

Abstract
The field of preimplantation genetic testing (PGT) is evolving fast, and best practice advice is essential for regulation and standardisation of diagnostic testing. The previous ESHRE guidelines on best practice for PGD, published in 2005 and 2011, are considered outdated, and the development of new papers outlining recommendations for good practice in PGT was necessary. The current paper provides recommendations on the technical aspects of PGT for chromosomal structural rearrangements (PGT-SR) and PGT for aneuploidies (PGT-A) and covers recommendations on array-based comparative genomic hybridisation (aCGH) and next-generation sequencing (NGS) for PGT-SR and PGT-A and on fluorescence in situ hybridisation (FISH) and single nucleotide polymorphism (SNP) array for PGT-SR, including laboratory issues, work practice controls, pre-examination validation, preclinical work-up, risk assessment and limitations. Furthermore, some general recommendations on PGT-SR/PGT-A are formulated around training and general risk assessment, and the examination and post-examination process. This paper is one of a series of four papers on good practice recommendations on PGT. The other papers cover the organisation of a PGT centre, embryo biopsy and tubing and the technical aspects of PGT for monogenic/single-gene defects (PGT-M).
Together, these papers should assist everyone interested in PGT in developing the best laboratory and clinical practice possible.
Keywords: ESHRE, preimplantation genetic testing, structural chromosomal aberrations, numerical chromosomal aberrations, aneuploidy, good practice
WHAT DOES THIS MEAN FOR PATIENTS?
The paper describes good practice recommendations for preimplantation genetic testing (or PGT). Similar documents have been published in 2011, but these needed updating to the new techniques used in IVF and genetics labs.
The recommendations should help laboratory personnel and geneticist to perform PGT according to the best laboratory and clinical practice possible. The current paper provides recommendations on the technical aspects of PGT for detection of chromosomal rearrangements and detection of aneuploidy (formerly called PGS). Chromosomal rearrangements are changes from the normal size or arrangement of chromosomes, which are the structures that hold our genetic material. The aim of the testing is to select the ‘normal’ embryos thereby improving the chance of a healthy pregnancy.
These technical recommendations are not directly relevant for patients, but they should ensure that PGT patients receive the best care possible.
Disclaimer
This Good Practice Recommendations (GPR) document represents the views of ESHRE, which are the result of consensus between the relevant ESHRE stakeholders and are based on the scientific evidence available at the time of preparation.
ESHRE GPRs should be used for information and educational purposes. They should not be interpreted as setting a standard of care or be deemed inclusive of all proper methods of care nor exclusive of other methods of care reasonably directed to obtaining the same results. They do not replace the need for application of clinical judgment to each individual presentation, nor variations based on locality and facility type.
Furthermore, ESHRE GPRs do not constitute or imply the endorsement or favour any of the included technologies by ESHRE.
Introduction
The previous terms of preimplantation genetic diagnosis (PGD) and preimplantation genetic screening (PGS) have been replaced by the term preimplantation genetic testing (PGT), following a revision of terminology used in infertility care (Zegers-Hochschild et al., 2017). PGT is defined as a test performed to analyse the DNA from oocytes (polar bodies) or embryos (cleavage stage or blastocyst) for HLA typing or for determining genetic abnormalities. This includes PGT for aneuploidy (PGT-A), PGT for monogenic/single gene defects (PGT-M) and PGT for chromosomal structural rearrangements (PGT-SR) (Zegers-Hochschild et al., 2017). PGT for chromosomal numerical aberrations of high genetic risk are included within PGT-SR in the data collections of the ESHRE PGT consortium.
PGT began as an experimental procedure in the 1990s with polymerase chain reaction (PCR)-based methods used for sex selection and the detection of monogenic diseases. Interphase fluorescence in situ hybridisation (FISH) was introduced a few years later and became the standard method for sexing embryos and for detecting numerical and structural chromosomal aberrations. Genome-wide technologies began to replace the gold standard methods of FISH and PCR over the last decade and this trend was most apparent for PGT-A. PGT-A has been carried out mainly for in vitro fertilisation (IVF) patients with original aims of increasing pregnancy rates per embryo transfer and decreasing miscarriage rates. Other outcome measures such as increasing elective single embryo transfer and reduced time to pregnancy have been added more recently. Cited indications for PGT-A include advanced maternal age (AMA), recurrent implantation failure (RIF) and severe male factor (SMF) and couples with normal karyotypes who have experienced recurrent miscarriage (RM). The value of the procedure for all IVF patients and/or appropriate patient selection remains an ongoing discussion, but this is outside the scope of this manuscript (Harper et al., 2018).
The goal of this series of papers is to bring forward best practices to be followed in all types of PGT services, offering PGT-A as well as PGT-M and PGT-SR.
In order to take PGT to the same high-quality level as routine genetic testing, guidelines for best practice have been designed by several societies. The PGD International Society has drafted guidelines (2008, 2004) while the American Society for Reproductive Medicine reviewed PGT practice in the USA (Practice Committee of the Society for Assisted Reproductive Technology and Practice Committee of the American Society for Reproductive Medicine, 2008) and published several opinion papers (on blastocyst culture, embryo transfer and on PGT-A). The first guidelines of the ESHRE PGT Consortium were published in 2005, as one of the missions of the Consortium was to bring overall standardisation and improve quality standards (Thornhill et al., 2005). In collaboration with the Cytogenetic European Quality Assessment (CEQA) and the UK National External Quality Assessment Service (UKNEQAS), now together in Genomics Quality Assessment (GenQA), the ESHRE PGT Consortium also initiated External Quality Assessment (EQA) schemes to provide an independent evaluation of laboratories and help them improving their techniques and reports. A review of the original guidelines yielded four sets of recommendations on different aspects of PGT: one on the organisation of PGT and three relating to the methods used: embryo biopsy, amplification-based testing and FISH-based testing (Harton et al., 2011a, Harton et al., 2011b, Harton et al., 2011c, Harton et al., 2011d). These four guidelines are now being updated and extended, taking into account the fast changes in the provision of PGT services. In these updated guidelines, the laboratory performing the diagnosis will be referred to as the PGT centre and the centre performing the IVF as the IVF centre.
General aspects of PGT, including patient selection, counselling, pregnancy and children follow-up and transport PGT, will be covered in the paper on organisation of PGT. Technical recommendations for embryo biopsy and tubing will be covered in the paper on embryo biopsy. Recommendations for genetic testing will be covered in the papers on detection of numerical and structural chromosomal aberrations and on detection of monogenic disorders. The content of the different papers is aligned with the IVF/PGT clinical procedure in Fig. 1.
Figure 1.
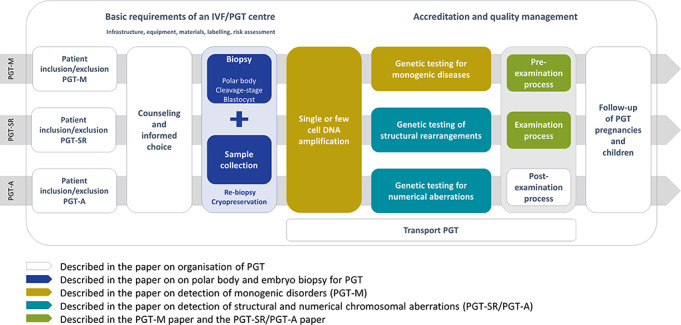
Overview of the IVF/PGT process, and how all aspects are covered by one of the four recommendations papers. IVF: in vitro fertilisation, PGT: preimplantation genetic testing.
The ESHRE PGT Consortium recognises that owing to variations in local or national regulations and specific laboratory practices, there will remain differences in the ways in which PGT is practiced (from initial referral through IVF treatment, genetic testing to follow-up of pregnancies, births and children). This does not preclude a series of consensus recommendations for best practice based on experience and available evidence. These recommendations are not intended as the only approved standard of practice nor are they legally binding. The unique needs of individual patients may justify deviation, and the recommendations must be applied according to individual patients’ needs using professional judgement. However, recommendations and opinions may be used to frame laws and regulations, and practitioners should ensure that they comply with statutory requirements or clinical practice guidelines in their own countries. To keep the papers concise, repetitions have been excluded as much as possible and many cross-references were included. Therefore, it is recommended to not consult the papers independently but always as a set when one is seeking guidance on a PGT issue.
Materials and Methods
The current paper was developed according to the published methodology for ESHRE Recommendations for good practice papers (Vermeulen et al., 2019). A working group was composed of people with hands-on expertise on the described techniques, aiming at a representation of different settings and nationalities. The working group members assessed the previous guidelines (Harton et al., 2011c) and deducted an outline for the current paper. As the aim was to provide technical guidance and support, it was not considered relevant to perform a formal literature search and as a result, no references were added, except for references to other guidance documents. All group members according to their expertise wrote a section that was later discussed in depth with the entire group until consensus was reached. Eleven online meetings were organised for discussion. The final draft of the paper was checked for consistency with the other papers of the series. The draft was then submitted for stakeholder review; it was published on the ESHRE website between 10 June and 10 July, and ESHRE members were invited to send in comments. All comments were checked by the working group, discussed in an online meeting and incorporated in the final version where relevant. A review report is published on the ESHRE website.
For easier use of the recommendations, terms in bold and italic are explained in a glossary (Supplementary Table SI) and abbreviations are listed (Supplementary Table SII).
Results/Recommendations
Introduction to PGT-SR/PGT-A techniques
This paper provides detailed technical recommendations for the most applied methods for PGT-SR, including fluorescence in situ hybridisation (FISH), array-based comparative genomic hybridisation (aCGH), next-generation sequencing (NGS) and single nucleotide polymorphism (SNP) array, and for PGT-A including whole genome amplification (WGA)-based aCGH and NGS. Detailed technical recommendations for SNP array are covered in the paper on detection of monogenic disorders (ESHRE PGT-M Working Group et al., 2020).
General recommendations for PGT-SR and PGT-A are formulated, independent of the testing method applied.
Training and personnel
Genetic testing procedures should be performed under the supervision of a (cyto)geneticist, competent or authorised to perform clinical diagnostics.
All personnel undertaking genetic testing should be trained adequately as required in a clinical molecular cytogenetic laboratory and should follow written standard operating procedures (SOPs).
-
Training for each technique should be documented.
-
°
Training for tubing is discussed in the paper on polar body (PB) and embryo biopsy for PGT (ESHRE PGT Consortium and SIG-Embryology Biopsy Working Group et al., 2020).
For FISH, training should be at least to the standard required for routine testing in a clinical cytogenetic laboratory. It is recommended that at least 30 samples are successfully spread or fixed and subjected to FISH by each trainee during preclinical training. Supervised clinical training should include at least an additional 20 samples.
For aCGH and NGS, it is recommended that at least 30 samples are subjected to WGA, followed by aCGH or NGS by each trainee during preclinical training. Supervised clinical training should include at least an additional 20 samples.
-
°
Laboratory infrastructure, equipment and materials
General aspects on infrastructure, equipment and materials are covered in the paper on organisation of PGT (see ESHRE PGT Consortium Steering committee. et al., 2020).
Labelling and witnessing
General guidance on labelling and witnessing is covered in the paper on organisation of PGT (ESHRE PGT Consortium Steering committee. et al., 2020).
Risk assessment
When suboptimal samples or samples not meeting the internal requirements (e.g. lysed cells, nucleus not seen) are received for testing, this should be documented and a procedure on how to further process these samples should be in place.
Specifically, for PGT-SR:
Risk assessment for the patient should include figures on the potential risk of a viable unbalanced offspring due to failure to detect any of the unbalanced segregation products.
In case only one of the two translocation segments can be detected, not all possible unbalanced segregation products can be identified. A test that cannot detect all the segments, and possibly some unbalanced products, may be less effective in decreasing the risk of a viable unbalanced offspring, first-trimester miscarriage and stillbirth. This should be mentioned in the preclinical work-up report.
Appropriate indications for specific tests
It is recommended that specific indications for PGT should remain within the scope of individual clinics.
FISH is not recommended for PGT-A as only a subset of chromosomes can be tested, and better comprehensive molecular approaches to detect aneuploidy for all 24 chromosomes are available.
Selection of embryos based on sex for social reasons is not acceptable.
PGT for chromosomal structural rearrangements
Structural chromosomal rearrangements form a major indication category for PGT. There are different types of structural chromosomal rearrangements: reciprocal and Robertsonian translocations, insertional translocations, deletions, duplications and inversions, all of which may be inheritable or occur de novo. Familial reciprocal and Robertsonian translocations constitute the most common indications for PGT for chromosomal structural rearrangement (PGT-SR).
In case of familial rearrangements, PGT-SR provides an opportunity to identify chromosomally unbalanced progeny at the earliest stages of embryo development. Preconception testing of PBs provides a means to indirectly identify chromosomally unbalanced oocytes.
Several methods are applied to perform PGT-SR, among which are FISH, aCGH and NGS. PGT-SR is mostly performed on embryonic biopsies taken at the cleavage stage (Day 3 post insemination) or the blastocyst stage (Day 5–7 post insemination). PGT-SR on PBs is less applied and involves a different kind of analysis as the genomic content of the oocyte (and corresponding embryo) is inferred from that of the first and second PB (indirect test). Detailed information on PB-based PGT is available in the paper by Magli et al. (2011) and Geraedts et al. (2011) (Geraedts et al., 2011, Magli et al., 2011).
FISH-based PGT-SR
FISH-based PGT is mainly applied for inherited chromosomal rearrangements but can also be used for embryo sexing in X-linked diseases (if direct mutation testing is not applicable) (PGT-M).
FISH enables enumeration of chromosomal loci that are involved in structural rearrangements or are indicative of sex chromosomes. Based on signal scoring, chromosomal imbalance or embryo sex can be established, and subsequently balanced embryos or embryos of the non-affected sex can be selected for transfer.
Disadvantages of the FISH technique constitute its technical nature: diagnosis is based on visual inspection of fluorescent signals, making loss of DNA integrity and overlapping signals two of the major problems. Furthermore, genomic information is limited to the loci targeted by the probes used.
Therefore, FISH-based PGT is acceptable for rearrangements involving small fragments or subtelomeric regions of chromosomes that are difficult or impossible to detect using other methods (e.g. < 10 Mb).
Laboratory issues
The principle of the FISH technology is based on the use of specific DNA probes that are labelled with distinctive fluorochromes (either direct or indirect via a hapten). The DNA probes and the target DNA, typically embryonic interphase nuclei, are (simultaneously) denatured and left to anneal. Following hybridisation, results are visualised via fluorescence microscopy.
Many variations in FISH methods have been published and all appropriately validated methods are acceptable. The method used should have been previously implemented, tested and validated in the PGT centre.
FISH protocol: structural rearrangements.
For structural rearrangements, it is recommended that the probe set contains at least sufficient probes to detect all expected unbalanced variants of the chromosomal rearrangement. The analysis of the predicting segregation outcomes for carriers of a structural rearrangement should include an assessment of the plausible mechanism for chromosome pairing and the products of disjunction following the first and second meiotic divisions.
It is recommended that a combination of three informative probes (two distal and one proximal, or two proximal and one distal probe relative to the translocation break points) be used to detect all unbalanced segregation products, as for more common two-way reciprocal translocation. For Robertsonian translocations and inversions, two probes are acceptable. For deletions, duplications and insertions, locus-specific probes for the target region should be used and a control probe should be included in the diagnostic cycle.
Where suitable probes are not available, it is acceptable to use probe combinations that cannot detect some unbalanced forms of a rearrangement, provided that they have been assessed to be non-viable in a recognisable pregnancy or to have a very low prevalence. It has to be mentioned in the (pre-validation) report that there are unbalanced forms that cannot be detected, and patients should be counselled to this effect. A cytogeneticist or suitably qualified person should determine which probe combination to use.
For cleavage-stage embryos, PGT diagnosis on a single mononucleate blastomere is acceptable for chromosomal rearrangements provided that there are informative probes for at least two unbalanced segments for those products considered likely to be prevalent or viable in a recognisable pregnancy. PGT diagnosis based on concordant results from two mononucleate blastomeres is recommended where there is only one informative probe available for the chromosome imbalance involved that is considered likely to be prevalent or viable in a recognisable pregnancy.
For preconception PGT diagnosis, both PBs are required for analysis and all unbalanced products of meiotic segregation should be detectable so that it is possible to know the contents of the oocyte. However, it is important to point out that PGT-SR performed on PBs carries a risk of misdiagnosis for the carriers of structural rearrangements due to an uneven number of crossovers that may occur in meiosis I, which may be undetectable through FISH. The presence of cumulus cells attached to the zona pellucida (ZP) could heavily affect the result of the PGT-SR analysis.
Blastocyst biopsy for a FISH-based PGT diagnosis is acceptable, provided that special care is taken to avoid overlapping cells. On average a trophectoderm (TE) sample contains 5–10 cells, which in theory allows for a more reliable diagnosis. However, the multi-cell nature bears the possibility of discordant results in the different cells because of a technical failure (suboptimal FISH conditions) or true chromosomal mosaicism. Reporting of discordant results should be regulated and genetic counselling should be provided to the couple to explain the possible impact on the reliability of the PGT diagnosis.
The use of additional probes to screen for aneuploidies of chromosomes not involved in the rearrangement is acceptable. If multiple rounds of FISH are being applied, the probes indicative of the rearrangement should be included in the first round.
FISH protocol: sexing in case of X-linked diseases (PGT-M).
For embryo sexing, it is recommended that the probe set contains at least probes specific for the centromere region of the X and Y chromosomes and one autosome.
The use of additional probes to screen for aneuploidies of autosomes is acceptable. If multiple rounds of FISH are being applied, the probes indicative of embryo sex (X and Y) should be included in the first round.
PGT diagnosis on a single mononucleate cell is acceptable for sexing.
It should be noted that FISH-based PGT for sexing to exclude transmission of X-linked diseases could be less advantageous when compared with amplification-based diagnosis of the disease-associated mutation alongside gender determination. A haplotyping-based diagnosis allows for identification of unaffected males as well as carrier females.
Turnaround time.
The turnaround time for FISH-based PGT-SR depends on the number of embryos analysed and the number of hybridisation rounds applied. According to recommendations from commercial probe manufacturers, the hybridisation time for each round should be at least 4 h, but laboratories may develop and validate their own protocol that will shorten the time for hybridisation while maintaining the intensity and brightness of the fluorescent signals. Thus, a clinical cycle report can be obtained within 4–48 h from sample fixation to signal scoring.
Documentation.
The patient’s file should include relevant laboratory documentation:
-
-
high-resolution (550–800 bands) parental karyotype, preferably with FISH verification of chromosome regions involved in structural rearrangements; also, it may include a karyotype of the affected child or other family member;
-
-
results of cytogenetic analysis of any previous unbalanced pregnancies or preimplantation embryos;
-
-
genetic counselling report with recommendations for PGT-SR, an indication of the testing method and the benefits and the limitations of the test;
-
-
the informed consent of the couple with risk assessment and indication of test limitations.
Laboratory infrastructure, equipment and materials
Infrastructure.
The following recommendations are for the laboratory space.
The laboratory should be well ventilated to minimise the effect of any noxious fumes. This is particularly important if cells are fixed using methanol and acetic acid. In this case the use of a fume cabinet for the fixation steps is recommended.
FISH outcomes, including cell spreading and fixation, are dependent on humidity. The humidity in the FISH laboratory should be controlled and stable. FISH protocols should be optimised in these conditions.
FISH signals may be bleached or weakened in bright light. It is recommended that the FISH laboratory be fitted with variable intensity incandescent lighting. Fluorescent lighting is acceptable. The slides should be stored cool and in light-tight storage boxes or folders.
Equipment.
A FISH-based PGT diagnosis requires the following equipment: a fluorescence microscope equipped with appropriate filters for the fluorescent dyes used, a water bath and a hybridisation device. A fluorescent image capture system is preferred for documenting FISH images.
Materials and reagents.
Required materials are glass slides and coverslips, and a probe set specific for the chromosomal structural rearrangement of interest.
Daylight should be avoided during hybridisation and post-hybridisation steps.
The use of commercial probes is recommended since they generally come with quality control (QC) and validation reports.
The use of homemade probes is acceptable with appropriate preclinical quality assurance (QA)/QC and validation.
It is recommended that all probe vials be tested before clinical application, to confirm that they contain the correct chromosome-specific probe and are labelled with the correct fluorochrome or hapten. Furthermore, it is recommended that they be informative for the intended PGT-SR couple and meet documented acceptance levels for signal specificity, brightness and discreteness. Batch numbers should be recorded to ensure continuous traceability.
It is recommended that only appropriately qualified personnel (as documented in written competency lists) authorise selection of probes for clinical use.
In case of a Robertsonian translocation, fluorescent probes for any locus on the long arm of the two acrocentric chromosomes involved in the rearrangement can be used. For reciprocal translocations, alpha-satellite probes, locus-specific probes or subtelomere probes indicative of the translocated regions may be used. For inversions, mostly locus-specific probes for the short and the long arm of the intended chromosome are used, possibly combined with alpha-satellite repeat probes. For the detection of deletions, duplications or insertions, it is preferable to use locus-specific probes indicative of the target chromosomal region, combined with a control probe (alpha-satellite or subtelomere probe) to discriminate between a true deletion/duplication and a whole chromosome copy number change.
It is recommended that for each round of FISH all probes be labelled with a different fluorochrome or combination of fluorochromes so that the colours of different probe signals can be distinguished from one another. The signals should be one domain apart.
When using prehybridisation steps, such as pepsin and paraformaldehyde, it is recommended that measures should be taken to ensure appropriate QC for these solutions. The temperature ranges and pH values of solutions should be verified before using in every round of FISH. Creation dates of solutions for all steps should be recorded and the solutions should be checked for possible cellular contamination prior to use.
Mounting medium containing antifade (with or without DAPI, depending on the probe combination) is recommended to allow maintenance of fluorescent signals. It is recommended that prior to each FISH procedure, denaturation and hybridisation, the pH values of solutions and wash temperatures be verified.
Work practice controls
Identification and witnessing.
The use of an adequate labelling system, written or barcoded (electronic), using two unique patient and embryo/cell(s) identifiers, is recommended.
-
Labelling and sample identification should be confirmed for critical and high-risk steps by an independent observer, preferably one who is trained in FISH. It is recommended that the unique patient identifier and embryo/cell number be witnessed and signed off by two operators during biopsy, sample collection and genetic testing [see also in the paper on organisation of PGT (see ESHRE PGT Consortium Steering committee. et al., 2020)]. Witnessing is also indicated at the following steps of the FISH procedure:
-
-
at probe preparation, to check that the correct FISH probes (patient-specific pre-validated probe mixes should be correctly labelled in advance) are used for the case,
-
-
when diagnostic FISH results are recorded to ensure that FISH images correspond to the correct cell and/or embryo.
-
-
The location of the fixed/spread cell on the slide may be recorded to facilitate tracing.
Intra-assay controls.
The use of positive and negative controls in a FISH-based PGT diagnosis may be considered.
Suitable positive controls are not readily available (i.e. unbalanced single human blastomeres, TE cells or other cell types to represent unbalanced human blastomeres or TE cells).
Normal or carrier human metaphase lymphocytes may serve as control to ascertain that the probes in the hybridisation mixture identify the expected chromosomes/chromosomal regions.
Pre-examination process
The pre-examination process includes preclinical work-up, test development and validation.
Preclinical work-up and test development.
It is recommended to perform a preclinical work-up to assess PGT-SR feasibility, identify informative probes and work on a clinical testing strategy. It is recommended to perform segregation analysis for the intended structural rearrangement to ensure that the testing strategy allows for the detection of all expected genotypes in the embryos.
It is acceptable to carry out FISH tests on sperm cells from male translocation carriers in an attempt to predict the efficacy of PGT-SR for these cases.
When using a probe set previously shown to have a very low polymorphism rate, it is acceptable to forego any preclinical work-up. Other probes may be more prone to polymorphism and preclinical testing of peripheral blood lymphocytes is then recommended. Sequences in the heterochromatin regions of chromosomes 1, 9, 16 and Y are closely related and therefore cross-hybridisation among those chromosomes is frequently observed. In addition, the D15Z1 region on the short arm of chromosome 15 cross-hybridises with the short arm regions of other acrocentric chromosomes, especially chromosome 14. Moreover, the centromeric probes D1Z7 (chromosome 1), D5Z2 (chromosome 5) and D19Z3 (chromosome 19) occasionally show cross-hybridisation. Finally, an overlap of signals generated by probes specific for the centromeres of chromosome 18 and chromosome 16 is frequently observed.
Following the fixation procedure and following each round of FISH the location and integrity of the cells should be checked.
Pre-examination validation.
It is recommended to perform the validation on both the carrier of the rearrangement and the partner, but it is acceptable to perform the validation on the carrier only.
It is acceptable to perform the validation on blastomeres and TE cells from embryos donated to research prior to clinical PGT-SR testing. It is also acceptable to perform the validation on other cell types such as peripheral blood lymphocytes and fibroblasts.
It is recommended that at least 10 metaphase spreads are examined: (i) to ensure that the probes are specific for the correct chromosomes; (ii) to assess chromosome polymorphism and signal cross-hybridisation; and (iii) with respect to carriers of a chromosome rearrangement, to ensure that the probes hybridise to the expected segments of the rearrangement.
In addition, it is recommended that at least 100 interphase nuclei are scored using appropriate scoring criteria (signal specificity, brightness and discreteness).
Acceptable ranges of FISH hybridisation efficiency should be determined in each laboratory for each FISH probe and combined probe set. Validation tests should at least confirm that the probes hybridise as expected, that they are informative for the rearrangement and that >95% of the cells shows the expected number of signals for each of the probes used.
It is recommended that scoring criteria are determined ahead of time (published or ‘in-house’) and should be adhered to as per written procedure.
Preclinical work-up report.
General guidance and recommendations on administration and patient information for the preclinical work-up report is given in the paper on organisation of PGT (see ESHRE PGT Consortium Steering committee. et al., 2020). A preclinical work-up report should also include a summary of the PGT-SR work-up with details on the protocol and validation steps. It should further describe the FISH probes used and the hybridisation efficiency, the false-positive rate and the false-negative rate of the probe set. Where applicable, the latest version of the International System for Human Cytogenetic Nomenclature (ISCN)/Human Genome Variation Society (HGVS) nomenclature can be used. Finally, the report should include potential limitations of the test.
Risk assessment
Risk assessment should cover:
-
-
risks caused by errors in sample tracking;
-
-
risks caused by handling biopsy samples prior to FISH analysis that, if not performed with care, may compromise DNA integrity;
-
-
risk of inconclusive or false results due to suboptimal experimental conditions; the reliability of the FISH diagnosis may be negatively influenced by the inability to accurately interpret signals, inconsistent fixation or suboptimal hybridisation; signal overlap may lead to an underestimation of the actual chromosome (region) copy number; and in addition, locus-specific and subtelomere probes produce less bright signals when compared with alpha-satellite probes and show a higher rate of split signals, which compromises correct signal scoring;
-
-
risk of inconclusive or false results due to biological reasons: (i) unbalanced segregations may arise from crossing over during meiosis I in the gametes of the carrier of the rearrangement; (ii) chromosomal mosaicism, either at cleavage stage or blastocyst stage, may lead to misinterpretation of the actual embryo karyotype;
-
-
patient’s risk of miscarriage, stillbirth, (viable) unbalanced offspring, mosaic offspring or offspring with a chromosomal imbalance that is unrelated to the test, whether biological or caused by a technical error.
Limitations of the test
The limitations of the FISH technique should be clearly mentioned in the preclinical work-up report and/or be discussed with the patients during genetic counselling.
FISH-based PGT-SR analysis does not allow for a distinction between embryos with a normal or a balanced karyotype.
FISH-based PGT-SR analysis does not allow for the detection of uniparental disomy (UPD).
FISH-based PGT analysis can only assess the copy number of the chromosomes targeted by the DNA probes used.
Due to the limited number of available fluorochromes, the number of chromosomes that can be simultaneously detected is also restricted. Sequential rounds of FISH may therefore be required, which negatively affects DNA integrity and signal quality.
Commercial probes are available for only a limited number of loci, which may complicate the selection of probes for the analysis of rare chromosomal rearrangements.
Impossibility to detect mosaicism if FISH is performed in a single cell biopsy.
Array-based PGT-SR
aCGH involves the competitive hybridisation of differentially labelled sample and reference DNA on a microscope slide with fixed DNA probes. DNA probes correspond to specific chromosomal regions and occupy discrete spots on the slide. Each spot has a colour that results from the fluorescence ratio of the two colours after hybridisation. The evaluation of fluorescence ratios is automated and indicative of chromosomal loss or gain.
Arrays are considered a more reliable approach for PGT-SR when compared with FISH since they provide multiple points of measure for each translocation segment. Furthermore, they allow for simultaneous copy number assessment of the chromosomes not involved in the rearrangement.
Currently, two types of commercial array platforms are being used. The first is an aCGH platform based on oligonucleotides providing a resolution of 5 to 10 Mb. The second is a single nucleotide polymorphism (SNP) array platform based on oligonucleotides providing a resolution of 2.4 to 5 Mb (see also the paper on detection of monogenic disorders (ESHRE PGT-M Working Group et al., 2020).
Laboratory issues
The aCGH workflow involves (i) sample cell lysis and WGA; (ii) labelling of sample and reference DNA with different fluorochromes (e.g. green and red); (iii) purification of labelled DNA; (iv) microarray processing (hybridisation of biopsied and reference DNA samples followed by washing of microarray slides); (v) scanning; and (vi) analysis of scanned microarray tiff images where data are extracted to fluorescence ratio. The resulting log2 of fluorescence ratios is computed by specific software to identify structural and numerical chromosome copy number aberrations.
aCGH protocol.
It is recommended that wet-laboratory experimental conditions be established for all steps in the aCGH workflow followed by a preclinical assessment of the accuracy of the test to detect a chromosome aberration.
It is acceptable to perform aCGH-based PGT-SR on PB biopsies, provided that both PBs can be analysed, and all unbalanced products of meiotic segregation can be detected so that it is possible to know the contents of the oocyte. However, it is important to point out that PGT-SR performed on PBs carries a higher risk of misdiagnosis for the carriers of structural rearrangements due to an uneven number of crossovers that may occur in meiosis I, which may be undetectable through aCGH. The presence of cumulus cells attached to the ZP could heavily affect the result of the PGT-SR analysis.
It is acceptable to perform aCGH-based PGT-SR on single-cell biopsies, although they present with an overall increased noise and step change chromosome artefacts in the aCGH profile. Acceptance criteria for noise level should be part of the QA/QC parameters.
Blastocyst biopsy for an aCGH-based PGT diagnosis allows for a more reliable diagnosis, as on average a TE sample contains 5–10 cells.
It is recommended to use a WGA protocol which is compatible with the specific aCGH platform that has been used for validation.
Turnaround time.
The net aCGH turnaround time from sample processing to comprehensive chromosome analysis is 24 h, although results can be obtained within 8–12 h. However, each laboratory needs to validate whether shorter hybridisation times affect hybridisation efficiency.
Documentation.
Relevant laboratory documentation should include:
-
-
a patients’ karyotype, preferably at high resolution (550–800 bands), if available with FISH verification of the breakpoints;
-
-
a report on any previous unbalanced products of conception;
-
-
genetic counselling report with possibly a recommendation for PGT-SR, an indication of the testing method, and the benefits and the limitations of the test;
-
-
the informed consent of the couple with risk assessment and indication of test limitation.
Laboratory infrastructure, equipment and materials
Infrastructure.
To prevent carry-over of amplified DNA, the laboratory space should be divided into pre- and post- amplification rooms that are physically separated.
Preferably the pre- and post-amplification rooms/areas should be equipped with UV-C light for DNA decontamination.
Positive air-pressure is recommended for the pre-amplification room. When positive and negative pressure rooms are present, they are preferably enclosed by a locked chamber.
A dedicated set of equipment, consumables and laboratory coats should be used for each designated area and not be exchanged between the pre- and post-amplification rooms.
Pre-amplification steps should be carried out in a laminar downflow cabinet. The workflow between the pre- and post-amplification areas should be unidirectional, from the pre-amplification room (clean room) to the post-amplification room.
Constant regulation of environmental conditions (ozone, temperature and humidity) is recommended during all steps to ensure efficient hybridisation results.
Equipment.
- Equipment required for WGA and aCGH analysis of biopsied samples includes:
-
-a class II safety cabinet, preferably equipped with UV-C light, to prevent contamination of samples at the pre-amplification stage;
-
-thermal cyclers with heated lids (one for the pre- and one for the post-amplification room);
-
-microcentrifuges (one for pre-amplification, one for all the following stages) and a benchtop swingout centrifuge;
-
-a magnetic stirrer, fume cabinet, hybridisation oven/incubator, water bath, gel electrophoresis equipment to check successful amplification and a vortex mixer;
-
-a scanner, equipped with the corresponding lasers to excite the hybridised fluorophores to read and store the resulting images of the hybridisations, placed in the post-amplification room in an atmosphere with low ozone parameters, regulated temperature and protected from daylight and validated and adjusted to the required resolution for the specific PGT protocols.
-
-
The use of a DNA quantification system (to determine the amount of amplified DNA after WGA) and a vacuum concentrator (to reduce the time required to process high numbers of samples) is optional.
Associated servers should be also allocated in proper conditions and instruments used in critical steps should be UPS-connected.
It is recommended that prior to each step of the protocol, the temperature ranges and/or pH values of equipment and solutions are verified. Specific temperature and thermocycler programmes should be validated in individual PGT centres for all equipment, and instruments should be serviced and calibrated regularly to ensure accuracy.
Software for automatic calling of structural aberrations is not always available and therefore segmental aneuploidies need to be manually called by the operator.
Materials.
Materials required for WGA and aCGH analysis of biopsied samples include:
-
-
cell lysis, pre-amplification, and amplification enzymes and buffers specific to each amplification method used;
-
-
DNA labelling reaction buffers, enzymes, dNTPs and fluorophore-marked dUTP that should be used under minimal light exposure since they are light sensitive;
-
-
hybridisation and washing buffers, human Cot-1 DNA, and DNase/RNase-free distilled water;
-
-
microarray slides.
Work practice controls
Identification and witnessing.
An adequate labelling system with two unique patient identifiers and embryo/cell(s) number is recommended.
- Labelling and sample identification should be confirmed for critical and high-risk steps by an independent observer, preferably one who is trained in molecular genetics. It is recommended that the unique patient identifiers plus the embryo/cell number be witnessed and signed off by two operators during biopsy, sample collection and genetic testing (see also the paper on organisation of PGT (see ESHRE PGT Consortium Steering committee. et al., 2020). Witnessing is also indicated at the following steps of the aCGH procedure:
-
-at the start of the WGA procedure to ensure that the correct volume of PCR master mixture is loaded into each tube;
-
-at the start of the labelling procedure to ensure that the correct volume of labelling mixture is loaded into each tube;
-
-at loading of the labelled DNA samples on array slides to ensure that each sample matches the sample identifier on the slide;
-
-and when recording aCGH results to ensure that aCGH files correspond to the correct cell and/or embryo.
-
-
Intra-assay controls.
Suitable positive controls are not readily available (i.e. unbalanced single human blastomeres, TE cells or other cell types to represent unbalanced human blastomeres or TE cells).
Negative controls serve to confirm that no contamination is present in the ‘no-template’ tube, which does not confirm the absence of contamination for the rest of reaction tubes carrying the biopsied samples.
Diluted genomic DNA is recommended for positive intra-assay controls to check successful amplification of single or few cells and a successful reaction, respectively.
Negative controls with sample collection buffer, biopsy media or washing media (based on the protocols of the PGT centre) are recommended to control for contamination for each biopsy sample cohort (i.e. the IVF laboratory negative control).
A minimum of one negative control with amplification mixture only is recommended to control for contamination during setting up of amplification reactions (i.e. the genetic laboratory negative control).
Pre-examination process
Internal quality control.
When using aCGH for PGT-SR, the challenge is to reliably call an unbalanced chromosomal rearrangement while avoiding false positives or false negatives.
The probability of detecting (small) unbalanced chromosomal segments depends on the performance parameters of the platform used.
- It is recommended to determine the effective resolution threshold as well as the percentage false-negative and false-positive results, and the specificity and sensitivity of the platform in a series of experiments using DNA from:
-
-isolated single cells from cell lines with established structural copy number changes;
-
-previous unbalanced pregnancies, when available;
-
-cells isolated from donated embryos from previously performed PGT-SR cases. Initial PGT results obtained with a validated technique should be used as a reference to determine the false positive/negative detection rate for the particular chromosome regions involved in the rearrangement.
-
-
It is recommended to test replicates of the same DNA sample in order to affirm that deviating ratios most likely represent a true copy number change.
Following DNA amplification, a clear agarose gel band should be visible and/or quantitative measurement of DNA concentration should at least be 20–50 ng/μl.
It is recommended to test the quality of each batch of arrays.
It is recommended to use hybridisation template forms to record sample tracking.
Barcoding of aCGH slides is mandatory to maintain the correlation between the sample and the array slide used for hybridisation.
It is acceptable to re-analyse unbalanced embryos for QA/QC purposes.
Test efficiency.
To check for amplification efficiency, it is recommended that samples and intra-assay controls (if used) be put on an agarose gel and/or quantified by Qubit Fluorometer.
The use of male and female reference DNA is recommended to assess hybridisation efficiency and interpret the results. Marked X/Y chromosome separation is indicative of a successful experiment in gender-mismatched samples, and the corresponding levels of gain for the X chromosome and loss for the Y chromosome are used as a reference to evaluate aneuploidy events for the autosomes.
Gender-matched samples must show consistently no change on chromosome X or Y and none of the probes in the array should report a change.
Negative amplification, negative intra-assay control or failed hybridisation should show a consistent noisy profile where no significant pattern is observed.
Storage time and temperature have an impact on the integrity of cells, DNA and/or solutions and laboratories should validate that the conditions used in their protocols are fit for purpose. Furthermore, it is not recommended to use repeatedly frozen-thawed solutions containing DNA or enzymes.
Hybridisation bias due to drying out of the microarray surface could lead to signal loss, degradation of fluorophore-marked dUTP and suboptimal scanned images.
It is recommended to stringently wash the aCGH slides with minimum light exposure and under controlled ozone concentration, temperature and humidity. The use of laboratory carbon-loaded non-woven filters is recommended in case of high ozone levels.
It is recommended to avoid the use of detergents to clean the wash equipment, as this may interfere with signal intensity.
Washing and scanning of slides in small batches (two to three slides) is recommended to minimise the exposure of slides and of labelling dyes to air.
It is critical that slides are dried by centrifugation shortly after the final washing step, to avoid drying through evaporation.
Scan images should have defined features with red and green images well registered and the colours evenly balanced.
The assay signal to background noise ratio (SBR) should be sufficiently high for the log2 ratio change to be observed. In case of low SBR, additional washing of the slides and rescanning are acceptable.
It is recommended to calculate the acceptable and optimum ranges of QCs for every array experiment. The QC measures of array data for every experiment are extrapolated by specific software and are indicative for the successful calling of all target probes. The QC measures will vary between array types and different scanners.
Preclinical work-up and report
Preclinical work-up.
Karyotype reports should be obtained for both partners from an accredited/certified cytogenetics laboratory.
A case-specific work-up is not required when performing aCGH for structural rearrangements, unless the carrier has an unbalanced karyotype.
It is recommended to upfront ensure that all unbalanced products of the specific rearrangement can be identified with the platform used. The ability to detect an unbalanced product depends on the effective resolution and the coverage of the array used. This needs to be established prior to clinical application by using DNA from cell lines with well-established segmental aneuploidy to validate the presence and the number of all (consecutive) clones/probes representing the respective chromosome regions.
It is acceptable that three out of four segments for two-way reciprocal translocations are detected to reliably identify unbalanced segregation products.
It is not acceptable to perform a clinical PGT-SR test if the size of the translocation segments, inferred from the karyotype, is below the threshold of resolution of the platform used.
It is acceptable to forego any additional work-up when performing aCGH for structural rearrangements.
Preclinical work-up report.
A case-specific preclinical wet-laboratory work-up report is not required, provided that no particularities have come to light during the work-up. However, a report on the theoretical evaluation of the preclinical work-up should be available.
Risk assessment
Risk assessment should cover:
-
-
risks caused by errors in sample tracking;
-
-
risks caused by handling biopsy samples prior to aCGH analysis (tubing, washing) that, if not performed with care, may compromise DNA integrity and lead to failed or poor WGA;
-
-
risks that the size of the structural rearrangement is different from the one expected based on non-uniform reporting of parental karyotypes and therefore may remain undetected by the aCGH protocol (if they are below the resolution of the platform used);
-
-
risk of inconclusive or false results due to suboptimal experimental conditions,
-
-
risk of inconclusive or false results due to biological reasons: (i) unbalanced segregations may arise from crossing-over during meiosis I in the gametes of the carrier of the rearrangement; (ii) chromosomal mosaicism, either at cleavage stage or blastocyst stage, may lead to misinterpretation of the actual embryo karyotype; (iii) embryos of poor morphology are at risk of containing cells with degraded DNA;
-
-
patient’s risk of miscarriage, stillbirth, (viable) unbalanced offspring, mosaic offspring or offspring with a chromosomal imbalance that is below the resolution of the test, whether biological or caused by a technical error.
Limitations of the test
Detection of translocation segments is limited by the resolution of the platform. If the size of more than one out of the four translocated segments is below this resolution limit, aCGH-based PGT is not possible.
Detection of unbalanced segregations that have breakpoints near the telomere or in the subtelomere region is not always possible, since the probe coverage in these regions is low. For each aCGH-based PGT-SR case, limitations should be investigated during preclinical work-up.
aCGH-based PGT-SR analysis does not allow for a distinction between embryos with a normal or a balanced karyotype.
aCGH-based PGT-SR analysis does not allow for the detection of UPD. There is an increased risk of UPD in carriers of chromosomal rearrangements when clinically relevant chromosomes (i.e. 6,7,11,14,15,20) are involved in the imbalance or a Robertsonian translocation, which involves chromosomes 14 or 15 (Kotzot, 2008). Prenatal diagnosis for UPD is acceptable but should be assessed critically on an individual basis.
Array-based PGT-SR analysis is less sensitive to detect mosaicism than NGS.
SNP array
SNP array-based PGT-SR is not based on the detection of the actual chromosomes. The embryo karyotype is merely inferred from the haplotypes detected in DNA from the embryo biopsy.
SNP array-based PGT-SR requires a preclinical work-up to phase the imbalance. Phasing is performed using DNA from the couple and one reference (a balanced reference is recommended, but an unbalanced is acceptable). If no reference is available, diagnosis can be performed during the clinical cycle and requires at least one unbalanced embryo or well-defined breakpoints to distinguish unbalanced embryos.
All samples need to be subjected to WGA prior to SNP array analysis.
In case of PGT-SR for carriers of inherited balanced rearrangements, an added value of the approach is that, based on haplotype information, embryos carrying the balanced form of the rearrangement can be distinguished from normal diploid non-carrier embryos.
Depending on the size of the involved segments, aberrant intensity ratios may or may not be detectable for the region(s) of interest. If detectable, it is recommended that the diagnosis is supported by log ratio and B allele frequency values.
Further recommendations on SNP array are covered in the paper on detection of monogenic disorders (ESHRE PGT-M Working Group et al., 2020).
Laboratory issues
Protocol.
The protocol can vary significantly depending on the platform used. Independent of the platform, it includes: (i) sample pre-processing; (ii) hybridisation on the slides; (iii) SNP staining and detection; and (iv) data analysis.
Sample pre-processing and hybridisation generally includes any or all of the following processes: handling of biopsy samples (PB, single blastomere or TE cells); cell lysis and WGA; and loading of the sample on the slides. Generation of reliable SNP calls is crucial, and the process for generating them can vary depending on the platform.
WGA material of insufficient quality and/or quantity as well as contamination of starting material can lead to poor genotyping data.
Raw data produced after reading the SNP calls from the array are further processed by computational analyses and bioinformatics using a variety of algorithms to optimise genotyping and enable haplotyping.
As these processes may vary depending on the platform, it is recommended to optimise and validate each step individually (including the entire wet-bench process as well as the bioinformatic analyses) to empirically determine optimal assay conditions and analysis settings. For each platform, the SNP calling threshold and minimum SNP call rate should be defined with validation experiments (see Pre-examination process section).
Turnaround time.
The turnaround time from sample processing to data analysis can vary from 24 h to several days, depending on the setting and the platform of choice. It is recommended that each laboratory validates in-house whether the implementation of shortened protocols has an effect on hybridisation efficiency and data quality.
With the aim of accumulating samples for a SNP array run, biopsy samples can be stored short term (weeks), and WGA samples can be stored long term (years) at −20 or −80°C.
Documentation.
Relevant laboratory documentation should include:
-
-
a karyotype, preferably at high resolution (550–800 bands), if available with FISH verification of the breakpoints from the patient and the phasing reference;
-
-
a report on any previous unbalanced products of conception;
-
-
genetic counselling report with possibly a recommendation for PGT-SR, an indication of the testing method and the benefits and the limitations of the test;
-
-
the informed consent of the couple with risk assessment and indication of test limitation.
Laboratory infrastructure, equipment and materials
Infrastructure.
General aspects on infrastructure are covered in the paper on organisation of PGT (see ESHRE PGT Consortium Steering committee. et al., 2020), and in section ‘Array-based PGT-SR’.
Equipment.
SNP array platforms differ, among others, in price, resolution (number of SNPs on the arrays) and chemistry. Initial set-up should follow manufacturer’s instructions, and it is recommended to collaborate with the manufacturer to ensure that the laboratory space has been optimised to meet the requirements. In addition, it is recommended to involve informaticians with relevant expertise to make sure all required elements (hardware, servers, data storage, internet) are in place.
- Equipment required for WGA and SNP array analysis of biopsied samples includes:
-
-a class II safety cabinet, preferably equipped with UV-C light, to prevent contamination of samples during WGA;
-
-thermal cycler with heated lid;
-
-fume cabinet, hybridisation oven/incubator, water bath, gel electrophoresis equipment to check successful amplification and vortex mixers for plates and tubes;
-
-a scanner, equipped with the corresponding lasers and suitable for the specific slide type, to excite the hybridised fluorophores to read and store the resulting images of the hybridisations, placed in the post-amplification room in an atmosphere with low ozone parameters, regulated temperature and protected from daylight.
-
-
The use of a DNA quantification system (to determine the amount of amplified DNA after WGA) is optional.
Associated servers should also be allocated in proper conditions and instruments used in critical steps should be UPS-connected.
It is recommended that prior to each step of the protocol, the temperature ranges and/or pH values of equipment and solutions are verified. Specific temperature and thermocycler programmes should be validated in individual PGT centres for all equipment, and instruments should be serviced and calibrated regularly to ensure accuracy.
Haplotyping analysis software is not always commercially available; therefore, close collaboration with bioinformaticians needs to be guaranteed.
Materials.
For all reagents employed in the different steps of the protocol, the lot numbers and expiration dates should be recorded.
Depending on the platform used and the manufacturer, materials required for WGA and SNP array analysis can vary substantially and may include one or more of the following constituents:
-
-
cell lysis, amplification enzymes and buffers;
-
-
DNA fragmentation buffers and enzymes, fluorophores and modified dNTPs that should be used under minimal light exposure since they are light-sensitive;
-
-
hybridisation and washing buffers;
-
-
microarray slides.
Work practice controls
Identification and witnessing.
An adequate labelling system with two unique patient identifiers and embryo/cell(s) number is recommended.
- Labelling and sample identification should be confirmed for critical and high-risk steps by an independent observer, preferably one who is trained in molecular genetics. It is recommended that the unique patient identifiers plus the embryo/cell number be witnessed and signed off by two operators during biopsy, sample collection and genetic testing (see also the paper on organisation of PGT (ESHRE PGT Consortium Steering committee. et al., 2020)). Witnessing is also indicated at the following steps of the WGA/SNP array procedure:
-
-at the start of the WGA procedure to ensure that the correct volume of reaction master mixture is loaded into each tube;
-
-at the start of the SNP array protocol to ensure that that the correct volume of sample is transferred to the correct reaction tube/plate;
-
-at loading of the DNA samples on SNP array slides to ensure that each sample matches the sample identifier on the slide (slide number and position per sample should be monitored and registered);
-
-and when recording SNP array results to ensure that scanned raw files correspond to the correct cell and/or embryo.
-
-
Intra-assay controls.
It is recommended to use negative and positive controls alongside the test samples to check if contamination or amplification failure has occurred.
Suitable positive controls are not readily available (i.e. unbalanced single human blastomeres, TE cells or other cell types to represent unbalanced human blastomeres or TE cells).
Diluted genomic DNA inputs are recommended as positive intra-assay controls to check successful amplification of single/few cells and a successful reaction, respectively.
Negative controls serve to confirm that no contamination is present in the ‘no-template’ tube, which does not confirm the absence of contamination for the rest of reaction tubes carrying the biopsied samples.
At least one ‘no-template’ reaction tube with washing buffer only (i.e. the IVF laboratory negative control) and one negative control with amplification mixture only (i.e. the genetic laboratory negative control) are recommended to exclude DNA contamination of these media.
Pre-examination process
Internal quality control.
QC parameters define the overall quality profile of the samples. Depending on the platform, QCs should be defined by the user lab regarding the acceptable call rate and level of noise of the samples. When using SNP arrays for PGT-SR, depending on the quality parameters, the chromosomal localisation of the aberration(s) and the size of the involved segments, aberrant intensity ratios may or may not be detectable for the region(s) of interest. If detectable, it is recommended that the diagnosis is supported by log ratio and B allele frequency values.
It is recommended to validate the protocol using single cells from cell lines with a known karyotype, or the same WGA products from embryos containing known deletions or duplications diagnosed with a previously validated technique.
It is recommended to perform accuracy assessment, including both normal and abnormal samples. As different chromosome regions may have different SNP coverage, the series of abnormal samples should represent the range of structural rearrangements that the test is required to detect. It is recommended to use a minimum of three positive samples for each rearrangement type.
Following DNA amplification, a clear agarose gel pattern should be visible and/or quantitative measurement of DNA concentration should be sufficient for further testing.
Following accuracy assessment tests, it is recommended to calculate the performance (sensitivity, specificity, positive predictive value and negative predictive value) of the protocol.
It is recommended to test the quality of each batch of SNP arrays.
It is recommended to use hybridisation template forms to record sample tracking.
Barcoding of SNP array slides is mandatory to maintain the correlation between the sample and the SNP array slide used for hybridisation.
Test efficiency.
It is recommended that the WGA procedure be performed in the same tube that the sample was collected in.
To check for amplification efficiency, it is recommended that samples and intra-assay controls (if used) be run on an agarose gel and/or quantified by Qubit Fluorometer.
Negative amplification, negative intra-assay control or failed hybridisation should show a consistent noisy profile where no significant pattern is observed.
Storage time and temperature have an impact on the integrity of cells, DNA and/or solutions, and laboratories should validate that the conditions used in their protocols are fit for purpose. Furthermore, it is not recommended to use repeatedly frozen-thawed solutions containing DNA or enzymes.
It is recommended to calculate the acceptable and optimum ranges of QCs for every SNP array experiment. The QC measures will vary between array types and different scanners. It is recommended to perform an internal validation to establish a test-specific threshold for the overall noise value.
Preclinical work-up and report
Preclinical work-up.
It is recommended that the following steps are taken during preclinical work-up:
It is recommended to check whether the chromosomal segments involved in the rearrangement are adequately covered on the SNP array of interest.
Parental and phasing reference karyotypes may facilitate testing and genetic counselling.
Preclinical work-up report.
General guidance and recommendations on administration and patient information for the preclinical work-up report are provided in the paper on organisation of PGT (see ESHRE PGT Consortium Steering committee. et al., 2020). For PGT-SR using SNP array, the preclinical work-up report should also include a summary of the work-up.
It is recommended that the following are clearly stated in the report:
indication and karyotype of the patient (ISCN nomenclature can be used);
test limitations and residual risk of PGT misdiagnosis, including a figure.
Risk assessment
Risk assessment should cover the following:
-
-
risks caused by errors in sample tracking;
-
-
risks caused by handling biopsy samples prior to SNP array analysis (tubing, washing) which, if not performed with care, may compromise DNA integrity and lead to failed or poor WGA;
-
-
risk of inconclusive or false results due to suboptimal experimental conditions at WGA or high background noise;
-
-
risk of inconclusive results due to homologous recombination events in the vicinity of the fragments of interest;
-
-
patient’s risk of miscarriage, stillbirth, (viable) unbalanced offspring, mosaic offspring or offspring with a chromosomal imbalance that is below the resolution of the test, whether biological or caused by a technical error;
-
-
risk of incidental findings.
Limitations of the test
SNP array haplotyping requires at least one first degree relative of the partner carrying the rearrangement of interest for phase determination.
Next-generation sequencing
NGS allows for direct reading of sequenced DNA fragments and their quantification based on sequence read numbers. Depending on the sequencing read depth, NGS can be applied in different assays, from whole chromosome aneuploidy to medium size deletions or insertions in chromosomes and detection of single gene disorders. Compared with aCGH, chromosomal copy number assessment based on NGS may offer several advantages including: (i) reduced DNA sequencing cost made possible by high throughput sequencing technologies and the larger number of samples that can be simultaneously sequenced during a single experiment (the latter requires adding a unique tag); (ii) enhanced detection of deletions and duplications because of the potential increase in resolution (as assessed in the pre-examination validation); (iii) increased dynamic range enabling enhanced detection of chromosomal mosaicism in TE samples; (iv) the potential automation of the sequencing library preparation to minimise human errors, reduce hands-on time and enable higher throughput and consistency.
Laboratory issues
NGS protocol.
The sequencing by NGS protocol comprises five steps: (i) sample processing; (ii) initial quality analysis; (iii) library preparation; (iv) sequencing; and (v) data analysis.
The sample processing and sequencing generally includes any or all of the following processes: handling of biopsy samples (PB, single blastomere or TE cells); cell lysis; barcoding (molecular indexing) of samples; adapter ligation; amplification; library preparation; flow cell loading; and generation of sequence reads. It is recommended to perform initial quality analysis of DNA; contamination of starting material can lead to poor sequencing data quality.
DNA sequence generation by NGS platforms is almost entirely automated and the output consists of millions to billions of short sequence-reads. Raw data produced after sequencing are further processed by computational analyses and bioinformatics using a variety of algorithms to map and align the short sequence reads to a linear reference human genome sequence.
As these processes may vary depending on the platform, it is recommended to optimise and validate each step individually (including the entire wet-bench process as well as the bioinformatic analyses) to empirically determine optimal assay conditions and analysis settings.
For each platform, the genome coverage, average read depth and minimum number of reads should be defined with validation experiments (see pre-examination process section).
Turnaround time.
The turnaround time of NGS (from DNA amplification to reporting) can vary according to the platform, but currently it is at least 12 h. Turnaround time is expected to significantly decrease in the future.
With the aim of accumulating samples for an NGS run, biopsy samples can be stored short term (weeks), and WGA samples can be stored long term (years) at −20 or −80°C.
Documentation.
Relevant laboratory documentation should include:
-
-
a patients’ karyotype, preferably at high resolution (550–800 bands), if available with verified breakpoints from an accredited/certified cytogenetics laboratory; often, the rearrangement breakpoints are defined based on GTG-banded chromosomes and, as the resolution of this technique is quite low, there is a potential risk that the actual translocation segments are (much) smaller than expected and hence the probability of detection of all the unbalanced segregation products of the structural rearrangement (much) lower;
-
-
a report on any previous unbalanced products of conception;
-
-
genetic counselling report with possibly recommendations for PGT-SR, an indication of the testing method, and the benefits and the limitations of the test;
-
-
the informed consent of the couple with risk assessment and indication of test limitation.
Laboratory infrastructure, equipment and materials
Infrastructure.
General aspects on infrastructure are covered in the paper on organisation of PGT (ESHRE PGT Consortium Steering committee. et al., 2020), and in section ‘Array-based PGT-SR’.
Equipment.
NGS platforms differ, among others in price, capacity, chemistry and read length. Initial set-up of an NGS system should follow manufacturer’s instructions, and it is recommended to collaborate with the manufacturer to ensure that the laboratory space has been optimised to meet the requirements. In addition, it is recommended to involve informaticians with relevant expertise to make sure all required elements (hardware, servers, data storage, internet) are in place.
NGS-based PGT requires the following equipment:
A DNA quantitation instrument: it is crucial to accurately determine the amount of starting DNA for library preparation. There are several options that give highly accurate quantitation of low amounts of DNA. Among those is the Qubit high-sensitivity double-stranded DNA (HS dsDNA) fluorometer, which measures dsDNA. The HS dsDNA fluorometer has been found to give a much more accurate estimation of the amount of DNA present in the sample compared with standard spectrophotometry. The ratio of absorbance at 260 nm to absorbance at 280 nm is used as an indication of sample purity. It is recommended to use DNA with absorbance ratio values ranging from 1.8 to 2.0.
Thermocyclers: DNA amplification and labelling are necessary steps during the library preparation, therefore requiring the use of a thermocycler.
Pipettors or pipetting robots: dedicated multi-channel and single-channel pipettes are a necessity for NGS.
Multichannel pipette or automated systems are recommended to minimise the risks of mislabelling or misallocation of samples during the different steps of the protocol.
Sequencers should be allocated to a specifically designed room, with modulated light exposure and regulated temperature according to manufacturers´ instructions. Associated servers should also be kept under proper conditions and instruments used in critical steps should be UPS-connected.
Sequencers should be validated for the specific PGT protocols and incorporate the latest version of the specified software, allowing proper performance of the PGT protocol.
It is recommended that prior to each step of the protocol, the temperature ranges and or pH values of equipment and solutions are verified. Specific temperature and thermocycler programmes should be validated in individual PGT centres for all equipment, and instruments serviced and calibrated regularly to ensure accuracy.
Software for automatic calling of structural aberrations is not always available, and therefore, segmental aneuploidies need to be manually called by the operator.
Materials.
For all reagents employed in the different steps of the protocol, the lot numbers and expiration dates should be recorded.
Depending on the manufacturer, NGS kits may include one or more of the following constituents:
-
-
cell lysis and DNA extraction media: lysis buffer and specific enzymes for DNA extraction;
-
-
DNA amplification media: some WGA protocols are PCR-based whereas others are not, and it is recommended to use a WGA protocol which is compatible with the specific NGS platform that has been validated;
-
-
library preparation media: although many methods are available, some preparation procedures are specific for a particular NGS platform, and therefore, it is recommended to pay attention to the compatibility of the libraries with the sequencing platforms.
Work practice controls
Identification and witnessing.
An adequate labelling system with two unique patient identifiers and embryo/cell(s) number is recommended.
- Labelling and sample identification should be confirmed for critical and high-risk steps by an independent observer, preferably one who is trained in molecular genetics. It is recommended that the unique patient identifier and embryo/cell number be witnessed and signed off by two operators during biopsy, sample collection and genetic testing (see also the paper on organisation of PGT (ESHRE PGT Consortium Steering committee. et al., 2020)). Witnessing is also indicated at the following steps of the WGA/NGS procedure:
-
-at the start of the WGA procedure to ensure that the correct volume of PCR master mixture is loaded into each reaction tube;
-
-at the start of the library preparation to ensure that embryo identification corresponds to a dedicated barcode or index primers;
-
-at pooling, to make sure that all barcoded libraries are included in the pool before the start of the NGS run;
-
-during NGS run preparation; data input for each sample should be checked to ensure that samples match their identifier on the plate.
-
-
Intra-assay controls.
It is recommended to use negative and positive controls alongside the test samples to check if contamination or amplification failure has occurred.
As suitable positive controls are not readily available, it is recommended to use validated samples containing deletions or duplications (from very small size 5 to 20 Mb), and a diploid control sample.
Diluted genomic DNA inputs are recommended as positive intra-assay controls to check successful amplification of single/few cells and a successful reaction, respectively.
One ‘no-template’ reaction tube with washing buffer only (i.e. the IVF laboratory negative control) and one negative control with amplification mixture only (i.e. the genetic laboratory negative control) are recommended to exclude DNA contamination of these media.
Pre-examination process
Internal quality control.
QC parameters define the overall quality profile of the samples. Platforms have proper QCs defined as the minimum reading value and the lowest noise value needed to detect a copy number variation. Because the genomic resolution of NGS for PGT-SR can be an issue for small segmental abnormalities, NGS platforms may have already been validated for sensitivity, specificity and negative and positive predictive values. Despite the information provided by the manufacturer, an implementation validation with respect to the resolution is necessary. These values may vary between NGS platforms depending on coverage, insert size, WGA methodology and single versus paired-end sequencing.
Before testing patient samples, the analytical validity of the intended tests needs to be established with appropriate QC/QA.
It is recommended to validate the protocol using single cells from cell lines with a known karyotype, or the same WGA products from embryos containing known deletions or duplications diagnosed with a previously validated technique.
It is recommended to perform accuracy assessment, including both normal and abnormal samples. As different chromosome regions may have different coverage, the series of abnormal samples should represent the range of structural rearrangements that the test is required to detect. It is recommended to use a minimum of three positive samples for each rearrangement type.
Following amplification, it is recommended to quantify DNA. DNA concentration should be at least 20–50 ng/μl.
In general, poor-quality or failed WGA products should be excluded from further analysis as these samples may affect the sequencing read distribution per sample after library pooling and sequencing.
Following accuracy assessment tests, it is recommended to calculate the performance (sensitivity, specificity, positive predictive value and negative predictive value) of the protocol.
As the presence of chromosomal mosaicism is an issue when analysing TE biopsy samples, it is recommended to include mosaic samples (i.e. a mixture of cells with known segmental aneuploidies and euploid cells) in the validation study (see also section ‘Array-based and NGS-based PGT-A’).
Test efficiency.
For checking amplification efficiency, gel electrophoresis is recommended for samples and intra-assay controls using proper standards.
It is recommended that the WGA procedure be performed in the same tube that the sample was collected in.
After preparation the library should be quantified and normalised for each sample before creating the library pool.
It is recommended to have high coverage for the region of interest and ascertain that the expected translocation is covered by a sufficient number of sequenced fragments.
Sequencing by NGS comprises a series of steps that uniquely contribute to the overall quality of the data set. Thus, each individual step needs to be controlled to ensure high-quality results.
NGS run parameters (coverage, number of reads, noise) should be monitored before the analysis of raw sequencing data to ascertain that the overall and individual run parameters for each sample correspond to the platform-specific required criteria. These sequencing quality metrics can provide important information about the accuracy of each step in this process, including library preparation, base calling and read alignment.
From the total number of reads, 70–80% should align to the genome. Lower percentages indicate contamination in the DNA sample, degraded DNA or suboptimal WGA.
Each run should have an acceptable, previously established level of noise. It is recommended to perform an internal validation to establish a test-specific threshold for the overall noise value.
Various amplification protocols are in use, which may be affected by single-cell artefacts, such as allele drop-out (ADO), amplification bias or allele drop-in (ADI), that might affect the accuracy of the diagnostic test, and therefore, extensive validation of WGA is required.
It is recommended to calculate the acceptable and optimum ranges of QCs for every NGS experiment. The QC measures of NGS data for every experiment are extrapolated by specific software and are indicative for the successful calling of all target DNA sequencing. The QC measures will vary between NGS platforms and different software versions.
Preclinical work-up and report
Preclinical work-up.
It is recommended to check whether the chromosomal segments involved in the rearrangement are adequately covered, in terms of the number of sequence reads.
Parental karyotypes may facilitate testing and genetic counselling.
It is acceptable that at least three out of four segments for two-way reciprocal translocations can be detected to reliably identify unbalanced segregation products.
It is not acceptable to perform a clinical PGT-SR test if the size of the translocation segments, inferred from the karyotype, is below the threshold of resolution of the platform used.
It is acceptable to adjust the lower detection limit provided by the platform’s manufacturer, based on a feasibility study using DNA from previous unbalanced products of conception.
It is acceptable to forego any additional work-up when performing NGS for structural rearrangements.
Preclinical work-up report.
A case-specific preclinical work-up report is not required, provided that no particularities have come to light during the work-up. However, a report on the theoretical evaluation of the work-up should be available.
Risk assessment
Risk assessment should cover the following:
-
-
risks caused by errors in sample tracking;
-
-
risks caused by handling biopsy samples prior to NGS analysis (tubing, washing) which, if not performed with care, may compromise DNA integrity and lead to failed or poor WGA;
-
-
risk of inconclusive or false results due to suboptimal experimental conditions at WGA or high background noise or low coverage;
-
-
risk that the size of the deletion or duplication is different from the one based on the karyotypes in the parents, and therefore they may remain undetected by the NGS protocol (if they are below the resolution of the test);
-
-
risk of misinterpretation of the actual embryo karyotype due to the presence of chromosomal mosaicism, either at cleavage-stage or at blastocyst stage.
Limitations of the test
Limitations of the standard NGS protocols for PGT-SR without genotyping are based on the fact that the analysis cannot:
-
-
detect whole ploidy changes;
-
-
discriminate balanced from normal results;
-
-
detect low level chromosomal mosaicism;
-
-
detect abnormalities below the predefined resolution.
Preimplantation testing for numerical aberrations
Applications of PGT-A comprise former PGS in IVF couples with normal karyotype and PGT-A in couples with chromosomal numerical aberrations such as Klinefelter syndrome and other sex chromosome abnormalities. Both applications share the same techniques, but reporting may be different.
FISH is not recommended for PGT-A, as only a subset of chromosomes can be tested and better comprehensive molecular approaches to detect aneuploidy for all 24 chromosomes are available.
Real-time quantitative PCR (qPCR) is used for PGT-A, but the limits of the technique, such as the low resolution in the detection of chromosomal mosaicism, have led to its disuse in favour of techniques such as NGS. For this reason, real-time qPCR will not be addressed in this paper.
Array-based and NGS-based PGT-A
aCGH was clinically applied for PGT of whole chromosome abnormalities and has revolutionised the field by providing accurate identification of comprehensive chromosome copy numbers and rapid analysis.
aCGH platforms utilizing bacterial artificial chromosomes (BACs), chromosome-specific libraries, oligonucleotides and SNPs have been clinically applied and all succeed in detecting aneuploidies in PBs, single blastomeres and TE samples.
The application of NGS for the detection of copy number variation differs from aCGH by using direct reads of genomic sequencing fragments and their quantification according to sequence read numbers instead of a signal intensity comparison between fluorescently labelled test and reference DNA samples. NGS has been extensively validated using cells of a known genotype and is now used for detecting aneuploidies in PBs, single blastomeres and TE samples.
Laboratory issues
Information on protocols, turnaround time and documents for aCGH and NGS is presented in the sections ‘Array-based PGT-SR’ and ‘Next-generation sequencing’, respectively.
Laboratory infrastructure, equipment and materials
Information on infrastructure, equipment and materials for aCGH and NGS is presented in the sections ‘Array-based PGT-SR’ and ‘Next-generationsequencing’, respectively.
Work practice controls
Information on identification and witnessing for aCGH and NGS is presented is presented in the sections ‘Array-based PGT-SR’ and ‘Next-generation sequencing’, respectively.
Use of intra-assay controls for aCGH.
Information on using intra-assay controls for aCGH is presented in section ‘Array-based PGT-SR’.
Use of intra-assay controls for NGS.
For an intra-assay control in each routine test, it is recommended to use negative and positive controls in the same NGS run with separate barcodes with the aim to monitor if the section has contamination or amplification failure.
It is recommended to perform an intra-assay control using isolated samples composed of single cells containing known whole-chromosome aneuploidies diagnosed with a previously validated technique.
Pre-examination process
Information on test efficiency materials for aCGH and NGS is presented in the sections ‘Array-based PGT-SR’ and ‘Next-generationsequencing’, respectively.
Internal quality control.
Effective resolution of the aCGH and NGS platform and protocol should be internally validated in each laboratory prior to clinical application for PGT-A.
- It is recommended to validate aCGH and NGS for aneuploidy testing with a series of positive controls that should include DNA from:
-
-single cells from cell lines with established numerical copy number changes (aneuploidy);
-
-previous aneuploid pregnancies, when available;
-
-blastomeres or TE biopsies isolated from donated embryos from previously performed PGT-A cases analysed with an established technique, when available. Pre-clinical testing on PBs is not straightforward, as it would deprive the couple of valuable embryos that could also be used for clinical treatment.
-
-
It is recommended to determine false-negative, false-positive and specificity and sensitivity rates of the specific platform to be used.
When using aCGH and NGS for aneuploidy testing in TE biopsy samples, the possibility of misdiagnosis due to chromosomal mosaicism represents the main issue relating to copy number variation (CNV) and log2 ratio value threshold detection by NGS and aCGH, respectively.
It is recommended to perform validation studies with true aneuploid and euploid cell lines and mosaic models by using cell mixtures (ratios from 10 to 90%) to establish thresholds for chromosomal mosaicism detection rates (i.e. the minimum ratio of aneuploid to euploid cells that is needed to detect a chromosomal CNV) and quantification of mosaicism levels. After statistical analysis, the results of these experiments can be used as a reference to determine the mosaicism level of analysed samples. In the first step of the validation process, it is recommended to analyse a wide number of euploid samples (including six to eight cells from euploid cell lines), in order to determine the standard deviation from the euploidy baseline value (two chromosome copy number and log2 ratio for NGS and aCGH, respectively) and thus define the ‘euploidy’-threshold values. Similarly, threshold values should be defined for trisomy and monosomy.
It is recommended to test replicates of the same DNA sample to perform accuracy and variability assessment in independent aCGH experiments and NGS runs.
To mimic a blastocyst biopsy, a sample size of 8–10 cells is recommended for all mosaicism cell mixture models. Although validation experiments will set euploid/aneuploid parameters, it is important to mention that limitations still exist when analysing biopsy samples with few cells, where it will be almost impossible to detect changes that represent less than 20–30% of the biopsy.
In order to define the detection threshold, the quality (intrinsic DNA sample quality, QC) of the experiments, the noise and technical artefacts should also be considered.
As different chromosomes might have a different resolution, the series of aneuploid samples should represent the range of aneuploidies that the test is required to detect.
Sensitivity and specificity of the mosaicism detection specifically apply for each aCGH and NGS platform (hardware and protocol for WGA, or library preparation for NGS) and software or bioinformatics paradigm used to analyse the data. These cannot be exchanged among platforms.
During the validation of aCGH and NGS for PGT-A, de novo segmental chromosome aberrations are also encountered.
It is recommended to establish the true resolution and specificity of the aCGH and NGS platform to detect segmental aneuploidy through a validation study that has been already mentioned in sections ‘Array-based PGT-SR’ and ‘Next-generation sequencing’ for PGT-SR.
Preclinical work-up and report
Information on the preclinical work-up and report related to aCGH and NGS is presented in the sections ‘Array-based PGT-SR’ and ‘Next-generation sequencing’, respectively.
Preclinical work-up. Case-specific preclinical work-up or specific genetic documentation is not required when performing aCGH and NGS for aneuploidy testing (high-risk and low-risk).
Preclinical work-up report.
A case-specific preclinical wet-laboratory work-up report is not required for aCGH and NGS.
Risk assessment
Information on risk assessment related to aCGH and NGS is presented in the sections ‘Array-based PGT-SR’ and ‘Next-generation sequencing’, respectively, and additional issues related to aCGH and NGS for PGT-A are listed here.
The clinical significance of transferring embryos with mosaicism and/or de novo segmental abnormalities (full or in mosaic state) is under current investigation and therefore unknown. The transfer of such embryos could potentially carry a risk of first-trimester miscarriage or of a viable unbalanced offspring.
-
aCGH and NGS can detect chromosomal mosaicism and segmental aneuploidies. However, both biological limitations and technical artefacts may affect the accuracy of the test and this should be discussed during patient counselling.
-
°
Biological limitations may include non-specific chromosome gain or loss due to cells being in S-phase, the biopsy being non-representative of the embryo, failure to detect chromosomal mosaicism due to non-disjunction, and apoptotic or dead cells in the biopsy sample that can generate profiles resembling mosaicism.
-
°
Technical artefacts may include WGA artefacts, contamination, cells damaged during biopsy and cell lysed during tubing.
-
°
Limitations of the test
aCGH and standard NGS cannot reliably detect all variants of polyploidy (they can detect polyploidy with unbalanced sex chromosome ratios as 69,XXY and 69,XYY) and haploidy.
The currently used aCGH platforms for PGT-A are unable to detect small microdeletions or microduplications, such as the 22q11.2 microdeletion syndrome (DiGeorge/velocardiofacial syndrome).
Due to the intrinsic nature of chromosomal mosaicism, the chromosomal make-up achieved from a biopsy may only represent a picture of a small part of the embryo and may not necessarily reflect the chromosomal content of the entire embryo. Also, the mosaicism level inferred from a multi-cell TE biopsy might not unequivocally represent the exact chromosomal mosaicism percentage of the TE cells or the inner cell mass constitution.
NGS and aCGH are currently able to detect mosaicism down to 20–30% when no noise is present in the sample and after proper validation. Array-based PGT-SR analysis is less sensitive to detect mosaicism than NGS.
As the number of cells in a TE biopsy is unknown, the exact level of mosaicism in the sample cannot be determined.
aCGH cannot analyse aneuploidy and gene defects simultaneously, whereas NGS can.
Based on the embryo biopsy, aCGH cannot identify the nature (meiotic or mitotic) and/or the parental origin of aneuploidy whereas genotyping-based NGS can, provided that phasing references are available.
Noisy profiles are difficult to evaluate and to appropriately score the chromosome copy number.
Strengths and limitations
Technical strengths and limitations of FISH, aCGH and NGS (without genotyping) are outlined in Table I.
Table I.
Overview of the strengths and limitations of the methods applied for PGT-SR and PGT-A.
| PGT-SR | PGT-SR/PGT-A | ||
|---|---|---|---|
| FISH | aCGH | NGS (without genotyping) | |
| Number of chromosomes | Information is limited to chromosomes and/or targeted loci for which probes are used. | All 24 chromosomes analysed. | All 24 chromosomes analysed. |
| Minimal resolution | Limited by the availability of (commercial) probes. Commercial probes are available for only a limited number of loci, which may complicate the selection of probes for the analysis of rare chromosomal rearrangements. | Limited by the empirical resolution of the platform established in each laboratory after proper validation of wet-laboratory protocol and analysis software. | Limited by the empirical resolution of the platform established in each laboratory after proper validation of wet-laboratory protocol and analysis software. |
| Whole ploidy changes | Inferred from the number of hybridisation signals from multiple probes. | Not all variants of polyploidy and haploidy can be detected. | Not all variants of polyploidy and haploidy can be detected. |
| No conclusive results | As a result of improper fixation, overlapping cells or signals. Rebiopsy is an option. | As a result of cell lysis during tubing, cells with degraded DNA, cell loss or poor experimental conditions. Re-analysis or rebiopsy is an option. | As a result of cell lysis during tubing, cell loss or poor experimental conditions. Re-analysis or rebiopsy is an option. |
| Abnormalities not diagnosed | FISH-based PGT-SR diagnosis of biopsied material from cleavage stage or blastocyst embryos does not allow for a distinction between embryos with a normal or a balanced karyotype. | aCGH-based PGT-SR diagnosis of biopsied material from cleavage stage or blastocyst embryos does not allow for a distinction between embryos with a normal or a balanced karyotype. | NGS-based PGT-SR diagnosis of biopsied material from cleavage stage or blastocyst embryos does not allow for a distinction between embryos with a normal or a balanced karyotype. |
| Mosaicism-related issues | Chromosomal mosaicism, either at cleavage stage or blastocyst stage, may lead to misinterpretation of the actual embryo karyotype. | Chromosomal mosaicism, either at cleavage or blastocyst stage, may lead to misinterpretation of the actual embryo karyotype. | Chromosomal mosaicism, either at cleavage or blastocyst stage, may lead to misinterpretation of the actual embryo karyotype. |
| Uniparental disomy (UPD) | FISH analysis does not allow for the detection of UPD. | aCGH analysis does not allow for the detection of UPD. | NGS analysis does not allows for the detection of UPD. |
| Risk of misdiagnosis | Contamination with cumulus cells. Visual inspection allows for the identification of sperm cells, Incomplete nucleus, or presence of nuclear fragments. | Contamination with remaining cumulus cells after ICSI. | Contamination with remaining cumulus cells after ICSI. |
| Impact of biopsy on the results | Cells (DNA) damaged during biopsy may have a negative impact on the reliability of the test result. Analysis of a multi-cell biopsy is less favourable compared to a single cell biopsy. |
Cells (DNA) damaged during biopsy may have a negative impact on the reliability of the test result. Analysis of a multi-cell biopsy is more efficient than of a single cell biopsy. |
Cells (DNA) damaged during biopsy may have a negative impact on the reliability of the test result. Analysis of a multi-cell biopsy is more efficient than of a single cell biopsy. |
| Simultaneous detection of chromosome copy number and single gene disorder(s) | Not feasible. | Not feasible. | Feasible. |
| Origin of aneuploidy | Cannot identify the nature (meiotic or mitotic) and/or the parental origin of aneuploidy when based on the analysis of biopsied material from cleavage stage or blastocyst embryos. | Cannot identify the nature (meiotic or mitotic) and/or the parental origin of aneuploidy when based on the analysis of biopsied material from cleavage stage or blastocyst embryos | Cannot identify the nature (meiotic or mitotic) and/or the parental origin of aneuploidy when based on the analysis of biopsied material from cleavage stage or blastocyst embryos. |
PGT-SR: preimplantation genetic testing for chromosomal structural rearrangements, PGT-A: PGT for aneuploidies, FISH: fluorescence in situ hybridisation, aCGH: array-based comparative genomic hybridisation, NGS: next-generation sequencing, ICSI: intracytoplasmic sperm injection.
The most important limitations include the following:
Based on the embryo biopsy alone, FISH, aCGH and NGS cannot discriminate between samples that are carrying the rearrangement (i.e. balanced) and those that are not (i.e. normal) and this should be clearly stated in the report. Although there is no expected difference in the phenotype of embryos with a ‘normal ‘or a ‘balanced’ karyotype, many couples wish to know whether the structural rearrangement is being transferred to their offspring, to be aware of possible future reproductive problems related to the rearrangement. When PBs are used for PGT analysis, discrimination between oocytes carrying the rearrangement and those that are not, is feasible.
FISH and aCGH cannot, but NGS can, analyse aneuploidy and gene defects simultaneously in the same diagnostic sample.
Based on the embryo biopsy alone, FISH, aCGH and NGS without genotyping cannot identify the nature (meiotic or mitotic) or the parental origin of aneuploidies. When PBs are used for PGT analysis, inferred errors in the oocyte are always of maternal, meiotic origin.
Based on the embryo biopsy alone, FISH, aCGH and NGS without genotyping cannot detect UPD.
Examination process
Clinical testing protocols should include scoring criteria and reporting procedures as well as a framework for counselling patients in the presence of diagnostic results.
General recommendations on the PGT examination process are included in the paper on organisation of PGT (see ESHRE PGT Consortium Steering committee. et al., 2020). The sections below highlight specific issues relevant to PGT-SR and PGT-A.
Scoring of clinical results
FISH results
FISH signals should be scored according to brightness, size and distance. The signals should have approximately the same brightness and size (depending on the probes used) and should be at least one signal in diameter apart. Two signals that are in close proximity and have approximately the same size, but are not connected by a visible link, are considered as two signals. A diffuse signal should be scored as one if the signal is continuous and of expected size. Two small signals connected by a visible link are counted as one signal.
Signal scoring criteria should be established in a written protocol and adhered to for the interpretation of signals.
It is recommended that signals are analysed by two independent observers and that discrepancies adjudicated (where possible) by a third observer. If no consensus is reached the embryo should not be recommended for transfer, i.e. should be given the diagnosis of uninterpretable or inconclusive result.
It is acceptable to score signals from probes labelled with fluorochromes not detectable to the human eye using an image capture system.
All fluorescent images should be captured and filed for QC purposes. If possible, the position and co-ordinates of the embryonic cells on the slide can be recorded.
‘No result rescue’ for embryos without a clear diagnosis is acceptable. An additional hybridisation round should be performed with probes indicative of the same chromosome(s) but a different region or, if not available, at least with probes in a different colour scheme. A second biopsy can also be performed, followed by the full FISH protocol.
When there is a combination of chromatid gain/loss in the first PB, which is balanced by the second PB, a normal chromosome copy number in the corresponding oocyte is predicted and reported, and the resulting embryo can be considered for transfer after discussion with the patient.
aCGH and NGS results
Software analysis and copy number scoring criteria should be established in a written protocol and adhered to for the interpretation of whole chromosome and segmental-chromosome gains and/or losses.
Interpretation of raw data or profiles resulting after specific software analysis by a single observer is acceptable. Additional confirmation by an independent observer is recommended.
All files resulting from the scanning and sequencing, as well as profiles after specific software analysis, should be stored and filed for QC purposes.
‘No result rescue’ for embryos without a clear diagnosis is acceptable. This could imply a second analysis of the existing WGA as well as a second biopsy followed by WGA, full aCGH/NGS processing and analysis.
When there is a combination of chromatid gain/loss in the first PB, which is balanced by the second PB, a normal chromosome copy number in the corresponding oocyte is predicted and reported, and the resulting embryo can be recommended for transfer.
Issuing a PGT report
General items required in PGT preclinical work-up or clinical cycle reports have been listed in the paper on organisation of PGT (see ESHRE PGT Consortium Steering committee. et al., 2020). The ISCN reporting is acceptable for PGT-SR and PGT-A. It is recommended to add the following technical or interpretation items to the clinical report:
If the profile is noisy or QCs are not sufficient, re-analysis is acceptable to try and obtain a result and this should be included in the report to the IVF centre.
In the absence of any amplification or when contamination is suspected, rebiopsy is acceptable to try and obtain a result and this should be included in the report to the IVF centre.
Each centre should decide whether or not to report mosaicism based on internal validation and recent literature.
The clinical significance of transferring mosaic embryos is currently unknown. The centre’s policy about the identification and transfer of embryos with mosaicism or segmental aneuploidy should be documented and shared with the patient during genetic counselling.
In case of an embryo with chromosomal mosaicism or segmental aneuploidy, genetic counselling should be offered to the couple and if transfer is decided and pregnancy occurs, it should receive appropriate follow-up (ESHRE PGT Consortium Steering committee. et al., 2020)) section ‘Follow-up of PGT pregnancies and children’).
Post-examination process
Recommendations on PGT follow-up, baseline IVF live birth rates for PGT and misdiagnosis are covered in the paper on organisation of PGT (see ESHRE PGT Consortium Steering committee. et al., 2020).
Supplementary Material
Acknowledgements
The authors would like to thank everyone that contributed to the stakeholder review for the constructive remarks that improved the quality of the paper. The list of reviewers is available in Supplementary Table SIII.
Footnotes
†ESHRE Pages content is not externally peer reviewed. The manuscript has been approved by the Executive Committee of ESHRE.
Authors’ roles
E.C. and C.R.L. chaired the working group. All authors contributed to the conception and design, drafting the content and discussing it. All authors approved the final version.
Funding
The work and meetings were supported by ESHRE.
Conflict of interest
The authors have nothing to disclose.
References
- ESHRE PGT-M Working Group, Carvalho F, Moutou C, Dimitriadou E, Dreesen J, Giménez C, Goossens V, Kakourou G, Vermeulen N, Zuccarello D et al. ESHRE PGT consortium good practice recommendations for the detection of monogenic disorders. Hum Reprod Open 2020. doi: 10.1093/hropen/hoaa018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ESHRE PGT Consortium and SIG-Embryology Biopsy Working Group, Kokkali G, Coticchio G, Bronet F, Celebi C, Cimadomo D, Goossens V, Liss J, Sofia Nunes S, Sfontouris I et al. ESHRE PGT Consortium and SIG Embryology good practice recommendations for polar body and embryo biopsy for preimplantation genetic testing. Hum Reprod Open 2020. doi: 10.1093/hropen/hoaa020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ESHRE PGT Consortium Steering committee, Carvalho F, Coonen E, Goossens V, Kokkali G, Rubio C, Meijer-Hoogeveen M, Moutou C, Vermeulen N, De Rycke M. ESHRE PGT consortium good practice recommendations for the organisation of preimplantation genetic testing. Hum Reprod Open 2020. doi: 10.1093/hropen/hoaa021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geraedts J, Montag M, Magli MC, Repping S, Handyside A, Staessen C, Harper J, Schmutzler A, Collins J, Goossens V et al. Polar body arrayCGH for prediction of the status of the corresponding oocyte. Part I: clinical results. Hum Reprod 2011;26:3173–3180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper JC, Aittomaki K, Borry P, Cornel MC, Wert G, Dondorp W, Geraedts J, Gianaroli L, Ketterson K, Liebaers I et al. Recent developments in genetics and medically assisted reproduction: from research to clinical applications. Eur J Hum Genet 2018;26:12–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harton G, Braude P, Lashwood A, Schmutzler A, Traeger-Synodinos J, Wilton L, Harper JC. ESHRE PGD consortium best practice guidelines for organization of a PGD centre for PGD/preimplantation genetic screening. Hum Reprod 2011a;26:14–24. [DOI] [PubMed] [Google Scholar]
- Harton GL, De Rycke M, Fiorentino F, Moutou C, SenGupta S, Traeger-Synodinos J, Harper JC. ESHRE PGD consortium best practice guidelines for amplification-based PGD. Hum Reprod 2011b;26:33–40. [DOI] [PubMed] [Google Scholar]
- Harton GL, Harper JC, Coonen E, Pehlivan T, Vesela K, Wilton L. ESHRE PGD consortium best practice guidelines for fluorescence in situ hybridization-based PGD. Hum Reprod 2011c;26:25–32. [DOI] [PubMed] [Google Scholar]
- Harton GL, Magli MC, Lundin K, Montag M, Lemmen J, Harper JC. ESHRE PGD Consortium/Embryology Special Interest Group--best practice guidelines for polar body and embryo biopsy for preimplantation genetic diagnosis/screening (PGD/PGS). Hum Reprod 2011d;26:41–46. [DOI] [PubMed] [Google Scholar]
- Kotzot D. Prenatal testing for uniparental disomy: indications and clinical relevance. Ultrasound Obstet Gynecol 2008;31:100–105. [DOI] [PubMed] [Google Scholar]
- Magli MC, Montag M, Koster M, Muzi L, Geraedts J, Collins J, Goossens V, Handyside AH, Harper J, Repping S et al. Polar body array CGH for prediction of the status of the corresponding oocyte. Part II: technical aspects. Hum Reprod 2011;26:3181–3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Practice Committee of the Society for Assisted Reproductive Technology, Practice Committee of the American Society for Reproductive Medicine. Preimplantation genetic testing: a Practice Committee opinion. Fertil Steril 2008;90:S136–S143. [DOI] [PubMed] [Google Scholar]
- Preimplantation Genetic Diagnosis International Society. Guidelines for good practice in PGD: programme requirements and laboratory quality assurance. Reprod Biomed Online 2008;16:134–147. [DOI] [PubMed] [Google Scholar]
- The Preimplantation Genetic Diagnosis International Society (PGDIS). Guidelines for good practice in PGD. Reprod Biomed Online 2004;9:430–434. [DOI] [PubMed] [Google Scholar]
- Thornhill AR, Die-Smulders CE, Geraedts JP, Harper JC, Harton GL, Lavery SA, Moutou C, Robinson MD, Schmutzler AG, Scriven PN et al. ESHRE PGD Consortium 'Best practice guidelines for clinical preimplantation genetic diagnosis (PGD) and preimplantation genetic screening (PGS)'. Hum Reprod 2005;20:35–48. [DOI] [PubMed] [Google Scholar]
- Vermeulen N, Le Clef N, Veleva Z, D'Angelo A, Tilleman K. European Recommendations for good practice in addition to an evidence-based guidelines programme: rationale and method of development. BMJ Evid Based Med 2019;24:30–34. [DOI] [PubMed] [Google Scholar]
- Zegers-Hochschild F, Adamson GD, Dyer S, Racowsky C, Mouzon J, Sokol R, Rienzi L, Sunde A, Schmidt L, Cooke ID et al. The international glossary on infertility and fertility care 2017. Hum Reprod 2017;32:1786–1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.