

Abstract
In eukaryotes, chromatin remodeling and post-translational modifications (PTMs) shape the local chromatin landscape to establish permissive and repressive regions within the genome, orchestrating transcription, replication, and DNA repair in concert with other epigenetic mechanisms. While cellular nutrient signaling encompasses a huge number of pathways, recent attention has turned to the hypothesis that the metabolic state of the cell is communicated to the genome through the type and concentration of metabolites in the nucleus that are cofactors for chromatin-modifying enzymes. Importantly, both epigenetic and metabolic dysregulation are hallmarks of a range of diseases, and this metabolism-chromatin axis may yield a well of new therapeutic targets. In this Perspective, we highlight emerging themes in the inter-regulation of the genome and metabolism via chromatin, including nonenzymatic histone modifications arising from chemically reactive metabolites, the expansion of PTM diversity from cofactor-promiscuous chromatin-modifying enzymes, and evidence for the existence and importance of sub-nucleocytoplasmic metabolite pools.
Genomic DNA in eukaryotes resides within the nucleoprotein complex called chromatin, which packages and spatially organizes the genome.1 Chromatin consists of nucleosomes, which are stabilized by electrostatic interactions between the DNA and the four canonical histones H2A, H2B, H3, and H4. These histones are highly post-translationally modified by a diverse array of chemotypes,2,3 which serve as a mechanism to dynamically regulate the genome.2–4 Aberrant regulation of histone PTMs is associated with diseases such as cancer and developmental and neurological disorders, creating an impetus for elucidating the downstream effects of these PTMs on chromatin as well as the mechanisms that dictate their deposition, removal, and recognition by binding proteins.5,6 The enzymes responsible for carrying out chromatin PTM installation and removal utilize small molecule cofactors to perform these functions (Figure 1a–c). Thus, histone status acts as a link between DNA-mediated processes and cellular metabolism. When the levels of these cellular metabolites fluctuate around the KM of a chromatin-modifying enzyme, the abundance of the corresponding PTM is affected (Figure 1a).7–9 Similarly, the nuclear localization of metabolite-producing enzymes has been shown to affect the activity of the corresponding metabolite-consuming enzyme. For example, nuclear localization of S-adenosylmethionine (SAM)- and acetyl-CoA-producing enzymes impacts histone methylation and acetylation, respectively.10,11 Conversely, some metabolites are endogenous inhibitors of chromatin-modifying enzymes that can accumulate to inhibitory concentrations under certain cellular conditions and disease states (Figure 1b).12 Since histones are such abundant and extensively modified proteins,1,2 they are hypothesized to play a role in maintaining the levels of metabolites like SAM and acetyl-CoA (Figure 1c).13,14 Clearly, metabolism and genome regulation are central to cellular homeostasis and are inextricably linked by a massive number of interconnected pathways (for comprehensive reviews see the following references15–17). Nonetheless, the development of sophisticated chemical biology and analytical tools are constantly revealing new and unexpected modes of interregulation. In this Perspective, we focus on select recent advances in the rapidly growing metabolism-chromatin axis, highlighting the emerging roles of nonenzymatic histone PTMs, cofactor promiscuity in histone-modifying enzymes, and subcellular metabolite pools.
Figure 1. Metabolites regulate chromatin modifications.
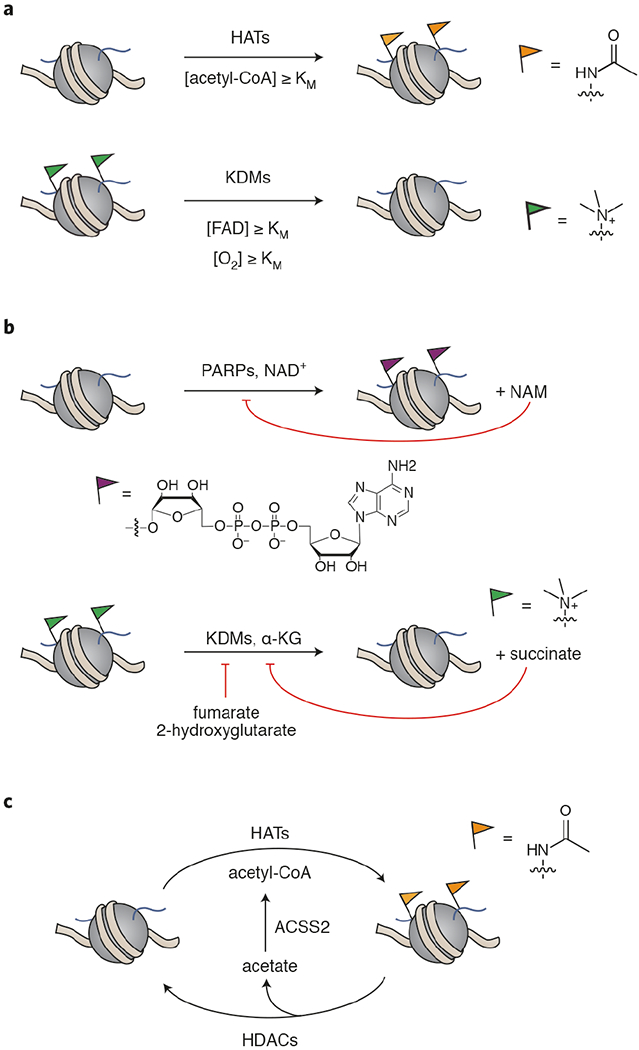
a) The concentration of cofactor with respect to KM of the chromatin-modifying enzyme affects that enzyme’s activity. HAT = histone acetyltransferase, KDM = lysine demethylase, FAD = flavin adenine dinucleotide. b) Metabolites can be endogenous inhibitors of chromatin modifiers. PARP = poly-ADP-ribose polymerase, KDM = lysine demethylase, NAM = nicotinamide. c) Chromatin may serve as a storage depot for metabolic capacity. For example, acetyl-CoA can be stored as histone acetylation that is mobilized by the action of histone deacetylases and ACSS2. HAT = histone acetyltransferase, HDAC = histone deacetylase, ACSS2- acetyl-CoA synthetase short-chain family member 2.
Nonenzymatic histone modifications
While histones are well-known for being post-translationally modified by a variety of different enzymes, they also undergo nonenzymatic modification.18 Adducts arise from the reaction of nucleophilic amino acids in histones, typically lysine and arginine, with endogenous or exogenous electrophiles. These electrophiles include acyl-CoAs,19,20 lipid peroxidation products,21 DNA peroxidation products,22 glycolytic by-products,23 and environmental toxins such as acrolein, 1,3-butadiene, and diepoxybutane.18 Since these adducts are often measured by immunoblotting- or qualitative mass spectrometry-based methods, a more quantitative approach will be necessary to address the question of abundance in cells.24,25 While it is difficult to imagine that these nonenzymatic PTMs could be deposited in a gene-specific manner akin to canonical enzymatic histone PTMs,2–4 it is possible that the marks may concentrate in euchromatic regions due simply to histone substrate accessibility.4 Regardless, there is still the potential for these marks to influence chromatin in other ways, such as by affecting nucleosome structure and stability, the subsequent enzymatic deposition of PTMs, and binding of reader proteins to nearby histone PTMs, particularly if a mark is chronically elevated in disease.26–29 Detailed mechanistic studies will be key to identifying specific mechanisms by which these marks perturb chromatin structure and function. Since these electrophilic metabolites are produced in pathways such as acyl-CoA synthesis,19,20 lipid degradation,26 and glycolysis,30 the corresponding modifications may represent a mechanism by which metabolic activity perturbs chromatin regulation. Nonenzymatic histone modifications are frequently characterized as undesirable by-products of reactive species accumulation,18 which is particularly relevant in the context of diseases that alter metabolism such as cancer31 and diabetes.32 To what extent these adducts are merely a symptom of disease or also themselves a cause of dysregulation via epigenetic mechanisms remains to be determined. In the latter case, modulation of these chemical marks could be an important therapeutic goal, particularly in post-mitotic cells such as neurons where histone proteins are long-lived33 and possibly more susceptible to the accumulation of adducts.
Chemical acylation by acyl-CoA
Acyl-CoAs alone are sufficiently electrophilic to acylate proteins on the ε-amines of lysine residues, perhaps most commonly in the high pH environment of the mitochondria.34,35 To explore nonenzymatic histone acylations, a biochemical characterization using histone substrates found that the site and acyl group specificities of nonenzymatic modification differed from those determined for GCN5-catalyzed acylations.20 In the enzymatic reactions, acetylation (Kac), propionylation (Kpr), and butyrylation (Kbu) were favored primarily on the histone tail residues, while malonylation (Kmal), succinylation (Ksuc), glutarylation (Kglu), and β-hydroxybutyrylation (Kbhb) (Figure 2a) occurred with similar frequency in the enzymatic and nonenzymatic reactions and generally favored the core residues.20 Since denatured histone substrates were used, it is unclear if these reactivity profiles would be recapitulated in a nucleosome context, although the authors point out that the major sites of Kmal and Ksuc they identified agree with the findings in a separate study of these modifications in human cells.36 While the authors’ results would seem to suggest that Ksuc, Kglu, and Kbhb are largely nonenyzmatic,20 these modifications have been shown to be catalyzed by p30036–38 and by GCN5 in the case of succinylation and glutarylation.39,40 Thus, an important goal for this field is to comprehensively delineate the relative contribution of nonenzymatic and enzymatic mechanisms to the acylation of histones, ideally in a cellular context, and to determine whether nonenzymatic pathways produce functional consequences for chromatin. Histone malonylation36 remains the only one of the histone acylations shown in Figure 2a for which no acyltransferase has been identified to date.41 Indeed, malonyl-CoA is more electrophilic compared to other acyl-CoAs,19,20 leaving the possibility that this acylation is primarily or exclusively nonenzymatic. Although histone malonylation has not yet been extensively characterized, H2AK119mal has been shown to affect chromosome segregation via a crosstalk with phosphorylation at H2AS121.41 Since malonyl-CoA is a key player in fatty acid synthesis,42 this study may point to a connection between metabolism and the coordination of cell division, although this link remains to be investigated.
Figure 2. Non-enzymatic histone modifications.
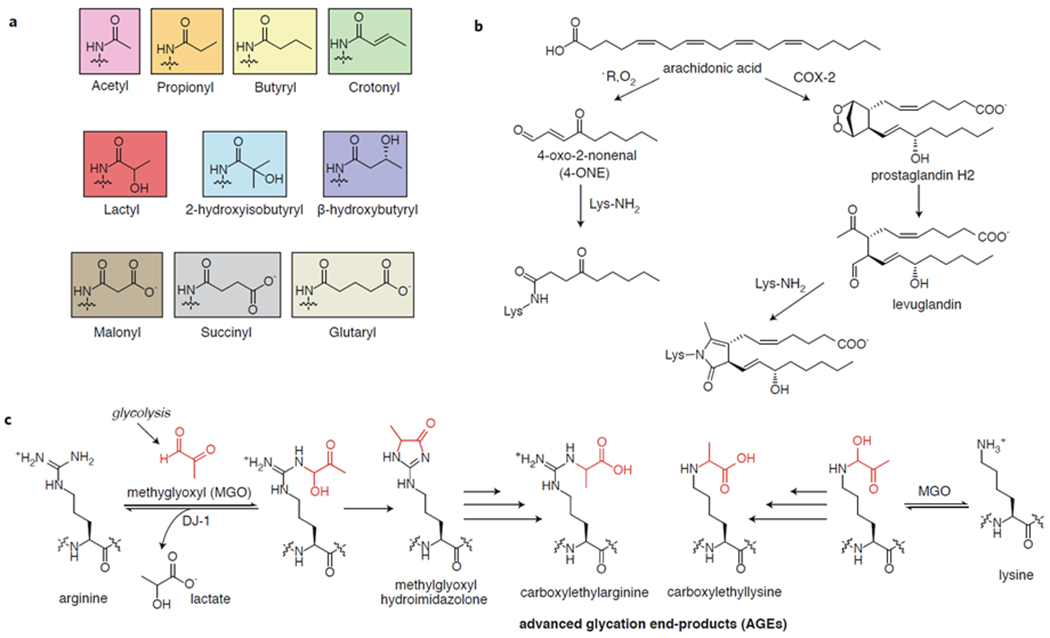
a) Structures of histone acylations from the corresponding acyl-CoA. b) Oxidation of unsaturated fatty acids leads to reactive species that react with histone lysines. COX2 = cyclooxygenase 2. c) Methylglyoxyl (MGO) generated from glycolysis reacts with lysine and arginine residues, eventually leading to advanced glycation products (AGEs) that stimulate inflammatory pathways. DJ-1 = protein/nucleic acid deglycase DJ-1.
Ketoamidation
Lipid oxidation by free radicals, reactive oxygen species (ROS), or endogenous enzymes leads to the generation of aldehydes such as malondialdehyde (MDA), 4-hydroxyl-2-nonenal (4-HNE), and 4-oxo-2-nonenal (4-ONE) that readily form adducts with DNA and proteins (Figure 2b).26 While 4-HNE reacts primarily with nucleophiles by a 1,4-addition, 4-ONE can form stable adducts by 1,2-addition to lysine residues43 and reacts preferentially with the lysine-rich histones to form ketoamide products.21 In colon carcinoma RKO cells, H3K23, H3K27, H2BK116, and H4K79 were found by mass spectrometry to be modified with 4-ONE, although the abundance of the mark was not measured.21 In the same work, in vitro nucleosome assembly assays revealed that modification of H3 or H4 inhibited nucleosome formation similarly to histone acetylation. Furthermore, RAW264.7 macrophages enriched with arachidonic acid displayed modification of H3K27, an important hotspot for histone PTMs, by 4-ONE upon treatment with a pro-inflammatory lipopolysaccharide mimic,21 suggesting a role for this histone modification in inflammation. Importantly, the deacetylase SIRT2 has been shown to remove this adduct from histone in cells,44 indicating that cells can reverse this type of oxidative damage to histones.
Aldehyde products of arachidonic acid peroxidation called levuglandins have also been shown to react with lysines in histones.25,45 Cyclooxygenase-2 (COX-2), an enzyme expressed during inflammation and associated with the development of multiple cancers, produces prostaglandin H2, which can spontaneously rearrange to the highly reactive levuglandins.46 Levuglandin adducts (Figure 2b) were identified by mass spectrometry on histones at levels reaching nearly 100 picograms of lactam per milligram of histone in RAW264.7 macrophages and lung carcinoma A549 cells upon COX-2 expression and arachidonic acid treatment.25 An immunoblot assay showed that H4 appears to be preferentially modified over H2A, H2B, and H3, although no specific sites of modification were identified. Using salt fractionation of nuclei in stimulated and unstimulated cells, the authors observed that the modification appears to disrupt H4-DNA interactions, pointing to a potential functional consequence of this mark.25 To date, it is unknown if these adducts are removed enzymatically or only through histone turnover.
Glycation
Reducing sugars (e.g. fructose, glucose-6-phosphate) and glycolytic by-products (e.g. glyoxal, methylglyoxal, 3-deoxyglucosone) containing aldehydes are susceptible to nucleophilic addition by lysine or arginine side chains of proteins (Figure 2c).30 These aldehydes are produced in multiple metabolic pathways but are made primarily by spontaneous decomposition of glycolytic intermediates and thus accumulate under increased glycolytic flux or in deficiencies that impair steps of glycolysis.28,30 For example, one pathway for methylglyoxal (MGO) formation is by fragmentation of glyceraldehyde-3-phosphate and dihydroxyacetone phosphate.28 Once these aldehydes react with proteins, the adducts become advanced glycation end-products (AGEs, e.g., carboxylmethyllysine, carboxylethyllysine, pentosidine) through a series of rearrangements. AGEs accumulate in the process of aging as well as in many diseases such as diabetes, cancer, neurodegeneration, and cardiovascular disease.28,30,47 These glycated proteins bind to the Receptor for AGE (RAGE), which initiates a signaling cascade leading to NFκB and pro-inflammatory gene activation.47 The cell possesses some endogenous mechanisms for mitigating the harmful effects of AGEs, including glyoxalase 1 (GLO-1), which isomerizes the product between MGO and glutathione (GSH) to form an inert species,28 and AGER1, which inhibits RAGE signaling.48
Initial work in this area has shown that histones are susceptible to glycation by reducing sugars both in vitro and in vivo.49,50 Subsequent studies focused on histone modification by the more reactive electrophiles 3-deoxyglucosone (3-DG) and MGO. 3-DG was shown to readily glycate histones and to progress to AGE products.51,52 Two recent studies undertook a detailed characterization of the glycation of histones by MGO, showing significant adducts on H3, H4, and H2B both in vitro and in cells.24,53 The observed modifications included the methylglyoxal hydroimidazolone and N7-carboxylethyl arginine (Figure 2c), which were identified on histones from cells grown in low glucose (5 mM) conditions.24 These modifications were each detected at levels (~1-4 pmol per nmol Leu) comparable to those measured for asymmetric dimethylated arginine in histones (~3-7 pmol per nmol Leu) from the same cell lines.24 N-ε-(carboxylethyl)lysine was also found on lysine residues (Figure 2c), although the abundance was an order of magnitude lower compared to the arginine adducts. Glycation of histones H3 and H4, but not of H2A or H2B, was found to disrupt nucleosome formation.53 This modification was further shown to affect nucleosome array structure, having a decompaction effect akin to acetylation at lower MGO concentrations and eventually leading to increased compaction at high MGO concentrations, which was attributed to the formation of MGO-mediated chemical crosslinks. The two studies reported quite different effects of MGO adducts on other histone PTMs in cells: the authors of one study found significant decreases in H2B acetylation and ubiquitylation but not on the other histones,24 while another study found that several H3 and H4 marks decreased, while H2B ubiquitylation was unaffected.53 Knocking out GLO-1 or DJ-1, a deglycase shown to remove MGO lesions from DNA,54 or increasing the glucose supplementation of the cultured cells increased the abundance of the MGO-histone adducts, showing that GLO-1 and DJ-1 protect histones from MGO-glycation and that glycolytic flux directly impacts levels of MGO adducts.24,53 Indeed, cancer cells rely on aerobic glycolysis-- the so-called Warburg effect31-- suggesting that glycation may play an important role in pathogenesis. Even though DJ-1 expression is upregulated in breast cancer cells,55 histone glycation levels are still high, leading the authors to propose that these cells depend on DJ-1 to hold glycation in check to prevent cell death.53 When DJ-1 was knocked down in these cells, survival was quickly compromised, indicating that DJ-1 may be a promising therapeutic target in cancer. Importantly, protein glycation is also a hallmark of diabetes, and reduced GLO-1 activity is associated with diabetic nephropathy.56 These two studies have established the importance of glycation as a nonenzymatic histone PTM and laid the foundation for further investigation into the role of this adduct in epigenetic misregulation across multiple disease states.
Cofactor promiscuity in histone-modifying enzymes
Histones are decorated with a variety of PTMs that act as a complex signaling code for genome regulation.2,3 Diversity in these biochemical signals is achieved through different PTM chemotypes and sites of modification, each with its own unique function. For most modifications, a class of enzymes utilizes a specific cofactor (e.g., methyltransferases use SAM, ADP-ribosyltransferases use NAD+). However, some histone-modifying enzymes can use multiple cofactors, leading to multiple possible chemotypes of PTMs from the same enzyme.57,58 Thus, the levels of the acceptable cofactors and how effectively the enzyme can use them in catalysis are expected to influence the abundance of a modification.
Acylation
Although acetyl-CoA represents the canonical cofactor of histone acetyltransferases (HATs), some HATs will accept other acyl-CoAs. The HAT p300 has been shown to transfer propionyl,59 butyryl,59 succinyl,36 glutaryl,37 β-hydroxybutyryl,38 2-hydroxyisobutyryl,60 crotonyl,61 and lactyl62 groups to histone lysines, while GCN5 can propionylate,63 butyrylate,63 glutarylate,39 and succinylate histones.40 There is even recent evidence that GCN5 may prefer succinyl-CoA over acetyl-CoA.40 To date, these different acylations have been studied biochemically and in cells to varying extents to try to address questions including how their deposition is regulated (e.g., identity of acyltransferases, deacylases), how abundant particular modifications are, what is the genomic distribution of these modifications, and what are the functional consequences of each PTM in chromatin regulation. While some of these questions have been answered for some modifications, a great deal of work remains to be done to fully characterize the ever-growing list of histone acylations. From a biochemical standpoint, this field would benefit from a rigorous analysis of the deposition and removal of these acylations on physiologically-relevant substrates such as mononucleosomes or nucleosome arrays. With regard to abundance, there is increasing evidence that the high glucose environment of standard cell culture conditions may artificially elevate acetyl-CoA levels compared to that of other acyl-CoAs, suppressing histone acylations.61,64,65 Thus, there are many experimental considerations that must be taken into account in both biochemical and cell-based studies of histone acylations in order for a clear understanding of these modifications to emerge. We refer the reader to recent reviews57,66,67 on this topic for a more comprehensive discussion of the current state of the field. In this section, we will highlight some recent works that focus on connections to metabolism (Figure 3).
Figure 3. Metabolic pathways that produce and interconvert acyl-CoAs involved in histone acylations.
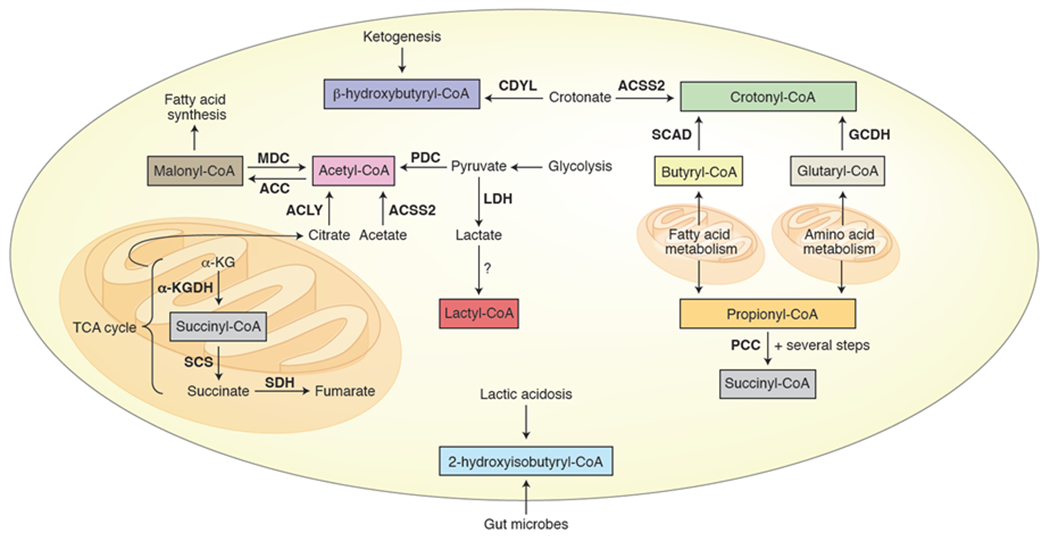
Bold = enzyme, italics = pathway, ACLY = ATP citrate lyase, ACSS2 = Acyl-CoA synthetase short-chain family member 2, PDC = pyruvate dehydrogenase complex, TCA cycle = tricarboxylic acid cycle, CDYL = chromodomain Y-like transcription corepressor, MDC = malonyl-CoA decarboxylase, ACC = acetyl-coA carboxylase, SCAD = short-chain acyl-CoA dehydrogenase, GCDH = glutaryl-CoA dehydrogenase, PCC = propionyl-CoA carboxylase, α-KG = α-ketoglutarate, α-KGDH = α-ketoglutarate dehydrogenase, SCS = succinyl-CoA synthetase, SDH = succinate dehydrogenase, LDH = lactate dehydrogenase.
Propionylation and butyrylation of histones are activating marks akin to acetylation, although the bulkier Kbu has been shown to bind poorly to some bromodomains compared to Kac and Kpr, pointing to a distinct functional role for histone butyrylation.68,69 From a metabolic perspective, propionyl-CoA derives from odd-chain fatty acid and amino acid catabolism, while butyryl-CoA is produced during β-oxidation of fatty acids70 and also enters intestinal cells from microbial fermentation (Figure 3).71 Importantly, the levels of these short chain acyl-CoAs can fluctuate as a function of cellular and physiological conditions.20,69,72 Indeed, knocking out propionyl-CoA carboxylase (PCC), which degrades propionyl-CoA, led to increased histone propionylation in mouse livers.69 Conversely, knocking out short-chain acyl-CoA dehydrogenase (SCAD), which degrades butyryl-CoA, did not lead to changes in butyrylated histone levels even though butyryl-CoA levels and nonhistone protein butyrylation did increase.69 Such evidence indicates that there are other regulatory mechanisms for controlling histone butyrylation beyond the simple abundance of cofactor. While histone glutarylation was first reported several years ago,37 there is still relatively little known about this histone mark. p30037 and GCN539 have been reported to glutarylate histones, while SIRT537 and SIRT739 can remove this PTM. In a recent study, H4K91glu was found to be the most abundant histone glutarylation site in Hela cells and to disrupt nucleosome structure in biochemical assays.39 In cells, deglutarylation of this site was associated with chromatin condensation, with levels of the mark peaking during S phase and dropping during the mitosis.
While protein succinylation is perhaps most widely associated with the mitochondria,34 histones are also known be succinylated.36 This activating mark localizes to promoters and transcription start sites40,73 and was even found in one study to occur on H4K79 in a third of nucleosomes in cells.73 The authors further observed that levels of succinyl-CoA and histone succinylation increased in a TCA cycle defect resulting in loss of succinate dehydrogenase (SDH), which oxidizes succinate to fumarate, linking transcription to the TCA cycle via histone succinylation. Additionally, the succinyl-CoA-producing enzyme α-ketoglutarate dehydrogenase (α-KGDH), which is also involved in the TCA cycle, has been shown to localize to the nucleus and associate with GCN5 to promote histone succinylation.40,74 Interestingly, succinate inhibits α-ketoglutarate-dependent demethylases,12 suggesting a possible role for histone succinylation in buffering extramitochondrial succinate levels to prevent accumulation to inhibitory concentrations.
Histone β-hydroxybutyrylation has been linked to metabolism by the accumulation of the ketone body β-hydroxybutyrate through ketogenesis during starvation or diabetes.65 In particular, increased levels of H3K9bhb during starvation were found to be associated with upregulation of genes involved in metabolism, such as amino acid catabolism and oxidative phosphorylation. Lysine 2-hydroxyisobutyrylation occurs on histones75,76 as well as other proteins, notably glycolytic enzymes.60,76 The precursor metabolite 2-hydroxyisobutyrate was found to be upregulated in the urine of morbidly obese patients77 and pregnant women with gestational diabetes mellitus78 and has been shown to be produced by symbiotic gut microbes.77,79 Together this evidence suggests a possible connection between the microbiome, histone acylation, and metabolic disease.
Histone crotonylation80 is a mark of active transcription81 that is recognized by distinct reader domains (e.g., YEATS domain in AF9 and DPF domain in the BAF complex).57,81 Crotonyl-CoA is present in cells at ~1000-fold lower concentration than acetyl-CoA, well below the KM of p300, and is produced from butyryl-CoA, glutaryl-CoA, or directly from crotonate by ACSS2 (Figure 3).61 Cells cultured in standard high glucose media exhibit much lower levels of crotonyl-CoA and protein crotonylation compared to cells grown under lower-glucose conditions, suggesting that histone crotonylation may be artificially repressed in many cell culture systems.61 Supplementation of sodium crotonate or knockdown of ATP-citrate lyase (ACLY) or the pyruvate dehydrogenase complex (PDC) leads to an increase in histone crotonylation, while knockdown of ACSS2 leads to decreases in these levels.61 While all three enzymes can produce acetyl- and other acyl-CoAs, this finding indicates that only ACSS2 produces crotonyl-CoA, providing a potential regulatory mechanism for controlling crotonyl-CoA levels separately from other acyl-CoAs. Crotonyl-CoA can be converted to β-hydroxybutyryl-CoA by the hydratase activity of the chromodomain Y-like transcription corepressor (CDYL) negatively regulating histone crotonylation at the substrate level as well.82
Recently, lactylation (Kla) was discovered as a new histone acylation.62 The authors identified lysine lactylation on all four of the canonical histones in human HeLa cells and mouse bone marrow-derived macrophages (BMDMs) by mass spectrometry. They further used a pan-lysine lactylation antibody to show that titrating exogenous lactate leads to a dose-dependent change in histone lactylation levels. Since lactate is produced from pyruvate by lactate dehydrogenase (LDH), high levels of cellular lactate are associated with the increased glycolytic activity known as the Warburg effect found in cancer and other diseases.31 Indeed, the authors observed an increase in histone lactylation as human cell lines were exposed to conditions that increase cellular lactate levels including 1) glucose supplementation, 2) the inhibitor rotenone, which promotes glycolysis, 3) hypoxia, and 4) M1 macrophage polarization. In contrast, treatment with inhibitors of LDH led to a decrease in histone lactylation.62 With clear evidence in hand that cellular lactate levels influence histone lactylation, the authors next determined the effects of this PTM on gene expression. ChIP-seq experiments with H3K18la and H3K18ac antibodies revealed that the PTMs are both enriched at promoters, with H3K18Kla enriched at specific genes. Furthermore, the authors demonstrated that upon stimulation of M1 polarization in macrophages genes specifically marked by H3K18la were upregulated, indicating that histone lactylation may be directly regulating these genes as a function of lactate. In particular, increased histone lactylation during M1 polarization was found at the promoter for the M2-like homeostatic gene Arg1. Importantly, histone lactylaton was found to exhibit different temporal dynamics in this M1 macrophage polarization system than histone acetylation, bolstering the evidence that histone lactyation is functionally distinct. Overexpression or deletion of p300 in cells attenuated histone lactylation levels, suggesting that p300 is a promising candidate for the histone lactyltransferase. Overall this work establishes the importance of lactylation as a non-canonical histone acylation that is metabolically regulated and leads to distinct transcriptional outcomes from acetylation. Further investigation of this modification will elucidate more details about how histone lactylation is deposited in a gene-specific manner, how the mark regulates transcription, as well as the identities of the lactyl-CoA-producing enzyme/s and of an eraser/s of the mark.
Monoaminylation
A new histone PTM was recently reported on H3Q5 that results from monoaminylation by transglutaminase 2 (TGM2).58 TGM2 is a ubiquitously expressed protein that catalyzes the transamidation between glutamine residues and a variety of primary amine substrates, including serotonin, dopamine, norepinephrine, and lysine (Figure 4a).83 Indeed, prior to this report, TGM2 was shown to form histone crosslinks between glutamine and lysine residues,84 so it was hypothesized that histones might be monoaminylated by TGM2 using nucleocytoplasmic serotonin (Figure 4b).58 By delivering propargylated serotonin to Hela cells, the authors enriched for serotonylated proteins, revealing that H3 is modified. Exogenous expression of TGM2 in 293T cells, which do not express the enzyme, revealed that H3 serotonylation is TGM2-dependent. Using mass spectrometric analysis and later mutagenesis, the authors identified H3Q5 as a substrate for serotonylation by TGM2. Based on the proximity of H3Q5 to H3K4, the methylation of which is an important mark of transcriptional activation, they looked for the co-occurrence of these two marks on the same H3 tail and found that, in fact, they frequently occur together in mammalian cells and tissues that produce serotonin, including serotonergic neurons. Genomic distribution indicates that the H3K4me3Q5ser mark is enriched at promoters, specifically those associated with neuronal differentiation and development. H3K4me3Q5ser is enriched in euchromatin and correlates with permissive gene expression, possibly by enhancing binding of the general transcription factor complex TFIID to H3K4me3. A great deal of work remains to determine how histone monoaminylation is regulated and what effects different modifications have on chromatin. While histone serotonylation may dominate under high serotonin levels, TGM2 is able to transfer other monoamines as well,83 and other histone monoaminylations may occur in different cell types or conditions depending on the relative monoamine concentrations. For example, cell types that produce and store dopamine, such as dopaminergic neurons and certain endocrine cells,85 or histamine, such as histaminergic neurons or certain immune cells,86 may display significant histone dopaminylation or histaminylation. It is worth noting that gut microbiota are known to produce biogenic amines including serotonin,87 raising the intriguing possibility that bacterially-produced metabolites may find their way onto histones by TGM2-mediated monoaminylation.
Figure 4. Monoaminylation of histones.
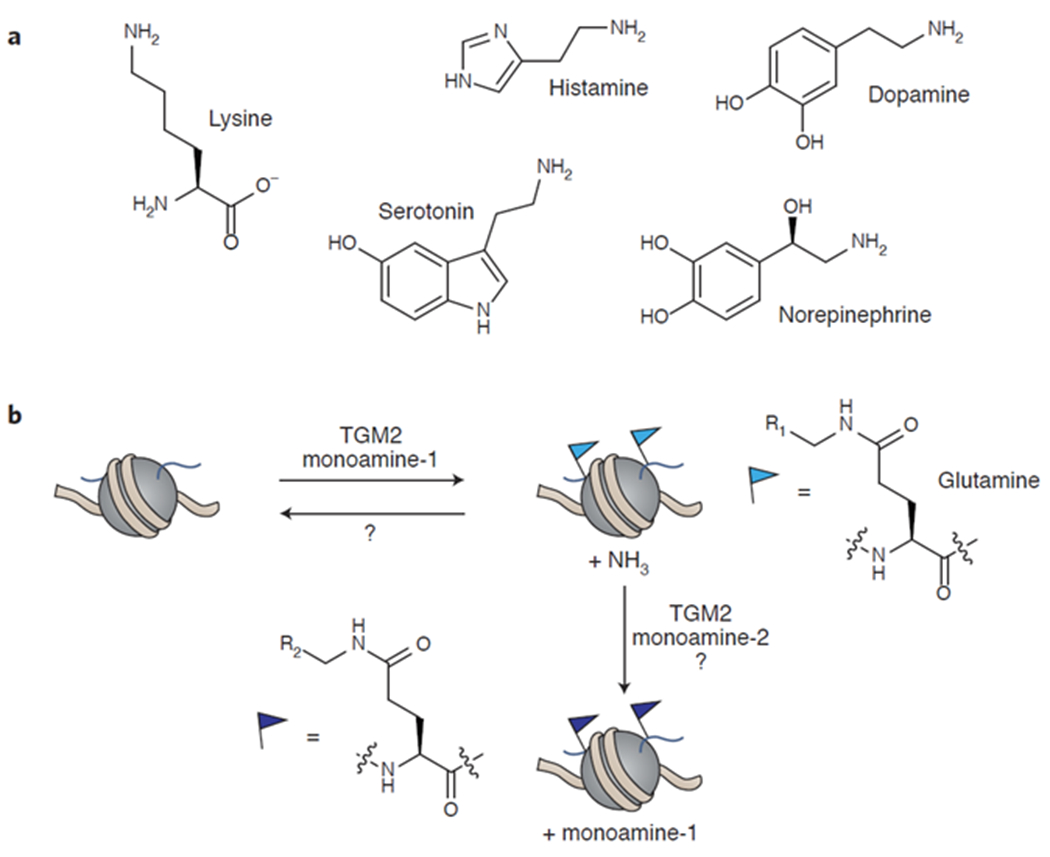
a) Examples of biogenic amines that TGM2 can use as cofactors. b) TGM2 transfers monoamines to glutamate residues in histones, releasing ammonia. While an enzyme has not yet been identified that can remove histone monoaminylation to restore the unmodified glutamine, TGM2 may be able to replace the first monoamine with a second depending on the relative abundance of the monoamines. TGM2 = tissue transglutaminase 2.
Sub-nucleocytoplasmic metabolite pools
The traditional view of the cellular environment is of a “bag of molecules”; that is, metabolites freely diffuse and thus are homogenously distributed.88,89 Of course, some membrane-organelles such as mitochondria and certain types of vesicles are well-known to restrict the diffusion of metabolites, requiring active mechanisms for transport across these membranes. Although the nucleus is also membrane-bound, its membrane contains pores that are sufficiently large to allow small molecules to passively diffuse through them.90 Thus, the entire cytoplasm and nucleus are frequently considered together to be one compartment without any barriers to metabolite diffusion within it. However, this notion has been challenged by both theoretical models and direct experimental evidence that suggest that certain factors may enable the formation of sub-nucleocytoplasmic metabolite domains.89,91,92 Far from being a dilute and homogenous solution, the nucleocytoplasm is dense and crowded with macromolecules, with estimates for cytosolic protein concentrations reaching 300 mg/mL.93 Small molecule diffusion may be reduced by an order of magnitude under these conditions. Moreover, these macromolecules are highly spatially organized within the nucleocytoplasm by their interactions with one another (e.g., protein-protein, protein-membrane, protein-DNA), enabling metabolite micro-compartmentalization and channeling by restricting diffusion and by binding metabolites.91
With regard to chromatin regulation, the possibility of distinct nuclear and even sub-nuclear metabolite pools is an exciting idea. There is now overwhelming evidence that chromatin state and metabolism are inter-regulated by metabolites,15–17 and local generation and consumption of small molecules within the nucleus may represent another important piece of the puzzle. While direct observation of these metabolite domains in cells has proved challenging with existing analytical techniques,89 perhaps the most compelling evidence in support of them is indirect. There now exist many examples of metabolite-producing enzymes either found localized to the nucleus or translocated there from other cellular compartments under certain conditions, and this localization has been shown to impact histone modification levels and other aspects of chromatin state.17,94 For example, the acetyl-CoA-producing enzymes ACSS2,14 ACLY,11 and PDC95 all translocate to the nucleus under specific cellular conditions, leading to increased histone acetylation and gene upregulation (Figure 5). Under conditions of metabolic stress like those found in tumors, ACSS2, which converts acetate to acetyl-CoA, enters the nucleus and appears to use acetate generated from deacetylation of histones by HDACs as a way of redistributing histone acetylation in the genome.14 In addition to serving as a major source of nucleocytoplasmic acetyl-CoA under normal cellular conditions, ACLY promotes H4 acetylation at nucleosomes proximal to DNA double-strand breaks, leading to the recruitment of BRCA1 and resultant homologous recombination (HR) at the site.96 PDC, which is normally found in the mitochondria, enters the nucleus at certain stages of embryonic development95 and under other conditions to provide acetyl-CoA from pyruvate.97 In fact, nuclear PDC forms a complex with p300 and the transcription factor arylhydrocarbon receptor (ArH),97 again demonstrating how the cell spatially organizes metabolite production for efficient generation of metabolites close to where they are consumed. What is particularly exciting about this example is that the involvement of a transcription factor provides genomic targeting of these activities, promoting deposition of H3K9ac at a specific target locus of ArH and subsequent enhancement of transcription of the target gene. Similarly, α-ketoglutarate dehydrogenase (α-KGDH), which converts α-KG to succinyl-CoA, localizes to the nucleus and binds to GCN5 (also known as KAT2A) to promote succinylation of H3K79.40 While persuasive, the evidence described above for nuclear and sub-nuclear compartmentation of metabolites is largely circumstantial. Clearly, if these subcellular metabolite domains could be observed directly it would allow questions related to the distribution and overall dynamics of the putative metabolite pools to be addressed in further detail, including whether there exist any spatiotemporal correlations with chromatin structure and organization in the nucleus.
Figure 5. Spatial control of cellular acetyl-CoA.
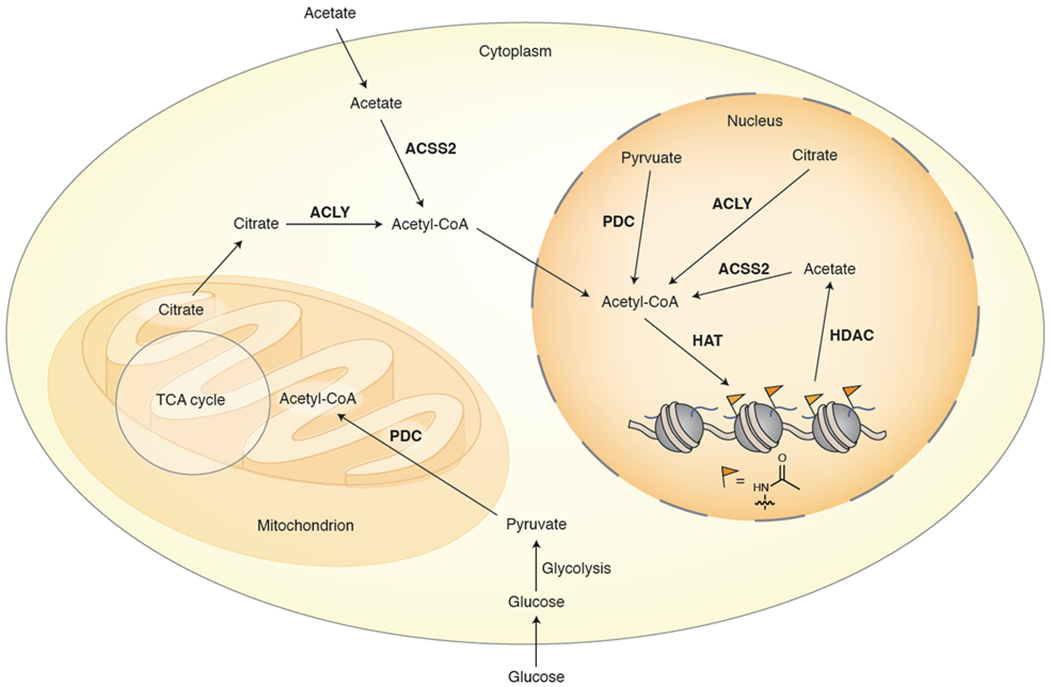
ACLY, ACSS2, and PDC use citrate, acetate, and pyruvate, respectively, to generate acetyl-CoA that is available for histone acetylation. Under certain conditions these enzymes translocate to the nucleus where they can provide localized production of the cofactor, potentially contributing to chromatin regulation through histone acetylation pathways. ACLY = ATP citrate lyase, ACSS2 = Acyl-CoA synthetase short-chain family member 2, PDC = pyruvate dehydrogenase complex, HAT = histone acetyltransferase, HDAC = histone deacetylase, TCA cycle = tricarboxylic acid cycle.
Since precise sub-nucleocytoplasmic fractionation is challenging, methods that can measure metabolite levels in situ in live cells represent a promising approach for directly observing different metabolite domains.98 Genetically-encoded fluorescent metabolite sensors have the potential to perform this task with the necessary spatiotemporal resolution and have already been developed for a number of different metabolites. A recent example of this approach utilized a genetically-encoded NAD+ sensor to measure changes in cytoplasmic and nuclear NAD+ levels during adipocyte differentiation.9 By measuring the levels in these cellular compartments separately using a ratiometric sensor, the authors were able to quantitate the respective NAD+ concentrations and determine that the nuclear levels fluctuate during differentiation below the KM of PARPs and SIRTs, which are NAD+-dependent enzymes, while cytoplasmic levels increased. By bringing the nuclear NAD+ concentration below the KM of PARP1, its ADP-ribosylation transferase activity is abrogated, which decreases PARylation of the adipogenic transcription factor C/EBPβ (CCAAT-enhancer-binding protein β) and regulates the factor’s binding to its target genes (Figure 6). By further investigation of the expression and activity of nicotinamide mononucleotide adenylyltransferase 1 (NMNAT-1, nuclear) and NMNAT-2 (cytoplasmic), which synthesize NAD+ from nicotinamide mononucleotide (NMN), the authors conclude that increased NMNAT-2 expression during differentiation leads to the increased cytosolic NAD+ as well as a decrease in cellular NMN, which regulates the activity of NMNAT-1. In other words, the two enzymes compete for cellular NMN, and, by tuning the expression levels of the two enzymes, the cell controls the compartmentalized NAD+ levels. The induction of NMNAT-2 expression is linked to increased glucose metabolism, providing a critical link between central metabolism and adipogenesis. Since many genetically-encoded sensors for a variety of metabolites already exist and sensor technologies are constantly improving,98 the approach used for NAD+ will likely be quickly extended to other metabolites and the corresponding histone PTMs.
Figure 6. Compartmentalized NAD+ synthesis coordinates glucose metabolism and transcription.
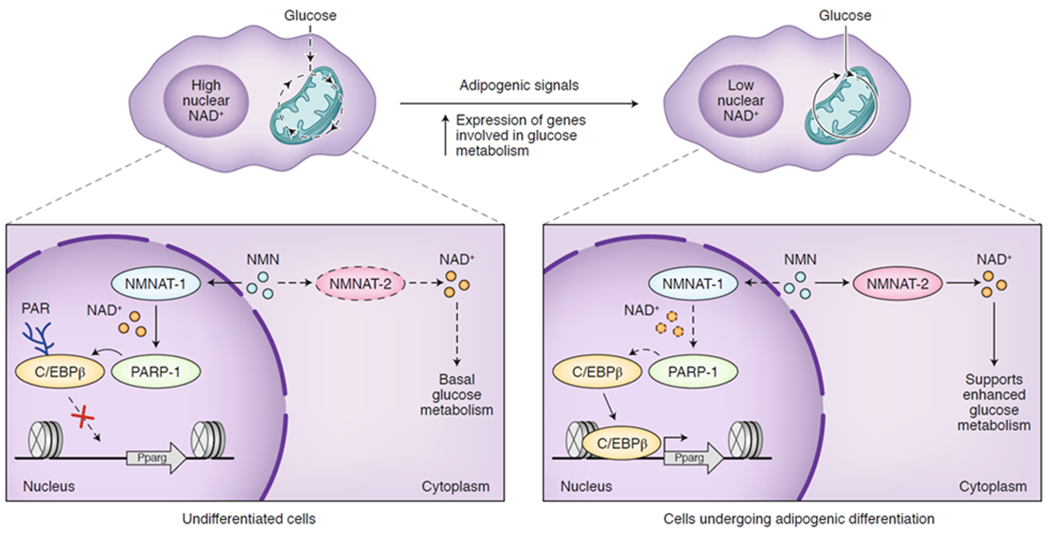
In an undifferentiated state (left), NMN is used by the nuclear NMNAT-1 to produce NAD+, promoting PARP1-dependent ADP-ribosylation of the transcription factor C/EBPβ to hold the cell in this undifferentiated state. Once proper adipogenic signals are received (top), cytoplasmic NMNAT-2 is induced to produce NAD+ for glycolysis, depleting the nuclear NAD+ pool and initiating C/EBPβ-mediated adipogenic differentiation (right). PARP1 = poly-ADP-ribose polymerase 1, C/EBPβ = CCAAT-enhancer-binding protein β, NMN = nicotinamide mononucleotide, NMNAT = nicotinamide mononucleotide adenylyltransferase, Pparg = peroxisome proliferator activated receptor gamma.
Conclusion and outlook
The cell must coordinate metabolic, signaling, and epi/genetic pathways for survival and growth. While nutrient sensing in higher eukaryotes is traditionally thought of as occurring by kinases such as AMPK, GCN2, and TOR99 as well as by nuclear receptors,100 the hypothesis that chromatin senses metabolic status and converts that information to specific epigenetic outputs has emerged as an additional mechanism. Chromatin remodeling and modification require a variety of core metabolites, suggesting that the concentration of specific metabolites in the nucleus will impact chromatin state, which in turn regulates gene expression. Nevertheless, the characterization of specific mechanisms that connect metabolism and chromatin has remained challenging and many fundamental questions persist regarding how this is orchestrated, if and how specificity at the modification and gene level is accomplished, and how metabolite sensing by chromatin is integrated with the other known nutrient-sensing mechanisms. With the identification of new histone PTMs, such as nonenzymatic adducts, novel acylations, and monoaminylations discussed here, the picture has become even more complex. Spatiotemporal control of nuclear metabolites is on track to be an important piece of this puzzle, but there is a long way left to go toward understanding precisely how this is accomplished both biochemically and biophysically. Ultimately, the goal of teasing apart these mechanisms is to identify therapeutically targetable pathways, particularly ones that can be modulated selectively to effect epigenetic regulation while preserving core metabolism in the cytoplasm and mitochondria.
Acknowledgments
We thank current and former members of the Muir laboratory for discussions and comments. Some of the work discussed herein was performed in the author’s laboratory and was supported by National Institutes of Health (NIH) Grants R37 GM086868, R01 GM107047 and P01 CA196539. K.L.D. was supported by an NIH Research Service Awards (5F32CA206418).
Footnotes
Competing Financial Interests Statement
The authors declare no competing interests.
References
- 1.Kornberg RD Structure of Chromatin. Annu. Rev. Biochem 46, 931–954 (1977). [DOI] [PubMed] [Google Scholar]
- 2.Kouzarides T Chromatin Modifications and Their Function. Cell 128, 693–705 (2007). [DOI] [PubMed] [Google Scholar]
- 3.Jenuwein T & Allis CD Translating the Histone Code. Science 293, 1074–1080 (2001). [DOI] [PubMed] [Google Scholar]
- 4.Allis CD, Caparros ML, Jenuwein T & Reinberg D Epigenetics. (Cold Spring Harbor Laboratory Press, 2015). [Google Scholar]
- 5.Zoghbi HY & Beaudet AL Epigenetics and Human Disease. Cold Spring Harb. Perspect. Biol 8, a019497 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chi P, Allis CD & Wang GG Covalent histone modifications — miswritten, misinterpreted and mis-erased in human cancers. Nat. Rev. Cancer 10, 457–469 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cai L, Sutter BM, Li B & Tu BP Acetyl-CoA Induces Cell Growth and Proliferation by Promoting the Acetylation of Histones at Growth Genes. Mol. Cell 42, 426–437 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shyh-Chang N et al. Influence of Threonine Metabolism on S-Adenosylmethionine and Histone Methylation. Science 339, 222–226 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ryu KW et al. Metabolic regulation of transcription through compartmentalized NAD+ biosynthesis. Science 360, eaan5780 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper details the subcellular compartmentalization of NAD+ synthesis and how these nuclear and cytoplasmic NAD+ pools regulate adipogenic transcriptional programs.
- 10.Li S et al. Serine and SAM Responsive Complex SESAME Regulates Histone Modification Crosstalk by Sensing Cellular Metabolism. Mol. Cell 60, 408–421 (2015). [DOI] [PubMed] [Google Scholar]
- 11.Wellen KE et al. ATP-Citrate Lyase Links Cellular Metabolism to Histone Acetylation. Science 324, 1076–1080 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang M, Soga T & Pollard PJ Oncometabolites: linking altered metabolism with cancer. J. Clin. Invest 123, 3652–3658 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ye C, Sutter BM, Wang Y, Kuang Z & Tu BP A Metabolic Function for Phospholipid and Histone Methylation. Mol. Cell 66, 180–193 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bulusu V et al. Acetate Recapturing by Nuclear Acetyl-CoA Synthetase 2 Prevents Loss of Histone Acetylation during Oxygen and Serum Limitation. Cell Rep. 18, 647–658 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schvartzman JM, Thompson CB & Finley LWS Metabolic regulation of chromatin modifications and gene expression. J. Cell Biol 217, 2247–2259 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Suganuma T & Workman JL Chromatin and Metabolism. Annu. Rev. Biochem 87, 27–49 (2018). [DOI] [PubMed] [Google Scholar]
- 17.Li X, Egervari G, Wang Y, Berger SL & Lu Z Regulation of chromatin and gene expression by metabolic enzymes and metabolites. Nat. Rev. Mol. Cell Biol 19, 563–578 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Galligan JJ & Marnett LJ Histone Adduction and Its Functional Impact on Epigenetics. Chem. Res. Toxicol 30, 376–387 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kulkarni RA et al. Discovering Targets of Non-enzymatic Acylation by Thioester Reactivity Profiling. Cell Chem. Biol 24, 231–242 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Simithy J et al. Characterization of histone acylations links chromatin modifications with metabolism. Nat. Commun 8, 1141–1153 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Galligan JJ et al. Stable Histone Adduction by 4-Oxo-2-nonenal: A Potential Link between Oxidative Stress and Epigenetics. J. Am. Chem. Soc 136, 11864–11866 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper identifies 4-oxo-2-nonenal adducts on histones both in vitro and in cells and showed that treatment of macrophages with lipopolysaccharide enhanced this modification on H3K27.
- 22.Jiang T, Zhou X, Taghizadeh K, Dong M & Dedon PC N-formylation of lysine in histone proteins as a secondary modification arising from oxidative DNA damage. Proc. Natl. Acad. Sci 104, 60–65 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guedes S, Vitorino R, Domingues MRM, Amado F & Domingues P Glycation and oxidation of histones H2B and H1: in vitro study and characterization by mass spectrometry. Anal. Bioanal. Chem 399, 3529–3539 (2011). [DOI] [PubMed] [Google Scholar]
- 24.Galligan JJ et al. Methylglyoxal-derived posttranslational arginine modifications are abundant histone marks. Proc. Natl. Acad. Sci 115, 9228–9233 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; Along with ref. 53, these papers characterize glycation as an abundant histone PTM modulated by glycolytic activity and regulated by the enzymes GLO-1 and DJ-1.
- 25.Carrier EJ, Zagol-Ikapitte I, Amarnath V, Boutaud O & Oates JA Levuglandin Forms Adducts with Histone H4 in a Cyclooxygenase-2-Dependent Manner, Altering Its Interaction with DNA. Biochemistry 53, 2436–2441 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper identifies adducts of levuglandin, a by-product of fatty acid peroxidation, on histones as a function of arachidonic acid treatment and COX-2 expression and describes how these modifications affect nucleosome structure.
- 26.Long EK, Olson DM & Bernlohr DA High-fat diet induces changes in adipose tissue trans-4-oxo-2-nonenal and trans-4-hydroxy-2-nonenal levels in a depot-specific manner. Free Radic. Biol. Med 63, 390–398 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Preidis GA et al. The undernourished neonatal mouse metabolome reveals evidence of liver and biliary dysfunction, inflammation, and oxidative stress. J. Nutr 144, 273–281 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Allaman I, Bélanger M & Magistretti PJ Methylglyoxal, the dark side of glycolysis. Front. Neurosci 9, 23–34 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vinodkumar P & Ajith TA Advanced Glycation End Products: Association with the Pathogenesis of Diseases and the Current Therapeutic Advances. Curr. Clin. Pharmacol 11, 118–127 (2016). [DOI] [PubMed] [Google Scholar]
- 30.Singh R, Barden A, Mori T & Beilin L Advanced glycation end-products: a review. Diabetologia 44, 129–146 (2001). [DOI] [PubMed] [Google Scholar]
- 31.Palsson-McDermott EM & O’Neill LAJ The Warburg effect then and now: From cancer to inflammatory diseases. BioEssays 35, 965–973 (2013). [DOI] [PubMed] [Google Scholar]
- 32.Bonadonna RC Alterations of Glucose Metabolism in Type 2 Diabetes Mellitus. An Overview. Rev. Endocr. Metab. Disord 5, 89–97 (2004). [DOI] [PubMed] [Google Scholar]
- 33.Maze I et al. Critical Role of Histone Turnover in Neuronal Transcription and Plasticity. Neuron 87, 77–94 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wagner GR & Payne RM Widespread and Enzyme-independent Nϵ-Acetylation and Nϵ-Succinylation of Proteins in the Chemical Conditions of the Mitochondrial Matrix. J. Biol. Chem 288, 29036–29045 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Baeza J, Smallegan MJ & Denu JM Site-Specific Reactivity of Nonenzymatic Lysine Acetylation. ACS Chem. Biol 10, 122–128 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xie Z et al. Lysine Succinylation and Lysine Malonylation in Histones. Mol. Cell. Proteomics 11, 100–107 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tan M et al. Lysine Glutarylation Is a Protein Posttranslational Modification Regulated by SIRT5. Cell Metab. 19, 605–617 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kaczmarska Z et al. Structure of p300 in complex with acyl-CoA variants. Nat. Chem. Biol 13, 21–29 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bao X et al. Glutarylation of Histone H4 Lysine 91 Regulates Chromatin Dynamics. Mol. Cell 76, 660–675 (2019). [DOI] [PubMed] [Google Scholar]
- 40.Wang Y et al. KAT2A coupled with the α-KGDH complex acts as a histone H3 succinyltransferase. Nature 552, 273–277 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper reports KAT2A (also called GCN5) as a histone succinyltransferase that binds to the α-KGDH enzyme in the nucleus to utilize the locally-produced succinyl-CoA for depositing the mark on H3K79 at gene promoters.
- 41.Ishiguro T et al. Malonylation of histone H2A at lysine 119 inhibits Bub1-dependent H2A phosphorylation and chromosomal localization of shugoshin proteins. Sci. Rep 8, 7671 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Röhrig F & Schulze A The multifaceted roles of fatty acid synthesis in cancer. Nat. Rev. Cancer 16, 732–749 (2016). [DOI] [PubMed] [Google Scholar]
- 43.Zhu X & Sayre LM Long-Lived 4-Oxo-2-enal-Derived Apparent Lysine Michael Adducts Are Actually the Isomeric 4-Ketoamides. Chem. Res. Toxicol 20, 165–170 (2007). [DOI] [PubMed] [Google Scholar]
- 44.Jin J, He B, Zhang X, Lin H & Wang Y SIRT2 Reverses 4-Oxononanoyl Lysine Modification on Histones. J. Am. Chem. Soc 138, 12304–12307 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mont S et al. Accumulation of isolevuglandin-modified protein in normal and fibrotic lung. Sci. Rep 6, 24919 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Boutaud O, Andreasson KI, Zagol-Ikapitte I & Oates JA Cyclooxygenase-Dependent Lipid-Modification of Brain Proteins. Brain Pathol. 15, 139–142 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schröter D & Höhn A Role of Advanced Glycation End Products in Carcinogenesis and their Therapeutic Implications. Curr. Pharm. Des 24, 5245–5251 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lu C et al. Advanced glycation endproduct (AGE) receptor 1 is a negative regulator of the inflammatory response to AGE in mesangial cells. Proc. Natl. Acad. Sci. U. S. A 101, 11767–11772 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Talasz H, Wasserer S & Puschendorf B Nonenzymatic glycation of histones in vitro and in vivo. J. Cell. Biochem 85, 24–34 (2002). [PubMed] [Google Scholar]
- 50.Gugliucci A & Bendayan M Histones from Diabetic Rats Contain Increased Levels of Advanced Glycation End Products. Biochem. Biophys. Res. Commun 212, 56–62 (1995). [DOI] [PubMed] [Google Scholar]
- 51.Ashraf JM et al. 3-Deoxyglucosone: A Potential Glycating Agent Accountable for Structural Alteration in H3 Histone Protein through Generation of Different AGEs. PLoS One 10, e0116804 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ashraf JM et al. Physicochemical analysis of structural alteration and advanced glycation end products generation during glycation of H2A histone by 3-deoxyglucosone. IUBMB Life 66, 686–693 (2014). [DOI] [PubMed] [Google Scholar]
- 53.Zheng Q et al. Reversible histone glycation is associated with disease-related changes in chromatin architecture. Nat. Commun 10, 1289 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Richarme G et al. Guanine glycation repair by DJ-1/Park7 and its bacterial homologs. Science 357, 208–211 (2017). [DOI] [PubMed] [Google Scholar]
- 55.Cao J, Lou S, Ying M & Yang B DJ-1 as a human oncogene and potential therapeutic target. Biochem. Pharmacol 93, 241–250 (2015). [DOI] [PubMed] [Google Scholar]
- 56.Qi W et al. Pyruvate kinase M2 activation may protect against the progression of diabetic glomerular pathology and mitochondrial dysfunction. Nat. Med 23, 753–762 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sabari BR, Zhang D, Allis CD & Zhao Y Metabolic regulation of gene expression through histone acylations. Nat. Rev. Mol. Cell Biol 18, 90–101 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Farrelly LA et al. Histone serotonylation is a permissive modification that enhances TFIID binding to H3K4me3. Nature 567, 535–539 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper reports serotonylation of H3Q5 and details its impact on transcription in serotonergic neurons through this PTM’s co-occurrence with H3K4me3.
- 59.Chen Y et al. Lysine Propionylation and Butyrylation Are Novel Post-translational Modifications in Histones. Mol. Cell. Proteomics 6, 812–819 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Huang H et al. p300-Mediated Lysine 2-Hydroxyisobutyrylation Regulates Glycolysis. Mol. Cell 70, 663–678 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sabari BR et al. Intracellular Crotonyl-CoA Stimulates Transcription through p300-Catalyzed Histone Crotonylation. Mol. Cell 58, 203–215 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper characterizes histone crotonylation as a function of the ratio of acetyl-CoA:crotonyl-CoA showing that this modification is more abundant under conditions of low acetyl-CoA and modulates gene expression in a manner distinct from histone acetylation.
- 62.Zhang D et al. Metabolic regulation of gene expression by histone lactylation. Nature 574, 575–580 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper characterizes lactylation of histone lysines as an epigenetic modification that directly stimulates gene transcription and is regulated by lactate production.
- 63.Ringel AE & Wolberger C Structural basis for acyl-group discrimination by human Gcn5L2. Acta Crystallogr. Sect. D, Struct. Biol 72, 841–848 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lee JV et al. Akt-Dependent Metabolic Reprogramming Regulates Tumor Cell Histone Acetylation. Cell Metab. 20, 306–319 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Xie Z et al. Metabolic Regulation of Gene Expression by Histone Lysine β-Hydroxybutyrylation. Mol. Cell 62, 194–206 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper reports lysine β-hydroxybutyrylation as a new histone modification that is enriched at active gene promoters and is upregulated under starvation conditions in mice that produce high levels of the ketone body β-hydroxybutyrate.
- 66.Barnes CE, English DM & Cowley SM Acetylation & Co: an expanding repertoire of histone acylations regulates chromatin and transcription. Essays Biochem. 63, 97–107 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhao S, Zhang X & Li H Beyond histone acetylation—writing and erasing histone acylations. Curr. Opin. Struct. Biol 53, 169–177 (2018). [DOI] [PubMed] [Google Scholar]
- 68.Goudarzi A et al. Dynamic Competing Histone H4 K5K8 Acetylation and Butyrylation Are Hallmarks of Highly Active Gene Promoters. Mol. Cell 62, 169–180 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kebede AF et al. Histone propionylation is a mark of active chromatin. Nat. Struct. Mol. Biol 24, 1048–1056 (2017). [DOI] [PubMed] [Google Scholar]; This paper characterizes histone propionylation and butyrylation, finding the H3K14pr and H3K14bu marks enriched at active gene promoters. While H3K14bu was unaffected by butyryl-CoA levels, H3K14pr levels were modulated at a function of cellular propionyl-CoA.
- 70.Nelson DL & Cox MM Lehninger Principles of Biochemistry. (W. H. Freeman, 2017). [Google Scholar]
- 71.Stilling RM et al. The neuropharmacology of butyrate: The bread and butter of the microbiota-gut-brain axis? Neurochem. Int 99, 110–132 (2016). [DOI] [PubMed] [Google Scholar]
- 72.Liu X et al. High-Resolution Metabolomics with Acyl-CoA Profiling Reveals Widespread Remodeling in Response to Diet. Mol. Cell. Proteomics 14, 1489–1500 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Smestad J, Erber L, Chen Y & Maher LJ Chromatin Succinylation Correlates with Active Gene Expression and Is Perturbed by Defective TCA Cycle Metabolism. iScience 2, 63–75 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wang Y, Guo YR, Xing D, Tao YJ & Lu Z Supramolecular assembly of KAT2A with succinyl-CoA for histone succinylation. Cell Discov. 4, 47 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Dai L et al. Lysine 2-hydroxyisobutyrylation is a widely distributed active histone mark. Nat. Chem. Biol 10, 365–370 (2014). [DOI] [PubMed] [Google Scholar]
- 76.Huang H et al. Landscape of the regulatory elements for lysine 2-hydroxyisobutyrylation pathway. Cell Res. 28, 111–125 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Calvani R et al. Gut microbiome-derived metabolites characterize a peculiar obese urinary metabotype. Int. J. Obes 34, 1095–1098 (2010). [DOI] [PubMed] [Google Scholar]
- 78.Diaz SO et al. Metabolic Biomarkers of Prenatal Disorders: An Exploratory NMR Metabonomics Study of Second Trimester Maternal Urine and Blood Plasma. J. Proteome Res 10, 3732–3742 (2011). [DOI] [PubMed] [Google Scholar]
- 79.Li M et al. Symbiotic gut microbes modulate human metabolic phenotypes. Proc. Natl. Acad. Sci 105, 2117–2122 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Tan M et al. Identification of 67 Histone Marks and Histone Lysine Crotonylation as a New Type of Histone Modification. Cell 146, 1016–1028 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Li Y et al. Molecular Coupling of Histone Crotonylation and Active Transcription by AF9 YEATS Domain. Mol. Cell 62, 181–193 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Liu S et al. Chromodomain Protein CDYL Acts as a Crotonyl-CoA Hydratase to Regulate Histone Crotonylation and Spermatogenesis. Mol. Cell 67, 853–866 (2017). [DOI] [PubMed] [Google Scholar]
- 83.Hummerich R, Thumfart J-O, Findeisen P, Bartsch D & Schloss P Transglutaminase-mediated transamidation of serotonin, dopamine and noradrenaline to fibronectin: Evidence for a general mechanism of monoaminylation. FEBS Lett. 586, 3421–3428 (2012). [DOI] [PubMed] [Google Scholar]
- 84.Kim J-H et al. Histone cross-linking by transglutaminase. Biochem. Biophys. Res. Commun 293, 1453–1457 (2002). [DOI] [PubMed] [Google Scholar]
- 85.Rubí B & Maechler P Minireview: New Roles for Peripheral Dopamine on Metabolic Control and Tumor Growth: Let’s Seek the Balance. Endocrinology 151, 5570–5581 (2010). [DOI] [PubMed] [Google Scholar]
- 86.Maintz L & Novak N Histamine and histamine intolerance. Am. J. Clin. Nutr 85, 1185–1196 (2007). [DOI] [PubMed] [Google Scholar]
- 87.Sudo N Biogenic Amines: Signals Between Commensal Microbiota and Gut Physiology. Front. Endocrinol. (Lausanne). 10, 504 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Barros LF & Martínez C An Enquiry into Metabolite Domains. Biophys. J 92, 3878–3884 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zecchin A, Stapor PC, Goveia J & Carmeliet P Metabolic pathway compartmentalization: an underappreciated opportunity? Curr. Opin. Biotechnol 34, 73–81 (2015). [DOI] [PubMed] [Google Scholar]
- 90.Kabachinski G & Schwartz TU The nuclear pore complex--structure and function at a glance. J. Cell Sci 128, 423–429 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Saks V, Beraud N & Wallimann T Metabolic Compartmentation – A System Level Property of Muscle Cells. Int. J. Mol. Sci 9, 751–767 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kekenes-Huskey PM, Scott CE & Atalay S Quantifying the Influence of the Crowded Cytoplasm on Small Molecule Diffusion. J. Phys. Chem. B 120, 8696–8706 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Fulton AB How crowded is the cytoplasm? Cell 30, 345–347 (1982). [DOI] [PubMed] [Google Scholar]
- 94.Sivanand S, Viney I & Wellen KE Spatiotemporal Control of Acetyl-CoA Metabolism in Chromatin Regulation. Trends Biochem. Sci 43, 61–74 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Nagaraj R et al. Nuclear Localization of Mitochondrial TCA Cycle Enzymes as a Critical Step in Mammalian Zygotic Genome Activation. Cell 168, 210–223 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Sivanand S et al. Nuclear Acetyl-CoA Production by ACLY Promotes Homologous Recombination. Mol. Cell 67, 252–265 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Matsuda S et al. Nuclear pyruvate kinase M2 complex serves as a transcriptional coactivator of arylhydrocarbon receptor. Nucleic Acids Res. 44, 636–647 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Greenwald EC, Mehta S & Zhang J Genetically Encoded Fluorescent Biosensors Illuminate the Spatiotemporal Regulation of Signaling Networks. Chem. Rev 118, 11707–11794 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Chantranupong L, Wolfson RL & Sabatini DM Nutrient-Sensing Mechanisms across Evolution. Cell 161, 67–83 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Evans RM & Mangelsdorf DJ Nuclear Receptors, RXR, and the Big Bang. Cell 157, 255–266 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]