ADO rate per locus should be <5%. An ADO rate less than 10% is acceptable, but more markers need to be included.
-
∘
Every new test based on targeted amplification should be validated.
For targeted amplification, validation assays should be performed in 50 single- or few-cell samples, in two or three separate runs prior to clinical use. It is acceptable to validate updated protocols (i.e. adaptations of existing protocols) with fewer samples.
No validation is needed on few-cell samples when the protocol has been previously validated on single cells.
- The following criteria apply for generic strategies such as SNP arrays with prior WGA:
-
∘When using commercially available SNP arrays or NGS-based protocols, which already have been validated by the manufacturer, it is still recommended to carry out an implementation validation of the complete wet- and dry-laboratory workflow prior to clinical use.
- It is recommended to perform the validation assay with WGA products from single- or few-cell samples of known genotype and/or WGA products from embryonic cell(s) diagnosed with a formerly validated method.
- The validation assay should be performed with a minimum of 50 WGA samples, ideally covering various indications.
- Validation assays will determine amplification efficiency, accuracy and minimum of genetic markers in the region of interest required for diagnosis.
- The amplification efficiency should be >95% for good quality samples (this may not be achievable for biopsy samples from embryos donated for research/training). Accuracy should be >99% for WGA samples from single or few cells of known genotype. Similarly, for WGA products from embryonic cell(s) formerly diagnosed, concordance with another validated method should be >99%.
- If both single- and few-cell analyses are to be performed clinically, it is necessary to validate each separately.
-
∘
For combined PGT-M and PGT-A
It is necessary to validate both indications. Again, validation criteria are dependent on the number of cells biopsied (single cell at cleavage stage, or few cells at blastocyst stage) and on the type of strategy used. For PGT-M, the above-mentioned recommendations apply. For PGT-A, recommendations for validation are described in the paper on detection of structural and numerical chromosomal aberrations (ESHRE PGT-SR/PGT-A Working Group et al., 2020).
Once validated, preclinical work-up and testing of PGT-M conditions on five WGA products is sufficient.
Risk assessment
Assessment of the risk of misdiagnosis with PGT-M depends on the analysis strategy followed. The residual risk of a protocol with targeted amplification of genetic markers and pathogenic variant(s) has to take into account the genetic distance of the flanking markers to the variant or gene of interest and the ADO rate of the pathogenic variant(s). Undetected recombination or double recombination and ADO of the pathogenic variant(s) may result in a misdiagnosis. Recombination may go unnoticed when using partially informative markers and imply an elevated residual risk. If a marker-only protocol is used, an undetected recombination or double recombination may also result in a misdiagnosis.
For the genome-wide SNP array- or NGS-based haplotyping strategies, the residual risk may be lower compared with the conventional targeted amplification strategies. This is due to the presence of multiple SNPs flanking a gene or locus of interest, thereby eliminating the effect of ADO of an individual marker. Also, by using multiple SNP markers the effect of a recombination event may less frequently result in an inconclusive result. Still, the distances of the used informative SNP markers to the gene are crucial for the residual recombination risk.
Risk assessment should also cover:
-
-
risks caused by errors in sample tracking;
risks caused by handling biopsy samples prior to DNA analysis which, if not performed with care, may compromise DNA integrity;
risk of inconclusive or false results due to suboptimal experimental conditions (contamination, ADO, ADI) or biological reasons (recombination, double recombination, meiotic or mitotic chromosomal aberrations);
risk of incidental findings;
risk of test failure (i.e. insufficient markers and/or sequencing to produce a diagnosis).
Preclinical work-up report
General guidance and recommendations on administration and patient information for the preclinical work-up report are provided in the paper on organisation of PGT (ESHRE PGT-SR/PGT-A Working Group et al., 2020). For PGT-M, the preclinical work-up report should also include a summary of the work-up and specify the test strategy for the clinical cycle.
It is recommended that the following are clearly stated in the report:
Indication and gene (with OMIM number when possible), pathogenic variant(s) nomenclature using Human Genome Variation Society (HGVS) recommendations;
Gene reference sequence, genome build, inheritance mode, polymorphic marker selection when using STRs, number of informative SNPs, distance from marker to gene or pathogenic variant(s), results from informativity testing on all available family members, pathogenic variant(s) detection and linkage analysis (depending on the strategy chosen for the PGT cycle);
Test limitations and residual risk of PGT misdiagnosis, including a graphic presentation.
Special cases
De novo pathogenic variant(s)
In case of a de novo pathogenic variant(s) in one partner or in a child, it is mandatory to include mutation detection in the test strategy. Determination of high-risk and low-risk haplotypes or phasing may be completed only during PGT cycle(s).
De novo pathogenic variant(s) in a prospective parent
If DNA samples from affected offspring are available, the case can be dealt with as a usual PGT-M request. If no DNA samples from affected offspring are available, the following recommendations apply:
It is mandatory to include the mutation detection in the test strategy, and diagnosis will depend on the presence or absence of the mutation. Amplification failure at the mutation locus will yield no diagnosis.
It is recommended to try to, when possible, establish the high-risk and low-risk haplotypes prior to clinical application. In case of a de novo pathogenic variant(s) in the male partner, it is recommended to establish phasing from single sperm analysis. Establishing phase from PBs for a de novo pathogenic variant(s) in the female partner is also an option, but it may be much more complex (requires an extra biopsy procedure and haplotypes in the oocytes are deduced from haplotypes in the PBs where recombinations may be present). Phasing can also be deduced by long-read sequencing by NGS of disease-specific amplicons from the affected partner and his/her parents. This will indicate the grandparental haplotype on which the de novo pathogenic variant(s) arose in the prospective parent. High-risk and low-risk haplotypes should be confirmed in the clinical cycles.
Alternatively, it is acceptable to establish genetic marker haplotypes using DNA from the affected partner and his/her parents prior to the clinical cycle, and then complete phasing during the PGT cycle(s). In the scenario when only DNA samples of the prospective parents are available, establishing the haplotypes and phasing needs to be based on the genotypes of the embryos.
When phasing is unknown at the start of the clinical cycle, the following recommendations apply:
It is mandatory to include pathogenic variant(s) detection in the test strategy.
TE biopsy is recommended to limit the risk for ADO at the pathogenic variant(s) locus. If cleavage-stage biopsy is performed, two independent cells should be tested.
When too few embryos are available for biopsy, it is recommended to biopsy and analyse unfertilised oocytes (if pathogenic variant in the female partner) and/or embryos that are non-suitable for biopsy, to support phasing.
Ideally, at least one affected embryo and one unaffected embryo are needed to establish the correct phase and detect recombination events. The pathogenic variant(s) should be consistently detected in the presence of the same parental haplotype. If this is not possible, it is recommended to cryopreserve the embryos and to wait for the analysis of embryos from the next cycle(s). Couples should be counselled upfront about this possibility. Alternatively, it is acceptable to transfer embryos after extended counselling and to strongly recommend confirmation by prenatal diagnosis.
As germline mosaicism due to post-zygotic de novo pathogenic variant(s) in the prospective parent cannot be excluded, it is not recommended to use an unaffected child/prenatal/embryo sample as phasing reference. If mosaicism has been detected, the transmission risk has to be evaluated.
Germline mosaicism detected in the prospective parents can be an indicator of somatic mosaicism and vice versa. In the single-cell validation of the PGT protocol for the ADO rate of the pathogenic variant(s) tested, using single cells of such an individual can lead to increased ADO rates of the mutant allele, depending of the degree of mosaicism.
Following single-cell analysis, it is not recommended to transfer embryos that carry the wild-type allele for the pathogenic variant(s) locus and the high-risk haplotype because of the ADO risk. It is acceptable to transfer such embryos following analysis of TE samples. Prenatal diagnosis is then strongly recommended.
De novo pathogenic variant in an affected child
When a de novo pathogenic variant(s) is detected in a child, it is important to thoroughly counsel the couple with regards to the possibility of recurrence and to assist them in making a well-informed reproductive choice. Achieving a pregnancy and performing prenatal diagnosis should be considered in all cases prior to initiating a PGT-M procedure.
The decision on whether PGT-M is permitted for cases of a de novo pathogenic variant(s) in a child may vary depending on local regulations.
It is recommended to exclude a post-zygotic origin of the de novo pathogenic variant(s) in the previously affected child of the couple. In this case, the recurrence risk is a minimum and the option of IVF treatment with PGT should be carefully evaluated. An initial evaluation of the couple’s reproductive history may provide evidence of potential germline mosaicism in the parents, for example through evidence of recurrent transmission in previous pregnancies. If DNA from previous terminated cases is available, the case can be dealt with as a usual PGT-M protocol. Further evidence of potential germline mosaicism in the parents may come from evaluation of the pathogenic variant(s) in various parental tissues. If germline mosaicism is detected, the recommendations from the above section on a de novo pathogenic variant(s) in a prospective parent apply.
Consanguineous families
It may be necessary to adapt the testing strategy when consanguineous relationships are present in the pedigree, especially in case of targeted amplification.
Consanguineous grandparents
A prospective parent may have two identical haplotypes in the region of interest because of a consanguinity between his/her parents, and it may be difficult to find informative genetic markers within the 1–2-Mb flanking region. In case of autosomal dominant disease, the pathogenic variant(s) analysis should be included in the test strategy and ADO rates for the pathogenic variant(s) locus after validation should be low (TE sample analysis is preferable to a two-cell analysis at Day 3). In case of autosomal recessive disease, diagnosis should be based on the low-risk haplotype of both partners.
Consanguineous couple
In case the prospective parents share the high-risk haplotype for an autosomal recessive disorder, parental contamination (most often maternal) in a homozygous affected embryo cannot be distinguished from a carrier embryo, and this may lead to adverse misdiagnosis with transfer of an affected embryo. It is recommended to adapt the testing strategy by either including analysis of unlinked informative polymorphic marker(s) or by performing the analysis on two independent biopsy samples. If this is not done, it is acceptable to prioritise the transfer of healthy embryos compared with carrier embryos.
When SNP markers are used after WGA, parental contamination can be detected.
HLA typing
The aim of HLA testing of preimplantation embryos is to establish a pregnancy with an embryo that is HLA-compatible with an affected child in need of haematopoietic stem cell transplantation. Haematopoietic stem cells are collected from the umbilical cord blood or the bone marrow of the HLA-matched donor sibling born (or a combination of both sources) and are used for transplantation to, and cure of, the affected sibling. Recommendations on counselling and important considerations prior to embarking on the PGT-HLA procedure are discussed in the paper on organisation of PGT (ESHRE PGT Consortium Steering committee et al., 2020). The following recommendations are relevant as well.
Test strategy
The preferred PGT methodology is indirect HLA haplotyping (using STR markers or genome-wide SNP haplotyping), which involves linkage analysis of genetic markers flanking the HLA-A, HLA-B, HLA-C, HLA-DR and HLA-DQ regions, to identify matching haplotypes between the tested embryos and the affected child.
The PGT protocol must include a minimum of one fully informative marker located at each of the following regions: telomeric to the HLA-A, between HLA-A and HLA-B, between HLA-B and HLA-DRA, between HLA-DRA and HLA-DQB1 and downstream to HLA-DQB1. It must be noted that some difficulty in finding markers between HLA-DRA and DQB1 has been encountered. In this case, two fully informative markers flanking the HLA-DQB1 must be included.
If a fully informative marker is not available for each of the regions above, a combination of partially informative markers must be included to provide adequate information on the parental HLA haplotypes.
A highly multiplexed protocol for the amplification of selected STRs or genome-wide SNP must be preferred where possible (i.e. more than one marker per region) to make the test more robust and to assist in detecting potential recombination that can occur throughout the whole major histocompatibility complex (MHC) region.
As HLA haplotyping may be compromised by genetic recombination, it is recommended that during the preclinical PGT work-up, protocol testing includes testing of any additional available first degree family members, aside from the parents and the affected child, to be able to detect recombination having occurred in the affected child.
In case recombination is detected in the affected child or in case recombination is detected in embryos during the PGT cycle, any decision on PGT and selection of embryos for transfer must be carefully discussed with the haematologist and transplantation experts, as it may be that a certain mismatch is permissive of haematopoietic stem cell transplantation. The location of the recombination event is of major significance for this purpose. This further highlights the importance of a highly multiplexed protocol.
HLA typing of preimplantation embryos can be performed as a sole indication when the affected child requires transplantation to treat an acquired disease, or in combination with PGT-M when there is a need to concurrently avoid transmission of an inherited disease in the family. This requires combining in one protocol the HLA typing approach with the recommended PGT-M strategies. For this purpose, the most comprehensive approaches, NGS and SNP haplotyping, are advantageous by allowing whole genome haplotyping from a single data set.
Exclusion testing
In families with a history of late-onset diseases, individuals at risk who want to avoid presymptomatic testing but wish for their own biological children to be free of the disease may opt for PGT. Exclusion testing is preferred over PGT with non-disclosure of the direct test results to the couple.
It is recommended to apply indirect testing with selection of embryos carrying the haplotype of the unaffected prospective grandparent for transfer. Haplotyping can be performed with STR or with SNP markers, relying on targeted amplification at the single or few-cell level or on targeted amplification following WGA.
Preclinical informativity/segregation testing is applied to DNA samples of the couple and the grandparents (parents of the partner at risk) only; other relatives of the partner at risk should not be tested.
Mitochondrial DNA disorders
Maternally inherited mtDNA mutations are a frequent cause of mitochondrial disorders. The great majority of pathogenic mtDNA mutations show heteroplasmy, a coexistence of wild-type and mutated mtDNA. PGT based on quantifying mutation load is an acceptable reproductive option for female carriers of heteroplasmic mtDNA point mutations, which requires case-by-case counselling, considering the uncertainties linked to this risk-reduction strategy. The key factor is selecting embryos with a mutation load below the threshold of phenotypic expression. For common mutations (e.g. m.3243A>G and m.8993T>G), a mutation-specific heteroplasmy threshold can be established based on available data. For rare or private mutations, the correlation between mutation load and phenotype should be investigated on a case-by-case basis, and literature should be reviewed in order to establish an acceptable expression threshold (see also in the paper on organisation of PGT (ESHRE PGT-SR/PGT-A Working Group et al., 2020).
Next to the recommendations for targeted amplification of nuclear monogenic disorders, the following recommendations are relevant for the quantitative analysis of monogenic mtDNA disorders by restriction enzyme digestion (see also section ‘Basic methods for allele discrimination’):
PCR master mix should be decontaminated by restriction enzyme treatment prior to PCR amplification to eliminate external mtDNA contamination.
Complete restriction enzyme digestion should be checked by spiking all the amplification products with an amplicon containing the restriction site of interest if no control site is present in the amplification product.
Reproducibility of the mtDNA mutation protocol based on restriction enzyme digestion should be validated preclinically, using replicates of mixes of wild-type and mutant mtDNA molecules in a broad range from 0 to 100% mutant and especially for the mutation load around the expression threshold.
In addition to the controls described in section ‘Single- or few-cell targeted amplification’ (diluted and/or undiluted genomic DNA, IVF and genetic laboratory negative controls and single cell control samples), control samples with a known mutation load (preferably around the expression threshold) should be used.
Biopsy at the cleavage stage is recommended. It is acceptable to biopsy one blastomere. Biopsy at the preconception stage (first and second PB) is not recommended. For biopsy at the blastocyst stage, sufficient data on the representability of the TE biopsy for the embryo as a whole is currently lacking to make recommendations at this point (ESHRE PGT Consortium Steering committee et al., 2020).
It should be taken into account that the cytoplasm of lysed blastomeres may no longer fully represent the embryo mutation load.
Examination process
General recommendations on the PGT examination process are described in the paper on organisation of PGT (ESHRE PGT Consortium Steering committee et al., 2020). The sections below highlight specific issues relevant to PGT-M.
Scoring of clinical results
When suboptimal samples or samples not meeting the preferred requirements (e.g. mislabelled samples, lysed cells or when a nucleus is not observed) are received for testing, this should be documented by the PGT lab and a procedure for how to further process and interpret these samples should be in place.
It is recommended that results are analysed by two independent observers and discrepancies adjudicated by a third observer (where possible). If no consensus is reached the embryo should not be recommended for transfer and should therefore be given the diagnosis of uninterpretable or inconclusive.
Haplotyping scoring criteria should be established in a written protocol and adhered to for the interpretation of the results.
It is acceptable to attempt to reduce the number of embryos with no result or no clear diagnosis following PGT, by adopting a ‘no result rescue’ approach. This could involve either a second biopsy step or repetition of the analysis after WGA. For practical recommendations regarding the re-biopsy procedure please refer to the PGT biopsy paper. In case of targeted amplification following cleavage-stage biopsy, a second biopsy (at the blastocyst stage) may be performed, followed by a second analysis. In case of genome-wide testing, a second analysis of the existing WGA and/or a second biopsy, followed by WGA and a second analysis, may be performed.
Clinical cycle report
General items required on PGT work-up and clinical cycle reports are included in the paper on organisation of PGT (ESHRE PGT Consortium Steering committee et al., 2020).
It is recommended that the following nomenclature is used in the clinical cycle report:
unaffected carrier or affected when reporting for monogenic disease;
low-risk or high-risk when reporting for mitochondrial disorder;
at risk or not at risk (for exclusion testing);
HLA compatible or HLA not compatible (for HLA typing);
no amplification (no result);
inconclusive (results but no diagnosis due to AF, ADO or recombination events);
abnormal (or aberrant): when a numerical or structural abnormality involves the chromosome(s) carrying the disease locus.
Post-examination process
Recommendations on PGT follow-up, baseline IVF live birth rates for PGT and misdiagnosis are covered in the paper on organisation of PGT (ESHRE PGT Consortium Steering committee et al., 2020).
Supplementary Material
Acknowledgements
The authors would like to thank everyone that contributed to the stakeholder review for the constructive remarks that improved the quality of the paper. The list of reviewers is available in Supplementary Table SIII.
Footnotes
†ESHRE Pages content is not externally peer reviewed. The manuscript has been approved by the Executive Committee of ESHRE.
Supplementary data are available at Human Reproduction online.
Authors’ roles
F.C. and C.M. chaired the working group. All authors contributed to conception and design, drafting the content and discussing it. All authors approved the final version.
Funding
European Society of Human Reproduction and Embryology.
Conflict of interest
D.Z. reports personal fees from Igenomix Italia, outside the submitted work. All other authors have nothing to disclose.
References
- ESHRE PGT-SR/PGT-A Working Group, Coonen E, Rubio C, Christopikou D, Dimitriadou E, Gontar J, Goossens V, Maurer M, Spinella F, Vermeulen N et al. ESHRE PGT Consortium good practice recommendations for the detection of structural and numerical chromosomal aberrations. Hum Reprod Open 2020. doi: 10.1093/hropen/hoaa017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ESHRE PGT Consortium and SIG-Embryology Biopsy Working Group, Kokkali G, Coticchio G, Bronet F, Celebi C, Cimadomo D, Goossens V, Liss J, Sofia Nunes S, Sfontouris I et al. ESHRE PGT Consortium and SIG Embryology good practice recommendations for polar body and embryo biopsy for preimplantation genetic testing. Hum Reprod Open 2020. doi: 10.1093/hropen/hoaa020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ESHRE PGT Consortium Steering committee, Carvalho F, Coonen E, Goossens V, Kokkali G, Rubio C, Meijer-Hoogeveen M, Moutou C, Vermeulen N, De Rycke M. ESHRE PGT Consortium good practice recommendations for the organisation of preimplantation genetic testing. Hum Reprod Open 2020. doi: 10.1093/hropen/hoaa021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper JC, Aittomaki K, Borry P, Cornel MC, de G, Dondorp W, Geraedts J, Gianaroli L, Ketterson K, Liebaers I et al. Recent developments in genetics and medically assisted reproduction: from research to clinical applications. Eur J Hum Genet 2018;26:12–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harton G, Braude P, Lashwood A, Schmutzler A, Traeger-Synodinos J, Wilton L, Harper JC. ESHRE PGD consortium best practice guidelines for organization of a PGD centre for PGD/preimplantation genetic screening. Hum Reprod 2011a;26:14–24. [DOI] [PubMed] [Google Scholar]
- Harton GL, De Rycke M, Fiorentino F, Moutou C, SenGupta S, Traeger-Synodinos J, Harper JC. ESHRE PGD consortium best practice guidelines for amplification-based PGD. Hum Reprod 2011b;26:33–40. [DOI] [PubMed] [Google Scholar]
- Harton GL, Harper JC, Coonen E, Pehlivan T, Vesela K, Wilton L. ESHRE PGD consortium best practice guidelines for fluorescence in situ hybridization-based PGD. Hum Reprod 2011c;26:25–32. [DOI] [PubMed] [Google Scholar]
- Harton GL, Magli MC, Lundin K, Montag M, Lemmen J. Harper JC. ESHRE PGD Consortium/Embryology Special Interest Group--best practice guidelines for polar body and embryo biopsy for preimplantation genetic diagnosis/screening (PGD/PGS). Hum Reprod 2011d;26:41–46. [DOI] [PubMed] [Google Scholar]
- Practice Committee of the Society for Assisted Reproductive Technology, Practice Committee of the American Society for Reproductive Medicine. Preimplantation genetic testing: a Practice Committee opinion. Fertil Steril 2008;90:S136–S143. [DOI] [PubMed] [Google Scholar]
- Preimplantation Genetic Diagnosis International Society. Guidelines for good practice in PGD: programme requirements and laboratory quality assurance. Reprod Biomed Online 2008;16:134–147. [DOI] [PubMed] [Google Scholar]
- The Preimplantation Genetic Diagnosis International Society (PGDIS). Guidelines for good practice in PGD. Reprod Biomed Online 2004;9:430–434. [DOI] [PubMed] [Google Scholar]
- Thornhill AR, de Bie-Smulders CE, Geraedts JP, Harper JC, Harton GL, Lavery SA, Moutou C, Robinson MD, Schmutzler AG, Scriven PN et al. ESHRE PGD Consortium ‘Best practice guidelines for clinical preimplantation genetic diagnosis (PGD) and preimplantation genetic screening (PGS)’. Hum Reprod 2005;20:35–48. [DOI] [PubMed] [Google Scholar]
- Vermeulen N, Le Clef N, Veleva Z, D'Angelo A, Tilleman K. European Recommendations for good practice in addition to an evidence-based guidelines programme: rationale and method of development. BMJ Evid Based Med 2019;24:30–34. [DOI] [PubMed] [Google Scholar]
- Zegers-Hochschild F, Adamson GD, Dyer S, Racowsky C, de J, Sokol R, Rienzi L, Sunde A, Schmidt L, Cooke ID et al. The international glossary on infertility and fertility care, 2017. Hum Reprod 2017;32:1786–1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.