

Abstract
Fragile X syndrome (FXS) is associated with deficits in various types of learning, including those that require the hippocampus. Relatedly, hippocampal long-term potentiation (LTP) is impaired in the Fmr1 knockout (KO) mouse model of FXS. Prior research found that infusion of brain-derived neurotrophic factor (BDNF) rescues LTP in the KOs. Here, we tested if, in Fmr1 KO mice, up-regulating BDNF production or treatment with an agonist for BDNF’s TrkB receptor restores synaptic plasticity and improves learning. In hippocampal slices, bath infusion of the TrkB agonist 7,8-dihydroxyflavone (7,8-DHF) completely restored otherwise impaired hippocampal field CA1 LTP of Fmr1 KOs without effect in wild types (WTs). Similarly, acute, semi-chronic, or chronic treatments with 7,8-DHF rescued a simple hippocampus-dependent form of spatial learning (object location memory: OLM) in Fmr1 KOs without effect in WTs. The agonist also restored object recognition memory, which depends on cortical regions. Semi-chronic, but not acute, treatment with the ampakine CX929, which up-regulates BDNF expression, lowered the training threshold for OLM in WT mice and rescued learning in the KOs. Positive results were also obtained in a test for social recognition. An mGluR5 antagonist did not improve learning. Quantification of synaptic immunolabeling demonstrated that 7,8-DHF and CX929 increase levels of activated TrkB at excitatory synapses. Moreover, CX929 induced a robust synaptic activation of the TrkB effector ERK1/2. These results suggest that enhanced synaptic BDNF signaling constitutes a plausible strategy for treating certain aspects of the cognitive disabilities associated with FXS.
Keywords: Fragile X syndrome; TrkB; Ampakine; Object location memory; 7,8-dihydroxyflavone; Social approach
1. Introduction
Fragile X syndrome (FXS) is the most common cause of inherited intellectual disability (Turner et al., 1996; Fombonne, 2006). No mechanism-based therapeutics exist for the condition’s cognitive component, although aspects of the FXS phenotype are treated symptomatically (Hagerman and Hagerman, 2002). The Fmr1 knockout (KO) mouse model is widely used for investigations into neurobiological defects underlying cognitive and behavioral abnormalities in FXS. The studies describing disturbances in neuronal biochemistry, circuitry, and cellular phenotypes in Fmr1 KOs have exploded in recent years (Pfeiffer and Huber, 2009; Bhakar et al., 2012; Gross et al., 2012), but progress in developing treatment strategies for cognitive disabilities in FXS is still limited. Recent work has, however, identified synaptic defects in the hippocampus that are plausibly related to the disorder’s characteristic learning problems. Although baseline synaptic transmission is not significantly disturbed in the mutants, long-term potentiation (LTP), an effect intimately related to the encoding of many forms of memory, is impaired (Wang et al., 2018). For hippocampal field CA1, the pertinent defects involve an increase in the amount of stimulation needed to induce LTP and some of the complex signaling cascades that stabilize the potentiated state (Lauterborn et al., 2007; Chen et al., 2010c; Lee et al., 2011; Seese et al., 2012). These observations describe potential targets for mechanism-based treatment strategies.
Brain-derived neurotrophic factor (BDNF) is a releasable neurotrophin that plays a central role in the production of hippocampal LTP and in the formation of hippocampus-dependent spatial memory (Mizuno et al., 2000). Acting through synaptic TrkB receptors, BDNF promotes signaling that regulates the assembly and stabilization of subsynaptic actin networks, a process required for LTP consolidation. Multiple studies have shown that learning activates TrkB and LTP-related actin management (Bramham, 2008; Chen et al., 2010b; Bekinschtein et al., 2014). Up-regulating the neurotrophin with semi-chronic treatment with a positive AMPA receptor modulator (ampakine) rescues synaptic plasticity and learning in rodent models of cognitive impairment (Rex et al., 2006; Simmons et al., 2009, 2011; Baudry et al., 2012; Kramar et al., 2012; Lauterborn et al., 2016). Other studies have suggested that modulating TrkB signaling reduces abnormalities in the Fmr1 KO brain (Osterweil et al., 2010; Louhivuori et al., 2011; Uutela et al., 2012). Along these lines, infused BDNF restores LTP in Fmr1 KO hippocampal slices (Lauterborn et al., 2007). Importantly, in vivo activation of TrkB with a selective agonist has been reported to rescue learning in Fmr1 KOs (Tian et al., 2015) and decreasing BDNF expression in Fmr1 KOs has been shown to induce a robust impairment in contextual fear conditioning (Uutela et al., 2012). These earlier studies employed behavioral paradigms that rely on an animal’s ability to avoid stressful and anxiety-provoking circumstances. Fmr1 KOs exhibit increased anxiety-like behaviors and are particularly sensitive to stress (Kazdoba et al., 2014). Thus, given the possibility of treatment effects on the stress response, we extended the above studies to test if two alternative strategies leading to increased TrkB activation – systemic treatment with a TrkB agonist or with an ampakine – normalize hippocampus-dependent learning in Fmr1 KOs using paradigms that instead rely on rodents’ inherent preference for novelty. Additionally, it is noteworthy that ampakine effects are somewhat more naturalistic in that they rely on increased BDNF expression and thus increased TrkB signaling specifically within regions engaged during learning.
Previous work has demonstrated that Fmr1 KOs have a robust impairment in object location memory (OLM) (Seese et al., 2014b), a simple form of spatial learning that relies on the preference for novelty and is dependent on hippocampal field CA1 (Haettig et al., 2011). This deficiency in the Fmr1 KOs was associated with a failure of training to activate the TrkB effector ERK1/2 at hippocampal synapses, a response that is pronounced in wild type mice (Seese et al., 2014b). ERK1/2 contributes to the dynamic modification of both actin- and microtubule-based cytoskeletal systems (Sanchez et al., 2000; Harrison and Turley, 2001; Cosen-Binker and Kapus, 2006) and, as expected from this, plays a prominent role in the stabilization of LTP. We accordingly tested for the effects of TrkB signaling up-regulation on activation of the kinase at hippocampal synapses in Fmr1 KO mice.
2. Materials and methods
2.1. Animals
Adult (3 to 5-month-old) male Fmr1 KO mice on the FVB/129 background (Irwin et al., 2002; Seese et al., 2012) and age-, sex-, and background-matched wild type (WT) mice were group housed with littermates (2–5 mice per cage) in rooms maintained at 22 °C and 55% humidity on a 12-hours-on/12-hours-off light cycle and with food and water available ad libitum. Experiments were performed on the animal’s light cycle and in accordance with National Institutes of Health Guide for the Care and Use of Laboratory Animals and protocols approved by the University of California, Irvine Institutional Animal Care and Use Committee.
2.2. Slice electrophysiology
Adult (3–4 months of age) male Fmr1 KO and WT FVB/129 mice were used for extracellular field recordings from acute hippocampal slices. Transverse 360-μm thick slices were prepared from the middle third of hippocampus and were promptly transferred to an interface recording chamber maintained at 31 ± 1 °C with a constant 60–70 ml/h infusion of oxygenated artificial cerebrospinal fluid (aCSF) containing 124 mM NaCl, 3 mM KCl, 1.25 mM KH2PO4, 1.5 mM MgSO4, 26 mM NaHCO3, 2.5 mM CaCl2, and 10 mM dextrose. Experiments began 1.5 h after slice preparation. To monitor responses to stimulation of the Schaffer-commissural projection from field CA3 to field CA1, a bipolar stimulating electrode was placed in CA1a or CA1c stratum radiatum, and a glass pipette electrode filled with 2 M NaCl was placed in CA1b stratum radiatum; both electrodes were placed equidistant from the pyramidal cell layer. For baseline recordings, stimulation was applied at 0.05 Hz with pulses that were set to elicit field EPSPs (fEPSPs) that were 50% of the maximum pop-spike free response. The responses were collected using NAC Gather 2.0 software (Theta Burst Corp., Irvine, CA, USA).
For studies of LTP, 7,8-dihydroxyflavone (7,8-DHF; 1 μM; Tokyo Chemical Industry Co., Tokyo, Japan) or vehicle was infused for 30 min prior to induction of LTP, which was done by using a single short train of theta burst stimulation (TBS: 5 bursts of four pulses at 100 Hz, with 200 ms between bursts, at baseline stimulation intensity). After TBS, low frequency (0.05 Hz) stimulation was resumed and responses were collected for 60 min. To assess the level of LTP, the mean fEPSP response during the last 5 min of recordings was compared to the mean response during the last 5 min of baseline recordings.
The 7,8-DHF was dissolved in dimethyl sulfoxide (DMSO) and diluted so that DMSO was at < 0.001% in the aCSF bath.
2.3. Object recognition and object location memory tasks
Behavioral analyses were performed as described (Stefanko et al., 2009; Haettig et al., 2011; Seese et al., 2014b). Briefly, animals were handled for 2 min daily for 5 days and then habituated to a white chamber measuring 24 × 30 × 30 cm and containing sawdust bedding for 5 min daily for 5 days. On the following day, mice were subjected to a 5-min training episode in which they were placed in an experimental chamber containing two identical 100 ml glass beakers placed in two corners along the same wall, approximately 1 in. from the perimeter.
Mice were then tested for either OLM or object recognition memory (ORM). For OLM testing, this involved a 5-min retention trial 1 day after the training trial. One beaker remained in the original training location and one was moved towards the center of the apparatus. For ORM testing, one beaker was replaced with a novel object; both objects remained in the same positions as the objects in the training trial. Animals were video recorded and tracked with ANY-Maze software (Stoelting, Wood Dale, IL, USA).
Videos were scored offline by raters blind to genotype and experimental group. Mice were scored as interacting with an object when they were sniffing and their nose touched or was within 0.5 cm of the object. Interaction was not scored when the animal was within this radius while grooming or digging or if the animal fell within this zone when turning. Total exploration time was quantified as the time interacting with both objects. A discrimination index was calculated as (tnovel – tfamiliar)/(tnovel + tfamiliar) x 100. “Familiar” refers to the object that was in the same location during training or the object that remained identical in the training and testing sessions. “Novel” refers to the object that was moved to the new location (OLM) or the object that was replaced (ORM). Thus, a positive discrimination index represents a preference for the new location or new object. Total distance traveled was calculated using ANY-Maze.
2.4. Social approach and recognition tasks
The social approach task was performed as described previously (Yang et al., 2011). Briefly, behaviorally naïve wild type C57BL/6 mice, to be used as ‘targets’, were habituated to the inside of inverted wire mesh cups for 10 min at a time until they no longer urinated or defecated. Experimental mice (FVB/129 background) were then habituated to the room (60 min), the center chamber of the three-chambered apparatus (10 min), and then all three chambers (10 min). Following habituation, experimental animals were placed in the three-chambered apparatus for 10 min, with one chamber containing an empty inverted cup and the other chamber containing a target mouse inside the inverted cup (“social approach”). To assess “social recognition”, experimental mice were removed from the chamber and then 5 min later, they were reintroduced to the arena for a total of 10 min, with one chamber containing (under the cup) the familiar target mouse from the social approach task and the other chamber containing (under the cup) a novel C57BL/6 WT target mouse.
Videos were recorded and then scored by raters blind to genotype. Exploration was defined as time in seconds that the experimental mouse was sniffing or directly facing the mouse inside the cup. A discrimination index for the social approach task was calculated as (tmouse − tobject)/(tmouse + tobject) × 100, and a discrimination index for the social recognition task was similarly calculated as (tnovel − tfamiliar)/(tnovel + tfamiliar) × 100.
2.5. In vivo drug administration
The TrkB agonist 7,8-DHF (Jang et al., 2010) was dissolved in 100% DMSO before 0.1 M sterile phosphate-buffered saline (PBS) was added to reach a diluted solution of 17.5% DMSO at a final concentration of 0.67 mg/ml 7,8-DHF. Animals received twice daily (9 am and 4 pm) intraperitoneal (IP) injections of sterile saline (2 days), 17.5% DMSO (4 days), and then either 17.5% DMSO or 7,8-DHF (5 mg/kg; 4 days). The first of these injections corresponded with the first day of handling. An episode of handling or habituation followed each injection. A final 7,8-DHF injection was given 1 h before training. For acute injection studies, mice were injected once daily with sterile saline (2 days), 17.5% DMSO (4 days), and then either 17.5% DMSO or 7,8-DHF (5 mg/kg; 1 day). The first of these injections corresponded with the fifth day of handling such that 7,8-DHF was injected 1 h prior to the training episode. We chose this time point and dose based on prior studies (Jang et al., 2010; Andero et al., 2011; Devi and Ohno, 2012). In some experiments, 7,8-DHF was given in the drinking water (Johnson et al., 2012). To do so, 7,8-DHF was dissolved in DMSO to generate a stock solution of 50 mg/ml, and 160 μl of this stock was added to every 100 ml of tap water containing 1% sucrose. Mice were provided this 7,8-DHF solution or vehicle (1% sucrose in tap water) continuously for 1 month prior to training. Drinking bottles containing these solutions were replaced every 3–4 days. Because animals were group housed, the total volume of water containing 7,8-DHF consumed by each animal could not be directly measured.
The ampakine CX929 was prepared as a stock solution at 7.5 mg/ml in sterile 30% cyclodextrin (CDX) before being diluted with sterile 0.9% sodium chloride to a working concentration of 2.5 mg/ml in 10% CDX (Simmons et al., 2011; Baudry et al., 2012). In some studies, animals were given two daily (9 am and 4 pm) IP injections containing equivalent volumes of sterile saline (first 2 days), 10% CDX (next 4 days), and then either 10% CDX or CX929 (5 mg/kg; next 4 days) (Rex et al., 2006; Simmons et al., 2011). The beginning of these injections corresponded with the first day of handling such that the final injection occurred on the afternoon of the fifth day of habituation (e.g., approximately 17 h prior to training). In another set of experiments, mice were given once daily IP injections of sterile saline (2 days), 10% CDX (4 days), and then either 10% CDX or CX929 (5 mg/kg; 1 day). This dose of CX929 was chosen as it has been shown to increase BDNF and promote learning in other rodent models of neurocognitive and neurodevelopmental disorders (Rex et al., 2006; Simmons et al., 2011; Baudry et al., 2012). The beginning of these injections corresponded with the fifth day of handling such that CX929 was injected 15 min prior to the training episode.
The metabotropic glutamate receptor 5 (mGluR5) antagonist 2-Methyl-6-(phenylethynyl)-pyridine (MPEP; gift of FRAXA, Newburyport, MA, USA) was dissolved in sterile saline to a final concentration of 2.5mg/ml. Mice were given once daily IP injections with saline (4 days) and then with saline or MPEP (5mg/kg; 1 day). The first injection corresponded with the second day of habituation and the final injection containing MPEP was administered 30min before training. This dose and protocol were selected based on prior studies (Seese et al., 2014a).
2.6. Tissue collection and immunohistochemistry
Mice were euthanized 2 min after a 5-min training episode with deep isoflurane anesthesia followed by decapitation. Brains were harvested, cryostat sectioned (20 μm, coronal), and fixed in −20 °C methanol as described (Seese et al., 2012). After being air dried, the tissue was incubated (48 h at room temperature) in a primary antisera cocktail containing mouse anti-PSD95 (1:1000; Thermo Scientific, Waltham, MA, USA) and with either rabbit anti-phosphorylated (p-) TrkB Y515 (1:250; Abcam, Cambridge, MA, USA) or p-ERK1/2 Thr202/Tyr204 (1:500; Cell Signaling, Danvers, MA, USA). We specifically analyzed the Y515 site of TrkB, as this TrkB phosphorylation site promotes ERK1/2 activation (Minichiello, 2009), which is dysregulated in Fmr1 KOs (Hou et al., 2006; Price et al., 2007; Michalon et al., 2012; Seese et al., 2012; Osterweil et al., 2013; Seese et al., 2014b). Slides were rinsed in 0.1 M phosphate buffer (PB) and then incubated with donkey anti-mouse IgG and donkey anti-rabbit IgG tagged with AlexaFluor 488 and 594, respectively (each 1:1000; Invitrogen, Carlsbad, CA, USA), for 1 h. The diluent for both primary and secondary cocktails included 4% bovine serum albumin and 0.1% Triton X-100 in PB. Following the second incubation, slides were washed in PB, air dried, and cover slipped using Vectashield containing DAPI (Vector Laboratories, Burlingame, CA, USA).
2.7. Fluorescence deconvolution tomography
Using a Leica DM6000 epifluorescence microscope, a 63× oil-immersion objective (numerical aperture 1.4), and an ORCA-ER camera (Hamamatsu, Shizuoka, Japan), digital image z-stacks were collected through 3 μm (0.2 μm steps) for 136 × 105 μm sample fields that were centered on stratum radiatum of hippocampal field CA1. For each section, 2–3 z-stacks were collected and processed for restorative deconvolution (Volocity 5.0, Perkin Elmer, Waltham, MA, USA). Automated systems were used to normalize background fluorescence intensity, construct three-dimensional (3D) montages of each 42,840 μm3 sample field (Rex et al., 2009; Seese et al., 2012, 2013), and measure the size, fluorescence intensities, and numbers of each single-and double-labeled element that satisfied the size and eccentricity constraints of synapses. To be included in the analysis, each of the double-labeled elements had to be detected across multiple density thresholds and fully contained within the image z-stack. Elements were classified as double-labeled if there was any overlap in their 3D immunolabeled boundaries as assessed in three dimensions. Counts of double-labeled postsynaptic densities (PSDs; i.e., objects immunoreactive to PSD95) were normalized to the total number of PSD95-immunopositive (+) elements within a given sample field; these normalized values from each z-stack were averaged to obtain a mean value for each brain.
Data were expressed as frequency distributions for density of immunolabeling for phosphorylated target proteins (TrkB, ERK1/2) co-localized with synapse-sized clusters of PSD95 (% of all doubled-labeled contacts at each density bin in an ascending series). Variability in the skewedness of individual curves within a group was low (i.e., the coefficient of variation was < 10%) and comparable between control (vehicle) and drug-treated groups. We removed one animal with the greatest skew for a single experiment in which these conditions were not met.
2.8. Statistical analysis
Values in text and figures are group means ± standard error of the mean (SEM). A single n was an animal for behavioral tests and immunohistochemical analyses and a hippocampal slice for electrophysiological studies. For electrophysiological studies, each group included slices from at least 3 mice. Two-way analyses of variance (ANOVAs) with Bonferroni post-hoc tests and two-tailed Student’s t-tests were used to assess statistical significance (considered as P < .05). For LTP analyses, one-way ANOVAs with Tukey’s post hoc tests were used.
3. Results
Prior studies have shown that LTP in hippocampal field CA1 is abnormal in Fmr1 KO mice: the threshold for induction is elevated and potentiation does not undergo normal stabilization (Lauterborn et al., 2007; Chen et al., 2010c). Infusion of BDNF, which is known to facilitate LTP in WT mice, normalizes Fmr1 KO hippocampal LTP (Lauterborn et al., 2007; Chen et al., 2010c). Does increasing TrkB signaling reduce the amount of training necessary for field CA1-dependent long-term memory in the mutants? To address this question, we used two peripherally administered agents to increase signaling through BDNF’s TrkB receptor: the agonist 7,8-DHF that selectively increases brain TrkB activation following systemic administration (Jang et al., 2010) and the ampakine CX929, a short half-life positive modulator of AMPA-type glutamate receptors that increases BDNF expression (Rex et al., 2006; Simmons et al., 2009, 2011). Relative efficacy of the treatments was assessed with the OLM task in which long-term encoding is sensitive to the duration of training and dependent on field CA1 (Stefanko et al., 2009; Haettig et al., 2011).
3.1. Activation of hippocampal BDNF signaling by the TrkB agonist 7,8-DHF
A single dose of vehicle or 7,8-DHF was given by IP injection to WT mice, and brains were harvested and processed for fluorescence deconvolution tomography 1 h later (Seese et al., 2013) to quantify levels of phosphorylated TrkB in association with the postsynaptic density protein PSD95 (Fig. 1A and B). The TrkB agonist increased the number of synapse-sized PSD95+ elements that were densely labeled for TrkB phosphorylated at its Y515 auto-activation site in CA1 stratum radiatum of WT mice. Specifically, the frequency distribution for the density of immunolabeling for p-TrkB colocalized with PSD95 had a greater rightward skew, towards greater densities, in mice given 7,8-DHF relative to the distribution for the vehicle group (F28, 392 = 2.998, P < .0001; two-way ANOVA; Fig. 1E).
Fig. 1.
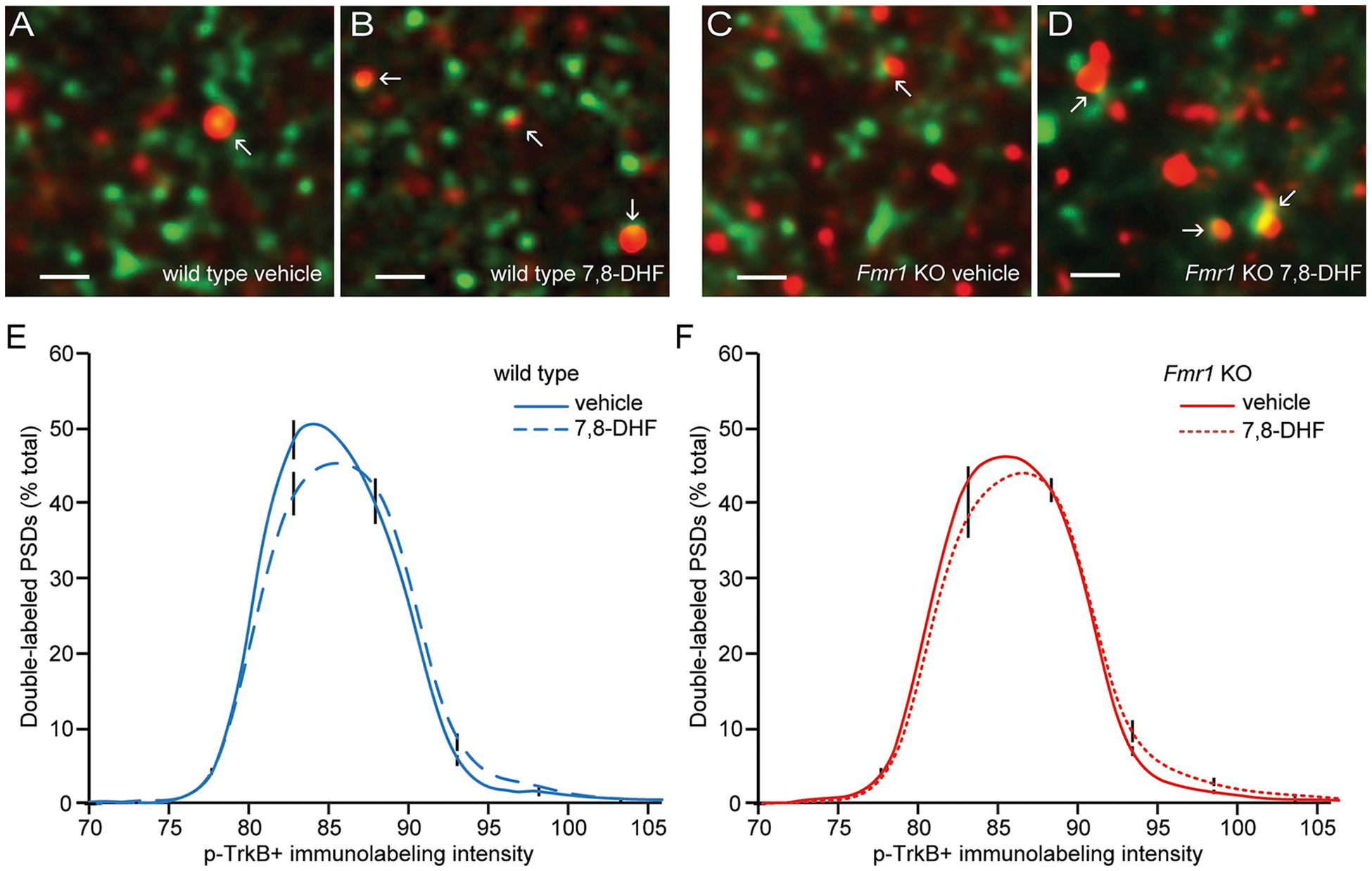
7,8-dihydroxyflavone (7,8-DHF) increases synaptic TrkB activation in WT and Fmr1 KO CA1 stratum radiatum.
A single intraperitoneal injection of vehicle or 7,8-DHF was given to WT and Fmr1 KO mice, and brains were processed for fluorescence deconvolution tomography 1 h later. A-D, Deconvolved images (bar = 1 μm) show merged immunoreactivity for PSD95 (green) and p-TrkB Y515 (red) in field CA1b from WT and Fmr1 KO mice that received either vehicle or 7,8-DHF. E–F, Frequency distributions for the density of p-TrkB immunolabeling co-localized with PSD95 in wild type (E) and Fmr1 KO (F) mice treated with vehicle or 7,8-DHF (means ± SEMs; n = 8/group). Shown is the percentage of the total double-labeled population that fell within each bin of an ascending series of p-TrkB immunolabeling density values. The rightward skews towards higher densities in the 7,8-DHF groups relative to the vehicle groups were significant for both genotypes (WT: two-way ANOVA: F28,392 = 2.998, P < .0001; Fmr1 KO: two-way ANOVA: F28,420 = 1.571, P = .034).
Immunolabeling for total TrkB and PSD95 in the Fmr1 KO mice was comparable to that for the WTs and numbers of double-labeled elements per 105 × 136 × 3 μm sample field were not detectably different (WT: 13,667 ± 674, n = 16 animals; KO: 13,020 ± 744, n = 18. P > .50, Student’s t-test). As in the WTs, treatment with 7,8-DHF increased synaptic levels of p-TrkB activation in Fmr1 KO mice (Fig. 1C and D). To quantify this effect, we compared each group’s frequency distribution curves for all p-TrkB immunolabeling intensities. One hour following 7,8-DHF treatment, the intensity frequency distribution curve for synaptic p-TrkB in the mutants had a greater rightward skew relative to that for vehicle-treated animals (F28,420 = 1.57; P = .034; Fig. 1F).
3.2. The TrkB agonist 7,8-DHF normalized long-term potentiation in Fmr1 KO field CA1
Given that BDNF application restores LTP in Fmr1 KO hippocampal slices (Lauterborn et al., 2007), we next tested if bath infusion of the TrkB agonist 7,8-DHF similarly facilitates LTP induced by a short train of TBS using field recordings from acute hippocampal slices. Using a perfusion system, 7,8-DHF (1 μM) was bath applied to WT or Fmr1 KO slices. As shown in Fig. 2A, in slices from WT mice, infusion of the agonist or vehicle had no effect on baseline responses. For WT slices, delivery of five theta bursts produced stable potentiation of comparable magnitude in the presence of 7,8-DHF or vehicle (Fig. 2A). In contrast, in Fmr1 KO slices treated with vehicle, potentiation was weak and failed to stabilize, whereas in those treated with 7,8-DHF, LTP appeared indistinguishable from that observed in slices from WTs. Analysis of responses during the last 5 min of recording shows that the level of potentiation was significantly greater for KO slices treated with 7,8-DHF as compared to those treated with vehicle (P < .01; one-way ANOVA with Tukey’s post hoc) and was comparable for KO and WT slices treated with 7,8-DHF (P > .05) and for WT slices treated with 7,8-DHF or vehicle (P > .05; Fig. 2B). Moreover, compared to responses in slices that were treated with vehicle alone, the TrkB agonist did not alter the burst response profile (Fig. 2C) or the facilitation of burst responses during the TBS train for either genotype (P > .05 in all groups; repeated measures ANOVA; Fig. 2D).
Fig. 2.
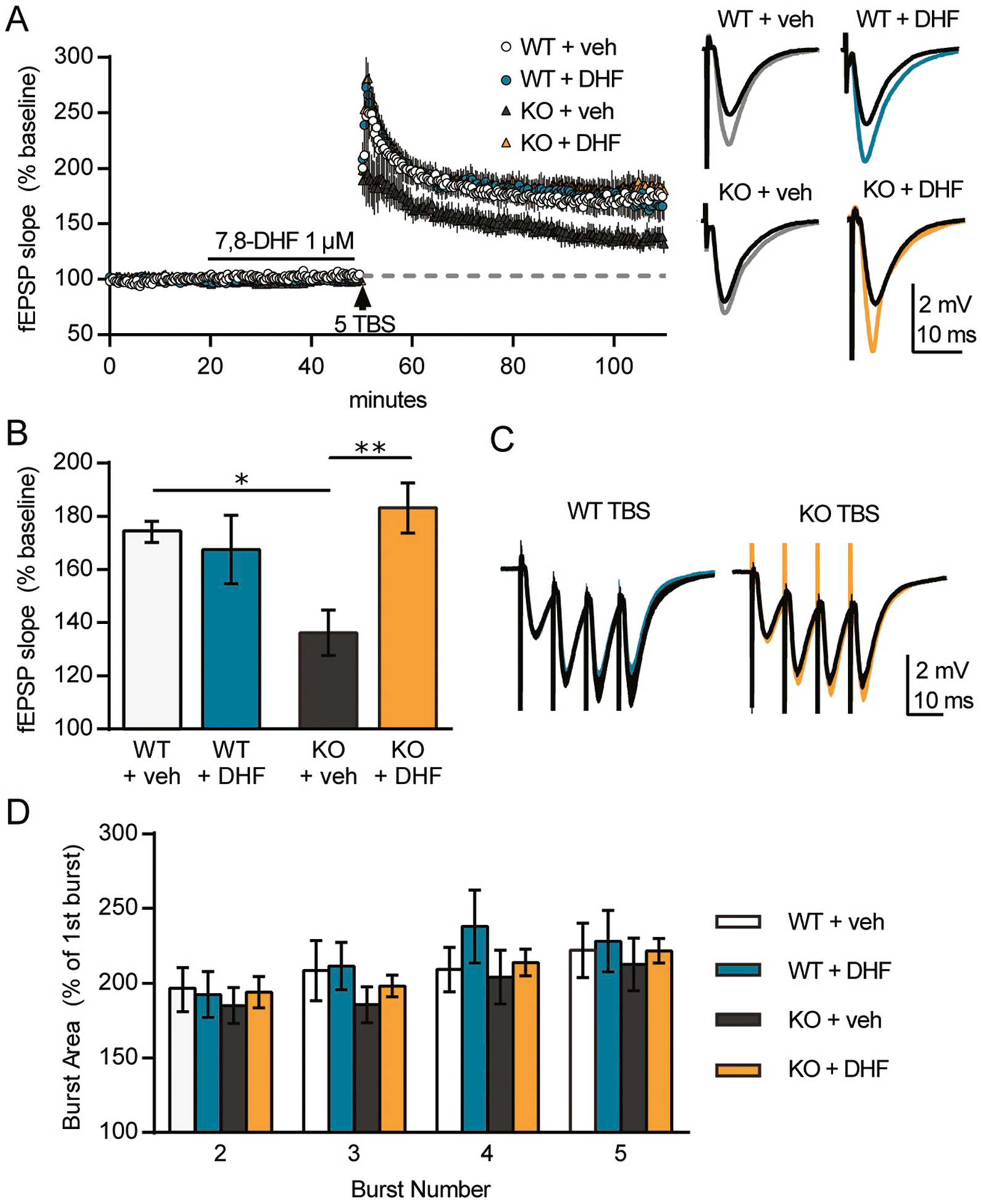
The TrkB agonist 7,8-DHF rescues field CA1 long-term potentiation (LTP) in Fmr1 KO mice.
For all groups, stable baseline responses were collected for 20 min, 7,8-DHF (1 μM) was infused for 30 min, and then a single train of 5 theta bursts was applied to Schaffer-commissural projections and field EPSPs (fEPSPs) were recorded from CA1b apical dendrites for 60 min (n ≥ 5/group). A, left, Plots of fEPSP slopes, normalized to the mean baseline responses, show no effect of 7,8-DHF on baseline responses in any group. Theta burst stimulation (TBS) induced robust potentiation in WTs with no effect of 7,8-DHF (DHF) vs. vehicle (veh) treatment. In slices from Fmr1 KOs receiving vehicle, the responses show a gradual decline towards baseline over the 60 min post-TBS, whereas TBS induced robust and stable potentiation in KO slices receiving 7,8-DHF. A, right, Plots show overlays of the mean of 5 fEPSP responses at the end of the baseline period (black lines) and the mean of 5 fEPSPs at the end of recordings (gray or colour). Scale bar: 2 mV, 10 ms. B, Mean fEPSP slopes from the last 5 min of the recordings (from panel A) show that 7,8-DHF treatment enhanced KO LTP to WT levels but had no significant effect on WT LTP (P = .0066, F(3, 20) = 5.457; one-way ANOVA with Tukey’s post-hoc: *WT + veh vs. KO + veh: P < .05, **KO + veh vs. KO + DHF: P < .01). C, Overlay of average responses to the first theta burst (black: vehicle, colour: 7,8-DHF) shows that the waveforms are comparable across all groups. Scale bar: 2 mV, 10 ms. D, The area of theta burst responses 2 through 5 normalized to the area of the first burst response shows no effect of genotype or 7,8-DHF treatment.
The present responses to 7,8-DHF align with those of BDNF, which similarly rescued LTP in Fmr1 KOs without effect on potentiation in WTs (Lauterborn et al., 2007). Our results also align with reports that 7,8-DHF facilitates otherwise impaired hippocampal LTP in rodent models of post-traumatic stress disorder (Sanz-Garcia et al., 2016) and age-related cognitive disability (Zeng et al., 2012a,b). This is, however, the first report that 7,8-DHF restores potentiation in Fmr1 KO mice, a model that is unique in having both a heightened threshold for induction (Lauterborn et al., 2007; Lee et al., 2011) and impaired consolidation of potentiation (Seese et al., 2012).
3.3. A TrkB agonist rescues memory in Fmr1 KO mice
We next tested if previously described OLM impairments in Fmr1 KOs are reduced or eliminated by 7,8-DHF treatment. Mice received injections of 7,8-DHF or vehicle twice daily for 4 days and then one final injection 1 h prior to training. Mice were tested for retention without 7,8-DHF present 1 day after training (Fig. 3A). Vehicle-treated KOs exhibited marked OLM impairments compared to vehicle-injected WTs (two-way ANOVA: genotype, F1,32 = 4.8, P = .05; treatment, F1,32 = 4.02, P = .05; genotype x treatment, F1,32 = 2.63, P = .11; Bonferroni post-test: vehicle-treated WT vs. KO, P < .05), consistent with previous findings (Seese et al., 2014b). In contrast, after semi-chronic pre-treatment with 7,8-DHF, this form of memory was completely restored in the mutants with discrimination indices being similar to those of WTs (Bonferroni post-test: 7,8-DHF-treated WT vs. KO, P > .05) but much greater than those of mutants that received vehicle (Bonferroni post-test: vehicle-treated KO vs. 7,8-DHF-treated KO, P < .05; Fig. 3B). There were no significant interactions between treatment and genotype for total exploration times during either training or retention trials (two-way ANOVAs, genotype x treatment: F1,33 = 2.94, P = .10 for the training trial; F1,33 = 0.77, P = .39 for the retention trial; Fig. 3C, circles). Total distance traveled was not affected by 7,8-DHF treatment in both genotypes during either the training (two-way ANOVA: genotype F1,30 = 4.28, P < .05; treatment, F1,30 = 0.21, P = .64; genotype x treatment, F1,30 = 0.02, P = .88) or retention trials (two-way ANOVA: genotype, F1,31 = 13.92, P < .001; treatment, F1,31 = 0.27, P = .61; genotype x treatment, F1,31 = 0.04, P = .85; Fig. 3C, squares).
Fig. 3.
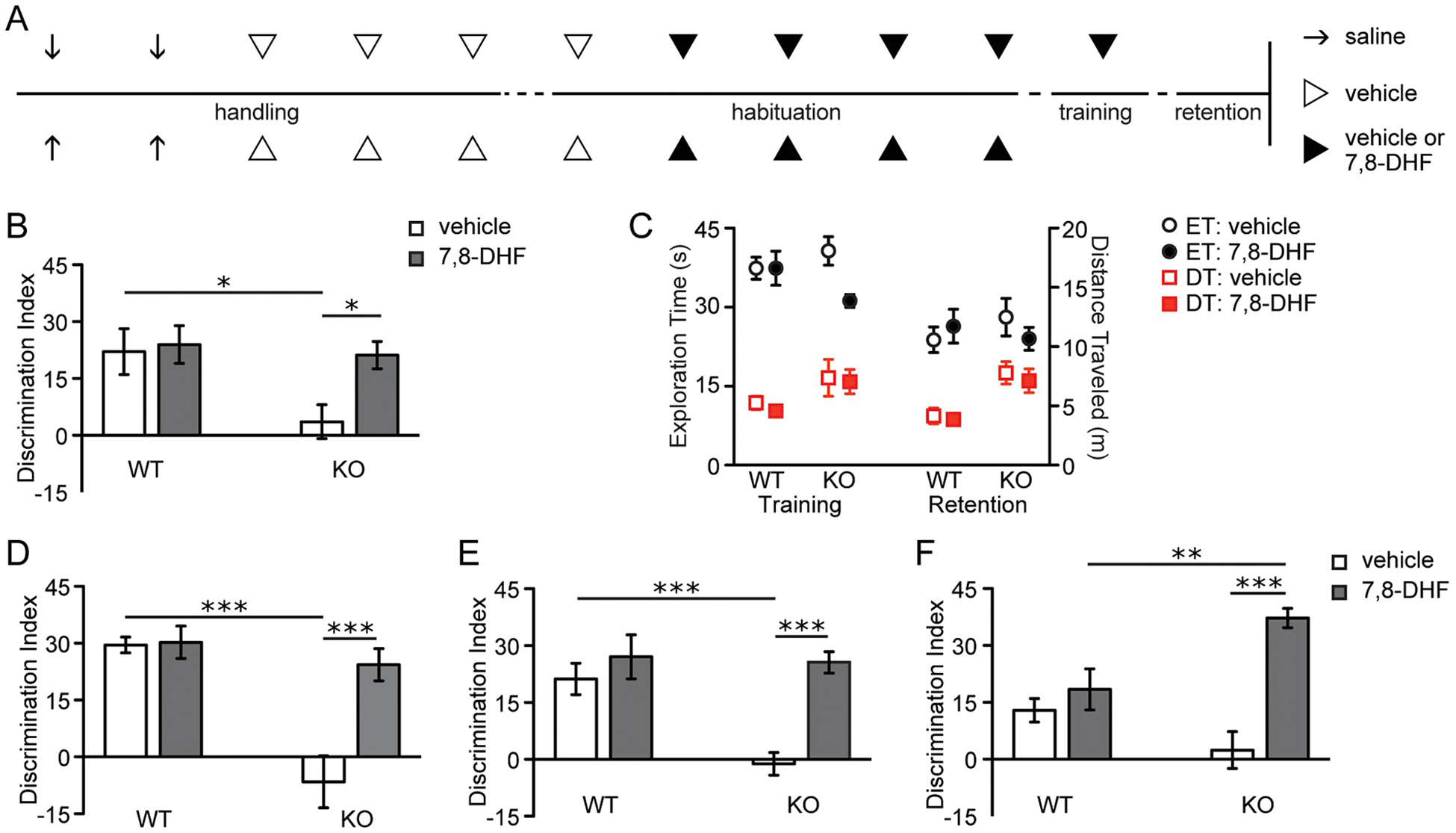
The TrkB agonist 7,8-DHF promotes long-term object location memory (OLM) and object recognition memory (ORM) in Fmr1 KOs.
A, Schematic of experimental paradigm for B and C. Throughout handling and habituation, WT and KO animals were injected with saline (arrows), vehicle (open arrowheads), or either vehicle or 7,8-DHF (closed arrowheads) twice daily. One hour prior to training, animals received a final injection of vehicle or 7,8-DHF. Retention was tested 24 h later. B, Discrimination indices of 7,8-DHF-treated WTs were indistinguishable from those that received vehicle. Vehicle-treated KOs performed significantly worse than vehicle-treated WTs (F1,32 = 2.63; Bonferroni post-test, *P < .05), but KOs that received 7,8-DHF exhibited similarly robust OLM as 7,8-DHF-treated WTs. This effect was significant when compared to mutants that received vehicle (Bonferroni post-test, *P < .05; n = 9/group). C, Exploration time (ET; circles) and distance traveled (DT; red squares) for both genotypes that received vehicle (open shapes) and 7,8-DHF (closed shapes) during training and retention trials are shown (n = 9/group). D, In animals given 7,8-DHF orally for one month prior to training, OLM discrimination indices for vehicle-treated KOs were significantly less than those for vehicle-treated WTs (F1,36 = 10.89; Bonferroni post-test, ***P < .001), but 7,8-DHF normalized mutant performance to that of WTs (n ≥ 9/group), an effect that was significantly greater than mutants that received vehicle (Bonferroni post-test, ***P < .001). E, When vehicle or 7,8-DHF was given once one hour prior to training in experimentally-naïve animals and retention was tested one day later, vehicle-treated KOs performed significantly worse than vehicle-treated WTs (F1,31 = 6.95; Bonferroni post-test, ***P < .001), but 7,8-DHF robustly restored their memory (Bonferroni post-test, ***P < .001; n ≥ 8/group). F, Vehicle or 7,8-DHF was again given prior to training; retention for ORM was tested one day later by changing the identity, rather than the location, of one of the objects. Vehicle-treated KOs showed an impairment in this type of memory compared to vehicle-treated WTs, though this difference was not significant (F1,36 = 1.00; Bonferroni post-test, P > .05). 7,8-DHF robustly potentiated mutant ORM such that their discrimination indices were significantly higher than both WTs that received the compound (Bonferroni post-test, **P < .01) and KOs that received vehicle (***P < .001; n = 10/group).
Interestingly, 7,8-DHF reportedly increases TrkB activity following oral administration (Johnson et al., 2012). To test if OLM rescue could be obtained with this more clinically relevant treatment strategy, WT and Fmr1 KO mice were given 7,8-DHF or vehicle in drinking water continuously for 30 days prior to training. Well-handled mice were then trained in the OLM paradigm using a single 5-min trial and tested for retention 1 day later. Again, vehicle-treated KOs performed significantly worse than vehicle-treated WTs (Bonferroni post-test: P < .001); 7,8-DHF treatment markedly improved retention scores in the mutants (two-way ANOVA for agonist vs. vehicle: genotype, F1,36 = 20.99, P < .0001; treatment, F1,36 = 11.88, P < .01; genotype x treatment, F1,36 = 10.86, P < .01) to a level that was indistinguishable from the 7,8-DHF-treated WT group (Bonferroni post-test: 7,8-DHF-treated WT vs. KO, P > .05) but much greater than vehicle-treated mutants (Bonferroni post-test: vehicle-treated KO vs. 7,8-DHF-treated KO, P < .001; Fig. 3D).
To test if the 7,8-DHF-associated rescue in OLM reflected acute agonist-mediated TrkB activation at the time of training and was not an effect of TrkB activation over the course of the several days of treatments (i.e., either daily injections or chronic oral administration), additional well-handled and sham-injected mice were given a single IP dose of 7,8-DHF or vehicle 1 h prior to training. At retention testing 1 day later, vehicle-treated Fmr1 KOs performed significantly worse than vehicle-treated WTs (Bonferroni post-test: P < .001), whereas 7,8-DHF-treated KOs performed no differently than 7,8-DHF-treated WTs (Bonferroni post-test: P > .05). However, 7,8-DHF-treated KOs did perform significantly better than vehicle-treated mutants (Bonferroni post-test: P < .001). By these measures, acute TrkB activation during the training period restored OLM in the mutants (two-way ANOVA: genotype, F1,31 = 9.02, P < .01; treatment, F1,31 = 16.89, P < .001; genotype x treatment, F1,31 = 6.95, P = .01; Fig. 3E).
As noted above, the OLM task depends on hippocampal field CA1 for memory retrieval (Stefanko et al., 2009; Haettig et al., 2011; Babayan et al., 2012). To test if acute 7,8-DHF treatment also restores learning in a cortex-dependent task, we tested the effects of 7,8-DHF treatment on ORM, a task that depends on perirhinal and insular cortices for retrieval (Balderas et al., 2008; Haettig et al., 2011) and is impaired in Fmr1 KOs (Ventura et al., 2004; Seese et al., 2014b). Previous work demonstrated that when tested for retention 24 h after 5 min of training, WTs exhibit robust ORM but Fmr1 KOs do not. As shown in Fig. 3F, acute treatment with 7,8-DHF 1 h prior to training potently promoted enduring ORM in the KOs when tested 1 day later (two-way ANOVA: genotype, F1,36 = 1.00, P = .32; treatment, F1,36 = 23.55, P < .0001; genotype x treatment, F1,36 = 12.44, P = .001) such that the discrimination indices of agonist-treated mutants were significantly greater than those of both WTs receiving 7,8-DHF (Bonferroni post-test, P < .01) and KOs receiving vehicle (Bonferroni post-test, P < .001).
3.4. Semi-chronic ampakine treatment activates TrkB signaling
Semi-chronic (twice daily) ampakine treatment causes a sustained increase in hippocampal and cortical BDNF protein content, and with cessation of treatment, the increased amount of BDNF protein declines towards normal levels within about 72 h (Lauterborn et al., 2003; Rex et al., 2006; Lynch et al., 2008; Lauterborn et al., 2009). Here, we tested if an ampakine treatment schedule expected to increase BDNF during the period of training (Simmons et al., 2009) affects levels of synaptic p-TrkB immunolabeling in Fmr1 KOs. WT and KO mice were given IP injections with the short half-life (15 min) ampakine CX929 twice daily for 4 days, as in the earlier studies (Simmons et al., 2009; Kramar et al., 2012). To prevent confounding effects of acute AMPA receptor modulation, the last CX929 treatment was given on the afternoon before sacrifice. As was the case with 7,8-DHF treatment, CX929 caused a greater right-ward skew in the frequency distribution for the density of synaptic (PSD95 colocalized) p-TrkB relative to mutants given vehicle (F22,352 = 1.767, P = .019, two-way ANOVA).
TrkB phosphorylation activates multiple signaling cascades (Yoshii and Constantine-Paton, 2010). Normally, OLM training activates ERK1/2 at excitatory synapses in field CA1 (Seese et al., 2014b). Specifically, phosphorylation of TrkB’s Y515 site promotes activation (phosphorylation) of ERK1/2 (Minichiello, 2009), a kinase that is necessary for both LTP and multiple forms of memory (Kelly et al., 2003; Thomas and Huganir, 2004). We accordingly tested if the above CX929 treatment increases synaptic levels of ERK1/2 phosphorylated at its Thr202/Tyr204 activation sites in Fmr1 KOs. The dual immunofluorescence analysis focused on dorsal field CA1b stratum radiatum, a region implicated in OLM and previously shown to exhibit LTP-related synaptic signaling in association with spatial learning (Chen et al., 2010a; Seese et al., 2014b). Quantification of sections immunolabeled for PSD95 and p-ERK1/2 (Fig. 4A–D) indicated that the ampakine clearly shifted the frequency distribution for the density of PSD95 co-localized p-ERK1/2 towards higher values, relative to vehicle-treated wild type mice (F23,291 = 3.288, P < .0001. two-way ANOVA; Fig. 4E). A comparable drug vs. vehicle effect was found with ampakine treatment in Fmr1 KO mice (F23,391 = 3.804, P < .0001; Fig. 4F). There were no evident differences between the WT and KO groups. These results confirm that an ampakine treatment that causes a days-long increase in BDNF protein content also increases activation of synaptic TrkB and related downstream signaling in association with learning.
Fig. 4.
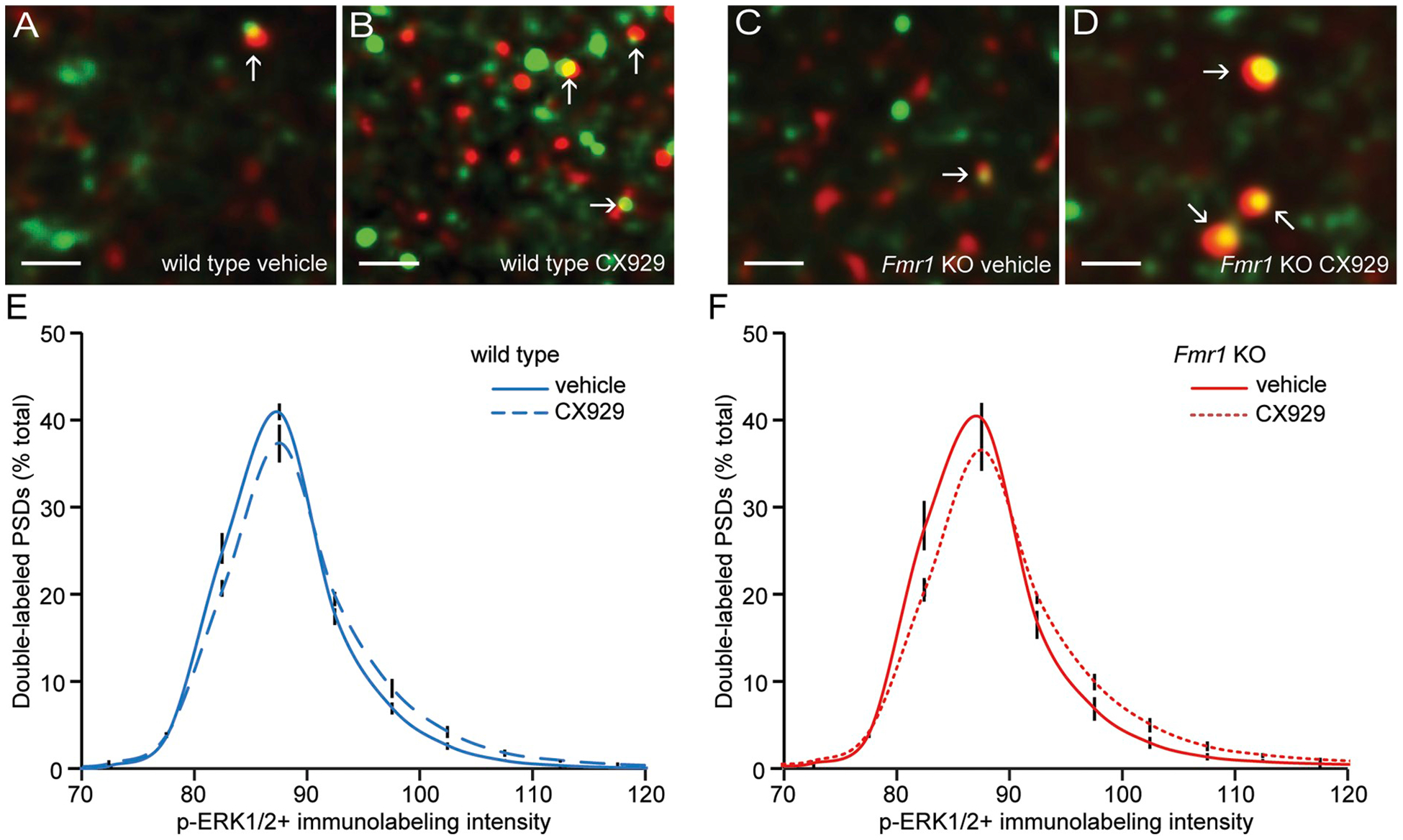
The ampakine CX929 increases synaptic p-ERK1/2 in CA1 stratum radiatum in WT and Fmr1 KO mice.
Fmr1 KO mice were injected with vehicle or CX929 daily for four days and hippocampal sections were processed for dual immunolabeling and fluorescence deconvolution tomography analysis of PSD95 and p-ERK1/2 Thr202/Tyr204 17 h after the final injection (a time point corresponding to the time at which the mice would otherwise be exposed to training). A-D, Deconvolved images (bar = 1 μm) show merged immunolabeling for PSD95 (green) and p-ERK1/2 Thr202/Tyr204 (red) in the CA1b stratum radiatum from WT and Fmr1 KO mice that received vehicle or CX929. E-F, Frequency distributions for the density of p-ERK1/2 immunolabeling co-localized with PSD95 in wild type (E) and Fmr1 KO mice (F) treated with vehicle or CX929 (means ± SEMs; n = 9/group). Shown is the percentage of the total synapse-sized, double-labeled elements that fell within each bin of an ascending series of p-ERK1/2+ values. The greater rightward skew towards higher densities in the CX929 group relative to vehicle controls was significant for both genotypes (WT: two-way ANOVA: F23,391 = 3.288, P < .0001; Fmr1 KO: two-way ANOVA: F23,391 = 3.804, P < .0001).
3.5. Positive AMPA receptor modulation rescues memory in Fmr1 KOs
Wild type and Fmr1 KO mice were treated with the ampakine CX929 for 4 days, as above, and approximately 17 h following the last injection, they were exposed to OLM training and then were tested for retention 1 day later (Fig. 5A). As in prior sets, vehicle-treated KOs did not discriminate the novel object location while vehicle-treated WTs did (Bonferroni post-test: P < .001). CX929 treatment completely rescued long-term OLM in the mutants (two-way ANOVA: genotype, F1,19 = 10.29, P < .01; treatment, F1,19 = 27.48, P < .0001; genotype x treatment, F1,19 = 16.22, P < .001) without affecting discrimination indices in WTs (Fig. 5B). The memory rescue in the mutants was not associated with significant interactions of genotype with treatment for the total time exploring objects during training (two-way ANOVA: genotype x treatment, F1,18 = 0.87, P = .09) or retention (two-way ANOVA: genotype x treatment, F1,19 = 0.87, P = .36) trials (Fig. 5C, circles). Moreover, ampakine treatment did not affect the total distance traveled by either genotype during either the training (two-way ANOVA: genotype, F1,18 = 0.02, P = .88; treatment, F1,18 = 1.93, P = .18; genotype x treatment, F1,18 = 0.11, P = .74) or retention trials (two-way ANOVA: genotype, F1,19 = 0.91, P = .35; treatment, F1,19 = 1.60, P = .22; genotype x treatment, F1,19 = 0.36, P = .55; Fig. 5C, squares).
Fig. 5.
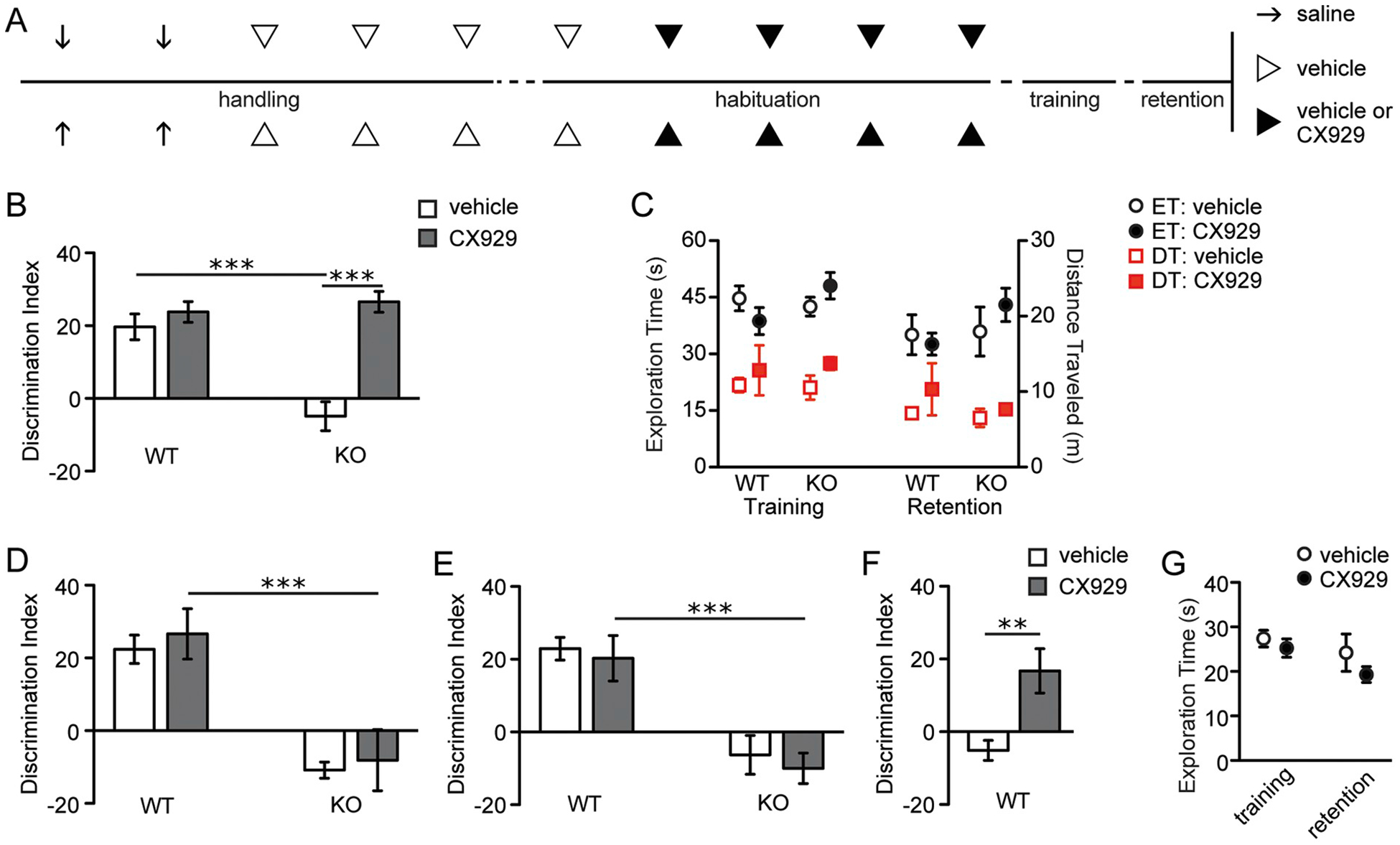
The ampakine CX929 rescues long-term object location memory in Fmr1 KOs and decreases the learning threshold in WTs.
A, Schematic of experimental paradigm for panels B and C. Throughout handling and habituation, WT and Fmr1 KO animals were injected with saline alone (arrows), vehicle alone (open arrowheads), or either vehicle or CX929 (closed arrowheads) twice daily. Animals were then subjected to a 5-min training trial for OLM and retention was tested 24 h later. B, Vehicle-treated mutants performed significantly worse than vehicle-treated WTs (F1,19 = 16.22; Bonferroni post-test, ***P < .001), but CX929 completely restored long-term memory in the mutants (n ≥ 5/group). C, Exploration time (ET; black circles) and distance traveled (DT; red squares) for both genotypes that received vehicle (open shapes) and CX929 (closed shapes) during training and retention trials are shown (n ≥ 5/group). D, When vehicle or CX929 was given just once 15 min prior to training in experimentally-naïve, sham-injected animals and retention was tested 1 day later, no effect of treatment on discrimination indices was appreciated in either genotype (F1,27 = 0.02; Bonferroni post-test, ***P < .001; n ≥ 7/group). E, Animals described in panel B were tested 2 weeks after completion of their first OLM testing using a new context and a new set of objects. Long-term OLM discrimination indices showed that mutants previously treated with CX929 no longer performed at levels of CX929-treated WTs two weeks after the completion of treatment (F1,18 = 0.01; Bonferroni post-test, ***P < .001; n ≥ 5/group). F, Experimentally naïve WT mice were subjected to the same paradigm as depicted in A but with a training trial lasting 3 min. When tested one day later, discrimination indices were significantly higher in CX929-treated WTs as compared to vehicle-treated WTs (Student’s t-test, **P < .01; n = 9/group). G, Exploration times for the WT mice described in F were similar in training and retention trials (n = 9/group).
The effects of semi-chronic ampakine treatments on long-term memory are likely independent of acute AMPA receptor modulation during training given the compound’s short half-life (Simmons et al., 2011). Specifically, since CX929’s half-life is 15 min, over sixty half-lives passed between the time of the last administration of the compound and the time of training. Given that AMPA receptor function is needed for acquiring OLM (Barker and Warburton, 2015), we tested for acute ampakine effects on this form of memory by injecting a second set of WT and KO mice with a single dose of 5 mg/kg CX929 15 min prior to training. With this timing, the compound would be expected to directly modulate AMPA receptors during the training session. At retention testing 1 day later, the KO mice that received acute CX929 treatment performed no differently than KOs injected with vehicle. Moreover, the discrimination indices for both of these groups were lower than those for WT mice injected with vehicle or CX929 just before training (two-way ANOVA: genotype, F1,27 = 34.32, P < .0001; treatment, F1,27 = 0.36, P = .55; genotype x treatment, F1,27 = 0.02, P = .90; Bonferroni post-test, WT CX929 vs. KO CX929, P < .001; Fig. 5D).
The above results indicate that the effects of semi-chronic CX929 administration are likely secondary to changes in gene expression rather than to direct AMPA receptor modulation. Either way, to test if CX929-mediated facilitation of learning is long lasting, we retested the same mice used in the semi-chronic CX929 experiment 2 weeks after their initial retention trial. We did not expect the effects of CX929 on gene expression to be evident after this delay given that neuronal activity-dependent changes in the expression of BDNF, and other genes related to synaptic signaling, are most pronounced within the first 12 h following increases in synaptic activity (Hansen et al., 2014) and BDNF protein levels have largely declined by 72–96 h thereafter (Nawa et al., 1995; Lauterborn et al., 2003). Fourteen days after the initial retention trial, during which no injections were administered, mice were habituated to a novel context (black chamber) for 5 min daily for 5 days; we chose to use a novel context to avoid interference from object-in-context memory (Dix and Aggleton, 1999). On the sixth day, animals were trained in a single 5 min session using a novel set of objects. The following day, one object was moved to a novel location and retention testing was performed a day later. As anticipated, the WT mice showed a preference for the novel location object in the retention trial (previous vehicle group: 22.9 ± 3.1; previous CX929 group: 20.3 ± 6.2). In contrast, Fmr1 KO mice previously treated with either chronic CX929 or vehicle showed no preference for the novel location object on retention testing (Bonferroni post-test, WT CX929 vs. KO CX929, P < .001; Fig. 5E). Specifically, there was a robust difference between genotypes (two-way ANOVA: F1,18 = 37.07, P < .0001), no interaction between genotype and treatment (F1,18 = 0.01, P = .91), and no effect of treatment (F1,18 = 0.42, P = .52). Thus, while the facilitation of learning with a 4-day period of CX929 treatment extends beyond the drug’s immediate modulation of AMPA receptors, the benefit of ampakine treatment dissipated by 2 weeks following treatment cessation.
In the above studies, WT mice that received CX929 did not perform better than WTs that received vehicle, at least when given training that is otherwise sufficient for their learning. However, it is possible that treatments that facilitate TrkB signaling might lower the threshold for learning in WTs. Therefore, we tested if semi-chronic CX929 treatment facilitates object location learning in WT mice given a 3-min long training session that is otherwise insufficient for enduring OLM (Haettig et al., 2011; Seese et al., 2014b). Adult WT mice were injected with CX929 twice daily for 4 days, with the last dose administered approximately 17 h prior to a 3 min training trial. Mice that received the ampakine performed significantly better than those pretreated with vehicle (10% CDX) on the retention trial performed 1 day later (Student’s t-test: P = .005; Fig. 5F). This facilitation of learning object location was not associated with ampakine effects on object exploration times during either the training (Student’s t-test: P = .46) or retention trials (Student’s t-test: P = .30; Fig. 5G).
3.6. Metabotropic glutamate receptor 5 antagonism does not rescue Fmr1 KO OLM
Antagonism of mGluR5 using MPEP normalizes a number of abnormalities in Fmr1 KOs, including spine morphology, mGluR-driven long-term depression, seizure threshold, and other behavioral phenotypes (Krueger and Bear, 2011). To determine if mGluR5 antagonism normalizes OLM, Fmr1 KO and WT mice were given IP injections of saline or 5 mg/kg MPEP 30 min before a 5-min OLM training trial and tested for retention 1 day later (Fig. 6A). Discrimination indices for KOs treated with MPEP were not different than for those treated with vehicle (two-way ANOVA: genotype, F1,32 = 29.16, P < .0001; treatment, F1,32 = 0.21, P = .65; genotype x treatment, F1,32 = 0.59, P = .45) and were significantly smaller than those for WTs treated with MPEP (Bonferroni post-test: MPEP-treated WT vs. KO, P < .01; Fig. 6B). This was not associated with any influences on the distance traveled (two-way ANOVA, genotype x treatment: F1,30 = 1.60, P = .22 for training trial; F1,31 = 0.001, P = .97 for retention trial; Fig. 6C, squares) or on times of object exploration (two-way ANOVA, genotype x treatment: F1,30 = 2.81, P = .10 for training trial; F1,32 = 1.44, P = .24 for retention trial; Fig. 6C, circles). Thus, acute administration of the mGluR5 antagonist MPEP did not rescue this form of memory in the Fmr1 KOs.
Fig. 6.
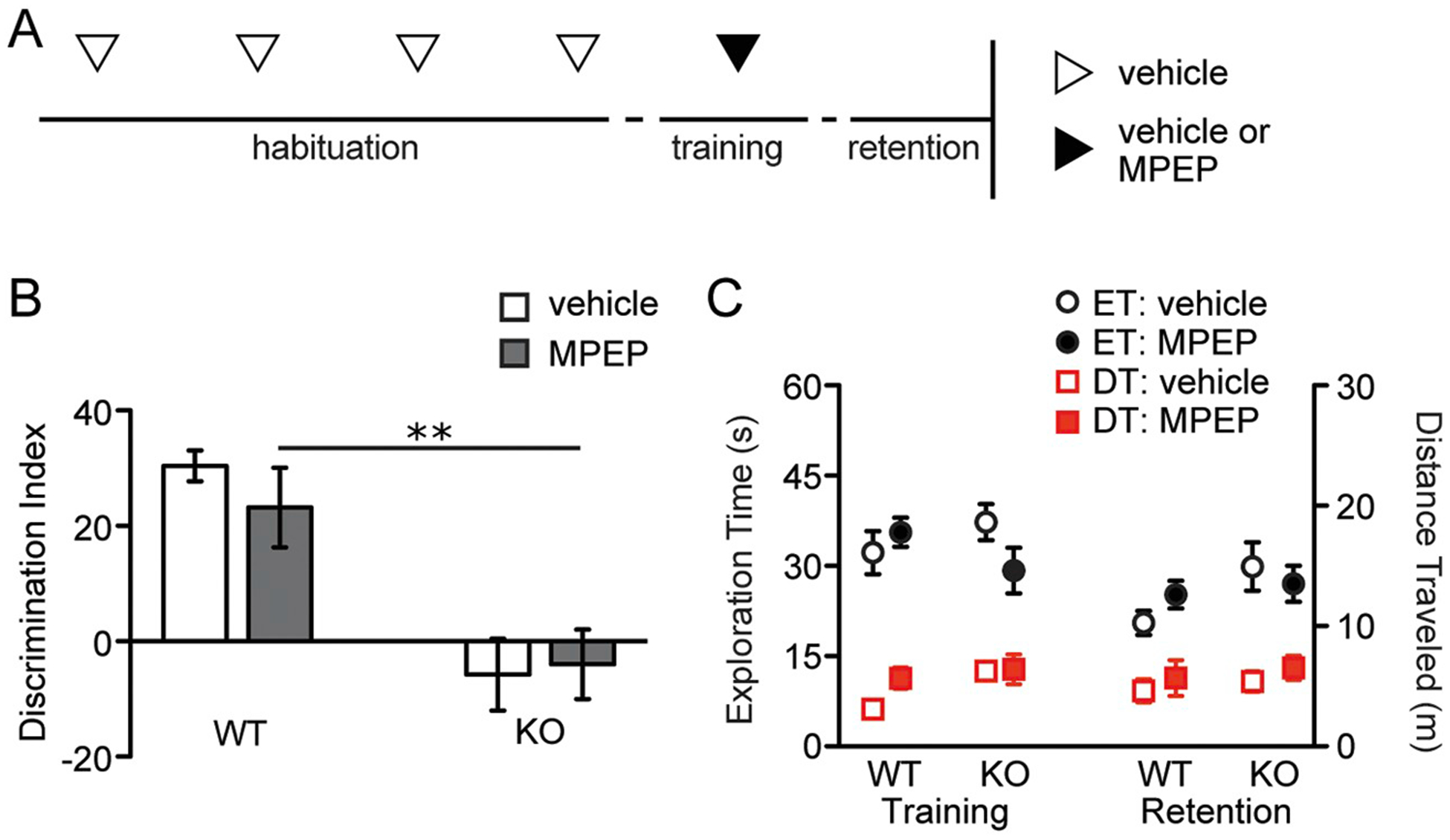
The mGluR5 antagonist MPEP does not restore Fmr1 KO long-term OLM.
A, Schematic of experimental paradigm for panels B and C. Starting on the second day of habituation, WT and Fmr1 KO mice were injected with vehicle alone (open arrowheads) once daily for four days. Thirty minutes prior to training, animals were injected with either vehicle or MPEP (closed arrowhead). Retention was tested 24 h later. B, MPEP-treated KOs, like vehicle-treated KOs, continued to exhibit impaired OLM retention indices, an effect that was statistically significant when compared to MPEP-treated WTs (F1,32 = 0.59; Bonferroni post-test, **P < .01; n ≥ 8/group). C, Exploration time (ET; black circles) and distance traveled (DT; red squares) for both genotypes that received vehicle (open shapes) and MPEP (closed shapes) during training and retention trials are shown (n ≥ 8/group).
3.7. AMPAR modulation rescues social recognition in Fmr1 KOs
Finally, we tested if the pharmacological manipulations that restored memory in the above paradigms also influence the social recognition abnormalities that characterize FXS and Fmr1 KOs. Mice were tested in the three-chambered social approach and recognition paradigm described by Crawley and colleagues (Yang et al., 2011). The social approach test involves a choice between sampling, within chamber A, an empty inverted wire mesh cup versus, within chamber B, an identical cup containing a novel target mouse; after a 5 min delay, this is followed by a social recognition test in which the animal is exposed to the previously encountered mouse in chamber A vs. a novel mouse in chamber B (Fig. 7A). Prior studies found that WT but not Fmr1 KO mice have a strong preference for the novel mouse in the recognition test (Budimirovic and Kaufmann, 2011). We first evaluated the total time spent exploring the two choices, a measure that would be sensitive to arousal level and overall locomotion. The total time spent exploring the two choices was not detectably different for Fmr1 KOs treated with vehicle as compared to treatment with 7,8-DHF or CX929 during both approach (Fig. 7B) and recognition (Fig. 7C) phases of testing; this was the case for both acute and semi-chronic treatment schedules (Fig. 7B and C). Fmr1 KOs treated with vehicle showed a strong preference for exploration of the target mouse (vs. empty cup) in the approach phase of the task and this was unaffected by treatment with 7,8-DHF (Fig. 7D, left). Across groups, the mutants spent 6 to 8 times more time exploring the mouse than the empty cup (P values ranged from 0.002 to 0.00001 in paired t-tests). In the social recognition test, the Fmr1 KOs given vehicle did not exhibit preferential exploration of the novel vs. familiar mouse (P > .20, paired t-tests), as expected from earlier studies (Budimirovic and Kaufmann, 2011). In contrast, KOs given either acute or semi-chronic treatment with 7,8-DHF spent considerably more time investigating the novel mouse (Fig. 7D, right; P = .021 and 0.002 vs. vehicle, respectively). In a second study of KOs given ampakine or vehicle treatment, the acute and semi-chronic vehicle groups again had high discrimination indices for the approach phase of the task. These were associated with robust differences between the total exploration times of the target mouse and empty cup (P < .00001 in both groups). During the recognition phase, the vehicle-treated KOs demonstrated no evident preference for the novel mouse. Ampakine treatment did not influence the otherwise high discrimination indices of Fmr1 KOs in the approach phase of the task, whereas semi-chronic, but not acute, treatment with CX929 rescued social recognition, increasing the discrimination index to levels seen in the approach phase (acute: P > .30; semi-chronic: P < .00001 vs. vehicle; Fig. 7E). In all, stimulation of TrkB receptors directly with the agonist 7,8-DHF or indirectly via ampakine treatment and associated increases in BDNF levels had a normalizing effect on the response to social novelty in Fmr1 KO mice.
Fig. 7.
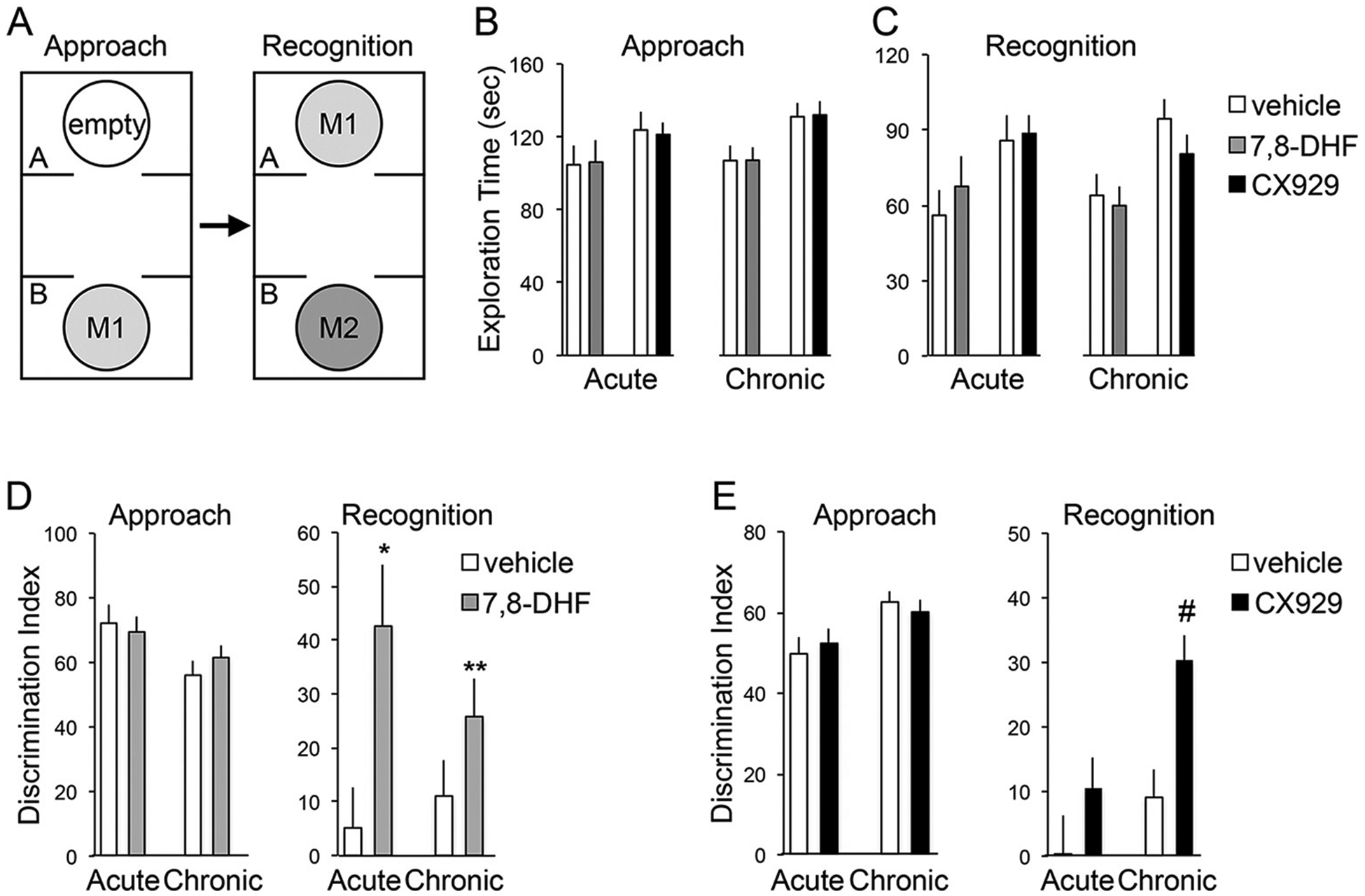
The TrkB agonist 7,8-DHF and the ampakine CX929 independently restore social recognition in Fmr1 KO mice.
A, Schematic for the experimental paradigm. Social recognition tested for discrimination between a novel C57BL/6 wild type mouse (“M2”) from a familiar mouse (“M1”) that was previously introduced during the social approach phase. B–C, Total exploration times of Fmr1 KOs for both cups during the social approach (B) and social recognition (C) phases are shown. As described in Methods, 7,8-DHF (gray bars) or vehicle (white bars) was injected acutely (n ≥ 5/group) or given through drinking water chronically (n = 19/group), whereas CX929 (black bars) or vehicle was injected for both acute (n ≥ 11/group) and chronic (n ≥ 19/group) treatment experiments. D, Fmr1 KOs treated with either vehicle or 7,8-DHF showed a strong preference for exploration of the target mouse (vs. empty cup) in the approach phase. KOs injected with 7,8-DHF spent significantly more time with the novel mouse than with the familiar mouse during social recognition when compared to KOs injected with vehicle (*P < .05; **P < .01; unpaired t-tests). E, Chronic CX929 treatment did not influence the strong positive discrimination indices of KOs interacting with the novel mouse over the inanimate object during the social approach phase. However, KOs treated with CX929 chronically, as compared to those treated with vehicle, spent far more time interacting with the novel mouse than the familiar mouse in the social recognition phase (#P < .00001; unpaired t-test).
4. Discussion
The present studies show that enhanced TrkB signaling lowers the learning threshold in WTs and restores long-term potentiation as well as two forms of otherwise impaired learning in Fmr1 KO mice, a model for the most common form of inherited intellectual disability (Hagerman and Hagerman, 2002). Specifically, the TrkB agonist 7,8-DHF and the ampakine CX929 both promoted long-term memory. Moreover, training-associated signaling known to be impaired in the KOs – namely, synaptic ERK1/2 activation – was enhanced following semi-chronic ampakine administration.
As demonstrated previously for other models of cognitive impairment (Simmons et al., 2009; Baudry et al., 2012), we show here that a semi-chronic ampakine treatment regimen known to increase endogenous BDNF protein levels (Rex et al., 2006; Lauterborn et al., 2009; Simmons et al., 2009) rescued memory encoding in the Fmr1 KOs. Additionally, we show that ampakine treatment reduced the previously established threshold (Stefanko et al., 2009; Seese et al., 2014b) for encoding simple spatial memory in WT mice. This result aligns with slice electrophysiology results showing that ampakines lower the induction threshold and increase the magnitude of LTP in rat hippocampus (Lynch, 2002; Lynch and Gall, 2006). Nevertheless, effects of acute ampakine treatment were not sufficient to rescue either OLM or social recognition, indicating that second-order actions of positive AMPA receptor modulation, such as changes in gene expression, underlie the efficacy of treatment.
How might increasing endogenous BDNF with an ampakine or increasing signaling through the TrkB receptor with an agonist alter learning thresholds (see Fig. 8)? LTP requires reorganization of the actin cytoskeleton (Krucker et al., 2000; Kramar et al., 2006; Rex et al., 2009, 2010), and stabilization of filamentous actin has been implicated in different forms of learning (Fedulov et al., 2007; Cox et al., 2014; Rudy, 2015; Lamprecht, 2016). Along these lines, hippocampus-dependent learning induces robust TrkB signaling at hippocampal synapses (Chen et al., 2010b). Prior studies demonstrated that activity-induced signaling leading to new filamentous actin is not impaired in the Fmr1 KOs (Lauterborn et al., 2007), a feature that distinguishes these mice from other rodent models of cognitive disability (Simmons et al., 2009; Baudry et al., 2012). We show here that semi-chronic ampakine treatment significantly enables training-induced phosphorylation of ERK1/2, a kinase downstream of TrkB Y515 phosphorylation that contributes to actin stabilization through phosphorylating actincrosslinking proteins (Kruchten et al., 2008; Seese et al., 2012). Synaptic ERK1/2 is hyper-phosphorylated in the mutants (Hou et al., 2006; Price et al., 2007; Michalon et al., 2012; Seese et al., 2012; Osterweil et al., 2013), an effect that is associated with multiple measures of kinase dysfunction, including a lack of responsiveness to synaptic activity (Seese et al., 2012) and training (Seese et al., 2014b) as well as impaired associations with upstream (Ras and protein phosphatase 2A) and downstream (cortactin) substrates (Seese et al., 2012 and unpublished results). Further increasing phosphorylation of this kinase to offer therapeutic benefits seems counterintuitive in these conditions. However, the ERK1/2 measurements in the present study were collected after several daily ampakine treatments and so reflect an extended period over which BDNF levels and TrkB signaling were increased. The neurotrophin activates multiple small GTPase-initiated pathways and downstream effectors leading to rapid structural modifications to synapses and gene expression (Minichiello, 2009). While these processes are expected to have been influenced in ampakine-treated KOs, we propose that the combination of events results in a more normal baseline ERK1/2 activity prior to learning. This could allow for successful, task-specific regulation of cytoskeletal events (e.g., cortactin phosphorylation and actin network stabilization) critical for memory encoding that are otherwise impaired in Fmr1 KO synapses (Chen et al., 2010a; Seese et al., 2012).
Fig. 8.
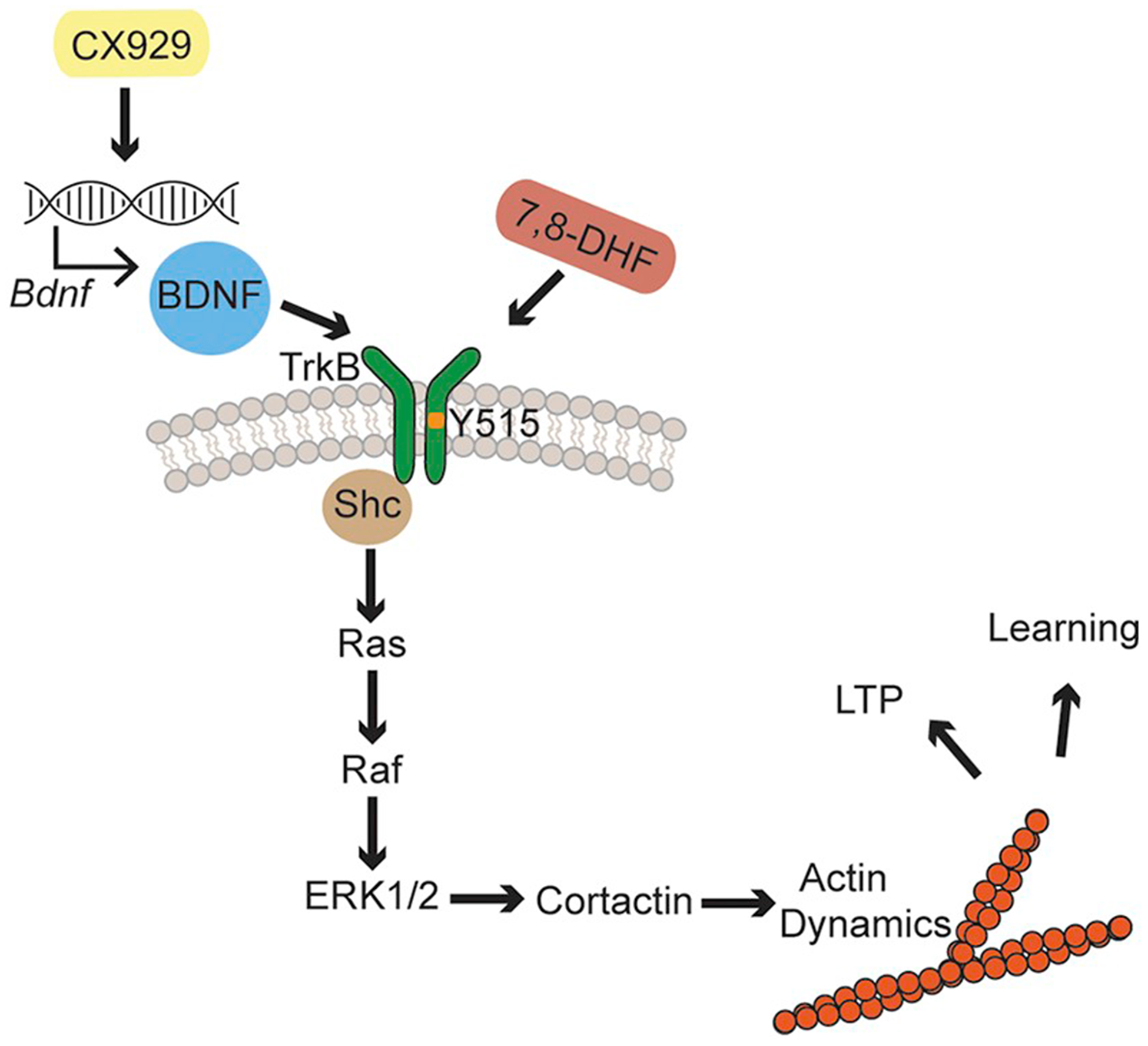
Phosphorylation of TrkB at Y515 promotes synaptic signaling that modifies the actin cytoskeleton and promotes memory.
This report tests the effects of two compounds in Fmr1 KO mice: 7,8-DHF and CX929. 7,8-DHF is a small molecular agonist of the TrkB receptor (Jang et al., 2010) and CX929 has been demonstrated to increase endogenous BDNF, TrkB’s major ligand (Simmons et al., 2009; Simmons et al., 2011). The TrkB receptor is phosphorylated at many different residues, including Y515, Y816, and Y490. The Y515 site has been demonstrated to be crucial for activation of the canonical Ras-ERK1/2 signaling pathway (Minichiello, 2009). ERK1/2, in turn, has many effectors, many of which are related to the modification of the actin cytoskeleton. One of these effectors is cortactin, which plays an important role in stabilization of actin (Sanchez et al., 2000; Harrison and Turley, 2001; Cosen-Binker and Kapus, 2006). Actin dynamics are crucial for both LTP and many different forms of learning (Krucker et al., 2000; Kramar et al., 2006; Fedulov et al., 2007; Rex et al., 2009, 2010; Cox et al., 2014; Rudy, 2015; Lamprecht, 2016).
The present results build on existing literature implicating ERK1/2 as a therapeutic target in Fmr1 KOs. ERK1/2 inhibition normalizes excessive protein synthesis and abolishes audiogenic seizures in the Fmr1 KOs (Osterweil et al., 2010; Wang et al., 2012). Spaced training both rescues topographic increases in synaptic phosphorylated ERK1/2 levels in hippocampal field CA1 of Fmr1 KOs and restores long-term memory in an ERK1/2-dependent manner (Seese et al., 2014b). Interestingly, in the BTBR T(+) Itpr3(tf)/J mouse model of autism spectrum disorders, increased synaptic ERK1/2 phosphorylation in hippocampal field CA1 successfully predicted the subset of animals with poorer OLM performance (Seese et al., 2014a). With high-throughput fluorescence deconvolution tomography, we confirmed here that in vivo CX929 decreases Fmr1 KO object location learning threshold while promoting activation of ERK1/2 within synapses of the same region. Although other studies have demonstrated that the mGluR5 antagonist MPEP normalizes many aspects of the Fmr1 KO phenotype (Krueger and Bear, 2011), the present results show that mGluR5 antagonism does not exert the same potent effects as does promoting TrkB signaling in restoring hippocampus-dependent learning in the mutants.
Evidence that the TrkB agonist 7,8-DHF facilitated long-term memory in all paradigms tested supports the hypothesis that CX929’s therapeutic effects involve enhanced TrkB signaling. This aligns with a rapidly growing literature demonstrating that 7,8-DHF rescues many cognitive functions in different models of intellectual disability (Devi and Ohno, 2012; Johnson et al., 2012; Zeng et al., 2012a, 2012b). Importantly, Tian and colleagues showed that 7,8-DHF promotes fear conditioning and performance in the Morris water maze in Fmr1 KOs (Tian et al., 2015) and Uutela and colleagues demonstrated that decreasing BDNF expression in Fmr1 KOs induces a significant impairment in contextual fear conditioning (Uutela et al., 2012). To build on this work, we confirmed the compound’s utility in less anxiety-provoking tasks while also demonstrating the compound’s efficacy through oral administration, a paradigm that may be more clinically relevant. One surprising result in our study was that KOs that received 7,8-DHF performed even better than treated WTs on ORM, a task that depends on cortical regions for retrieval (Balderas et al., 2008; Haettig et al., 2011). Further studies are needed to test if there are regional differences in TrkB expression or activation following learning and if these differences are affected by genotype. However, one clue to help explain this effect of genotype comes from work showing that pilocarpine-induced seizures increases BDNF mRNA in the proximal dendrites of layer V cortical neurons significantly more in KOs than in WTs, while the degree of increase of BDNF mRNA in hippocampal proximal dendrites is not different between the genotypes (Louhivuori et al., 2011). Thus, it is plausible that learning-induced BDNF increases may be more pronounced in KO cortex than in WT cortex, resulting in more robust ORM once exogenous 7,8-DHF activates a baseline amount of required TrkB receptors. Nonetheless, our observations further support the conclusion that BDNF signaling enhancement leading to cytoskeletal modifications produces cognitive improvement in the Fmr1 KOs. Future studies can test this hypothesis more directly by assessing effects of 7,8-DHF on OLM in Fmr1 KOs when actin dynamics have been disrupted by infusion of local treatments with agents that specifically target actin assembly and/or stabilization (Rex et al., 2010).
Notably, results for the two TrkB-related treatments on a social recognition test in which Fmr1 KOs are markedly different from wild types paralleled those for object location learning: acute 7,8-DHF or semi-chronic CX929 treatment increased the extent to which the mutants interacted with a novel mouse. These effects could not be ascribed to global changes in behavior because the compounds did not noticeably influence the total time exploring the choices in either phase of testing. Our findings do not address the question of whether the positive actions of the drugs reflect enhanced discrimination between a familiar and a stranger mouse or instead are due to normalization of the response to social novelty. One interesting possibility as to how increasing TrkB signaling produces the shift towards the WT behavior is that the KOs may have problems rapidly familiarizing themselves with a stranger and so do not spend the greater part of their time interacting with this stranger when given the choice between a recently encountered mouse and a novel one. Enhanced BDNF signaling could, via actions like those underlying its effects on spatial learning, enable a more effective and rewarding encoding of a novel, complex encounter.
The beneficial effects of up-regulating TrkB signaling may come hand-in-hand with adverse effects. A third of FXS patients exhibit some degree of seizure disorder (Hagerman and Stafstrom, 2009), Fmr1 KOs exhibit increased audiogenic seizures (Yan et al., 2005), and BDNF is reported to promote seizures in the epileptic brain (Binder et al., 2001; Heinrich et al., 2011; McNamara and Scharfman, 2012). Moreover, the potential contributions of BDNF signaling to epilepsy in FXS is complicated by the fact that different TrkB phosphorylation sites normally promote different seizure-related processes. Phosphorylation of TrkB residue Y515 promotes recruitment of Shc to TrkB and ultimately protects against status epilepticus-induced hippocampal injury (Huang et al., 2019). Phosphorylation of TrkB residue Y816, on the other hand, promotes the receptor’s association with PLC-γ1, and disrupting this association prevents the development of epilepsy following seizures (Gu et al., 2015). Whether pharmacological up-regulation of BDNF production or signaling with either CX929 or 7,8-DHF promotes seizures in Fmr1 KOs is an unresolved issue. We did not observe evidence for such an effect in the sizable number of mice tested in the present studies or with rats and mice in prior studies using CX929 (Rex et al., 2006; Simmons et al., 2009, 2011; Baudry et al., 2012). Further studies will need to directly test the effects of these compounds on the TrkB Y816 phosphorylation site in Fmr1 KOs.
Decreasing BDNF expression in Fmr1 KO mice has been demonstrated to ameliorate many phenotypes in the mouse model, including locomotor hyperactivity and rotarod-based motor coordination (Uutela et al., 2012). Given that the present studies used treatments that increase TrkB signaling, we tested if either compound influenced the total distance traveled or interaction with objects during the behavioral trials (see Figs. 3C and 5C). We observed no significant effects of treatment on these measures, during training or retention trials, for 7,8-DHF or CX929. However, further in-depth examination of these issues is warranted and should be conducted in conjunction with conventional preclinical toxicology. Limitations aside, the results presented here suggest strong translatable approaches for the treatment of cognitive and social impairments in FXS and possibly other neurodevelopmental disabilities.
Acknowledgements
We thank Lucy Yao, Jihua Liu, and Michael Weinberg for their excellent technical assistance.
Funding sources
This work was supported by National Institutes of Health grants HD089491, NS085709, and NS04260. R.R.S. was supported by training grant T32-GM0862 and NIMH predoctoral fellowship FMH095432A.
Footnotes
Declaration of Competing Interest
The authors report no conflict of interests.
References
- Andero R, Heldt SA, Ye K, Liu X, Armario A, Ressler KJ, 2011. Effect of 7,8-dihydroxyflavone, a small-molecule TrkB agonist, on emotional learning. Am. J. Psychiatry 168, 163–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babayan AH, Kramar EA, Barrett RM, Jafari M, Haettig J, Chen LY, Rex CS, Lauterborn JC, Wood MA, Gall CM, Lynch G, 2012. Integrin dynamics produce a delayed stage of long-term potentiation and memory consolidation. J. Neurosci 32, 12854–12861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balderas I, Rodriguez-Ortiz CJ, Salgado-Tonda P, Chavez-Hurtado J, McGaugh JL, Bermudez-Rattoni F, 2008. The consolidation of object and context recognition memory involve different regions of the temporal lobe. Learn. Mem 15, 618–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker GR, Warburton EC, 2015. Object-in-place associative recognition memory depends on glutamate receptor neurotransmission within two defined hippocampal-cortical circuits: a critical role for AMPA and NMDA receptors in the hippocampus, perirhinal, and prefrontal cortices. Cereb. Cortex 25, 472–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baudry M, Kramar E, Xu X, Zadran H, Moreno S, Lynch G, Gall C, Bi X, 2012. Ampakines promote spine actin polymerization, long-term potentiation, and learning in a mouse model of Angelman syndrome. Neurobiol. Dis 47, 210–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekinschtein P, Cammarota M, Medina JH, 2014. BDNF and memory processing. Neuropharmacology 76 (Pt C), 677–683. [DOI] [PubMed] [Google Scholar]
- Bhakar AL, Dolen G, Bear MF, 2012. The pathophysiology of fragile X (and what it teaches us about synapses). Annu. Rev. Neurosci 35, 417–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binder DK, Croll SD, Gall CM, Scharfman HE, 2001. BDNF and epilepsy: too much of a good thing? Trends Neurosci. 24, 47–53. [DOI] [PubMed] [Google Scholar]
- Bramham CR, 2008. Local protein synthesis, actin dynamics, and LTP consolidation. Curr. Opin. Neurobiol 18, 524–531. [DOI] [PubMed] [Google Scholar]
- Budimirovic DB, Kaufmann WE, 2011. What can we learn about autism from studying fragile X syndrome? Dev. Neurosci 33, 379–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen LY, Rex CS, Pham DT, Lynch G, Gall CM, 2010a. BDNF signaling during learning is regionally differentiated within hippocampus. J. Neurosci 30, 15097–15101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen LY, Rex CS, Sanaiha Y, Lynch G, Gall CM, 2010b. Learning induces neurotrophin signaling at hippocampal synapses. Proc. Natl. Acad. Sci. U. S. A 107, 7030–7035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen LY, Rex CS, Babayan AH, Kramar EA, Lynch G, Gall CM, Lauterborn JC, 2010c. Physiological activation of synaptic Rac > PAK (p-21 activated kinase) signaling is defective in a mouse model of fragile X syndrome. J. Neurosci 30, 10977–10984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosen-Binker LI, Kapus A, 2006. Cortactin: the gray eminence of the cytoskeleton. Physiology (Bethesda) 21, 352–361. [DOI] [PubMed] [Google Scholar]
- Cox CD, Rex CS, Palmer LC, Babayan AH, Pham DT, Corwin SD, Trieu BH, Gall CM, Lynch G, 2014. A map of LTP-related synaptic changes in dorsal hippocampus following unsupervised learning. J. Neurosci 34, 3033–3041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devi L, Ohno M, 2012. 7,8-dihydroxyflavone, a small-molecule TrkB agonist, reverses memory deficits and BACE1 elevation in a mouse model of Alzheimer’s disease. Neuropsychopharmacology 37, 434–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dix SL, Aggleton JP, 1999. Extending the spontaneous preference test of recognition: evidence of object-location and object-context recognition. Behav. Brain Res 99, 191–200. [DOI] [PubMed] [Google Scholar]
- Fedulov V, Rex CS, Simmons DA, Palmer L, Gall CM, Lynch G, 2007. Evidence that long-term potentiation occurs within individual hippocampal synapses during learning. J. Neurosci 27, 8031–8039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fombonne E, 2006. Past and future perspectives on autism epidemiology. In: Moldin S, Rubenstein J (Eds.), Understanding Autism: From Basic Neuroscience to Treatment. CRC Press, pp. 25–48. [Google Scholar]
- Gross C, Berry-Kravis EM, Bassell GJ, 2012. Therapeutic strategies in fragile X syndrome: dysregulated mGluR signaling and beyond. Neuropsychopharmacology 37, 178–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu B, Huang YZ, He XP, Joshi RB, Jang W, McNamara JO, 2015. A peptide uncoupling BDNF receptor TrkB from phospholipase Cgamma1 prevents epilepsy induced by status epilepticus. Neuron 88, 484–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haettig J, Stefanko DP, Multani ML, Figueroa DX, McQuown SC, Wood MA, 2011. HDAC inhibition modulates hippocampus-dependent long-term memory for object location in a CBP-dependent manner. Learn. Mem 18, 71–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagerman RJ, Hagerman PJ, 2002. Fragile X Syndrome: Diagnosis, Treatment, and Research. Johns Hopkins University Press, Baltimore. [Google Scholar]
- Hagerman PJ, Stafstrom CE, 2009. Origins of epilepsy in fragile X syndrome. Epilepsy Curr. 9, 108–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen KF, Sakamoto K, Pelz C, Impey S, Obrietan K, 2014. Profiling status epilepticus-induced changes in hippocampal RNA expression using high-throughput RNA sequencing. Sci. Rep 4, 6930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison RE, Turley EA, 2001. Active erk regulates microtubule stability in H-ras-transformed cells. Neoplasia 3, 385–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinrich C, Lahteinen S, Suzuki F, Anne-Marie L, Huber S, Haussler U, Haas C, Larmet Y, Castren E, Depaulis A, 2011. Increase in BDNF-mediated TrkB signaling promotes epileptogenesis in a mouse model of mesial temporal lobe epilepsy. Neurobiol. Dis 42, 35–47. [DOI] [PubMed] [Google Scholar]
- Hou L, Antion MD, Hu D, Spencer CM, Paylor R, Klann E, 2006. Dynamic translational and proteasomal regulation of fragile X mental retardation protein controls mGluR-dependent long-term depression. Neuron 51, 441–454. [DOI] [PubMed] [Google Scholar]
- Huang YZ, He XP, Krishnamurthy K, McNamara JO, 2019. TrkB-Shc signaling protects against hippocampal injury following status epilepticus. J. Neurosci 39, 4624–4630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin SA, Idupulapati M, Gilbert ME, Harris JB, Chakravarti AB, Rogers EJ, Crisostomo RA, Larsen BP, Mehta A, Alcantara CJ, Patel B, Swain RA, Weiler IJ, Oostra BA, Greenough WT, 2002. Dendritic spine and dendritic field characteristics of layer V pyramidal neurons in the visual cortex of fragile-X knockout mice. Am. J. Med. Genet 111, 140–146. [DOI] [PubMed] [Google Scholar]
- Jang SW, Liu X, Yepes M, Shepherd KR, Miller GW, Liu Y, Wilson WD, Xiao G, Blanchi B, Sun YE, Ye K, 2010. A selective TrkB agonist with potent neurotrophic activities by 7,8-dihydroxyflavone. Proc. Natl. Acad. Sci. U. S. A 107, 2687–2692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson RA, Lam M, Punzo AM, Li H, Lin BR, Ye K, Mitchell GS, Chang Q, 2012. 7,8-dihydroxyflavone exhibits therapeutic efficacy in a mouse model of Rett syndrome. J. Appl. Physiol 112, 704–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazdoba TM, Leach PT, Silverman JL, Crawley JN, 2014. Modeling fragile X syndrome in the Fmr1 knockout mouse. Intractable Rare Dis. Res 3, 118–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly A, Laroche S, Davis S, 2003. Activation of mitogen-activated protein kinase/extracellular signal-regulated kinase in hippocampal circuitry is required for consolidation and reconsolidation of recognition memory. J. Neurosci 23, 5354–5360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramar EA, Lin B, Rex CS, Gall CM, Lynch G, 2006. Integrin-driven actin polymerization consolidates long-term potentiation. Proc. Natl. Acad. Sci. U. S. A 103, 5579–5584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramar EA, Chen LY, Lauterborn JC, Simmons DA, Gall CM, Lynch G, 2012. BDNF upregulation rescues synaptic plasticity in middle-aged ovariectomized rats. Neurobiol. Aging 33, 708–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruchten AE, Krueger EW, Wang Y, McNiven MA, 2008. Distinct phospho-forms of cortactin differentially regulate actin polymerization and focal adhesions. Am. J. Phys. Cell Phys 295, C1113–C1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krucker T, Siggins GR, Halpain S, 2000. Dynamic actin filaments are required for stable long-term potentiation (LTP) in area CA1 of the hippocampus. Proc. Natl. Acad. Sci. U. S. A 97, 6856–6861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger DD, Bear MF, 2011. Toward fulfilling the promise of molecular medicine in fragile X syndrome. Annu. Rev. Med 62, 411–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamprecht R, 2016. The role of actin cytoskeleton in memory formation in amygdala. Front. Mol. Neurosci 9, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauterborn JC, Truong GS, Baudry M, Bi X, Lynch G, Gall CM, 2003. Chronic elevation of brain-derived neurotrophic factor by ampakines. J. Pharmacol. Exp. Ther 307, 297–305. [DOI] [PubMed] [Google Scholar]
- Lauterborn JC, Rex CS, Kramar E, Chen LY, Pandyarajan V, Lynch G, Gall CM, 2007. Brain-derived neurotrophic factor rescues synaptic plasticity in a mouse model of fragile X syndrome. J. Neurosci 27, 10685–10694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauterborn JC, Pineda E, Chen LY, Ramirez EA, Lynch G, Gall CM, 2009. Ampakines cause sustained increases in brain-derived neurotrophic factor signaling at excitatory synapses without changes in AMPA receptor subunit expression. Neuroscience 159, 283–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauterborn JC, Palmer LC, Jia Y, Pham DT, Hou B, Wang W, Trieu BH, Cox CD, Kantorovich S, Gall CM, Lynch G, 2016. Chronic ampakine treatments stimulate dendritic growth and promote learning in middle-aged rats. J. Neurosci 36, 1636–1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HY, Ge WP, Huang W, He Y, Wang GX, Rowson-Baldwin A, Smith SJ, Jan YN, Jan LY, 2011. Bidirectional regulation of dendritic voltage-gated potassium channels by the fragile X mental retardation protein. Neuron 72, 630–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louhivuori V, Vicario A, Uutela M, Rantamaki T, Louhivuori LM, Castren E, Tongiorgi E, Akerman KE, Castren ML, 2011. BDNF and TrkB in neuronal differentiation of Fmr1-knockout mouse. Neurobiol. Dis 41, 469–480. [DOI] [PubMed] [Google Scholar]
- Lynch G, 2002. Memory enhancement: the search for mechanism-based drugs. Nat. Neurosci 5, 1035–1038 Suppl. [DOI] [PubMed] [Google Scholar]
- Lynch G, Gall CM, 2006. Ampakines and the threefold path to cognitive enhancement. Trends Neurosci. 29, 554–562. [DOI] [PubMed] [Google Scholar]
- Lynch G, Rex CS, Chen LY, Gall CM, 2008. The substrates of memory: defects, treatments, and enhancement. Eur. J. Pharmacol 585, 2–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNamara JO, Scharfman HE (2012) Temporal lobe epilepsy and the BDNF receptor, TrkB. Jasper’s Basic Mechanisms of the Epilepsies, 4th Edition (Noebels JL, Avoli M, Rogawski MA, Olsen RW, Delgado-Escueta AV). (Bethesda (MD)). [PubMed] [Google Scholar]
- Michalon A, Sidorov M, Ballard TM, Ozmen L, Spooren W, Wettstein JG, Jaeschke G, Bear MF, Lindemann L, 2012. Chronic pharmacological mGlu5 inhibition corrects fragile X in adult mice. Neuron 74, 49–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minichiello L, 2009. TrkB signalling pathways in LTP and learning. Nat. Rev. Neurosci 10, 850–860. [DOI] [PubMed] [Google Scholar]
- Mizuno M, Yamada K, Olariu A, Nawa H, Nabeshima T, 2000. Involvement of brain-derived neurotrophic factor in spatial memory formation and maintenance in a radial arm maze test in rats. J. Neurosci 20, 7116–7121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nawa H, Carnahan J, Gall C, 1995. BDNF protein measured by a novel enzyme immunoassay in normal brain and after seizure: partial disagreement with mRNA levels. Eur. J. Neurosci 7, 1527–1535. [DOI] [PubMed] [Google Scholar]
- Osterweil EK, Krueger DD, Reinhold K, Bear MF, 2010. Hypersensitivity to mGluR5 and ERK1/2 leads to excessive protein synthesis in the hippocampus of a mouse model of fragile X syndrome. J. Neurosci 30, 15616–15627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osterweil EK, Chuang SC, Chubykin AA, Sidorov M, Bianchi R, Wong RK, Bear MF, 2013. Lovastatin corrects excess protein synthesis and prevents epileptogenesis in a mouse model of fragile X syndrome. Neuron 77, 243–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeiffer BE, Huber KM, 2009. The state of synapses in fragile X syndrome. Neuroscientist 15, 549–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price TJ, Rashid MH, Millecamps M, Sanoja R, Entrena JM, Cervero F, 2007. Decreased nociceptive sensitization in mice lacking the fragile X mental retardation protein: role of mGluR1/5 and mTOR. J. Neurosci 27, 13958–13967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rex CS, Lauterborn JC, Lin CY, Kramar EA, Rogers GA, Gall CM, Lynch G, 2006. Restoration of long-term potentiation in middle-aged hippocampus after induction of brain-derived neurotrophic factor. J. Neurophysiol 96, 677–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rex CS, Chen LY, Sharma A, Liu J, Babayan AH, Gall CM, Lynch G, 2009. Different rho GTPase-dependent signaling pathways initiate sequential steps in the consolidation of long-term potentiation. J. Cell Biol 186, 85–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rex CS, Gavin CF, Rubio MD, Kramar EA, Chen LY, Jia Y, Huganir RL, Muzyczka N, Gall CM, Miller CA, Lynch G, Rumbaugh G, 2010. Myosin IIb regulates actin dynamics during synaptic plasticity and memory formation. Neuron 67, 603–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudy JW, 2015. Actin dynamics and the evolution of the memory trace. Brain Res. 1621, 17–28. [DOI] [PubMed] [Google Scholar]
- Sanchez C, Diaz-Nido J, Avila J, 2000. Phosphorylation of microtubule-associated protein 2 (MAP2) and its relevance for the regulation of the neuronal cytoskeleton function. Prog. Neurobiol 61, 133–168. [DOI] [PubMed] [Google Scholar]
- Sanz-Garcia A, Knafo S, Pereda-Perez I, Esteban JA, Venero C, Armario A, 2016. Administration of the TrkB receptor agonist 7,8-dihydroxyflavone prevents traumatic stress-induced spatial memory deficits and changes in synaptic plasticity. Hippocampus 26, 1179–1188. [DOI] [PubMed] [Google Scholar]
- Seese RR, Babayan AH, Katz AM, Cox CD, Lauterborn JC, Lynch G, Gall CM, 2012. LTP induction translocates cortactin at distant synapses in wild-type but not Fmr1 knock-out mice. J. Neurosci 32, 7403–7413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seese RR, Chen LY, Cox CD, Schulz D, Babayan AH, Bunney WE, Henn FA, Gall CM, Lynch G, 2013. Synaptic abnormalities in the infralimbic cortex of a model of congenital depression. J. Neurosci 33, 13441–13448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seese RR, Maske AR, Lynch G, Gall CM, 2014a. Long-term memory deficits are associated with elevated synaptic ERK1/2 activation and reversed by mGluR5 antagonism in an animal model of autism. Neuropsychopharmacology 39, 1664–1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seese RR, Wang K, Yao YQ, Lynch G, Gall CM, 2014b. Spaced training rescues memory and ERK1/2 signaling in fragile X syndrome model mice. Proc. Natl. Acad. Sci. U. S. A 111, 16907–16912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmons DA, Rex CS, Palmer L, Pandyarajan V, Fedulov V, Gall CM, Lynch G, 2009. Up-regulating BDNF with an ampakine rescues synaptic plasticity and memory in Huntington’s disease knockin mice. Proc. Natl. Acad. Sci. U. S. A 106, 4906–4911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmons DA, Mehta RA, Lauterborn JC, Gall CM, Lynch G, 2011. Brief ampakine treatments slow the progression of Huntington’s disease phenotypes in R6/2 mice. Neurobiol. Dis 41, 436–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefanko DP, Barrett RM, Ly AR, Reolon GK, Wood MA, 2009. Modulation of long-term memory for object recognition via HDAC inhibition. Proc. Natl. Acad. Sci. U. S. A 106, 9447–9452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas GM, Huganir RL, 2004. MAPK cascade signalling and synaptic plasticity. Nat. Rev. Neurosci 5, 173–183. [DOI] [PubMed] [Google Scholar]
- Tian M, Zeng Y, Hu Y, Yuan X, Liu S, Li J, Lu P, Sun Y, Gao L, Fu D, Li Y, Wang S, McClintock SM, 2015. 7, 8-Dihydroxyflavone induces synapse expression of AMPA GluA1 and ameliorates cognitive and spine abnormalities in a mouse model of fragile X syndrome. Neuropharmacology 89, 43–53. [DOI] [PubMed] [Google Scholar]
- Turner G, Webb T, Wake S, Robinson H, 1996. Prevalence of fragile X syndrome. Am. J. Med. Genet 64, 196–197. [DOI] [PubMed] [Google Scholar]
- Uutela M, Lindholm J, Louhivuori V, Wei H, Louhivuori LM, Pertovaara A, Akerman K, Castren E, Castren ML, 2012. Reduction of BDNF expression in Fmr1 knockout mice worsens cognitive deficits but improves hyperactivity and sensor-imotor deficits. Genes Brain Behav. 11, 513–523. [DOI] [PubMed] [Google Scholar]
- Ventura R, Pascucci T, Catania MV, Musumeci SA, Puglisi-Allegra S, 2004. Object recognition impairment in Fmr1 knockout mice is reversed by amphetamine: involvement of dopamine in the medial prefrontal cortex. Behav. Pharmacol 15, 433–442. [DOI] [PubMed] [Google Scholar]
- Wang X, Snape M, Klann E, Stone JG, Singh A, Petersen RB, Castellani RJ, Casadesus G, Smith MA, Zhu X, 2012. Activation of the extracellular signal-regulated kinase pathway contributes to the behavioral deficit of fragile x-syndrome. J. Neurochem 121, 672–679. [DOI] [PubMed] [Google Scholar]
- Wang W, Cox BM, Jia Y, Le AA, Cox CD, Jung KM, Hou B, Piomelli D, Gall CM, Lynch G, 2018. Treating a novel plasticity defect rescues episodic memory in Fragile X model mice. Mol. Psychiatry 23, 1798–1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan QJ, Rammal M, Tranfaglia M, Bauchwitz RP, 2005. Suppression of two major Fragile X Syndrome mouse model phenotypes by the mGluR5 antagonist MPEP. Neuropharmacology 49, 1053–1066. [DOI] [PubMed] [Google Scholar]
- Yang M, Silverman JL, Crawley JN, 2011. Automated three-chambered social approach task for mice. Curr. Protoc. Neurosci 8, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshii A, Constantine-Paton M, 2010. Postsynaptic BDNF-TrkB signaling in synapse maturation, plasticity, and disease. Dev. Neurobiol 70, 304–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng Y, Liu Y, Wu M, Liu J, Hu Q, 2012a. Activation of TrkB by 7,8-dihydroxyflavone prevents fear memory defects and facilitates amygdalar synaptic plasticity in aging. J. Alzheimers Dis 31, 765–778. [DOI] [PubMed] [Google Scholar]
- Zeng Y, Lv F, Li L, Yu H, Dong M, Fu Q, 2012b. 7,8-dihydroxyflavone rescues spatial memory and synaptic plasticity in cognitively impaired aged rats. J. Neurochem 122, 800–811. [DOI] [PubMed] [Google Scholar]