

Abstract
Sodium-glucose co-transporter 2 (SGLT2) inhibitors immediately reduce the glomerular filtration rate (GFR) in patients with type 2 diabetes mellitus. When given chronically, they confer benefit by markedly slowing the rate at which chronic kidney disease progresses and are the first agents to do so since the advent of angiotensin-converting enzyme inhibitors (ACEIs) and angiotensin receptor blockers (ARBs). Salutary effects on the kidney were first demonstrated in cardiovascular outcomes trials and have now emerged from trials enriched in subjects with type 2 diabetes mellitus and chronic kidney disease. A simple model that unifies the immediate and long-term effects of SGLT2 inhibitors on kidney function is based on the assumption that diabetic hyperfiltration puts the kidney at long-term risk and evidence that hyperfiltration is an immediate response to a reduced signal for tubuloglomerular feedback, which occurs to the extent that SGLT2 activity mediates a primary increase in sodium and fluid reabsorption by the proximal tubule. This model will likely continue to serve as a useful description accounting for the beneficial effect of SGLT2 inhibitors on the diabetic kidney, similar to the hemodynamic explanation for the benefit of ACEIs and ARBs. A more complex model will be required to incorporate positive interactions between SGLT2 and sodium-hydrogen exchanger 3 in the proximal tubule and between sodium-glucose co-transporter 1 (SGLT1) and nitric oxide synthase in the macula densa. The implication of these latter nuances for day-to-day clinical medicine remains to be determined.
Keywords: Diabetic kidney disease, Glomerular filtration, Proximal tubule, Tubuloglomerular feedback
SODIUM-GLUCOSE CO-TRANSPORTER BASICS
Glucose and the Proximal Tubule
The daily glomerular filtrate of a healthy person contains ~1 mol (~180 g) of glucose.1 If excreted into the urine, this would constitute a loss of energy equivalent to ~30% of the body’s total caloric expenditure. Instead, nearly all the filtered glucose is reclaimed by sodium-glucose co-transporters (SGLT) 1 and 2 in the proximal tubule, which has capacity to reabsorb ~2.5 mol glucose per day. Gibbs free energy to drive SGLT1 and SGLT2 is stored in the electrochemical sodium potential, which is generated by the basolateral Na/K-ATPase. SGLT1, which reabsorbs 2 sodium per glucose, uses twice as much energy per glucose as SGLT2, which reabsorbs 1 sodium per glucose. The concentration ratio of tubular fluid to plasma glucose has a theoretical minimum, which is determined by laws of thermodynamics. This minimum varies exponentially with the Gibbs free energy available for transport. The kidney makes most efficient use of energy by positioning SGLT2 in the early proximal tubule to perform the bulk of glucose reabsorption and positioning SGLT1 in the late proximal tubule where the tubular fluid glucose concentration passes below the line where 1:1 stoichiometry of SGLT2 can provide sufficient free energy to lower it further.
SGLT2 reabsorbs sodium along with glucose, and important “off-target” effects of SGLT2 inhibitors stem from their effect on proximal tubular sodium transport. Here, we put this in perspective by comparing the amount of sodium reabsorbed with glucose with that reabsorbed with bicarbonate, the benchmark for co-transported sodium in the proximal tubule (Figure 1). If the filtered glucose concentration is 5 mM and 80% of the reabsorption is via SGLT2, then 6 mEq of sodium per liter of glomerular filtrate will pass through SGLT2 or SGLT1. This is equivalent to ~25% of the sodium directly linked to bicarbonate reabsorption, which is the main driver of net proximal reabsorption. If filtered glucose rises to the transport maximum, then the amount of sodium passing through SGLT2 or SGLT1 increases to ~19 mmol/L of filtrate, which is equivalent to ~80% of the sodium directly linked to bicarbonate. Like sodium-bicarbonate reabsorption, sodium-glucose reabsorption draws water from the proximal tubule, which elevates the tubular fluid chloride concentration and drives additional passive sodium chloride reabsorption. Hence, SGLT2 contributes both directly and indirectly to proximal tubular fluid and sodium chloride reabsorption, and the net contribution to overall proximal reabsorption may approach that of bicarbonate as blood glucose rises.
Figure 1.
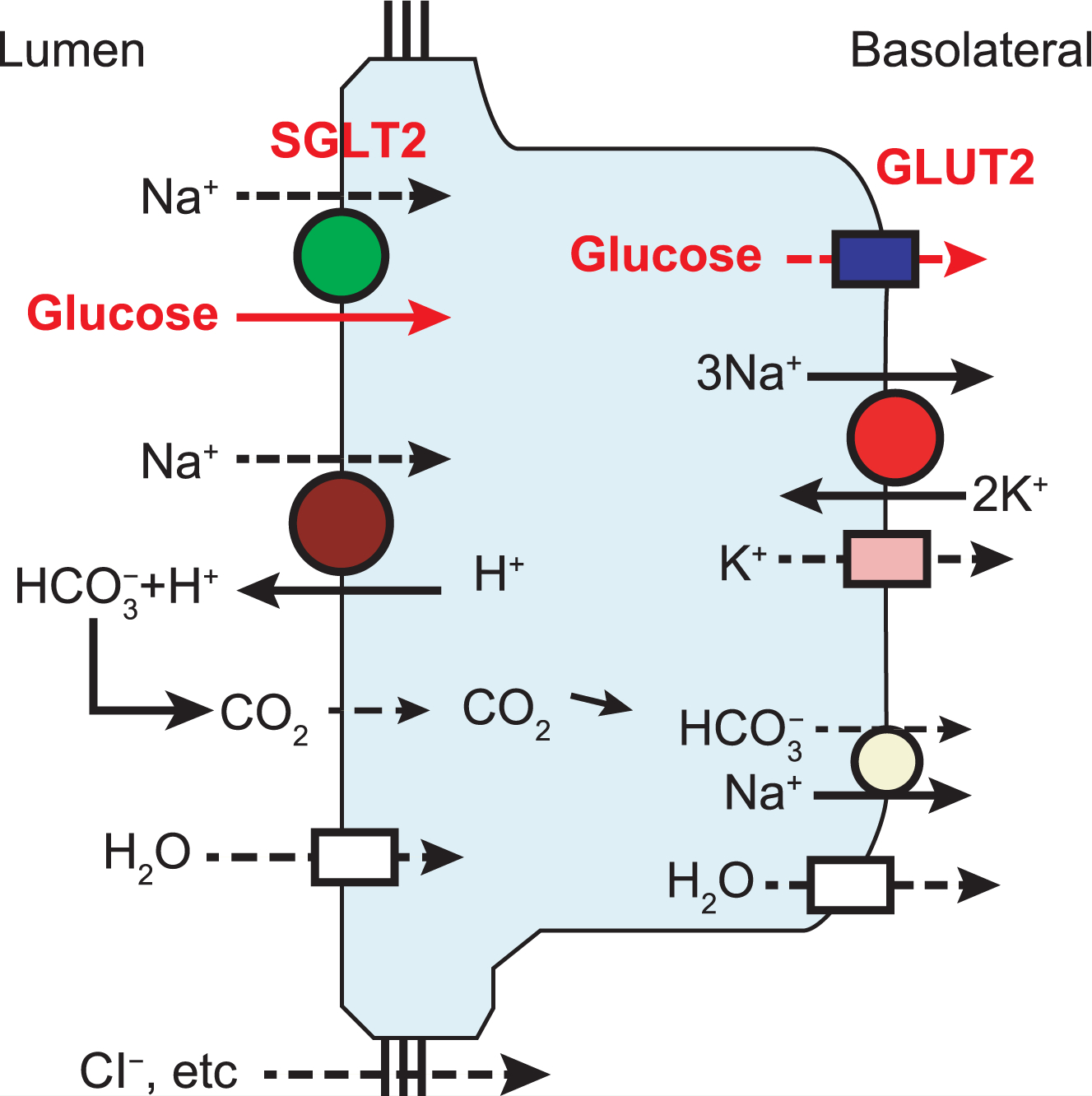
SGLT2 mechanism in early proximal tubule. SGLT2, bicarbonate reabsorption, and sodium-potassium ATPase, which provides sodium potential to drive both, are shown.
GLUT2 = glucose transporter 2; SGLT2 = sodium-glucose co-transporter 2.
PHLORIZIN AND ITS DERIVATIVES
Phlorizin is a botanical that was discovered in the 19th century and shown to cause glucosuria.2 During the 1960s, phlorizin became an essential tool for scientists developing the concept of secondary active glucose transport. The 1980s brought the cloning of SGLTs and confirmation that phlorizin blocks both SGLT1 and SGLT2.3 In 1987, Rosetti et al presented proof of concept by using phlorizin to lower blood glucose in experimental diabetes.4 However, phlorizin lacks therapeutic potential as treatment for patients with diabetes because of its poor oral absorption and because SGLT1 is the intestinal SGLT and blocking it causes diarrhea. In the current century, phlorizin derivatives have been generated that are bioavailable selective inhibitors of the SGLT2 isoform. In the last 5 years, several SGLT2 inhibitors have been approved as glucose-lowering agents for patients with type 2 diabetes mellitus. SGLT2 inhibitors have other effects tied to their mechanism of action that are not shared by other glucose-lowering drugs. Six effects that distinguish them from other diabetes drugs are that SGLT2 inhibitors 1) cause weight loss by exporting calories from the body to the urine5; 2) reduce blood pressure commensurate with their natriuretic effect6; 3) are uricosuric as a result of inhibition of the tubular urate transporter URAT17 or because tubular fluid glucose transstimulates uric acid secretion by the facilitative glucose transporter, GLUT98,9; 4) should not cause hypoglycemia by themselves because their glucosuric effect disappears when blood glucose levels decline below ~4 mM (~70 mg/dL) as a result of transport capacity residual in SGLT110,11 and because the drugs leave metabolic counter-regulation intact12; 5) may increase the risk of genitourinary infections because patients taking SGLT2 inhibitors excrete more glucose in their urine13; and 6) reversibly lessen glomerular filtration rate (GFR) by activating tubuloglomerular feedback (TGF),14 an effect that will be discussed in more detail.
In the 1930s, Shannon measured inulin clearance before and after administering phlorizin to normal human subjects, thereby providing the first demonstration that SGLT blockade acutely lowers GFR.15 In the modern era, Cherney et al documented decreases in inulin and para-aminohippurate clearances and in filtration fraction when hyperfiltering patients with type 1 diabetes were treated for 8 weeks with an SGLT2 inhibitor.16 Animal studies beginning in the 1990s have pin-pointed the renal hemodynamic effects of SGLT inhibitors to the TGF system, which will be addressed herein.
RENAL RESULTS IN CARDIOVASCULAR OUTCOMES TRIALS (CVOTS)
Since 2008, the US Food and Drug Administration (FDA) has required proof of cardiovascular safety for new glucose-lowering therapies.17 Hence, there are data from large-scale clinical trials designed to measure cardiovascular disease outcomes in patients randomized to receive SGLT2 inhibitors and incretin-based treatments,18–20 2 classes of diabetes drug that were in development when the guidance came into effect. The investigators who designed these trials were required to prove the noninferiority of a novel agent with respect to cardiovascular disease outcomes. Because the studies were designed to meet the same objective and are similar in basic design, their findings are amenable to comparison, with few caveats.
Although the primary objective of these trials was to confirm cardiovascular safety, some also acquired data on microvascular complications, including diabetic kidney disease, which were reported as secondary outcomes. Reports of renal outcomes in large trials with incretin-based drugs cite statistics on a composite renal outcome that combines some measure of albuminuria, declining estimated GFR (eGFR), and incident end-stage renal disease (ESRD). In each instance in which the composite renal outcome was improved, the effect was solely attributable to reduced albuminuria. None of the major trials with dipeptidyl peptidase-4 (DPP-4) inhibitors or glucagon-like peptide-1 (GLP-1) receptor agonists suggested lesser rates of decline in eGFR.21,22
The SGLT2 inhibitors empagliflozin, canagliflozin, and dapagliflozin have been evaluated for cardiovascular safety in 3 large-scale, multicenter clinical trials: Empagliflozin Cardiovascular Outcome Event Trial in Type 2 Diabetes Mellitus Patients–Removal of Excess Glucose (EMPA-REG OUTCOME),23,24 Canagliflozin Cardiovascular Assessment Study (CANVAS Program),25 and Dapagliflozin Effect on Cardiovascular Events–Thrombolysis in Myocardial Infarction 58 (DECLARE-TIMI 58),26 respectively.
In addition to showing a lesser incidence of heart failure, each of these trials also discovered salutary effects of SGLT2 blockade on the kidney, including lower hazard ratios (HRs) for major decline in eGFR. The relative risk reduction was approximately 40%−50% in EMPA-REG OUTCOME23,24 and CANVAS.25 The effect was smaller but still significant in DECLARE-TIMI 58 (HR reduced by approximately 25%).26 Thus, data from multiple large-scale randomized trials uniformly indicate SGLT2 blockade, but not incretin-based treatment, mitigates the loss of kidney function in patients with type 2 diabetes mellitus and cardiovascular disease. Because the primary objective of these trials was to document cardiovascular safety, enrollment was targeted toward subjects at high cardiovascular risk, and the studies included relatively few patients with established chronic kidney disease. Furthermore, because SGLT2 inhibitors are predictably less effective at lowering blood glucose levels when GFR is low, there was no initial incentive to include individuals with chronic kidney disease in these CVOTs. Those with chronic kidney disease were excluded from DECLARE-TIMI 58, which may explain why this trial yielded a smaller signal for renal sparing than EMPA-REG OUTCOME or CANVAS.
The improved renal outcomes in these trials brought about the suggestion that SGLT2 inhibitors might also improve renal outcomes for patients with established diabetic kidney disease, even though the glucose-lowering effect will be attenuated in these patients. The first demonstration that SGLT2 blockade slows the progression of established diabetic kidney disease was recently provided by the Canagliflozin and Renal Events in Diabetes With Established Nephropathy Clinical Evaluation (CREDENCE) trial.27 CREDENCE enrolled 4,401 subjects with type 2 diabetes mellitus, urine protein-to-creatinine ratio >0.3 mg/g, and eGFR 30 to 90 mL/min/1.73 m2 who were already receiving angiotensin-converting enzyme inhibitors (ACEIs) or angiotensin receptor blockers (ARBs) and randomized them to receive canagliflozin or placebo. The study was terminated early when planned interval analysis showed several end points had been reached. Over 2.6 median years of follow-up, the relative risk of a renal-specific composite including progression to ESRD, a doubling of the creatinine level, or death from renal causes was lower by 34% in those receiving the SGLT2 inhibitor (p<0.001).
SGLT2 inhibitors are the first class of drugs to show promise with respect to hard renal outcomes since the renin-angiotensin-aldosterone system (RAAS) blockers were introduced in the 1980s. As such, it is natural to compare the renal outcomes of recent SGLT2 inhibitor trials and older RAAS blocker trials. Two notable similarities are highlighted in Figures 2 and 3. Figure 2 juxtaposes figures from EMPA-REG OUTCOME in 201624 and CREDENCE in 2019,27 in which SGLT2 blockade was superimposed on angiotensin blockade with a figure from the iconic captopril trial performed by the Collaborative Study Group in 1993.28 By design, the Collaborative Study Group focused on subjects at high risk for renal failure whereas EMPA-REG OUTCOME did not. Hence, the ordinates are scaled differently. Otherwise, the results of these trials are highly similar, both yielding a gradual improvement in HR, which was reduced by ~50% after 3 to 4 years.
Figure 2.
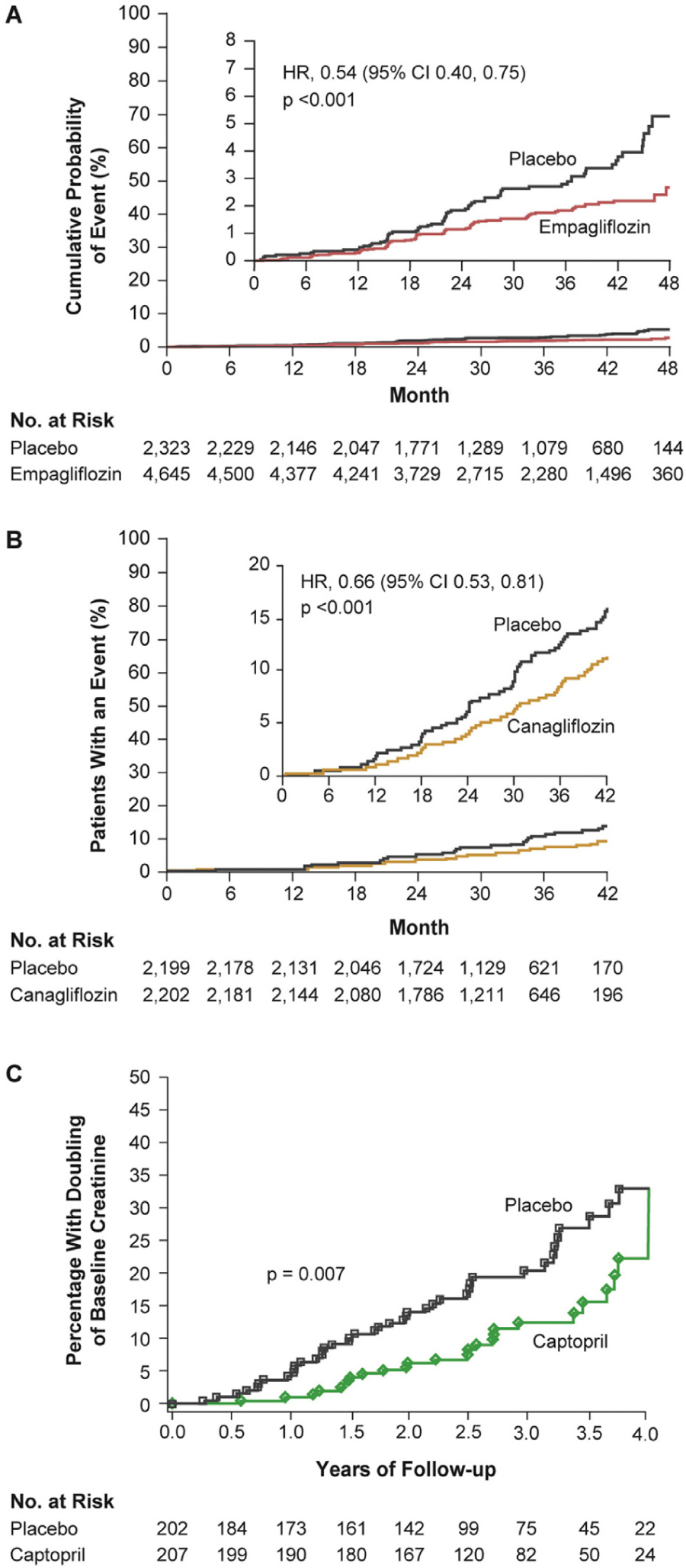
Renal outcomes in SGLT2 inhibitor trials of (A) empagliflozin (EMPA-REG OUTCOME)24 and (B) canagliflozin (CREDENCE)27 compared with (C) an earlier RAAS blocker (captopril) trial.28 (A) Renal-specific composite outcome of doubling of SCr level, initiation of renal replacement therapy, or death from renal disease in the EMPA-REG OUTCOME trial.24 From New England Journal of Medicine, Wanner C, et al., Empagliflozin and progression of kidney disease in type 2 diabetes, 375, 323–334. Copyright © 2016 Massachusetts Medical Society. Reprinted with permission from Massachusetts Medical Society. (B) Renal-specific composite outcome of ESRD, doubling of SCr level, or renal death in the CREDENCE trial.27 From New England Journal of Medicine, Perkovic V, et al. for the CREDENCE Trial Investigators, Canagliflozin and renal outcomes in type 2 diabetes and nephropathy, 380, 2295–2306. Copyright © 2019 Massachusetts Medical Society. Reprinted with permission from Massachusetts Medical Society. The placebo group was already receiving an ACEI or ARB and their rate of poor outcomes is remarkably similar to those who received captopril in the 1993 trial (~15% at 3.5 years28). (C) Renal end point of doubling of SCr level to ≥2.0 mg/dL in the 1993 captopril trial.28 From New England Journal of Medicine, Lewis EJ, et al., The effect of angiotensin-convertingenzyme inhibition on diabetic nephropathy, 329, 1456–1462, Copyright © 1993 Massachusetts Medical Society. Reprinted with permission from Massachusetts Medical Society. ACEI = angiotensin-converting enzyme inhibitor; ARB = angiotensin receptor blocker; RAAS = renin-angiotensin-aldosterone system; SCr = serum creatinine; SGLT2 = sodium-glucose co-transporter 2.
Figure 3.
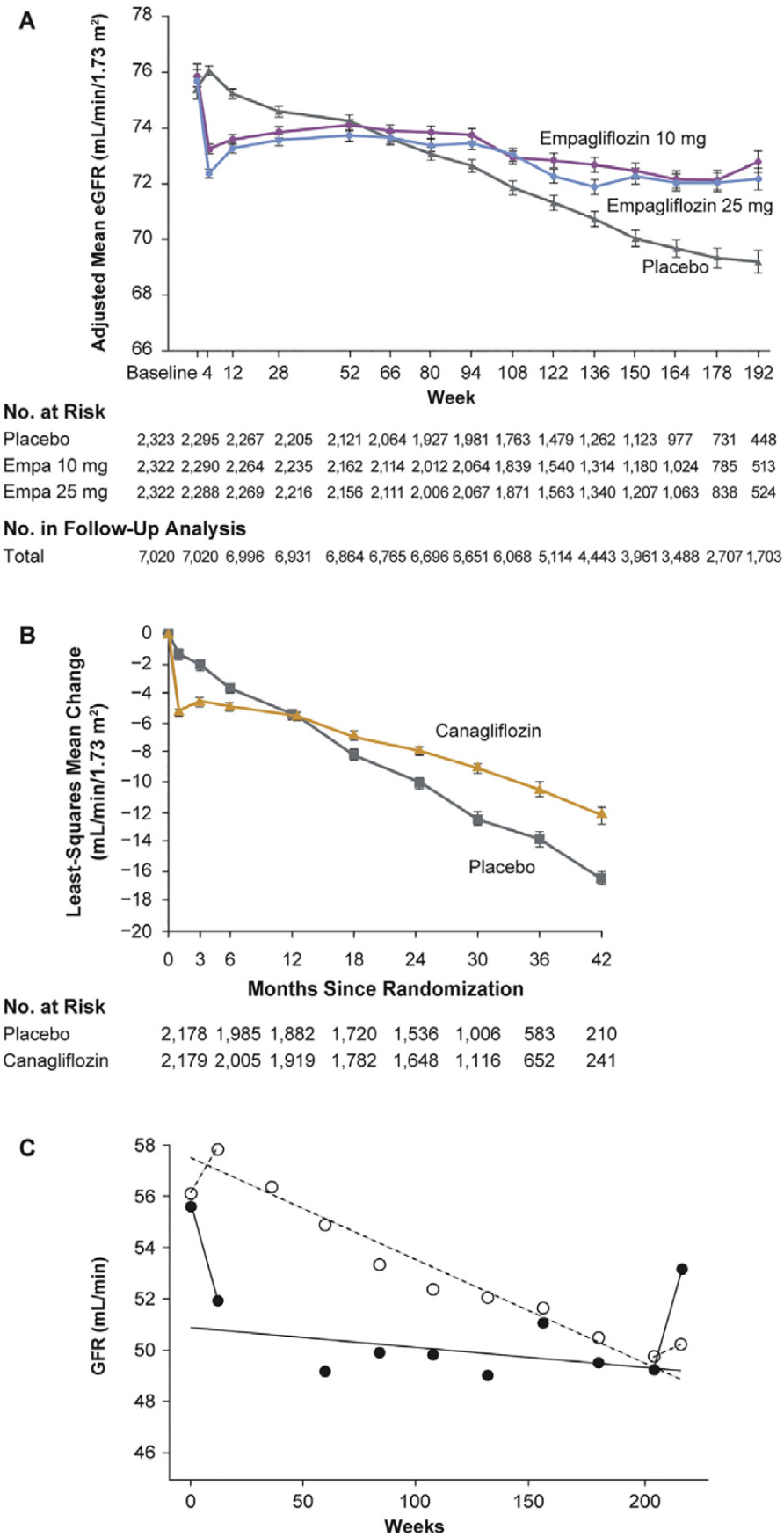
Similarity of eGFR outcomes in SGLT2 inhibitor clinical trials of (A) empagliflozin (EMPA-REG OUTCOME)24 and (B) canagliflozin (CREDENCE)27 compared with (C) an earlier RAAS blocker (enalapril) trial.29 Patients destined to benefit from SGLT2 inhibitor or ACEI treatment show an immediate decline in eGFR, which remains stable thereafter, whereas eGFR continues to decline in the comparator group. (A) Adjusted means for the eGFR over a period of 192 weeks in the EMPA-REG OUTCOME trial.24 From New England Journal of Medicine, Wanner C, et al., Empagliflozin and progression of kidney disease in type 2 diabetes, 375, 323–334. Copyright © 2016 Massachusetts Medical Society. Reprinted with permission from Massachusetts Medical Society. (B) Change from the screening level in eGFR in the on-treatment population in the CREDENCE trial.27 The I bars indicate the standard error. From New England Journal of Medicine, Perkovic V, et al. for the CREDENCE Trial Investigators, Canagliflozin and renal outcomes in type 2 diabetes and nephropathy, 380, 2295–2306. Copyright © 2019 Massachusetts Medical Society. Reprinted with permission from Massachusetts Medical Society. (C) Time course of eGFR before, during, and after withdrawal of antihypertensive treatment: in group A (•), patients who initially showed a distinct fall in GFR, and in group B (°), patients in whom eGFR did not fall after start of treatment in the enalapril trial.23 This figure shows a rebound increase in eGFR on cessation of enalapril treatment; this rebound also occurs with cessation of treatment with SGLT2 inhibitors (data not shown). Reprinted from Kidney International, 51 (3), Apperloo AJ, et al., A short-term antihypertensive treatment-induced fall in glomerular filtration rate predicts long-term stability of renal function, 793–797, Copyright 1997, with permission from Elsevier and International Society of Nephrology. ACEI = angiotensin-converting enzyme inhibitor; eGFR = estimated glomerular filtration rate; Empa = empagliflozin; RAAS = renin-angiotensin-aldosterone system; SGLT2 = sodium-glucose co-transporter 2.
Another analogy between SGLT2 and RAAS blockade is made in Figure 3.24,27,29 SGLT2 blockade causes an immediate and reversible decline in GFR, but subsequent protection from the indolent decline in GFR that otherwise occurs.24 The same phenomenon has been reported for enal-april, which confers long-term stability in kidney function to a subset of patients whose GFR declines on immediate exposure.29
FIRST-ORDER THEORIES TO EXPLAIN RENAL BENEFIT OF SGLT2 BLOCKADE
Glomerular Hemodynamics
At the onset of diabetes, the kidney grows large and GFR increases. Individuals with the greatest initial increase in GFR are more likely to develop kidney disease. Hence, hyperfiltration appears to be harmful.30 A large body of work in experimental diabetes has isolated the root cause of hyperfiltration to a primary increase in proximal reabsorption, which reduces the sodium chloride content of tubular fluid reaching the macula densa and leads to hyperfiltration through the normal action of TGF (reviewed in Thomson 2004 and Vallon 2011).31,32 Net sodium and fluid reabsorption by the proximal tubule increases with the amount of fluid available for reabsorption. This phenomenon, known as glomerulotubular balance (GTB), is inherently unable to yield an increase in reabsorption exceeding the increase in GFR that drives it. Therefore, if GFR increases and distal delivery decreases, a primary increase in tubular reabsorption independent of GTB is implied. The association of high GFR with low macula densa delivery is a consistent observation among animals and humans with hyperfiltering diabetes.33–35 Combining this state of the diabetic kidney with concise logic of control theory would lead to the inference that whatever direct effect diabetes may have on glomerular microcirculation, the dominant influence over GFR comes from the proximal tubule via TGF. If this is correct, then normalizing proximal reabsorption should eliminate diabetic hyperfiltration.
Initial proof of concept that SGLT contributes to the primary increase in proximal reabsorption and leads to hyperfiltration via TGF was provided by micropuncture experiments in diabetic rats.35 In these experiments, phlorizin was delivered directly into Bowman’s space of individual nephrons, while single-nephron GFR (SNGFR) and ionic content of the early distal tubular fluid were measured. In hyperglycemic rats the basal amount of sodium chloride reaching the macula densa was low, despite increased SNGFR, confirming a primary increase in proximal reabsorption. This was reversed by phlorizin, which suppressed proximal reabsorption, normalized delivery to the early distal nephron, and eliminated hyperfiltration. When phlorizin was given to nondiabetic nephrons the effect was smaller (Figure 4).35
Figure 4.
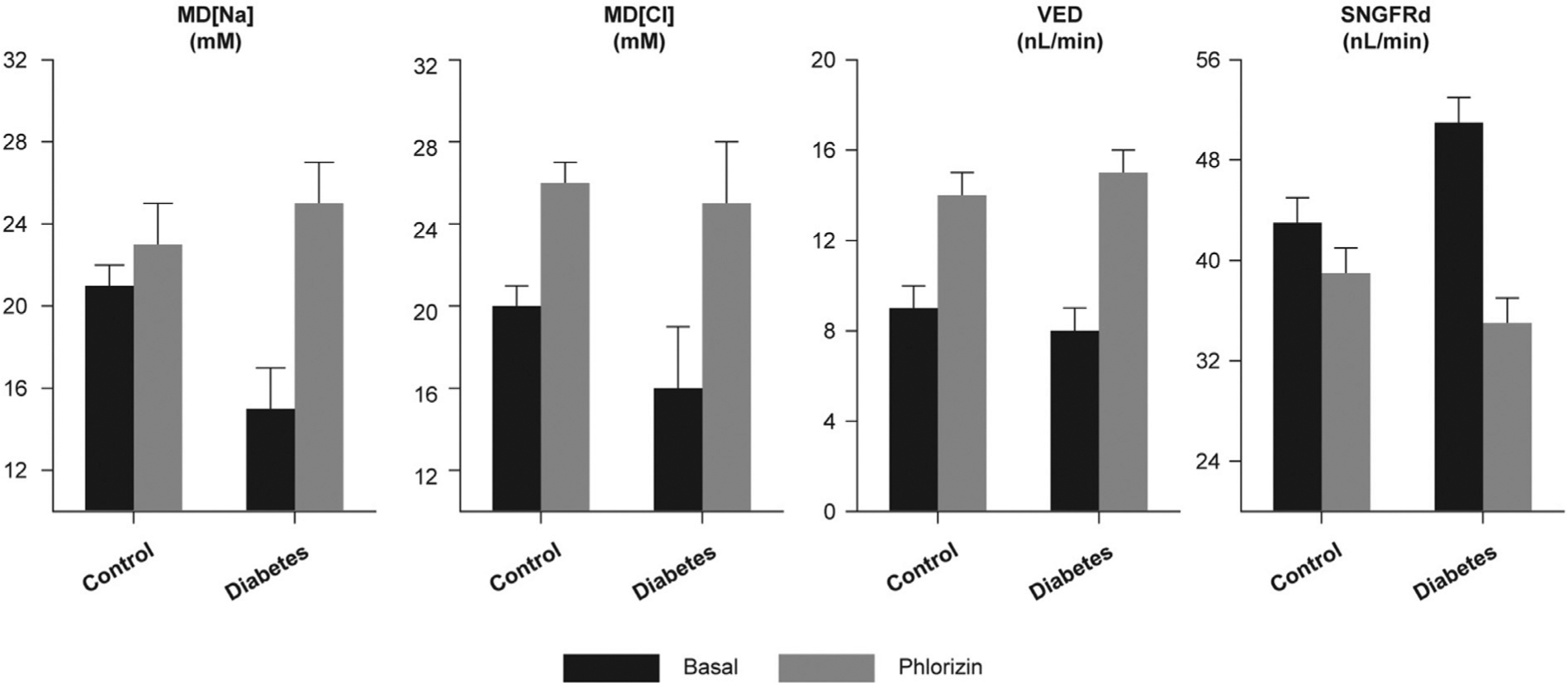
Immediate effects of phlorizin on macula densa delivery of Na, Cl, fluid volume, and SNGFR measured downstream of the macula densa to allow TGF to operate. Phlorizin was delivered to Bowman’s space.33 Cl = chloride; MD = macula densa; Na = sodium; SNGFRd = single-nephron glomerular filtration rate, determined by distal tubular collection; TGF = tubuloglomerular feedback; VED = early distal tubular flow rate. Data from Vallon V et al. Glomerular hyperfiltration in experimental diabetes mellitus: potential role of tubular reabsorption. J Am Soc Nephrol. 1999;10:2569–76.
TGF is typically studied in micropuncture experiments in which the system is manipulated on a scale of minutes. Because TGF is capable of resetting itself to accommodate long-term changes in delivery to the distal nephron, it should not be inferred from acute phlorizin microperfusion experiments that SGLT inhibitors can exert long-term control over SNGFR by way of TGF. The TGF response to SGLT blockade was eventually shown to be durable in micropuncture experiments using diabetic rats treated acutely or chronically with the SGLT2 inhibitor dapagliflozin.14 There was no tachyphylaxis to the effect of dapagliflozin on net fluid reabsorption by the proximal tubule and the TGF response remained intact. However, the chronic effect of dapagliflozin on SNGFR was attenuated by about half because of a compensatory increase in reabsorption by Henle’s loop (Figure 5).
Figure 5.
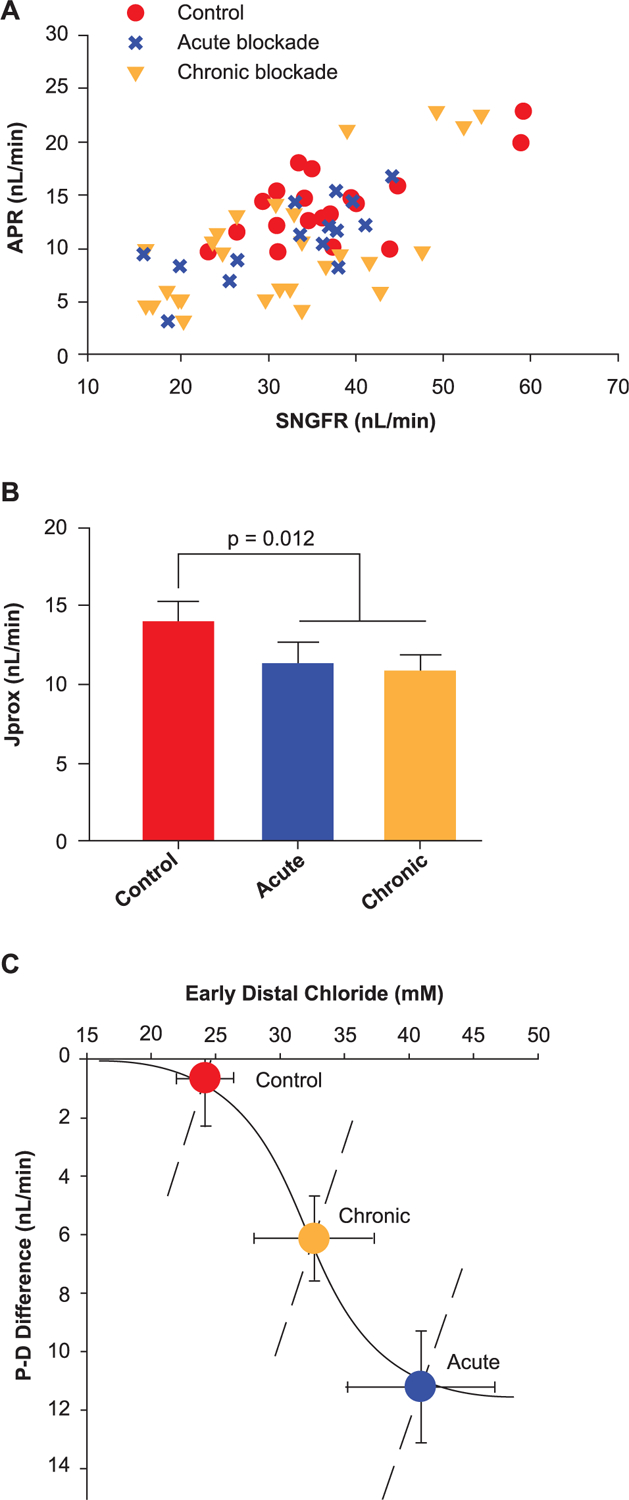
Acute effects and chronic effects with SGLT2 blockade on tubular reabsorption in Wistar Froemter rats with early diabetes.14 (A) Individual late proximal tubular fluid collections showing APR as a function of SNGFR. (B) Proximal reabsorption adjusted for SNGFR by ANCOVA (APR). Collections were made without perfusing Henle’s loop, so each point represents the shoulder of a TGF curve. (C) Degree of TGF activation (P-D difference [proximal minus distal]) as a function of the TGF stimulus (CED). The solid curve is a hyperbolic tangent parameterized to represent the acute TGF response. The dashed lines represent the forward effects of SNGFR on CED as a result of glomerulotubular balance. Superimposed on this graph are the operating points (mean § SE) for the 3 phases of SGLT2 blockade in Wistar Froemter rats. This figure illustrates 2 salient features of the transition from acute to chronic SGLT2 blockade. ANCOVA = analysis of covariance; APR = absolute proximal reabsorption; CED = early distal chloride concentration; SE = standard error; SGLT2 = sodium-glucose co-transporter 2; SNGFR = single-nephron glomerular filtration rate; TGF = tubuloglomerular feedback. Adapted from Thomson SC, et al. Acute and chronic effects of SGLT2 blockade on glomerular and tubular function in the early diabetic rat. Am J Physiol Regul Integr Comp Physiol. 2012; 302: R75–R83. Copyright © 2012, The American Physiological Society.
What is known about both SGLT2 inhibitors and RAAS blockers is that they have particular effects on glomerular hemodynamics and improve hard renal end points. In neither case is there sufficient knowledge to prove that the renal end points are the result of the associated hemodynamic effects. Nonetheless, the causal relation has been inferred for RAAS blockers since the 1980s, and no better explanation has emerged. It is, therefore, reasonable to make the same case for SGLT2 inhibitors.
EMERGING BASIC SCIENCE
SGLT2 and Sodium-Hydrogen Exchange
According to the first-order model presented, SGLT2 and sodium-bicarbonate reabsorption occur in parallel. Recently, co-localization and positive interference have been shown between SGLT2 and sodium-hydrogen exchanger (NHE) 3.36–39 Inhibiting SGLT2 suppresses the activity of NHE3 and knocking out tubular NHE3 reduces expression of SGLT2 and the natriuretic effect of SGLT2 blockade. Theories are in development to explain why this arrangement, which further increases the impact of SGLT2 on sodium reabsorption and ties it to acid-base balance, is advantageous in normal physiology. The coupling may simply facilitate the glomerulotubular balance of sodium, glucose, and bicarbonate when GFR is increasing. It is also noteworthy that acid-base balance and glucose metabolism are already coupled through phosphoenolpyruvate carboxy-kinase (PEPCK), a key enzyme for both gluconeogenesis and the renal compensatory response for systemic acidosis.40 Thus, blood glucose will tend to rise whenever tubular PEPCK is stimulated by acidosis and systemic pH will rise when PEPCK is stimulated to perform gluconeogenesis. This confounding could be mitigated by having SGLT2 and NHE3 change in parallel because increasing SGLT2 to raise cell glucose above its equilibrium concentration would suppress PEPCK and increasing NHE3 expression would sustain ammonia secretion, thereby allowing the proximal tubule to perform its function as a regulator of systemic pH with less reliance on PEPCK.
It has also been observed that empagliflozin suppresses NHE1 in cardiac myocytes, which do not express SGLT2.41 Suppression of overactive NHE in the heart by a mechanism that does not involve glucose transport has been proposed to explain the cardioprotective effects of SGLT2 inhibitors in CVOTs.42
Metabolic Theory
Another theory has been proposed that by depleting the body of calories, SGLT2 inhibitors are ketogenic and thereby improve cardiac function because the heart requires less oxygen when generating adenosine triphosphate (ATP) from ketones. It has been suggested that SGLT2 could also benefit the diabetic kidney by this same mechanism, effectively reducing regional hypoxia.43,44 This theory awaits further testing.
SGLT1 and Macula Densa Signaling
Blocking the macula densa nitric oxide synthase 1 (NOS1) eliminates hyperfiltration in diabetic rats while having minimal impact on GFR in nondiabetic animals.45,46 It has recently been noted that macula densa cells express SGLT1 and that activating SGLT1 in macula densa triggers NOS1 activity.47 New studies show knockout of SGLT1 prevents diabetes-induced upregulation of NOS1 in the macula densa and mitigates glomerular hyperfiltration.48 Absence of SGLT1 also attenuates upregulation of macula densa NOS1 expression in response to SGLT2 inhibition in nondiabetic mice. Furthermore, the effects of SGLT1 and SGLT2 inhibition on diabetic glomerular hyperfiltration were additive.48
ONGOING CLINICAL TRIALS
As noted previously, renal outcomes reported for SGLT2 inhibitors were first reported as secondary end points from CVOTs in which kidney disease was uncommon. However, the findings raised the possibility that SGLT2 inhibitors may improve renal outcomes for patients with kidney disease, even though the glucose-lowering effect will be attenuated in these patients. The first demonstration of this came in the CREDENCE trial, as noted. Several other studies are now underway to test for renal-sparing effects of SGLT2 inhibitors in patients with kidney disease, as are smaller studies directed toward mechanism of action. These trials are described in the online Supplemental Table. One large study, Study to Evaluate the Effect of Dapagliflozin on Renal Outcomes and Cardiovascular Mortality in Patients With Chronic Kidney Disease (DAPA-CKD), is designed along the same lines as CREDENCE to determine whether SGLT2 blockade improves hard renal outcomes in patients with chronic kidney disease and macroalbuminuria who are already RAAS blocked with or without diabetes. Another large study, Study of Heart and Kidney Protection With Empagliflozin (EMPA KIDNEY), will evaluate the effect of empagliflozin on renal and cardiovascular outcomes on top of standard of care in subjects with chronic kidney disease with or without diabetes.
CONCLUSIONS
SGLT2 inhibitors reversibly lower GFR because they reduce proximal tubular reabsorption, which leads to activation of TGF. SGLT2 inhibitors also mitigate the progression of diabetic kidney disease and are the first agents to do so convincingly since the advent of ACEIs and ARBs. A concise theory to explain both observations is that the salutary effect of SGLT2 inhibitors on long-term kidney function owes to reduced hyperfiltration, which puts physical stress on the glomerulus and metabolic demands on the tubule. A more complex model is required to incorporate recent observations that SGLT2 is coupled to NHE3 in the proximal tubule and that SGLT1 triggers NOS1 activity in the macula densa.
Supplementary Material
CLINICAL SIGNIFICANCE.
Sodium-glucose co-transporter 2 (SGLT2) inhibitors slow the progression of chronic kidney disease in patients with type 2 diabetes mellitus.
SGLT2 inhibitors are the first class of drugs to improve hard renal outcomes in diabetes since the advent of angiotensin blockers in the 1980s.
The beneficial effect of SGLT2 inhibitors and angiotensin blockers on kidney outcomes are additive.
Among the proposed mechanisms for the renal-sparing effect of SGLT2 inhibitors, a hemodynamic theory whereby SGLT2 inhibitors mitigate diabetic hyperfiltration through tubuloglomerular feedback has the most evidence to support it.
Funding:
This work was supported by NIH RO1 DK112042, R01DK106102, R01HL139836 and O’Brien Center for Acute Kidney Injury Research Grant P30DK079337. This supplement was supported by Boehringer Ingelheim Pharmaceuticals, Inc. Manuscript and figure formatting support was provided by Marissa Buttaro, MPH, RPh, of Elevate Scientific Solutions, which was contracted and funded by Boehringer Ingelheim Pharmaceuticals, Inc. The authors entirely drafted the manuscript and received no direct or indirect compensation related to the development of the manuscript.
Footnotes
Conflicts of Interest: SCT has received research support from Merck and Pfizer. VV has served as a consultant and received honoraria from AstraZeneca, Boehringer Ingelheim, Intarcia Therapeutics, Janssen, Eli Lilly, and Merck and received grant support for investigator-initiated research from Boehringer Ingelheim, Astra-Zeneca, Fresenius, Janssen, and Bayer.
Authorship: The authors meet criteria for authorship as recommended by the International Committee of Medical Journal Editors (ICMJE). The authors were fully responsible for all content and editorial decisions, were involved at all stages of manuscript development, and approved the final version that reflects the authors’ interpretations and conclusions. Boehringer Ingelheim was given the opportunity to review the manuscript for medical and scientific accuracy as well as intellectual property considerations.
SUPPLEMENTARY DATA
Supplementary data to this article can be found online at https://doi.org/10.1016/j.amjmed.2019.08.005.
References
- 1.Abdul-Ghani MA, DeFronzo RA, Norton L. Novel hypothesis to explain why SGLT2 inhibitors inhibit only 30–50% of filtered glucose load in humans. Diabetes 2013;62:3324–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stiles P, Lusk G. On the action of phlorizin. Am J Phys 1903;10:61–79. [Google Scholar]
- 3.Wright EM, Loo DD, Hirayama BA. Biology of human sodium glucose transporters. Physiol Rev 2011;91:733–94. [DOI] [PubMed] [Google Scholar]
- 4.Rossetti L, Smith D, Shulman GI, Papachristou D, DeFronzo RA. Correction of hyperglycemia with phlorizin normalizes tissue sensitivity to insulin in diabetic rats. J Clin Invest 1987;79:1510–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Leiter LA, Yoon KH, Arias P, et al. Canagliflozin provides durable glycemic improvements and body weight reduction over 104 weeks versus glimepiride in patients with type 2 diabetes on metformin: a randomized, double-blind, phase 3 study. Diabetes Care 2015;38:355–64. [DOI] [PubMed] [Google Scholar]
- 6.Weber MA, Mansfield TA, Cain VA, Iqbal N, Parikh S, Ptaszynska A. Blood pressure and glycaemic effects of dapagliflozin versus placebo in patients with type 2 diabetes on combination antihypertensive therapy: a randomised, double-blind, placebo-controlled, phase 3 study. Lancet Diabetes Endocrinol 2016;4:211–20. [DOI] [PubMed] [Google Scholar]
- 7.Novikov A, Fu Y, Huang W, et al. SGLT2 inhibition and renal urate excretion: role of luminal glucose, GLUT9, and URAT1. Am J Physiol Renal Physiol. 2019;316:F173–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chino Y, Samukawa Y, Sakai S, et al. SGLT2 inhibitor lowers serum uric acid through alteration of uric acid transport activity in renal tubule by increased glycosuria. Biopharm Drug Dispos 2014;35:391–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhao Y, Xu L, Tian D, et al. Effects of sodium-glucose co-transporter 2 (SGLT2) inhibitors on serum uric acid level: a meta-analysis of randomized controlled trials. Diabetes Obes Metab 2018;20:458–62. [DOI] [PubMed] [Google Scholar]
- 10.Calado J, Santer R, Rueff J. Effect of kidney disease on glucose handling (including genetic defects). Kidney Int 2011;79:S7–S13. [DOI] [PubMed] [Google Scholar]
- 11.Rieg T, Masuda T, Gerasimova M, et al. Increase in SGLT1-mediated transport explains renal glucose reabsorption during genetic and pharmacological SGLT2 inhibition in euglycemia. Am J Physiol Renal Physiol. 2014;306:F188–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vallon V, Thomson SC. Targeting renal glucose reabsorption to treat hyperglycaemia: the pleiotropic effects of SGLT2 inhibition. Diabetologia 2017;60:215–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ferrannini E, Berk A, Hantel S, et al. Long-term safety and efficacy of empagliflozin, sitagliptin, and metformin: an active-controlled, parallel-group, randomized, 78-week open-label extension study in patients with type 2 diabetes. Diabetes Care 2013;36:4015–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thomson SC, Rieg T, Miracle C, et al. Acute and chronic effects of SGLT2 blockade on glomerular and tubular function in the early diabetic rat. Am J Physiol Regul Integr Comp Physiol. 2012;302:R75–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shannon JA, Smith HW. The excretion of inulin, xylose and urea by normal and phlorizinized man. J Clin Invest 1935;14:393–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cherney DZ, Perkins BA, Soleymanlou N, et al. Renal hemodynamic effect of sodium-glucose cotransporter 2 inhibition in patients with type 1 diabetes mellitus. Circulation 2014;129:587–97. [DOI] [PubMed] [Google Scholar]
- 17.US Food and Drug Administration. Guidance for industry: diabetes mellitus—evaluating cardiovascular risk in new antidiabetic therapies to treat type 2 diabetes. Available at: http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm071627.pdf. Accessed on March 26, 2019.
- 18.Schnell O, Standl E, Catrinoiu D, et al. Report from the 3rd cardiovascular outcome trial (CVOT) summit of the diabetes & cardiovascular disease (D&CVD) EASD study group. Cardiovasc Diabetol 2018;17:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schnell O, Standl E, Catrinoiu D, et al. Report from the 2nd cardiovascular outcome trial (CVOT) summit of the diabetes and cardiovascular disease (D&CVD) EASD study group. Cardiovasc Diabetol 2017;16:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schnell O, Standl E, Catrinoiu D, et al. Report from the 4th cardiovascular outcome trial (CVOT) summit of the diabetes & cardiovascular disease (D&CVD) EASD study group. Cardiovasc Diabetol 2019;18:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Thomson SC, Vallon V. Renal effects of incretin-based diabetes therapies: pre-clinical predictions and clinical trial outcomes. Curr Diab Rep 2018;18:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rosenstock J, Perkovic V, Johansen OE, et al. Effect of linagliptin vs placebo on major cardiovascular events in adults with type 2 diabetes and high cardiovascular and renal risk: the CARMELINA randomized clinical trial. JAMA 2019;321:69–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zinman B, Wanner C, Lachin JM, et al. Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N Engl J Med 2015;373:2117–28. [DOI] [PubMed] [Google Scholar]
- 24.Wanner C, Inzucchi SE, Zinman B. Empagliflozin and progression of kidney disease in type 2 diabetes. N Engl J Med 2016;375:1801–2. [DOI] [PubMed] [Google Scholar]
- 25.Neal B, Perkovic V, Mahaffey KW, et al. Canagliflozin and Cardiovascular and Renal Events in Type 2 Diabetes. N Engl J Med 2017;377:644–57. [DOI] [PubMed] [Google Scholar]
- 26.Wiviott SD, Raz I, Bonaca MP, et al. Dapagliflozin and cardiovascular outcomes in type 2 diabetes. N Engl J Med 2019;380:347–57. [DOI] [PubMed] [Google Scholar]
- 27.Perkovic V, Jardine MJ, Neal B, et al. Canagliflozin and renal outcomes in type 2 diabetes and nephropathy. N Engl J Med 2019;380:2295–306. [DOI] [PubMed] [Google Scholar]
- 28.Lewis EJ, Hunsicker LG, Bain RP, Rohde RD. The effect of angiotensin-converting-enzyme inhibition on diabetic nephropathy. The Collaborative Study Group. N Engl J Med 1993;329:1456–62. [DOI] [PubMed] [Google Scholar]
- 29.Apperloo AJ, de Zeeuw D, de Jong PE. A short-term antihypertensive treatment-induced fall in glomerular filtration rate predicts long-term stability of renal function. Kidney Int 1997;51:793–7. [DOI] [PubMed] [Google Scholar]
- 30.Mogensen CE. Early glomerular hyperfiltration in insulin-dependent diabetics and late nephropathy. Scand J Clin Lab Invest 1986;46: 201–6. [DOI] [PubMed] [Google Scholar]
- 31.Thomson SC, Vallon V, Blantz RC. Kidney function in early diabetes: the tubular hypothesis of glomerular filtration. Am J Physiol Renal Physiol. 2004;286:F8–F15. [DOI] [PubMed] [Google Scholar]
- 32.Vallon V The proximal tubule in the pathophysiology of the diabetic kidney. Am J Physiol Regul Integr Comp Physiol 2011;300:R1009–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pruijm M, Wuerzner G, Maillard M, et al. Glomerular hyperfiltration and increased proximal sodium reabsorption in subjects with type 2 diabetes or impaired fasting glucose in a population of the African region. Nephrol Dial Transplant 2010;25:2225–31. [DOI] [PubMed] [Google Scholar]
- 34.Pollock CA, Lawrence JR, Field MJ. Tubular sodium handling and tubuloglomerular feedback in experimental diabetes mellitus. Am J Phys 1991;260:F946–52. [DOI] [PubMed] [Google Scholar]
- 35.Vallon V, Richter K, Blantz RC, Thomson S, Osswald H. Glomerular hyperfiltration in experimental diabetes mellitus: potential role of tubular reabsorption. J Am Soc Nephrol 1999;10:2569–76. [DOI] [PubMed] [Google Scholar]
- 36.Pessoa TD, Campos LC, Carraro-Lacroix L, Girardi AC, Malnic G. Functional role of glucose metabolism, osmotic stress, and sodium-glucose cotransporter isoform-mediated transport on Na+/H+ exchanger isoform 3 activity in the renal proximal tubule. J Am Soc Nephrol 2014;25:2028–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fu Y, Gerasimova M, Mayoux E, Masuda T, Vallon V. SGLT2 inhibitor empagliflozin increases renal NHE3 phosphorylation in diabetic Akita mice: possible implications for the prevention of glomerular hyperfiltration. Diabetes 2014;63:A132. [Google Scholar]
- 38.Huang W, Patel R, Onishi A, et al. Tubular NHE3 is a determinant of the acute natriuretic and chronic blood pressure lowering effect of the SGLT2 inhibitor empagliflozin. FASEB J 2018;32:620. [Google Scholar]
- 39.Onishi A, Fu Y, Darshi M, et al. Effect of renal tubule-specific knockdown of the Na+/H+ exchanger NHE3 in Akita diabetic mice. Am J Physiol Renal Physiol. 2019;317:F419–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Curthoys NP, Moe OW. Proximal tubule function and response to acidosis. Clin J Am Soc Nephrol 2014;9:1627–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Uthman L, Nederlof R, Eerbeek O, et al. Delayed ischemic contracture onset by empagliflozin associates with NHE-1 inhibition and is dependent on insulin in isolated mouse hearts. Cardiovasc Res 2019; 115:1533–45. [DOI] [PubMed] [Google Scholar]
- 42.Packer M, Anker SD, Butler J, Filippatos G, Zannad F. Effects of sodium-glucose cotransporter 2 inhibitors for the treatment of patients with heart failure: proposal of a novel mechanism of action. JAMA Cardiol 2017;2:1025–9. [DOI] [PubMed] [Google Scholar]
- 43.Ferrannini E, Mark M, Mayoux E. CV protection in the EMPA-REG OUTCOME trial: a “thrifty substrate” hypothesis. Diabetes Care 2016;39:1108–14. [DOI] [PubMed] [Google Scholar]
- 44.Mudaliar S, Alloju S, Henry RR. Can a shift in fuel energetics explain the beneficial cardiorenal outcomes in the EMPA-REG OUTCOME study? A unifying hypothesis. Diabetes Care 2016;39:1115–22. [DOI] [PubMed] [Google Scholar]
- 45.Thomson SC, Deng A, Komine N, Hammes JS, Blantz RC, Gabbai FB. Early diabetes as a model for testing the regulation of juxtaglomerular NOS I. Am J Physiol Renal Physiol. 2004;287:F732–8. [DOI] [PubMed] [Google Scholar]
- 46.Komers R, Lindsley J, Oyama T, Allison K, Anderson S. Role of neuronal nitric oxide synthase (NOS1) in the pathogenesis of renal hemodynamic changes in diabetes. Am J Physiol Renal Physiol 2000;279: F573–83. [DOI] [PubMed] [Google Scholar]
- 47.Zhang J, Wei J, Jiang S, et al. Macula densa SGLT1-NOS1-tubuloglomerular feedback pathway, a new mechanism for glomerular hyperfiltration during hyperglycemia. J Am Soc Nephrol 2019;30:578–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Song P, Huang W, Onishi A, et al. Knockout of Na-glucose-cotransporter SGLT1 mitigates diabetes-induced upregulation of nitric oxide synthase-1 in macula densa and glomerular hyperfiltration. Am J Physiol Renal Physiol 2019;317:F207–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.