

Abstract
Schizophrenia (SZ) is a neurodevelopmental disorder with cognitive deficits manifesting during early stages of the disease. Evidence suggests that genetic factors in combination with environmental insults lead to complex changes to glutamatergic, GABAergic, and dopaminergic systems. In particular, the N-methyl-D-aspartate receptor (NMDAR), a major glutamate receptor subtype, is implicated in both the disease progression and symptoms of SZ. NMDARs are critical for synaptic plasticity and cortical maturation, as well as learning and memory processes. In fact, any deviation from normal NMDAR expression and function can have devastating consequences. Surprisingly, there is little evidence from human patients that direct mutations of NMDAR genes contribute to SZ. One intriguing hypothesis is that epigenetic changes, which could result from early insults, alter protein expression and contribute to the NMDAR hypofunction found in SZ. Epigenetics is referred to as modifications that alter gene transcription without changing the DNA sequence itself. In this review, we first discuss how epigenetic changes to NMDAR genes could contribute to NMDAR hypofunction. We then explore how NMDAR hypofunction may contribute to epigenetic changes in other proteins or genes that lead to synaptic dysfunction and symptoms in SZ. We argue that NMDAR hypofunction occurs in early stage of the disease, and it may consequentially initiate GABA and dopamine deficits. Therefore, targeting NMDAR dysfunction during the early stages would be a promising avenue for prevention and therapeutic intervention of cognitive and social deficits that remain untreatable. Finally, we discuss potential questions regarding the epigenetic of SZ and future directions for research.
Keywords: NMDA receptor hypofunction, prefrontal cortex, development, schizophrenia, epigenetics
Introduction
Schizophrenia (SZ) is a severe mental illness that affects approximately 1% of the population and confers devastating consequences for the affected person as well as their family members. Patients with SZ suffer from a combination of positive, negative, and cognitive symptoms that severely impact interpersonal relationships as well as the ability to function in society. Despite decades of intense basic and clinical research, the exact etiology of SZ remains unknown. Still, SZ is recognized as a neurodevelopmental disorder, and the pathophysiological process involves multiple disrupted brain circuits and neurotransmitter systems that are closely related to the onset of symptoms (Jaaro-Peled et al., 2010; Lewis and Gonzalez-Burgos, 2008; Weinberger, 1987). Although psychosis emerges late in adolescence or early adulthood, the earliest cognitive symptoms are evident much earlier (Costa et al., 2006; Crow, 2007; Niwa et al., 2010; Walsh et al., 2008).
For over the past 50 years, the dopamine (DA) model has been the leading neurochemical hypothesis of SZ. This model has proven heuristically valuable, with all current medications for SZ functioning to primarily block DA receptor subtype D2 (Gray and Roth, 2007; Tamminga and Lahti, 1996). However, cognitive dysfunction is a core feature of SZ that strongly correlates with functional outcome, and current therapeutics do not adequately address this aspect of the disease. Therefore, it is unlikely that dopaminergic dysfunction, on its own, can fully account for the wide range of symptoms and neurocognitive deficits seen in SZ. The key to forestalling the disorder is to detect and prevent early stages of risk and prodrome with novel therapeutic targets for early treatment (Insel, 2010; Lieberman et al., 2006). It is thus critical to understand the early developmental changes that contribute to the progression of SZ.
The N-methyl-D-aspartate (NMDA) receptor-mediated glutamatergic model provides a critical alternate approach for conceptualizing the brain abnormalities associated with SZ (Harrison and Weinberger, 2005; Lewis and Moghaddam, 2006; Lisman et al., 2008). Although it remains unclear what changes induce the onset of cognitive dysfunction, NMDAR hypofunction appears to be a convergence point for progression and symptoms of SZ, especially for cognitive deficits (Cohen et al., 2015; Coyle, 2006; Lisman et al., 2008; Marek, 2010; Moghaddam, 2003; Nakazawa et al., 2017; Snyder and Gao, 2013). NMDARs are crucial in synaptic plasticity, learning and memory, and proper neuronal functioning. Importantly, NMDARs contribute to circuit formation for early postnatal stages of brain development, which is otherwise known as the “critical developmental window.” Numerous studies have indicated that the maturation of brain circuitry is usually coincident with the NMDAR subunit switch (e.g., GluN2B-to-GluN2A and GluN3A-to-GluN3B) that occurs at the onset of the critical period of development (Monaco et al., 2015; Monyer et al., 1994; Quinlan et al., 1999; Roberts et al., 2009; Sheng et al., 1994; Snyder et al., 2013; Wang and Gao, 2009; Wang et al., 2008). The NMDAR subunit shift marks the transition from juvenile to “adult” neural processing (Dumas, 2005; Henson et al., 2010) and the switch makes the NMDARs extremely vulnerable to genetic and environmental risk factors (Brenhouse et al., 2008; Spear, 2000). It is likely that convergent mechanisms target NMDAR, which in turn contribute to negative symptoms and neurocognitive dysfunction directly (Lau and Zukin, 2007), as well as to positive symptoms via dysregulation of brain DA systems indirectly.
Still the question remains, how do early life experiences influence genes to alter the course of normal brain development. SZ is most likely due to a complex interaction between environmental risk factors and genetic susceptibility (Harrison and Weinberger, 2005). The high heritability rate of SZ points to a strong genetic component for the disease (Cardno and Gottesman, 2000). Indeed, recent genome-wide association studies (GWAS) have identified 108 genetic risk loci in post-mortem SZ tissue (Ripke, 2014) that have an important role for many patients (Birnbaum and Weinberger, 2017; Coelewij and Curtis, 2018; Weinberger, 2017). Yet, the genetic mutations discovered are unlikely to account for all SZ cases or for the complex symptomology present (Rodriguez-Murillo et al., 2012). Further, GWAS studies indicate only a familial liability close to 20%, suggesting that some other mechanisms aside from simple genetic mutation contribute to the heritability rate for SZ (Agerbo et al., 2015). The answer may be epigenetics, which has been gaining attention for its role in mental illness. During the past two decades, understanding of epigenetic processes in chromatin stability, gene regulation, response to environmental factors, and disease states has grown rapidly (Allis and Jenuwein, 2016). Epigenetic mechanisms tightly control gene expression and repression without any alteration to the DNA sequence, and importantly may provide a mechanism for the missed heritability in SZ (Akbarian, 2014; Trerotola et al., 2015).
Epigenetic mechanisms for SZ - epigenetic changes to NMDAR genes contribute to NMDAR hypofunction
Epigenetics is a complex biological process that regulates access to DNA and thus the ease with which genes can be transcribed. Negatively charged DNA is packaged around an octamer of positively charged histones (H), termed H2A, H2B, H3, and H4 to form a nucleosome, a unit of chromatin. Nucleosomes are connected by linker DNA which is stabilized by H1. In addition to packaging DNA, chromatin provides a scaffold for proteins to interact with DNA, including transcriptional machinery as well as repressor proteins. While once thought to be static, it is now known that chromatin in and of itself is a dynamic structure contributing to the accessibility of DNA (Maeshima et al., 2016). Chromatin is further regulated by alterations to DNA through methylation and post-translational modification of histones.
DNA methylation is a process involving the addition of a methyl group to cytosines within DNA. This occurs most frequently at CpG dinucleotides, regions of DNA where a cytosine nucleotide is followed by a guanine nucleotide in the linear sequence of bases (Bayraktar and Kreutz, 2018). DNA methylation is catalyzed by related DNA methyl transferases (DNMTs) that include DNMT1, DNMT3A, DNMT3B, and DNMT3L (Guidotti et al., 2011; Zhubi et al., 2009). These modifications alter gene transcription and can be highly stable and heritable. However, evidence also suggests that DNA methylation patterns can be dynamically regulated in response to learning and memory or life experience (Day and Sweatt, 2011). In contrast, histone modification is a more dynamic and complex process with a myriad of post-translational modifications occurring in concert (Peterson and Laniel, 2004). These include ubiquitylation, acetylation, methylation, and sumoylation of lysines, phosphorylation of serines and threonines, and acetylation and methylation of arginines. This multitude of modifications on chromatin either constrict or loosen the physical attraction/interaction between DNA and histone amino-acid tails, leading to changes in gene transcription. Regulation of histone tails is more readily reversible than DNA methylation and likely plays an important role in plasticity processes and diseases (Christopher et al., 2017). However, a fundamental question raised is when and how epigenetic changes contribute to SZ, and more specifically how they result in NMDAR hypofunction during the early stages of the disease.
There are many ways in which epigenetics could contribute to the SZ disease process. Firstly, there could be global alterations to epigenetic machinery such as histone deacetylases (HDACs) or DNMTs. Data suggest alterations in the expression of various methyltransferases and demethylases in SZ (Morishita et al., 2015). Further, several studies examining DNA methylation in SZ patients found evidence for hypomethylation in specific human populations (Melas et al., 2012; Shimabukuro et al., 2007). However, there is also evidence for hypermethylation in distinct cellular populations (Veldic et al., 2004; Zhubi et al., 2009), supporting the notion that global methylation changes could occur in a brain region or neuronal population-specific manner. For example, one report found that DNMT1 mRNA and protein levels are significantly increased in the telencephalic GABAergic interneurons of SZ patients (Veldic et al., 2004), while another showed overexpression of DNMT3A in distinct GABAergic interneuron populations, and overexpression of both DNMT1 and DNMT3A in peripheral blood lymphocytes (Zhubi et al., 2009). Interestingly, levels of DNMT3A are altered in the prefrontal cortex (PFC) following chronic defeat stress (Elliot and Chapman, 2016; Hammels et al., 2015a; Hammels et al., 2015b). Moreover, prenatal stress, a risk factor in SZ, alters DNMT1 and DNMT3a protein levels, leading to a reduced number of GABAergic interneurons and altered circuit function (Matrisciano et al., 2013). Similarly, early life stress altered plasticity of GABAergic synapses onto ventral tegmental area dopamine neurons via epigenetic mechanisms involving HDACs (Authement et al., 2015). Bahari-Javan and colleagues demonstrated that early life stress increased HDAC1 levels, resulting in altered PFC development and SZ-like phenotypes (Bahari-Javan et al., 2017).
Interestingly, a recent neuroimaging study found reduced expression of HDACs in the dorsolateral prefrontal cortex of SZ patients and this correlated with cognitive deficits (Gilbert et al., 2018). The same group previously showed reduced HDAC2 mRNA levels in the dorsal lateral prefrontal cortex (PFC) of SZ patients (Schroeder et al., 2017). These findings are especially interesting because in mice, HDAC2 expression has been shown to regulate dendritic spines, long-term potentiation, and performance on cognitive tasks (Guan et al., 2009), and HDAC2 was enriched in the promoter of NMDAR subunits compared to HDAC1. However, in this study, HDAC2 knockout mice had increased long-term potentiation (LTP) and improved performance on certain cognitive tasks, which is contradictory to the human study. Future studies are needed to better understand changes to HDACs in normal development and disease models. Still together, these data demonstrate that life experiences can alter epigenetics, impacting the development of brain regions important in SZ and targeting epigenetic machinery may hold therapeutic value (Collier et al., 2016; Ovenden et al., 2018).
Another possibility is that specific SZ-related genes may be epigenetically regulated. SZ human post-post-mortem studies demonstrate epigenetic modifications on several important genes. This includes hypermethylation of the REELIN promoter (Abdolmaleky et al., 2005; Grayson et al., 2005) and DNA methylation changes in the HTR2A, encoding the serotonin receptor type 2A (Cheah et al., 2017) and catechol-O-methyltransferase (COMT) promoter (Gao et al., 2017). Further, SZ patients had a reduction in the activating histone marker H3K4me3 on the glutamate decarboxylase (GAD) 1 promoter, which regulates GAD67 γ-aminobutyric acid (GABA) synthesis enzyme expression and this corresponded to reduced GAD1 mRNA levels (Huang et al., 2007). Of note, this change was found only in females while the methylation changes for COMT were driven by male patients. Together these data point to the possibility of sex differences in SZ epigenetics.
Given the importance of NMDARs both for normal cognitive function and their role in SZ, understanding if and how they are epigenetically regulated is of particular interest. Several studies have demonstrated that NMDAR epigenetic regulation contributes to normal brain development and plasticity processes. Specifically, histone methylation at gene promoters is associated with developmental regulation and region-specific expression of ionotropic and metabotropic glutamate receptors in human brain (Stadler et al., 2005). As listed in Table 1, there is a close association with HDAC2 and the promoter of Grin2a and Grin2b (Graff et al., 2012; Guan et al., 2009; Qin et al., 2018). Further, in the prefrontal cortex, distal regulatory Grin2b sequences are controlled by H3K4me and H3K9me and this is highly associated with the maintenance of working memory (Bharadwaj et al., 2014) and affective and motivational behaviors via setbd1, which encodes an H3K9 methyltransferase (Jiang et al., 2010).
Table 1.
Epigenetic regulation of NMDA receptor subunits.
| Treat ment |
NMDAR subunit expression | Epigenetic effect |
Reference |
|---|---|---|---|
| Human brain development | H3K4me2 causes GRIN1, 2A and 2C increase in PFC, but no change in CB; H3K4me2 causes GRIN2B increase for 27 folds in CB; H3K27me3 induces no change in GRIN1, 2A-C in CB; GRIN2D not detectable; H3K4me2/3 is high at GRIN2B promotor in fetal/infant vs adult in CB. | Open: H3K4me2 and 3 Closed: H3K27me3 H4K20me3 | (Stadler et al., 2005) |
| Treatment | Effector | NMDAR subunit expression |
Epigenetic effect |
Effect on gene expression |
Gene altered |
Reference |
|---|---|---|---|---|---|---|
| Chronic stress | GRIN1 increase | H3K27ac decrease | Gene expression | P300 | (Nasca et al., 2015) | |
| MEF2C | GRIN1 increase | H3K4me3 | Gene expression | (Mitchell et al., 2018) | ||
| REST | GRIN1, GRIN2A | H3K9/14me, H3K9/14ac | Gene silencing | mSin3A,CoREST, HDACs-1/2, G9a, MeCP2 | (Noh et al., 2012) | |
| HDAC2 | GRIN2A decrease | H4K12ac | Gene silencing | (Graff et al., 2012); (Guan et al., 2009) | ||
| HDAC2 | GRIN2A decrease | Gene silencing | (Qin et al., 2018) | |||
| Glutamate in cultured hippocampal neurons | GRIN2A increase | H4ac, H3K4me3, H3K9me3, H3K27me3 | Gene expression | (Kiese et al., 2017) | ||
| GRIN2A increase | DNA hypermethylation | Gene expression | OTX2, LYNX1, GPR | (Kaut et al., 2015) | ||
| GRIN2A increase | H3K27ac | Gene expression | (Sun et al., 2016) | |||
| SETDB1 | GRIN2B decrease | H3K9me2/3 | Gene silencing | HP-1a | (Jiang et al., 2010) | |
| REST | GRIN2B decrease | H3K27me3 increase, H3K4me3 decrease | Gene silencing | G9a | (Rodenas-Ruano et al., 2012) (Tamminga and Zukin, 2015) | |
| REST | GRIN2B decrease | H3K27me3 | Gene silencing | RE-1 | (Gulchina et al., 2017) | |
| Kmt2a/Mll1 | GRIN2B decrease | H3K4me1/2/3 | Gene silencing | (Bharadwaj et al., 2014) | ||
| Kmt1e/SETDB1 | GRIN2B decrease | H3K9me3 | Gene silencing | HP-1 | (Bharadwaj et al., 2014) | |
| GRIN2B increase | H3K27ac | Gene expression | AP-1, NRF-1 | (Bharadwaj et al., 2014) | ||
| Imipramine | GRIN2B increase | H3K9ac increase, H3K27ac increase, HDAC3 & 4 decrease | Gene expression | (Nghia et al., 2015) | ||
| Chronic ethanol (CIE) and TCA | GRIN2B increase | DNA methylation, H3K9ac | Gene expression | AP1, CRE | (Qiang et al., 2010), (Qiang et al., 2014) | |
| ERK | GRIN2B increase | H3K9me2 increase | Gene expression | H/Kmts-G9a, KDM-LSD1 | (Gupta-Agarwal et al., 2014) | |
| Prenatal Bisphenol A exposure | GRIN2B increase | DNA methylation | Gene expression | (Alavian-Ghavanini et al., 2018) | ||
| Vorinostat | HDAC | GRIN2B increase | HDAC inhibition | Gene expression | p-CREB | (Fujita et al., 2012) |
Epigenetic regulation of NMDAR has been found with environmental insults and in mental illness. Prenatal Bisphenol A (a chemical used to make plastic for food container) exposure, which is linked to neurodevelopment disorders, alters Grin2b methylation in both mice and humans (Alavian-Ghavanini et al., 2018b), suggesting that environmental exposures could impact epigenetic regulation of NMDARs. Interestingly, these results are sex-specific and suggest that epigenetic regulation may differ in a sex-specific and cellular specific manner. Epigenome-wide methylation analysis in major depressive disorder, found Grin2a to be hypermethylated in both human PFC and hippocampus (Kaut et al., 2015). Yet, analysis of NMDAR mRNA levels in post-mortem brain tissue of patients with SZ has resulted in conflicting findings with some studies reporting increases, decreases, or no change in transcript levels (Coyle et al., 2012). This inconsistency hinders our ability to identify whether the pathophysiological mechanisms underlying SZ are a direct result of diminished NMDAR subunit proteins (Weickert et al., 2013). Still, while the use of post-mortem brain tissue provides a specific link to SZ, it only allows for analysis of a very specific examination of adult tissue and misses any potential changes that occur earlier in brain development. Nevertheless, evidence suggests that NMDA receptor subunits, especially GRIN2B, are bidirectionally regulated by different epigenetic markers under various conditions (Table 1), including DNA methylation and histone methylation and acetylation (Table 1, Figure 1).
Figure 1.
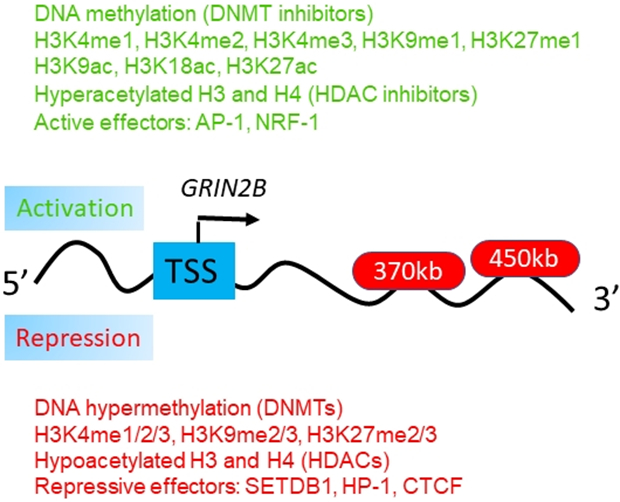
Epigenetic markers and their functions in the regulation of NMDA receptors. Epigenetic markers that could potentially regulate the genes associated with NMDA receptor subunits, e.g., the GRIN2B gene locus (Bharadwaj et al., 2014). Multiple intronic and intergenic DNA sequences, up to 450kb downstream from the GRIN2B/Grin2b transcription start site (TSS), compete for access to the TSS. These sequences are loaded with multiple transcription factors and are in physical proximity to the GRIN2B/Grin2b transcription start site. It was proposed that transcriptional regulation at the GRIN2B/Grin2b locus involves a dynamic and competitive interplay of multiple effectors, each of which could engage with the GRIN2B promoter [Modified from Bharadwaj et al, 2014, Neuron (Bharadwaj et al., 2014). Copyright requested through the Copyright Clearance Center’s RightsLink® service](Rajarajan et al., 2018).
In particular, the higher order of chromatin relevant to the GRIN2B gene locus was recently reported (Bharadwaj et al., 2014). As shown in Figure 1, it is possible that multiple intronic and intergenic DNA sequences, up to 450kb downstream from the GRIN2B/Grin2b transcription start site (TSS), for example, could compete for access to the TSS. These sequences are loaded with multiple transcription factors and are in physical proximity to the GRIN2B/Grin2b transcription start site (Bharadwaj et al., 2014). Interestingly, the sequences interacting with the GRIN2B promoter appeared to harbor single nucleotide polymorphisms (SNP) and are implicated with liability for working memory and SZ (Bharadwaj et al., 2014). Therefore, epigenetic markers associated with DNA methylation or histone modification may target the GRIN2B promoter and carry disease-associated sequence polymorphisms. This consequently may either facilitate or repress expression in a highly dynamic and activity-dependent manner for access to the GRIN2B promoter sequences (Bharadwaj et al., 2014). Importantly, this GRIN2B higher order chromatin study and others (Table 1) have provided a deeper insight into the understanding of the neurobiology of and novel treatment avenues for common psychiatric disease (Rajarajan et al., 2018).
Interestingly and importantly, epigenetic regulation of NMDARs is known to be an important factor in the critical developmental period of the NMDAR subunit switch through repressor element 1-silencing transcription factor (REST). Historically, REST was thought to only play a critical role early in neuronal differentiation, brain development, and neurodegenerative diseases (Hwang and Zukin, 2018; Tamminga and Zukin, 2015). However, REST is now known to be involved in synaptic plasticity, normal aging, and disease processes (Lu et al., 2014; Rodenas-Ruano et al., 2012). REST binds within the promoter region of target genes and recruits co-repressor CoREST and mSin3 that recruit multiple proteins and possess enzymes that catalyze the posttranslational modifications of histone tails (Andres et al., 1999). These include, but are not limited to, multiple HDACs, histone methyltransferases, and DNMTs (Qureshi and Mehler, 2009). While it has been known that REST functions during embryogenesis to epigenetically regulate many genes, including Grin2b, Zukin and colleagues discovered an important role for REST in mature neurons (Rodenas-Ruano et al., 2012; Schoenherr and Anderson, 1995). They found that REST is activated at a critical window of time in hippocampal neurons, possibly through the actions of the prion protein, and acts to repress Grin2b expression via epigenetic remodeling (Rodenas-Ruano et al., 2012; Song et al., 2018). Thus, under normal physiological conditions, epigenetics plays a fundamental role in regulating NMDAR levels leaving open the possibility that this process could be dysregulated in SZ.
Studies from SZ animal models indicate that NMDAR subunit levels are reduced in a brain region and developmental time window specific manner (Gulchina et al., 2017; Snyder and Gao, 2013). Using the prenatal methylazoxymethanol acetate (MAM) prenatal exposure model, we found reduced expression of NMDAR subunits in the juvenile hippocampus that altered NMDAR function during this critical stage of development. We also recently demonstrated that GluN2B expression and function are compromised in PFC of this SZ model. Further, this protein loss during the juvenile period is correlated with an aberrant increase in the epigenetic transcriptional repressor REST and the repressive histone marker H3K27me3 at the Grin2b promoter, as assayed by chromatin immunoprecipitation (ChIP)-quantitative polymerase chain reaction (Gulchina et al., 2017). These studies provide evidence that epigenetic regulation of NMDARs could contribute to the disease process of SZ (Figure 2).
Figure 2.

A schematic representation of epigenetic changes to NMDARs on pyramidal neurons in Schizophrenia (SZ). A. Under normal conditions, NMDARs are subjected to both activating (green hexagons) and repressive epigenetic markers (red ovals). This balance allows for the proper expression of NMDARs. However prenatal insult, early life stress, or inherited epigenetic marks, switch the balance creating an over-abundance of repressive epigenetic markers. These modifications lead to a repression of NMDAR transcription and the NMDAR hypofunction found in SZ. B. Top panel, NR2B-NMDAR levels are persistently high in the developing mPFC and play a critical role in synaptic plasticity and PFC-dependent cognitive functions. However, in the MAM SZ model, animals are vulnerable to cognitive dysfunction due to the significant loss of synaptic NR2B-NMDARs in the critical juvenile period. Bottom panel, NR2B-NMDAR loss in juvenile MAM rat mPFC is due to epigenetic modifications within the promoter of Grin 2b. REST (red ovals) and H3K27me3 (black triangles) enrichment levels in saline mPFC are higher at the Grin2b response element (RE1) site compared to the Grin1 and Grin2a RE1 sites, demonstrating selective REST regulation of Grin2b is an endogenous repressor mechanism. In the MAM mPFC, REST and H3K27me3 enrichment levels are greatly elevated compared to the saline group, demonstrating the hyper-repression of the proximal promoter region of Grin2b (Modified from Gulchina et al, 2017, J. Neurochem. Copyright requested through Copyright Clearance Center’s RightsLink® service).
NMDAR hyopfunctionality induces epigenetic changes in other proteins and/or genes that lead to synaptic dysfunction and symptoms in SZ
In addition to epigenetic regulation of NMDARs themselves, it is important to consider the consequence of NMDAR hypofunction to epigenetic processes. Neuronal activity is translated into transcription alterations in a variety of genes, dependent upon the type and duration of stimulation (Flavell and Greenberg, 2008). These changes are critical both for the normal development of neuronal networks but also for incorporating sensory experiences in postnatal life. Kyrke-Smith and Williams recently proposed that neuronal network function, particularly for synaptic plasticity, is dependent upon proper expression of a wide variety of proteins regulated through epigenetic changes (Kyrke-Smith and Williams, 2018). The mechanism(s) by which this occurs is a very active area of research, but data suggest that epigenetic changes together with NMDARs play a critical role (Jarome and Lubin, 2014; Tyssowski et al., 2018). Calcium influx through NMDARs activates a calcium calmodulin-dependent kinase II (CaMKII)- cAMP response element-binding protein (CREB)-P signalling pathway that alters levels of H3K27ac to control dendritogenesis (Bustos et al., 2017). In another example, contextual fear conditioning led to a rapid, time-dependent increase in histone H3 phosphorylation in hippocampal area CA1. Interestingly, this was blocked by MK801, an NMDAR antagonist (Chwang et al., 2006). Similarly, NMDAR activation in hippocampal slices increased ERK2 and acetylation of histone 3 (Levenson et al., 2004). Neuronal stimulation induced long-term potentiation, which requires NMDAR activation, increased expression of HDAC1 as well as the HDAC interacting protein DnaJ Heat Shock Protein Family (Hsp40) Member B5 (Ryan et al., 2012).
Based on these data, it is reasonable to hypothesize that alterations to glutamatergic synaptic transmission, due to NMDAR hypofunction as in SZ, could contribute to a disruption in network functioning and/or improper network formation. For example, ablation of NMDARs during postnatal development led to epigenetic repression of Kv1.1-type potassium channels, altering neuronal excitability and impairing dendritic maturation (Frangeul et al., 2017). Further, inhibiting NMDAR activity prevented increases in histone H3 acetylation and phosphorylation in striatal tissue extracts (Li et al., 2004). Conversely, NMDAR blockade with MK-801 increased phosphorylation of histone 3 (H3) at serine 10 in the medial PFC, which is associated with chromatin relaxation and increased probability of gene expression (Mackowiak et al., 2013). Together these data suggest that any increase or decrease in NMDAR function will impact epigenetic processes, resulting in an alteration in normal synapse and circuit function.
One of the most important properties of the NMDA receptor is their calcium permeability. Along with changes to cellular excitability, NMDAR hypofunction also leads to drastic alterations in calcium influx and cellular signalling. Evidence indicates that the activity of epigenetic machinery could be impacted by the changes to calcium influx. Calcium is the critical second messenger that allows synapse to nucleus communication to alter gene transcription (Bading, 2013). NMDAR mediated calcium influx, along with calcium from voltage-gated calcium channels (VGCC) engages a variety of signalling cascades to transmit information about neuronal activity in the dendrites to the nucleus (Bading, 2013). Data suggests a high fidelity between synaptic activation and the nuclear calcium signal which can be disrupted by NMDAR or VGCC blockade {Bengtson, 2010 #39226}. Once in the nucleus, calcium can lead to activation of CREB-CREB binding protein (CBP) dependent transcription. Interestingly, CBP partially functions as a histone acetyltransferase (HAT) as well as associates with many HATs to regulate chromatin and thus gene transcription (Chan and La Thangue, 2001). Additionally, epigenetic machinery itself can be targeted by calcium-induced signaling cascades. For example, DNMTs undergo post-translational modifications, which likely regulate their functional activity (Denis et al., 2011). DNMTs are phosphorylated by protein kinase C and protein kinase B (AKT) among others (Denis et al., 2011). Interestingly, calcium entry through NMDARs can trigger activation of CaMKII and subsequently AKT (Sutton and Chandler, 2002). Thus, it is possible that reduced NMDAR function, as seen in SZ, will affect protein kinase activation that may, in turn, alter DNMT functionality or expression. In fact, NMDAR mediated calcium entry is critical for both short-term and long-term synaptic plasticity. Therefore, alteration of NMDAR function could consequentially regulate other proteins or genes that are critically involved in synaptic functions (Figure 3).
Figure 3.

Mechanism of NMDAR hypofunction and its consequences. A. Mechanisms of NMDA hypofunction in excitatory pyramidal neurons. Genetic and other risk factors lead to epigenetic alterations with the promoter regions of NMDARs. These, in turn, lead to reduced mRNA and protein expression with pyramidal neurons culminating in NMDAR hypofunction. B. NMDAR hypofunction within excitatory pyramidal neurons induces a cascade of downstream effects that alter synaptic development and sub-sequential GABAergic and dopaminergic dysfunction. Together these changes to neuronal network formation and function induce the learning, cognitive, and social deficit found in schizophrenia.
Brain-derived neurotrophic factor (BDNF) is a neurotrophic that is involved in neurodevelopment, synaptogenesis, and neuroplasticity (Angelucci et al., 2005; Poo, 2001). Recent work confirmed that reduced BDNF levels in humans are significantly associated with SZ (Rodrigues-Amorim et al., 2018). Interestingly, epigenetic regulation of BDNF itself is suggested to play a role in psychiatric disorders, including SZ (Ikegame et al., 2013; Mitchelmore and Gede, 2014). In the hippocampus, BDNF secretion in response to activity requires activation of calcium-responsive transcription factors and is at least partially dependent on calcium influx through NMDARs (Kolarow et al., 2007; Tao et al., 2002). In hippocampal cultures, NMDA stimulation and resulting calcium influx, increased phosphorylation of MeCp2 leading to increased BDNF mRNA levels (Zhou et al., 2006). Work by Lipsky’s group expanded these findings with CHIP to show that NMDA activation results in multiple epigenetic changes to the BDNF promoter, including a decrease in histone H3 at lysine 9 dimethylation (H3K9me2) and increases in H3K4me2 and H3K9/14 acetylation (H3AcK9/14) (Tian et al., 2009). Furthermore, Lau et al. show that BDNF-induced neuroprotection is mediated by synaptic NMDA-receptor-dependent nuclear calcium signals activating transcription of inhibin b-A (activin A). Activin A, in turn, reduces toxic extrasynaptic NMDAR-mediated calcium influx, shields neurons from mitochondrial dysfunction, and protects against stroke-induced brain damage (Lau et al., 2015). Therefore, NMDAR dysfunction could significantly impact BDNF levels that in turn could negatively impact other systems involved in the pathophysiology of SZ. For example, an interaction between BDNF and dopamine receptor type 3 is implicated in SZ (Gourion et al., 2005). A clearer link exists between BDNF and GABA deficits (Hashimoto et al., 2005; Lu et al., 2009), as well as BDNF and DA dysfunction (Krebs et al., 2000), but the correlation between NMDAR hypofunction and BDNF, and whether NMDAR hypofunction can affect BDNF secretion in the brain, remain to be explored.
Targeting NMDAR dysfunction during early stage is a promising avenue for prevention and therapeutic intervention of cognitive and social deficits
Early NMDAR hypofunction is a promising proposed mechanism for the development of SZ. However, it is critical to progress from a better understanding of disease mechanisms to promising therapeutic interventions. Unfortunately, less information can be gathered from the developing human SZ brain. Diagnosis is often in late adolescence and early adulthood and patients have already developed neuropathophysiological deficits that underlie cognitive and social impairments before treatment begins. It is unknown if treatment could fully reverse alterations to neuronal network development. Intervention during PFC development in patients at-risk for SZ presents a therapeutic window that is currently unopened.
As reviewed above, mounting evidence indicates that NMDAR hypofunction occurs during the early stages of postnatal development and is proceeded by disruption of the dopaminergic systems. The current therapeutic approaches target this later dopaminergic dysfunction to minimize the symptoms of SZ (Yang and Tsai, 2017). Further, current antipsychotic medications demonstrate minimal therapeutic efficacy in treating cognitive and social deficits in SZ (Kim et al., 2017). However, therapeutic agents that have previously failed to demonstrate efficacy in clinical trials may be more effective when employed during the earlier stages of development, specifically during the initial stages of NMDAR hypofunction (Li et al., 2015b). Indeed, treatment during juvenile development (Li et al., 2017b; Xing et al., 2018) with a metabotropic glutamate receptors (mGluR)2 agonist/mGluR3 antagonist or mGluR2/3 agonist has demonstrated ameliorative effects on NMDAR-mediated neurotransmission, working memory function, and social behaviors, as well synaptic morphology in the mPFC in animal models.
The mGluR2/3 subtypes of glutamate receptors have long been linked with SZ, and mGluR2/3 agonism has been proposed to target the dysfunctional glutamatergic system, particularly NMDAR hypofunction (Li et al., 2015b; Maksymetz et al., 2017; Moghaddam, 2004; Walker and Conn, 2015). In a series of recent studies using the MAM SZ model, we found that targeting NMDAR hypofunction during the early stage of development is effective to prevent SZ-like cognitive and social phenotypes in adult. In juvenile MAM animals, synaptic GluN2B protein levels are significantly decreased resulting in reduced NMDA-excitatory postsynaptic currents. Further, this protein loss was correlated with an aberrant increase in the enrichment of the epigenetic transcriptional repressor REST and the repressive histone marker H3K27me3 at the Grin2b promoter, as assayed by ChIP-qPCR (Gulchina et al., 2017). In adulthood, MAM animals exhibited PFC-dependent learning and memory deficits. Excitingly, both prefrontal NMDAR hypofunction and cognitive deficits were prevented by treatment with either mGluR2/3 modulators mGluR2 agonist/mGluR3 antagonist LY395756 (Li et al., 2017a) and mGluR2/3 agonist LY379268 (Xing et al., 2018) in the juvenile, but not adult (Li et al., 2015a) rats. Both LY395756 LY379268 prevented NMDA dysfunction, learning deficits, and cognitive flexibility. We also found that the preventive effects of LY379268 were mediated by improving GluN2B-NMDAR function via inhibiting GSK3β (Xing et al., 2018).
Our work confirmed that NMDAR hypofunction is a feature of early postnatal development, with epigenetic hyper-repression of the Grin2b promoter being a contributing factor. The selective loss of GluN2B protein and subsequent synaptic dysfunction weakens the PFC function during development and may underlie early cognitive impairments in SZ models and patients. The latter is supported by the efficacy of glutamatergic modulators to prevent both prefrontal NMDAR hypofunction and cognitive deficits, highlighting the importance of targeting glutamatergic dysfunction as a potential early intervention for SZ. Altogether, these findings emphasize the importance of developmental NMDAR function in the maturation of cognitive functions, a process which is disrupted and can be subsequently recovered in the MAM model for SZ by a brief pharmacological intervention during juvenile development.
Another avenue of therapeutic intervention is targeting HDACs. HDACs compact chromatin structure and repress gene transcription. Clinical studies have demonstrated that HDAC inhibitors are efficacious when given in combination with atypical antipsychotics in the treatment of SZ (Fischer et al., 2010; Kurita et al., 2012). Specifically, chronic atypical antipsychotics downregulated the transcription of mGluR2 by decreasing histone acetylation at its promoter in mouse and human frontal cortex. Conversely, HDAC inhibitors prevented the repressive histone modifications induced at the mGluR2 promoter by atypical antipsychotics and augmented their therapeutic-like effects, suggesting that HDAC2 is a promising new target to improve SZ treatment (Kurita et al., 2013a). Furthermore, it has been reported that HDAC2 regulates atypical antipsychotic responses through the modulation of mGluR2 promoter activity in adult mice (Kurita et al., 2012). Kurita et al also reported repressive epigenetic changes at the mGluR2 promoter in the frontal cortex of 5-HT2A knockout mice (Kurita et al., 2013b). Specifically, disruption of 5-HT2A receptor-dependent signaling in mice was associated with decreased acetylation of histone H3 (H3ac) and H4 (H4ac) and increased H3K27me3 enrichment at the mGlu2 promoter, suggesting transcriptional repression of mGluR2 expression and possible indirect regulation of NMDAR function. However, despite the importance of the therapeutic potential of HDAC inhibition, it remains unclear exactly if and how HDAC inhibitors, mGluR, and NMDARs are related. More research is needed.
One question raised by these studies is whether positive allosteric modulators of mGluR2 would work better for SZ treatment and if this is related to NMDAR regulation (Ellaithy et al., 2015). Allosteric signaling through a mGluR2 and 5-HT2A heteromeric receptor complex has shown its potential contribution to SZ (Moreno et al., 2016). Ibi et al reported that antipsychotic-induced HDAC2 transcription via nuclear factor kappa-light-chain-enhancer of activated B cells (NF-kB) leads to synaptic and cognitive side effects (Ibi et al., 2017), suggesting a protective effect on cognition from antipsychotics. In addition, Mitchell et al found that MEF2C transcription factor is associated with genetic and epigenetic risk architectures of SZ and improves cognition in mice (Mitchell et al., 2018). However, whether these effects have anything to do with glutamatergic dysfunction is unknown. A recent study reported that vorinostat, a histone deacetylase inhibitor, facilitates fear extinction and enhances expression of the hippocampal GluN2B-containing NMDA receptor gene (Fujita et al., 2012). In addition, it has been shown that basal, but not evoked, NMDAR activity regulates gene expression in part through HDAC4, and, that HDAC4 has neuroprotective functions under conditions of low NMDAR activity, at least in hippocampal neuronal cultures (Chen et al., 2014). However, whether HDAC inhibitors would have side effects in the juvenile brain by regulating NMDAR activity directly is another concern that is worth exploring.
Potential questions regarding the epigenetics of SZ and future directions for research
SZ is a polygenic condition whose exact etiology remains unknown. Recent research highlights NMDAR hypofunctioning as an attractive hypothesis of its molecular basis (Snyder and Gao, 2013); however, NMDAR hypofunction itself could not explain the other anatomical and clinical abnormalities such as GABAergic deficits (Gonzalez-Burgos and Lewis, 2008) and dopaminergic dysfunction (Howes and Kapur, 2009) found in SZ. Although SZ is a neurodevelopmental disorder, the visible onset traditionally appears in later adolescence and is preceded by a period of apparent abnormality. Understanding exactly when and how brain development moves off course is of critical importance. Most likely there is more than one molecular mechanism involved and they are all governed by common factors. Reelin, Nrg1, and BDNF have been linked to SZ and they have three common features: their involvement in the GABAergic interneuron migration, in the proper functioning of the NMDA receptors, and they are all subject to similar epigenetic control (Moreau and Kullmann, 2013). Considering all of these aspects for treatment and how they are intertwined should improve the chances of successful therapeutic intervention. Future directions for treatment include mGluR2 positive allosteric modulators (PAM), selective mGluR2 agonist and mGluR effects through regulation of epigenetic mechanism, and treatment in different animal models, especially genetic models during early stage and multiple behavioral evaluations (Ellaithy et al., 2015; Griebel et al., 2016; Maksymetz et al., 2017; Matrisciano et al., 2016).
Operating under the hypothesis that SZ is a disorder of synaptic dysfunction, one may believe a single treatment, or combination of therapeutic agents will be able to completely recover cognitive deficits in this disorder. However, we must not forget the overt structural damage to the SZ brain. Decreases in grey and white matter, as observed by fractional anisotropy, demonstrate reductions in myelination (Green et al., 2004). Therefore, we must consider cognitive impairments, and even positive and negative symptoms, can likely only recover to a certain degree if treatment begins in the later stages, given our current inability to treat these structural alterations (Green et al., 2004). Nonetheless, functional improvement for patients will lessen the economic burden of this debilitating illness as well as the psychosocial burden on family and caregivers. These steps to independence for SZ patients are the ultimate goal of our research endeavors.
There are many outstanding questions for epigenetic changes in the developing brain, particularly the effects on behaviors associated with cognitive function and sociability, as recently pointed by Kenerne et al (Keverne et al., 2015) in the Sackler Colloquium. For example, what are the roles of DNA methylation, including CpG islands, in NMDAR hypofunction? How does DNA methylation affect NMDAR subunits change during development? What are the relations among different epigenetic marks with NMDAR subunit composition and function during development in both normal brain and animal models for SZ? What are the roles of long noncoding RNAs in the modification of neuronal nuclear architecture, especially on NMDAR hypofunction occurrence? A large body of behavioral epigenetic studies attempts to correlate epigenetic marker changes (e.g., acetyl-histone H3) at global levels and in mixed populations of cells with phenotypic changes. However, specific changes at gene and single cell levels correlating with behavioral changes remain largely unknown. This is particularly true for NMDAR function in GABAergic interneurons and glial cells. The role of glial in normal and disease conditions is rapidly expanding and cannot be ignored for SZ. What are the epigenetic mechanisms for NMDAR subunit changes during the early development that would eventually be triggered to initiate the onset of SZ symptoms, especially early cognitive and social deficits? Answering these questions will provide not only new insights into the understanding of neuropathophysiology but also eventual early interventions for SZ.
Acknowledgments
This paper is supported by the NIH R01MH085666 to W.J. Gao. We would like to thank Ms Sarah Monaco for comments on the manuscript.
Abbreviations
- CREB-CBP
cAMP response element-binding protein (CREB)-CREB binding protein
- DA
dopamine
- DISC1
disrupted-in-schizophrenia 1
- DNMT
DNA methyltransferase
- DTNBP1
dystrobrevin binding protein 1
- GABA
gamma-aminobutyric acid
- GWAS
genome-wide association study
- HAT
acetyltransferase
- HDAC
histone deactelyases
- DNA
methyl transferases
- NRG1
neuregulin 1
- MAM
methylazoxymethanol acetate
- NMDA
N-methyl-D-aspartate
- PFC
prefrontal cortex
- REST
repressor element 1-silencing transcription factor
- SZ
schizophrenia
- VGCC
voltage-gated calcium channels
Footnotes
Conflicts of Interests
The authors claim no financial and other conflicts of interest.
References
- Abdolmaleky HM, Cheng KH, Russo A, Smith CL, Faraone SV, Wilcox M, Shafa R, Glatt SJ, Nguyen G, Ponte JF, Thiagalingam S, Tsuang MT, 2005. Hypermethylation of the reelin (RELN) promoter in the brain of schizophrenic patients: a preliminary report. American journal of medical genetics. Part B, Neuropsychiatric genetics : the official publication of the International Society of Psychiatric Genetics 134b(1), 60–66. [DOI] [PubMed] [Google Scholar]
- Agerbo E, Sullivan PF, Vilhjalmsson BJ, Pedersen CB, Mors O, Borglum AD, Hougaard DM, Hollegaard MV, Meier S, Mattheisen M, Ripke S, Wray NR, Mortensen PB, 2015. Polygenic Risk Score, Parental Socioeconomic Status, Family History of Psychiatric Disorders, and the Risk for Schizophrenia: A Danish Population-Based Study and Meta-analysis. JAMA psychiatry 72(7), 635–641. [DOI] [PubMed] [Google Scholar]
- Akbarian S, 2014. Epigenetic mechanisms in schizophrenia. Dialogues in Clinical Neuroscience 16(3), 405–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alavian-Ghavanini A, Lin P-I, Lind PM, Risén Rimfors S, Halin Lejonklou M, Dunder L, Tang M, Lindh C, Bornehag C-G, Rüegg J, 2018a. Prenatal Bisphenol A Exposure is Linked to Epigenetic Changes in Glutamate Receptor Subunit Gene Grin2b in Female Rats and Humans. Scientific Reports 8(1), 11315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allis CD, Jenuwein T, 2016. The molecular hallmarks of epigenetic control. Nature reviews. Genetics 17(8), 487–500. [DOI] [PubMed] [Google Scholar]
- Andres ME, Burger C, Peral-Rubio MJ, Battaglioli E, Anderson ME, Grimes J, Dallman J, Ballas N, Mandel G, 1999. CoREST: a functional corepressor required for regulation of neural-specific gene expression. Proceedings of the National Academy of Sciences of the United States of America 96(17), 9873–9878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angelucci F, Brene S, Mathe AA, 2005. BDNF in schizophrenia, depression and corresponding animal models. Mol Psychiatry 10(4), 345–352. [DOI] [PubMed] [Google Scholar]
- Authement ME, Kodangattil JN, Gouty S, Rusnak M, Symes AJ, Cox BM, Nugent FS, 2015. Histone Deacetylase Inhibition Rescues Maternal Deprivation-Induced GABAergic Metaplasticity through Restoration of AKAP Signaling. Neuron 86(5), 1240–1252. [DOI] [PubMed] [Google Scholar]
- Bading H, 2013. Nuclear calcium signalling in the regulation of brain function. Nature reviews. Neuroscience 14(9), 593–608. [DOI] [PubMed] [Google Scholar]
- Bahari-Javan S, Varbanov H, Halder R, Benito E, Kaurani L, Burkhardt S, Anderson-Schmidt H, Anghelescu I, Budde M, Stilling RM, Costa J, Medina J, Dietrich DE, Figge C, Folkerts H, Gade K, Heilbronner U, Koller M, Konrad C, Nussbeck SY, Scherk H, Spitzer C, Stierl S, Stöckel J, Thiel A, von Hagen M, Zimmermann J, Zitzelsberger A, Schulz S, Schmitt A, Delalle I, Falkai P, Schulze TG, Dityatev A, Sananbenesi F, Fischer A, 2017. HDAC1 links early life stress to schizophrenia-like phenotypes. Proceedings of the National Academy of Sciences 114(23), E4686–E4694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayraktar G, Kreutz MR, 2018. Neuronal DNA Methyltransferases: Epigenetic Mediators between Synaptic Activity and Gene Expression? The Neuroscientist : a review journal bringing neurobiology, neurology and psychiatry 24(2), 171–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bharadwaj R, Peter CJ, Jiang Y, Roussos P, Vogel-Ciernia A, Shen EY, Mitchell AC, Mao W, Whittle C, Dincer A, Jakovcevski M, Pothula V, Rasmussen TP, Giakoumaki SG, Bitsios P, Sherif A, Gardner PD, Ernst P, Ghose S, Sklar P, Haroutunian V, Tamminga C, Myers RH, Futai K, Wood MA, Akbarian S, 2014. Conserved higher-order chromatin regulates NMDA receptor gene expression and cognition. Neuron 84(5), 997–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birnbaum R, Weinberger DR, 2017. Genetic insights into the neurodevelopmental origins of schizophrenia. Nature reviews. Neuroscience 18(12), 727–740. [DOI] [PubMed] [Google Scholar]
- Brenhouse HC, Sonntag KC, Andersen SL, 2008. Transient D1 dopamine receptor expression on prefrontal cortex projection neurons: relationship to enhanced motivational salience of drug cues in adolescence. J. Neurosci. 28(10), 2375–2382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bustos FJ, Jury N, Martinez P, Ampuero E, Campos M, Abarzua S, Jaramillo K, Ibing S, Mardones MD, Haensgen H, Kzhyshkowska J, Tevy MF, Neve R, Sanhueza M, Varela-Nallar L, Montecino M, van Zundert B, 2017. NMDA receptor subunit composition controls dendritogenesis of hippocampal neurons through CAMKII, CREB-P, and H3K27ac. Journal of cellular physiology 232(12), 3677–3692. [DOI] [PubMed] [Google Scholar]
- Cardno AG, Gottesman II 2000. Twin studies of schizophrenia: from bow-and-arrow concordances to star wars Mx and functional genomics. American journal of medical genetics 97(1), 12–17. [PubMed] [Google Scholar]
- Chan HM, La Thangue NB, 2001. p300/CBP proteins: HATs for transcriptional bridges and scaffolds. Journal of cell science 114(Pt 13), 2363–2373. [DOI] [PubMed] [Google Scholar]
- Cheah SY, Lawford BR, Young RM, Morris CP, Voisey J, 2017. mRNA Expression and DNA Methylation Analysis of Serotonin Receptor 2A (HTR2A) in the Human Schizophrenic Brain. Genes (Basel) 8(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Wang Y, Modrusan Z, Sheng M, Kaminker JS, 2014. Regulation of Neuronal Gene Expression and Survival by Basal NMDA Receptor Activity: A Role for Histone Deacetylase 4. The Journal of Neuroscience 34(46), 15327–15339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christopher MA, Kyle SM, Katz DJ, 2017. Neuroepigenetic mechanisms in disease. Epigenetics & chromatin 10(1), 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chwang WB, O'Riordan KJ, Levenson JM, Sweatt JD, 2006. ERK/MAPK regulates hippocampal histone phosphorylation following contextual fear conditioning. Learning & memory; (Cold Spring Harbor, N.Y.) 13(3), 322–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coelewij L, Curtis D, 2018. Mini-review: Update on the genetics of schizophrenia. Annals of human genetics 82(5), 239–243. [DOI] [PubMed] [Google Scholar]
- Cohen SM, Tsien RW, Goff DC, Halassa MM, 2015. The impact of NMDA receptor hypofunction on GABAergic interneurons in the pathophysiology of schizophrenia. Schizophrenia research 167(0), 98–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collier DA, Eastwood BJ, Malki K, Mokrab Y, 2016. Advances in the genetics of schizophrenia: toward a network and pathway view for drug discovery. Ann N Y Acad Sci 1366(1), 61–75. [DOI] [PubMed] [Google Scholar]
- Costa E, Dong E, Grayson DR, Ruzicka WB, Simonini MV, Veldic M, Guidotti A, 2006. Epigenetic targets in GABAergic neurons to treat schizophrenia. Adv Pharmacol 54, 95–117. [DOI] [PubMed] [Google Scholar]
- Coyle JT, 2006. Glutamate and schizophrenia: beyond the dopamine hypothesis. Cellular and Molecular Neurobiology 26(4-6), 365–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coyle JT, Basu A, Benneyworth M, Balu D, Konopaske G, 2012. Glutamatergic synaptic dysregulation in schizophrenia: therapeutic implications. Handb Exp Pharmacol(213), 267–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crow TJ, 2007. How and why genetic linkage has not solved the problem of psychosis: review and hypothesis. Am J Psychiatry 164(1), 13–21. [DOI] [PubMed] [Google Scholar]
- Day JJ, Sweatt JD, 2011. Epigenetic modifications in neurons are essential for formation and storage of behavioral memory. Neuropsychopharmacology : official publication of the American College of Neuropsychopharmacology 36(1), 357–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denis H, Ndlovu MN, Fuks F, 2011. Regulation of mammalian DNA methyltransferases: a route to new mechanisms. EMBO reports 12(7), 647–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumas TC, 2005. Developmental regulation of cognitive abilities: modified composition of a molecular switch turns on associative learning. Prog Neurobiol 76(3), 189–211. [DOI] [PubMed] [Google Scholar]
- Ellaithy A, Younkin J, Gonzalez-Maeso J, Logothetis DE, 2015. Positive allosteric modulators of metabotropic glutamate 2 receptors in schizophrenia treatment. Trends Neurosci 38(8), 506–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliot AJ, Chapman BP, 2016. Socioeconomic status, psychological resources, and inflammatory markers: Results from the MIDUS study. Health psychology : official journal of the Division of Health Psychology, American Psychological Association 35(11), 1205–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer A, Sananbenesi F, Mungenast A, Tsai L-H, 2010. Targeting the correct HDAC(s) to treat cognitive disorders. Trends in Pharmacological Sciences 31(12), 605–617. [DOI] [PubMed] [Google Scholar]
- Flavell SW, Greenberg ME, 2008. Signaling mechanisms linking neuronal activity to gene expression and plasticity of the nervous system. Annu Rev Neurosci 31, 563–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frangeul L, Kehayas V, Sanchez-Mut JV, Fievre S, Krishna KK, Pouchelon G, Telley L, Bellone C, Holtmaat A, Graff J, Macklis JD, Jabaudon D, 2017. Input-dependent regulation of excitability controls dendritic maturation in somatosensory thalamocortical neurons. Nat Commun 8(1), 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita Y, Morinobu S, Takei S, Fuchikami M, Matsumoto T, Yamamoto S, Yamawaki S, 2012. Vorinostat, a histone deacetylase inhibitor, facilitates fear extinction and enhances expression of the hippocampal NR2B-containing NMDA receptor gene. Journal of psychiatric research 46(5), 635–643. [DOI] [PubMed] [Google Scholar]
- Gao S, Cheng J, Li G, Sun T, Xu Y, Wang Y, Du X, Xu G, Duan S, 2017. Catechol-O-methyltransferase gene promoter methylation as a peripheral biomarker in male schizophrenia. European psychiatry : the journal of the Association of European Psychiatrists 44, 39–46. [DOI] [PubMed] [Google Scholar]
- Gilbert TM, Zürcher NR, Wu CJ, Bhanot A, Hightower BG, Kim M, Albrecht DS, Wey H-Y, Schroeder FA, Rodriguez-Thompson A, Morin TM, Hart KL, Pellegrini AM, Riley MM, Wang C, Stufflebeam SM, Haggarty SJ, Holt DJ, Loggia ML, Perlis RH, Brown HE, Roffman JL, Hooker JM, 2018. PET neuroimaging reveals histone deacetylase dysregulation in schizophrenia. The Journal of clinical investigation 129(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Burgos G, Lewis DA, 2008. GABA Neurons and the Mechanisms of Network Oscillations: Implications for Understanding Cortical Dysfunction in Schizophrenia. Schizophrenia bulletin 34(5), 944–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gourion D, Goldberger C, Leroy S, Bourdel MC, Olie JP, Krebs MO, 2005. Age at onset of schizophrenia: interaction between brain-derived neurotrophic factor and dopamine D3 receptor gene variants. Neuroreport 16(12), 1407–1410. [DOI] [PubMed] [Google Scholar]
- Graff J, Rei D, Guan J-S, Wang W-Y, Seo J, Hennig KM, Nieland TJF, Fass DM, Kao PF, Kahn M, Su SC, Samiei A, Joseph N, Haggarty SJ, Delalle I, Tsai L-H, 2012. An epigenetic blockade of cognitive functions in the neurodegenerating brain. Nature 483(7388), 222–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray JA, Roth BL, 2007. The pipeline and future of drug development in schizophrenia. Mol Psychiatry 12(10), 904–922. [DOI] [PubMed] [Google Scholar]
- Grayson DR, Jia X, Chen Y, Sharma RP, Mitchell CP, Guidotti A, Costa E, 2005. Reelin promoter hypermethylation in schizophrenia. Proceedings of the National Academy of Sciences of the United States of America 102(26), 9341–9346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green MF, Nuechterlein KH, Gold JM, Barch DM, Cohen J, Essock S, Fenton WS, Frese F, Goldberg TE, Heaton RK, Keefe RS, Kern RS, Kraemer H, Stover E, Weinberger DR, Zalcman S, Marder SR, 2004. Approaching a consensus cognitive battery for clinical trials in schizophrenia: the NIMH-MATRICS conference to select cognitive domains and test criteria. Biological psychiatry 56(5), 301–307. [DOI] [PubMed] [Google Scholar]
- Griebel G, Pichat P, Boulay D, Naimoli V, Potestio L, Featherstone R, Sahni S, Defex H, Desvignes C, Slowinski F, Vige X, Bergis OE, Sher R, Kosley R, Kongsamut S, Black MD, Varty GB, 2016. The mGluR2 positive allosteric modulator, SAR218645, improves memory and attention deficits in translational models of cognitive symptoms associated with schizophrenia. Sci Rep 6, 35320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan JS, Haggarty SJ, Giacometti E, Dannenberg JH, Joseph N, Gao J, Nieland TJ, Zhou Y, Wang X, Mazitschek R, Bradner JE, DePinho RA, Jaenisch R, Tsai LH, 2009. HDAC2 negatively regulates memory formation and synaptic plasticity. Nature 459(7243), 55–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guidotti A, Auta J, Chen Y, Davis JM, Dong E, Gavin DP, Grayson DR, Matrisciano F, Pinna G, Satta R, Sharma RP, Tremolizzo L, Tueting P, 2011. Epigenetic GABAergic targets in schizophrenia and bipolar disorder. Neuropharmacology 60(7-8), 1007–1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulchina Y, Xu SJ, Snyder MA, Elefant F, Gao WJ, 2017. Epigenetic mechanisms underlying NMDA receptor hypofunction in the prefrontal cortex of juvenile animals in the MAM model for schizophrenia. Journal of neurochemistry 143(3), 320–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta-Agarwal S, Jarome TJ, Fernandez J, Lubin FD, 2014. NMDA receptor- and ERK-dependent histone methylation changes in the lateral amygdala bidirectionally regulate fear memory formation. Learning & memory; (Cold Spring Harbor, N.Y.) 21(7), 351–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammels C, Pishva E, De Vry J, van den Hove DL, Prickaerts J, van Winkel R, Selten JP, Lesch KP, Daskalakis NP, Steinbusch HW, van Os J, Kenis G, Rutten BP, 2015a. Defeat stress in rodents: From behavior to molecules. Neuroscience and biobehavioral reviews 59, 111–140. [DOI] [PubMed] [Google Scholar]
- Hammels C, Prickaerts J, Kenis G, Vanmierlo T, Fischer M, Steinbusch HW, van Os J, van den Hove DL, Rutten BP, 2015b. Differential susceptibility to chronic social defeat stress relates to the number of Dnmt3a-immunoreactive neurons in the hippocampal dentate gyrus. Psychoneuroendocrinology 51, 547–556. [DOI] [PubMed] [Google Scholar]
- Harrison PJ, Weinberger DR, 2005. Schizophrenia genes, gene expression, and neuropathology: on the matter of their convergence. Mol Psychiatry 10(1), 40–68. [DOI] [PubMed] [Google Scholar]
- Hashimoto T, Bergen SE, Nguyen QL, Xu B, Monteggia LM, Pierri JN, Sun Z, Sampson AR, Lewis DA, 2005. Relationship of brain-derived neurotrophic factor and its receptor TrkB to altered inhibitory prefrontal circuitry in schizophrenia. J. Neurosci. 25(2), 372–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henson MA, Roberts AC, Pérez-Otaño I, Philpot BD, 2010. Influence of the NR3A subunit on NMDA receptor functions. Progress in Neurobiology 91(1), 23–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howes OD, Kapur S, 2009. The dopamine hypothesis of schizophrenia: version III--the final common pathway. Schizophrenia bulletin 35(3), 549–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang HS, Matevossian A, Whittle C, Kim SY, Schumacher A, Baker SP, Akbarian S, 2007. Prefrontal dysfunction in schizophrenia involves mixed-lineage leukemia 1-regulated histone methylation at GABAergic gene promoters. The Journal of neuroscience : the official journal of the Society for Neuroscience 27(42), 11254–11262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang JY, Zukin RS, 2018. REST, a master transcriptional regulator in neurodegenerative disease. Current opinion in neurobiology 48, 193–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibi D, delaFuenteRevenga M, Kezunovic N, Muguruza, Saunders JM, Gaitonde SA, Moreno JL, Ijaz MK, Santosh V, Kozlenkov A, Holloway T, Seto J, Garcia-Bea A, Kurita M, Mosley GE, Jiang Y, Christoffel DJ, Callado LF, Russo SJ, Dracheva S, Lopez-Gimenez JF, Ge Y, Escalante CR, Meana JJ, Akbarian S, Huntley GW, Gonzalez-Maeso J, 2017. Antipsychotic-induced Hdac2 transcription via NF-kappaB leads to synaptic and cognitive side effects. Nat Neurosci 20(9), 1247–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikegame T, Bundo M, Murata Y, Kasai K, Kato T, Iwamoto K, 2013. DNA methylation of the BDNF gene and its relevance to psychiatric disorders. Journal of human genetics 58(7), 434–438. [DOI] [PubMed] [Google Scholar]
- Insel TR, 2010. Rethinking schizophrenia. Nature 468(7321), 187–193. [DOI] [PubMed] [Google Scholar]
- Jaaro-Peled H, Ayhan Y, Pletnikov MV, Sawa A, 2010. Review of pathological hallmarks of schizophrenia: comparison of genetic models with patients and nongenetic models. Schizophrenia bulletin 36(2), 301–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarome TJ, Lubin FD, 2014. Epigenetic mechanisms of memory formation and reconsolidation. Neurobiology of learning and memory 115, 116–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y, Jakovcevski M, Bharadwaj R, Connor C, Schroeder FA, Lin CL, Straubhaar J, Martin G, Akbarian S, 2010. Setdb1 histone methyltransferase regulates mood-related behaviors and expression of the NMDA receptor subunit NR2B. J. Neurosci. 30(21), 7152–7167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaut O, Schmitt I, Hofmann A, Hoffmann P, Schlaepfer TE, Wullner U, Hurlemann R, 2015. Aberrant NMDA receptor DNA methylation detected by epigenome-wide analysis of hippocampus and prefrontal cortex in major depression. European archives of psychiatry and clinical neuroscience 265(4), 331–341. [DOI] [PubMed] [Google Scholar]
- Keverne EB, Pfaff DW, Tabansky I, 2015. Epigenetic changes in the developing brain: Effects on behavior. Proceedings of the National Academy of Sciences 112(22), 6789–6795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiese K, Jablonski J, Hackenbracht J, Wrosch JK, Groemer TW, Kornhuber J, Blumcke I, Kobow K, 2017. Epigenetic control of epilepsy target genes contributes to a cellular memory of epileptogenesis in cultured rat hippocampal neurons. Acta neuropathologica communications 5(1), 79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YK, Choi J, Park SC, 2017. A Novel Bio-Psychosocial-Behavioral Treatment Model in Schizophrenia. International journal of molecular sciences 18(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolarow R, Brigadski T, Lessmann V, 2007. Postsynaptic secretion of BDNF and NT-3 from hippocampal neurons depends on calcium calmodulin kinase II signaling and proceeds via delayed fusion pore opening. The Journal of neuroscience : the official journal of the Society for Neuroscience 27(39), 10350–10364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krebs MO, Guillin O, Bourdell MC, Schwartz JC, Olie JP, Poirier MF, Sokoloff P, 2000. Brain derived neurotrophic factor (BDNF) gene variants association with age at onset and therapeutic response in schizophrenia. Mol Psychiatry 5(5), 558–562. [DOI] [PubMed] [Google Scholar]
- Kurita M, Holloway T, Garcia-Bea A, Kozlenkov A, Friedman AK, Moreno JL, Heshmati M, Golden SA, Kennedy PJ, Takahashi N, Dietz DM, Mocci G, Gabilondo AM, Hanks J, Umali A, Callado LF, Gallitano AL, Neve RL, Shen L, Buxbaum JD, Han M-H, Nestler EJ, Meana JJ, Russo SJ, Gonzalez-Maeso J, 2012. HDAC2 regulates atypical antipsychotic responses through the modulation of mGlu2 promoter activity. Nat Neurosci 15(9), 1245–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurita M, Holloway T, Gonzalez-Maeso J, 2013a. HDAC2 as a new target to improve schizophrenia treatment. Expert Rev Neurother 13(1), 1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurita M, Moreno JL, Holloway T, Kozlenkov A, Mocci G, Garcia-Bea A, Hanks JB, Neve R, Nestler EJ, Russo SJ, Gonzalez-Maeso J, 2013b. Repressive epigenetic changes at the mGlu2 promoter in frontal cortex of 5-HT2A knockout mice. Mol Pharmacol 83(6), 1166–1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyrke-Smith M, Williams JM, 2018. Bridging Synaptic and Epigenetic Maintenance Mechanisms of the Engram. Front Mol Neurosci 11, 369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau CG, Zukin RS, 2007. NMDA receptor trafficking in synaptic plasticity and neuropsychiatric disorders. Nature reviews. Neuroscience 8(6), 413–426. [DOI] [PubMed] [Google Scholar]
- Lau D, Bengtson CP, Buchthal B, Bading H, 2015. BDNF Reduces Toxic Extrasynaptic NMDA Receptor Signaling via Synaptic NMDA Receptors and Nuclear-Calcium-Induced Transcription of inhba/Activin A. Cell reports 12(8), 1353–1366. [DOI] [PubMed] [Google Scholar]
- Levenson JM, O'Riordan KJ, Brown KD, Trinh MA, Molfese DL, Sweatt JD, 2004. Regulation of histone acetylation during memory formation in the hippocampus. The Journal of biological chemistry 279(39), 40545–40559. [DOI] [PubMed] [Google Scholar]
- Lewis DA, Gonzalez-Burgos G, 2008. Neuroplasticity of neocortical circuits in schizophrenia. Neuropsychopharmacol 33(1), 141–165. [DOI] [PubMed] [Google Scholar]
- Lewis DA, Moghaddam B, 2006. Cognitive dysfunction in schizophrenia: convergence of gamma-aminobutyric acid and glutamate alterations. Arch Neurol 63(10), 1372–1376. [DOI] [PubMed] [Google Scholar]
- Li J, Guo Y, Schroeder FA, Youngs RM, Schmidt TW, Ferris C, Konradi C, Akbarian S, 2004. Dopamine D2-like antagonists induce chromatin remodeling in striatal neurons through cyclic AMP-protein kinase A and NMDA receptor signaling. Journal of neurochemistry 90(5), 1117–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M-L, Yang S-S, Xing B, Ferguson BR, Gulchina Y, Li Y-C, Li F, Hu X-Q, Gao W-J, 2015a. LY395756, an mGluR2 agonist and mGluR3 antagonist, enhances NMDA receptor expression and function in the normal adult rat prefrontal cortex, but fails to improve working memory and reverse MK801-induced working memory impairment. Experimental neurology 273, 190–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li ML, Gulchina Y, Monaco SA, Xing B, Ferguson BR, Li YC, Li F, Hu XQ, Gao WJ, 2017a. Juvenile treatment with a novel mGluR2 agonist/mGluR3 antagonist compound, LY395756, reverses learning deficits and cognitive flexibility impairments in adults in a neurodevelopmental model of schizophrenia. Neurobiol Learn Mem 140, 52–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li ML, Gulchina Y, Monaco SA, Xing B, Ferguson BR, Li YC, Li F, Hu XQ, Gao WJ, 2017b. Juvenile Treatment with a Novel mGluR2 Agonist/mGluR3 Antagonist Compound, LY395756, Reverses Learning Deficits and Cognitive Flexibility Impairments in Adults in A Neurodevelopmental Model of Schizophrenia. Neurobiology of learning and memory, 52–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li ML, Hu XQ, Li F, Gao WJ, 2015b. Perspectives on the mGluR2/3 agonists as a therapeutic target for schizophrenia: Still promising or a dead end? Progress in neuro-psychopharmacology & biological psychiatry 60, 66–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman JA, Jarskog LF, Malaspina D, 2006. Preventing clinical deterioration in the course of schizophrenia: the potential for neuroprotection. J Clin Psychiatry 67(6), 983–990. [DOI] [PubMed] [Google Scholar]
- Lisman JE, Coyle JT, Green RW, Javitt DC, Benes FM, Heckers S, Grace AA, 2008. Circuit-based framework for understanding neurotransmitter and risk gene interactions in schizophrenia. Trends in Neurosci 31(5), 234–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu B, Wang KH, Nose A, 2009. Molecular mechanisms underlying neural circuit formation. Current opinion in neurobiology 19(2), 162–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu T, Aron L, Zullo J, Pan Y, Kim H, Chen Y, Yang T-H, Kim H-M, Drake D, Liu XS, Bennett DA, Colaiacovo MP, Yankner BA, 2014. REST and stress resistance in ageing and Alzheimer/'s disease. Nature 507(7493), 448–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackowiak M, Guzik R, Dudys D, Bator E, Wedzony K, 2013. MK-801, a NMDA receptor antagonist, increases phosphorylation of histone H3 in the rat medial prefrontal cortex. Pharmacological reports : PR 65(5), 1112–1123. [DOI] [PubMed] [Google Scholar]
- Maeshima K, Ide S, Hibino K, Sasai M, 2016. Liquid-like behavior of chromatin. Current opinion in genetics & development 37, 36–45. [DOI] [PubMed] [Google Scholar]
- Maksymetz J, Moran SP, Conn PJ, 2017. Targeting metabotropic glutamate receptors for novel treatments of schizophrenia. Molecular brain 10(1), 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marek GJ, 2010. Metabotropic glutamate2/3 (mGlu2/3) receptors, schizophrenia and cognition. European Journal of Pharmacology 639(1-3), 81–90. [DOI] [PubMed] [Google Scholar]
- Matrisciano F, Panaccione I, Grayson DR, Nicoletti F, Guidotti A, 2016. Metabotropic glutamate 2/3 receptors and epigenetic modifications in psychotic disorders: a review. Curr Neuropharmacol 14(1), 41–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matrisciano F, Tueting P, Dalal I, Kadriu B, Grayson DR, Davis JM, Nicoletti F, Guidotti A, 2013. Epigenetic modifications of GABAergic interneurons are associated with the schizophrenia-like phenotype induced by prenatal stress in mice. Neuropharmacology 68, 184–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melas PA, Rogdaki M, Osby U, Schalling M, Lavebratt C, Ekstrom TJ, 2012. Epigenetic aberrations in leukocytes of patients with schizophrenia: association of global DNA methylation with antipsychotic drug treatment and disease onset. FASEB journal : official publication of the Federation of American Societies for Experimental Biology 26(6), 2712–2718. [DOI] [PubMed] [Google Scholar]
- Mitchell AC, Javidfar B, Pothula V, Ibi D, Shen EY, Peter CJ, Bicks LK, Fehr T, Jiang Y, Brennand KJ, Neve RL, Gonzalez-Maeso J, Akbarian S, 2018. MEF2C transcription factor is associated with the genetic and epigenetic risk architecture of schizophrenia and improves cognition in mice. Mol Psychiatry 23(1), 123–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchelmore C, Gede L, 2014. Brain Derived Neurotrophic Factor: epigenetic regulation in psychiatric disorders. Brain research 1586, 162–172. [DOI] [PubMed] [Google Scholar]
- Moghaddam B, 2003. Bringing order to the glutamate chaos in schizophrenia. Neuron 40(5), 881–884. [DOI] [PubMed] [Google Scholar]
- Moghaddam B, 2004. Targeting metabotropic glutamate receptors for treatment of the cognitive symptoms of schizophrenia. Psychopharmacol (Berl) 174(1), 39–44. [DOI] [PubMed] [Google Scholar]
- Monaco SA, Gulchina Y, Gao W-J, 2015. NR2B subunit in the prefrontal cortex: A double-edged sword for working memory function and psychiatric disorders. Neuroscience & Biobehavioral Reviews 56(0), 127–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monyer H, Burnashev N, Laurie DJ, Sakmann B, Seeburg PH, 1994. Developmental and regional expression in the rat brain and functional properties of four NMDA receptors. Neuron 12(3), 529–540. [DOI] [PubMed] [Google Scholar]
- Moreau AW, Kullmann DM, 2013. NMDA receptor-dependent function and plasticity in inhibitory circuits. Neuropharmacology 74, 23–31. [DOI] [PubMed] [Google Scholar]
- Moreno JL, Miranda-Azpiazu P, Garcia-Bea A, Younkin J, Cui M, Kozlenkov A, Ben-Ezra A, Voloudakis G, Fakira AK, Baki L, Ge Y, Georgakopoulos A, Moron JA, Milligan G, Lopez-Gimenez JF, Robakis NK, Logothetis DE, Meana JJ, Gonzalez-Maeso J, 2016. Allosteric signaling through an mGlu2 and 5-HT2A heteromeric receptor complex and its potential contribution to schizophrenia. Sci Signal 9(410), ra5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morishita H, Kundakovic M, Bicks L, Mitchell A, Akbarian S, 2015. Interneuron epigenomes during the critical period of cortical plasticity: Implications for schizophrenia. Neurobiology of learning and memory 124, 104–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakazawa K, Jeevakumar V, Nakao K, 2017. Spatial and temporal boundaries of NMDA receptor hypofunction leading to schizophrenia. NPJ Schizophr 3, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasca C, Zelli D, Bigio B, Piccinin S, Scaccianoce S, Nistico R, McEwen BS, 2015. Stress dynamically regulates behavior and glutamatergic gene expression in hippocampus by opening a window of epigenetic plasticity. Proceedings of the National Academy of Sciences of the United States of America 112(48), 14960–14965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nghia NA, Hirasawa T, Kasai H, Obata C, Moriishi K, Mochizuki K, Koizumi S, Kubota T, 2015. Long-term imipramine treatment increases N-methyl-d-aspartate receptor activity and expression via epigenetic mechanisms. Eur J Pharmacol 752, 69–77. [DOI] [PubMed] [Google Scholar]
- Niwa M, Kamiya A, Murai R, Kubo K, Gruber AJ, Tomita K, Lu L, Tomisato S, Jaaro-Peled H, Seshadri S, Hiyama H, Huang B, Kohda K, Noda Y, O'Donnell P, Nakajima K, Sawa A, Nabeshima T, 2010. Knockdown of DISC1 by in utero gene transfer disturbs postnatal dopaminergic maturation in the frontal cortex and leads to adult behavioral deficits. Neuron 65(4), 480–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noh KM, Hwang JY, Follenzi A, Athanasiadou R, Miyawaki T, Greally JM, Bennett MV, Zukin RS, 2012. Repressor element-1 silencing transcription factor (REST)-dependent epigenetic remodeling is critical to ischemia-induced neuronal death. Proceedings of the National Academy of Sciences of the United States of America 109(16), E962–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ovenden ES, McGregor NW, Emsley RA, Warnich L, 2018. DNA methylation and antipsychotic treatment mechanisms in schizophrenia: Progress and future directions. Progress in Neuro-Psychopharmacology and Biological Psychiatry 81, 38–49. [DOI] [PubMed] [Google Scholar]
- Peterson CL, Laniel MA, 2004. Histones and histone modifications. Current biology : CB 14(14), R546–551. [DOI] [PubMed] [Google Scholar]
- Poo MM, 2001. Neurotrophins as synaptic modulators. Nature reviews. Neuroscience 2(1), 24–32. [DOI] [PubMed] [Google Scholar]
- Qiang M, Denny A, Chen J, Ticku MK, Yan B, Henderson G, 2010. The site specific demethylation in the 5'-regulatory area of NMDA receptor 2B subunit gene associated with CIE-induced up-regulation of transcription. PLoS ONE 5(1), e8798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiang M, Li JG, Denny AD, Yao JM, Lieu M, Zhang K, Carreon S, 2014. Epigenetic mechanisms are involved in the regulation of ethanol consumption in mice. The international journal of neuropsychopharmacology 18(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin L, Ma K, Wang ZJ, Hu Z, Matas E, Wei J, Yan Z, 2018. Social deficits in Shank3-deficient mouse models of autism are rescued by histone deacetylase (HDAC) inhibition. Nat Neurosci. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan EM, Olstein DH, Bear MF, 1999. Bidirectional, experience-dependent regulation of N-methyl-D-aspartate receptor subunit composition in the rat visual cortex during postnatal development. Proceedings of the National Academy of Sciences of the United States of America 96(22), 12876–12880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qureshi IA, Mehler MF, 2009. Regulation of non-coding RNA networks in the nervous system--what's the REST of the story? Neuroscience letters 466(2), 73–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajarajan P, Jiang Y, Kassim BS, Akbarian S, 2018. Chromosomal Conformations and Epigenomic Regulation in Schizophrenia. Progress in molecular biology and translational science 157, 21–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ripke Se.a., 2014. Biological insights from 108 schizophrenia-associated genetic loci. Nature 511(7510), 421–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts AC, Díez-García J, Rodriguiz RM, López IP, Luján R, Martínez-Turrillas R, Picó E, Henson MA, Bernardo DR, Jarrett TM, Clendeninn DJ, López-Mascaraque L, Feng G, Lo DC, Wesseling JF, Wetsel WC, Philpot BD, Pérez-Otaño I, 2009. Downregulation of NR3A-containing NMDARs is required for synapse maturation and memory consolidation. Neuron 63(3), 342–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodenas-Ruano A, Chavez AE, Cossio MJ, Castillo PE, Zukin RS, 2012. REST-dependent epigenetic remodeling promotes the developmental switch in synaptic NMDA receptors. Nat Neurosci 15(10), 1382–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigues-Amorim D, Rivera-Baltanas T, Bessa J, Sousa N, Vallejo-Curto MC, Rodriguez-Jamardo C, de Las Heras ME, Diaz R, Agis-Balboa RC, Olivares JM, Spuch C, 2018. The neurobiological hypothesis of neurotrophins in the pathophysiology of schizophrenia: A meta-analysis. Journal of psychiatric research 106, 43–53. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Murillo L, Gogos JA, Karayiorgou M, 2012. The genetic architecture of schizophrenia: new mutations and emerging paradigms. Annual review of medicine 63, 63–80. [DOI] [PubMed] [Google Scholar]
- Ryan MM, Ryan B, Kyrke-Smith M, Logan B, Tate WP, Abraham WC, Williams JM, 2012. Temporal profiling of gene networks associated with the late phase of long-term potentiation in vivo. PloS one 7(7), e40538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoenherr CJ, Anderson DJ, 1995. The neuron-restrictive silencer factor (NRSF): a coordinate repressor of multiple neuron-specific genes. Science 267(5202), 1360–1363. [DOI] [PubMed] [Google Scholar]
- Schroeder FA, Gilbert TM, Feng N, Taillon BD, Volkow ND, Innis RB, Hooker JM, Lipska BK, 2017. Expression of HDAC2 but Not HDAC1 Transcript Is Reduced in Dorsolateral Prefrontal Cortex of Patients with Schizophrenia. ACS chemical neuroscience 8(3), 662–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng M, Cummings J, Roldan LA, Jan YN, Jan LY, 1994. Changing subunit composition of heteromeric NMDA receptors during development of rat cortex. Nature 368(6467), 144–147. [DOI] [PubMed] [Google Scholar]
- Shimabukuro M, Sasaki T, Imamura A, Tsujita T, Fuke C, Umekage T, Tochigi M, Hiramatsu K, Miyazaki T, Oda T, Sugimoto J, Jinno Y, Okazaki Y, 2007. Global hypomethylation of peripheral leukocyte DNA in male patients with schizophrenia: a potential link between epigenetics and schizophrenia. Journal of psychiatric research 41(12), 1042–1046. [DOI] [PubMed] [Google Scholar]
- Snyder MA, Adelman AE, Gao W-J, 2013. Gestational methylazoxymethanol exposure leads to NMDAR dysfunction in hippocampus during early development and lasting deficits in learning. Neuropsychopharmacology : official publication of the American College of Neuropsychopharmacology 38(2), 328–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder MA, Gao W-J, 2013. NMDA hypofunction as a convergence point for progression and symptoms of schizophrenia. Frontiers in cellular neuroscience 7, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Z, Yang W, Cheng G, Zhou X, Yang L, Zhao D, 2018. Prion protein is essential for the RE1 silencing transcription factor (REST)-dependent developmental switch in synaptic NMDA receptors. Cell death & disease 9(5), 541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spear LP, 2000. The adolescent brain and age-related behavioral manifestations. Neuroscience and biobehavioral reviews 24(4), 417–463. [DOI] [PubMed] [Google Scholar]
- Stadler F, Kolb G, Rubusch L, Baker SP, Jones EG, Akbarian S, 2005. Histone methylation at gene promoters is associated with developmental regulation and region-specific expression of ionotropic and metabotropic glutamate receptors in human brain. Journal of neurochemistry 94(2), 324–336. [DOI] [PubMed] [Google Scholar]
- Sun W, Poschmann J, Cruz-Herrera del Rosario R, Parikshak NN, Hajan HS, Kumar V, Ramasamy R, Belgard TG, Elanggovan B, Wong CCY, Mill J, Geschwind DH, Prabhakar S, 2016. Histone Acetylome-wide Association Study of Autism Spectrum Disorder. Cell 167(5), 1385–1397.e1311. [DOI] [PubMed] [Google Scholar]
- Sutton G, Chandler LJ, 2002. Activity-dependent NMDA receptor-mediated activation of protein kinase B/Akt in cortical neuronal cultures. Journal of neurochemistry 82(5), 1097–1105. [DOI] [PubMed] [Google Scholar]
- Tamminga CA, Lahti AC, 1996. The new generation of antipsychotic drugs. Int Clin Psychopharmacol 11 Suppl 2, 73–76. [DOI] [PubMed] [Google Scholar]
- Tamminga CA, Zukin RS, 2015. Schizophrenia: Evidence implicating hippocampal GluN2B protein and REST epigenetics in psychosis pathophysiology. Neuroscience 309, 233–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao X, West AE, Chen WG, Corfas G, Greenberg ME, 2002. A calcium-responsive transcription factor, CaRF, that regulates neuronal activity-dependent expression of BDNF. Neuron 33(3), 383–395. [DOI] [PubMed] [Google Scholar]
- Tian F, Hu XZ, Wu X, Jiang H, Pan H, Marini AM, Lipsky RH, 2009. Dynamic chromatin remodeling events in hippocampal neurons are associated with NMDA receptor-mediated activation of Bdnf gene promoter 1. Journal of neurochemistry 109(5), 1375–1388. [DOI] [PubMed] [Google Scholar]
- Trerotola M, Relli V, Simeone P, Alberti S, 2015. Epigenetic inheritance and the missing heritability. Human genomics 9, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyssowski KM, DeStefino NR, Cho J-H, Dunn CJ, Poston RG, Carty CE, Jones RD, Chang SM, Romeo P, Wurzelmann MK, Ward JM, Andermann ML, Saha RN, Dudek SM, Gray JM, 2018. Different neuronal activity patterns induce different gene expression programs. Neuron 98(3), 530–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veldic M, Caruncho HJ, Liu WS, Davis J, Satta R, Grayson DR, Guidotti A, Costa E, 2004. DNA-methyltransferase 1 mRNA is selectively overexpressed in telencephalic GABAergic interneurons of schizophrenia brains. Proceedings of the National Academy of Sciences of the United States of America 101(1), 348–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker AG, Conn PJ, 2015. Group I and group II metabotropic glutamate receptor allosteric modulators as novel potential antipsychotics. Curr Opin Pharmacol 20, 40–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh T, McClellan JM, McCarthy SE, Addington AM, Pierce SB, Cooper GM, Nord AS, Kusenda M, Malhotra D, Bhandari A, Stray SM, Rippey CF, Roccanova P, Makarov V, Lakshmi B, Findling RL, Sikich L, Stromberg T, Merriman B, Gogtay N, Butler P, Eckstrand K, Noory L, Gochman P, Long R, Chen Z, Davis S, Baker C, Eichler EE, Meltzer PS, Nelson SF, Singleton AB, Lee MK, Rapoport JL, King MC, Sebat J, 2008. Rare structural variants disrupt multiple genes in neurodevelopmental pathways in schizophrenia. Science 320(5875), 539–543. [DOI] [PubMed] [Google Scholar]
- Wang HX, Gao WJ, 2009. Cell type-specific development of NMDA receptors in the interneurons of rat prefrontal cortex. Neuropsychopharmacol 34(8), 2028–2040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HX, Stradtman G.G.r., Wang XJ, Gao WJ, 2008. A specialized NMDA receptor function in layer 5 recurrent microcircuitry of the adult rat prefrontal cortex. Proc. Nat. Acad. Sci. U. S. A 105(43), 16791–16796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weickert CS, Fung SJ, Catts VS, Schofield PR, Allen KM, Moore LT, Newell KA, Pellen D, Huang XF, Catts SV, Weickert TW, 2013. Molecular evidence of N-methyl-D-aspartate receptor hypofunction in schizophrenia. Mol Psychiatry 18(11), 1185–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberger DR, 1987. Implications of normal brain development for the pathogenesis of schizophrenia. Arch Gen Psychiatry 44(7), 660–669. [DOI] [PubMed] [Google Scholar]
- Weinberger DR, 2017. Future of Days Past: Neurodevelopment and Schizophrenia. Schizophrenia bulletin 43(6), 1164–1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing B, Han G, Wang M-J, Snyder MA, Gao W-J, 2018. Juvenile treatment with mGluR2/3 agonist prevents schizophrenia-like phenotypes in adult by acting through GSK3β. Neuropharmacology 137, 359–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang AC, Tsai SJ, 2017. New Targets for Schizophrenia Treatment beyond the Dopamine Hypothesis. International journal of molecular sciences 18(8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Hong EJ, Cohen S, Zhao WN, Ho HY, Schmidt L, Chen WG, Lin Y, Savner E, Griffith EC, Hu L, Steen JA, Weitz CJ, Greenberg ME, 2006. Brain-specific phosphorylation of MeCP2 regulates activity-dependent Bdnf transcription, dendritic growth, and spine maturation. Neuron 52(2), 255–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhubi A, Veldic M, Puri NV, Kadriu B, Caruncho H, Loza I, Sershen H, Lajtha A, Smith RC, Guidotti A, Davis JM, Costa E, 2009. An upregulation of DNA-methyltransferase 1 and 3a expressed in telencephalic GABAergic neurons of schizophrenia patients is also detected in peripheral blood lymphocytes. Schizophrenia research 111(1-3), 115–122. [DOI] [PMC free article] [PubMed] [Google Scholar]