

Abstract
Alzheimer’s disease (AD) is one of the most prevalent neurodegenerative diseases, characterized by impaired cognitive function due to progressive loss of neurons in the brain. Under the microscope, neuronal accumulation of abnormal tau proteins and amyloid plaques are two pathological hallmarks in affected brain regions. Although the detailed mechanism of the pathogenesis of AD is still elusive, a large body of evidence suggests that damaged mitochondria likely play fundamental roles in the pathogenesis of AD. It is believed that a healthy pool of mitochondria not only supports neuronal activity by providing enough energy supply and other related mitochondrial functions to neurons, but also guards neurons by minimizing mitochondrial related oxidative damage. In this regard, exploration of the multitude of mitochondrial mechanisms altered in the pathogenesis of AD constitutes novel promising therapeutic targets for the disease. In this review, we will summarize recent progress that underscores the essential role of mitochondria dysfunction in the pathogenesis of AD and discuss mechanisms underlying mitochondrial dysfunction with a focus on the loss of mitochondrial structural and functional integrity in AD including mitochondrial biogenesis and dynamics, axonal transport, ER-mitochondria interaction, mitophagy and mitochondrial proteostasis.
Keywords: Alzheimer’s disease, Mitochondrial dysfunction, Bioenergetics, mtDNA, Mitochondrial dynamics, Axonal transport, Mitochondrial biogenesis, Mitochondrial quality control, ER-mitochondria association, Mitochondrial proteostasis
Background
Alzheimer’s disease (AD) is one of the most prevalent neurodegenerative diseases in the world [1–3]. The major symptom presents as deterioration of cognition and memory functions due to the progressive and selective loss of neurons in forebrain and other brain areas. The debilitating neurological conditions of the disease cause severe disability in AD patients with progression of the disease. Unfortunately, none of the current therapies cure the disease and all are very limited in their ability to slow down its progression. More importantly, there is increased prevalence and incidence of AD in the world and by 2050, 1 in 85 persons worldwide will be living with the disease and 43% of those afflicted need a high level of care [4, 5]. Considering the huge social and economic burdens devoted to the care of AD patients, enormous effort is focused on exploring effective interventions to alleviate the symptoms or even cure the disease. During the past decades, studies suggested that multiple factors [6] including biological factors (e.g., aging, gender, body weight, etc.), environmental factors (e.g., lifestyle, toxins, brain injury, etc.), and genetic factors (e.g., APP, PS1, and PS2 genetic mutation in familial AD and susceptibility genetic polymorphisms in sporadic cases) contribute to the pathogenesis of AD. Although accumulated knowledge greatly expanded our understanding of AD, the underlying mechanism of AD pathogenesis remains elusive.
Mitochondria are conserved organelles that carry out multiple essential functions in different cellular processes [7]: Many neuronal activities are energy-taxing and human nervous system consumes a great deal of energy, and mitochondria are the major energy source providing ATP through oxidative phosphorylation to maintain the normal neuronal homeostasis and function. Mitochondria are essential for the biosynthesis of essential iron-sulfur center and heme in neurons, and are involved in the presynaptic transmitter synthesis in synapses. Mitochondria provide important buffering machinery to regulate calcium concentration during signal transduction, which is of particular importance to excitable cells such as neurons. Neurons are long-lived cells with the same life span as the organism. As the essential hub in regulating cell survival and death under various stresses, mitochondria safeguard neuronal survival through a variety of stresses during long neuronal lives. In order to perform these various functions, mitochondria are dynamically interacting with one another and with other cellular organelles to coordinate mitochondrial stress response under physiological and pathological conditions. It is therefore not surprising that disturbances of mitochondrial function are closely associated with the mechanisms underlying nervous system abnormalities including neurodegenerative diseases [7].
To date, a large body of research has shown extensive mitochondria abnormalities in the brain of AD patients [8]. Consistent with the observation that impaired energy metabolism invariantly precedes the clinical onset of AD, mitochondrial dysfunction has been established as an early and prominent feature of the disease [8, 9], suggesting a critical role in the pathogenesis of AD. Here we will review evidence of mitochondrial abnormalities in the brain of human AD patients and discuss in more detail recent advances in the understanding of mechanisms underlying mitochondrial dysfunction in AD which may offer novel targets for future therapeutic development.
Impaired energy metabolism implicates mitochondrial dysfunction in AD
Brain constitutes on average 2% of the total body weight, but utilizes 25% of total body glucose and 20% of body oxygen consumption in resting awake state. As one of the high-energy consuming organs, brain is vulnerable to impaired energy metabolism such that even mild changes in energy metabolism in human brain closely associates with the disturbance in nervous function. In fact, impaired energy metabolism is one of the earliest and most consistent features in AD.
Glucose is the predominant substrate for the human adult brain under physiological conditions, and its utilization is widely used as one primary measure to assess energy metabolism in the brain. A large body of evidence demonstrated significantly reduced glucose utilization as an early and consistent feature in AD, which actually occurs decades before the onset of disease [10–13]. Using fluoro-2-deoxyglucose positron-emission tomography (FDG-PET), greater decline in glucose utilization was consistently found in the hippocampus and cortex in AD brain as compared to individuals without dementia. Among many brain regions, the posterior cingulate cortex is metabolically affected in the earliest clinical stages of AD [10]. Glucose hypometabolism, to a lesser extent in terms of magnitude or spatial distribution, was also observed in patients with mild cognitive impairment (MCI), a prodromal stage of AD, suggesting an early role in the course of disease [12]. An 84-months longitudinal study clearly demonstrated an ApoE4-associated brain-region specific longitudinally declined glucose metabolism pattern in the context of MCI [14]. Such an early role is further supported by the finding of abnormally low rates of glucose metabolism in the vulnerable brain regions in young adults carrying apoE4 allele in their 20s, several decades before the possible onset of dementia [15]. The extent and topography of glucose hypometabolism correlated with symptom severity and also reflected the regional distribution of impaired synaptic activity and density in AD [16, 17]. Accepted as a hallmark of the disease, cerebral glucose hypometabolism assessed with FDG-PET is now used as a common biomarker for early detection of AD and can predict the conversion from MCI to AD with reasonable sensitivity and accuracy [18, 19], which underscores the critical role of impaired energy metabolism in the course of AD.
Multimodal imaging studies, using both FDG PET and amyloid PET biomarkers, have investigated the relationship between amyloid plaque deposition and glucose metabolism. In autosomal-dominant AD mutations carriers, longitudinal Aβ depositions increase in nearly every cortical region 15–25 years before the estimated age of onset, followed by reduced glucose metabolism in selective cortical region approximately 5–10 years later (i.e., amyloid-first biomarker profile pathway), suggesting that glucose hypometabolism could be a secondary event after Aβ depositions in AD pathogenesis in these cases [13, 20, 21]. Although reductions in regional glucose metabolism are associated with global amyloid pathology, there is poor association between regional amyloid pathology and regional hypometabolism when they are compared side-by-side in the same subjects: only 1 out of 404 regions of interest showed a negative association between amyloid plaque deposition and glucose metabolism [22]. This study suggests that glucose hypometabolism, even as a secondary event in the cases of autosomal-dominant AD, may play an essential role in the ensuing clinical onset of the disease. Repeated failure of Aβ-centered clinical trials suggests that it may be too late to target Aβ in AD patients or even MCI patients when some toxic cascade of events have been initiated after adecade of presence of amyloid pathology. These critical secondary pathogenic events such as impaired energy metabolism may provide an extended time window for therapeutic intervention. Therefore, it is still of paramount significance to understand the essential role and mechanisms underlying impaired energy metabolism in AD, even as a secondary event.
On the other hand, energy hypometabolism may play a primary role in a subset of sporadic AD patients. In a population study, while the majority (60%) incident amyloid-positive subjects followed the amyloid-first biomarker profile pathway, 27% had an abnormal FDG-PET at baseline, suggesting the existence of “hypometabolism-first” biomarker profile pathway to preclinical AD [23]. FDG PET studies have identified AD-like glucose hypometabolism in ApoE4 carriers without amyloid deposition who probably also followed this hypometabolism-first biomarker profile pathway since these carriers are likely to develop amyloid pathology in the future [24, 25]. While it is possible that Aβ dysmetabolism prior to amyloid plaque formation or other pathophysiologies such as tau or TDP43 may underlie glucose hypometabolism in these cases, the hypometabolism-first biomarker profile pathway to preclinical AD lends strong support to a primary role for energy hypometabolism in the pathogenesis of at least a subset of sporadic AD patients.
Glucose metabolism is a multi-step process involving glucose transportation and intracellular glucose metabolism. Abnormalities involving almost all of these steps from glucose transportation abnormalities including insulin resistance, abnormal glucose transporter [26] and/or blood flow [27], to intracellular glucose metabolism disturbance including abnormalities in cytosolic processes (i.e., glycolysis and pentose phosphate pathway), in addition to abnormal mitochondria-dependent processes (TCA cycle and oxidative phosphorylation as discussed in details in the next section), were identified in AD brain, which all could contribute to glucose hypometabolism. It is perhaps not impossible that one or more of these factors may play a more important role than other factors in causing cerebral glucose hypometabolism in AD, but it would be challenging to identify such major player(s) given the complex interactions among these factors and the possibility that different AD variants may have differences in the major factor(s) involved.
Nevertheless, glucose hypometabolism in AD brain was generally interpreted as impaired energy metabolism through oxidative phosphorylation, which thus strongly implicates the involvement of mitochondrial dysfunction early in the course of AD. Along this line, glucose hypometabolism in frontal, temporal, and parietal cortices of patients with AD were closely correlated with the reduced levels of blood thiamine diphosphate (TDP), a critical coenzyme of pyruvate dehydrogenase (PDHC) and α-ketoglutarate dehydrogenase (KGDHC) in the Krebs cycle and transketolase in the pentose phosphate pathway [28]. Similar to glucose hypometabolism, TDP reduction and reduced activities of thiamine-dependent enzymes are also a significant and common feature in patients with AD [29, 30]. Oxygen metabolism measured by positron-emission tomography (PET) detection of Oxygen-15 is another primary measure for brain energy metabolism, which provides direct evidence for mitochondrial function through electron transport chain (ETC) in the brain. Cerebral metabolic rate of oxygen was significantly decreased in the frontal, parietal and temporal cortex in AD, which showed significant correlation with severity of dementia [31–33]. Another study found significant correlation between reduced oxygen metabolism and electroencephalogram slowing in the parieto-temporal regions of AD brain [34]. In fact, multiple lines of evidence demonstrated impaired bioenergetics machinery, especially the tricarboxylic acid (TCA) cycle and the ETC chain, in the mitochondria in AD. Collectively, these studies strongly suggest that mitochondrial dysfunction likely plays a critical role in glucose hypometabolism and energy impairment in AD.
Mitochondrial deficits in AD
Disrupted mitochondrial bioenergetics in AD
Consistent with impaired energy metabolism in AD, gene expression studies repeatedly identified defects in mitochondrial related metabolic pathways in AD, which provided direct evidence for impaired bioenergetic machinery in mitochondria of AD. For example, a genome-wide transcriptome study in laser-capture micro-dissected neurons found significantly greater proportion of underexpressed nuclear genes encoding mitochondrial ETC subunits in the posterior cingulate cortex than those in the primary visual cortex, a region that is relatively spared metabolically in AD vs. control [35]. A microarray analysis and quantitative RT-PCR studies found 15 out of 51 members of the glycolytic, TCA cycle, oxidative phosphorylation, and associated pathways were significantly downregulated in AD [36]. More recent microarray data confirmed significant downregulation in nuclear-encoded but not mitochondria-encoded OXPHOS genes in the hippocampus of AD patients, which however was puzzlingly increased in the hippocampus from MCI patients [37]. Complex I of OXPHOS was downregulated while complexes III and IV showed increased mRNA expressions in both early and definite AD brain specimens [38]. A bioinformatics analysis of four transcriptome datasets for the hippocampus of AD patients identified OXPHOS pathway as one of most significant pathways involved in AD [39]. Gene set enrichment analysis demonstrated that mitochondrial oxidative phosphorylation (OXPHOS) downregulation and mitochondrial import pathways disruption were hallmarks of AD [40].
Proteomic and protein expression studies also confirmed underexpressed proteins in OXPHOS pathway as one of the most affected processes in the cortex of AD patients [41]. Quantitative proteomics approaches revealed differentially altered mitochondriomes in AD brain are different from aging-associated changes suggesting that dysregulated mitochondrial complexes (i.e., ETC complexes and ATP-synthase) are the potential driver for pathology of the AD [42]. Significantly decreased immunocytochemical staining of various complexes including Complex I and IV in various brain regions in AD was also reported [43, 44]. Given that the activity of some enzymes is regulated by posttranslational modification, it would be of importance to determine the alterations in the enzymatic activities of these enzymes in AD brain. The activity of enzymes involved in TCA cycle changed in AD following a distinct pattern: the dehydrogenases/decarboxylases (including PDHC, ICDH and KGDHC) were reduced while dehydrogenases (including SDH and MDH) were increased and all of these changes correlated with the clinical state [45]. Biochemical studies of enzyme activities in mitochondria isolated from autopsied AD brain demonstrated a generalized depression of activities of all ETC complexes with most dramatic reduction in COX activity [46]. Some studies reported more specific defect in the COX activity [47–49]. For example, careful histochemical quantification of cytochrome c oxidase activity in the metabolically affected posterior cingulate cortex and less affected primary motor cortex between AD and age-matched control revealed significantly lower COX activity in the former but not in the latter [43]. More recent studies demonstrated that mitochondrial ATP synthase activity is impaired in the brain of AD patients due to loss of oligomycin sensitive conferring protein subunit [50] and/or changes in the O-GlcNAcylation of ATP synthase subunit α [51].
However, contradictory results were reported where several groups failed to find difference or even increased expression of OXPHOS genes in AD compared to control, some of which may be attributed to different brain regions and/or the widespread heterogeneity of sporadic AD brain samples used. For example, decreased expression of complexes I, II, IV and V was found in the entorhinal cortex but not in the frontal cortex in AD Braak stages V-VI compared with stages I-II [52]. Cell-type specific effects may also contribute to the variance as evidenced by a recent study demonstrating significantly reduced nuclear-encoded OXPHOS genes in laser-captured hippocampal pyramidal neurons from AD compared to that of control cases despite increased expression of these genes in the whole homogenates from these same cases [53].
Overall, these gene and protein expression studies along with biochemical studies clearly demonstrated extensive mitochondria bioenergetics defects in AD brains, which makes it a valuable therapeutic target. It remains to pinpoint specific alterations in oxidative metabolism and to generalize the role of mitochondrial dysfunction in regional vulnerability and pathogenesis of AD, which requires consistent measurement of these parameters across different brain regions. Furthermore, it has been demonstrated that reductions of activity of complex I, III and IV that reach certain thresholds such as greater than 70% in some studies were necessary to produce a significant decrease in ATP production [54, 55]. It thus remains unresolved whether and how chronic but mild impairment in the ETC complexes (i.e., around 15–50% reduction in complex I or IV [8]) is sufficient to cause bioenergetics impairment seen in AD brain. Nevertheless, it should be noted that such threshold appears widely varied (for example, a threshold of 25% [56], 35% [55] or 70% reduction [54] for complex I were suggested by different studies) which could be tissue specific and influenced by other factors such as antioxidant status. For example, glutathione depletion eliminates the complex I threshold in PC12 cells [56]. A combination of mild impairments in different individual complexes may also effectively lower the threshold to produce bioenergetic dysfunction.
Increased oxidative stress in AD
Reactive oxygen species (ROS) are unavoidable byproducts during electron transport of aerobic respiration in the mitochondria due to electron leaks at complex I and complex III and it is estimated that mitochondria contribute approximately 90% of the cellular ROS [57]. While ROS serve important signaling roles, when in excess, they lead to oxidative stress with extensive damage. Mitochondria are susceptible to oxidative damage despite the presence of an antioxidant system and damaged mitochondria are less efficient producers of ATP and more efficient producers of ROS. Therefore, increased oxidative stress could be both the cause and consequence of mitochondrial dysfunction.
A large body of evidence demonstrated increased oxidative damage to almost all types of macromolecules in the brain of AD patients including proteins, sugar, lipid and nucleic acids [58]. For example, significant increase in protein carbonyls and 3-nitrotyrosine modification as protein oxidation markers and elevated glycation and glycooxidation marking oxidative modifications to sugars were widely reported in the brains of patients with AD and MCI [9]. Lipid peroxidation products such as reactive aldehydes including 4-hydroxynonal, malondialdehyde (MDA), and acrolein were increased in multiple brain regions affected in AD and MCI [59]. AD brains demonstrated significant increase of 8-hydroxydeoxyguanosine (8-OHdG) and 8- hydroxyguanosine (8-OHG) respectively in DNA (including mtDNA, which will be discussed in more detail in the next session) and RNA [60]. On the other hand, significantly decreased antioxidant levels and/or changes in the expression and activities of antioxidant enzymes were also reported in AD brain. Recent in vivo imaging studies confirmed AD-dependent reduction of glutathione levels in affected brain regions in AD and MCI patients [61, 62], which strongly correlated with declines in cognitive function. The increase in oxidative stress markers strongly associated with significant loss of synaptic proteins in the brains of MCI and pre-AD patients too [63]. Detailed analysis of stable oxidation modifications such as lipid peroxidation and protein glycation revealed widely distributed cumulative oxidative damage in neurons both with and without AD-associated pathology; interestingly, short-lived oxidative damage such as oxidized DNA/RNA and 3-nitrosylation was prominent in neurons without pathology but reduced in cells with pathology [9]. These studies suggest that oxidative stress occurs earlier than the formation of AD-related pathology and AD-associated pathology could play a protective role in fighting against ROS production/damage.
Redox proteomics studies to identify oxidatively modified proteins contributed a great deal to the understanding of the diseased proteome in the various stages of AD and shed light on potential molecular pathways involved [58]. These studies found that many antioxidant enzymes were oxidized which likely compromised their functions and contributed to increased oxidative stress in AD. For example, glutathione-S-transferase Mu, peroxiredoxin 6, multidrug-resistant protein 1 or 3, and GSH were all found to be HNE-modified and/or nitrated in various brain regions of MCI and AD patients [64]. This unbiased approach also revealed that many proteins involved in the energy metabolic processes were posttranslationally modified either by lipid conjugation or by reaction with ROS in the brains from patients with AD or MCI. For example, ATP synthase, the enzyme responsible for the final step of ATP production, was found to be HNE-modified and nitrated in AD and MCI hippocampus [64]. HNE-modification or carbonylation was found in aconitase in the TCA cycle and creatine kinase in ATP maintenance [64]. These data suggest that increased oxidative stress contributed to mitochondrial dysfunction and impaired energy metabolism in AD.
Disturbed mitochondrial genomic homeostasis in AD
Mitochondria maintain their own DNA called mtDNA, which is a multicopy (1–10 copies per mitochondrion), extrachromosomal genome that codes for 13 mitochondrial core proteins of the ETC complexes and 2 rRNA and 22 tRNAs necessary for mitochondrial protein synthesis [65, 66]. While mtDNA is critical to the proper function of mitochondria, it is prone to oxidative damage due to its proximity to the site of ROS generation and relative lack of DNA-protective histones and efficient DNA repair mechanisms, which gives rise to mutations [67, 68]. Mutations in mtDNA, whether through inheritance or gradual somatic accumulation, propagate through clonal expansion, which may eventually gain momentous deleterious effects after exceeding critical threshold, and compromise mitochondrial function and result in cell death and disease [8].
Many patients with primary mtDNA mutations demonstrated pronounced cognitive deficits quite similar to those commonly seen in AD [69], which supports a critical role of mtDNA in proper cognitive function. Interestingly, it is reported that in families with a history of dementia, a consistently identified risk factor for AD, maternal transmission is significantly more frequent than paternal transmission. Along this line, maternal family history of AD is associated with increased atrophy in AD-vulnerable brain regions [70], a pattern of progressive reduction of brain glucose metabolism [71, 72] and higher white matter hyperintensity load in temporal and occipital lobes [73] in cognitively normal individuals, pointing towards family of origin effects. Given the maternal inheritance of mtDNA, this implicated a potential role of inherited mtDNA variability in AD. Indeed, while no primary mtDNA mutations were associated with AD, multiple studies have found that mtDNA SNPs and germline variants (i.e., haplogroups) likely play a role in AD (Table 1): for example, haplogroup UK is associated with higher risk of AD while haplogroup T is protective [74, 75]. Interaction between mtDNA inherited variability and other factors such as gender or apoE alleles may change the susceptibility to AD: some mtDNA haplogroups (K and U) seem to neutralize the harmful effect of the apoE4 allele [76]; haplogroup U is associated with higher risk for men but reduced risk for women [77]. However, an association between mtDNA inherited variability and the development of AD remains inconclusive at this time since a large-scale mtDNA haplogroup study from three Caucasian populations failed to replicate these findings [78]. Interestingly, this study noticed some evidence for association between individual mtDNA SNPs previously implicated in AD within subsets of samples, dependent on geographic locations, suggesting that geographic difference in the fine details of the sub-haplogroup structure of mtDNA could contribute to the inconsistency between studies.
Table 1.
mtDNA changes in AD
| Mutation type | Affected mtDNA region | Analysis Method | Changes in AD | Reference |
|---|---|---|---|---|
| mtDNA Δ4977 | I, III and V | PCR | Increased | Corral-Debrinski et al. [80] |
| mtDNA Δ4977 | I, III and V | PCR | No changes | Blanchard et al. [79] |
| mtDNA Δ4977 | I, III and V | In situ hybridization | Increased | Hirai et al. [87] |
| mtDNA Δ4977 | I, III and V | PCR | No changes | Bender et al. [90] |
| mtDNA Δ4977 | I, III and V | PCR | Increased | Krishnan et al. [88] |
| mtDNA Δ4977 | I, III and V | Realtime PCR | No changes | Strobel et al. [89] |
| DNA Rearrangement | Mitochondrial genome | Next generation sequencing | Increased | Chen et al. [81] |
| Point mutation | D-loop region | PCR/Sanger sequencing | Increased | Coskun et al. [82] |
| Point mutation | Mitochondrial genome | Random mutation capture | No changes | Soltys et al. [83] |
| Point mutation | Mitochondrial genome | PCR-cloning-sequencing | Increased | Lin et al. [84] |
| Point mutation | Mitochondrial genome | Next generation sequencing | Increased | Hoekstra et al. [91] |
| DNA methylation | D-loop region | TaqMan PCR | Increased | Blanch et al. [96] |
| DNA methylation | D-loop region | Realtime PCR | Decreased | Stoccoro et al. [97] |
Involvement of somatic mtDNA mutations were also extensively studied in AD with a focus on the common 5-kb deletion (mtDNA Δ4977) occurring between positions 8470–8482 and 13,447–13,459, which presumably affects the expression of ETC complex I, III and V in AD. Earlier quantitative PCR studies found age-related accumulation of this deletion in frontal cortex [79] and a striking 15 fold increase of this deletion in AD patients younger than 75 years of age [80]. A comprehensive assessment of mtDNA rearrangement events found significantly higher levels F-type and R-type rearrangements, in addition to deletion, in AD brain [81]. AD brains had an average 63% increase in heteroplasmic mtDNA point mutations in the control-region (CR) and certain AD brains harbored the disease-specific CR mutations at levels up to 70–80% heteroplasmy, which preferentially altered regulatory elements of known mtDNA and suppressed transcription and replication of mtDNA [82].
However, conflicting results were reported since some groups found no changes in the aggregate burden of brain mtDNA point mutations between AD and control [83–85], which was probably owing to the small sample size and approach difference [86]. Lack of distinction of cell-specific mtDNA may also contribute to the variability since more sensitive studies by in situ hybridization [87] and laser capture microdissection in single hippocampus neurons or glial cells followed by a multiplex real-time qPCR method [88, 89] demonstrated increased neuronal but not glial occurrence of mtDNA Δ4977 in AD. More detailed study revealed markedly increased ratio of mtDNA Δ4977 over normal mtDNA in COX negative neurons that were selectively enriched in AD [90]. Different disease stage may also contribute to the variability. A recent study on enriched neuronal mtDNA using more accurate next generation sequencing methodology, which eliminates sequencing errors associated with PCR and DNA damage, revealed significantly elevated frequency of mtDNA point mutation in the hippocampus of patients with early stage AD (i.e., individuals who were not demented, but had high Braak staging characteristic of AD dementia), but not in patients with pathologically confirmed AD dementia [91]. This study not only suggested an early role of mtDNA mutations in AD, but also demonstrated that mutated mtDNA may be lost as neurons die when disease progresses. It also needs to be noted that ancient accumulated polymorphisms and somatic mutations are not mutually exclusive but their combined effects are not studied.
It was believed that increased mtDNA mutations are due to increased oxidative damage found in AD brain [8]. Indeed, mtDNA had approximately 10-fold higher levels of oxidized bases than nuclear DNA and mtDNA underwent an average of threefold increase in oxidative damage in the brain from AD patients compared to age-matched controls [92, 93]. In fact, levels of oxidized nucleic acids in mtDNA were found to be significantly elevated in preclinical Alzheimer’s disease (PCAD) and MCI patients [94], suggesting that this is an early event during the course of disease. More recent studies demonstrated decreased OGG1 activity [95] and impaired base-excision repair (BER) activity in both AD and MCI patients [83], suggesting significant contribution of replication error to increased mtDNA mutations in AD [91]. Other modifications to mtDNA may also impact its transcription and function. For example, increased 5-methylcytosine levels are found in the D-loop region of mtDNA in brain samples with AD-related pathology [96]. On the contrary, there is a decreased methylation of the D-loop region in peripheral blood mtDNA from LOAD patients [97]. The implication of these findings to human AD pathogenesis remains to be explored.
Overall, these studies suggest a likely critical role of mtDNA variabilities, mutations and modification in the pathogenesis of AD. Indeed, it has been proposed by the mitochondrial cascade hypothesis that inherited mtDNA variants determine one’s vulnerability and the accumulation of brain somatic mtDNA modifications and mutations reflecting the influence of the environment along aging determines the manifestation of the phenotype [8]. However, specific mtDNA alterations, if any, and a potential causal role of mtDNA alterations in AD pathogenesis are yet to be proven. Additionally, mtDNA alterations are found in other neurodegenerative diseases but not specific to AD, and how they specifically relate to AD-type changes await further exploration.
Mechanisms underlying mitochondrial dysfunction in AD
The existence of hypometabolism-first biomarker profile pathway to preclinical AD along with the extensive evidence that mitochondrial abnormalities could lead to AD-related deficits in model organisms suggest that mitochondrial dysfunction could play a primary role at least in a subset of sporadic AD patients (Fig. 1). As to the autosome dominant AD mutations carriers where amyloid-first biomarker profile pathway to AD plays the primary role, impaired energy metabolism is an invariant feature preceding clinical onset of the disease suggesting that mitochondrial dysfunction likely plays an upstream role, although secondary to other fundamental AD events, in mediating and amplifying neuronal dysfunction and neurodegeneration in AD (Fig. 1). Regardless whether mitochondrial dysfunction plays a primary or secondary role, impaired mitochondrial bioenergetics, increased oxidative stress and disturbed mitochondrial genome are consistent features of mitochondrial abnormalities in AD, all which interact with each other to form a deleterious downward spiral. While the relative importance of these abnormalities in triggering mitochondrial dysfunction may vary among patients with AD depending on the unique biological, environmental and genetic factors of each individual, any of these abnormalities could induce the other two to complete the downward spiral to mediate and amplify neuronal dysfunction and neurodegeneration (Fig. 1). Considering the essential role mitochondrial dysfunction plays in the pathogenesis of AD, the mechanisms underlying mitochondrial impairments and related neuronal loss in AD have been extensively studied, which generated novel insights that may offer new targets for future therapeutic development.
Fig. 1.

Critical role of mitochondrial dysfunction in AD. Mitochondrial dysfunction plays a critical role in AD either as a primary or secondary event. In either case, impaired mitochondrial bioenergetics, increased oxidative stress and disturbed mitochondrial genome are consistent features of mitochondrial abnormalities in AD, all which interact with each other to form a deleterious downward spiral. While the relative importance of these abnormalities in triggering mitochondrial dysfunction may vary among patients with AD depending on the unique biological, environmental and genetic factors of each individual, any of these abnormalities could induce the other two to complete the downward spiral to mediate and amplify neuronal dysfunction and neurodegeneration. Recent studies revealed mechanisms underlying the loss of integrity of mitochondria, which provides mechanistic link among these abnormalities and offers multiple novel intervention sites to improve mitochondrial function in AD
Abnormal mitochondrial fusion and fission in AD
Mitochondria are highly dynamic organelles undergoing continuous fusion and fission in the cytoplasm, a process that is essential for maintaining a healthy pool of mitochondria with proper distribution [98]. The molecular mechanisms of the fusion and fission are still under intensive exploration but accumulating evidence suggest that a group of large GTPase domain-containing proteins play critical roles, which either enhance mitochondrial fission such as DLP1(also referred to as Drp1), or promote mitochondrial fusion such as Mfn1, Mfn2 and OPA1 [99] (Fig. 2). Deficits in either fission or fusion cause human neurological disorders, which underscores the importance of balance of mitochondrial fission and fusion in neuronal function and brain health [99].
Fig. 2.
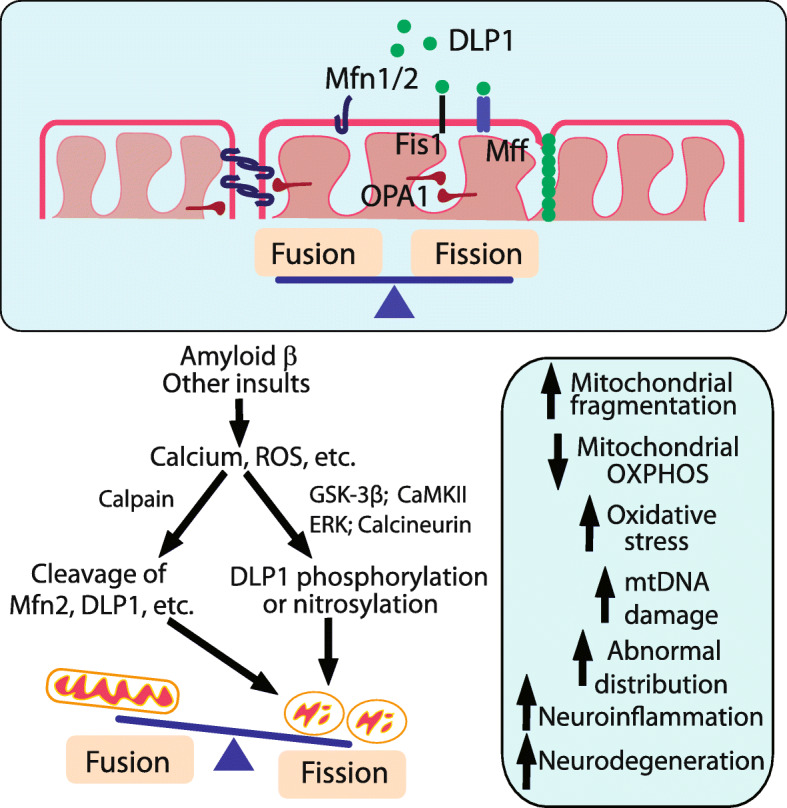
Abnormal mitochondrial fusion and fission in AD. (Top) A family of large GTPases regulate balanced mitochondrial fusion (e.g. Mfn1/2, OPA1) and fission (DLP1). For mitochondrial fission, mitochondrial outer membrane proteins (e.g. Fis1, Mff and others) recruit cytosolic DLP1 protein to mitochondria that oligomerize and form a ring structure around fission site. (Bottom) Amyloid-β and other AD related insults cause neuronal calcium influx and increased ROS production that activate downstream proteases (calpain) and protein kinase that act on mitochondrial fission/fusion GTPases and disturb mitochondrial fusion and fission in AD
Early studies demonstrated ultrastructural damage to the susceptible pyramidal neurons in the biopsied brain tissues of AD [87]. More detailed analysis revealed altered size and number and reduced aspect ratio of mitochondria in these neurons, suggestive of a potential fragmented mitochondrial network in AD brain [100, 101]. Fragmented mitochondria could cause mitochondrial bioenergetics deficits either through negative impact to the proper complex assembly critical for ETC function [102, 103] or enhance ROS generation [104] (Fig. 2). Moreover, it also caused reduced exchange of mitochondrial contents exacerbating mtDNA deficits [99], all prominent features found in AD brain (Fig. 2).
Indeed, biochemical evaluation of AD brains demonstrated significantly reduced protein expression levels of all the large dynamin-related GTPases involved in fission and fusion including DLP1, OPA1, Mfn1, and Mfn2 along with significantly increased levels of fission factor, Fis1, in AD brain [101, 105]. Given that DLP1 and Mfn2 are substrates of calpain and cleaved by calpain activation induced by multiple AD-relevant insults in vitro [106], the fact that reduced levels of these GTPases correlated with calpain activation in AD brain suggests that calpain-mediated degradation could at least contribute to the reduction of these fission/fusion GTPases in AD [106]. Despite some controversial reports on the expression of DLP1 in the AD brain [105], studies from multiple groups demonstrated significant changes in the post-translational modifications of DLP1 consistent with its increased translocation to mitochondria in AD: Cho et al. reported that Aβ induced S-nitrosylation of DLP1 (forming SNO-Drp1) triggered mitochondrial fission, synaptic loss, and neuronal damage in AD [107]. Wang et al. found significantly increased phosphorylation at the Ser616 sites along with increased S-nitrosylation of DLP1 associated with increased mitochondrial DLP1 in AD brain [101]. Interestingly, Manczack et al. reported interactions between oligomeric Aβ and DLP1 as well as hyperphosphorylated tau and DLP1 in AD brain [105, 108]. While it was suggested that these abnormal interactions likely facilitate mitochondrial fragmentation, it remains to be solved where these interactions occur and how they enhance fission activity of DLP1.
Impaired mitochondrial fusion and fission balance having an essential role in mitochondrial dysfunction and pathogenesis of AD was corroborated by studies in both in vitro and in vivo experimental models of AD [9, 87, 100, 101, 109]. Overexpression of wild type or mutant APP caused mitochondrial fragmentation in M17 neuroblastoma cells and primary neurons, which was blocked by beta-APP cleaving enzyme (BACE1) inhibitor, suggesting that Aβ induced this effect [100, 101]. Indeed, exposure to soluble Aβ oligomers caused time- and dose-dependent changes in the expression of mitochondrial fission and fusion proteins along with significant mitochondrial fragmentation and dysfunction [101, 110]. Importantly, inhibition of mitochondrial fission rescued APP- or Aβ-induced mitochondrial deficits and neuronal deficits [100, 101], which established a critical role of mitochondrial dynamics in these models. It has been suggested that calcium signaling-dependent and/or oxidative stress-induced posttranslational modifications to DLP1 mediate Aβ-induced mitochondrial fragmentation: Aβ-induced calcium influx led to increased DLP1 phosphorylation at Ser616 sites through CaMKII-dependent Akt activation that stimulate DLP1 translocation to mitochondria and fission activity [111]. Increased oxidative stress induced by Aβ activates ERK, which in turn leads to DLP1 phosphorylation and mediates downstream toxic effects on mitochondria and neurons [112]. An artificial polypeptide, TAT-Drp1-SpS, could specifically block GSK3β-induced Drp1 phosphorylation and rescue Aβ toxicity related mitochondrial dysfunction in vivo and in vitro [113]. Aβ induced increased S-nitrosylation of DLP1 at Cys644 through enhanced nitric oxide production also enhanced its dimerization and fission activity [107] although this notion was challenged by a later study [114].
Aβ induced abnormal mitochondrial dynamics is an early event during neurodegeneration in vivo in Drosophila models [115, 116]. A 3D electron microscopy study demonstrated a peculiar “beads-on-the-string” morphology of mitochondria in the pyramidal neurons in two different APP transgenic mouse models [117], which likely represents an excessive fission process that is stalled at the last step. Indeed, an in vivo multiphoton imaging study confirmed fragmented mitochondria near amyloid plaques in vivo in an APP transgenic model [118]. Wang et al. further reported mitochondrial fragmentation and ultrastructural damage in the brain of CRND8 APP transgenic mice by both confocal microscopy and electron microscopy studies, accompanying mitochondrial functional deficits at 3 months of age, well before any noticeable amyloid deposition, which suggest that mitochondrial dynamic abnormalities occur early in the course of AD-related changes [109]. Collectively, these studies suggest a tipped mitochondrial dynamics balance towards excessive fission in vivo in various AD mouse models, but falls short in demonstrating a causal role of mitochondrial dynamic abnormalities in neurodegeneration in vivo. Notably, recent studies reported that mitochondrial fragmentation in the cortex and hippocampus caused by unopposed fission due to Mfn2 knockout led to mitochondrial ultrastructural damage and functional deficits along with extensive oxidative stress followed by dramatic neuroinflammation that eventually caused significant neuronal loss [119, 120]. This study demonstrated that mitochondrial fragmentation could initiate a degenerating cascade culminating neurodegeneration that replicates many of the pathological features during the course of AD, thus establishing a causal role of mitochondrial dynamic abnormalities in vivo.
These studies pave the road to pursue abnormal mitochondrial fission as a potential therapeutic target for AD, and so far specific attention focused on the extensively studied mitochondrial dynamic protein DLP1 which adds more evidence of a critical role of mitochondrial dynamics in the course of AD. Chemical inhibition of mitochondrial fission by DLP1 specific inhibitor Mdivi-1 and genetic reduction of DLP1 proteins were explored in AD models in vivo and in vitro. By inhibition of ERK-DLP1 signaling, Gan et al. reported a protective effect of DLP1 inhibitor mdivi-1 on maintenance of normal mitochondrial structure and function in AD cybrid cell [121]. Consistently, two groups demonstrated the rescue effects of DLP1 inhibitor midivi-1 on mitochondria morphology and movements at early stage in APP transgenic mice which alleviated amyloid pathology likely through reducing Aβ production and improved cognitive deficits [109, 122]. The DLP1 inhibitor midivi-1 presumably prevents DLP1 assembly and inhibits DLP1-dependent mitochondrial fission [123]. However, one recent study challenged the specificity of mdivi-1 on mitochondrial fission inhibition and suggested that mdivi-1 might perform as a reversible mitochondrial complex I inhibitor rather than as a specific DLP1 inhibitor [124]. It is important to note that the reported non-DLP1 dependent effects of mdivi-1 were only noticeable when cells received relative high doses of the inhibitor in vitro [124]. Previous in vitro studies did not apply such high concentration of mdivi-1 in neuronal cultures and conclusions should remain valid. However, due to the difficulty in manipulating chemical concentration in in vivo system such as mouse brain, the underlying mechanism of the protective effects of midivi-1 in AD models in vivo thus need to be interpreted carefully [125]. Therefore, more specific methods to inhibit fission or enhance fusion are needed in AD research. In this regard, using real-time PCR and western blot, Manczak et al. explored the protective effect of reduced DLP1 expression in APP mice and found that DLP1 haplodifficiency leads to the restoration of the expression of proteins related to the mitochondrial dynamics, mitochondrial biogenesis and synapses and rescued mitochondrial function in APP transgenic mice as compared to APP transgenic mice alone (Tg2576 line) [126]. Unfortunately, it had not been determined whether DLP1 haploinsufficiency has any beneficial effects on cognitive function and pathological changes in this study. Given that DLP1 knockdown depletes mitochondria from neuronal process and causes synaptic deficits [127, 128], it may have unwanted effects. Perhaps a better approach to correct mitochondrial dynamics abnormalities in AD models is to enhance mitochondrial fusion in vivo instead [129].
Mitochondrial axonal trafficking deficits and abnormal mitochondrial distribution in AD
In addition to mitochondrial morphological abnormality, mitochondrial distribution was also disturbed in AD brain: mitochondria become less abundant in the neuronal processes in the susceptible pyramidal neurons in AD [101]. This uneven mitochondria distribution in the processes leaves large axonal or dendritic segments devoid of mitochondria, as clearly demonstrated by a recent electron microscopy study [130]. Kinesin-based anterograde transport of mitochondria populates axons with fresh mitochondria, and dynein-based retrograde transport of mitochondria facilitates the recycling of damaged mitochondria and maintains a healthy mitochondrial population in the processes [131, 132]. Therefore, disruption of either anterograde or retrograde transport or both of these processes, either due to faulty mitochondria or impaired mitochondrial transport system, leads to decreased proportion of healthy mitochondria or increased proportion of damaged mitochondria that impaired the integrity and function of mitochondria. It also could significantly affect mitochondrial distribution, which have profound impacts on synaptic and neuronal function [133]. Abnormal changes in mitochondrial transport in AD are under intensive studies.
Mutations in presenilin 1 impair kinesin-based axonal transport through GSK3β activation, which phosphorylates kinesin light chain and releases kinesin from the cargo at sites of membrane insertion [134]. Primary neurons isolated from APP transgenic mice also demonstrated impaired axonal transport of mitochondria [135], which is likely caused by Aβ. Indeed, overexpression of Aβ42 caused mitochondria mislocalization with reduction in axons and dendrites and accumulation in the soma, which contribute to Aβ42-induced neuronal dysfunction in a transgenic Drosophila model in vivo, and this is exacerbated by genetic reductions in mitochondrial transport [115]. Similarly, exposure of neuronal cultures to Aβ oligomers reduces motile mitochondria in axons using live imaging [136–138]. A recent study suggested that amyloid peptides with higher propensity to aggregate also inhibit mitochondrial trafficking [135].
How may Aβ affect the axonal transport of mitochondria? The finding that Cyclophilin D deficiency rescues Aβ-induced axonal mitochondrial transport deficit [139] implicated the potential involvement of calcium and its downstream signaling. Calcium elevation could modulate mitochondrial transport by directly impacting the adaptor proteins involved in mitochondrial transport such as calcium-sensing protein Miro1 or by influencing downstream calcium signaling molecules such as calcineurin and GSK3β [140]. The rescuing effect of mitochondria-targeted antioxidant peptide SS31 on Aβ-induced impaired anterograde axonal transport of mitochondria suggests that oxidative stress could be involved [135]. Motor proteins in both anterograde and retrograde transport can be impacted: Aβ caused reduced expression of anterograde motor proteins KIF5A and restoration of KIF5A corrects Aβ-induced impaired anterograde transport of mitochondria [141]. On the retrograde transport side, oligomeric Aβ interacts with dynein intermediate chain and disrupts the coupling of dynein-Snapin which could potentially impact mitochondrial transport [142]. Microtubule tracks could also be impacted, as Kim et al. suggested that the HDAC6-dependent regulation of α-tubulin acetylation status was essential for Aβ-induced impairment of mitochondrial transport in hippocampus neuronal cultures [143]. Their follow-up study further identified peroxiredoxin1 as another substrate of HDAC6, which is involved in Aβ-induced disruption of ROS, calcium homeostasis and axonal transport in 5xFAD AD model mice and AD patients [144]. Aβ may also impact mitochondrial transport through changes in mitochondrial dynamics through reduction of DLP1 or Mfn2 since reduced DLP1 or Mfn2 cause reduced mitochondrial distribution in the processes [101] and Mfn2 interacts with Miro/Milton complex and is required for axonal transport of mitochondria [145].
Overexpression and/or phosphorylation of tau is another negative regulator for mitochondrial movement in neurons. Earlier studies demonstrated that tau controls the balance of axonal transport through locally differential modulation of dynein and kinesin motor proteins [146], predicting tau accumulation in the somatodendritic compartments compromise axonal anterograde transport. Indeed, tau overexpression preferentially impairs kinesin-dependent anterograde axonal transport of mitochondria and other vesicles through enhanced microtubule binding [147]. Perhaps more relevant to conditions in AD, Shahpasand et al. found that tau phosphorylated at the AT8 sites inhibited mitochondrial movement in the neurite processes of PC12 cells as well as the axons in mouse cortical neurons due to impaired microtubule spacing [148]. Consistent with a critical role in tau phosphorylation in the regulation of mitochondrial transport, neurons from a tau P301L mutant knock-in mouse model had reduced levels of phosphorylated tau but displayed increased anterograde mitochondrial transport in axons [149]. As a result, mitochondrial distribution is progressively disrupted with age in rTg4510 brain and in Alz50-positive neurons in AD brain [150] which probably contributes to significant mitochondrial loss in the tau positive neurons in AD [151]. Interestingly, the effects of Aβ species on mitochondrial movement was subject to the presence of tau proteins. Quintanilla et al. demonstrated that Aβ treatment combined with expression of truncated tau significantly increases the stationary mitochondrial population and the levels of oxidative stress in cortical neurons [152]. Consistently, tau reduction prevented Aβ-induced deficits in the anterograde axonal transport of mitochondria in primary neurons by blocking the activation of GSK3β [153].
Despite the consensual view that Aβ and tau alterations impaired mitochondrial transport, it is unclear whether they specifically impaired mitochondrial transport or also affected other organelles. There is also debate on whether Aβ preferentially impacted anterograde axonal transport of mitochondria or retrograde axonal transport of mitochondria or both [136, 137, 141, 142, 154]. The accumulation of damaged mitochondria at synapses could be the consequence of an impaired retrograde axonal transport of mitochondria [137]. Furthermore, changes in other aspects such as mitochondrial docking that may impact mitochondrial distribution have not been studied [155].
Impaired mitochondrial biogenesis in AD
There are more than 1000 proteins in neuronal mitochondria, 13 of which are encoded by mitochondrial genome and are hydrophobic proteins that form the core parts of the oxidative phosphorylation complexes of the inner membrane of mitochondria, while the remainder are encoded by nuclear genome [156]. Therefore, mitochondria biogenesis involves coordinated expression between both nuclear and mitochondrial genomes. PGC-1α is considered the master regulator of mitochondrial biogenesis and coordinates/regulates energy metabolism and respiration through interactions with different transcription factors, including nuclear respiratory factor 1 (NRF 1) and nuclear respiratory factor 2 (NRF 2) [157]. NRF-1/2 controls the expression of many nuclear-encoded mitochondrial proteins including mitochondrial transcription factor A (TFAM) which drives the transcription and replication of mtDNA [157]. A complex and multifaceted ROS defense system is linked by PGC-1α to mitochondrial oxidative metabolism, enabling cells to maintain normal redox status in response to changing oxidative capacity [158]. Obviously, mitochondrial biogenesis plays a critical role in maintaining mitochondrial homeostasis during the life cycle of mitochondria.
As discussed earlier, multiple studies demonstrated reduced levels of critical components of the electron transport chain in the brain tissues of AD, which not only underlies the well-documented energy hypometabolism in AD, but may also suggest impaired mitochondrial biogenesis or enhanced mitochondrial clearance. However, mitochondrial clearance through mitophagy is actually impaired in AD (discussed in more detail later). Therefore, an impaired mitochondrial biogenesis is implicated. PGC-1α is abundantly expressed in tissues with high energy demand including the brain. Qin et al. first demonstrated the reduced expression of PGC-1α in AD patients and transgenic mouse model of AD [159]. mtDNA copy numbers were significantly reduced and mitochondrial biogenesis transcriptome signaling is disrupted in laser-capture microdissected pyramidal neurons from AD hippocampus compared to that of control hippocampus [160]. Decreased PGC-1α levels are also associated with abnormal brain insulin signaling, providing one possible mechanism for obesity being a risk factor for AD [161]. Sheng et al. demonstrated that expression of APP Swedish mutant caused reduced expression of PGC-1α and impaired mitochondrial biogenesis likely through a PKA-dependent pathway and restored PGC-1α expression rescued mitochondrial and neuronal functions in cell models of AD [162]. Interestingly, Presenilin 1 is also involved in the regulation of PGC-1α expression through the production of APP intracellular domain (AICD) peptide, the APP processing product after γ-secretase cleavage, and PS1-FAD mutations lost the ability to enhance PGC-1α mRNA levels due to the impaired cleavage of APP proteins in AD [163]. Reciprocally, exogenous expression of PGC-1α in N2a neuroblastoma cells could regulate APP processing by downregulating the transcription of BACE1, which effected decreased secreted Aβ and increased non-amyloidogenic soluble APPα [164]. Another study also suggested that PGC-1α reciprocally regulated BACE1 in vitro and in vivo, in collaboration with SIRT1-mediated deacetylation of PPARγ constituting essential mechanisms for regulation Aβ production in AD [165].
Considering the essential role of PGC-1α impairment on mitochondrial dysfunction and Aβ production in AD, it was attractive to investigate how restoration of PGC-1α expression or its activity would affect mitochondrial and neuronal functions in models of AD. Katsouri et al. studied the potential therapeutic effect of PGC-1α by generating a lentiviral vector to express human PGC-1α in hippocampus and cortex of APP23 transgenic mice [166] and found this abrogated neuronal loss and Aβ aggregation likely through inhibition of BACE-1. In contrast, Dumont et al. crossed the Tg19959 mouse model of AD with transgenic mice overexpressing human PGC-1α protein [167], which unexpectedly exacerbated amyloid and tau accumulation accompanied by an impairment of proteasome activity. The discrepancy of these transgenic animal studies underscores the need to manipulate the expression levels of exogenous PGC-1α proteins in vivo with care because abnormal PGC-1α levels induced toxic effects in some peripheral organs such as in the heart [168].
Another strategy to restore mitochondrial biogenesis was to enhance PGC-1α activity by chemical stimulation [169]. The first evidence came from a study by Dumont et al. in which administration of the PGC-1α agonist bezafibrate exerted neuroprotective effects in a mouse model of tauopathy, as shown by decreased tau pathology and behavioral improvement, which suggested beneficial effect by increased activity in a non-APP model of AD [170]. In an Aβ toxicity mouse model, Gong et al. demonstrated that nicotinamide adenine dinucleotide (NAD+) promoted PGC-1α expression coinciding with enhanced degradation of BACE1 and the reduction of Aβ production in Tg2576 mice in the brain [171]. Furthermore, supplementation of melatonin in drinking water, which enhanced PGC-1α activity in vivo, increased mitochondrial biogenesis and alleviated mitochondrial impairment which led to improved spatial learning and memory deficits, and reduced Aβ deposition and soluble Aβ levels [172].
An alternative strategy is to focus on mitochondrial biogenesis effectors downstream of PGC-1α. Oka et al. examined the effects of human mitochondrial transcriptional factor A (hTFAM) on the pathology of a mouse model of AD (3xTg-AD). They found that expression of hTFAM significantly improved cognitive function, reduced oxidative stress and intracellular Aβ in 3xTg-AD mice and increased expression of transthyretin, known to inhibit Aβ aggregation [173].
Given that 99% of mitochondrial proteins are encoded by nuclear genome and must be imported into mitochondria, mitochondrial biogenesis is heavily dependent on proper mitochondrial protein import [174, 175], which is regulated by protein import machinery in the mitochondrial outer and inner membrane. The translocase of the outer membrane consisting of a pore-forming protein TOM44 and three receptor proteins on the cytosolic side (i.e., TOM20, TOM22, and TOM70) is the main entry gate [175]. Interestingly, Alan Roses reported an association of a polymorphic poly-T variant, rs10524523, in the TOMM40 gene with the age of onset of late-onset AD [176]. While this finding remains controversial [177], it provided the first hint of a possible involvement of mitochondrial import alteration in AD pathogenesis. Indeed, mitochondrial protein import is inhibited by oxidative stress, suggesting that mitochondrial import could be impacted in AD where extensive oxidative damage was documented in susceptible neurons in AD [178]. Gene set enrichment analysis of datasets from patients with AD archived in GeneNetwork showed disruption of mitochondrial import pathway as a hallmark of AD [40]. This was confirmed by investigation of protein expression in the brain tissue which revealed reduction of Tom20 and Tom70, as well as components of OXPHOS complex I and III in AD hippocampus [179].
Several groups had pursued the potential role of APP or Aβ on mitochondrial import machinery [180–184]. Anandatheerthavarada first identified mitochondrial-targeting signal in APP proteins and demonstrated mitochondria APP in cortical neuronal culture and in select regions of the brain of a transgenic mouse model for AD [183]. The follow-up study demonstrated APP is incompletely translocated to mitochondria and forms stable complex with mitochondrial outer and inner membrane translocase in AD brain, which likely blocked mitochondrial import machinery and caused mitochondrial dysfunction [180]. Similarly, Hansson et al. showed that Aβ is translocated to mitochondria through interaction with TOM import machinery and localized to mitochondrial cristae [181]. Aβ also impaired the import competence of mitochondrial precursor proteins although through an extramitochondrial coaggregation mechanism with the inhibitory potency positively correlating with the amyloidogenic capacity [181, 184]. Accumulation of mitochondrial Aβ correlates with early synaptic deficits in AD mouse models [137, 185].
Abnormal endoplasmic reticulum-mitochondrial interaction in AD
Both endoplasmic reticulum (ER) and mitochondria are continuous tubular networks of membranes in the cytoplasm. Approximately 5–20% of the mitochondrial surface is closely apposed at 10–30 nm distance to ER membrane and form a specialized structure called ER-mitochondria contact sites which provide a stable platform to synergize the function of these two organelles [186, 187]. An expanding number of crucial physiological functions has been ascribed to ER-mitochondria contact sites which include regulation of phospholipid synthesis and metabolism, calcium exchange between ER and mitochondria, regulation of mitochondrial dynamics and autophagy, inflammasome activation, and apoptosis [187–189], all of which are essential for proper mitochondrial function. Emerging evidence demonstrated a crucial role of ER-mitochondria contact sites in neuronal function and survival and disturbed MAM signaling and function is increasingly implicated in neurodegenerative diseases including AD [190].
The potential involvement of MAM dysfunction in AD was first implicated by Eric Schon’s finding of the MAM localization of presenilin 1 and 2 as well as the gamma-secretase activity [191, 192]. Later studies demonstrated that APP and β-secretases are also present and harbor APP processing activities in MAMs [193], this is consistent with the notion that APP processing occurs at lipid raft domains which is present in MAMs. Indeed, a considerable amount of Aβ was produced at mitochondria-ER contact sites in wild type mouse brain [194] which makes ER-mitochondria contact sites a likely focal point for toxic effects of Aβ. Importantly, Hedskog et al. found up-regulated MAM-associated proteins in the AD brain, and demonstrated dysregulated MAM occurs during the course of disease in a transgenic AD mouse model [195], although direct evidence of specific alterations in the ER-mitochondria contact sites in AD patient are still lacking. Mutations in the C. elegans gene encoding a PSEN homolog, sel-12 resulted in elevated endoplasmic reticulum (ER)-mitochondrial Ca2+ signaling and an increase in mitochondrial superoxide production [196]. Molecular changes of MAM components occurred in the cerebral cortex of 3 months old APP/PS1 mice assayed using label-free LC-MS/MS which suggest that MAM dysregulation is likely an early event in vivo [197].
Consistent with the in vivo findings, multiple groups demonstrated aberrantly increased ER-mitochondria contacts and/or enhanced MAM function in various cell models of AD which enables more detailed mechanistic studies: for example, overexpression of APP mutants or exposure to nanomolar concentrations of Aβ increases ER-mitochondria contact points and mitochondrial calcium concentrations [193, 195]. Enhanced cholesteryl ester and phospholipid synthesis were found in presenilin-1 and -2 double knockout mouse embryonic fibroblast cells and in fibroblasts from patients with both the familial and sporadic forms of AD, suggesting an aberrant upregulation of MAM function and ER-mitochondria crosstalk in these cell models [198]. Follow-up studies from this same group demonstrated that presenilins likely regulates ER-connectivity and function through APP processing since accumulation of C99, the 99-aa C-terminal fragment of APP after beta-secretase cleavage and a substrate of gamma-secretase, at MAM caused elevated sphingolipid turnover and increased ceramide which altered lipid composition and thus impacted the ER-mitochondria contacts, resulted in metabolic disturbance and reduced mitochondrial respiration [199]. Consistent with an enhanced ER-mitochondria connectivity and function, increased C99 levels and ceramide levels were found in MAM fractions in cell and animal models of AD and in fibroblasts from AD patients carrying PS2 mutations [199]. Neuronal MAM was also subject to the regulation of extraneuronal apolipoprotein that is associated with increased AD risk. Tambini et al. suggested apolipoprotein E (ApoE4) secreted by astrocytes significantly increased ER-mitochondrial communication and MAM function as measured by the synthesis of phospholipids and of cholesteryl esters [200]. Consistently, disrupted cholesterol homeostasis and related neurotoxicity in AD were shown to be mediated by ER-mitochondria stress triggered by Aβ that promoted cholesterol synthesis and mitochondrial cholesterol influx [201].
However, there is controversy on the effects of presenilins on MAM structure and function since in tissues from AD patients carrying PS1 E280A mutation, ER-mitochondria tethering was impaired, a result further confirmed by in vitro studies [202]. Pizzo’s group also reported that PS2 expression, but not its ablation, enhanced both physical interaction and function coupling of ER-mitochondria likely through modulation of Mfn2 antagonism of ER-mitochondria interactions [203]. While further studies are needed to resolve the discrepancy, it should be noted that both enhanced and disturbed ER-mitochondria tethering could lead to ER and mitochondrial dysfunction and cause mitochondrial dysfunction. In fact, it is not without precedence that both enhanced and disturbed ER-mitochondria tethering could contribute to the same neurodegenerative disease as in the case of Parkinson’s disease where Parkin mutations enhanced [204], but alpha synuclein and DJ-1 mutations disturbed [205, 206], ER-mitochondrial tethering. Overall, these studies collectively demonstrated that abnormalities in ER-mitochondria tethering contributes to mitochondrial dysfunction in AD. Now, it would be of importance to understand whether and how such abnormalities in ER-mitochondria tethering relates to other pathological changes such as impaired mitophagy and inflammasome activation in AD.
Impaired mitophagy in AD
As metabolic active organelles where more than 90% of reactive oxygen species (ROS) are produced [57], mitochondria develop a sophisticated mitochondrial quality control system to cope with unavoidable damage to its contents as well as the organelles as a whole. At the organelle level, damaged mitochondria are degraded through mitophagy. The most well characterized mitophagy pathway involves stabilization/activation of PINK1 at the outer mitochondrial membrane by impaired mitochondrial membrane potential characterizing damaged mitochondria [207]. PINK1 not only phosphorylates and recruits the E3-ubiquitin ligase, Parkin, to mitochondria, but also phosphorylates ubiquitin to feed Parkin mediated ubiquitination of mitochondrial outer membrane proteins which labels the damaged mitochondria for degradation through mitophagy pathway [208]. It is of importance to note that mutations in either PINK1 or PARKIN are associated with early-onset familial Parkinson disease, the second most common neurodegenerative disease after AD [209], suggesting the critical role of this pathway in the CNS.
Mitochondria are key targets of autophagic degradation in the brain of AD patients [210, 211] and strong evidence suggests autophagy/lysosome failure in AD [212]. Accumulation of damaged mitochondria as evidenced by swollen appearance with distorted cristae have been identified by electron microscopy studies both in biopsy of human AD cases and in transgenic animal models of AD [87, 109, 213]. Increased PINK1, Parkin and/or increased ubiquitination of mitochondrial proteins were found in the accumulated mitochondria in pyramidal neurons in AD hippocampus, APP transgenic mice and cell models expressing mutant APP or PS1 or isolated from human AD patients, implicating an activated but stalled mitophagy process [213–215]. It is likely that inadequate mitophagic capacity in eliminating increased number of damaged mitochondria [214] or impairment in the later steps in mitophagy involving lysosomal degradation [40, 213, 216, 217] that resulted in the accumulation of damaged mitochondria and disturbance in mitochondrial homeostasis.
Mechanistically, PS1 promotes PINK1 promoter transactivation, mRNA and protein expression through AICD, the cleavage product of APP by gamma-secretase yet PS1 mutations disrupted lysosomal acidification and proteolysis due to the failure of v-ATPase targeting to lysosomes during autophagy [218]. Corsetti et al. described enhanced mitophagy triggered by a specific form of tau protein in vitro AD model. They reported a 20–22 kDa NH2-tau fragment that contributed to synaptic deterioration in AD by aberrantly recruiting Parkin and UCHL-1 to mitochondria which made them more prone to detrimental autophagic clearance [219]. However, Hu et al. found increased tau protein might increase mitochondrial membrane potential that prevents mitochondrial recruitment of Parkin by PINK1 in AD [220]. A most recent study found significantly less mitophagy events in AD hippocampus and in human neurons generated from iPS cells from AD patients bearing APP mutation or two copies of ApoE4 and pinpoint the impaired orchestration of mitophagy at earlier steps of initiation in AD due to decreased levels of activated mitophagic proteins, such as p-TBK1 and p-ULK1 [215].
While detailed mechanisms underlying impaired mitophagy in AD remains to be worked out, enhancing mitophagy by either genetic manipulation or pharmaceutical methods appears beneficial across different AD models. Overexpression of PARK2 reversed mitophagy failure and led to the recovery of mitochondrial membrane potential in sAD fibroblasts [213]. Overexpression of PINK1, by activating mitophagy signaling in APP transgenic mice, restores mitochondrial function, reduces Aβ production and amyloid pathology, and alleviates synaptic function as well as cognitive/behavioral functions [221]. Treatment with actinonin or urolithin A, compounds that enhance mitophagy, restored normal memory to C. elegans models overexpressing Aβ1–42 or tau in a PINK1-dependent manner [215]. Importantly, these compounds similarly enhanced mitophagy and restored normal mitochondria in APP/PS1 mice, which resulted in alleviation of amyloid pathology and improved cognitive/behavioral functions. Interestingly, the clearance of amyloid plaques appears due to increased phagocytic efficiency of microglia whose mitophagy is also impaired in APP/PS1 mice and enhanced by these treatments. This underscores an intertwined role of defective mitophagy in the development of amyloid pathology, which makes mitophagy as an important node for intervention. Recent studies demonstrated enhanced mitophagy protects against inflammasome-mediated neuroinflammation, a prominent feature of AD, which makes it a more promising target [222]. In this regard, NAD+-boosting compounds such as nicotinamide riboside (NR) potently induce mitophagy [223, 224]. NR treatment restored mitochondrial function and robustly decreased amyloid pathology and improved context-dependent memory in the APP/PS1 mice [40].
Impaired mitochondrial proteostasis in AD
At protein level of mitochondrial quality control mechanisms, a process called mitochondria proteostasis monitors mitochondrial protein damages through an interconnected network consisting of chaperones and proteases in each compartment of mitochondria: Mitochondrial chaperones are involved in protein translocation and folding reactions while ATP-dependent proteases are responsible for directly removing damaged or misfolded proteins from mitochondria [225–229]. Defects in these proteins impair the capability of mitochondria to monitor, repair and remove damaged proteins which eventually induces accumulation of protein aggregation within mitochondria and causes mitochondrial dysfunction [230–233]. Importantly, genetic mutations in all these mitochondrial chaperones and proteases cause human diseases with severe neurological symptoms [229, 234–238] which underscores the significance of mitochondrial proteostasis regulated by mitochondrial protease and chaperones in mitochondrial function/dysfunction in the nervous system.
At the suborganelle level, abundant evidence demonstrated accumulation of damaged mitochondrial contents including mtDNA and proteins in AD [239], which suggest mitochondrial proteostasis may also be impaired. In general, maturation of imported mitochondrial proteins and degradation of damaged proteins remain in balance. However, one study actually found mitochondrial proteases and chaperones are upregulated in AD patients [50] possibly as an insufficient protective response. Upregulation of these mitochondrial proteases and chaperones in the brain of MCI patients and in the 3XTgAD mice precedes amyloid and tau pathology, suggestive of an early event during the course of disease [40]. Moreover, enhanced mitochondrial proteostasis might reduce Aβ proteotoxicity in AD animal models [40].
Current studies focused on two mitochondrial proteases related to the processing and metabolism of APP or Aβ. One is the mitochondrial peptidasome, PreP, located in mitochondria matrix which is involved in the cleavage/maturation of presequence of mitochondrial matrix proteins after its import [182]. Falkevall et al. first identified that the PreP was capable of degrading Aβ40 and Aβ42 in vitro, revealing the likely Aβ degradation mechanism in mitochondria [240]. Consistent with this biochemical study, Alikhani et al. reported decreased activity of PreP in AD patients and transgenic AD mice in which increased oxidative stress likely underlies the decreased PreP activity in AD [241]. In addition to detrimental effects of oxidative stress on PreP activity, one study suggested a feedback mechanism of Aβ on PreP activity that triggered imbalanced mitochondrial proteome due to the rapid degradation of impaired preprotein maturation [242]. These studies were further corroborated in vivo utilizing PreP transgenic mice. In PreP-overexpressed AD transgenic mice, increased expression of human PreP in cortical neurons attenuated mitochondrial amyloid pathology and synaptic mitochondrial dysfunction [243]. It is interesting to note that genetic mutations in PreP associated with an autosomal recessive, slowly progressive syndrome characterized by mental retardation, spinocerebellar ataxia, cognitive decline and psychosis [244]. Although it was impractical to analyze the pathological changes in the brains of these patients in this study, PreP (+/−) heterozygous mouse showed progressive ataxia associated with brain degenerative lesions, including accumulation of Aβ-positive amyloid deposits thus providing a mechanistic demonstration of the mitochondrial involvement in amyloidotic neurodegeneration.
The other mitochondrial protease of more interest in AD field is HtrA2/Omi, a serine protease in the mitochondrial intermembrane space (IMS). A weak association between HtrA2 A141S and AD was found in Swedish case control studies and specific protease activity of HtrA2 was found to be significantly increased in AD patients [245]. Using yeast-two hybrid assay, it was found that HtrA2/Omi interacts with Aβ through the PDZ domain at C-terminus, which was further confirmed in HEK392 cells by co-immunoprecipitation assay [246]. Aβ was found in matrix, but there is no consensus on its localization in mitochondrial compartments. It is possible that Aβ may have access to HtrA2 in the intermembrane space, but how such interaction impacts HtrA2 activity is unknown. A later study demonstrated that HtrA2/Omi performs a chaperone function and significantly delays the aggregation of Aβ1–42 peptide but independent of the PDZ domain in vitro [247]. Densely accumulated HtrA2 immunoreactivity was identified extracellularly in the cortex and hippocampus of AD patients [248], suggesting changes in HtrA2 may affect amyloid deposition extracellularly which is consistent with the partial localization of HtrA2 immunoreactivity in amyloid plaques. However, the physiological significance of these observations in vivo as it relates to mitochondria is unclear. APP is localized to mitochondria and partially colocalizes with HtrA2. Interestingly, mitochondrial APP is directly and efficiently cleaved by the HtrA2 both in vitro and in vivo, which releases C161 fragments into cytosol [249]. It is postulated that HtrA2 cleavage of APP may alleviate APP accumulation induced mitochondrial dysfunction. However, the fate of C161 related to amyloidogenic- and non- amyloidogenic pathways were not determined. Adding more complexity to the role of HtrA2, it also interacts with presenilin in active γ-secretase complexes located to mitochondria and modulates the cleavage of APP [250]. Interestingly, such interaction also impacts the HtrA2 protease activity since the C-terminus of PS1 is an active peptide ligand for the PDZ domain of HtrA2 and induces HtrA2-dependent cell death [251].
Overall, despite ample evidence demonstrating accumulation of damage mitochondrial proteins, there were sparse studies on alterations in mitochondrial proteostasis in AD. Whether and how specific mitochondrial chaperones and proteases in different mitochondrial compartments are involved is not clear. Importantly, recent studies demonstrated “mitochondria as guardian in cytosol” where mitochondria proteases degrade aggregation-prone cytosolic proteins after their importation [252, 253], suggesting that mitochondrial proteostasis could also regulate cytosolic protein homeostasis and neuronal integrity. However, this important aspect remains to be explored in AD.
Conclusion
Mitochondrial dysfunction plays a critical role in AD either as a primary or secondary event, which could represent promising therapeutic targets (Fig. 1). One therapeutic strategy targeting mitochondria directly focuses on modulating mitochondrial activities such as bioenergetics. For example, oxaloacetate (OAA), a Krebs cycle and gluconeogenesis intermediate that enhanced bioenergetic fluxes and upregulated some brain bioenergetic infrastructure-related parameters, is in phase I clinical trial [254]. NAD, an intermediate common to several mitochondrial metabolic pathways such as glycolysis, TCA cycle, and oxidative phosphorylation, is in phase II trial for AD [255]. Interestingly, recent studies demonstrated that mild inhibition of complex I at the FMN subunit (i.e., NDUFA1) by CP2 compound augmented respiratory capacity and reduced proton leaks. CP2 could alleviate cognitive and pathological deficits in various animal models of AD and is also being developed to treat AD [256], suggesting the potential use of bioenergetics modulators in AD treatment. However, it must be noted that partial complex I deficiency induced by neuronal ablation of another complex I subunit, NDUFA5, caused mild chronic encephalopathy in mice [257], suggesting that selection of safe bioenergetics modulators for AD treatment is likely a considerable challenge due to our incomplete understanding of the mechanisms behind these differential outcomes.
Alternatively, given that the loss of mitochondrial structural and functional integrity is likely causally associated with impaired energy metabolism and increased oxidative stress during the course of AD, it probably holds greater promise to consider restoration of the integrity of mitochondria for therapeutic development, which requires better understanding of the underlying mechanisms. Indeed, recent advances in the field demonstrated the involvement of impaired mitochondrial dynamics and transport, biogenesis and mitophagy, proteostasis and quality control as well as its interactions with other organelles such as ER, which offers multiple novel intervention sites to target the integrity of mitochondria. Most of the mechanistic studies rely on genetic models of AD in vitro and in vivo, which demonstrated how APP or Aβ causes impaired mitochondrial integrity and dysfunction by impacting these mitochondrial regulatory mechanisms separately. However, the relationship between these deficits and their relative contribution to APP- or Aβ-induced mitochondrial dysfunction and neuronal dysfunction was not elucidated. It is possible that they act together in a downward spiral manner to impair mitochondrial integrity and cause mitochondrial dysfunction that mediates neurotoxic effect of APP/Aβ. This implies that restoration of one deficit may have an overall beneficial effect on mitochondrial integrity and function as repeatedly demonstrated in these studies.
More studies are needed to address how mitochondria become dysfunctional in the case when mitochondrial dysfunction plays the primary role in the pathogenesis. Because aging is the only known risk factor for this subset of AD, given the multifaceted nature of biological aging, the lack of specific models is the biggest hurdle. Nevertheless, as stipulated by the “mitochondrial cascade hypothesis”, the accumulation of somatic mtDNA mutations with advancing aging influences brain function. Many of these mitochondrial regulation mechanisms become loose during aging. For example, mitochondrial biogenesis and mitophagy decline while mitochondria fission enhances along aging. It is possible that mild deficit in one or several of these mitochondrial regulation mechanisms determined by one’s genetic background could set the motion of a downward spiral and all of these mitochondrial deficits could come into play at some points that impairs mitochondrial integrity, which causes damage, affects repair and/or replication of mtDNA and thus accelerates the accumulation of mtDNA changes, leading to mitochondrial dysfunction and eventually, the disease (Fig. 1). Therefore, the better understanding of how these mitochondrial regulation mechanisms are involved in AD, either as a primary or secondary event, could potentially provide multiple novel therapeutic targets that benefit all AD patients.
Acknowledgements
Not applicable.
Abbreviations
- AD
Alzheimer’s disease
- Aβ
Amyloid-β
- ApoE4
Apolipoprotein E
- BACE1
Beta-APP cleaving enzyme
- ETC
Electron transfer chain
- ER
Endoplasmic reticulum
- hTFAM
human mitochondrial transcriptional factor A
- MAM
Mitochondria-associated membrane
- mtDNA
mitochondrial DNA
- TFAM
Mitochondrial transcription factor A
- MDVs
Mitochondrial-derived vesicles
- NRF 1/2
Nuclear respiratory factor 1 and 2
- PGC-1α
Peroxisome proliferator activator receptor gamma-coactivator 1α
- ROS
Reactive oxygen species
- SOD2
Superoxide dismutase 2
- TIM23
Translocase of the inner mitochondrial membrane 23
- TOM40
Translocase of the outer mitochondrial membrane 40
Authors’ contributions
WW, FZ, XM and GP wrote the manuscript. XZ conceived and wrote the manuscript. All authors read and approved the final manuscript.
Funding
The work was supported in part by the Dr. Robert M. Kohrman Memorial Fund, the National Institutes of Health (NS083385, AG049179 and AG056363 to XZ and AG058015 and AG063362 to WW) and the Alzheimer’s Association (AARG-16-443584). WW was a program participant in the Research Education Component of the Cleveland Alzheimer’s Disease Research Center supported by NIA P30 AG062428.
Availability of data and materials
Not applicable.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Wenzhang Wang, Email: wenzhang.wang@case.edu.
Fanpeng Zhao, Email: fanpeng.zhao@case.edu.
Xiaopin Ma, Email: xiaopin.ma@case.edu.
George Perry, Email: george.perry@utsa.edu.
Xiongwei Zhu, Email: xiongwei.zhu@case.edu.
References
- 1.Coyle JT, Price DL, DeLong MR. Alzheimer’s disease: a disorder of cortical cholinergic innervation. Science. 1983;219(4589):1184–1190. doi: 10.1126/science.6338589. [DOI] [PubMed] [Google Scholar]
- 2.Selkoe DJ. Alzheimer’s disease: genes, proteins, and therapy. Physiol Rev. 2001;81(2):741–766. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- 3.GBD 2016 Neurology Collaborators Global, regional, and national burden of neurological disorders, 1990–2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019;18(5):459–480. doi: 10.1016/S1474-4422(18)30499-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brookmeyer R, Johnson E, Ziegler-Graham K, Arrighi HM. Forecasting the global burden of Alzheimer’s disease. Alzheimers Dement. 2007;3(3):186–191. doi: 10.1016/j.jalz.2007.04.381. [DOI] [PubMed] [Google Scholar]
- 5.Cornutiu G. The epidemiological scale of Alzheimer's disease. J Clin Med Res. 2015;7(9):657–666. doi: 10.14740/jocmr2106w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Qiu C, Kivipelto M, von Strauss E. Epidemiology of Alzheimer’s disease: occurrence, determinants, and strategies toward intervention. Dialogues Clin Neurosci. 2009;11(2):111–128. doi: 10.31887/DCNS.2009.11.2/cqiu. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443(7113):787–795. doi: 10.1038/nature05292. [DOI] [PubMed] [Google Scholar]
- 8.Swerdlow RH. Mitochondria and mitochondrial cascades in Alzheimer’s disease. J Alzheimers Dis. 2018;62(3):1403–1416. doi: 10.3233/JAD-170585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang X, Wang W, Li L, Perry G, Lee HG, Zhu X. Oxidative stress and mitochondrial dysfunction in Alzheimer’s disease. Biochim Biophys Acta. 2014;1842(8):1240–1247. doi: 10.1016/j.bbadis.2013.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kapogiannis D, Mattson MP. Disrupted energy metabolism and neuronal circuit dysfunction in cognitive impairment and Alzheimer's disease. Lancet Neurol. 2011;10(2):187–198. doi: 10.1016/S1474-4422(10)70277-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Crane PK, Walker R, Hubbard RA, Li G, Nathan DM, Zheng H, et al. Glucose levels and risk of dementia. N Engl J Med. 2013;369(6):540–548. doi: 10.1056/NEJMoa1215740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Croteau E, Castellano CA, Fortier M, Bocti C, Fulop T, Paquet N, et al. A cross-sectional comparison of brain glucose and ketone metabolism in cognitively healthy older adults, mild cognitive impairment and early Alzheimer’s disease. Exp Gerontol. 2018;107:18–26. doi: 10.1016/j.exger.2017.07.004. [DOI] [PubMed] [Google Scholar]
- 13.Gordon BA, Blazey TM, Su Y, Hari-Raj A, Dincer A, Flores S, et al. Spatial patterns of neuroimaging biomarker change in individuals from families with autosomal dominant Alzheimer's disease: a longitudinal study. Lancet Neurol. 2018;17(3):241–250. doi: 10.1016/S1474-4422(18)30028-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Paranjpe MD, Chen X, Liu M, Paranjpe I, Leal JP, Wang R, et al. The effect of ApoE epsilon4 on longitudinal brain region-specific glucose metabolism in patients with mild cognitive impairment: a FDG-PET study. Neuroimage Clin. 2019;22:101795. doi: 10.1016/j.nicl.2019.101795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Reiman EM, Chen K, Alexander GE, Caselli RJ, Bandy D, Osborne D, et al. Functional brain abnormalities in young adults at genetic risk for late-onset Alzheimer's dementia. Proc Natl Acad Sci U S A. 2004;101(1):284–289. doi: 10.1073/pnas.2635903100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen Z, Zhong C. Decoding Alzheimer’s disease from perturbed cerebral glucose metabolism: implications for diagnostic and therapeutic strategies. Prog Neurobiol. 2013;108:21–43. doi: 10.1016/j.pneurobio.2013.06.004. [DOI] [PubMed] [Google Scholar]
- 17.Weise CM, Chen K, Chen Y, Kuang X, Savage CR, Reiman EM, et al. Left lateralized cerebral glucose metabolism declines in amyloid-beta positive persons with mild cognitive impairment. Neuroimage Clin. 2018;20:286–296. doi: 10.1016/j.nicl.2018.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Arbizu J, Festari C, Altomare D, Walker Z, Bouwman F, Rivolta J, et al. Clinical utility of FDG-PET for the clinical diagnosis in MCI. Eur J Nucl Med Mol Imaging. 2018;45(9):1497–1508. doi: 10.1007/s00259-018-4039-7. [DOI] [PubMed] [Google Scholar]
- 19.Sorensen A, Blazhenets G, Rucker G, Schiller F, Meyer PT, Frings L, et al. Prognosis of conversion of mild cognitive impairment to Alzheimer’s dementia by voxel-wise cox regression based on FDG PET data. Neuroimage Clin. 2019;21:101637. doi: 10.1016/j.nicl.2018.101637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McDade E, Wang G, Gordon BA, Hassenstab J, Benzinger TLS, Buckles V, et al. Longitudinal cognitive and biomarker changes in dominantly inherited Alzheimer disease. Neurology. 2018;91(14):e1295–ee306. doi: 10.1212/WNL.0000000000006277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Benzinger TL, Blazey T, Jack CR, Jr, Koeppe RA, Su Y, Xiong C, et al. Regional variability of imaging biomarkers in autosomal dominant Alzheimer’s disease. Proc Natl Acad Sci U S A. 2013;110(47):E4502–E4509. doi: 10.1073/pnas.1317918110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Altmann A, Ng B, Landau SM, Jagust WJ, Greicius MD. Alzheimer’s disease neuroimaging I. regional brain hypometabolism is unrelated to regional amyloid plaque burden. Brain. 2015;138(Pt 12):3734–3746. doi: 10.1093/brain/awv278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jack CR, Jr, Wiste HJ, Weigand SD, Knopman DS, Lowe V, Vemuri P, et al. Amyloid-first and neurodegeneration-first profiles characterize incident amyloid PET positivity. Neurology. 2013;81(20):1732–1740. doi: 10.1212/01.wnl.0000435556.21319.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Filippini N, MacIntosh BJ, Hough MG, Goodwin GM, Frisoni GB, Smith SM, et al. Distinct patterns of brain activity in young carriers of the APOE-epsilon4 allele. Proc Natl Acad Sci U S A. 2009;106(17):7209–7214. doi: 10.1073/pnas.0811879106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jagust WJ, Landau SM. Alzheimer’s disease neuroimaging I. apolipoprotein E, not fibrillar beta-amyloid, reduces cerebral glucose metabolism in normal aging. J Neurosci. 2012;32(50):18227–18233. doi: 10.1523/JNEUROSCI.3266-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Szablewski L. Glucose transporters in brain: in health and in Alzheimer's disease. J Alzheimers Dis. 2017;55(4):1307–1320. doi: 10.3233/JAD-160841. [DOI] [PubMed] [Google Scholar]
- 27.Aliev G, Smith MA, Obrenovich ME, de la Torre JC, Perry G. Role of vascular hypoperfusion-induced oxidative stress and mitochondria failure in the pathogenesis of Azheimer disease. Neurotox Res. 2003;5(7):491–504. doi: 10.1007/BF03033159. [DOI] [PubMed] [Google Scholar]
- 28.Sang S, Pan X, Chen Z, Zeng F, Pan S, Liu H, et al. Thiamine diphosphate reduction strongly correlates with brain glucose hypometabolism in Alzheimer’s disease, whereas amyloid deposition does not. Alzheimers Res Ther. 2018;10(1):26. doi: 10.1186/s13195-018-0354-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pan X, Fei G, Lu J, Jin L, Pan S, Chen Z, et al. Measurement of blood thiamine metabolites for Alzheimer’s disease diagnosis. EBioMedicine. 2016;3:155–162. doi: 10.1016/j.ebiom.2015.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gibson GE, Sheu KF, Blass JP, Baker A, Carlson KC, Harding B, et al. Reduced activities of thiamine-dependent enzymes in the brains and peripheral tissues of patients with Alzheimer’s disease. Arch Neurol. 1988;45(8):836–840. doi: 10.1001/archneur.1988.00520320022009. [DOI] [PubMed] [Google Scholar]
- 31.Ishii K, Kitagaki H, Kono M, Mori E. Decreased medial temporal oxygen metabolism in Alzheimer’s disease shown by PET. J Nucl Med. 1996;37(7):1159–1165. [PubMed] [Google Scholar]
- 32.Tohgi H, Yonezawa H, Takahashi S, Sato N, Kato E, Kudo M, et al. Cerebral blood flow and oxygen metabolism in senile dementia of Alzheimer’s type and vascular dementia with deep white matter changes. Neuroradiology. 1998;40(3):131–137. doi: 10.1007/s002340050553. [DOI] [PubMed] [Google Scholar]
- 33.Lajoie I, Nugent S, Debacker C, Dyson K, Tancredi FB, Badhwar A, et al. Application of calibrated fMRI in Alzheimer’s disease. Neuroimage Clin. 2017;15:348–358. doi: 10.1016/j.nicl.2017.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Buchan RJ, Nagata K, Yokoyama E, Langman P, Yuya H, Hirata Y, et al. Regional correlations between the EEG and oxygen metabolism in dementia of Alzheimer’s type. Electroencephalogr Clin Neurophysiol. 1997;103(3):409–417. doi: 10.1016/s0013-4694(97)00015-5. [DOI] [PubMed] [Google Scholar]
- 35.Liang WS, Reiman EM, Valla J, Dunckley T, Beach TG, Grover A, et al. Alzheimer’s disease is associated with reduced expression of energy metabolism genes in posterior cingulate neurons. Proc Natl Acad Sci U S A. 2008;105(11):4441–4446. doi: 10.1073/pnas.0709259105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brooks WM, Lynch PJ, Ingle CC, Hatton A, Emson PC, Faull RL, et al. Gene expression profiles of metabolic enzyme transcripts in Alzheimer’s disease. Brain Res. 2007;1127(1):127–135. doi: 10.1016/j.brainres.2006.09.106. [DOI] [PubMed] [Google Scholar]
- 37.Mastroeni D, Khdour OM, Delvaux E, Nolz J, Olsen G, Berchtold N, et al. Nuclear but not mitochondrial-encoded oxidative phosphorylation genes are altered in aging, mild cognitive impairment, and Alzheimer’s disease. Alzheimers Dement. 2017;13(5):510–519. doi: 10.1016/j.jalz.2016.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Manczak M, Park BS, Jung Y, Reddy PH. Differential expression of oxidative phosphorylation genes in patients with Alzheimer’s disease: implications for early mitochondrial dysfunction and oxidative damage. NeuroMolecular Med. 2004;5(2):147–162. doi: 10.1385/NMM:5:2:147. [DOI] [PubMed] [Google Scholar]
- 39.Zhang L, Guo XQ, Chu JF, Zhang X, Yan ZR, Li YZ. Potential hippocampal genes and pathways involved in Alzheimer’s disease: a bioinformatic analysis. Genet Mol Res. 2015;14(2):7218–7232. doi: 10.4238/2015.June.29.15. [DOI] [PubMed] [Google Scholar]
- 40.Sorrentino V, Romani M, Mouchiroud L, Beck JS, Zhang H, D'Amico D, et al. Enhancing mitochondrial proteostasis reduces amyloid-beta proteotoxicity. Nature. 2017;552(7684):187–193. doi: 10.1038/nature25143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Minjarez B, Calderon-Gonzalez KG, Rustarazo ML, Herrera-Aguirre ME, Labra-Barrios ML, Rincon-Limas DE, et al. Identification of proteins that are differentially expressed in brains with Alzheimer’s disease using iTRAQ labeling and tandem mass spectrometry. J Proteome. 2016;139:103–121. doi: 10.1016/j.jprot.2016.03.022. [DOI] [PubMed] [Google Scholar]
- 42.Adav SS, Park JE, Sze SK. Quantitative profiling brain proteomes revealed mitochondrial dysfunction in Alzheimer’s disease. Mol Brain. 2019;12(1):8. doi: 10.1186/s13041-019-0430-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Valla J, Berndt JD, Gonzalez-Lima F. Energy hypometabolism in posterior cingulate cortex of Alzheimer’s patients: superficial laminar cytochrome oxidase associated with disease duration. J Neurosci. 2001;21(13):4923–4930. doi: 10.1523/JNEUROSCI.21-13-04923.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cottrell DA, Blakely EL, Johnson MA, Ince PG, Turnbull DM. Mitochondrial enzyme-deficient hippocampal neurons and choroidal cells in AD. Neurology. 2001;57(2):260–264. doi: 10.1212/wnl.57.2.260. [DOI] [PubMed] [Google Scholar]
- 45.Bubber P, Haroutunian V, Fisch G, Blass JP, Gibson GE. Mitochondrial abnormalities in Alzheimer brain: mechanistic implications. Ann Neurol. 2005;57(5):695–703. doi: 10.1002/ana.20474. [DOI] [PubMed] [Google Scholar]
- 46.Parker WD, Jr, Parks J, Filley CM, Kleinschmidt-DeMasters BK. Electron transport chain defects in Alzheimer’s disease brain. Neurology. 1994;44(6):1090–1096. doi: 10.1212/wnl.44.6.1090. [DOI] [PubMed] [Google Scholar]
- 47.Maurer I, Zierz S, Moller HJ. A selective defect of cytochrome c oxidase is present in brain of Alzheimer disease patients. Neurobiol Aging. 2000;21(3):455–462. doi: 10.1016/s0197-4580(00)00112-3. [DOI] [PubMed] [Google Scholar]
- 48.Wang P, Guan PP, Wang T, Yu X, Guo JJ, Wang ZY. Aggravation of Alzheimer’s disease due to the COX-2-mediated reciprocal regulation of IL-1beta and Abeta between glial and neuron cells. Aging Cell. 2014;13(4):605–615. doi: 10.1111/acel.12209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Woodling NS, Colas D, Wang Q, Minhas P, Panchal M, Liang X, et al. Cyclooxygenase inhibition targets neurons to prevent early behavioural decline in Alzheimer’s disease model mice. Brain. 2016;139(Pt 7):2063–2081. doi: 10.1093/brain/aww117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Beck JS, Mufson EJ, Counts SE. Evidence for mitochondrial UPR gene activation in familial and sporadic Alzheimer’s disease. Curr Alzheimer Res. 2016;13(6):610–614. doi: 10.2174/1567205013666151221145445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cha MY, Cho HJ, Kim C, Jung YO, Kang MJ, Murray ME, et al. Mitochondrial ATP synthase activity is impaired by suppressed O-GlcNAcylation in Alzheimer’s disease. Hum Mol Genet. 2015;24(22):6492–6504. doi: 10.1093/hmg/ddv358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Armand-Ugon M, Ansoleaga B, Berjaoui S, Ferrer I. Reduced mitochondrial activity is early and steady in the entorhinal cortex but it is mainly unmodified in the frontal cortex in Alzheimer’s disease. Curr Alzheimer Res. 2017;14(12):1327–1334. doi: 10.2174/1567205014666170505095921. [DOI] [PubMed] [Google Scholar]
- 53.Rice AC, Ladd AC, Bennett JP., Jr Postmortem Alzheimer’s disease hippocampi show oxidative phosphorylation gene expression opposite that of isolated pyramidal neurons. J Alzheimers Dis. 2015;45(4):1051–1059. doi: 10.3233/JAD-142937. [DOI] [PubMed] [Google Scholar]
- 54.Letellier T, Heinrich R, Malgat M, Mazat JP. The kinetic basis of threshold effects observed in mitochondrial diseases: a systemic approach. Biochem J. 1994;302(Pt 1):171–174. doi: 10.1042/bj3020171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Barrientos A, Moraes CT. Titrating the effects of mitochondrial complex I impairment in the cell physiology. J Biol Chem. 1999;274(23):16188–16197. doi: 10.1074/jbc.274.23.16188. [DOI] [PubMed] [Google Scholar]
- 56.Davey GP, Peuchen S, Clark JB. Energy thresholds in brain mitochondria. Potential involvement in neurodegeneration. J Biol Chem. 1998;273(21):12753–12757. doi: 10.1074/jbc.273.21.12753. [DOI] [PubMed] [Google Scholar]
- 57.Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell. 2005;120(4):483–495. doi: 10.1016/j.cell.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 58.Butterfield DA, Halliwell B. Oxidative stress, dysfunctional glucose metabolism and Alzheimer disease. Nat Rev Neurosci. 2019;20(3):148–160. doi: 10.1038/s41583-019-0132-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhu X, Castellani RJ, Moreira PI, Aliev G, Shenk JC, Siedlak SL, et al. Hydroxynonenal-generated crosslinking fluorophore accumulation in Alzheimer disease reveals a dichotomy of protein turnover. Free Radic Biol Med. 2012;52(3):699–704. doi: 10.1016/j.freeradbiomed.2011.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nunomura A, Tamaoki T, Motohashi N, Nakamura M, McKeel DW, Jr, Tabaton M, et al. The earliest stage of cognitive impairment in transition from normal aging to Alzheimer disease is marked by prominent RNA oxidation in vulnerable neurons. J Neuropathol Exp Neurol. 2012;71(3):233–241. doi: 10.1097/NEN.0b013e318248e614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mandal PK, Saharan S, Tripathi M, Murari G. Brain glutathione levels--a novel biomarker for mild cognitive impairment and Alzheimer’s disease. Biol Psychiatry. 2015;78(10):702–710. doi: 10.1016/j.biopsych.2015.04.005. [DOI] [PubMed] [Google Scholar]
- 62.Shukla D, Mandal PK, Tripathi M, Vishwakarma G, Mishra R, Sandal K. Quantitation of in vivo brain glutathione conformers in cingulate cortex among age-matched control, MCI, and AD patients using MEGA-PRESS. Hum Brain Mapp. 2020;41(1):194–217. doi: 10.1002/hbm.24799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Scheff SW, Ansari MA, Mufson EJ. Oxidative stress and hippocampal synaptic protein levels in elderly cognitively intact individuals with Alzheimer’s disease pathology. Neurobiol Aging. 2016;42:1–12. doi: 10.1016/j.neurobiolaging.2016.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Swomley AM, Butterfield DA. Oxidative stress in Alzheimer disease and mild cognitive impairment: evidence from human data provided by redox proteomics. Arch Toxicol. 2015;89(10):1669–1680. doi: 10.1007/s00204-015-1556-z. [DOI] [PubMed] [Google Scholar]
- 65.Taanman JW. The mitochondrial genome: structure, transcription, translation and replication. Biochim Biophys Acta. 1999;1410(2):103–123. doi: 10.1016/s0005-2728(98)00161-3. [DOI] [PubMed] [Google Scholar]
- 66.D’Souza AR, Minczuk M. Mitochondrial transcription and translation: overview. Essays Biochem. 2018;62(3):309–320. doi: 10.1042/EBC20170102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yan MH, Wang X, Zhu X. Mitochondrial defects and oxidative stress in Alzheimer disease and Parkinson disease. Free Radic Biol Med. 2013;62:90–101. doi: 10.1016/j.freeradbiomed.2012.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Boczonadi V, Ricci G, Horvath R. Mitochondrial DNA transcription and translation: clinical syndromes. Essays Biochem. 2018;62(3):321–340. doi: 10.1042/EBC20170103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Inczedy-Farkas G, Trampush JW, Perczel Forintos D, Beech D, Andrejkovics M, Varga Z, et al. Mitochondrial DNA mutations and cognition: a case-series report. Arch Clin Neuropsychol. 2014;29(4):315–321. doi: 10.1093/arclin/acu016. [DOI] [PubMed] [Google Scholar]
- 70.Honea RA, Swerdlow RH, Vidoni ED, Burns JM. Progressive regional atrophy in normal adults with a maternal history of Alzheimer disease. Neurology. 2011;76(9):822–829. doi: 10.1212/WNL.0b013e31820e7b74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mosconi L, Berti V, Swerdlow RH, Pupi A, Duara R, de Leon M. Maternal transmission of Alzheimer’s disease: prodromal metabolic phenotype and the search for genes. Hum Genomics. 2010;4(3):170–193. doi: 10.1186/1479-7364-4-3-170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mosconi L, Brys M, Switalski R, Mistur R, Glodzik L, Pirraglia E, et al. Maternal family history of Alzheimer’s disease predisposes to reduced brain glucose metabolism. Proc Natl Acad Sci U S A. 2007;104(48):19067–19072. doi: 10.1073/pnas.0705036104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Salvado G, Brugulat-Serrat A, Sudre CH, Grau-Rivera O, Suarez-Calvet M, Falcon C, et al. Spatial patterns of white matter hyperintensities associated with Alzheimer’s disease risk factors in a cognitively healthy middle-aged cohort. Alzheimers Res Ther. 2019;11(1):12. doi: 10.1186/s13195-018-0460-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lakatos A, Derbeneva O, Younes D, Keator D, Bakken T, Lvova M, et al. Association between mitochondrial DNA variations and Alzheimer’s disease in the ADNI cohort. Neurobiol Aging. 2010;31(8):1355–1363. doi: 10.1016/j.neurobiolaging.2010.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chagnon P, Gee M, Filion M, Robitaille Y, Belouchi M, Gauvreau D. Phylogenetic analysis of the mitochondrial genome indicates significant differences between patients with Alzheimer disease and controls in a French-Canadian founder population. Am J Med Genet. 1999;85(1):20–30. doi: 10.1002/(sici)1096-8628(19990702)85:1<20::aid-ajmg6>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 76.Carrieri G, Bonafe M, De Luca M, Rose G, Varcasia O, Bruni A, et al. Mitochondrial DNA haplogroups and APOE4 allele are non-independent variables in sporadic Alzheimer’s disease. Hum Genet. 2001;108(3):194–198. doi: 10.1007/s004390100463. [DOI] [PubMed] [Google Scholar]
- 77.van der Walt JM, Dementieva YA, Martin ER, Scott WK, Nicodemus KK, Kroner CC, et al. Analysis of European mitochondrial haplogroups with Alzheimer disease risk. Neurosci Lett. 2004;365(1):28–32. doi: 10.1016/j.neulet.2004.04.051. [DOI] [PubMed] [Google Scholar]
- 78.Hudson G, Sims R, Harold D, Chapman J, Hollingworth P, Gerrish A, et al. No consistent evidence for association between mtDNA variants and Alzheimer disease. Neurology. 2012;78(14):1038–1042. doi: 10.1212/WNL.0b013e31824e8f1d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Blanchard BJ, Park T, Fripp WJ, Lerman LS, Ingram VM. A mitochondrial DNA deletion in normally aging and in Alzheimer brain tissue. Neuroreport. 1993;4(6):799–802. doi: 10.1097/00001756-199306000-00051. [DOI] [PubMed] [Google Scholar]
- 80.Corral-Debrinski M, Horton T, Lott MT, Shoffner JM, McKee AC, Beal MF, et al. Marked changes in mitochondrial DNA deletion levels in Alzheimer brains. Genomics. 1994;23(2):471–476. doi: 10.1006/geno.1994.1525. [DOI] [PubMed] [Google Scholar]
- 81.Chen Y, Liu C, Parker WD, Chen H, Beach TG, Liu X, et al. Mitochondrial DNA rearrangement spectrum in brain tissue of Alzheimer’s disease: analysis of 13 cases. PLoS One. 2016;11(6):e0154582. doi: 10.1371/journal.pone.0154582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Coskun PE, Beal MF, Wallace DC. Alzheimer’s brains harbor somatic mtDNA control-region mutations that suppress mitochondrial transcription and replication. Proc Natl Acad Sci U S A. 2004;101(29):10726–10731. doi: 10.1073/pnas.0403649101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Soltys DT, Pereira CPM, Rowies FT, Farfel JM, Grinberg LT, Suemoto CK, et al. Lower mitochondrial DNA content but not increased mutagenesis associates with decreased base excision repair activity in brains of AD subjects. Neurobiol Aging. 2019;73:161–170. doi: 10.1016/j.neurobiolaging.2018.09.015. [DOI] [PubMed] [Google Scholar]
- 84.Lin MT, Simon DK, Ahn CH, Kim LM, Beal MF. High aggregate burden of somatic mtDNA point mutations in aging and Alzheimer’s disease brain. Hum Mol Genet. 2002;11(2):133–145. doi: 10.1093/hmg/11.2.133. [DOI] [PubMed] [Google Scholar]
- 85.Pienaar IS, Howell N, Elson JL. MutPred mutational load analysis shows mildly deleterious mitochondrial DNA variants are not more prevalent in Alzheimer’s patients, but may be under-represented in healthy older individuals. Mitochondrion. 2017;34:141–146. doi: 10.1016/j.mito.2017.04.002. [DOI] [PubMed] [Google Scholar]
- 86.Phillips NR, Simpkins JW, Roby RK. Mitochondrial DNA deletions in Alzheimer’s brains: a review. Alzheimers Dement. 2014;10(3):393–400. doi: 10.1016/j.jalz.2013.04.508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hirai K, Aliev G, Nunomura A, Fujioka H, Russell RL, Atwood CS, et al. Mitochondrial abnormalities in Alzheimer’s disease. J Neurosci. 2001;21(9):3017–3023. doi: 10.1523/JNEUROSCI.21-09-03017.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Krishnan KJ, Ratnaike TE, De Gruyter HL, Jaros E, Turnbull DM. Mitochondrial DNA deletions cause the biochemical defect observed in Alzheimer’s disease. Neurobiol Aging. 2012;33(9):2210–2214. doi: 10.1016/j.neurobiolaging.2011.08.009. [DOI] [PubMed] [Google Scholar]
- 89.Strobel S, Grunblatt E, Heinsen H, Riederer P, Espach T, Meder M, et al. Astrocyte- and microglia-specific mitochondrial DNA deletions levels in sporadic Alzheimer's disease. J Alzheimers Dis. 2019;67(1):149–157. doi: 10.3233/JAD-180661. [DOI] [PubMed] [Google Scholar]
- 90.Bender A, Schwarzkopf RM, McMillan A, Krishnan KJ, Rieder G, Neumann M, et al. Dopaminergic midbrain neurons are the prime target for mitochondrial DNA deletions. J Neurol. 2008;255(8):1231–1235. doi: 10.1007/s00415-008-0892-9. [DOI] [PubMed] [Google Scholar]
- 91.Hoekstra JG, Hipp MJ, Montine TJ, Kennedy SR. Mitochondrial DNA mutations increase in early stage Alzheimer disease and are inconsistent with oxidative damage. Ann Neurol. 2016;80(2):301–306. doi: 10.1002/ana.24709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wang J, Xiong S, Xie C, Markesbery WR, Lovell MA. Increased oxidative damage in nuclear and mitochondrial DNA in Alzheimer’s disease. J Neurochem. 2005;93(4):953–962. doi: 10.1111/j.1471-4159.2005.03053.x. [DOI] [PubMed] [Google Scholar]
- 93.Mecocci P, MacGarvey U, Beal MF. Oxidative damage to mitochondrial DNA is increased in Alzheimer’s disease. Ann Neurol. 1994;36(5):747–751. doi: 10.1002/ana.410360510. [DOI] [PubMed] [Google Scholar]
- 94.Lovell MA, Soman S, Bradley MA. Oxidatively modified nucleic acids in preclinical Alzheimer’s disease (PCAD) brain. Mech Ageing Dev. 2011;132(8–9):443–448. doi: 10.1016/j.mad.2011.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Shao C, Xiong S, Li GM, Gu L, Mao G, Markesbery WR, et al. Altered 8-oxoguanine glycosylase in mild cognitive impairment and late-stage Alzheimer’s disease brain. Free Radic Biol Med. 2008;45(6):813–819. doi: 10.1016/j.freeradbiomed.2008.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Blanch M, Mosquera JL, Ansoleaga B, Ferrer I, Barrachina M. Altered mitochondrial DNA methylation pattern in Alzheimer disease-related pathology and in Parkinson disease. Am J Pathol. 2016;186(2):385–397. doi: 10.1016/j.ajpath.2015.10.004. [DOI] [PubMed] [Google Scholar]
- 97.Stoccoro A, Siciliano G, Migliore L, Coppede F. Decreased methylation of the mitochondrial D-loop region in late-onset Alzheimer’s disease. J Alzheimers Dis. 2017;59(2):559–564. doi: 10.3233/JAD-170139. [DOI] [PubMed] [Google Scholar]
- 98.Zhu X, Perry G, Smith MA, Wang X. Abnormal mitochondrial dynamics in the pathogenesis of Alzheimer’s disease. J Alzheimers Dis. 2013;33(Suppl 1):S253–S262. doi: 10.3233/JAD-2012-129005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Mishra P, Chan DC. Mitochondrial dynamics and inheritance during cell division, development and disease. Nat Rev Mol Cell Biol. 2014;15(10):634–646. doi: 10.1038/nrm3877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Wang X, Su B, Siedlak SL, Moreira PI, Fujioka H, Wang Y, et al. Amyloid-beta overproduction causes abnormal mitochondrial dynamics via differential modulation of mitochondrial fission/fusion proteins. Proc Natl Acad Sci U S A. 2008;105(49):19318–19323. doi: 10.1073/pnas.0804871105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wang X, Su B, Lee HG, Li X, Perry G, Smith MA, et al. Impaired balance of mitochondrial fission and fusion in Alzheimer’s disease. J Neurosci. 2009;29(28):9090–9103. doi: 10.1523/JNEUROSCI.1357-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Liu W, Acin-Perez R, Geghman KD, Manfredi G, Lu B, Li C. Pink1 regulates the oxidative phosphorylation machinery via mitochondrial fission. Proc Natl Acad Sci U S A. 2011;108(31):12920–12924. doi: 10.1073/pnas.1107332108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Zhou L, Wang W, Hoppel C, Liu J, Zhu X. Parkinson’s disease-associated pathogenic VPS35 mutation causes complex I deficits. Biochim Biophys Acta Mol basis Dis. 2017;1863(11):2791–2795. doi: 10.1016/j.bbadis.2017.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Yu T, Robotham JL, Yoon Y. Increased production of reactive oxygen species in hyperglycemic conditions requires dynamic change of mitochondrial morphology. Proc Natl Acad Sci U S A. 2006;103(8):2653–2658. doi: 10.1073/pnas.0511154103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Manczak M, Calkins MJ, Reddy PH. Impaired mitochondrial dynamics and abnormal interaction of amyloid beta with mitochondrial protein Drp1 in neurons from patients with Alzheimer’s disease: implications for neuronal damage. Hum Mol Genet. 2011;20(13):2495–2509. doi: 10.1093/hmg/ddr139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Jiang S, Shao C, Tang F, Wang W, Zhu X. Dynamin-like protein 1 cleavage by calpain in Alzheimer’s disease. Aging Cell. 2019;18:e12912. doi: 10.1111/acel.12912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Cho DH, Nakamura T, Fang J, Cieplak P, Godzik A, Gu Z, et al. S-nitrosylation of Drp1 mediates beta-amyloid-related mitochondrial fission and neuronal injury. Science. 2009;324(5923):102–105. doi: 10.1126/science.1171091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Manczak M, Reddy PH. Abnormal interaction between the mitochondrial fission protein Drp1 and hyperphosphorylated tau in Alzheimer’s disease neurons: implications for mitochondrial dysfunction and neuronal damage. Hum Mol Genet. 2012;21(11):2538–2547. doi: 10.1093/hmg/dds072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Wang W, Yin J, Ma X, Zhao F, Siedlak SL, Wang Z, et al. Inhibition of mitochondrial fragmentation protects against Alzheimer’s disease in rodent model. Hum Mol Genet. 2017;26(21):4118–4131. doi: 10.1093/hmg/ddx299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Park J, Choi H, Min JS, Kim B, Lee SR, Yun JW, et al. Loss of mitofusin 2 links beta-amyloid-mediated mitochondrial fragmentation and Cdk5-induced oxidative stress in neuron cells. J Neurochem. 2015;132(6):687–702. doi: 10.1111/jnc.12984. [DOI] [PubMed] [Google Scholar]
- 111.Kim DI, Lee KH, Gabr AA, Choi GE, Kim JS, Ko SH, et al. Abeta-induced Drp1 phosphorylation through Akt activation promotes excessive mitochondrial fission leading to neuronal apoptosis. Biochim Biophys Acta. 2016;1863(11):2820–2834. doi: 10.1016/j.bbamcr.2016.09.003. [DOI] [PubMed] [Google Scholar]
- 112.Kim B, Park J, Chang KT, Lee DS. Peroxiredoxin 5 prevents amyloid-beta oligomer-induced neuronal cell death by inhibiting ERK-Drp1-mediated mitochondrial fragmentation. Free Radic Biol Med. 2016;90:184–194. doi: 10.1016/j.freeradbiomed.2015.11.015. [DOI] [PubMed] [Google Scholar]
- 113.Yan J, Liu XH, Han MZ, Wang YM, Sun XL, Yu N, et al. Blockage of GSK3beta-mediated Drp1 phosphorylation provides neuroprotection in neuronal and mouse models of Alzheimer’s disease. Neurobiol Aging. 2015;36(1):211–227. doi: 10.1016/j.neurobiolaging.2014.08.005. [DOI] [PubMed] [Google Scholar]
- 114.Bossy B, Petrilli A, Klinglmayr E, Chen J, Lutz-Meindl U, Knott AB, et al. S-Nitrosylation of DRP1 does not affect enzymatic activity and is not specific to Alzheimer's disease. J Alzheimers Dis. 2010;20(Suppl 2):S513–S526. doi: 10.3233/JAD-2010-100552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Iijima-Ando K, Hearn SA, Shenton C, Gatt A, Zhao L, Iijima K. Mitochondrial mislocalization underlies Abeta42-induced neuronal dysfunction in a Drosophila model of Alzheimer’s disease. PLoS One. 2009;4(12):e8310. doi: 10.1371/journal.pone.0008310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Zhao XL, Wang WA, Tan JX, Huang JK, Zhang X, Zhang BZ, et al. Expression of beta-amyloid induced age-dependent presynaptic and axonal changes in Drosophila. J Neurosci. 2010;30(4):1512–1522. doi: 10.1523/JNEUROSCI.3699-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Trushina E, Nemutlu E, Zhang S, Christensen T, Camp J, Mesa J, et al. Defects in mitochondrial dynamics and metabolomic signatures of evolving energetic stress in mouse models of familial Alzheimer’s disease. PLoS One. 2012;7(2):e32737. doi: 10.1371/journal.pone.0032737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Xie H, Guan J, Borrelli LA, Xu J, Serrano-Pozo A, Bacskai BJ. Mitochondrial alterations near amyloid plaques in an Alzheimer’s disease mouse model. J Neurosci. 2013;33(43):17042–17051. doi: 10.1523/JNEUROSCI.1836-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Jiang S, Nandy P, Wang W, Ma X, Hsia J, Wang C, et al. Mfn2 ablation causes an oxidative stress response and eventual neuronal death in the hippocampus and cortex. Mol Neurodegener. 2018;13(1):5. doi: 10.1186/s13024-018-0238-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Han S, Nandy P, Austria Q, Siedlak SL, Torres S, Fujioka H, et al. Mfn2 ablation in the adult mouse hippocampus and cortex causes neuronal death. Cells. 2020;9(1):E116. doi: 10.3390/cells9010116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Gan X, Huang S, Wu L, Wang Y, Hu G, Li G, et al. Inhibition of ERK-DLP1 signaling and mitochondrial division alleviates mitochondrial dysfunction in Alzheimer's disease cybrid cell. Biochim Biophys Acta. 2014;1842(2):220–231. doi: 10.1016/j.bbadis.2013.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Baek SH, Park SJ, Jeong JI, Kim SH, Han J, Kyung JW, et al. Inhibition of Drp1 ameliorates synaptic depression, Abeta deposition, and cognitive impairment in an Alzheimer’s disease model. J Neurosci. 2017;37(20):5099–5110. doi: 10.1523/JNEUROSCI.2385-16.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Cassidy-Stone A, Chipuk JE, Ingerman E, Song C, Yoo C, Kuwana T, et al. Chemical inhibition of the mitochondrial division dynamin reveals its role in Bax/Bak-dependent mitochondrial outer membrane permeabilization. Dev Cell. 2008;14(2):193–204. doi: 10.1016/j.devcel.2007.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Bordt EA, Clerc P, Roelofs BA, Saladino AJ, Tretter L, Adam-Vizi V, et al. The putative Drp1 inhibitor mdivi-1 is a reversible mitochondrial complex I inhibitor that modulates reactive oxygen species. Dev Cell. 2017;40(6):583–94.e6. doi: 10.1016/j.devcel.2017.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Smith G, Gallo G. To mdivi-1 or not to mdivi-1: is that the question? Dev Neurobiol. 2017;77(11):1260–1268. doi: 10.1002/dneu.22519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Manczak M, Kandimalla R, Fry D, Sesaki H, Reddy PH. Protective effects of reduced dynamin-related protein 1 against amyloid beta-induced mitochondrial dysfunction and synaptic damage in Alzheimer’s disease. Hum Mol Genet. 2016;25(23):5148–5166. doi: 10.1093/hmg/ddw330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Li Z, Okamoto K, Hayashi Y, Sheng M. The importance of dendritic mitochondria in the morphogenesis and plasticity of spines and synapses. Cell. 2004;119(6):873–887. doi: 10.1016/j.cell.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 128.Wakabayashi J, Zhang Z, Wakabayashi N, Tamura Y, Fukaya M, Kensler TW, et al. The dynamin-related GTPase Drp1 is required for embryonic and brain development in mice. J Cell Biol. 2009;186(6):805–816. doi: 10.1083/jcb.200903065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Zhao F, Wang W, Wang C, Siedlak SL, Fujioka H, Tang B, et al. Mfn2 protects dopaminergic neurons exposed to paraquat both in vitro and in vivo: implications for idiopathic Parkinson’s disease. Biochim Biophys Acta Mol basis Dis. 2017;1863(6):1359–1370. doi: 10.1016/j.bbadis.2017.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Pickett EK, Rose J, McCrory C, McKenzie CA, King D, Smith C, et al. Region-specific depletion of synaptic mitochondria in the brains of patients with Alzheimer's disease. Acta Neuropathol. 2018;136(5):747–757. doi: 10.1007/s00401-018-1903-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Lin MY, Cheng XT, Xie Y, Cai Q, Sheng ZH. Removing dysfunctional mitochondria from axons independent of mitophagy under pathophysiological conditions. Autophagy. 2017;13(10):1792–1794. doi: 10.1080/15548627.2017.1356552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Sheng ZH, Cai Q. Mitochondrial transport in neurons: impact on synaptic homeostasis and neurodegeneration. Nat Rev Neurosci. 2012;13(2):77–93. doi: 10.1038/nrn3156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Cagin U, Duncan OF, Gatt AP, Dionne MS, Sweeney ST, Bateman JM. Mitochondrial retrograde signaling regulates neuronal function. Proc Natl Acad Sci U S A. 2015;112(44):E6000–E6009. doi: 10.1073/pnas.1505036112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Pigino G, Morfini G, Pelsman A, Mattson MP, Brady ST, Busciglio J. Alzheimer’s presenilin 1 mutations impair kinesin-based axonal transport. J Neurosci. 2003;23(11):4499–4508. doi: 10.1523/JNEUROSCI.23-11-04499.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Calkins MJ, Manczak M, Mao P, Shirendeb U, Reddy PH. Impaired mitochondrial biogenesis, defective axonal transport of mitochondria, abnormal mitochondrial dynamics and synaptic degeneration in a mouse model of Alzheimer’s disease. Hum Mol Genet. 2011;20(23):4515–4529. doi: 10.1093/hmg/ddr381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Wang X, Perry G, Smith MA, Zhu X. Amyloid-beta-derived diffusible ligands cause impaired axonal transport of mitochondria in neurons. Neurodegener Dis. 2010;7(1–3):56–59. doi: 10.1159/000283484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Du H, Guo L, Yan S, Sosunov AA, McKhann GM, Yan SS. Early deficits in synaptic mitochondria in an Alzheimer’s disease mouse model. Proc Natl Acad Sci U S A. 2010;107(43):18670–18675. doi: 10.1073/pnas.1006586107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Calkins MJ, Reddy PH. Amyloid beta impairs mitochondrial anterograde transport and degenerates synapses in Alzheimer’s disease neurons. Biochim Biophys Acta. 2011;1812(4):507–513. doi: 10.1016/j.bbadis.2011.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Guo L, Du H, Yan S, Wu X, McKhann GM, Chen JX, et al. Cyclophilin D deficiency rescues axonal mitochondrial transport in Alzheimer’s neurons. PLoS One. 2013;8(1):e54914. doi: 10.1371/journal.pone.0054914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Shaw JL, Chang KT. Nebula/DSCR1 upregulation delays neurodegeneration and protects against APP-induced axonal transport defects by restoring calcineurin and GSK-3beta signaling. PLoS Genet. 2013;9(9):e1003792. doi: 10.1371/journal.pgen.1003792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Wang Q, Tian J, Chen H, Du H, Guo L. Amyloid beta-mediated KIF5A deficiency disrupts anterograde axonal mitochondrial movement. Neurobiol Dis. 2019;127:410–418. doi: 10.1016/j.nbd.2019.03.021. [DOI] [PubMed] [Google Scholar]
- 142.Tammineni P, Ye X, Feng T, Aikal D, Cai Q. Impaired retrograde transport of axonal autophagosomes contributes to autophagic stress in Alzheimer’s disease neurons. Elife. 2017;6:e21776. doi: 10.7554/eLife.21776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Kim C, Choi H, Jung ES, Lee W, Oh S, Jeon NL, et al. HDAC6 inhibitor blocks amyloid beta-induced impairment of mitochondrial transport in hippocampal neurons. PLoS One. 2012;7(8):e42983. doi: 10.1371/journal.pone.0042983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Choi H, Kim HJ, Kim J, Kim S, Yang J, Lee W, et al. Increased acetylation of Peroxiredoxin1 by HDAC6 inhibition leads to recovery of Abeta-induced impaired axonal transport. Mol Neurodegener. 2017;12(1):23. doi: 10.1186/s13024-017-0164-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Misko A, Jiang S, Wegorzewska I, Milbrandt J, Baloh RH. Mitofusin 2 is necessary for transport of axonal mitochondria and interacts with the Miro/Milton complex. J Neurosci. 2010;30(12):4232–4240. doi: 10.1523/JNEUROSCI.6248-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Dixit R, Ross JL, Goldman YE, Holzbaur EL. Differential regulation of dynein and kinesin motor proteins by tau. Science. 2008;319(5866):1086–1089. doi: 10.1126/science.1152993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Ebneth A, Godemann R, Stamer K, Illenberger S, Trinczek B, Mandelkow E. Overexpression of tau protein inhibits kinesin-dependent trafficking of vesicles, mitochondria, and endoplasmic reticulum: implications for Alzheimer’s disease. J Cell Biol. 1998;143(3):777–794. doi: 10.1083/jcb.143.3.777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Shahpasand K, Uemura I, Saito T, Asano T, Hata K, Shibata K, et al. Regulation of mitochondrial transport and inter-microtubule spacing by tau phosphorylation at the sites hyperphosphorylated in Alzheimer’s disease. J Neurosc. 2012;32(7):2430–2441. doi: 10.1523/JNEUROSCI.5927-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Gilley J, Seereeram A, Ando K, Mosely S, Andrews S, Kerschensteiner M, et al. Age-dependent axonal transport and locomotor changes and tau hypophosphorylation in a “P301L” tau knockin mouse. Neurobiol Aging. 2012;33(3):621.e1–621e15. doi: 10.1016/j.neurobiolaging.2011.02.014. [DOI] [PubMed] [Google Scholar]
- 150.Kopeikina KJ, Carlson GA, Pitstick R, Ludvigson AE, Peters A, Luebke JI, et al. Tau accumulation causes mitochondrial distribution deficits in neurons in a mouse model of tauopathy and in human Alzheimer’s disease brain. Am J Pathol. 2011;179(4):2071–2082. doi: 10.1016/j.ajpath.2011.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Wee M, Chegini F, Power JHT, Majd S. Tau positive neurons show marked mitochondrial loss and nuclear degradation in Alzheimer’s disease. Curr Alzheimer Res. 2018;15(10):928–937. doi: 10.2174/1567205015666180613115644. [DOI] [PubMed] [Google Scholar]
- 152.Quintanilla RA, Dolan PJ, Jin YN, Johnson GV. Truncated tau and Abeta cooperatively impair mitochondria in primary neurons. Neurobiol Aging. 2012;33(3):619.e25–619.e35. doi: 10.1016/j.neurobiolaging.2011.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Vossel KA, Xu JC, Fomenko V, Miyamoto T, Suberbielle E, Knox JA, et al. Tau reduction prevents Abeta-induced axonal transport deficits by blocking activation of GSK3beta. J Cell Biol. 2015;209(3):419–433. doi: 10.1083/jcb.201407065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Rui Y, Tiwari P, Xie Z, Zheng JQ. Acute impairment of mitochondrial trafficking by beta-amyloid peptides in hippocampal neurons. J Neurosci. 2006;26(41):10480–10487. doi: 10.1523/JNEUROSCI.3231-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Cai Q, Sheng ZH. Mitochondrial transport and docking in axons. Exp Neurol. 2009;218(2):257–267. doi: 10.1016/j.expneurol.2009.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Hallberg BM, Larsson NG. Making proteins in the powerhouse. Cell Metab. 2014;20(2):226–240. doi: 10.1016/j.cmet.2014.07.001. [DOI] [PubMed] [Google Scholar]
- 157.Ventura-Clapier R, Garnier A, Veksler V. Transcriptional control of mitochondrial biogenesis: the central role of PGC-1alpha. Cardiovasc Res. 2008;79(2):208–217. doi: 10.1093/cvr/cvn098. [DOI] [PubMed] [Google Scholar]
- 158.Choi HI, Kim HJ, Park JS, Kim IJ, Bae EH, Ma SK, et al. PGC-1alpha attenuates hydrogen peroxide-induced apoptotic cell death by upregulating Nrf-2 via GSK3beta inactivation mediated by activated p38 in HK-2 cells. Sci Rep. 2017;7(1):4319. doi: 10.1038/s41598-017-04593-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Qin W, Haroutunian V, Katsel P, Cardozo CP, Ho L, Buxbaum JD, et al. PGC-1alpha expression decreases in the Alzheimer disease brain as a function of dementia. Arch Neurol. 2009;66(3):352–361. doi: 10.1001/archneurol.2008.588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Rice AC, Keeney PM, Algarzae NK, Ladd AC, Thomas RR, Bennett JP., Jr Mitochondrial DNA copy numbers in pyramidal neurons are decreased and mitochondrial biogenesis transcriptome signaling is disrupted in Alzheimer’s disease hippocampi. J Alzheimers Dis. 2014;40(2):319–330. doi: 10.3233/JAD-131715. [DOI] [PubMed] [Google Scholar]
- 161.Sajan M, Hansen B, Ivey R, 3rd, Sajan J, Ari C, Song S, et al. Brain insulin signaling is increased in insulin-resistant states and decreases in FOXOs and PGC-1alpha and increases in Abeta1-40/42 and phospho-tau may abet Alzheimer development. Diabetes. 2016;65(7):1892–1903. doi: 10.2337/db15-1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Sheng B, Wang X, Su B, Lee HG, Casadesus G, Perry G, et al. Impaired mitochondrial biogenesis contributes to mitochondrial dysfunction in Alzheimer’s disease. J Neurochem. 2012;120(3):419–429. doi: 10.1111/j.1471-4159.2011.07581.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Robinson A, Grosgen S, Mett J, Zimmer VC, Haupenthal VJ, Hundsdorfer B, et al. Upregulation of PGC-1alpha expression by Alzheimer’s disease-associated pathway: presenilin 1/amyloid precursor protein (APP)/intracellular domain of APP. Aging Cell. 2014;13(2):263–272. doi: 10.1111/acel.12183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Katsouri L, Parr C, Bogdanovic N, Willem M, Sastre M. PPARgamma co-activator-1alpha (PGC-1alpha) reduces amyloid-beta generation through a PPARgamma-dependent mechanism. J Alzheimers Dis. 2011;25(1):151–162. doi: 10.3233/JAD-2011-101356. [DOI] [PubMed] [Google Scholar]
- 165.Wang R, Li JJ, Diao S, Kwak YD, Liu L, Zhi L, et al. Metabolic stress modulates Alzheimer’s beta-secretase gene transcription via SIRT1-PPARgamma-PGC-1 in neurons. Cell Metab. 2013;17(5):685–694. doi: 10.1016/j.cmet.2013.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Katsouri L, Lim YM, Blondrath K, Eleftheriadou I, Lombardero L, Birch AM, et al. PPARgamma-coactivator-1alpha gene transfer reduces neuronal loss and amyloid-beta generation by reducing beta-secretase in an Alzheimer's disease model. Proc Natl Acad Sci U S A. 2016;113(43):12292–12297. doi: 10.1073/pnas.1606171113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Dumont M, Stack C, Elipenahli C, Jainuddin S, Launay N, Gerges M, et al. PGC-1alpha overexpression exacerbates beta-amyloid and tau deposition in a transgenic mouse model of Alzheimer’s disease. FASEB J. 2014;28(4):1745–1755. doi: 10.1096/fj.13-236331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Russell LK, Mansfield CM, Lehman JJ, Kovacs A, Courtois M, Saffitz JE, et al. Cardiac-specific induction of the transcriptional coactivator peroxisome proliferator-activated receptor gamma coactivator-1alpha promotes mitochondrial biogenesis and reversible cardiomyopathy in a developmental stage-dependent manner. Circ Res. 2004;94(4):525–533. doi: 10.1161/01.RES.0000117088.36577.EB. [DOI] [PubMed] [Google Scholar]
- 169.Tsunemi T, La Spada AR. PGC-1alpha at the intersection of bioenergetics regulation and neuron function: from Huntington’s disease to Parkinson’s disease and beyond. Prog Neurobiol. 2012;97(2):142–151. doi: 10.1016/j.pneurobio.2011.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Dumont M, Stack C, Elipenahli C, Jainuddin S, Gerges M, Starkova N, et al. Bezafibrate administration improves behavioral deficits and tau pathology in P301S mice. Hum Mol Genet. 2012;21(23):5091–5105. doi: 10.1093/hmg/dds355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Gong B, Pan Y, Vempati P, Zhao W, Knable L, Ho L, et al. Nicotinamide riboside restores cognition through an upregulation of proliferator-activated receptor-gamma coactivator 1alpha regulated beta-secretase 1 degradation and mitochondrial gene expression in Alzheimer’s mouse models. Neurobiol Aging. 2013;34(6):1581–1588. doi: 10.1016/j.neurobiolaging.2012.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Song C, Li M, Xu L, Shen Y, Yang H, Ding M, et al. Mitochondrial biogenesis mediated by melatonin in an APPswe/PS1dE9 transgenic mice model. Neuroreport. 2018;29(18):1517–1524. doi: 10.1097/WNR.0000000000001139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Oka S, Leon J, Sakumi K, Ide T, Kang D, LaFerla FM, et al. Human mitochondrial transcriptional factor a breaks the mitochondria-mediated vicious cycle in Alzheimer’s disease. Sci Rep. 2016;6:37889. doi: 10.1038/srep37889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Harbauer AB, Zahedi RP, Sickmann A, Pfanner N, Meisinger C. The protein import machinery of mitochondria-a regulatory hub in metabolism, stress, and disease. Cell Metab. 2014;19(3):357–372. doi: 10.1016/j.cmet.2014.01.010. [DOI] [PubMed] [Google Scholar]
- 175.Wiedemann N, Pfanner N. Mitochondrial machineries for protein import and assembly. Annu Rev Biochem. 2017;86:685–714. doi: 10.1146/annurev-biochem-060815-014352. [DOI] [PubMed] [Google Scholar]
- 176.Roses AD, Lutz MW, Amrine-Madsen H, Saunders AM, Crenshaw DG, Sundseth SS, et al. A TOMM40 variable-length polymorphism predicts the age of late-onset Alzheimer’s disease. Pharmacogenomics J. 2010;10(5):375–384. doi: 10.1038/tpj.2009.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Chiba-Falek O, Gottschalk WK, Lutz MW. The effects of the TOMM40 poly-T alleles on Alzheimer’s disease phenotypes. Alzheimers Dement. 2018;14(5):692–698. doi: 10.1016/j.jalz.2018.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Wright G, Terada K, Yano M, Sergeev I, Mori M. Oxidative stress inhibits the mitochondrial import of preproteins and leads to their degradation. Exp Cell Res. 2001;263(1):107–117. doi: 10.1006/excr.2000.5096. [DOI] [PubMed] [Google Scholar]
- 179.Chai YL, Xing H, Chong JR, Francis PT, Ballard CG, Chen CP, et al. Mitochondrial translocase of the outer membrane alterations may underlie dysfunctional oxidative phosphorylation in Alzheimer’s disease. J Alzheimers Dis. 2018;61(2):793–801. doi: 10.3233/JAD-170613. [DOI] [PubMed] [Google Scholar]
- 180.Devi L, Prabhu BM, Galati DF, Avadhani NG, Anandatheerthavarada HK. Accumulation of amyloid precursor protein in the mitochondrial import channels of human Alzheimer’s disease brain is associated with mitochondrial dysfunction. J Neurosci. 2006;26(35):9057–9068. doi: 10.1523/JNEUROSCI.1469-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Hansson Petersen CA, Alikhani N, Behbahani H, Wiehager B, Pavlov PF, Alafuzoff I, et al. The amyloid beta-peptide is imported into mitochondria via the TOM import machinery and localized to mitochondrial cristae. Proc Natl Acad Sci U S A. 2008;105(35):13145–13150. doi: 10.1073/pnas.0806192105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Pinho CM, Teixeira PF, Glaser E. Mitochondrial import and degradation of amyloid-beta peptide. Biochim Biophys Acta. 2014;1837(7):1069–1074. doi: 10.1016/j.bbabio.2014.02.007. [DOI] [PubMed] [Google Scholar]
- 183.Anandatheerthavarada HK, Biswas G, Robin MA, Avadhani NG. Mitochondrial targeting and a novel transmembrane arrest of Alzheimer’s amyloid precursor protein impairs mitochondrial function in neuronal cells. J Cell Biol. 2003;161(1):41–54. doi: 10.1083/jcb.200207030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Cenini G, Rub C, Bruderek M, Voos W. Amyloid beta-peptides interfere with mitochondrial preprotein import competence by a coaggregation process. Mol Biol Cell. 2016;27(21):3257–3272. doi: 10.1091/mbc.E16-05-0313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Du H, Guo L, Yan SS. Synaptic mitochondrial pathology in Alzheimer's disease. Antioxid Redox Signal. 2012;16(12):1467–1475. doi: 10.1089/ars.2011.4277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Csordas G, Weaver D, Hajnoczky G. Endoplasmic reticulum-mitochondrial contactology: structure and signaling functions. Trends Cell Biol. 2018;28(7):523–540. doi: 10.1016/j.tcb.2018.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Kornmann B. The molecular hug between the ER and the mitochondria. Curr Opin Cell Biol. 2013;25(4):443–448. doi: 10.1016/j.ceb.2013.02.010. [DOI] [PubMed] [Google Scholar]
- 188.Marchi S, Patergnani S, Pinton P. The endoplasmic reticulum-mitochondria connection: one touch, multiple functions. Biochim Biophys Acta. 2014;1837(4):461–469. doi: 10.1016/j.bbabio.2013.10.015. [DOI] [PubMed] [Google Scholar]
- 189.Paillusson S, Stoica R, Gomez-Suaga P, Lau DHW, Mueller S, Miller T, et al. There’s something wrong with my MAM; the ER-mitochondria axis and neurodegenerative diseases. Trends Neurosci. 2016;39(3):146–157. doi: 10.1016/j.tins.2016.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Liu Y, Zhu X. Endoplasmic reticulum-mitochondria tethering in neurodegenerative diseases. Transl Neurodegener. 2017;6:21. doi: 10.1186/s40035-017-0092-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Area-Gomez E, de Groof AJ, Boldogh I, Bird TD, Gibson GE, Koehler CM, et al. Presenilins are enriched in endoplasmic reticulum membranes associated with mitochondria. Am J Pathol. 2009;175(5):1810–1816. doi: 10.2353/ajpath.2009.090219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Area-Gomez E, de Groof A, Bonilla E, Montesinos J, Tanji K, Boldogh I, et al. A key role for MAM in mediating mitochondrial dysfunction in Alzheimer disease. Cell Death Dis. 2018;9(3):335. doi: 10.1038/s41419-017-0215-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Del Prete D, Suski JM, Oules B, Debayle D, Gay AS, Lacas-Gervais S, et al. Localization and processing of the amyloid-beta protein precursor in mitochondria-associated membranes. J Alzheimers Dis. 2017;55(4):1549–1570. doi: 10.3233/JAD-160953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Schreiner B, Hedskog L, Wiehager B, Ankarcrona M. Amyloid-beta peptides are generated in mitochondria-associated endoplasmic reticulum membranes. J Alzheimers Dis. 2015;43(2):369–374. doi: 10.3233/JAD-132543. [DOI] [PubMed] [Google Scholar]
- 195.Hedskog L, Pinho CM, Filadi R, Ronnback A, Hertwig L, Wiehager B, et al. Modulation of the endoplasmic reticulum-mitochondria interface in Alzheimer’s disease and related models. Proc Natl Acad Sci U S A. 2013;110(19):7916–7921. doi: 10.1073/pnas.1300677110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Sarasija S, Laboy JT, Ashkavand Z, Bonner J, Tang Y, Norman KR. Presenilin mutations deregulate mitochondrial ca (2+) homeostasis and metabolic activity causing neurodegeneration in Caenorhabditis elegans. Elife. 2018;7:e33052. doi: 10.7554/eLife.33052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Volgyi K, Badics K, Sialana FJ, Gulyassy P, Udvari EB, Kis V, et al. Early presymptomatic changes in the proteome of mitochondria-associated membrane in the APP/PS1 mouse model of Alzheimer’s disease. Mol Neurobiol. 2018;55(10):7839–7857. doi: 10.1007/s12035-018-0955-6. [DOI] [PubMed] [Google Scholar]
- 198.Area-Gomez E, Del Carmen Lara Castillo M, Tambini MD, Guardia-Laguarta C, de Groof AJ, Madra M, et al. Upregulated function of mitochondria-associated ER membranes in Alzheimer disease. EMBO J. 2012;31(21):4106–4123. doi: 10.1038/emboj.2012.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Pera M, Larrea D, Guardia-Laguarta C, Montesinos J, Velasco KR, Agrawal RR, et al. Increased localization of APP-C99 in mitochondria-associated ER membranes causes mitochondrial dysfunction in Alzheimer disease. EMBO J. 2017;36(22):3356–3371. doi: 10.15252/embj.201796797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Tambini MD, Pera M, Kanter E, Yang H, Guardia-Laguarta C, Holtzman D, et al. ApoE4 upregulates the activity of mitochondria-associated ER membranes. EMBO Rep. 2016;17(1):27–36. doi: 10.15252/embr.201540614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Barbero-Camps E, Fernandez A, Baulies A, Martinez L, Fernandez-Checa JC, Colell A. Endoplasmic reticulum stress mediates amyloid beta neurotoxicity via mitochondrial cholesterol trafficking. Am J Pathol. 2014;184(7):2066–2081. doi: 10.1016/j.ajpath.2014.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Sepulveda-Falla D, Barrera-Ocampo A, Hagel C, Korwitz A, Vinueza-Veloz MF, Zhou K, et al. Familial Alzheimer’s disease-associated presenilin-1 alters cerebellar activity and calcium homeostasis. J Clin Invest. 2014;124(4):1552–1567. doi: 10.1172/JCI66407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Zampese E, Fasolato C, Kipanyula MJ, Bortolozzi M, Pozzan T, Pizzo P. Presenilin 2 modulates endoplasmic reticulum (ER)-mitochondria interactions and Ca2+ cross-talk. Proc Natl Acad Sci U S A. 2011;108(7):2777–2782. doi: 10.1073/pnas.1100735108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Gautier CA, Erpapazoglou Z, Mouton-Liger F, Muriel MP, Cormier F, Bigou S, et al. The endoplasmic reticulum-mitochondria interface is perturbed in PARK2 knockout mice and patients with PARK2 mutations. Hum Mol Genet. 2016;25(14):2972–2984. doi: 10.1093/hmg/ddw148. [DOI] [PubMed] [Google Scholar]
- 205.Guardia-Laguarta C, Area-Gomez E, Rub C, Liu Y, Magrane J, Becker D, et al. Alpha-Synuclein is localized to mitochondria-associated ER membranes. J Neurosci. 2014;34(1):249–259. doi: 10.1523/JNEUROSCI.2507-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Liu Y, Ma X, Fujioka H, Liu J, Chen S, Zhu X. DJ-1 regulates the integrity and function of ER-mitochondria association through interaction with IP3R3-Grp75-VDAC1. Proc Natl Acad Sci U S A. 2019;116(50):25322–25328. doi: 10.1073/pnas.1906565116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Nguyen TN, Padman BS, Lazarou M. Deciphering the molecular signals of PINK1/Parkin mitophagy. Trends Cell Biol. 2016;26(10):733–744. doi: 10.1016/j.tcb.2016.05.008. [DOI] [PubMed] [Google Scholar]
- 208.Fischer F, Hamann A, Osiewacz HD. Mitochondrial quality control: an integrated network of pathways. Trends Biochem Sci. 2012;37(7):284–292. doi: 10.1016/j.tibs.2012.02.004. [DOI] [PubMed] [Google Scholar]
- 209.Pickrell AM, Youle RJ. The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson's disease. Neuron. 2015;85(2):257–273. doi: 10.1016/j.neuron.2014.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Moreira PI, Siedlak SL, Wang X, Santos MS, Oliveira CR, Tabaton M, et al. Autophagocytosis of mitochondria is prominent in Alzheimer disease. J Neuropathol Exp Neurol. 2007;66(6):525–532. doi: 10.1097/01.jnen.0000240476.73532.b0. [DOI] [PubMed] [Google Scholar]
- 211.Moreira PI, Siedlak SL, Wang X, Santos MS, Oliveira CR, Tabaton M, et al. Increased autophagic degradation of mitochondria in Alzheimer disease. Autophagy. 2007;3(6):614–615. doi: 10.4161/auto.4872. [DOI] [PubMed] [Google Scholar]
- 212.Nixon RA, Yang DS. Autophagy failure in Alzheimer’s disease--locating the primary defect. Neurobiol Dis. 2011;43(1):38–45. doi: 10.1016/j.nbd.2011.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Martin-Maestro P, Gargini R, Perry G, Avila J, Garcia-Escudero V. PARK2 enhancement is able to compensate mitophagy alterations found in sporadic Alzheimer’s disease. Hum Mol Genet. 2016;25(4):792–806. doi: 10.1093/hmg/ddv616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Ye X, Sun X, Starovoytov V, Cai Q. Parkin-mediated mitophagy in mutant hAPP neurons and Alzheimer’s disease patient brains. Hum Mol Genet. 2015;24(10):2938–2951. doi: 10.1093/hmg/ddv056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Fang EF, Hou Y, Palikaras K, Adriaanse BA, Kerr JS, Yang B, et al. Mitophagy inhibits amyloid-beta and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat Neurosci. 2019;22(3):401–412. doi: 10.1038/s41593-018-0332-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Martin-Maestro P, Gargini R, Sproul A, Garcia E, Anton LC, Noggle S, et al. Mitophagy failure in fibroblasts and iPSC-derived neurons of Alzheimer’s disease-associated presenilin 1 mutation. Front Mol Neurosci. 2017;10:291. doi: 10.3389/fnmol.2017.00291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Kerr JS, Adriaanse BA, Greig NH, Mattson MP, Cader MZ, Bohr VA, et al. Mitophagy and Alzheimer’s disease: cellular and molecular mechanisms. Trends Neurosci. 2017;40(3):151–166. doi: 10.1016/j.tins.2017.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Lee JH, Yu WH, Kumar A, Lee S, Mohan PS, Peterhoff CM, et al. Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell. 2010;141(7):1146–1158. doi: 10.1016/j.cell.2010.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Corsetti V, Florenzano F, Atlante A, Bobba A, Ciotti MT, Natale F, et al. NH2-truncated human tau induces deregulated mitophagy in neurons by aberrant recruitment of Parkin and UCHL-1: implications in Alzheimer’s disease. Hum Mol Genet. 2015;24(11):3058–3081. doi: 10.1093/hmg/ddv059. [DOI] [PubMed] [Google Scholar]
- 220.Hu Y, Li XC, Wang ZH, Luo Y, Zhang X, Liu XP, et al. Tau accumulation impairs mitophagy via increasing mitochondrial membrane potential and reducing mitochondrial Parkin. Oncotarget. 2016;7(14):17356–17368. doi: 10.18632/oncotarget.7861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Du F, Yu Q, Yan S, Hu G, Lue LF, Walker DG, et al. PINK1 signalling rescues amyloid pathology and mitochondrial dysfunction in Alzheimer’s disease. Brain. 2017;140(12):3233–3251. doi: 10.1093/brain/awx258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Kesharwani R, Sarmah D, Kaur H, Mounika L, Verma G, Pabbala V, et al. Interplay between Mitophagy and Inflammasomes in neurological disorders. ACS Chem Neurosci. 2019;10(5):2195–2208. doi: 10.1021/acschemneuro.9b00117. [DOI] [PubMed] [Google Scholar]
- 223.Fang EF, Scheibye-Knudsen M, Brace LE, Kassahun H, SenGupta T, Nilsen H, et al. Defective mitophagy in XPA via PARP-1 hyperactivation and NAD(+)/SIRT1 reduction. Cell. 2014;157(4):882–896. doi: 10.1016/j.cell.2014.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Fang EF. Mitophagy and NAD(+) inhibit Alzheimer disease. Autophagy. 2019;15(6):1112–1114. doi: 10.1080/15548627.2019.1596497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Voos W. Mitochondrial protein homeostasis: the cooperative roles of chaperones and proteases. Res Microbiol. 2009;160(9):718–725. doi: 10.1016/j.resmic.2009.08.003. [DOI] [PubMed] [Google Scholar]
- 226.Voos W. Chaperone-protease networks in mitochondrial protein homeostasis. Biochim Biophys Acta. 2013;1833(2):388–399. doi: 10.1016/j.bbamcr.2012.06.005. [DOI] [PubMed] [Google Scholar]
- 227.Bohovych I, Chan SS, Khalimonchuk O. Mitochondrial protein quality control: the mechanisms guarding mitochondrial health. Antioxid Redox Signal. 2015;22(12):977–994. doi: 10.1089/ars.2014.6199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Quiros PM, Langer T, Lopez-Otin C. New roles for mitochondrial proteases in health, ageing and disease. Nat Rev Mol Cell Biol. 2015;16(6):345–359. doi: 10.1038/nrm3984. [DOI] [PubMed] [Google Scholar]
- 229.Martinelli P, Rugarli EI. Emerging roles of mitochondrial proteases in neurodegeneration. Biochim Biophys Acta. 2010;1797(1):1–10. doi: 10.1016/j.bbabio.2009.07.013. [DOI] [PubMed] [Google Scholar]
- 230.Bernstein SH, Venkatesh S, Li M, Lee J, Lu B, Hilchey SP, et al. The mitochondrial ATP-dependent Lon protease: a novel target in lymphoma death mediated by the synthetic triterpenoid CDDO and its derivatives. Blood. 2012;119(14):3321–3329. doi: 10.1182/blood-2011-02-340075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Ruan Y, Li H, Zhang K, Jian F, Tang J, Song Z. Loss of Yme1L perturbates mitochondrial dynamics. Cell Death Dis. 2013;4:e896. doi: 10.1038/cddis.2013.414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Tang H, Chen Y, Liu X, Wang S, Lv Y, Wu D, et al. Downregulation of HSP60 disrupts mitochondrial proteostasis to promote tumorigenesis and progression in clear cell renal cell carcinoma. Oncotarget. 2016;7(25):38822–38834. doi: 10.18632/oncotarget.9615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Deepa SS, Bhaskaran S, Ranjit R, Qaisar R, Nair BC, Liu Y, et al. Down-regulation of the mitochondrial matrix peptidase ClpP in muscle cells causes mitochondrial dysfunction and decreases cell proliferation. Free Radic Biol Med. 2016;91:281–292. doi: 10.1016/j.freeradbiomed.2015.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Unal Gulsuner H, Gulsuner S, Mercan FN, Onat OE, Walsh T, Shahin H, et al. Mitochondrial serine protease HTRA2 p.G399S in a kindred with essential tremor and Parkinson disease. Proc Natl Acad Sci U S A. 2014;111(51):18285–18290. doi: 10.1073/pnas.1419581111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Jenkinson EM, Rehman AU, Walsh T, Clayton-Smith J, Lee K, Morell RJ, et al. Perrault syndrome is caused by recessive mutations in CLPP, encoding a mitochondrial ATP-dependent chambered protease. Am J Hum Genet. 2013;92(4):605–613. doi: 10.1016/j.ajhg.2013.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Strauss KA, Jinks RN, Puffenberger EG, Venkatesh S, Singh K, Cheng I, et al. CODAS syndrome is associated with mutations of LONP1, encoding mitochondrial AAA+ Lon protease. Am J Human Genet. 2015;96(1):121–135. doi: 10.1016/j.ajhg.2014.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Hartmann B, Wai T, Hu H, MacVicar T, Musante L, Fischer-Zirnsak B, et al. Homozygous YME1L1 mutation causes mitochondriopathy with optic atrophy and mitochondrial network fragmentation. eLife. 2016;5:e16078. doi: 10.7554/eLife.16078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Goo H-G, Jung MK, Han SS, Rhim H, Kang S. HtrA2/Omi deficiency causes damage and mutation of mitochondrial DNA. Biochim Biophys Acta. 2013;1833(8):1866–1875. doi: 10.1016/j.bbamcr.2013.03.016. [DOI] [PubMed] [Google Scholar]
- 239.Sultana R, Butterfield DA. Oxidatively modified, mitochondria-relevant brain proteins in subjects with Alzheimer disease and mild cognitive impairment. J Bioenerg Biomembr. 2009;41(5):441–446. doi: 10.1007/s10863-009-9241-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Falkevall A, Alikhani N, Bhushan S, Pavlov PF, Busch K, Johnson KA, et al. Degradation of the amyloid beta-protein by the novel mitochondrial peptidasome, PreP. J Biol Chem. 2006;281(39):29096–29104. doi: 10.1074/jbc.M602532200. [DOI] [PubMed] [Google Scholar]
- 241.Alikhani N, Guo L, Yan S, Du H, Pinho CM, Chen JX, et al. Decreased proteolytic activity of the mitochondrial amyloid-beta degrading enzyme, PreP peptidasome, in Alzheimer’s disease brain mitochondria. J Alzheimers Dis. 2011;27(1):75–87. doi: 10.3233/JAD-2011-101716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Mossmann D, Vogtle FN, Taskin AA, Teixeira PF, Ring J, Burkhart JM, et al. Amyloid-beta peptide induces mitochondrial dysfunction by inhibition of preprotein maturation. Cell Metab. 2014;20(4):662–669. doi: 10.1016/j.cmet.2014.07.024. [DOI] [PubMed] [Google Scholar]
- 243.Fang D, Wang Y, Zhang Z, Du H, Yan S, Sun Q, et al. Increased neuronal PreP activity reduces Abeta accumulation, attenuates neuroinflammation and improves mitochondrial and synaptic function in Alzheimer disease’s mouse model. Hum Mol Genet. 2015;24(18):5198–5210. doi: 10.1093/hmg/ddv241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Brunetti D, Torsvik J, Dallabona C, Teixeira P, Sztromwasser P, Fernandez-Vizarra E, et al. Defective PITRM1 mitochondrial peptidase is associated with Abeta amyloidotic neurodegeneration. EMBO Mol Med. 2016;8(3):176–190. doi: 10.15252/emmm.201505894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Westerlund M, Behbahani H, Gellhaar S, Forsell C, Belin AC, Anvret A, et al. Altered enzymatic activity and allele frequency of OMI/HTRA2 in Alzheimer’s disease. FASEB J. 2011;25(4):1345–1352. doi: 10.1096/fj.10-163402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Park HJ, Seong YM, Choi JY, Kang S, Rhim H. Alzheimer’s disease-associated amyloid beta interacts with the human serine protease HtrA2/Omi. Neurosci Lett. 2004;357(1):63–67. doi: 10.1016/j.neulet.2003.11.068. [DOI] [PubMed] [Google Scholar]
- 247.Kooistra J, Milojevic J, Melacini G, Ortega J. A new function of human HtrA2 as an amyloid-beta oligomerization inhibitor. J Alzheimers Dis. 2009;17(2):281–294. doi: 10.3233/JAD-2009-1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Kawamoto Y, Ito H, Kobayashi Y, Suzuki Y, Takahashi R. Localization of HtrA2/Omi immunoreactivity in brains affected by Alzheimer’s disease. Neuroreport. 2010;21(17):1121–1125. doi: 10.1097/WNR.0b013e328340a731. [DOI] [PubMed] [Google Scholar]
- 249.Park HJ, Kim SS, Seong YM, Kim KH, Goo HG, Yoon EJ, et al. Beta-amyloid precursor protein is a direct cleavage target of HtrA2 serine protease. Implications for the physiological function of HtrA2 in the mitochondria. J Biol Chem. 2006;281(45):34277–34287. doi: 10.1074/jbc.M603443200. [DOI] [PubMed] [Google Scholar]
- 250.Behbahani H, Pavlov PF, Wiehager B, Nishimura T, Winblad B, Ankarcrona M. Association of Omi/HtrA2 with gamma-secretase in mitochondria. Neurochem Int. 2010;57(6):668–675. doi: 10.1016/j.neuint.2010.08.004. [DOI] [PubMed] [Google Scholar]
- 251.Gupta S, Singh R, Datta P, Zhang Z, Orr C, Lu Z, et al. The C-terminal tail of presenilin regulates Omi/HtrA2 protease activity. J Biol Chem. 2004;279(44):45844–45854. doi: 10.1074/jbc.M404940200. [DOI] [PubMed] [Google Scholar]
- 252.Liu W, Duan X, Fang X, Shang W, Tong C. Mitochondrial protein import regulates cytosolic protein homeostasis and neuronal integrity. Autophagy. 2018;14(8):1293–1309. doi: 10.1080/15548627.2018.1474991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Ruan L, Zhou C, Jin E, Kucharavy A, Zhang Y, Wen Z, et al. Cytosolic proteostasis through importing of misfolded proteins into mitochondria. Nature. 2017;543(7645):443–446. doi: 10.1038/nature21695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Swerdlow RH, Bothwell R, Hutfles L, Burns JM, Reed GA. Tolerability and pharmacokinetics of oxaloacetate 100 mg capsules in Alzheimer’s subjects. BBA Clin. 2016;5:120–123. doi: 10.1016/j.bbacli.2016.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Hara Y, McKeehan N, Fillit HM. Translating the biology of aging into novel therapeutics for Alzheimer disease. Neurology. 2019;92(2):84–93. doi: 10.1212/WNL.0000000000006745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Zhang L, Zhang S, Maezawa I, Trushin S, Minhas P, Pinto M, et al. Modulation of mitochondrial complex I activity averts cognitive decline in multiple animal models of familial Alzheimer’s disease. EBioMedicine. 2015;2(4):294–305. doi: 10.1016/j.ebiom.2015.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Peralta S, Torraco A, Wenz T, Garcia S, Diaz F, Moraes CT. Partial complex I deficiency due to the CNS conditional ablation of Ndufa5 results in a mild chronic encephalopathy but no increase in oxidative damage. Hum Mol Genet. 2014;23(6):1399–1412. doi: 10.1093/hmg/ddt526. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.