

Abstract
Autoimmune regulator (AIRE) mutations result in autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED) syndrome characterized by defective central T cell tolerance and the production of many auto-antibodies targeting tissue-specific antigens and cytokines. By studying CD3- and AIRE-deficient patients, we found that lack of either T cells or AIRE function resulted in the peripheral accumulation of autoreactive mature naïve B cells. Proteomic arrays and Biacore affinity measurements revealed that unmutated antibodies expressed by these autoreactive naïve B cells recognized soluble molecules and cytokines including insulin, IL-17A, and IL-17F, which are AIRE-dependent thymic peripheral tissue antigens targeted by autoimmune responses in APECED. AIRE-deficient patients also displayed decreased frequencies of regulatory T cells (Tregs) that lacked common TCRβ clones found instead in their conventional T cell compartment, thereby suggesting holes in the Treg TCR repertoire of these patients. Hence, AIRE-mediated T cell/Treg selection normally prevents the expansion of auto-reactive naïve B cells recognizing peripheral self-antigens.
INTRODUCTION
Patients with autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED) also named autoimmune polyglandular syndrome type 1 (APS-1) commonly suffer from a classic triad of condition and symptoms, including chronic mucocutaneous candidiasis, hypoparathyroidism, and adrenal insufficiency caused by mutations in the autoimmune regulator (AIRE) gene (1, 2). Similarly, AIRE-deficient mice also exhibit APECED-like features, such as multi-organ lymphocytic infiltrations and often circulating autoantibodies against tissue antigens (3–5). AIRE ensures immunologic tolerance by controlling the selection of the T cell receptor (TCR) repertoire through the ectopic expression of peripheral tissue antigens (PTAs) in medullary thymic epithelial cells (mTECs) (3, 6), the regulation of genes associated with antigen processing and presentation (7), and the expression of chemokines that mediate the accumulation of dendritic cells in the thymic medulla (8). AIRE is therefore responsible for the deletion of autoreactive thymocytes and for directing autoreactive T cell specificities into the regulatory T cell (Treg) lineage (7, 9–11).
Although the absence of functional AIRE results in abnormal T cell compartments with autoreactive clones found in conventional T cells (Tconv) instead of the Treg compartment, B cells also play a critical role in the initiation of autoimmunity in Aire−/− mice because Aire−/− B cell-deficient mice are protected from multi-organ auto-inflammation and lack lymphocytic infiltrates in some of the organs that are targeted by Aire-less autoimmunity (12, 13). The depletion of B cells with anti-hCD20 antibody in both the nonobese diabetic (NOD) and NOD Aire−/− mice delays the onset of diabetes, inhibits inflammation, and induces Treg expansion, suggesting that B cells may play an important early role in promoting T cell activation (12,14). In addition, B cell tolerance is broken in patients with APECED as illustrated by the wide variety of autoantibodies found in the sera of these individuals and that target tissue-specific antigens such as glutamic acid decarboxylase 1 (GAD1) and GAD2 or CYP21A2 as in patients with type 1 diabetes or Addison’s disease, respectively (15,16). AIRE deficiency in humans is also associated with the development of a distinct signature of anticytokine antibodies against interferon-ω (IFN-ω), IFN-α, and interleukin-17A (IL-17A), IL-17F, and IL-22, whereas only anti-IL-17A antibodies have been reported in Aire−/− BALB/c mice (4, 17–19). These autoantibodies are often present in young AIRE-deficient individuals even before they develop other typical APECED symptoms, suggesting that the break in B cell tolerance is a primary defect of the disease and not a secondary consequence of inflammation or therapy in patients with APECED (20). Although AIRE deficiency is associated with the emergence of auto-reactive B cells and autoantibody production, the stage at which B cell tolerance is breached has not yet been investigated.
Developing B cells with autoreactive B cell receptors (BCRs) are eliminated at two discrete early B cell tolerance checkpoints. A central checkpoint in the bone marrow removes most clones expressing polyreactive and antinuclear antibodies (ANAs), whereas the peripheral B cell tolerance checkpoint prevents the accumulation of autoreactive mature naïve B cells (21). This peripheral selection step appears to be controlled by B cell extrinsic factors such as Tregs because specific defects in this checkpoint has been associated with either decreased Treg frequencies or impaired Treg suppressive function in patients with adenosine deaminase (ADA), CD40L, DOCK8, major histocompatibility complex (MHC) class II, and Wiskott-Aldrich syndrome protein (WASP) deficiency (22–25). In addition, FOXP3-deficient, immune dysregulation, polyendocrinopathy, enteropathy, and X-linked (immune dysregulation, polyendocrinopathy, enteropathy, X-linked; IPEX) patients who lack functional Tregs also display a dysfunctional peripheral B cell tolerance checkpoint (26). The abnormal selection of T cells when AIRE functions are abrogated may therefore affect the removal of autoreactive B cells in the periphery.
We report here that either the absence of T cells in CD3-deficient patients or impaired AIRE expression in patients with APECED results in increased frequencies of circulating, autoreactive, mature naïve B cells in these individuals. The dysfunctional peripheral B cell tolerance checkpoint in AIRE-deficient patients is associated with decreased numbers of Tregs lacking TCRβ sequences found instead in their Tconv compartment that is abnormally selected in the absence of functional AIRE.
RESULTS
Central B cell tolerance is functional in CD3- and AIRE-deficient patients
Central B cell tolerance, which mediates the removal of developing B cells expressing highly polyreactive and ANAs in the bone marrow, appears to be regulated by intrinsic B cell pathways and is not affected by decreased Treg frequencies or suppressive function (22–28). To determine whether this checkpoint is established independently of T cells, we tested the reactivity by enzyme-linked immunosorbent assay (ELISA) of recombinant antibodies cloned from single CD19+CD27− CD10+IgMhiCD21lo new emigrant/transitional B cells from two CD3-deficient patients lacking T cells with either homozygous CD3D or homozygous CD3E gene mutations (fig. S1A) (21). In addition, we also assessed whether impaired expression of AIRE, which regulates T cell selection through the expression of peripheral tissue antigens in the thymus, may affect central B cell tolerance by studying four AIRE-deficient patients and three asymptomatic relatives carrying a heterozygous AIRE gene mutation (table S1). Most B cell subpopulations from CD3- and AIRE-deficient and heterozygous individuals were present within normal ranges of healthy donors (HDs), except CD19+CD27−CD21−/lo B cells that were expanded in AIRE-deficient patients like in other patients with autoimmune conditions (figs. S1B and S2) (29, 30). Immunoglobulin heavy chain gene segment usage, third complementarity-determining region (CDR3) length, and positively charged amino acid content in antibodies expressed by new emigrant/transitional B cells from CD3- and AIRE-deficient patients and AIRE heterozygous carriers were similar to HD counterparts, suggesting that mutations in CD3D, CD3E, or AIRE may not affect B cell development (fig. S3). In agreement with this hypothesis, the proportions of new emigrant/transitional polyreactive and anti-nuclear B cells in both CD3-deficient patients were low and comparable with those in HDs, which demonstrates that central B cell tolerance does not require CD3+ T cells to be properly established (Fig. 1, A to C; fig. S4; and table S2). Similarly, AIRE-deficient patients and heterozygous relatives displayed normal low frequencies of polyreactive and antinuclear new emigrant/transitional B cells, revealing an efficient removal of developing autoreactive B cells in the bone marrow of these individuals (Fig. 1, A to C; fig. S4; and table S2). Together, these findings show that human central B cell tolerance is established independently of T cells and their AIRE-dependent selection.
Fig. 1. Central B cell tolerance is functional in CD3- and AIRE-deficient patients.

(A) Antibodies from new emigrant B cells from HDs (n = 12), CD3-deficient patients (CD3-def., n = 2), AIRE-deficient patients (AIRE-def., n = 4), and AIRE+/− heterozygous relatives (AIRE+/−, n = 3) were tested by ELISA for reactivity against dsDNA, insulin, and LPS. Antibodies were considered polyreactive when they recognized all three analyzed antigens. Dotted lines show ED38-positive control. Horizontal lines show cut-off OD405 for positive reactivity. For each individual, the frequency of nonpolyreactive (open area) and polyreactive (filled area) clones is summarized in pie charts, with the total number of clones tested indicated in the centers. The frequencies of polyreactive and antinuclear new emigrant/transitional B cells are summarized in (B) and (C), respectively. Each symbol represents an individual. Solid lines show the mean, and dashed lines indicate the averaged mean value for HDs.
Impaired peripheral B cell tolerance checkpoint in CD3- and AIRE-deficient patients
Tregs have been suggested to prevent the accumulation of autoreactive clones in the mature naïve B cell compartment (22–26). To determine whether T cells control the peripheral selection of B cells, we analyzed the reactivity of recombinant antibodies cloned from single CD19+CD27−CD10−IgM+CD21+ B cells, which are mostly mature naïve B cells but may also include some late transitional T3 B cells and marginal zone B cell precursors (31, 32) from CD3-deficient patients who lack T cells (21). The studied CD3-deficient patients were pretransplanted and did not show any signs of infections or inflammation when blood samples were collected. Because CD3 is solely expressed in T cells, its loss does not affect the function of all other cell types. Although immunoglobulin heavy chain repertoire analysis did not reveal significant differences between patients and controls, analysis of CD3-deficient patients showed increased frequencies of HEp-2 reactive mature naïve B cells, which represented 46.1 ± 5.6% compared with 20.5 ± 3.3% in HD counterparts. This result reveals that the absence of T cells interferes with the establishment of the peripheral B cell tolerance checkpoint (P = 0.019; Fig. 2, A and B; figs. S5 and S6A; and table S2). Peripheral B cell selection defects in CD3 deficiency were further evidenced by the elevated proportions of mature naïve B cells that expressed polyreactive antibodies compared with HDs, whereas frequencies of clones producing ANAs remained low, similar to healthy controls (Fig. 2C and fig. S6, B and C).
Fig. 2. Defective peripheral B cell tolerance checkpoint in CD3- and AIRE-deficient patients.
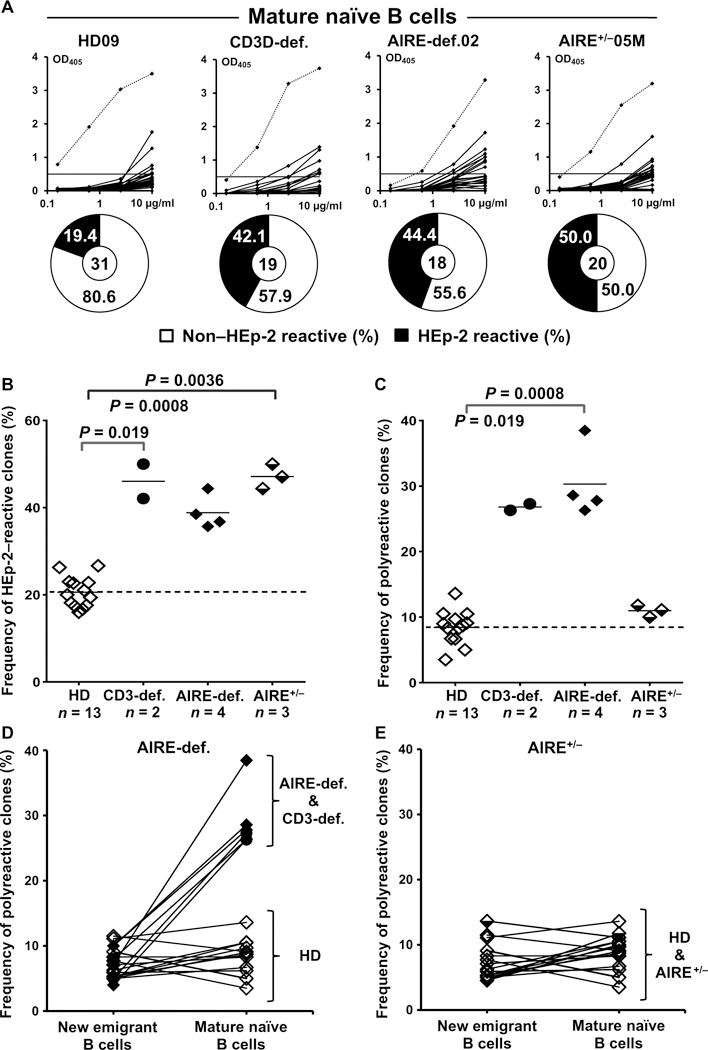
(A) Antibodies from mature naïve B cells from HDs (n = 12), CD3-deficient patients (n = 2), AIRE-deficient patients (n = 4), and AIRE+/− heterozygous relatives (n = 3) were tested by ELISA for anti-HEp-2 cell reactivity. Dotted lines show ED38-positive control. Horizontal lines show cut-off OD405 for positive reactivity. For each individual, the frequency of non-HEp-2 reactive (open area) and HEp-2-reactive (filled area) clones is summarized in pie charts, with the total number of clones tested indicated in the centers. The frequencies of HEp-2-reactive and polyreactive mature naïve B cells are summarized in (B) and (C), respectively. Each symbol represents an individual. Solid lines show the mean, and dashed line indicates the averaged mean value for HDs. The frequency of polyreactive B cells and their evolution between the new emigrant/ transitional and mature naïve B cell stages in (D) CD3- and AIRE-deficient patients and (E) AIRE+/− heterozygous relatives are shown in comparison with HDs.
We also assessed whether the lack of functional AIRE, which alters T cell selection, affects B cell selection in the periphery by analyzing the frequencies of autoreactive mature naïve B cells in AIRE-deficient patients and heterozygous relatives. We found that mature naïve B cells from both AIRE-deficient patients and heterozygous carriers often expressed HEp-2-reactive antibodies compared with control B cells, demonstrating that proper AIRE expression is required to inhibit self-reactive B cell accumulation in the periphery (AIRE-deficient: 38.9 ± 3.9%, P = 0.0008; AIRE-heterozygotes: 47.2 ± 2.8%, P = 0.0036; Fig. 2, A and B; fig. S6A; and table S2). AIRE deficiency was also associated with the reemergence of polyreactive clones during the transition between the new emigrant/transitional and mature naïve B cell stages, whereas AIRE+/− individuals exhibited low frequencies of polyreactive mature naïve B cells that were similar to those in HDs. Hence, the lack of AIRE function induces more severe defects in autoreactive B cell selection than putative partial loss in AIRE expression associated with a single functional allele (Fig. 2, C to E, and fig. S6B). These alterations in B cell reactivity in individuals with AIRE mutation(s) were not associated with obvious immunoglobulin heavy chain repertoire differences nor with the identification of clones expressing ANAs, which are also not found in the sera of patients with APECED (figs. S5 and S6C). We conclude that T cells and their AIRE-dependent selection are required to prevent the expansion of autoreactive B cells in the periphery.
Mature naïve B cells from AIRE-deficient patients show increased homeostatic expansion associated with elevated systemic cytokine concentrations
To investigate the mechanisms by which autoreactive clones were favored in the mature naïve B cell compartment in individuals with AIRE mutation(s), we first analyzed the phenotype of B cells from patients with APECED and heterozygous carriers. Reminiscent of B cells from IPEX patients, mature naïve B cells from individuals with one or two mutated AIRE alleles showed a significantly up-regulated expression of CD69, an activation marker that retards the egress of activated B cells from lymph nodes but lacked CD80 and CD86 costimulatory molecules (Fig. 3, A and B) (26). We then assessed the proliferative history of B cell subsets from patients and controls by measuring k-deleting recombination excision circles (KRECs), which provide an estimate of the number of divisions a B cell population has undergone (33). Similar to HDs, there was no expansion of transitional B cells that recently emigrated from the bone marrow in individuals with either one or two mutated AIRE alleles, further attesting to the unaltered early B cell development in these individuals (Fig. 3C). In contrast, mature naïve B cells from AIRE-deficient patients and, to a lesser extent, heterozygous carriers displayed increased homeostatic proliferation with an average of 3.5 and 2.4 divisions, respectively, compared with about 2.0 divisions in HD counterparts (Fig. 3C). The enhanced proliferation of mature naïve B cells in patients with APECED was correlated with the expansion of polyreactive clones and was associated with a specific increase in serum B cell activating factor (BAFF), an important B cell survival factor that regulates the number of peripheral B cells and may favor autoreactive B cell survival (Fig. 3D) (34, 35). In addition, patients with APECED also displayed elevated systemic plasma concentrations of IFN-γ, which promotes BAFF production (35) and IL-2, granulocyte-macrophage colony-stimulating factor (GM-CSF), IL-4, IL-5, and IL-10, TH1, and TH2 cytokines that favor B cell differentiation into plasma cells previously also identified in other patients harboring an impaired peripheral B cell tolerance checkpoint (Fig. 3E and fig. S7) (26, 36–38). Hence, the elevated plasma concentrations of cytokines favoring B cell survival and activation in patients with APECED likely promote the expansion of their autoreactive mature naïve B cells.
Fig. 3. Mature naïve B cells from AIRE-deficient patients show increased activation and homeostatic expansion associated with elevated serum BAFF and systemic cytokine concentrations. (A).
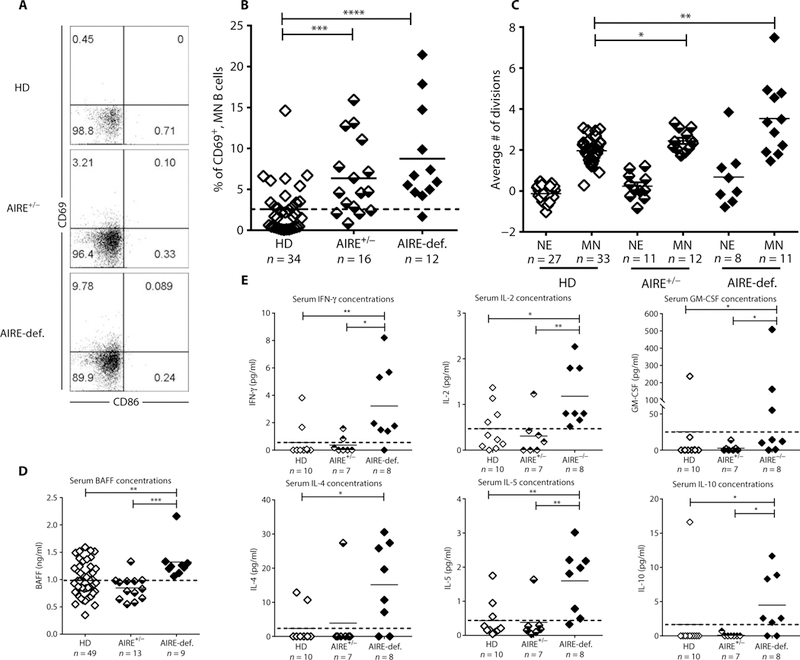
Representative CD69 and CD86 expression on CD19+CD27−CD21+CD10− mature naïve B cells from HDs, AIRE+/− heterozygous relatives, and AIRE-deficient patients. (B) Frequencies of CD69+ mature naïve B cells in indicated individuals. (C) Evaluation of the number of cell divisions undergone in vivo by KREC analysis of new emigrant (NE)/transitional and mature naïve (MN) B cells from HDs, AIRE+/− carriers, and AIRE-deficient patients. Serum concentrations of (D) BAFF and (E) IFN-γ, IL-2, GM-CSF, IL-4, IL-5, and IL-10 measured by ELISA and Luminex, respectively, in various individuals’ groups. Each symbol represents an individual in (B), (C), and (D). Solid lines show the mean, and dashed line indicates the averaged mean value for HDs. Statistically significant differences are indicated. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001.
Characterization of the antibody reactivity of naïve B cells from individuals with AIRE mutations
To further study the specificity of antibodies cloned from new emigrant/transitional and mature naïve B cells from these AIRE-deficient patients, we performed a FLEXMAP 3D Luminex multiplexing antibody assay, containing a wide array of cytokines and other self-antigens. Although central B cell tolerance was functional in HDs and individuals with AIRE mutation(s), about 10% of new emigrant/transitional B cells from these individuals recognized a large cadre of self-antigens that included actin, IFN-ω, IL-11, IL-17A, IL-17B, IL-17E, IL-17F, IL-20, IL-21, IL-28A, kallikrein-related peptidase 5 (KLK5), macrophage-derived chemokine (MDC/CCL22), matrix metallopeptidase 3 (MMP-3), threonyl-transfer RNA synthetase (PL-7), thyroid peroxidase (TPO), and tumor necrosis factor superfamily member 12/tumor necrosis factor-like weak inducer of apoptosis (TNFSF12/TWEAK) (fig. S8). These autoreactive clones often recognized more than one self-antigen on the protein array, and several were identified as polyreactive by ELISAs (Fig. 1 and figs. S4 and S9A). These data reveal that central B cell tolerance does not appear to be efficient at silencing clones, which express antibodies reacting with several soluble enzymes and cytokines that are likely only present at very low concentrations in the bone marrow (fig. S8). However, most of the clones that escaped central B cell tolerance were no longer identified in the mature naïve B cell compartment of HDs and asymptomatic AIRE+/− heterozygous carriers (Fig. 4). Hence, the peripheral B cell selection step is responsible for the counterselection of soluble self-antigens including many cytokines. In contrast, 16.7% of mature naïve B cells from AIRE-deficient patients continued to express anticytokine autoantibodies that targeted IFN-α2A, IFN-ω, IL-10, IL-11, IL-13, IL-17A, IL-17B, IL-17E, IL-17F, IL-20, IL-21, IL-28A/IFN-λ2 as well as actin, bone morphogenic protein 4 (BMP4), type VIII and X collagen, KLK5, MDC/CCL22, MMP-3, PL-7, TPO, and TNFSF12/TWEAK, further revealing an impaired peripheral B cell tolerance checkpoint in patients with APECED (Fig. 4). Notably, actin, BMP4, type VIII and X collagen, IL-17A, IL-17B, IL-17F, IL-21, KLK5, MDC/CCL22, MMP-3, and TPO are all AIRE-dependent PTAs expressed by mTECs in the thymus (table S3) (3, 39). Moreover, most of these autoreactive clones were previously identified as HEp-2 reactive and/or polyreactive by ELISAs, thereby demonstrating that traditional assays can effectively probe the functionality of the peripheral B cell tolerance checkpoint (fig. S9B). Thus, the peripheral naïve B cell selection normally prevents the accumulation of clones expressing antibodies that recognize many cytokine and other soluble molecules expressed in mTECs under the control of AIRE, further revealing that thymic T cell education plays an important role in the regulation of this peripheral naïve B cell tolerance checkpoint.
Fig. 4. Autoantigen array profiling of recombinant antibodies cloned from single-cell sorted mature naïve B cells from HDs, AIRE heterozygotes, and AIRE-deficient patients.
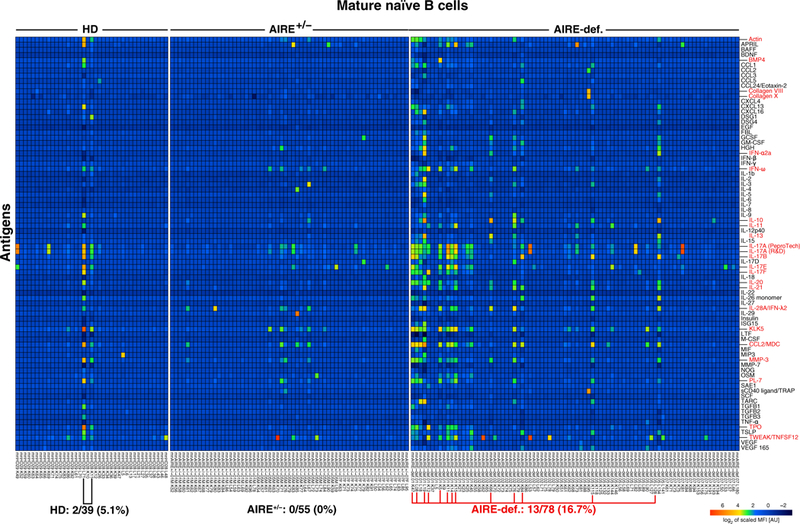
Heatmap displaying reactivity of individual mature naïve B cells from HDs (n = 2), AIRE+/− heterozygous individuals (n = 3), and AIRE-deficient patients (n = 4) against selected cytokines and other self-antigens. The frequency of self-reactive mature naïve B cells is elevated in AIRE-deficient patients compared with HDs and AIRE+/− heterozygous individuals, reflecting the accumulation of anticytokine and other autoreactive clones. MFI, mean fluorescent intensity; AU, arbitrary units.
Mature naïve B cells from AIRE-deficient patients often express unmutated autoantibodies with measurable affinity for PTAs
Candidate antibodies identified by either the autoantigen array profiling or the polyreactive and HEp-2 ELISAs were then analyzed by surface plasmon resonance (SPR) using Biacore T100 optical biosensor to evaluate their affinity to both AIRE-dependent (insulin, IL-17A, IL-17F, fibrinogen, collagen, MDC/CCL22, and KLK5) and AIRE-independent (IFN-α, IFN-ω, and TWEAK) self-antigens (39). The analysis of new emigrant/transitional B cells identified few clones that had low affinity, measurable in the micromolar range, for both AIRE-dependent (insulin, IL-17A, IL-17F, and MDC/CCL22) and AIRE-independent (TWEAK) self-antigens in patients and controls, confirming that developing autoreactive B cells targeting soluble self-antigens present at low concentrations in the bone marrow may escape central tolerance (Fig. 5, A and B; fig. S10; and tables S3 and S4) (40,41). In contrast, we identified a higher proportion of antibodies with a measurable equilibrium dissociation constant (KD) against the AIRE-dependent antigens insulin, IL-17A, and IL-17F, ranging from 10−5 to 10−7 M, cloned from the mature naïve B cells of AIRE-deficient patients compared with either HDs or asymptomatic AIRE+/− heterozygous relatives (Fig. 5, C and D, and table S4) (39). Although anti-type I IFN clones were identified by protein array in mature naïve B cells from patients with APECED, only one recombinant antibody had measurable affinity for IFN-α and IFN-ω by SPR (fig. S11B and table S4). One anti-fibrinogen clone was found in the mature naïve B cell compartment of an AIRE+/− heterozygous carrier and may reflect the altered peripheral B cell tolerance checkpoint in this individual (fig. S11A). Unexpectedly, we also identified several high-affinity anti-TWEAK antibodies, with a Kd ranging from 10−5 to 10−11 M expressed by mature naïve B cells from CD3- and AIRE-deficient patients (figs. S11B and S10 and table S4). Although the expression of TWEAK has not been previously linked with AIRE function, our observation suggests that the peripheral removal of TWEAK-reactive B cells could be affected by defects in the AIRE-mediated selection of T cells. We conclude that AIRE deficiency induces the accumulation of self-reactive B cells with enhanced affinity for AIRE-dependent antigens, which are normally counterselected by the peripheral B cell tolerance checkpoint.
Fig. 5. Elevated frequency of AIRE-dependent antigen-reactive mature naïve B cells in AIRE-deficient patients.
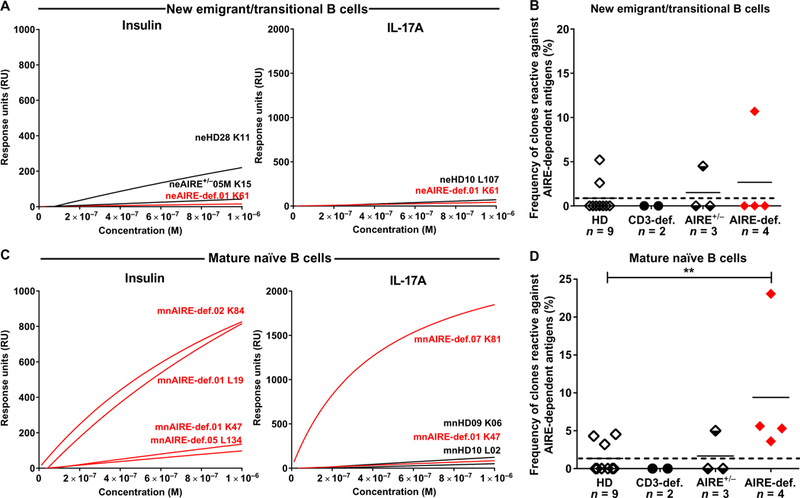
SPR analysis of binding to AIRE-dependent antigens insulin and IL-17A by recombinant antibodies cloned from single (A) new emigrant/transitional and (C) mature naïve B cells from nine HDs, two CD3-deficient patients, three AIRE+/− heterozygous individuals, and four AIRE-deficient patients. Frequencies of new emigrant/transitional and mature naïve B cells from HDs, CD3-deficient patients, AIRE+/− heterozygous individuals, and AIRE-deficient patients reactive against AIRE-dependent antigens are represented in (B) and (D), respectively. Each symbol represents an individual. **P ≤ 0.01.
Individuals with AIRE mutation(s) display decreased frequencies of Tregs with altered phenotype
Because alterations in Treg frequencies or suppressive function have been associated with specific defects of the peripheral B cell tolerance checkpoint in many patients with primary immunodeficiencies (23–26), we analyzed the Treg compartments of individuals with AIRE mutation(s). Circulating CD3+CD4+CD25hiCD127−/loFOXP3+ Treg frequencies were significantly decreased in AIRE+/− heterozygous relatives (3.79 ± 1.23%, P = 0.0016) and even lower in AIRE-deficient patients (3.26 ± 0.82%, P = 0.000014) compared with healthy controls (5.12 ± 1.47%) (Fig. 6, A and B). Similar observations were obtained with a CD3+CD4+FOXP3+Helios+ Treg gating strategy that has been useful to identify Treg when CD25 expression was altered (Fig. 6, A to C) (26). The reduction in Treg frequency was less severe in AIRE+/− individuals than in patients with APECED, suggesting an AIRE gene dosage effect on thymic Treg development potentially through differential PTA expression by mTECs (3, 42). Although decreased circulating Tregs may result in elevated frequencies of T follicular helper (TFH)-like cells (26, 36, 38), we found that TFH-like cell proportions were similar between HDs and individuals with AIRE mutation(s) (fig. S12), an observation that may correlate with normal suppressive function in Tregs both from AIRE+/− heterozygous carriers and AIRE-deficient patients (fig. S13). However, frequencies of highly suppressive human leukocyte antigen-DR+ (HLA-DR+) Tregs were decreased in the blood of AIRE-deficient patients compared with HDs and AIRE+/− heterozygous relatives (Fig. 6, D and E) (43). Additional phenotypic analysis of T cell subpopulations both from AIRE-deficient and heterozygous individuals revealed elevated proportions of circulating CD3+CD4+CD8−CD25loCD45RO+CCR6+CXCR3− T cells, which includes both TH17 and TH22 subsets; there was also a reduction in circulating CD3+CD4+CD8−CD25loCD45RO+CXCR3+CCR6− T cells, which are enriched for the TH1 subset, in these individuals compared with HDs (fig. S14). The activation of peripheral blood mononuclear cells (PBMCs) in vitro with phorbol 12-myristate 13-acetate (PMA)-ionomycin revealed significantly decreased frequencies in patients with APECED of naïve CD4+ T cells that produced IFN-γ, tumor necrosis factor-α (TNF-α), and IL-2 and expressed T-box transcription factor TBX21 (T-bet), further suggesting that TH1 differentiation is disfavored in AIRE deficiency (figs. S15 to S17). Of note, decreased IFN-γ, TNF-α, and T-bet expression was also observed in naïve CD8+ and TCRγδ+ T cells (figs. S15 and S16). Hence, AIRE-deficient patients display an altered T cell compartment characterized by decreased Treg and TH1 precursor frequencies.
Fig. 6. Altered Treg frequencies in AIRE-deficient patients and AIRE+/ heterozygous individuals.
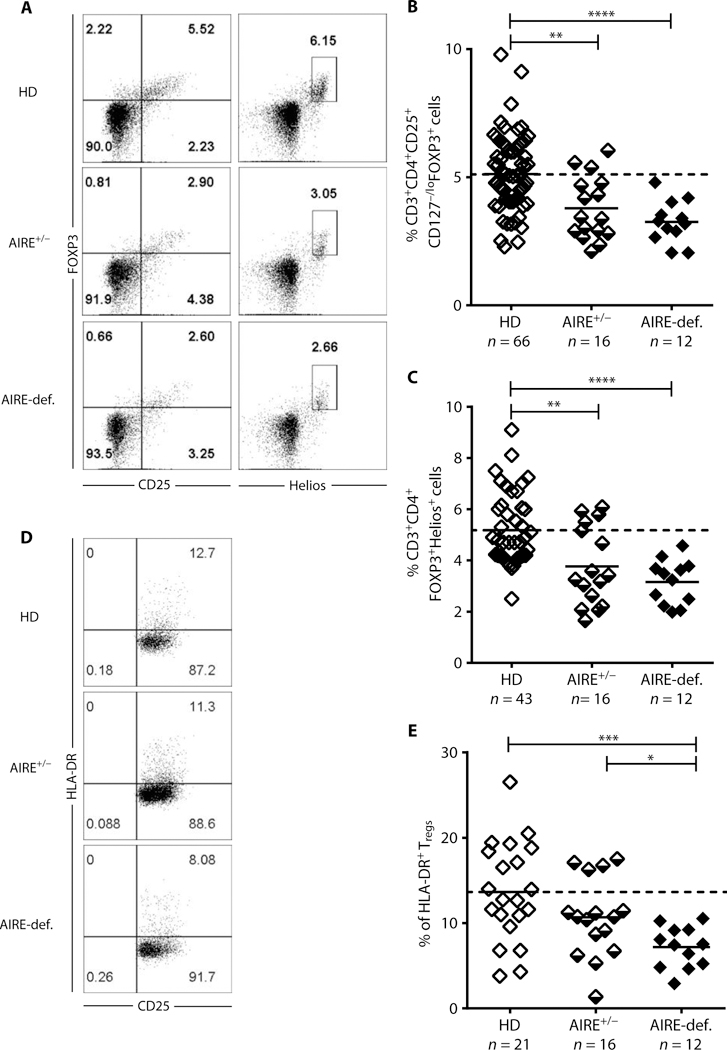
(A) Representative CD25 and FOXP3 (left) and Helios and FOXP3 (right) staining on CD3+CD4+ cells from an HD, AIRE+/− heterozygous individual, and AIRE-deficient patient. (B) CD3+CD4+CD25+CD127−/loFOXP3+ and (C) CD3+CD4+FOXP3+Helios+ Treg frequencies in HDs, AIRE+/− heterozygous individuals, and AIRE-def. patients. (D) Representative CD25 and HLA-DR staining on CD3+CD4+CD25+CD127−/lo T cells and (E) CD3+CD4+CD25+CD127loHLA-DR+ T cell frequencies in HDs, AIRE+/− heterozygous individuals, and AIRE-def. patients. Each symbol represents an individual and solid lines show the mean; dashed line indicates the averaged mean value for HDs. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001; **** P ≤ 0.0001.
Common Treg TCRBV rearrangements are diverted into the CD4+ Tconv cell compartment of AIRE-deficient patients
The absence of Aire in mice resulted in the diversion of autoreactive TCRs from the CD4+ Treg to the Tconv compartment (10). To study the impact of AIRE-dependent processes on the selection of CD4+ T cells, we analyzed the TCRβ chain [T cell receptor b variable (TCRBV)] repertoires ofCD4+CD127+ Tconv and CD4+CD25hiCD127−/lo Tregs isolated from the blood of five AIRE-deficient patients and five healthy controls using the ImmunoSEQ platform at Adaptive Biotechnologies. We obtained a total of 11.104 AIRE-deficient and 13.4.104 HD distinct in-frame TCRBV sequences for control Tconv (fig. S18A) and 5.65.104 and 9.56.104 Treg sequences from patients with APECED and HD, respectively (Fig. 7A), allowing us to survey the impact of AIRE expression on T cell repertoires. Tconv and Tregs from AIRE-deficient individuals displayed TCRBV gene segment usage, CDR3 length, and TCR diversity similar to HD counterparts (Fig. 7B and figs. S18B and S19).
Fig. 7. AIRE deficiency affects the peripheral CD4+ Treg and Tconv cell TCR repertoire.
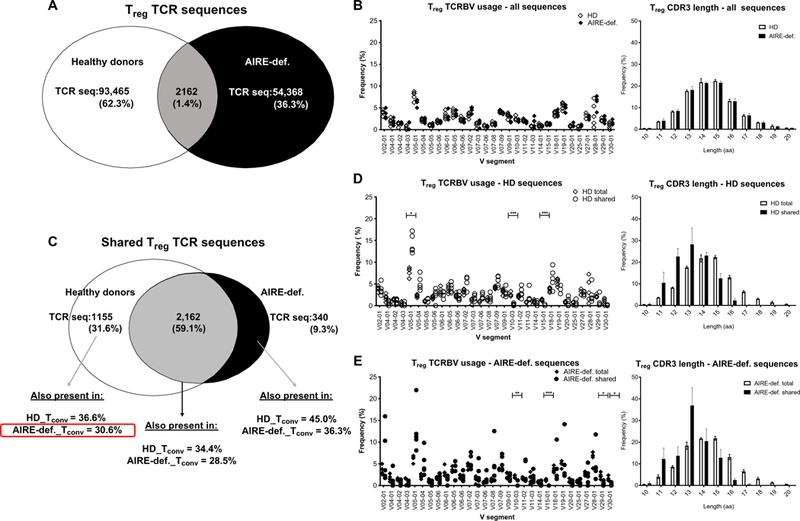
CD4+CD25hiCD127−/lo Treg and CD4+CD127+ Tconv cells were purified by fluorescence-activated cell sorting from an HD (n = 5) controls and AIRE-deficient patients (n = 5) and subjected to TCRBV sequencing. (A) Summary chart of total distinct Treg CDR3 TCR sequences obtained from pooled HD controls and AIRE-deficient patients. (B) TCRBV V gene segment usage and CDR3 length in Tregs from HDs and AIRE-deficient patients. (C) Treg TCRBV sequences common to HDs but absent in patients with APECED can be identified in the Tconv compartment of AIRE-deficient patients and HDs. Comparison of TCRBV V gene segment usage and CDR3 length in total versus shared Treg sequences in (D) HDs and (E) AIRE-deficient patients. aa, amino acids. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001.
Comparative analysis of Treg TCR representation revealed a total of 3657 Treg TCR sequences that were shared by two or more individuals. Although 2162 (59.1%) of these TCR sequences were found in Tregs both from HD and AIRE-deficient patients, we found that 1155 (31.6%) were only shared among HD Tregs, whereas only 340 (9.3%) were specific to Tregs from patients with APECED (Fig. 7C). This suggests that commonly shared Treg-biased TCRBV rearrangements are less efficiently selected in the Treg compartment in the absence of functional AIRE. In addition, out of a total of 1155 Treg TCRβ chains shared among HD, we identified 354 (30.6%) of these TCR sequences in the Tconv compartment of at least one AIRE-deficient patient, which reveals that AIRE-dependent Treg precursors expressing these TCRBV rearrangements may be diverted into the CD4+ Tconv subset of AIRE-deficient patients (Fig. 7C). Of note, 36.6% of the commonly shared Treg TCR sequences were also detected in Tconv from HDs, potentially due to alternative TCRα chains (Fig. 7C). These shared AIRE-dependent Treg clones favored TCRBV V05–01 gene segment usage, whereas TCRBV V10–03 and V15–01 were less used and displayed shorter CDR3s than those of distinct Treg sequences (Fig. 7D and fig. S20). This likely reflects the bias of identical sequences to be found within VDJ rearrangements with small nontemplate nucleotide insertions that increase CDR3 length. TCRBV sequences shared by only Tregs from patients with APECED also harbored short CDR3s and slightly different TCRBV gene usage and were found in HD Tconv, further illustrating altered TCR selection in the absence of functional AIRE (Fig. 7, C and E, and fig. S20). Together, defective AIRE-mediated T cell selection modifies the TCR repertoire of Tregs from AIRE-deficient patients.
DISCUSSION
We report that lack of T cells in severe combined immunodeficiency patients with mutations in CD3D or CD3E genes and impaired AIRE expression in AIRE-deficient patients result in the accumulation of autoreactive clones in the mature naïve B cell compartment, whereas central B cell tolerance remains functional. This T-independent regulation of central B cell tolerance was previously suggested by the analysis of Rag1−/− mice expressing a transgenic autoreactive BCRs and patients with diverse primary immunodeficiencies with altered T cell compartment or Treg suppressive function, all displaying normal low frequencies of autoreactive clones exiting the bone marrow (23, 24,26, 44). In contrast, mutations in molecules regulating BCR and potentially Toll-like receptor pathways such as Bruton’s tyrosine kinase, ADA, WASP, myeloid differentiation primary response 88, interleukin 1 receptor associated kinase 4, TNF receptor superfamily member 13B, and activation-induced cytidine deaminase (AID) affected central B cell tolerance, thereby revealing a B cell-intrinsic regulation of this checkpoint (22, 27, 28,36,45,46). Protein microarray and plasmon resonance analyses examining the reactivity of recombinant antibodies cloned from transitional B cells that recently emigrated from the bone marrow from control HDs, AIRE+/− heterozygous asymptomatic carriers, and AIRE-deficient patients revealed that self-reactive clones recognizing soluble enzymes and cytokines can escape central tolerance, which is therefore incomplete. However, anti-insulin and anticytokine clones were virtually absent from the mature naïve B cell compartment of HDs and asymptomatic AIRE+/− heterozygous carriers, revealing that the peripheral naïve B cell selection step is responsible for their elimination. Of note, few very-low-affinity anti-IL-17A mature naïve B cells were identified by Biacore in HDs, which suggests that, similarly to central tolerance, the peripheral counterselection of autoreactive B cells is also not absolute. In contrast, many mature naïve B cells isolated from AIRE-deficient patients were found to express antibodies that bound IFN-α2A, IFN-ω, IL-10, IL-11, IL-13, IL-17A, IL-17B, IL-17E, IL-17F, IL-20, IL-21, IL-28A/IFN-λ2 as well as actin, BMP4, type VIII and X collagen, KLK5, MDC/CCL22, MMP-3, PL-7, TPO, TNFSF12/TWEAK, and insulin. Because AIRE controls the expression of insulin, actin, BMP4, type VIII and X collagen, IL-17A, IL-17B, IL-17F, IL-21, KLK5, MDC/CCL22 MMP-3, and TPO in mTECs (39), PTA thymic expression may be necessary to prevent the accumulation of autoreactive naïve B cells that recognize these self-antigens in the periphery. In addition, mature naïve B cells from AIRE-deficient patients also included clones that expressed antibodies, which bound AIRE-independent self-antigens such as type I and type III IFNs, IL-10, IL-11, IL-13, IL-20, PL-7, and TWEAK. Although the mechanisms that normally prevent the accumulation of these clones are unclear, T cells are required because CD3-deficient patients displayed mature naïve B cells that expressed anti-TWEAK antibodies. Additional investigations are warranted to determine the significance of these anti-TWEAK clones and whether they neutralize or prolong the half-life of TWEAK, which in other studies, was shown to favor the production of proinflammatory cytokines including IL-17, and the development of autoimmune conditions (47).
Mature naïve B cells in AIRE-deficient patients that express un-mutated antibodies recognizing type I IFNs, IL-17A, and IL-17F likely contribute to the production of high-affinity anti–IFN-ω, anti–IFN-α, anti-IL-17A, and anti-IL-17F autoantibodies frequently detected in the serum of these individuals (17–19, 48). Because AIRE deficiency is often accompanied by fungal infections associated with increased IL-17 production by γS T cells from patients with APECED (17–19, 49), IL-17A secretion may therefore promote the activation of naïve anti-IL-17A B cells and lead to the production of high-affinity anti–IL-17A autoantibodies with the help of IL-17A–reactive Tconv cells (17–19, 48). By extension, production of type I IFN linked to autoimmune processes may also favor the activation of anti-IFN-α and anti-IFN-ω clones present in the mature naïve B cell compartment of AIRE-deficient patients. In addition, anti-cytokine autoantibodies are frequently found not only in patients with APECED but also in individual with hypomorphic RAG gene mutations that dampen the generation of thymocytes and TCR repertoire diversity (50). Because the decreased colonization of the thymus by thymocytes in patients with hypomorphic RAG mutation results in decreased AIRE expression by mTECs, the selection of the rare Tregs that develop in these patients may be altered similarly to AIRE deficiency and induce peripheral autoreactive B cell selection defects and the accumulation of naïve B cells expressing BCR with anticytokine reactivity (51). Hence, defects in early B cell tolerance checkpoints may be responsible for the production of autoreactive clones that may be activated by T cells and increase the specificity of their autoantibodies for self-antigens by acquiring somatic mutations.
How can AIRE expression normally control the peripheral selection of autoreactive naïve B cells? Previous studies of other patients with primary immunodeficiencies revealed that loss of function in molecules involved in Treg development or function interfered with the establishment of the peripheral B cell tolerance checkpoint. We previously reported that the mature naïve B cell compartment of FOXP3-deficient IPEX patients who lack functional Tregs is specifically enriched in autoreactive clones (26). Similarly, high frequencies of autoreactive mature naïve B cells were also observed in CD40L-, MHC class II-, and AID-deficient patients with decreased Treg numbers (23, 38,45); in DOCK8- and WASp-deficient patients who displayed defective Treg suppressive function (24–26); and in signaling lymphocytic activation molecule (SLAM)-associated protein-and AID-deficient patients who showed elevated serum cytokines that inhibited Treg function (37, 38). Our current analysis of CD3D- and CD3E-deficient patients, who lack virtually all T cells and showed an accumulation of autoreactive clones in their mature naïve B cell compartment, provides further evidence that T cells are key regulators of this peripheral B cell selection step. We and others also observed that AIRE mutations result in decreased Treg frequencies in patients with APECED and abrogate their peripheral B cell tolerance checkpoint, further suggesting an involvement of Tregs in the regulation of this selection step (11, 52). AIRE enforces immune tolerance via two complimentary mechanisms, i.e., eliminating self-reactive clonotypes from the Tconv lineage and diverting these cells into the FOXP3+ Treg compartment (7, 9,10,53,54). Our data studying AIRE-deficient patients are in agreement with this model because TCR sequences commonly found in the Treg compartment of HDs are missing from the Treg TCR repertoire selected in the absence of functional AIRE and are instead identified in the CD4+ Tconv subset of patients with APECED. Although these observations are currently limited to the analysis of non-HLA-matched control donors and patients, they may correspond to TCRβ sequences expressed by rare clones selected on common shared MHC II features among individuals. In addition, the absence of insulin, IL-17A, IL-17B, IL-17F, KLK5, and MDC/CCL22 PTA expression by mTECs in patients with APECED likely prevents the education of developing thymocytes into Tregs that produce TCRs specific for these molecules and result in the accumulation of mature naïve B cells expressing antibodies that bind these AIRE-dependent PTAs. Lack of AIRE in thymic B cells might also contribute to incomplete PTA expression in the thymus and abnormal Treg/Tconv differentiation (14, 55). Hence, decreased numbers and incomplete TCR repertoire of Tregs from AIRE-deficient patients may contribute to the escape of developing autoreactive mature naïve B cells with AIRE-dependent PTA reactivities. However, we cannot exclude that peripheral AIRE expression in extrathymic Aire-expressing cells and/or indirect effects of cytokine alterations may also play a role in the BCR repertoire selection of mature naïve B cells (56). It is unclear at this point whether Tregs may control the peripheral B cell tolerance checkpoint via direct interactions with autoreactive B cells as suggested (57–60) and potentially involving self-peptide presentation by MHC class II to their TCRs or whether they suppress other cell types that favor autoreactive B cell survival and which remain to be identified.
In summary, we showed that T cells and their AIRE-dependent selection normally control the peripheral selection of autoreactive B cells. However, defects in AIRE expression divert a subset of Treg-biased sequences into the Tconv compartment, creating holes in the Treg TCR repertoire of AIRE-deficient patients that are associated with the accumulation of mature naïve B cells reactive for AIRE-dependent PTAs. These autoreactive B cells may promote peripheral organ autoimmune manifestations in patients with APECED and produce autoantibodies after activation by diverted Tconv cells expressing autoreactive TCRs in the absence of specific Treg suppression.
MATERIALS AND METHODS
Patients and HD controls
We obtained peripheral blood or frozen PBMC from one CD3D-deficient patient with homozygous CD3D: c.[202C>T];[202C>T]; p.[Arg68X];[Arg68X] mutation [patient 5; (61)], one CD3E-deficient patient with homozygous CD3E: c.[173 del T];[173 del T]; p.[L58HfsX9];[L58HfsX9] mutations, 15 AIRE-deficient patients with homozygous or compound heterozygous autosomal recessive AIRE gene mutations, and 17 AIRE heterozygous relatives with a single autosomal recessive AIRE mutated allele (table S1). In addition, we collected additional samples from healthy unrelated donors. Frequencies of polyreactive and HEp-2–reactive and anti-nuclear new emigrant and mature naïve B cells from an HD were previously reported (26, 36, 37, 46) except for HD-Y541 who is a 59-year-old non-Hispanic Caucasian female (table S2).
Study design
The four AIRE-deficient patients enrolled at the time of the study for the analysis of their B cell reactivity did not receive any steroids, which could have interfered with our experiment readouts, as they did not suffer from adrenal insufficiency often associated with the disease. All samples were collected in accordance with institutional review board-reviewed protocols.
B cell staining and sorting
Peripheral B cells were purified from the blood of patients and control donors by positive selection using CD20 magnetic beads (Miltenyi Biotec). For sorting, enriched B cells were stained with FITC-anti-IgM, PE-anti-IgG, PE-Cy7-anti-CD10, APC-anti-CD21, Pacific Blue-anti-CD19, and PercP-Cy5.5-anti-CD27 (all from BioLegend) (gating strategy is shown in fig. S21). Single- or batch-sorted CD19+CD21loCD10+IgMhiCD27− new emigrant and CD19+CD21+CD10−IgM+CD27− mature naïve B cells from patients, heterozygous relatives, and HDs were sorted on a FACSAria flow cytometer (Becton Dickinson, Mountain View, CA) into 96-well polymerase chain reaction (PCR) plates or 5-ml round-bottom polystyrene test tubes, respectively. For phenotyping, enriched B cells were stained with FITC-anti-IgM, PE-anti-CD69, PE-Cy7-anti-CD10, APC-anti-CD86, PercP-Cy5.5-anti-CD27, APC-Cy7-anti-CD19 (all from BioLegend), and V450-anti-CD21 (BD Bioscience) (gating strategy is shown in fig. S22).
Complementary DNA, RT-PCR, antibody production, and purification
RNA from single cells was reverse-transcribed in the original 96-well plate in 12.5 ml reactions containing 100 U of Superscript II RT (Gibco BRL) for 45 min at 42°C. Reverse transcription (RT)–PCR reactions, primer sequences, cloning strategy, expression vectors, antibody expression, and purification were as described (21).
BCR repertoire analysis
Ig sequences and mutation status were analyzed by IgBLAST comparison with GenBank using the National Center for Biotechnology Information’s IgBLAST server (www.ncbi.nlm.nih.gov/igblast/). Heavy chain complementarity-determining region 3 was defined as the interval between amino acids at position 94 in the variable region of immunoglobulin heavy chain framework 3 and the conserved tryptophan at position 103 in JH segments. Sequences are shown in table S2.
ELISAs and IFAs
Antibody concentrations and reactivity were measured as described (21). Highly polyreactive ED38 was used as positive control in HEp-2 reactivity and polyreactivity ELISAs. Antibodies were considered polyreactive when they recognized all three analyzed antigens, which included double-stranded DNA (dsDNA), insulin, and lipopolysaccha-ride (LPS). For indirect immunofluorescence assays (IFAs), HEp-2 cell-coated slides (Bion Enterprises Ltd.) were incubated in a moist chamber at room temperature with purified recombinant antibodies at 50 to 100 μg/ml and detected with FITC-conjugated goat anti-human IgG.
KREC assay
The ratio of KREC joints (signal joint) to the Jκ-Cκ recombination genomic joints (coding joint) was determined as previously described (33). The number of cell divisions was calculated by subtracting the threshold cycle (CT) of the PCR detecting the coding joint from that detecting the signal joint.
Antigen arrays and analysis of cloned antibodies on the antigen arrays
Antigen arrays were generated, and assays on the arrays were performed as described previously (62, 63). The list of protein micro-array antigens and procedures are provided in table S5, data file S1, and Supplementary Materials and Methods.
Plasmon resonance analysis
Binding studies were performed at 25°C using a Biacore T100 optical biosensor (GE Healthcare, Biacore, Piscataway, NJ). The list of antigens and their immobilized on a CM5 research-grade sensor chip using amine coupling is provided in the Supplementary Materials and Methods. Antibodies were injected from threefold dilution series starting with either 3 or 1 μM stock using a single-cycle approach (64). The surfaces were regenerated by 30-s injections of 3 M MgCl2. The binding responses were double-referenced against the nonspecific binding to dextran alone and injections of buffer alone. Binding affinity was determined by either fitting the amplitudes observed during the steady-state phase, or kinetics of the binding reaction, to a simple 1:1 binding model using BioEvaluation software (GE Healthcare, Biacore, Piscataway, NJ).
Treg phenotype and in vitro suppression assay
Peripheral CD4+ T cells were enriched with the EasySep Human CD4+T cells kit (STEMCELL Technologies) from the blood of patients, heterozygous relatives, and control donors. Enriched CD4+ T cells were stained with the following antibodies: APC-Cy7-anti-CD4, PE-anti-CD25, PerCP-Cy5.5-anti-CD127, AlexaFluor700-anti-HLA-DR (all from BioLegend) and with AlexaFluor647-anti-Helios (BioLegend), Alexa Fluor 488-anti-FOXP3, and eV605-anti-CD3 (both from eBioscience) after fixation and permeabilization of T cells (gating strategy is shown in figs. S23 and S24). After staining enriched CD4+ T cells with APC-Cy7-anti-CD4, PE-anti-CD25, and Alexa Fluor 647-anti-CD127 (BioLegend), CD4+CD127+Tconv cells and CD4+CD25hiCD127−/lo Treg cells from patients, heterozygous relatives, and HDs were sorted using a FACSAria flow cytometer (Becton Dickinson, Mountain View, CA). CD4+CD25+CD127+ Tconv cells were labeled with CellTrace carboxyfluorescein diacetate succinimidyl ester (CFSE) (InVivogen). Cocultures of Treg and Tconv at 1:1, 1:4, 1:8, and 1:16 ratios were stimulated with the Treg Suppression Inspector Human kit (Miltenyi Biotec) at a 1 bead/1 cell ratio. Proliferation of the viable Tconv was analyzed by CFSE dilution at day 3.5.
Tconv and Treg TCRBV sequencing
Both CD4+CD127+ Tconv cells and CD4+CD25hiCD127lo/− Treg cells from patients and HDs were sorted on a FACSAria flow cytometer (Becton Dickinson, Mountain View, CA) into 5-ml round-bottom polystyrene test tubes. Dry pellets were then frozen at –80°C and sent for TCRBV sequencing at Adaptive Biotechnologies (https://clients.adaptivebiotech.com/pub/sng-2019-sciimmunol; DOI 10.21417/B7V926).
Cytokine measurements
Serum BAFF concentrations were determined by ELISA according to the manufacturer’s instruction (R&D Systems). Cytokines (GM-CSF, TNF-a, IFN-γ, IL-1B, IL-2, IL-4, IL-5, IL-6, IL-7, IL-8, IL-10, IL-12, and IL-13) in plasma were measured with the High Sensitivity Human Cytokine Magnetic Bead kit (Millipore), using a Luminex 200, according to the manufacturer’s instruction.
Statistics
Statistical analysis was performed using GraphPad Prism (version 7.03; GraphPad, San Diego, CA). Differences between groups of research individuals were analyzed for statistical significance with the Mann-Whitney U test. Multiple group comparisons were corrected for statistical significance with the Bonferroni-Dunn method. A P value of ≤0.05 was considered significant.
Supplementary Material
Acknowledgments:
We are very much indebted to the patients and their families and to J. Orange and D. Orange from the APS type 1 Foundation. We thank L. Devine and C. Wang for cell sorting and E. Folta-Stogniew for plasmon resonance analysis. Recombinant antibodies are available from E. Meffre under a material agreement with M. Lionakis (NIH).
Funding: This work was supported by NIH/NIAID grant AI-061093 to E.M. The T100 instrument is supported by grant 1S10RR026992-0110 from NIH to E.F.-S. P.J.U. was funded by a Donald E. and Delia B. Baxter Foundation Career Development Award, a gift from the Henry Gustav Floren Trust (R01 AI125197), and Stanford’s Autoimmunity Center of Excellence (ACE; U19-AI110491). The Autoimmunity Centers of Excellence is a research consortium supported by the National Institute of Allergy and Infectious disease (NIAID/NIH). J.S. was supported by the Agency for Science, Technology and Research National Science Scholarship Program. B.A. was supported by the Knut and Alice Wallenberg Foundation Postdoctoral Scholarship Program (KAW 2014.0412).
Footnotes
Competing interests: The authors declare that they have no competing interests.
Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper or in the Supplementary Materials.
REFERENCES AND NOTES
- 1.Nagamine K, Peterson P, Scott HS, Kudoh J, Minoshima S, Heino M, Krohn KJE, Lalioti MD, Mullis PE, Antonarakis SE, Kawasaki K, Asakawa S, Ito F, Shimizu N, Positional cloning of the APECED gene. Nat. Genet. 17, 393–398 (1997). [DOI] [PubMed] [Google Scholar]
- 2.Finnish-German APECED Consortium, An autoimmune disease, APECED, caused by mutations in a novel gene featuring two PHD-type zinc-finger domains. Nat. Genet. 17, 399–403 (1997). [DOI] [PubMed] [Google Scholar]
- 3.Anderson MS, Venanzi ES, Klein L, Chen Z, Berzins SP, Turley SJ, von Boehmer H, Bronson R, Dierich A, Benoist C, Mathis D, Projection of an immunological self shadow within the thymus by the aire protein. Science 298, 1395–1401 (2002). [DOI] [PubMed] [Google Scholar]
- 4.Kärner J, Meager A, Laan M, Maslovskaja J, Pihlap M, Remm A, Juronen E, Wolff ASB, Husebye ES, Podkrajsek KT, Bratanic N, Battelino T, Willcox N, Peterson P, Kisand K, Anti-cytokine autoantibodies suggest pathogenetic links with autoimmune regulator deficiency in humans and mice. Clin. Exp. Immunol. 171, 263–272 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ramsey C, Winqvist O, Puhakka L, Halonen M, Moro A, Kämpe O, Eskelin P, Pelto-Huikko M, Peltonen L, Aire deficient mice develop multiple features of APECED phenotype and show altered immune response. Hum. Mol. Genet. 11, 397–409 (2002). [DOI] [PubMed] [Google Scholar]
- 6.Derbinski J, Gäbler J, Brors B, Tierling S, Jonnakuty S, Hergenhahn M, Peltonen L, Walter J, Kyewski B, Promiscuous gene expression in thymic epithelial cells is regulated at multiple levels. J. Exp.Med. 202, 33–45 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Anderson MS, Venanzi ES, Chen Z, Berzins SP, Benoist C, Mathis D, The cellular mechanism of Aire control of T cell tolerance. Immunity 23, 227–239 (2005). [DOI] [PubMed] [Google Scholar]
- 8.Lei Y, Ripen AM, Ishimaru N, Ohigashi I, Nagasawa T, Jeker LT, Bösl MR, Holländer GA, Hayashi Y, de Waal Malefyt R, Nitta T, Takahama Y, Aire-dependent production of XCL1 mediates medullary accumulation of thymic dendritic cells and contributes to regulatory T cell development. J. Exp. Med. 208, 383–394 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.DeVoss J, Hou Y, Johannes K, Lu W, Liou GI, Rinn J, Chang H, Caspi RR, Fong L, Anderson MS, Spontaneous autoimmunity prevented by thymic expression of a single self-antigen. J. Exp.Med. 203, 2727–2735 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Malchow S, Leventhal DS, Lee V, Nishi S, Socci ND, Savage PA, Aire enforces immune tolerance by directing autoreactive T cells into the regulatory T cell lineage. Immunity 44, 1102–1113 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yang S, Fujikado N, Kolodin D, Benoist C, Mathis D, Immune tolerance. Regulatory T cells generated early in life play a distinct role in maintaining self-tolerance. Science 348, 589–594 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gavanescu I, Benoist C, Mathis D, B cells are required for Aire-deficient mice to develop multi-organ autoinflammation: A therapeutic approach for APECED patients. Proc. Natl. Acad. Sci. U.S.A. 105, 13009–13014 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Devoss JJ, Shum AK, Johannes KPA, Lu W, Krawisz AK, Wang P, Yang T, Leclair NP, Austin C, Strauss EC, Anderson MS, Effector mechanisms of the autoimmune syndrome in the murine model of autoimmune polyglandular syndrome type 1. J. Immunol. 181, 4072–4079 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hu CY, Rodriguez-Pinto D, du W, Ahuja A, Henegariu O, Wong FS, Shlomchik MJ, Wen L, Treatment with CD20-specific antibody prevents and reverses autoimmune diabetes in mice. J. Clin. Invest. 117, 3857–3867 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Peterson P, Pitkänen J, Sillanpää N, Krohn K, Autoimmune polyendocrinopathy candidiasis ectodermal dystrophy (APECED): A model disease to study molecular aspects of endocrine autoimmunity. Clin. Exp. Immunol. 135, 348–357 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Velloso LA, Winqvist O, Gustafsson J, Kämpe O, Karlsson FA, Autoantibodies against a novel 51 kDa islet antigen and glutamate decarboxylase isoforms in autoimmune polyendocrine syndrome type I. Diabetologia 37, 61–69 (1994). [DOI] [PubMed] [Google Scholar]
- 17.Kisand K, Boe Wolff AS, Podkrajsek KT, Tserel L, Link M, Kisand KV, Ersvaer E, Perheentupa J, Erichsen MM, Bratanic N, Meloni A, Cetani F, Perniola R, Ergun-Longmire B, Maclaren N, Krohn KJE, Pura M, Schalke B, Ströbel P, Leite MI, Battelino T, Husebye ES, Peterson P, Willcox N, Meager A, Chronic mucocutaneous candidiasis in APECED or thymoma patients correlates with autoimmunity to Th17-associated cytokines. J. Exp. Med. 207, 299–308 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Puel A, Döffinger R, Natividad A, Chrabieh M, Barcenas-Morales G, Picard C, Cobat A, Ouachée-Chardin M, Toulon A, Bustamante J, al-Muhsen S, al-Owain M, Arkwright PD, Costigan C, McConnell V, Cant AJ, Abinun M, Polak M, Bougnères PF, Kumararatne D, Marodi L, Nahum A, Roifman C, Blanche S, Fischer A, Bodemer C, Abel L, Lilic D, Casanova J-L, Autoantibodies against IL-17A, IL-17F, and IL-22 in patients with chronic mucocutaneous candidiasis and autoimmune polyendocrine syndrome type I. J. Exp.Med. 207, 291–297 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ferre EMN, Rose SR, Rosenzweig SD, Burbelo PD, Romito KR, Niemela JE, Rosen LB, Break TJ, Gu W, Hunsberger S, Browne SK, Hsu AP, Rampertaap S, Swamydas M, Collar AL, Kong HH, Lee CCR, Chascsa D, Simcox T, Pham A, Bondici A, Natarajan M, Monsale J, Kleiner DE, Quezado M, Alevizos I, Moutsopoulos NM, Yockey L, Frein C, Soldatos A, Calvo KR, Adjemian J, Similuk MN, Lang DM, Stone KD, Uzel G, Kopp JB, Bishop RJ, Holland SM, Olivier KN, Fleisher TA, Heller T, Winer KK, Lionakis MS, Redefined clinical features and diagnostic criteria in autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy. JCI Insight 1, e88782 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wolff ASB, Sarkadi AK, Marodi L, Kärner J, Orlova É, Oftedal BEV, Kisand K, Olah E, Meloni A, Myhre AG, Husebye ES, Motaghedi R, Perheentupa J, Peterson P, Willcox N, Meager A, Anti-cytokine autoantibodies preceding onset of autoimmune polyendocrine syndrome type I features in early childhood. J. Clin. Immunol. 33, 1341–1348 (2013). [DOI] [PubMed] [Google Scholar]
- 21.Wardemann H, Yurasov S, Schaefer A, Young JW, Meffre E, Nussenzweig MC, Predominant autoantibody production by early human B cell precursors. Science 301, 1374–1377 (2003). [DOI] [PubMed] [Google Scholar]
- 22.Sauer AV, Morbach H, Brigida I, Ng Y-S, Aiuti A, Meffre E, Defective B cell tolerance in adenosine deaminase deficiency is corrected by gene therapy. J. Clin. Invest. 122, 2141–2152 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hervé M, Isnardi I, Ng YS, Bussel JB, Ochs HD, Cunningham-Rundles C, Meffre E, CD40 ligand and MHC class II expression are essential for human peripheral B cell tolerance. J. Exp. Med. 204, 1583–1593 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Janssen E, Morbach H, Ullas S, Bannock JM, Massad C, Menard L, Barlan I, Lefranc G, Su H, Dasouki M, al-Herz W, Keles S, Chatila T, Geha RS, Meffre E, Dedicator of cytokinesis 8-deficient patients have a breakdown in peripheral B-cell tolerance and defective regulatory T cells. J. Allergy Clin. Immunol. 134, 1365–1374 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pala F, Morbach H, Castiello MC, Schickel J-N, Scaramuzza S, Chamberlain N, Cassani B, Glauzy S, Romberg N, Candotti F, Aiuti A, Bosticardo M, Villa A, Meffre E, Lentiviral-mediated gene therapy restores B cell tolerance in Wiskott-Aldrich syndrome patients. J. Clin. Invest. 125, 3941–3951 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kinnunen T, Chamberlain N, Morbach H, Choi J, Kim S, Craft J, Mayer L, Cancrini C, Passerini L, Bacchetta R, Ochs HD, Torgerson TR, Meffre E, Accumulation of peripheral autoreactive B cells in the absence of functional human regulatory T cells. Blood 121, 1595–1603 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ng Y-S, Wardemann H, Chelnis J, Cunningham-Rundles C, Meffre E, Bruton’s tyrosine kinase is essential for human B cell tolerance. J. Exp. Med. 200, 927–934 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Isnardi I, Ng YS, Srdanovic I, Motaghedi R, Rudchenko S, von Bernuth H, Zhang SY, Puel A, Jouanguy E, Picard C, Garty BZ, Camcioglu Y, Doffinger R, Kumararatne D, Davies G, Gallin JI, Haraguchi S, Day NK, Casanova JL, Meffre E, IRAK-4- and MyD88-dependent pathways are essential for the removal of developing autoreactive B cells in humans. Immunity 29, 746–757 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Isnardi I, Ng Y-S, Menard L, Meyers G, Saadoun D, Srdanovic I, Samuels J, Berman J, Buckner JH, Cunningham-Rundles C, Meffre E, Complement receptor 2/CD21− human naive B cells contain mostly autoreactive unresponsive clones. Blood 115, 5026–5036 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Saadoun D, Terrier B, Bannock J, Vazquez T, Massad C, Kang I, Joly F, Rosenzwajg M, Sene D, Benech P, Musset L, Klatzmann D, Meffre E, Cacoub P, Expansion of autoreactive unresponsive CD21−/low B cells in Sjögren’s syndrome-associated lymphoproliferation. Arthritis Rheum. 65, 1085–1096 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Palanichamy A, Barnard J, Zheng B, Owen T, Quach T, Wei C, Looney RJ, Sanz I, Anolik JH, Novel human transitional B cell populations revealed by B cell depletion therapy. J. Immunol. 182, 5982–5993 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Descatoire M, Weller S, Irtan S, Sarnacki S, Feuillard J, Storck S, Guiochon-Mantel A, Bouligand J, Morali A, Cohen J, Jacquemin E, Iascone M, Bole-Feysot C, Cagnard N, Weill J-C, Reynaud C-A, Identification of a human splenic marginal zone B cell precursor with NOTCH2-dependent differentiation properties. J. Exp. Med. 211, 987–1000 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.van Zelm MC, Szczepanski T, van der Burg M, van Dongen JJM, Replication history of B lymphocytes reveals homeostatic proliferation and extensive antigen-induced B cell expansion. J. Exp. Med. 204, 645–655 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mackay F, Schneider P, Cracking the BAFF code. Nat. Rev. Immunol. 9, 491–502 (2009). [DOI] [PubMed] [Google Scholar]
- 35.Lindmark E, Chen Y, Georgoudaki A-M, Dudziak D, Lindh E, Adams WC, Lore K, Winqvist O, Chambers BJ, Karlsson MCI, AIRE expressing marginal zone dendritic cells balances adaptive immunity and T-follicular helper cell recruitment. J. Autoimmun. 42, 62–70 (2013). [DOI] [PubMed] [Google Scholar]
- 36.Romberg N, Chamberlain N, Saadoun D, Gentile M, Kinnunen T, Ng YS, Virdee M, Menard L, Cantaert T, Morbach H, Rachid R, Martinez-Pomar N, Matamoros N, Geha R, Grimbacher B, Cerutti A, Cunningham-Rundles C, Meffre E, CVID-associated TACI mutations affect autoreactive B cell selection and activation. J. Clin. Invest. 123, 4283–4293 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Menard L, Cantaert T, Chamberlain N, Tangye SG, Riminton S, Church JA, Klion A, Cunningham-Rundles C, Nichols KE, Meffre E, Signaling lymphocytic activation molecule (SLAM)/SLAM-associated protein pathway regulates human B-cell tolerance. J. Allergy Clin. Immunol. 133, 1149–1161 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cantaert T, Schickel J-N, Bannock JM, Ng Y-S, Massad C, Delmotte FR, Yamakawa N, Glauzy S, Chamberlain N, Kinnunen T, Menard L, Lavoie A, Walter JE, Notarangelo LD, Bruneau J, al-Herz W, Kilic SS, Ochs HD, Cunningham-Rundles C, van der Burg M, Kuijpers TW, Kracker S, Kaneko H, Sekinaka Y, Nonoyama S, Durandy A, Meffre E, Decreased somatic hypermutation induces an impaired peripheral B cell tolerance checkpoint. J. Clin. Invest. 126, 4289–4302 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sansom SN, Shikama-Dorn N, Zhanybekova S, Nusspaumer G, Macaulay IC, Deadman ME, Heger A, Ponting CP, Holländer GA, Population and single-cell genomics reveal the Aire dependency, relief from Polycomb silencing, and distribution of self-antigen expression in thymic epithelia. Genome Res. 24, 1918–1931 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Goodnow CC, Cyster JG, Hartley SB, Bell SE, Cooke MP, Healy JI, Akkaraju S, Rathmell JC, Pogue SL, Shokat KP, Self-tolerance checkpoints in B lymphocyte development. Adv. Immunol. 59, 279–368 (1995). [DOI] [PubMed] [Google Scholar]
- 41.Nemazee D, Mechanisms of central tolerance for B cells. Nat. Rev. Immunol. 17, 281–294 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Oliveira EH, Macedo C, Donate PB, Almeida RS, Pezzi N, Nguyen C, Rossi MA, Sakamoto-Hojo ET, Donadi EA, Passos GA, Expression profile of peripheral tissue antigen genes in medullary thymic epithelial cells (mTECs) is dependent on mRNA levels of autoimmune regulator (Aire). Immunobiology 218, 96–104 (2013). [DOI] [PubMed] [Google Scholar]
- 43.Baecher-Allan C, Wolf E, Hafler DA, MHC class II expression identifies functionally distinct human regulatory T cells. J. Immunol. 176, 4622–4631 (2006). [DOI] [PubMed] [Google Scholar]
- 44.Spanopoulou E, Roman CAJ, Corcoran LM, Schlissel MS, Silver DP, Nemazee D, Nussenzweig MC, Shinton SA, Hardy RR, Baltimore D, Functional immunoglobulin transgenes guide ordered B cell differentiation in Rag-1-deficient mice. Genes Dev. 8, 1030–1042 (1994). [DOI] [PubMed] [Google Scholar]
- 45.Meyers G, Ng Y-S, Bannock JM, Lavoie A, Walter JE, Notarangelo LD, Kilic SS, Aksu G, Debre M, Rieux-Laucat F, Conley ME, Cunningham-Rundles C, Durandy A, Meffre E, Activation-induced cytidine deaminase (AID) is required for B-cell tolerance in humans. Proc. Natl. Acad. Sci. U.S.A. 108, 11554–11559 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cantaert T, Schickel JN, Bannock JM, Ng YS, Massad C, Oe T, Wu R, Lavoie A, Walter JE, Notarangelo LD, al-Herz W, Kilic SS, Ochs HD, Nonoyama S, Durandy A, Meffre E, Activation-induced cytidine deaminase expression in human B cell precursors is essential for central B cell tolerance. Immunity 43, 884–895 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Park JS, Park M-K, Lee S-Y, Oh H-J, Lim M-A, Cho W-T, Kim E-K, Ju J-H, Park Y-W, Park S-H, Cho M-L, Kim H-Y, TWEAK promotes the production of interleukin-17 in rheumatoid arthritis. Cytokine 60, 143–149 (2012). [DOI] [PubMed] [Google Scholar]
- 48.Meyer S, Woodward M, Hertel C, Vlaicu P, Haque Y, Karner J, Macagno A, Onuoha SC, Fishman D, Peterson H, Metskula K, Uibo R, Jantti K, Hokynar K, Wolff ASB, Krohn K, Ranki A, Peterson P, Kisand K, Hayday A, Meloni A, Kluger N, Husebye ES, Podkrajsek KT, Battelino T, Bratanic N, Peet A, AIRE-deficient patients harbor unique high-affinity disease-ameliorating autoantibodies. Cell 166, 582–595 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fujikado N, Mann AO, Bansal K, Romito KR, Ferre EMN, Rosenzweig SD, Lionakis MS, Benoist C, Mathis D, Aire inhibits the generation of a perinatal population of interleukin-17A-producing gd T cells to promote immunologic tolerance. Immunity 45, 999–1012 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Walter JE, Rosen LB, Csomos K, Rosenberg JM, Mathew D, Keszei M, Ujhazi B, Chen K, Lee YN, Tirosh I, Dobbs K, al-Herz W, Cowan MJ, Puck J, Bleesing JJ, Grimley MS, Malech H, de Ravin SS, Gennery AR, Abraham RS, Joshi AY, Boyce TG, Butte MJ, Nadeau KC, Balboni I, Sullivan KE, Akhter J, Adeli M, el-Feky RA, el-Ghoneimy DH, Dbaibo G, Wakim R, Azzari C, Palma P, Cancrini C, Capuder K, Condino-Neto A, Costa-Carvalho BT, Oliveira JB, Roifman C, Buchbinder D, Kumanovics A, Franco JL, Niehues T, Schuetz C, Kuijpers T, Yee C, Chou J, Masaad MJ, Geha R, Uzel G, Gelman R, Holland SM, Recher M, Utz PJ, Browne SK, Notarangelo LD, Broad-spectrum antibodies against self-antigens and cytokines in RAG deficiency. J. Clin. Invest. 126, 4389 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Poliani PL, Facchetti F, Ravanini M, Gennery AR, Villa A, Roifman CM, Notarangelo LD, Early defects in human T-cell development severely affect distribution and maturation of thymic stromal cells: Possible implications for the pathophysiology of Omenn syndrome. Blood 114, 105–108 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kekalainen E, Tuovinen H, Joensuu J, Gylling M, Franssila R, Pontynen N, Talvensaari K, Perheentupa J, Miettinen A, Arstila TP, A defect of regulatory T cells in patients with autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy. J. Immunol. 178, 1208–1215 (2007). [DOI] [PubMed] [Google Scholar]
- 53.Liston A, Lesage S, Wilson J, Peltonen L, Goodnow CC, Aire regulates negative selection of organ-specific T cells. Nat. Immunol. 4, 350–354 (2003). [DOI] [PubMed] [Google Scholar]
- 54.Cozzo Picca C, Simons DM, Oh S, Aitken M, Perng OA, Mergenthaler C, Kropf E, Erikson J, Caton AJ, CD4+CD25+Foxp3+ regulatory T cell formation requires more specific recognition of a self-peptide than thymocyte deletion. Proc. Natl. Acad. Sci. U.S.A. 108, 14890–14895 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yamano T, Nedjic J, Hinterberger M, Steinert M, Koser S, Pinto S, Gerdes N, Lutgens E, Ishimaru N, Busslinger M, Brors B, Kyewski B, Klein L, Thymic B cells are licensed to present self antigens for central T cell tolerance induction. Immunity 42, 1048–1061 (2015). [DOI] [PubMed] [Google Scholar]
- 56.Gardner JM, DeVoss JJ, Friedman RS, Wong DJ, Tan YX, Zhou X, Johannes KP, Su MA, Chang HY, Krummel MF, Anderson MS, Deletional tolerance mediated by extrathymic Aire-expressing cells. Science 321, 843–847 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ludwig-Portugall I, Hamilton-Williams EE, Gottschalk C, Kurts C, Cutting edge: CD25+ regulatory T cells prevent expansion and induce apoptosis of B cells specific for tissue autoantigens. J. Immunol. 181, 4447–4451 (2008). [DOI] [PubMed] [Google Scholar]
- 58.Zhao D-M, Thornton AM, DiPaolo R, Shevach EM, Activated CD4+CD25+ T cells selectively kill B lymphocytes. Blood 107, 3925–3932 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Iikuni N, Lourenco EV, Hahn BH, la Cava A, Cutting edge: Regulatory T cells directly suppress B cells in systemic lupus erythematosus. J. Immunol. 183, 1518–1522 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Weissler KA, Garcia V, Kropf E, Aitken M, Bedoya F, Wolf AI, Erikson J, Caton AJ, Distinct modes of antigen presentation promote the formation, differentiation, and activity of foxp3+ regulatory T cells in vivo. J. Immunol. 194, 3784–3797 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Marcus N, Takada H, Law J, Cowan MJ, Gil J, Regueiro JR, Plaza Lopez de Sabando D, Lopez-Granados E, Dalal J, Friedrich W, Manfred H, Hanson IC, Grunebaum E, Shearer WT, Roifman CM, Hematopoietic stem cell transplantation for CD35 deficiency. J. Allergy Clin. Immunol. 128, 1050–1057 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ayoglu B, Mitsios N, Kockum I, Khademi M, Zandian A, Sjoberg R, Forsstrom B, Bredenberg J, Lima Bomfim I, Holmgren E, Gronlund H, Guerreiro-Cacais AO, Abdelmagid N, Uhlen M, Waterboer T, Alfredsson L, Mulder J, Schwenk JM, Olsson T, Nilsson P, Anoctamin 2 identified as an autoimmune target in multiple sclerosis. Proc. Natl. Acad. Sci. U.S.A. 113, 2188–2193 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Degn SE, van der Poel CE, Firl DJ, Ayoglu B, Al Qureshah FA, Bajic G, Mesin L, Reynaud CA, Weill JC, Utz PJ, Victora GD, Carroll MC, Clonal evolution of autoreactive germinal centers. Cell 170, 913–926.e19 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Karlsson R, Katsamba PS, Nordin H, Pol E, Myszka DG, Analyzing a kinetic titration series using affinity biosensors. Anal. Biochem. 349, 136–147 (2006). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.