

Summary
Non-alcoholic steatohepatitis (NASH) is characterized by the accumulation of hepatic fat in an inflammatory/fibrotic background. Herein, we show that the hepatic high-activity glutaminase 1 isoform (GLS1) is overexpressed in NASH. Importantly, GLS1 inhibition reduced lipid content in choline and/or methionine deprivation-induced steatotic mouse primary hepatocytes, in human hepatocyte cell lines and in NASH mouse livers. We suggest that, under these circumstances, defective glutamine fueling of anaplerotic mitochondrial metabolism and concomitant reduction of oxidative stress promotes a reprogramming of serine metabolism, wherein serine is shifted from the generation of the antioxidant glutathione and channeled to provide one-carbon units to regenerate the methionine cycle. The restored methionine cycle can induce phosphatidylcholine synthesis from the phosphatidylethanolamine N-methyltransferase-mediated and CDP-choline pathways as well as by base-exchange reactions between phospholipids, thereby restoring hepatic phosphatidylcholine content and very-low density lipoprotein export. Overall, we provide evidence that hepatic GLS1 targeting is a valuable therapeutic approach in NASH.
Keywords: folates cycle, glutaminase, GLS1, GLS2, methionine cycle, NAFLD, NASH, phospholipids, TCA cycle, VLDL
Graphical Abstract

eTOC Blurb
Simon et al. show that the glutaminase GLS1 isoform is augmented in both NASH clinical biopsies and pre-clinical mouse models. GLS1 silencing significantly reduced steatosis and oxidative stress through complex metabolic reprogramming, involving increased VLDL export, indicating that GLS1 may be a valuable therapeutic target for the treatment of NASH.
Introduction
Non-alcoholic fatty liver disease (NAFLD) is a global health problem with an estimated worldwide prevalence of 30-40% of the adult population (Younossi et al., 2016). It includes a group of conditions characterized by the accumulation of fat in the liver resulting from a metabolic reprogramming which can include: i) increased fatty acid uptake; ii) reduced mitochondrial fatty acid beta-oxidation (FAO), the catabolic process by which fatty acids are metabolized; iii) increased de novo lipogenesis synthesis of fatty acids; iv) and/or disruptions in the very-low-density lipoprotein (VLDL) assembly and secretion, particles that account for the hepatic triglycerides, cholesterol, and apolipoproteins exported from the liver to peripheral tissues. Patients with simple steatosis, a condition usually considered rather benign on the NAFLD spectrum, have excellent prognosis from a liver standpoint. On the other hand, nonalcoholic steatohepatitis (NASH), a more aggressive form that is characterized by necroinflammation and fibrosis localized on the most harmful part of the spectrum of NAFLD progression, is an important risk factor for cirrhosis and liver cancer (Ascha et al., 2010), one of the deadliest and fastest-increasing types of cancer, as well as for cardiovascular diseases, a major cause of global mortality (Soderberg et al., 2010). Pharmacological approaches targeting NASH should ideally block progression and reverse liver injury. However, in spite of the huge investment by the pharmacological industry over the last years, there are still no approved therapies targeting NASH (Cassidy and Syed, 2016). Weight loss and exercise are the only effective therapeutic approaches, but compliance remains a problem.
Dysfunctional VLDL synthesis and secretion is a hallmark of NASH (Fujita et al., 2009). Hepatic VLDL assembly and secretion is a complex process involving apoliprotein B (ApoB), microsomal triglyceride transfer protein (MTP), and lipid mobilizing and synthesizing proteins, particularly phospholipids (Eisenberg and Olivecrona, 1979). Indeed, rare ApoB and MTP mutations are associated with progressive liver disease (Cefalu et al., 2013; Di Filippo et al., 2014). Likewise, feeding rodents a diet deficient in choline, a substrate of the synthesis of the main phospholipid forming the VLDL particle membranes, phosphatidylcholine (Ptd-Cho), results in the accumulation of fat in the liver (Caballero et al., 2010). Exclusively in the liver, Ptd-Cho can be additionally synthesized from the methylation of phosphatidylethanolamine (Ptd-Et), a reaction catalyzed by the enzyme phosphatidylethanolamine N-methyltransferase (PEMT) that uses S-adenosylmethionine (SAMe), an intermediate of the methionine cycle, as the methyl donor (Noga et al., 2002). On this basis, Pemt−/− mice present disrupted VLDL triglyceride export and are more prone to develop liver steatosis (Zhao et al., 2009). Likewise, both the methionine adenosyltransferase 1a (Mat1a−/−) mice and mice maintained on a methionine-deficient diet, have chronically low levels of SAMe and spontaneously develop NASH as a consequence of an impaired PEMT pathway and hepatic VLDL assembly (Caballero et al., 2010; Cano et al., 2011; Chen et al., 2010; Mato et al., 2013). This is relevant to human NAFLD as about 50% of NAFLD patients present a serum metabolic signature associated with Mat1a−/− mice (Alonso et al., 2017), indicating that impaired VLDL assembly may be present in a subtype of NAFLD patients.
The nutritional model of feeding a diet deficient in both methionine and choline (MCD) is characterized by macrovesicular steatosis, hepatocellular death, inflammation, oxidative stress, and fibrosis. Even though there are discrepancies between this model and human NASH, with weight loss, the lack of peripheral insulin resistance and hypoglycemia being the most confounding factors, this model is often used in NASH research as short-term dietary interventions are associated with NASH. Using this animal model, Li and colleagues have found significant changes in the serum levels of glutamine (Li et al., 2011). L-glutamine is the most abundant free amino acid in the human body and some reports have highlighted the beneficial protective effects of oral glutamine supplementation on the development of NASH (Sellmann et al., 2015). In the liver, glutaminase (EC 3.5.1.2) is the main regulator enzyme of glutamine metabolism and catalyzes the first step of glutamine catabolism through the conversion of glutamine to glutamate and ammonia. There are two different phosphate-activated glutaminase isoforms, GLS2 and GLS1. The GLS2 gene, located on chromosome 12, encodes two splice variants with a low activity and allosteric regulation, liver-type glutaminase (LGA, short transcript isoform), and glutaminase B (GAB, long transcript isoform), which are highly expressed in normal adult liver (Mates et al., 2013). Likewise, the GLS1 gene, located in chromosome 2, encodes two splice variants with a high activity and low Km, kidney-type glutaminase (KGA, long transcript isoform) and the glutaminase C (GAC, short transcript isoform), which is mainly expressed in the kidney under healthy conditions. Notably, a metabolic switch from the GLS2 to GLS1 isoform occurs in cirrhosis and liver cancer (Yu et al., 2015; Yuneva et al., 2012). More recently, Du and colleagues have shown that GLS1 is induced in fibrotic livers and inhibition of glutaminase blocked the activation of hepatic stellate cells (HSCs), the major hepatic cell type involved in fibrosis, halting fibrosis progression (Du et al., 2018). In a similar way, the scavenging of the toxic side product of glutaminase, ammonia, has been shown to be important for preventing fibrosis progression (Canbay and Sowa, 2019; De Chiara et al., 2019). In spite of this, the relevance of the expression of GLS1 isoform in NAFLD is poorly understood. A previous study has shown that treatment of hepatoma cells with phenylbutyrate, a glutamine scavenger, reduced the palmitate-mediated induction of triglycerides levels by decreasing endoplasmic reticulum (ER) stress (Rahman et al., 2009). In agreement with this, the hepatic metabolism of the amino acid glutamine has been previously implicated in the regulation of cellular redox balance in the pathophysiology of numerous diseases as glutamine can be used as an anaplerotic substrate to support mitochondrial metabolism (Alberghina and Gaglio, 2014; Faubert et al., 2014). In here, we show that GLS1 is overexpressed in NASH, both in the clinical setting and in pre-clinical mouse models and, more importantly, we show that specific GLS1-inhibition accounts for reduced liver steatosis and oxidative stress. Under conditions of methionine and choline deprivation, usually characterized by impaired VLDL export, GLS1 inhibition lowered oxidative stress and restored Ptd-Chol synthesis and VLDL triglyceride export, thereby reducing liver steatosis. Overall, hepatic GLS1 targeting is suggested as a valuable therapeutic approach in NASH.
Results
Glutaminase 1 (GLS1) is overexpressed in clinical non-alcoholic steatohepatitis (NASH)
High-throughput metabolomics is a widely used method to investigate metabolic phenotypes in specific conditions. Here, we screened serum levels of glutamine and the glutaminase reaction product, glutamate, in a large cohort of patients (Barr et al., 2012). Whereas there are no significant differences in serum glutamine levels between controls (n= 90) and NASH patients (n= 131), serum glutamate levels are significantly increased in NASH patients (Fig. 1A), suggesting that glutamine catabolism may be aberrant in NASH. Glutaminase is the main regulator enzyme of hepatic glutamine catabolism, catalyzing the conversion of glutamine to glutamate and ammonia (the latter is excreted by the urea cycle). Whereas GLS2 is the major isoform expressed in the healthy liver, a switch from GLS2 to GLS1 occurs in liver fibrosis (Du et al., 2018), cirrhosis and liver cancer (Yu et al., 2015; Yuneva et al., 2012). Herein, a group of patients diagnosed with NASH, characterized in Table S1, show increased hepatic GLS1 levels relative to healthy controls. Likewise, in another cohort of patients with NASH, mRNA levels of GLS1 were shown to be increased relative to healthy controls. Under these conditions, the isoform 2 of glutaminase, GLS2, usually distributed around the hepatic periportal compartment in healthy people, is decreased in the livers of NASH patients. In addition, glutamine synthetase, usually expressed in the perivenous hepatocytes of healthy livers, catalyzing the synthesis of glutamine from glutamate and elimination of residual ammonia that escapes from detoxification in the periportal hepatocytes, is induced (Fig. 1B,C).
Figure 1. Glutaminase 1 (GLS1) is overexpressed in clinical non-alcoholic steatohepatitis (NASH).
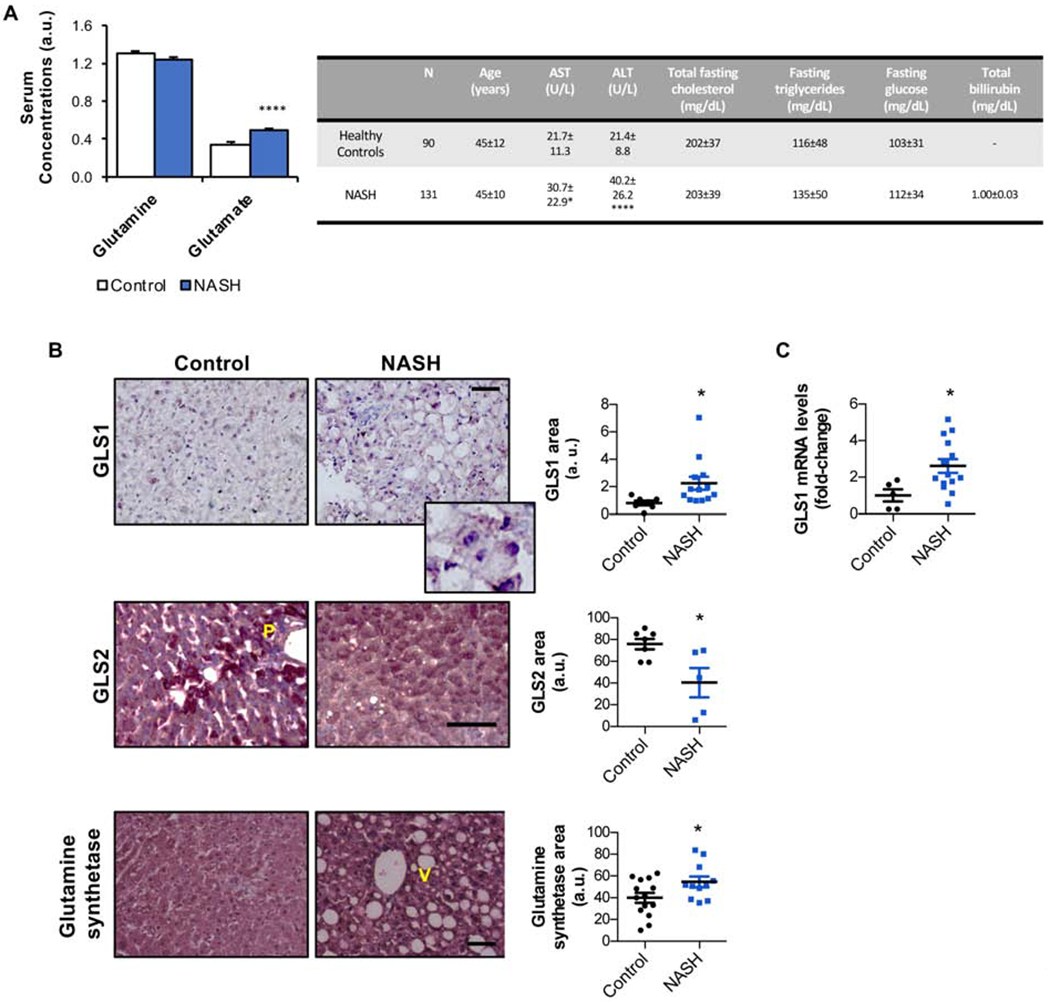
A. Serum levels of glutamine and the product of glutamine catabolism, glutamate, in a large cohort of patients diagnosed with NASH (n= 131) relative to a control group of healthy subjects (n= 90). A table showing the main serum biochemical parameters relative to these patients is shown. B. Liver immunohistochemical staining and respective quantification for the isoform 1 of glutaminase (GLS1), and inset zoom, the isoform 2 of glutaminase (GLS2) and glutamine synthetase in another cohort of NASH patients (n= 13) versus a control group of healthy subjects (n= 13). Scale bar corresponds to 100 μm. V-venous region; P-portal region. C. mRNA levels of GLS1 in a cohort of NASH patients (n=16) against a control group of age- and body-weight matched healthy controls (n=5). Data is shown as average ± SEM and Student’s t-test was used to compare groups. *p<0.05 and ****p<0.0001 against the control group are shown (See also Table S1).
Overall, we provide evidence that hepatic GLS1 expression is increased in NASH patients.
Glutaminase 1 (GLS1) is overexpressed in mouse models of non-alcoholic steatohepatitis (NASH)
Taking into consideration the relevance of GLS1 expression in clinical NASH, we have evaluated GLS1 expression in an in vivo pre-clinical mouse model of NASH, the mice fed a choline and methionine deficient diet. Even though this model presents some constraints it is one of the most often used models in NASH research. Herein, the choline deficient and 0.1% methionine diet (0.1%MCDD) was used as one of the limitations of a methionine and choline deficient diet is the rapid weight loss. Adding 0.1% methionine is able to prevent this (Iruarrizaga-Lejarreta et al., 2017). Animals fed a 0.1%MCDD rapidly accumulate hepatic fat in the form of macrovesicular steatosis and progress to inflammation and fibrosis, hallmarks of NASH, after two and six weeks of the diet, respectively (Suppl. Fig. 1A). To confirm that fibrosis is not significant at 4 weeks of 0.1%MCDD, we have measured gene expression of fibrotic markers, such as Acta2 (actin alpha 2) and Timp1 (tissue inhibitor of metalloproteinases-1), that were not significantly altered after 4 weeks of 0.1%MCDD (data not shown). Moreover, earlier evidence has highlighted that GLS1 expression is increased with advanced tumor grade and therefore dedifferentiation (Li et al., 2018). Herein, animals fed a 0.1%MCDD during four weeks do not present alterations in dedifferentiation parameters such as albumin, alpha-fetoprotein, and the transcription factor hepatocyte nuclear factor 4 (HNF4), at least as assessed by mRNA levels (Suppl. Fig. 1B).
Importantly, animals maintained on a 0.1%MCDD show a time-dependent increase in GLS1 expression, both at the protein and the mRNA level (Fig. 2A–C). Furthermore, staining of consecutive slides from liver biopsy of an animal fed a 0.1%MCDD for four weeks, shows that GLS1 staining does not overlap with F4/80 staining (a marker of KC) and fibrosis areas (Sirius red staining). On the other hand, after six weeks of 0.1%MCDD, liver biopsies show co-staining of GLS1 and Sirius red staining (Fig. 2D). In agreement, after four-weeks of 0.1%MCDD the accumulation of GLS1 is mainly localized in hepatocytes as shown by double immunofluorescence, where GLS1 co-localizes with Albumin, a hepatocyte marker, and not with αSMA, a HSC marker (Fig. 2E). As a positive control of GLS1 staining we show co-localization of GLS1 and Albumin in mouse liver tumors (Suppl. Fig. 2A,B). On the other hand, the expression of the GLS2 isoform of glutaminase, usually distributed around the periportal regions, is reduced in steatotic livers (Fig. 2A–C, Suppl. Fig. 2C). Likewise, in a NASH model of eight-month-old Mat1a−/− (Alonso et al., 2017; Cano et al., 2011), GLS1 liver expression is induced together with glutamine synthetase, whereas GLS2 is reduced (Suppl. Fig. 2D).
Figure 2. Glutaminase 1 (GLS1) is overexpressed in a mouse model of non-alcoholic steatohepatitis (NASH) of choline deficient and 0.1% methionine diet (0.1% MCDD)-fed rodents.

A. Hepatic Glutaminase 1 (GLS1), with higher magnification zoom shown in inset, and Glutaminase 2 (GLS2) immunostaining and respective quantifications. Scale bar corresponds to 100 μm. V-venous region; P-portal region; B. Hepatic GLS1 and GLS2 protein levels by Western blot analysis. Glyceraldehyde-3-phosphate (GAPDH) was used as a loading control; and C. Hepatic Gls1 and Gls2 mRNA levels in mice fed a choline deficient and 0.1% methionine diet (0.1% MCDD) against a standard chow diet (SC diet) during 2, 4 and 6 weeks. Data is shown as average ± SEM and Student’s t-test was used to compare groups. *p<0.05, **p<0.01 and ***p<0.001 are shown versus age- and gender-matched animals maintained on SC diet. D. Representative consecutive slides staining for GLS1, F4/80 and Sirius red staining in liver biopsies of animals maintained for four weeks or six weeks on 0.1% MCDD (fibrosis areas highlighted with white dashed line and GLS1 with yellow dashed line). E. Immunofluorescence double co-staining for GLS1 and albumin, a marker of hepatocytes, and alpha smooth muscle actin (aSMA), a marker of hepatic stellate cells (HSC) in 0.1% MCDD-fed mice for 4 weeks. (See also Supplemental Figure 1, Supplemental Figure 2, Supplemental Figure 3 and Supplemental Figure 4).
In addition, we have evaluated the expression of glutamine metabolism key regulators in a mouse model of mice fed a choline-deficient, high-fat diet (CD-HFD). Previous reports have shown that this mouse model develops NASH in a similar pattern to that observed in humans, showing hepatic ballooning and fibrosis, with concomitant obesity as well as dyslipidemia and insulin resistance (Wolf et al., 2014). After six weeks of CD-HFD, we observed increased body weight in these animals relative to the standard chow (SC) diet-fed age-matched rodents (Suppl. Fig. 3A). Hepatic triglycerides are increased, and hepatic inflammation is significant, although serum transaminases and triglycerides are not significantly altered (Suppl. Fig. 3BM–G). Finally, after six-weeks of CD-HFD, hepatic fibrosis is not significant (Suppl. Fig. 3B). Similar to what occurs in the 0.1%MCDD-fed rodents, CD-HFD fed animals present decreased VLDL triglyceride export (Suppl. Fig. 3H). Importantly, GLS1 levels are induced after as little as three weeks of CD-HFD (Suppl. Fig. 3I,J).
Overall, we provide evidence that hepatic GLS1 expression is increased in mouse models of NASH.
Targeting Glutaminase 1 (GLS1) in vitro resolves hepatocyte lipid accumulation
Based on recent reports suggesting the important role of GLS1 in the glutaminolysis of HSC during fibrosis progression (Du et al., 2018), particularly in animals treated with carbon tetrachloride (CCl4), we aimed at identifying the main hepatic cells where GLS1 expression is induced in mouse models of NASH as a result of choline and methionine deprivation. Thus, after four weeks of 0.1%MCDD, a time point where inflammation is present and fibrosis is not significant (Suppl. Fig. 1A), we perfused mouse livers to isolate hepatocytes as well as liver stroma cells, namely HSC and the liver-resident macrophages, the Kupffer cells (KC). We compared the expression of GLS1 in the different hepatic populations of 0.1%MCDD-fed rodents relative to animals fed a SC diet. We found that mRNA levels of Gls1 are significantly increased in isolated hepatocytes from animals maintained on a 0.1%MCDD relative to the controls, whereas no changes were observed in either HSC or KC (Suppl. Fig. 4A). Likewise, in cultured primary mouse HSC, that undergo activation in vitro, Gls1 mRNA levels are increased after 7 days of culture, corresponding to an increase in the activation of HSC (Zubiete-Franco et al., 2017). On the other hand, Gls1 mRNA levels are not significantly induced in cell cultures of primary mouse KC after stimulation with lipopolysaccharide (LPS) (Suppl. Fig. 4B,C). Overall, these results show that glutamine catabolism in the hepatocyte is relevant in our mouse model of NASH, although the relevance of other hepatic cell types for the net induction of GLS1 levels in the livers must be taken into consideration in NASH models presenting a higher degree of fibrosis.
In order to further assess the relevance of the high-activity glutaminase GLS1 isoform expression in the hepatocytes during NASH, primary mouse hepatocytes were isolated. Primary cultures of hepatocytes represent substantial limitations that include dedifferentiation. As expected and in agreement with previous evidence (Li et al., 2018), GLS1 is gradually increased during culture (Suppl. Fig. 4D). Isolated mouse hepatocytes were maintained on a methionine- and choline-deficient (MCD) medium that has been previously shown to induce steatosis and injury in the hepatocyte cell line AML-12 (Sahai et al., 2006) as well as in primary mouse hepatocytes (Iruarrizaga-Lejarreta et al., 2017). Under conditions of MCD treatment, GLS1 was inhibited in vitro by using either molecular approaches (siRNA) or the small pharmacological inhibitor BPTES (Bis-2-(5-phenylacetamido-1,3,4-thiadiazol-2-yl)ethyl sulfide). BPTES is an allosteric, time-dependent, and specific inhibitor of GLS1 that exhibits unique binding at the oligomerization interface of the glutaminase tetramer (DeLaBarre et al., 2011; Thangavelu et al., 2012; Thomas et al., 2013). Gls1 silencing in primary hepatocytes cultured for 48 h in MCD media reduced the accumulation of triglycerides (Fig. 3A–C, Suppl. Fig. 5A). Likewise, the pharmacological inhibition of GLS1 by using BPTES, as detected by the decreased cellular glutamate/glutamine ratio, significantly reduced the accumulation of triglycerides (Suppl. Fig. 5B–D). Under these conditions, cell viability, as assessed by caspase activity, was not significantly altered after Gls1 silencing or pharmacological inhibition by using BPTES (Suppl. Fig. 5E). In addition, GLS1 silencing in a human hepatocyte cell line, the THLE-2 cells, also reduces lipid accumulation after MCD treatment (Suppl. Fig. 5F) whereas in isolated mouse hepatocytes incubated in the presence of oleic acid, a cell model where hepatocytes accumulate lipids due to increase uptake and also inhibition of VLDL export (Nossen et al., 1986), both GLS1 silencing and pharmacological inhibition using BPTES, also reduces hepatocytes lipid content (Suppl. Fig. 5G).
Figure 3. Targeting Glutaminase 1 (GLS1) in vitro and in vivo resolves the accumulation of hepatic triglycerides and non-alcoholic steatohepatitis (NASH).
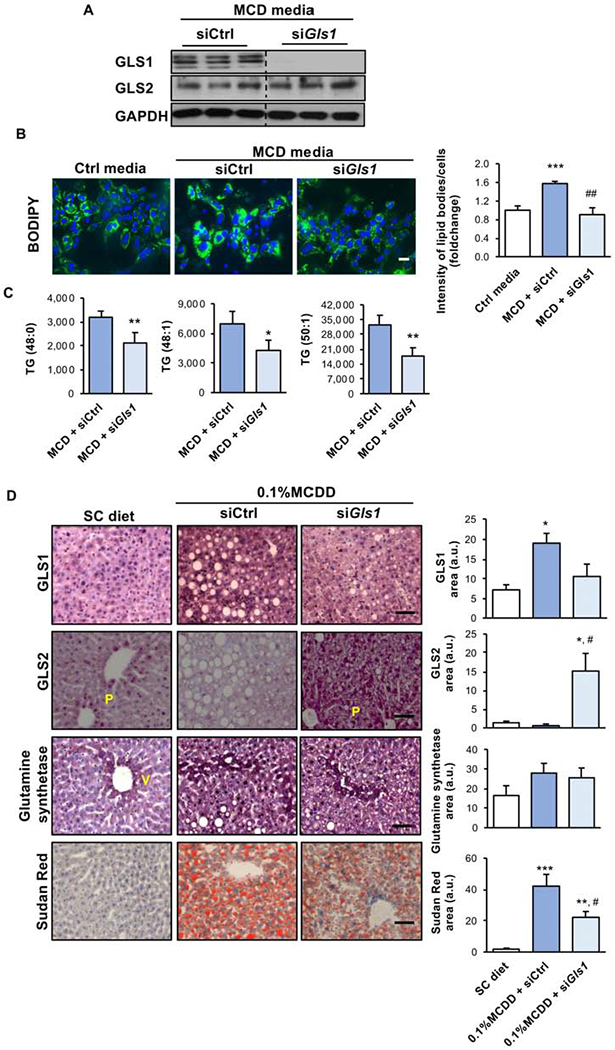
A. Western blot analysis of total protein levels of Glutaminase 1 (GLS1) and Glutaminase 2 (GLS2). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as a loading control; B. Representative BODIPY staining micrographs and respective quantification. Scale bar corresponds to 100 μm; and C. Mass-spectrometry analysis of different triglyceride (TG) isoforms in mouse isolated hepatocytes treated for 48 h with methionine- and choline-deficient media (MCD) after treatment with siRNA against Gls1 (siGls1) or unrelated control (siCtrl). At least triplicates were used for each experimental condition. Data is shown as average ± SEM and Student’s t-test was used to compare between the groups. *p<0.05 and **p<0.01 versus MCD + siCtrl and ##p<0.01 versus mcd + siCtrl are shown. D. GLS1, GLS2 and glutamine synthetase levels quantified by Immunohistochemistry and representative micrographs of Sudan Red staining and respective quantification in animals maintained on a choline deficient and 0.1% methionine diet (0.1% MCDD). From weeks two to four of diet, two different experimental groups were treated either with siCtrl or siGls1. Scale bar corresponds to 100 μm. At least n=5 were used for each experimental group. Data is shown as average ± SEM and one-way ANOVA followed by Bonferroni post-test was used to compare between multiple groups. *p<0.05, **p<0.01 and ***p<0.001 versus SC diet and #p<0.05 versus 0.1%MCDD + siCtrl are shown (See also Supplemental Figure 4, Supplemental Figure 5, Supplemental Figure 6 and Supplemental Table2).
Finally, when we silenced Gls1 in mouse hepatocytes treated with MCD media, dedifferentiation parameters, such as albumin, alpha-fetoprotein, and HNF4, were unaltered at the mRNA level. (Suppl. Fig. 5H).
Taken together, GLS1 silencing ameliorates steatosis in isolated hepatocytes.
Targeting Glutaminase 1 (GLS1) in vivo resolves non-alcoholic steatohepatitis (NASH)
Taking into consideration that GLS1 is increased in NASH, the potential therapeutic role of silencing Gls1 in vivo was assessed. Thus, we have evaluated the effects of Gls1 silencing in mice fed a 0.1%MCDD. Depriving mice of methionine and choline for four weeks caused an increase in hepatic content of fatty acids, cholesteryl esters, diglycerides, and triglycerides. As expected, the concentration of serum triglycerides and cholesterol was reduced, attributable to a phosphatidylcholine-related defect in hepatic VLDL secretion, as previously described (Rizki et al., 2006) (Table S2). Liver histology confirmed marked lipid accumulation after four weeks of 0.1%MCDD (Fig. 3D). In addition, four weeks of 0.1%MCDD lowered blood glucose (Table S2).
Hepatic Gls1 was silenced in 0.1%MCDD-fed rodents by using twice-a-week tail vein injections of Invivofectamine® conjugated to either Glsl-specific or Control siRNA, from weeks 2 to 4 of 0.1%MCDD. As a result of Gls1 silencing, GLS1 hepatic levels are reduced and GLS2 expression is increased, whereas glutamine synthetase expression is not significantly altered at the protein level (Fig. 3D, Suppl. Fig. 6A,B). Of relevance, the levels of hepatic ammonia, a secondary product of the glutaminase reaction, are not significantly altered after Gls1 silencing (Table S2). Under these circumstances, reduced glutaminase activity was confirmed by measuring the incorporation of 13C labeling from glutamine into glutamate, confirming that glutaminase activity is, on one hand, induced in the diet and on the other there is a tendency for decreased glutaminase activity after Gls1 silencing in vivo (Suppl. Fig. 6C). Importantly, GLS1 specific silencing in vivo, both at the protein and the mRNA level, significantly reduced liver steatosis, measured as Sudan red staining and by biochemical assay (Fig. 3D, Table S2). Furthermore, Gls1 silencing in vivo in 0.1%MCDD-fed rodents significantly increased hepatic phospholipid content whilst decreasing cholesteryl esters and restoring serum triglycerides levels (Table S2).
Finally, in animals fed a CD-HFD for six weeks, we have silenced GLS1 by using twice-a-week tail vein injections of Invivofectamine® conjugated to either GLS1-specific or control siRNA from weeks 3 to 6 of CD-HFD (Suppl. Fig. 6D,E). In these animals, GLS1 silencing was able to significantly reduce hepatic steatosis (Suppl. Fig. 6F,G) without changes to body weight and food intake (data not shown).
In summary, GLS1 inhibition ameliorates liver steatosis in pre-clinical mouse models of NASH.
Targeting Glutaminase 1 (GLS1) in vitro and in vivo restores very-low-density lipoprotein (VLDL) export after methionine and choline deprivation
As mentioned before, phospholipids are required for correct VLDL assembly and therefore methionine and choline deprivation results in impaired VLDL export. Taking that into account, the impact of Gls1 silencing in Ptd-Chol synthesis and VLDL export was evaluated both in vitro and in vivo. Firstly, hepatocytes grown for 48 h in complete and MCD media under conditions of Gls1 or control silencing were incubated with (U-13C)glucose for one hour. Using mass spectrometry we have measured the incorporation of the glucose tracer in intracellular Ptd-Chol species. As expected, under conditions of choline and methionine deprivation there is a tendency for reduced incorporation of carbon tracers in Ptd-Chol, reflecting impaired Ptd-Chol synthesis from glucose. On the other hand, Gls1 silencing under conditions of methionine and choline deprivation promoted the synthesis of some Ptd-Chol species (Suppl. Fig. 7A). Moreover, we treated isolated mouse hepatocytes with lomitapide, described previously to inhibit MTP and shown to hinder VLDL export (Sirtori et al., 2014). As expected, treatment with lomitapide caused the accumulation of cell lipids to a similar extent as in cells treated with complete and methionine- and choline-deficient media. Interestingly, under these conditions GLS1 silencing does not prevent cell lipid accumulation after lomitapide treatment, suggesting that Gls1 silencing-induced lipid lowering may somehow be related to VLDL export (Fig. 4A).
Figure 4. Targeting Glutaminase 1 (GLS1) in vitro and in vivo restores very-low-density lipoproteins (VLDL) triglyceride export after choline and methionine deprivation.
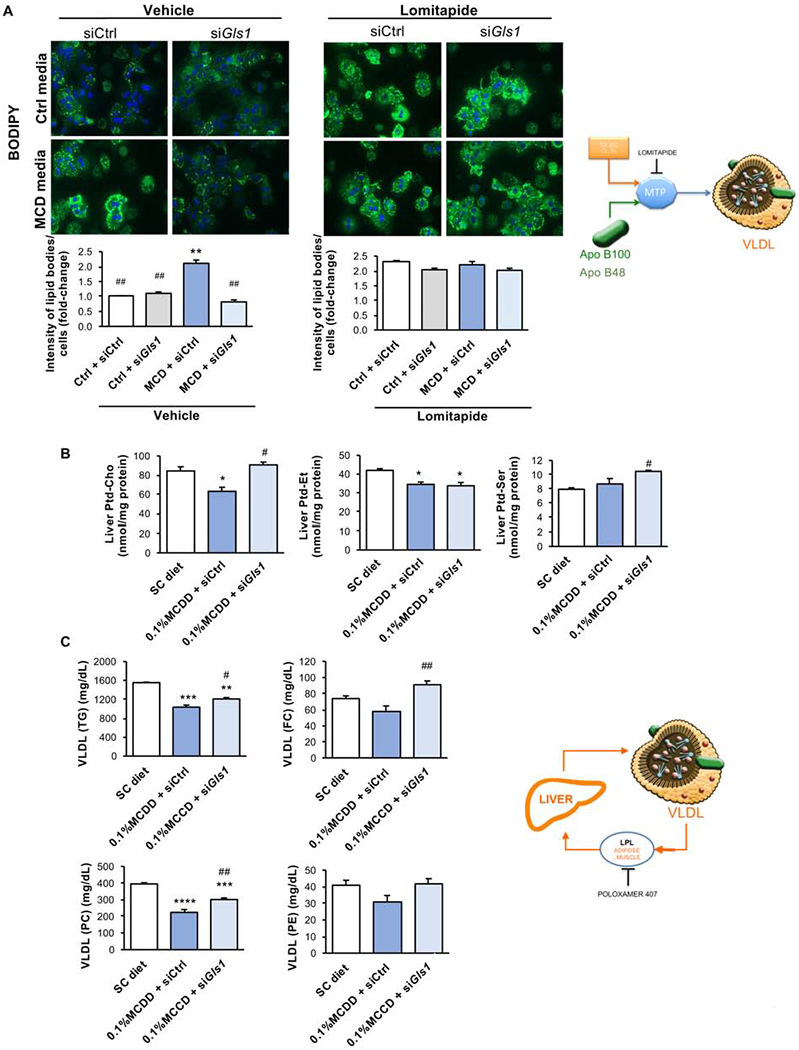
A. Representative BODIPY staining micrographs and respective quantification in mouse isolated hepatocytes treated with control media (Ctrl) or methionine- and choline -deficient media (MCD) for 24 h after overnight treatment with siRNA against Gls1 (siGls1) or unrelated control (siCtrl). In some experimental conditions lomitapide was added at 600 nM for 24 hours. Scale bar corresponds to 100 μm. At least triplicates were used for each experimental condition. Data is shown as average ± SEM and Student’s t-test was used to compare between groups. *p<0.05 and **p<0.01 versus Ctrl + siCtrl and ##p<0.01 versus MCD + siCtrl are shown. B. Liver phosphatidylcholine (Ptd-Cho), phosphatidylethanolamine (Ptd-Et) and phosphatidylserine (Ptd-Ser) hepatic levels and C. Serum very-low-density lipoprotein (VLDL) phospholipids and lipid content in mice fed either a standard chow (SC) diet or a diet deficient in choline with 0.1% methionine (0.1% MCDD) for four weeks. From weeks two to four of diet, two different experimental groups were treated either with siCtrl or siGls1. Animals were administered poloxamer 407 (P407) and serum VLDL isolated and analyzed at six hours after P407 administration. At least n=5 were used for each experimental group. Data is shown as average ± SEM and one-way ANOVA followed by Bonferroni post-test was used to compare between multiple groups. **p<0.01, ***p<0.001 and ****p<0.0001 versus SC diet and #p<0.05 and ##p<0.01 versus 0.1%MCDD + siCtrl are shown (See also Supplemental Figure 7).
In addition, hepatic phospholipid content was determined in mice maintained on a diet deprived of methionine and choline and after control or Gls1 silencing. We observed that, as a result of in vivo Gls1 silencing in 0.1%MCDD-fed rodents, hepatic Ptd-Cho and phosphatidylserine (Ptd-Ser) levels are augmented, whereas Ptd-Et levels remain unaltered (Fig. 4B). Thus, we decided to evaluate the composition of the secreted VLDL particles in mice fed a 0.1% MCDD and where we have silenced Gls1. For this, circulating VLDL catabolism was inhibited through the administration of poloxamer 407, a non-ionic detergent described to inhibit lipoprotein lipase (LPL) (Millar et al., 2005), an enzyme mostly abundant in tissues involved in fatty acid metabolism such as muscle and adipose tissue (Karpe et al., 1998). When we silence GLS1 in vivo in mice fed a 0.1%MCDD, the lipid content of the secreted VLDL particles was significantly enriched in lipids, such as triglycerides, phospholipids, and cholesterol derivatives (Fig. 4C). In spite of this, the number of VLDL particles, determined by the VLDL secretion rate and the molecules of apoB secreted, was not altered (Suppl. Fig. 7B,C). Likewise, in mice fed a CD-HFD for six weeks, Gls1-silencing results in a tendency for increased VLDL triglyceride content in VLDL isolated from vena cava serum after 2 h of fasting (Suppl. Fig. 7D).
Overall, we provide evidence that Gls1 silencing ameliorates liver steatosis by targeting VLDL assembly through mechanisms that have not been previously explored.
Targeting Glutaminase 1 (GLS1) in vitro and in vivo reduces oxidative stress
Oxidative stress plays a crucial role in the pathogenesis and progression of NASH. In agreement, we have observed that harvesting the primary mouse hepatocytes with media deprived of methionine and choline increases both total and mitochondrial reactive oxygen species (ROS) levels (Fig. 5A). Reduced glutathione (GSH) is considered to be one of the most important ROS scavengers. Importantly, the ratio between GSH and oxidized glutathione (GSSG) may be used as a marker of oxidative stress. Thus, four weeks of 0.1%MCDD is associated with augmented GSSG/GSH ratio and increased incorporation of labeled glutamine into GSH, highlighting that GSH synthesis is increased under these conditions and reverted under Gls1 silencing conditions(Fig. 5B). GSSG/GSH is also reduced under GLS1 silencing conditions in mice fed a CD-HFD (Fig. 5C). Likewise, the total levels of malondialdehyde (MDA), a marker of lipid peroxidation, were induced (Fig. 5D). Noteworthy, Gls1 silencing significantly reduces oxidative stress in primary hepatocytes and in the in vivo NASH models (Fig. 5A–D).
Figure 5. Targeting Glutaminase 1 (GLS1) in vitro and in vivo reduces oxidative stress.
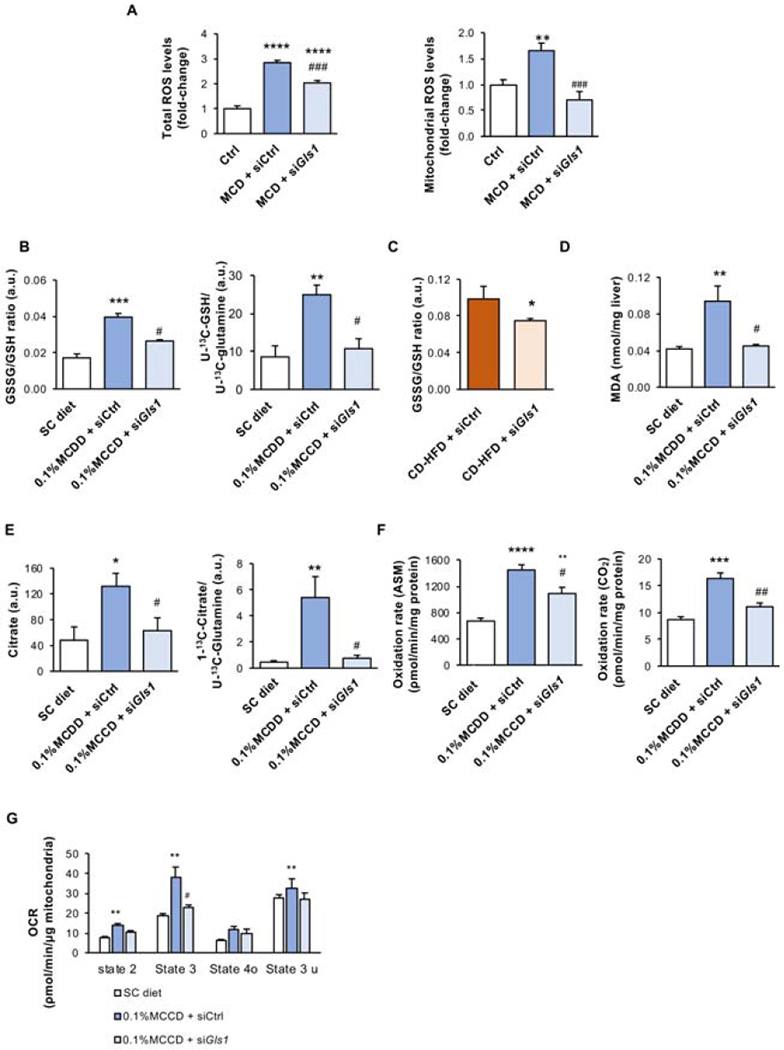
A. Total and mitochondrial reactive oxygen species (ROS) levels in mouse isolated hepatocytes treated with control media (Ctrl) or methionine and choline deficient media (MCD) for 48 h after overnight treatment with siRNA against Gls1 (siGls1) or unrelated control (siCtrl). At least triplicates were used for each experimental condition. Data is shown as average ± SEM and one-way ANOVA followed by Bonferroni post-test was used to compare between multiple groups. *p<0.05, **p<0.01, ***p<0.001 and ****p<0.0001 versus Ctrl media as well as ###p<0.001 versus MCD media + siCtrl are shown. B. Oxidized glutathione (GSSG) and reduced glutathione (GSH) ratio and incorporation of 13C carbons from U-13C(glutamine) carbons into 5-13C(GSH) in mice fed either a standard chow diet (SC) or a diet deficient in choline with 0.1% methionine (0.1% MCDD) for four weeks. From weeks two to four of diet, two different experimental groups were treated either with siCtrl or siGls1. At least n=5 were used for each experimental group. Data is shown as average ± SEM and one-way ANOVA followed by Bonferroni post-test was used to compare between multiple groups. *p<0.05, **p<0.01, ***p<0.001 and ****p<0.0001 versus SC diet and #p<0.05 and ##p<0.01 versus 0.1%MCDD+siCtrl are shown. C. Oxidized and reduced glutathione ratio (GSSG/GSH) in animals maintained on a choline deficient and high fat diet (CD-HFD) for 6 weeks. From week 3 to 6 of diet, two different experimental groups were treated either with siCtrl or siGls1 (CD-HFD + siCtrl or CD-HFD + siGls1). At least n=5 were used for each experimental group. Data is shown as average ± SEM and Student’s t-test was used to compare between groups. *p<0.05 versus CD-HFD + siCtrl is shown. D. Malondialdehyde (MDA) levels as a measurement of lipid peroxidation; E. Citrate and incorporation of U-13C(glutamine) carbons on 1-13C(citrate) levels; F. Fatty acid oxidation (FAO) rate quantified from the incorporation of 14C-palmitate into CO2 and in acid-soluble metabolites (ASM); and G. Mitochondrial Oxygen Consumption Rate (OCR) in different states of the respiration (State 2, State 3, State 4o, State 3u) in mice fed either a standard chow diet (SC) or a diet deficient in choline with 0.1% methionine (0.1% MCDD) for four weeks. From weeks two to four of diet, two different experimental groups were treated either with siCtrl or siGls1. At least n=5 were used for each experimental group. Data is shown as average ± SEM and one-way ANOVA followed by Bonferroni post-test was used to compare between multiple groups. *p<0.05, **p<0.01, ***p<0.001 and ****p<0.0001 versus SC diet and #p<0.05 and ##p<0.01 versus 0.1%MCDD+siCtrl are shown (See also Supplemental Figure 3).
Numerous causes of oxidative stress have been associated with NASH. Impaired tricarboxylic acid (TCA) cycle, FAO and oxidative phosphorylation (OXPHOS) originate ROS (Rosca et al., 2012; van de Wier et al., 2013). Here, we have measured hepatic TCA cycle activity, evaluated as the incorporation of U-13C-glutamine into 1-13C-citrate; hepatic FAO, measured using radioactive incorporation of labeled palmitate into CO2 and into incompletely oxidized acid-soluble metabolites (ASM); as well as mitochondrial oxidative phosphorylation (OXPHOS) as measured by oxygen consumption rate (OCR) using a Seahorse analyzer. All the analyzed parameters were significantly higher after four weeks of 0.1%MCDD and, importantly, Gls1 silencing during 0.1%MCDD restored TCA, β-oxidation, and OXPHOS pathway fluxes to control diet levels in association with reduced oxidative stress (Fig. 5E–G).
In summary, targeting hepatic GLS1 in vitro and in vivo reduces ROS levels by reducing oxidative metabolism.
Glutaminase 1 (GLSI)-mediated reduction of oxidative stress is associated with restored hepatic phospholipid content
Choline is essential for the de novo synthesis of Ptd-Chol, the major phospholipid component of plasma lipoproteins, via the cytidine-diphosphate pathway (CDP). Decreased hepatic Ptd-Chol reduces the levels of circulating VLDL (Cole et al., 2012). In the liver, Ptd-Cho can be additionally synthesized from the methylation of Ptd-Et, a reaction catalyzed by the enzyme PEMT and using SAMe, an intermediate of the methionine cycle, as a methyl donor (Noga et al., 2002). The other intermediate of the methionine cycle, homocysteine, at the crossroads of the metabolic pathways, is either degraded via the transsulfuration pathway to cysteine and then GSH or is remethylated back to methionine. Indeed, depletion of SAMe and GSH are early events in the MCD model of NASH (Caballero et al., 2010) (Table S3, Fig. 5B).
In one carbon metabolism, a carbon unit from serine or glycine is transferred to tetrahydrofolate (THF) to form methylene-THF (MTHF). MTHF either can be used for the synthesis of purines or reduced to methyl-THF, which can be used to methylate homocysteine to methionine. Likewise, serine, which has previously been shown to be able to transfer one-carbon units to recycle homocysteine to methionine in tumor cells (Maddocks et al., 2016), can be used in combination with homocysteine to synthesize GSH, or can be metabolized to Ptd-Ser by Ptd-ser synthase II (PTDSS2) (Kuge and Nishijima, 1997; Kuge et al., 1997). In mammals, Ptd-Ser can be further metabolized to Ptd-Et and later converted to Ptd-Chol by PEMT activity. Then, Ptd-Cho can undergo a base-exchange process with Ptd-Ser, releasing choline through the exchange with serine. Thus, even under conditions of choline deprivation, newly formed choline can be metabolized to Ptd-Cho through the CDP-choline pathway (Maldonado et al., 2014).
We have previously shown that Gls1 silencing on 0.1%MCDD-fed mice reduced oxidative stress and decreased GSH synthesis (Fig. 5B). In agreement, the expression of the genes involved in the transsulfuration pathway are reduced after Gls1 silencing, whereas expression of enzymes involved in the folate and methionine cycles is augmented (Fig. 6A). Likewise, the mRNA levels of the genes involved in the CDP-choline and CDP-ethanolamine pathways and the enzymes catalyzing the base-exchange among the different phospholipids are induced after Gls1 silencing (Fig. 6B). Although Pemt mRNA expression is not upregulated after Gls1 silencing, the increase in the Ptd-Cho/Ptd-Et ratio observed (1.4 ± 0.3 in 0.1%MCDD + siG/s1 vs. 1.0 ± 0.25 in 0.1%MCDD + siCtrl, p<0.01) can be indicative of increased PEMT activity that relies on the transfer of methylation units.
Figure 6. GLS1-mediated reduction of oxidative stress is associated with increased phospholipid synthesis and the activation of folate and methionine cycles.
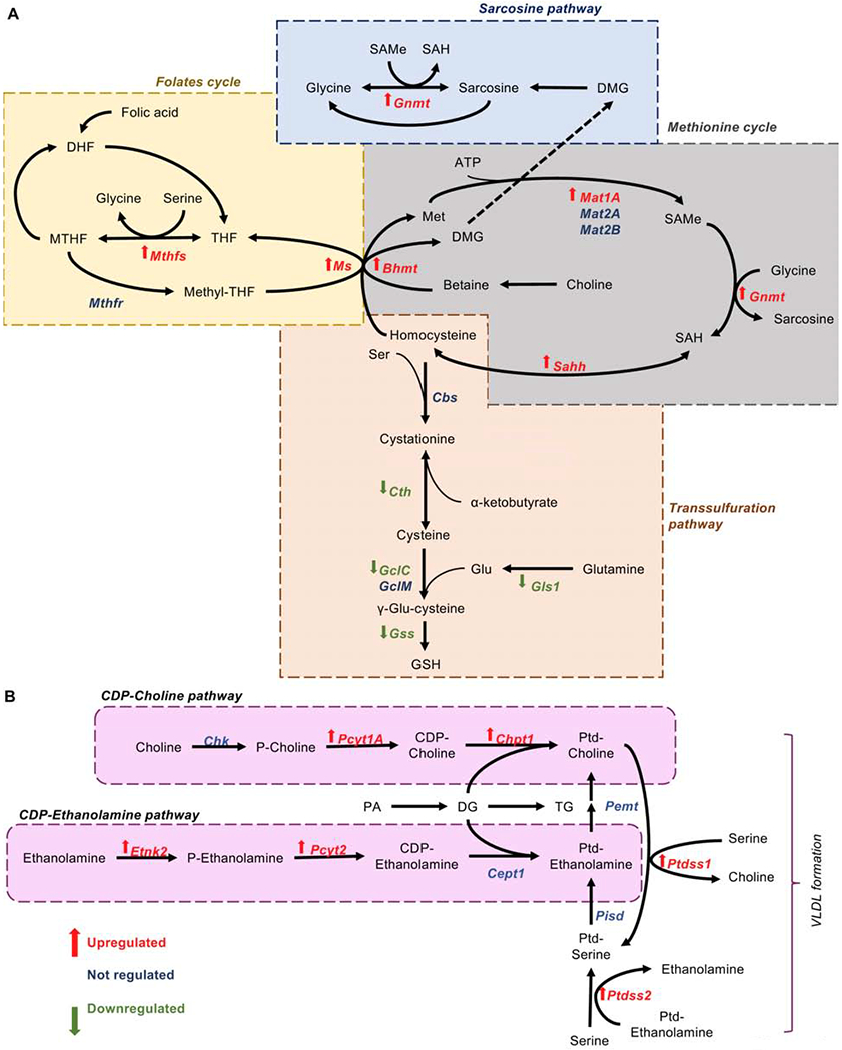
A. Differential expression of mRNA levels from genes significantly different involved either in glutathione (GSH) synthesis through the transsulfuration pathway, and the folates and methionine cycles in mice on a 0.1% MCDD and with Gls1 silencing (siGls1) versus control silencing (siCtrl) for four weeks. (Bhmt = betaine-homocysteine S-methyltransferase; Cbs = cysthationine-beta synthase; Cth = cysthathionine gamma-lyase; Gclc = glutamate-cysteine ligase, catalytic subunit; Gclm = glutamate-cysteine ligase, modifier subunit; Gls1 = glutaminase 1; GNMT = glycine N-methyltransferase; GSS = glutathione synthetase; Mat1a = methionine adenosyltransferase 1A; Mat2a = methionine adenosyltransferase 2A; Mat2b = methionine adenosyltransferase 2B; Ms = methionine synthetase; Mthfr = methylenetetrahydrofolate reductase; Mthfs = synthetase; Sahh = S-adenosyl-homocysteinase). (DMG = dimethyglycine; MTHF = L-methylfolate; SAMe = S-adenosylmethionine; SAH = S-adenosylhomocysteine; THF = tetrahydrofolate). B. Differential expression of mRNA levels from genes significantly different involved in phospholipid biosynthesis in mice on a 0.1%MCDD with siGls1 versus siCtrl for four weeks. (Cept1 = choline/ethanolamine phosphotransferase 1; Chk = choline kinase; Chpt1 = choline phosphotransferase 1; Etnk2 = ethanolamine kinase 2; Pcyt1a = phosphate cytidylyltransferase 1, choline; Pcyt2 = phosphate cytidylyltransferase 2, ethanolamine; Pemt = phosphatidylethanolamine methyltransferase; Pisd = phosphatidylserine decarboxylase; Ptdss1 = phosphatidylserine synthase 1; Ptdss2 = phosphatidylserine synthase 2). At least n=5 were used for each experimental group. Student’s t-test was used to compare the two groups and significance was set to p<0.05 (See also Supplemental Table 3).
These results indicate that GLS1-mediated reduction of oxidative stress is associated with restored hepatic phospholipid content.
Discussion
Therapeutic solutions to address the major health problem of NASH are a hot topic. Recent reports have highlighted the importance of glutamine catabolism in liver disease, namely in fibrosis, cirrhosis and HCC (Du et al., 2018; Yu et al., 2015; Yuneva et al., 2012). In the liver, glutaminase is the main regulator of glutamine catabolism, catalyzing the conversion of glutamine to glutamate and ammonia. In recent years, a metabolic switch from the low-activity GLS2 isoform to the high-activity GLS1 isoform was observed in end stages of chronic liver disease (Du et al., 2018; Yu et al., 2015; Yuneva et al., 2012). Herein, we have shown higher hepatic expression of GLS1 in NASH patients. Moreover, we have shown that, during NASH progression, the upregulation of GLS1 occurs at the mRNA level. Previously, other studies have shown that there are many factors related to the induction of GLS1 during cancer progression: i) c-Myc (Gao et al., 2009), ii) K-Ras mutations (Brunelli et al., 2014), iii) the splicing factor SLU7 (Elizalde et al., 2014), iv) elevated mitochondrial phosphorous levels under hypoxic conditions commonly found in the tumor microenvironment (Cassago et al., 2012), and v) oncogenic diffuse β-cell lymphoma protein in a NF-κB (nuclear factor kappa-light-chain-enhancer of activated B cells)-dependent manner (Wang et al., 2010). Related to this, further studies are necessary to better understand the mechanism underlying GLS1 upregulation in NASH.
Dysfunctional VLDL synthesis and secretion is a hallmark of NASH (Fujita et al., 2009). Methionine and choline are fundamental amino acids for the synthesis of Ptd-Col, an essential phospholipid for VLDL assembly and therefore hepatic triglyceride export. On this basis, mice fed a methionine- and/or choline-deficient diet present impaired VLDL export. Despite the limitations of the mouse models of methionine and choline-deprivation regarding human NASH, being the most obvious being a lack of weight gain, the methionine- and choline-deficient nutritional model is one of the most used mouse models of NASH. Importantly, in the nutritional mouse models of 0.1%MCDD- and CD-HFD we have observed increased hepatic GLS1 expression. Likewise, we have shown that in eight-month old Mat1a−/− mice, known to have impaired VLDL export (Cano et al., 2011), GLS1 hepatic expression is induced. Even though GLS1 has recently been shown to play an important role in fueling HSC glutaminolysis and proliferation during hepatic fibrosis (Du et al., 2018), in here we have focused on the earlier stages of NASH. Thus, in animals fed a 0.1%MCDD for four weeks, characterized by hepatic steatosis and inflammation, we have shown that GLS1 expression is specifically induced in hepatocytes and not in stromal cells.
GLS1 expression has been previously shown to be increased with advancing tumor grade and therefore dedifferentiation (Li et al., 2018). Herein, the augmented GLS1 expression in mouse hepatocytes from a NASH background even though is not associated with changes of dedifferentiation markers, such as HNF4a, at the mRNA levels, we cannot preclude that the regulation of this transcription factor is occurring at the protein level, as it known that post-transcriptional mechanisms play important roles in the regulation of factors such as HNF4a during hepatocyte differentiation/de-differentiation. Indeed, previous evidence from our group have shown that HNF4a was decreased in HCC derived from NASH and also in NASH patients (Frades et al., 2015). Thus, we could hypothesize that GLS1 is selectively up-regulated in dedifferentiated sub-populations of hepatocytes during NASH and could potentially drive HCC derived from NASH.
Taking into consideration the relevance of GLS1 during NASH pathogenesis, we have evaluated the therapeutic potential of GLS1 inhibition both in cell and in pre-clinical mouse models of NASH. Firstly, we have observed that GLS1 inhibition effectively reduced the accumulation of fat in mouse hepatocytes, in human hepatocyte cell line and mouse livers under methionine and/or choline deprivation. Under these circumstances, decreased steatosis was associated with augmented hepatic Ptd-Chol content that partly normalized VLDL assembly, increasing triglyceride and cholesterol export from the liver. It must be mentioned that, although secreted VLDL particles are lipid-enriched, serum levels of triglycerides and cholesterol after GLS1 -inhibition in 0.1%MCDD-fed mice are not higher than values from healthy age- and gender-matched control mice, suggesting that serum hypertriglyceridemia and hypercholesterolemia and associated atherosclerotic complications are not a potential concern when using GLS1-targeted therapies during NASH management. As a result of GLS1 inhibition in 0.1%MCDD-fed rodents, GLS2 levels around the portal region are increased. GLS2 has been recently shown to be an essential enzyme for glucagon-induced flux from glutamine to glucose in primary hepatocytes and Gls2−/− mice show reduced blood glucose levels after overnight fasting (Miller et al., 2018). These data are in agreement with our findings where 0.1%MCDD-fed rodents, with augmented GLS1 expression and lowering of GLS2 hepatic levels, have lower blood glucose. Lower blood glucose under these conditions can also be associated with the decreased hepatic glucose production caused by glycogen depletion, as previously reported (Leclercq et al., 2007). However, this raises concerns regarding the possible adverse effects of GLS1 inhibition and concomitant GLS2 increase in promoting hyperglycemia. In spite of this, in our mouse model of 0.1%MCDD, Gls1-silencing is not associated with fasting hyperglycemia.
Finally, as glutaminase accounts for the formation of glutamate and ammonia from glutamine, interventions targeting glutamine catabolism may potentially disturb ammonia homeostasis, a highly toxic by-product of this reaction that is excreted through the urea cycle. Neither four weeks of 0.1%MCDD nor Gls1 -silencing under these conditions is associated with accumulation of hepatic ammonia, in agreement with previous evidence (Gutierrez-de-Juan et al., 2017). Based on our evidence, we can speculate that as glutamine synthetase is not significantly altered in our NASH mouse model perhaps at this time point of disease progression the urea cycle enzymes are not decreased as occurs at later steps of the disease (De Chiara et al., 2018) and can therefore detoxify ammonia preventing its toxic accumulation.
Besides disruptions of VLDL assembly and export, methionine and/or choline deprivation in rodents also blunts hepatic lipogenesis and hypermetabolism characterized by enhanced FAO (Rizki et al., 2006). Under these conditions, augmented FAO increases the TCA cycle anaplerotic capacity, which accounts for the replenishment of the mitochondrial carbon pool. Under these circumstances, the excessive glutamine catabolism mediated by GLS1 accounts for increased levels of glutamate, which can be further deaminated to α-ketoglutarate in order to sustain augmented TCA cycle anaplerosis. Hepatic anaplerotic pathways are energetically backed by elevated oxidative metabolism in the liver through OXPHOS, as previously observed in mice fed a choline- and methionine-deficient diet (Romestaing et al., 2008). Altogether, rises in TCA cycle anaplerosis, FAO, and OXPHOS contribute to oxidative stress during NASH (Satapati et al., 2015). These results agree with earlier clinical data showing elevated FAO (Iozzo et al., 2010) as well as in vivo splanchnic oxygen consumption and mitochondrial respiration (Felig et al., 1974; Koliaki et al., 2015) and TCA cycle activity (Hyotylainen et al., 2016; Sunny et al., 2011) in NAFLD. Finally, Dasarathy and colleagues have shown that NASH subjects present a higher rate of synthesis of cystathionine, a precursor of GSH, from serine (Dasarathy et al., 2009). These results are in agreement with our data where we show that hepatic GSH synthesis is increased after four weeks of 0.1%MCDD. Serine is a metabolite that lies at the crossroads between the synthesis of phospholipids and total GSH synthesis. In the liver, serine can be metabolized to Ptd-Ser, and further metabolized to Ptd-Et and then methylated to Ptd-Chol by PEMT. Moreover, serine has been shown to be able to transfer one-carbon units to recycle homocysteine to methionine to support the methionine cycle (Maddocks et al., 2016). The methionine cycle, which is impaired after a choline- and/or methionine-deficient diet and in a subtype of NASH patients (Alonso et al., 2017), plays an important role in the biosynthesis of hepatic phospholipids by providing methyl groups for the reaction catalyzed by PEMT.
Herein, Gls1 silencing in 0.1%MCDD-fed rodents is associated with decreased TCA cycle activity, through the reduction of the replenishment of carbon units necessary for sustaining anaplerosis. Reduced TCA cycle and associated diminishment of NADH and FADH2 fueling of OXPHOS further reduce oxidative stress after GLS1 inhibition. Likewise, decreased oxidative stress is observed after Gls1 silencing in the CD-HFD rodents. Under these circumstances, reduced oxidative stress alleviates serine usage for GSH synthesis; instead, serine is recruited to fuel the folate and methionine cycle and synthetize phospholipids via PEMT. Increased Ptd-Chol is able to induce the assembly of lipid-enriched VLDL particles and, in this way, diminish the accumulation of triglycerides and cholesterol in the liver. Finally, a decreased triglyceride content is followed by a reduction in FAO, associated with diminished TCA cycle and OXPHOS and further promoting an improvement in NASH (Fig. 7).
Figure 7. Flowchart of Glutaminase 1 (GLS1)-inhibition resolution of non-alcoholic steatohepatitis (NASH).

Deprivation of methionine and choline induces NASH through the inhibition of very-low-density lipoprotein (VLDL) assembly/export. Under these circumstances, excessive glutamine catabolism mediated by GLS1 in NASH accounts for increased levels of glutamate that can be further deaminated to a-ketoglutarate in order to sustain augmented tricarboxylic acids (TCA) cycle anaplerosis and augmented fatty acid oxidation (FAO). Hepatic anaplerotic pathways are energetically backed by elevated oxidative metabolism in the liver through oxidative phosphorylation (OXPHOS). On the other hand, GLS1 inhibition under methionine and choline deprivation is associated with decreased TCA cycle activity, associated diminishment of mitochondrial OXPHOS, and reduced production of reactive oxygen species (ROS). Under these circumstances, serine demand for glutathione (GSH) synthesis is diminished and in alternative serine is channeled to phophatidylcholine synthesis, leading to the formation of lipid-enriched VLDL particles and decreased liver steatosis in a metabolic reprogramming where folates and methionine cycles are relevant. Overall, GLS1 inhibition is suggested as a novel therapeutic approach during NASH management.
Earlier therapeutic approaches targeting glutamine metabolism in cancer focused on scavenging glutamine usage, as for example using phenylbutyrate. Also, L-ornithine phenylacetate (OP) has been applied to the treatment of hyperammonemia and hepatic encephalopathy. The combination of L-ornithine (amino acid) with phenylacetate, reduces toxic levels of ammonia by (1) L-ornithine acting as a substrate for glutamine synthesis from ammonia in skeletal muscle and (2) phenylacetate excreting the ornithine-related glutamine as phenylacetylglutamine in the kidneys (Jalan et al., 2007). Currently, most therapeutic approaches regulating glutamine metabolism focus on the initial stage of glutaminolysis by inhibiting GLS1. GLS1 inhibitors such as UPGL00004 (Huang et al., 2018), 968 (Yuan et al., 2016), BPTES (Sappington et al., 2016), CB-839 or Telaglenastat (Calithera) (Gross et al., 2014), a family of thiourea derivatives (THDP17) (Diaz-Herrero et al., 2014), ebselen (Thomas et al., 2013) and 6-diazo-5-oxo-L-norleucine (Rahman et al., 1985) effectively inhibit GLS1 by different mechanism and reduce tumor growth in vitro and in vivo. Whereas BPTES’s poor aqueous solubility and unfavorable pharmacokinetic profile have hampered its clinical development (Shukla et al., 2012), BPTES-loaded polyethylenoglycol nanoparticles, a nanoencapsulation allowing the safe administration of this drug are currently being tested in pre-clinical mouse models (Elgogary et al., 2016). Also, Telaglenastat, an orally bioavailable inhibitor is in phase one clinical trials for the therapy in some types of cancer. In spite of this, the potential side effects of strategies impacting whole-body glutamine metabolism must be seriously considered and interventions targeting hepatic GLS1 inhibition are preferable for NASH management.
In summary, given the paramount importance of glutamine metabolism through GLS1 in the redox control and related liver fat build-up in NASH shown in here together with its role in the regulation of fibrosis progression, as previously described (Du et al., 2018), interfering with its function might represent a general mechanism for metabolic reprogramming under pathophysiological conditions and an attractive strategy for the treatment of NASH.
Limitations of the Study
The primary mouse hepatocyte model used in this study shows limitations associated with the dedifferentiation of these cells known to occur in culture. Moreover, herein, we show that GLS1 silencing is beneficial in animal models of choline and methionine-deprivation induced NASH, where NASH is attained as soon as after 4–6 weeks of dietary intervention. In our perspective, it will be important to validate these findings in other dietary models of NASH, specifically in chronic pre-clinical models of NASH, where timely disease progression resembles more to human pathology. This would be to assess efficacy and possible associated side-effects of hepatic GLS1-silencing in NASH.
STAR Methods text
Lead Contact and materials availability:
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Maria Luz Martínez-Chantar (mlmartinez@cicbiogune.es). This study did not generate new unique reagents.
Experimental Model and Subject details
Human Subjects:
The work described has been carried out in accordance with the code of ethics of the World Medical Association (Declaration of Helsinki) for experiments involving humans after obtaining informed consent. A total of 221 patients, from a study previously published elsewhere (Barr et al., 2012) were used. Inclusion criteria included (1) age 18–75 years; (2) no known acute or chronic disease except for obesity or type 2 diabetes based on medical history, physical examination, and standard laboratory tests; (3) alcohol consumption was less than 20 g/day for women and 30 g/day for men; (4) 76% of the patients (Control and NASH) were women. Exclusion criteria included viral- and drug-induced causes of liver disease. All of the subjects were of Caucasian origin. The institutional review board at each of the participating hospitals approved the study and written informed consent was obtained from all patients. For all the patients, diagnoses were established histologically in liver biopsy specimens. Following assessment, patients were classified by the pathologists into two histological groups: (1) Normal liver and (2) NASH (presence as determined by the pathologist). For all the subjects, blood was drawn on the morning a liver biopsy was performed, following a minimum 8 h, overnight fast. Serum was separated and stored at −80 C until analysis. An UPLC-single quadrupole-MS amino acid analysis system was combined with two separate UPLC-time-of-flight (TOF)-MS based platforms analyzing methanol serum extracts. 10 μL aliquots of the extracts prepared after methanol extract by adding 4 volumes of methanol were derivatized for amino acid analysison an Acquity-SQD system (Waters Corp., Manchester, UK). LCMS features (as defined by retention time, mass-to-charge ratio pairs, Rt-m/z), were associated with amino acids by comparison of their accurate mass spectra and chromatographic retention times in commercial serum metabolite extracts (Promocell inc., Germany) with those obtained using available reference standards. All data were processed using the TargetLynx application manager for MassLynx 4.1 (Waters Corp.) after intra- and inter-batch normalization, as previously described (Barr et al., 2012).
The immunohistochemical analysis of liver GLS1, GLS2 and glutamine synthetase expressions was assessed in a cohort of NASH patients and healthy controls recruited at the Department of Gastroenterology, Azienda Ospedaliero-Universitaria & University of Modena and Reggio Emilia, Modena, Italy. Patients show an average age of 42 ± 3 years and 38% of patients were female.
This study was approved by the Institutional Review Board of the Modena Province (n.2CEI-705; 08/04/2013) and was in accordance with the principles of the Declaration of Helsinki. Written informed consents were obtained from all participants, and characterized in Table S1. mRNA levels of GLS1 were analyzed in liver biopsies of a cohort of patients with NASH and controls recruited at the Hospital Marques de Valdecilla, Santander, Spain. Patients show an average age of 47 ± 3 years and 44% of patients were female. This study was approved by the Institutional Review Board of Cantabria (2012.095) and was in accordance with the principles of the Declaration of Helsinki. Written informed consents were obtained from all participants. Sample size estimation was made by using the “DSS Research sample size calculator”. Even though both females and males were used an analysis of the influence was not performed as this significantly reduces sample size number.
Animal maintenance:
Male adult (three-month old) C57BL/6 mice were acquired from Charles River (St Germain sur l’Arbresle, France) and maintained and closely monitored at the AAALAC -accredited CIC bioGUNE animal facility on a 12/12h light/dark cycle at 21±1°C and humidity of 45±10%. Eight-month male Mat1a−/− and their wild-type (WT) C57BL/6 littermates are bred at the animal facility of CIC bioGUNE. Animal procedures were approved by the CIC bioGUNE Institutional Animal Care and Use Committee and the Country Council of Bizkaia. Mice were maintained with ad libitum access to water and a diet devoid of choline with 0.1% methionine (0.1%MCDD) (A02082006i, Research Diets, Inc., New Jersey, USA) or a choline-deficient high-fat diet (CD-HFD) (D05010402, Research Diets Inc., New Jersey, USA). A control group was maintained on a regular diet with 1,030 mg/kg choline and 0.3% methionine (Teklad Global 14% Protein Rodent Maintenance diet; Envigo RMS Spain, Sant Feliu de Codines, Spain). In 0.1%MCDD and control diet, 17-20% of calories are derived from protein, 10-13% from fat and 63-67% from carbohydrates while in the CD-HFD, 20% of calories are derived from protein, 45% from fat and 35% of carbohydrates.
Isolation and culture of primary hepatocytes, hepatic stellate cells (HSC) and Kupffer cells (KC):
All animal experiments were performed according to the ARRIVE guidelines and carried out in accordance with the National Institutes of Health guide for the care and use of Laboratory animals (NIH Publications N0.8023, revised 1978) and the guidelines of European Research Council for animal care and use. Male mice were acquired from Charles River (St Germain sur l’Arbresle, France) and were maintained at the CIC bioGUNE Animal Facilities according to the criteria established by the European Union. In brief, mice were anesthetized with isoflurane (1.5% isoflurane in O2), the abdomen was opened and a catheter was inserted into the vena cava. Liver was perfused with buffer A [1X PBS, 5 mM EGTA] (37C, oxygenated) and portal vein was cut. Subsequently, liver was perfused with buffer B [1X PBS, 1mM CaCl2, collagenase type I (Worthington)] (37°C, oxygenated). After the perfusion, liver was placed in a petri dish containing buffer C [1X PBS, 2mM CaCl2, 0.6% bovine serum albumin (BSA)] and disaggregated with forceps. Digested liver was filtered through sterile gauze. Then, mouse primary hepatocytes, KC and HSC were isolated either from healthy control or mice fed during 4 weeks a 0.1%MCDD as previously described (Zubiete-Franco et al., 2017). Briefly, perfused livers were centrifuged at 48g for 5 min. Supernatant was removed and hepatocytes present in pellet were resuspended in fresh 10%-fetal bovine serum (FBS; Gibco) Minimun Essential Medium (MEM; Gibco) containing penicillin (100 U/ml), streptomycin (100 U/ml), and glutamine (2 mM) (PSG; Invitrogen). Hepatocytes were plated on collagen-coated plates. Cell viability was validated by trypan blue exclusion test and more than 70% of viability was considered acceptable to proceed with the experiments. KC and HSC were further isolated from the supernatant by gradient centrifugation with Percoll Plus (GE Healthcare, Little Chalfont, United Kindgom) and selective adherence. Purity of primary hepatocytes, HSC and KC was determined by using a DNA agarose gel after PCR to identify albumin (Alb), α-smooth-muscle actin (aSMA) and F4/80, respectively.
THLE-2 cell line:
The human liver epithelial cells acquired from ATCC® (isolated from adult from unknown sex) were cultured using BEGM from Lonza/Clonetics Corporation, Walkersville, MD 21793 (BEGM Bullet Kit; CC3170), where we have discarded the gentamycin/ Amphotericin (GA) and Epinephrine and to which we add extra 5 ng/mL EGF, 70 ng/mL Phosphoethanolamine and 10% fetal bovine serum. Cells were grown at 37°C and 5%CO2.
Method details
Treatments to primary mouse hepatocytes and THLE-2 cells:
Upon attachment, isolated mouse primary hepatocytes were transfected by overnight incubation with 100 nM Gls1 siRNA (Qiagen, Madrid, Spain) using DharmaFECT 1 reagent (Dharmacon). Controls were transfected with an unrelated siRNA (Qiagen, Madrid, Spain). Gene and protein knockdown were confirmed by RT-PCR and Western blotting, respectively. Once attached, primary hepatocytes were maintained at 0% FBS MEM (Gibco) overnight and were incubated during 24 or 48 h in Control media (MEM, Gibco) or methionine and choline-deficient DMEM/F-12 media (custom-made MCD media, Gibco). Isolated mouse hepatocytes were also treated with the GLS1 inhibitor Bis-2(5-phenylacetamido-1,3,4-thiadiazol-2-yl)ethyl sulfide (BPTES) (Sigma-Aldrich, St. QuentilFallavier , France). BPTES was dissolved in DMSO and used at a dose of 10μM. Lomitapide (N-(2,2,2-T rifluoroethyl)-9-[4-[4-[[[4′-(trifluoromethyl)[1,1′-biphenyl]2-yl]carbonyl]amino]-1 piperidinyl]butyl]9H-fluoren-9-carboxamde) (Sigma-Aldrich, St. QuentilFallavier, France), an inhibitor of VLDL export, that acts by inhibiting microsomal triglyceride transfer protein (MTP), was given to cells solubilized in DMSO at a concentration of 600 nM for a 24-h period. Oleic acid (Sigma-Aldrich, St. QuentilFallavier , France) conjugated to BSA at 400 mM was also given to hepatocytes during 6h. Cells were grown at 37°C and 5%CO2. Glutaminase inhibition after GLS1 pharmacological inhibitor treatment was confirmed by determining the amount of cellular glutamine and glutamate by UPLC-MS as explained below. Finally, cell viability after treatments was assessed using a caspase 3 activity assay as previously reported (Embade et al., 2012). Briefly, cells were lysed in caspase buffer and the protein content was determined by Bradford protein assay. 20 μl of 25x reaction buffer (PIPES pH 7.4 250 mM, EDTA 50 mM, 2.5% CHAPS, DTT 125 mM) were mixed with 2.5 μl of fluorogenic caspase-3 substrate (Enzo Life Sciences, USA) and with 10-50 μg of protein lysate in a total volume of 500 μΚ The mixture was incubated at 37°0 with gentle sh aking for 5 hours. Readings were taken at each hour using a Spectramax M3 spectrophotometer (excitation wavelength 390 nm, emission wavelength 510 nm). Caspase 3 activity was determined by calculating the increase in fluorescence per hour of incubation.
Treatments to primary HSC and KC:
HSC were cultured in 0% FBS RPMI-1640 Medium (Gibco) and collected at different times after plating (0, 3, 5 and 7 days). KC were cultured in 0% FBS MEM (Gibco) and stimulated with 200ng/ml lipopolyssacharide (LPS, Sigma) during 24 hours. Cells were grown at 37°C and 5%CO2.
Gls1 silencing in vivo in 0.1%MCDD-fed rodents:
After two weeks of 0.1% MCDD, mice were randomly divided into two experimental groups: in vivo silencing of Gls1 (siGls1) or unrelated siRNA control (siCtrl), receiving 150μl of 27μM Gls1-specific siRNA (Custom Ambion® In Vivo siRNA) or control siRNA using Invivofectamine® 3.0 Reagent (Thermo Fisher Scientific) by tail vein injection. Tail vein injection was performed each three-four day interval until four weeks of dietary intervention. The mice were analyzed for transaminases and biochemistry in blood at 0, 2, and 4 weeks of exposure to the 0.1% MCDD. Serum samples were analyzed using a Selectra Junior Spinlab 100 analyser (Vital Scientific, Dieren, Netherland) according to manufacturers’ suggested protocol. At the day of sacrifice, four weeks after 0.1%MCDD, two different experiments were carried out.
Experiment 1:
After overnight fast, animals were administered i.p. with (U-13C)-glutamine 99% (Cambridge Isotope Laboratories, Inc., Andover, MA, USA) at 350 mg/kg, 20 minutes before sacrifice. Depending on the final purpose of the sample, livers were removed and either snap frozen in liquid nitrogen, optical coherence tomography cryocompound (OCT-)embedded, formalin fixed, or immediately analyzed as far as the experiment regarding the quantification of fatty acid β-oxidation (FAO) and Seahorse Analyzer experiments.
Experiment 2:
In another group of animals, on the day of sacrifice mice were i.p. injected with poloxamer 407 (P-407) (Invitrogen, Carlsbad, CA, USA) in saline at 1 g/kg, as previously described (Millar et al., 2005). Blood was collected at baseline and six hours after P-407 injection and triglycerides were quantified using a commercially available kit (A. Menarini Diagnostics, Italy). Six hours after the P-407 injection, serum VLDL (d < 1.02 g/mL) were isolated by ultracentrifugation for 18-h at 35,000 rpm at 10 °C in a Beckman Coulter Type 50.4 Ti Rotor. Lipids content on VLDL were extracted as described previously (Folch et al., 1957) and separated by thin layer chromatography (Ruiz and Ochoa, 1997). From the total volume of extracted VLDL, 60% was used for lipid extraction. For lipid extraction, the correspondent volume of VLDL and up to 1.5 mL of dH2O were mixed with 8 mL of chloroform:methanol:HCl (2:1:0.0075, v/v) solution (Scharlau Chemicals, Spain). Tubes were vigorously shaken with a vortex for 2 minutes. Then, they were centrifuged at 1,000 xg for 15 minutes at 4 C to separate the aqueous phase from the organic phase. The organic phase was transferred to another extraction tube using a glass pasteur pipette. Lipids from the upper phase were reextracted. For reextraction, 4 mL of chloroform:methanol:HCl (2:1:0.0075, v/v) were added and the tubes were vigorously shaken for 2 minutes and centrifuged at 1,000 xg for 10 minutes at 4 C. The organic phase was mixed with the previously obtained one. For the removal of aqueous contaminant, the chloroform extract was washed with the third part of the total volume of KCl 0.88% solution. Each tube was shaken for 1 minute and centrifuged for 10 minutes at 1,000 xg and the lower phase was transferred to another extraction tube. Finally, the chloroform was evaporated in a concentrator-evaporator. Lipids were dissolved in 200 μL of toluene and kept at −20 °C in N2 atmosphere until processing. The lipid separation was performed by thin layer chromatography (TLC), performing a six chromatographic developments. First, the silica-gel plates of 20 x 20 cm (“Pre-coated TLC-plates SIL-G25”, Macherey-Nagel) were pre-treated with 1 mM EDTA-Na2. Silica-gel plates were left drying overnight and washed with chroloform:methanol:dH2O (60:40:10, v/v/v) overnight. The following day, the plates were activated at 100 °C for 30 minutes. Then, 3 μL of pure lipid mixtures (Avanti Polar Lipids, USA and Sigma-Aldrich) of known lipid concentrations and 1 or 3 μL of the lipid extracts from the samples were applied at 1.5 cm from the lower edge of the plate. Lipid quantities (nmol) for calibration were as next described: Cholesteryl ester (CE) (0.09-1.45), triglycerides (TG) (0.27-17), diglycerides (DG) (0.15-3), free cholesterol (FC) (0.36-3.6), phosphatidylethanolamine (PE) (0.33-3.91), phosphatidylinositol (PI) (0.1-1.72), phosphatidylserine (PS) (0.13-2.22) and phosphatidylcholine (PC) (0.63-7.63). Six chromatographic developments were carried out in solutions of decreasing polarity as presented in the Table S4. After each chromatographic development, the plates were dried using hot air. When the chromatographic separations were performed, the plate was stained immersing in a CuSO4 solution at 10% (p/v) in H3PO4 at 8% (v/v) for 10 seconds. After, plates were dried with hot air and plates were developed for 3 minutes at 200 °C. The image of the TLC plate was digitalized using the densitometer GS-800 and quantification was performed with Quantity One software. The integrated optic density (IOD) of each lipid, was interpolated in the IOD values of the calibration curves. For the analysis of the VLDL apoB, 0.3% of the secreted VLDL was used for western blotting. Since there are no proteins to use as loading control in the VLDL population, ponceau red staining was performed as a loading control.
Gls1 silencing in vivo in CD-HFD-fed rodents:
After three weeks of CD-HFD, mice were randomly divided into two experimental groups, siGls1 and siCtrl. Tail vein injection was performed each three-four day interval until six weeks of dietary intervention. For analysis of triglycerides content in circulating VLDL particles, serum VLDLs (d < 1.02 g/mL) were isolated from the cava vein after 2 hours fasting. VLDL isolation was performed as described above. Quantification of TG content in VLDL was assessed using a commercially available kit (A. Menarini Diagnostics, Italy).
RNA Isolation and Quantitative Real-Time Polymerase Chain Reaction:
Total RNA was isolated with Trizol (Invitrogen). One to two μg of total RNA was treated with DNAse (Invitrogen)and reverse transcribed into cDNA using M-MLV Reverse Transcriptase (Invitrogen). Quantitative real-time PCR (RT-PCR) was performed using SYBR® Select Master Mix (Applied Biosystems) and the Viia 7 Real-Time PCR System (Applied Biosystems). The Ct values were extrapolated to a standard curve, and data was then normalized to the housekeeping expression (Arp). Primers sequences are described in Table S5.
Immunohistochemistry:
Paraffin embedded liver samples were sectioned, dewaxed and hydrated. All procedures were performed according to standard protocols using the EnVision+ System HRP (Dako, Denmark). Samples were incubated with Vector Vip substrate (Vectorlabs, Burlingame, USA) for color development. Five to ten random images per sample were taken blinded using an AXIO Imager A1 microscope (Carl Zeiss AG, Jena, Germany). Stained area percentage of each sample was calculated using FIJI (ImageJ). Stainings was performed according to Table S5.
Immunofluorescence:
Paraffin-embedded liver samples were rehydrated, unmasked with the conditions described in Table S6, followed by the incubation with 2.5% goat serum for 30 min. The primary antibody and the corresponding secondary antibody were then incubated according to Table S5. Nuclei were counterstained with Fluoromount-G with DAPI (Southern Biotech). The slides were mounted with Fluoromount-G (Southern Biotech) containing DAPI and representative images were taken using an Upright fluorescent microscope (Axioimager D1).
H&E and Sirius Red staining:
We have stained for hematoxylin and eosin (H&E) and Sirius Red staining for collagen in paraffin-embedded sections of formalin-fixed liver samples. Samples were initially deparaffinised in Histo-Clear (Electron Microscopy Sciences, Hatfield. PA, USA) and rehydrated through graded alcohol solutions. After the deparaffinization and rehydration process, sections were subjected to conventional hematoxylin and eosin or Sirius red stainings. For H&E staining, sections were stained with Harris Hematoxylin (Bio-Optica, Milano, Italy) for 15 minutes followed by eosin staining (Sigma Aldrich) for 15 minutes. Washed in running tap water for 5 minutes and differentiated in 0.5% HCl for 1 second. Samples are then dehydrated in graded alcohol solutions until 100% and mounted in DPX mounting medium (Sigma Aldrich). For Sirius Red staining, rehydrated sections were then stained with Sirius red solution 1 (0.01% Fast Green FCF in picric acid, Sigma Aldrich) for 20 min and then with Sirius red solution 2 (0.04% Fast Green FCF/0.1% Sirius red in picric acid, Sigma Aldrich) for another 15 min. After the respective staining, sections were then dehydrated directly in 100% alcohol and mounted in DPX mounting medium (Sigma Aldrich). F4/80, a membrane macrophage marker, immunohistochemical staining was used. For a-SMA, a marker of activated HSC, immunofluorescence was used.
Liver Sudan Red staining:
For the histological quantification of hepatic lipids we have used Sudan Red staining. Briefly, cut liver cryostat section of 8 microms were incubated with freshly-prepared Sudan III stain (Sigma-Aldrich) and contrasted with Mayers hematoxylin. Hepatic ammonia content was quantified by using a methodology recently described by our group (Gutierrez-de-Juan et al., 2017). Five to ten random images per sample were taken blinded using an AXIO Imager A1 microscope (Carl Zeiss AG, Jena, Germany). Stained area percentage of each sample were calculated using FIJI (ImageJ).
Protein isolation and Western blotting:
Total protein extracts from primary hepatocytes and hepatic tissue were resolved in sodium dodecyl sulfate-polyacrylamide gels and transferred to nitrocellulose membranes. As secondary antibodies, we used anti-rabbit-IgG-HRP-linked (Cell Signaling) and anti-mouse IgG-HRP-linked (Cell Signaling). The antibodies and conditions used for Western Blotting are described in Table S6.
BODIPY staining in isolated mouse hepatocytes:
Hepatocytes in culture were collected in covers and incubated with boron-dipyromethene (BODIPY 493/503) (Molecular Probes, Thermo Fisher Scientific) at a concentration of 10μg/ml during 45 minutes after fixation in 4% paraformaldehyde for 10 minutes. Quantification of lipid bodies was performed using FIJI (ImageJ).
Quantification of hepatic lipids.
Liver triglycerides were quantified after extraction using a published method (Pitman et al., 2011). Briefly, pieces of mouse liver were weighed, then homogenized in 10X of PBS 1X. The homogenates were then extracted with a 2:1 (v:v) mixture of chloroform-methanol by vortexing thoroughly. Samples were then centrifuged at 4,200 g at 4°C for 10 min. The organic (lower) phase was air-d ried in a fresh tube then resuspended in 100 μl of 1% Triton X-100 in ethanol. This was then air dried and resuspended in 500 μl 1× PBS, for the final lipid extract. After extraction, aliquots of this extract were used to measure triglycerides using an automatized Selectra Junior Spinlab 100 analyser (Vital Scientific, Dieren, Netherland. For the quantification of fatty acids, liver tissues (30 mg) were homogenized with 10 volumes of ice-cold phosphate buffered saline (1XPBS) in a Potter homogenizer (20 strokes). Fatty acids were measured in homogenates using a kit from Wako Chemicals (Richmond, VA). Hepatic total phospholipids were quantified by using a commercially available kit from Sigma-Aldrich (St. QuentilFallavier, France). Families of phospolipids were extracted from 1.5 mg of protein from homogenates (Folch J et al, 1957) and were quantified as described previously (Ruiz and Ochoa, 1997). Lipid extraction from liver homogenates was performed as explained above but here from 1.5 mg of protein from the homogenate. 3 or 8 μL from the lipid extracts were separated by TLC as explained above.
Quantification of lipids in primary hepatocytes:
Triglycerides in primary hepatocytes were measured after extraction by following a modification of an already existing protocol (García-Ruiz I et al., 2014). Briefly, samples were washed in cold PBS and resuspended in 500 μl of 5% NP-40/ddH2O solution to be heated at 95°C during 5 minutes. The samples were analyzed using a Selectra Junior Spinlab 100 analyser (Vital Scientific) according to manufacturers’ suggested protocol.
Determination of cellular Reactive Oxygen Species (ROS):
Cellular ROS production in mouse primary hepatocytes was assessed using CellROX Deep Green Reagent (Thermo Fisher Scientific). Briefly, hepatocytes were loaded with 1.5 μM CellROX in 10% FBS MEM for 30 min at 37° C in a CO2 incubator. The hepatocytes were then carefully washed three times with 1XPBS, collected and analyzed by flow cytometry FACS Canto II (BD Biosciences).
Determination of mitochondrial Reactive Oxygen Species (ROS):
Mitochondrial ROS production in primary hepatocytes was assessed using MitoSOX Red mitochondrial superoxide indicator (Invitrogen, USA). The cells were loaded with 2 μM MitoSOX Red for 10 min at 37° C in a CO2 incubator. The cells were then washed three times with PBS. Fluorescence was read at 510 nm (excitation) and 595 (emission) using a plate reader SpectraMax M2 (bioNova).
Lipid peroxidation Assay kit:
Malondialdehyde (MDA), together with 4-hydroxynonenal (4-HNE), is a natural bi-product of lipid peroxidation and its quantification is generally used as marker for lipid peroxidation. The MDA in the sample reacts with thiobarbituric acid (TBA) to generate a MDA-TBA adduct. The MDA-TBA adduct can be easily quantified colorimetrically (OD = 532 nm) MDA content in liver samples was quantified by using a commercially available kit from Sigma-Aldrich (St. QuentilFallavier, France).
Hepatic Fatty Acid Oxidation (FAO):
Fatty acid β-oxidation was assessed as described before (Gao et al., 2015; Hirschey et al., 2010; Porteiro et al., 2017). Fresh liver pieces were homogenized in a Potter homogenizer (5 strokes) in cold buffer (25 mM Tris-HCl, 500 nM sucrose, 1 mM EDTA-Na2 pH 7,4) and sonicated for 10 s. Then, the homogenates were centrifuged at 500 xg for 10 min at 4 °C. Approximately 500 μg of protei n from the homogenates supernatant was used for the assay in a volume of 200 μl. The reaction started by adding 400 μl of assay mixture containing 0.5 μCi/ml [1-14C] palmitic acid to the samples and was incubated for 1 h at 37 °C in eppendorf tubes with a Whatman paper circle in the cap. The reaction was stopped by adding 300 μl of 3 M perchloric acid and 1 M NaOH was added to impregnate the whatman cap. After 2 h the Whatman caps were retired and the radioactivity associated was measured in a scintillation counter. The eppendorf tubes were centrifuged at 21,000 xg 10 min at 4 °C. 400 μl from the supernatant were collected and the radioactivity was counted in a scintillation counter. The supernatant contained the acid soluble metabolites (ASM) and the Whatman caps captured the released CO2.
Respiration studies in liver mitochondria:
The respiration of liver mitochondria was measured at 37°C by high-resolution respirometry with the Seahorse Bioscience XF24–3 Extracellular Flux Analyzer. Liver mitochondria were isolated as recommended by Agilent Seahorse Application note. Succinate (10 mM) and rotenone (2 μM) were used as substrates to quantify State 2. State 3 was initiated with ADP, State 4 induced with the addition of oligomycin (State 4o), and FCCP-induced maximal uncoupler-stimulated respiration (State 3u) were sequentially measured. The normalized data were expressed as pmol of O2 per minute or milli-pH units (mpH) per minute, per mg mitochondrial protein.
Glutaminase activity:
Glutaminase activity was determined in vivo from mouse liver tissue as well as in vitro in primary cultured hepatocytes. To assess glutaminase activity in mouse liver, we have calculated the ratio between 13C5-glutamate and 13C5-glutamine after i.p. injection of 13C5-glutamine (20 minutes before sacrifice. Semi-quantification of the labelled metabolites was performed by UPLC-MS. Glutaminase activity in hepatocytes was measured after 48 h incubation with methionine/choline deficient media (MCD) and treatment with siRNA against GLS1 (siGls1) or unrelated control (siCtrl) overnight. The concentration of glutamate and glutamine per μg protein was determined by LCMS and the ratio between the concentrations was taken as a measure for glutaminase activity. LCMS and extraction procedures are described below.
Tricarboxylic acid (TCA) cycle activity:
An i.p. injection of 13C5-glutamine was given to animals fed a 0.1%MCDD 20 minutes before sacrifice. Upon these conditions, accumulation of the citrate metabolite and more importantly the ratio between 13C1-citrate and 13C5-glutamine were measured by LCMS after extraction.
Incorporation of 13C6-glucose in Ptd-Cho in isolated mouse hepatocytes:
Isolated mouse hepatocytes (5x105 cells) were incubated with MCD medium, MCD medium combined with siRNA against Gls1 or normal medium for 24 h. To each treatment either labelled 13C6-glucose or non-labelled glucose was added in a concentration of 10 mM.
LCMS sample preparation:
Two types of biological material were analyzed with LCMS, namely liver tissue and primary cultured hepatocytes. All liver extractions were carried out in the same manner with a method that was optimized for polar metabolites. On the other hand, hepatocyte extraction methods depended on the metabolite type of interest i.e. being polar or apolar metabolites. The polar metabolite set consisted of GSH, GSSG, glutamine, glutamate and citrate. The apolar set consisted of the following panel of phosphatidylcholines: PC(32:0), PC(32:1), PC(34:1), PC(34:2) and PC(36:2). In short, liver tissues for the determination of glutaminase and TCA cycle activity and levels of GSSG and GSH (polar metabolite set) were homogenized in 500 μL of ice-cold extraction liquid (EL) with a tissue homogenizer (FastPrep) inb a 40 second cycle at 6000 rpm. The EL consisted of a mixture of methanol/water (50/50 %v/v) with 10 mM acetic acid. Next 400 μL of homogenate was transferred into a new aliquot and shaken for 30 minutes at 1400 rpm and centrifuged for 30 minutes at 14000 rpm at 4 °C. Next, 75 μL of the supernatant was transferred to a fresh aliquot and stored at −80 °C for 30 minutes. The chilled supernatants were evaporated with a speedvac in approximately 2h. The resulting pellets were resuspended in 250 μL water/acetonitrile(MeCN)/formic acid (FA) (39.9/60/0.1 %v/v/v, resuspension liquid RL). These resuspensions were transferred to glass vails for LCMS analysis.
Hepatocytes were extracted as follows. Cell pellets were collected and lysed in 500 μL EL. Homogenization was performed with a tissue homogenizer (Precellys) in two 30-second cycles at 6000 rpm. Subsequently 400 μL of the homogenate was transferred to a new aliquot and shaken at 1400 rpm for 30 minutes at 4 °C. Next the aliquots were centrifuged for 15’ at 14000 rpm at 4 °C. Three aliquots of 75 μL per sample (22 5 μL) of the supernatant were transferred to fresh aliquots and placed at −80 °C for 20’. The ch illed supernatants were evaporated with a speedvac in approximately 2h. The resulting pellets (3 per sample) were serially resuspended in 150 μL RL.
To extract Ptd-Cho from hepatocytes to 400 μL of homogenate, obtained as described above, another 400 μL of chloroform was added. The resulting solution was shaken at 1400 rpm for 1 hour at 4 °C. Next, the aliquots were centrifuged for 30 minutes at 14000 rpm at 4 °C in order to separate the phases. From the organic phase (chloroform) 200 μL per sample was transferred to fresh aliquots and placed at −80 °C for 20 minutes and evaporated with a speedvac. The resulting pellets were resuspended in 150 μL pure methanol.
Liquid chromatography and mass spectrometry methods:
Liquid chromatography-coupled mass spectrometry (LCMS) was used to determine activity of the tricarboxylic acid (TCA) cycle in liver, relative levels of reduced or oxidized glutathione (GSH and GSSG respectively) in liver, glutaminase activity in liver and hepatocytes and incorporation of 13C6-glucose in Ptd-Cho hepatocytes.
The LCMS instrumentation consisted of an Acquity-SQD UPLC system (Waters Corp., Manchester) coupled to a SYNAPT G2 HDMS Time of Flight mass spectrometer (Waters Corp.). For all performed assays the following MS settings were kept constant. The ion source temperature was 120 °C and capillary temperature 450 °C. The flow of the cone and desolvation gas (both nitrogen) were set to 5 L/h and 600 L/h, respectively. A 2 ng/mL leucine-enkephalin solution in RL was infused at 10 μL/min and used for a lock mass to automatically correct deviations in mass accuracy. This signal was measured approximately each 30 seconds for 0.5 seconds.
Two different LCMS methods were used to measure either polar or apolar metabolites. Metabolites from the polar set were separated prior to MS analysis under the following conditions. Chromatography was performed on a 2.1 x 100 mm, 1.7 μm BEH amide column (Waters Corp.), thermostated at 40 °C. Mobile phase solvent A (aqueous phase) containedf 99.5% water, 0.5% FA and 20 mM ammonium formate while solvent B (organic phase) contained 29.5% water, 70% MeCN, 0.5% FA and 1 mM ammonium formate. In order to obtain a good separation of the analytes, the following gradient was used: from 5% A to 50% A in 2.4 minutes in curved gradient (#8, as defined by Waters), from 50% A to 99.9% A in 0.2 minutes constant at 99.9% A for 1.2 minutes, back to 5% A in 0.2 minutes. The flow rate was 0.250 mL/min and the injection volume was 2 μL. All samples were injected randomly. After every 8 injections a QC low and QC high were injected. All samples were injected in duplicate. The MS was operated in positive and negative electrospray ionization (+ESI, −ESI) in full scan mode depending on the analyte. Citrate, glutamate and glutamine were measured in ESI- and glutathione in ESI+. The cone voltage was 25 V and capillary voltage was 250 V in +ESI and 500 V in −ESI.
As for the apolar set a 1 x 100 mm, 1.7 μm BEH C18 column (Waters Corp.), thermostated at 60 °C, was used to separate the analytes before entering the MS. Solvent A (aqueous phase) consisted of 40% water, 60% MeCN 10 mM ammonium formate while solvent B (organic phase) consisted of 10% MeCN, 90% isopropanol and 10 mM ammonium formate. The following gradient was used: from 50 %A to 1 %A in 4 minutes in a curved gradient (#8, as defined by Waters), constant at 1 %A for 0.8 minutes, back to 50 %A in 0.2 minutes. The flow rate was 0.120 mL/min and the injection volume was 2 μL. All samples were injected randomly. After every 8 injections a QC sample (pooled samples) was injected. The mass spectrometer was operated ESI+ with a capillary voltage of 250 V.
LCMS data analysis:
Extracted ion traces (XICs) were obtained and integrated for specific analytes in a 20 mDa integration window and subsequently smoothed and integrated with QuanLynx software (Waters Corp.). For GSH the XIC was determined at an m/z =308.0916, for GSSG at m/z =613.1598, for citric acid at m/z 191.0192, for 13C1-citric acid at m/z 192.0225, for glutamine at m/z 145.0613, for 13C5-glutamine at m/z 150.0781, for glutamate at m/z 146.0453, for 13C5-glutamate at m/z 151.0621.
As for the Ptd-Cho species it was found that labelling via 13C6-glucose introduced three 13C atoms in these phospholipids. Therefore the following XICs were extracted: PC(32:0) at m/z 734.57 and 737.58005, PC(32:1) at m/z 732.5543 and 735.56435, PC(34:1) at m/z 760.5864 and 763.59645 , PC(34:2) at m/z 758.57 and 761.58005, PC(36:2) at m/z 786.6013 and 789.61135.
As for labelling experiments, since the m/z values of 13C-labelled species can overlap with the natural occurring isotopes in a 20 mDa window, the signals of the 13C-labelled species needed to be corrected. Therefore, an isotope model of the endogenous metabolite was made with the Isotope Model tool in MassLynx software (Waters Corp.). Next, the absolute abundances of naturally occurring isotopes were estimated by taking the chromatographic peak intensities of the most abundant spectral peak representing the endogenous species and multiplying that by the relative abundances of the natural isotopes. These numbers were than subtracted from the integrated signal of the 13C-isotopes with the same mass as the naturally occurring ones. The following equation describes these calculations:
where Sm,isotope is the signal (area or height) of the 13C-isotope with mass m ± 20 mDa (integration window) of a specific analyte, Sm,total is the measured signal at mass m ± 20 mDa, dm,natural is the relative abundance of the natural occurring isotope with mass m (the abundance of the most abundant signal representing the analyte is set to 1) and S0 is the signal of the natural occurring metabolite with the highest spectral peak. Finally, the corrected signals were normalized on tissue weight or μg protein.
Quantification and Statistical Analysis:
Statistical significance was determined by using Prism 5 (GraphPad Software). The Grubb’s outlier test was performed by using the GraphPad, Prism8 software, and no value was excluded. One-way analysis of variance (ANOVA) followed by post hoc Bonferroni test was used in case 3 groups were compared or using the Student’s t-test in case 2 groups were compared. A p<0.05 was considered statistical different. Statistic parameters are reported in figure legends.
Data and Code availability:
This study did not generate any unique datasets or code.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Albumin antibody | Affinity Biologicals | #CatPA1-25673 RRID:AB_2539935 |
| Anti-Apolipoprotein B antibody | Millipore | #CatAB742 RRID:AB_92217 |
| GAPDH antibody [6C5] | Abcam | #CatAb8245 RRID:AB_2107448 |
| Anti-GLS1 | Agios Pharmaceuticals | N/A |
| Anti-GLS2 antibody | LifeSpan Biosciences | #CatLS-C80586-100 RRID: AB_1594624 |
| Glutamine Synthetase Antibody | Novus Biologicals | #CatNB110-41404 RRID:AB_790020 |
| Mouse Anti-Human HNF4 alpha Monoclonal Antibody, Unconjugated, Clone K9218 | Actif SCETI (Perseus Proteomics) | Cat# PP-K9218-00 RRID:AB_1964277 |
| Anti-mouse IgG, HRP-linked Antibody | Cell Signaling | #Cat7076S RRID:AB_330924 |
| Anti-rabbit IgG, HRP-linked Antibody | Cell Signaling | #Cat7074S RRID:AB_2099233 |
| Rat anti-mouse F4/80:ANTIGEN Monoclonal antibody, Biotin Conjugated, Clone CI:A3-1 | Abd Serotec | #CatMCA497-BB RRID:AB_323893 |
| Cy3-AffiniPure Goat Anti-Rabbit IgG (H+L) antibody | Jackson ImmunoResearch | #Cat111-165-003 RRID:AB_2338000 |
| Goat anti-chicken-Alexa fluor488 | Abcam | Abcam Cat# ab150169, RRID:AB_2636803 |
| Fluorescein (FITC)-AffiniPure Goat Anti-Rabbit IgG (H+L) antibody | Jackson ImmunoResearch | #Cat111-095-003 RRID:AB_2337972 |
| Bacterial and Virus Strains | ||
| Biological Samples | ||
| cDNA of NASH patients | Supplied by MD PhD Javier Crespo Garcia | Hospital Universitario Marqués de Valdecilla www.humv.es |
| Serum of NASH patients | Already published study | Barr et al., 2012 doi: 10.1021/pr201223p |
| Paraffin-embedded liver NASH patients | Supplied by MD PhD Erica Villa | Azienda Ospedaliero-Universitaria http://www.ao-pisa.toscana.it/ |
| Chemicals, Peptides, and Recombinant Proteins | ||
| 2-propanol | Sigma-Aldrich | #Cat 19516 CAS 67-63-0 |
| 30% Acrilamide/Bis Solution | Bio-Rad | #Cat161-0156 |
| Acetonitrile | Pierce | #Cat51101 |
| Ac-DEVD-AFC Caspase-3-substrate | Enzo | ALX-260-032 |
| Adenosine 5’-diphosphate sodium salt | Sigma-Aldrich | A2754 CAS 20398-34-9 |
| Agarose | Sigma-Aldrich | A9539 CAS 9012-36-6 |
| Bis-2(5-phenylacetamido-1,3,4-thiadiazol-2-yl)ethyl sulfide (BPTES) | Sigma-Aldrich | SML0601-25MG CAS 314045-39-1 |
| Bore-dipyromethene (BODIPY) | Molecular Probes (ThermoFischer) | D3922 |
| CHAPS | Sigma-Aldrich | CHAPS-RO |
| Chloroform | Sigma-Aldrich | C2431 CAS 67-66-1 |
| Corning® Collagen I, Rat Tail, 100 mg | Corning | #Cat354236 |
| Collagenase type IV, CLS-4 | Worthington | LS004188 |
| Dako EnVIsion system, Peroxidase | Dako | K5007 |
| 2-(4-Amidinophenyl)-6-indolecarbamidine dihydrochloride, 4′,6-Diamidino-2-phenylindole dihydrochlorid (DAPI) | Sigma-Aldrich | D9542 CAS 28718-90-3 |
| DharmaFECT 1 Transfection Reagent | Dharmacon | T-2001-01 |
| Direct Red 80 | Sigma-Aldrich | 365548-25G CAS 2610-10-8 |
| DMEM/F-12 methionine and choline-deficient media | Gibco (ThermoFischer) | ME120128L1 EDC11632701 |
| DPX | Sigma-Aldrich | #Cat06522 |
| DTT | Sigma-Aldrich | DTT-RO |
| EDTA | Sigma-Aldrich | E9884 |
| EnVision+ System HRP | Dako | K4001 |
| Eosin B | Sigma-Aldrich | #Cat2853 |
| Fast Green FCF | Sigma-Aldrich | F7258-25G CAS 2353-45-9 |
| Carbonyl cyanide-p-trifluoromethoxyphenylhydrazone (FCCP) | CaymanChem | #Cat15218 CAS 370-86-5 |
| caspase-3 (human), | Enzo Life Sciences | ALX-201059 |
| Fetal Bovine Serum | Gibco (ThermoFischer) | A38401 |
| Formic Acid | PanReac AppliChem | #Cat1002641000 |
| Glucose [U-13C] | Sigma-Aldrich | #Cat389374 CAS 110187-42-3 |
| Glutamine [U-13C] 99% | Cambridge Isotope Laboratoires | CLM-1822 CAS 184161-19-1 |
| Harris Hematoxylin | Bio-Optica | #Cat05-06005/L |
| Histo-Clear | Electron Microscopy Sciences | #Cat64110-004 |
| Hydrogen Peroxide 30% w/v | PanReac AppliChem | #Cat121076 CAS 7722-84-1 |
| Invivofectamine™ 3.0 Reagent | ThermoFischer | IVFs3001 |
| Isoflurane | Baxter SL | NR60378 |
| Leukine/encephalin acetate salt hydrate | Sigma-Aldrich | CAS 588-25-6 |
| L-glutamine | Gibco (ThermoFischer) | #Cat25030-024 |
| Lipopolysaccharide from Escherichia Coli O111 :B4 | Sigma-Aldrich | L3012-10MG |
| Methanol | PanReac AppliChem | #Cat1610911714 |
| Minimum Essential Media | Gibco (ThermoFischer) | #Cat31095029 |
| Mount Quick Aqueous | Bio-Optica | #Cat05-1740 |
| N-(2,2,2-Trifluoroethyl)-9-[4-[4-[[[4′-(trifluoromethyl)[1,1′-biphenyl]2-yl]carbonyl]amino]-1 piperidinyl]butyl]9H-fluoren-9-carboxamde (Lomitapide) | Sigma-Aldrich | SML1385 CAS 182431-12-5 |
| OCT | Pioneer | PRC/OCT |
| Oleic acid | Sigma-Aldrich | O1008 |
| Oligomycin | Sigma-Aldrich | O4876-5MG CAS1404-19-9 |
| Oxygen | Air Liquide SLU | ESCG101710 |
| Palmitic acid |1-14C] (56.1 mCi/mmol) | Perkin Elmer | NEC075H |
| Paraformaldehyde 4% solution in PBS | Santa Cruz | Sc-281692 CAS 30525-89-4 |
| Penicillin-Streptomycin-Glutamine (100x) | Gibco (ThermoFischer) | #Cat10378016 |
| Percoll® Plus | GE Healthcare (Sigma-Aldrich) | GE17-5445-01 |
| Picric Acid | PanReac AppliChem | #Cat141048-1610 CAS 88-89-1 |
| PIPES | Sigma-Aldrich | P6757 |
| Pluronic™ F-127, low UV absorbance (Poloxamer P-407) | Invitrogen (ThermoFischer) | P6867 |
| Protease Inhibitor Cocktail Tablets | Roche Diagnostics | #Cat11697498001 |
| Ponceau S Solution | Sigma-Aldrich | P7170-1L CAS 6226-79-5 |
| RPMI-1640 | Gibco (ThermoFischer) | #Cat1185-093 |
| Rotenone | Sigma-Aldrich | R8875 CAS 83-79-4 |
| Sodium chloride | PanReac AppliChem | #Cat1064041000 |
| Sodium dodecyl sulfate | Sigma-Aldrich | L5750 CAS 151-21-3 |
| Sodium orthovanadate | Sigma-Aldrich | S6508-50G |
| Sodium fluoride | Sigma-Aldrich | S7920-100G |
| Succinate | Sigma-Aldrich | S9512 |
| Sudan-III | PanReac AppliChem | #Cat251731 CAS 85-86-9 |
| Triton X-100 | Sigma-Aldrich | #Cat100-500mL |
| Tris Base | Fisher | BP152-1 |
| Trizol | Invitrogen (ThermoFischer) | #Cat15596026 |
| XF-Calibrant (pH 7.4) | Seahorse Bioscience | #Cat100840-000 |
| Critical Commercial Assays | ||
| CellROX Deep Green Reagent | ThermoFischer | C104444 |
| DNase I, amplification Grade | Invitrogen (ThermoFischer) | #Cat18068015 |
| Fatty acids Kit | Wako Chemicals | #Cat434-91975 |
| Lipid Peroxidation (MDA) Assay Kit | Sigma-Aldrich | MAK085 |
| M-MLV Reverse Transcriptase (200 U/μL) | Invitrogen (ThermoFischer) | #Cat28025013 |
| MitoSOX™ Red MItochondrial Superoxide Indicator | Invitrogen (ThermoFischer) | M36008 |
| Phospholipid Assay Kit | Sigma-Aldrich | MAK122 |
| SYBR® Select Master Mix | Applied Biosystems (ThermoFischer) | #Cat44720903 |
| Protein Assay Dye Reagent Concentrate (Bradford) | Bio-Rad | #Cat500-0006 |
| Triglyceride Assay Kit | Abcam | ab65336 |
| Vector® VIP Peroxidase Substrate Kit | Vector Laboratories | SK-4600 |
| Deposited Data | ||
| Experimental Models: Cell Lines | ||
| Human: THLE-2 cells | ATCC® | THLE-2 (ATCC® CRL-2706™) |
| Experimental Models: Organisms/Strains | ||
| 0.1% methionine and choline-deficient diet (0.1%MCDD) | Research Diets | A02082006i |
| Choline-deficient high-fat diet (CD-HFD) | Research Diets | D05010402 |
| C57BL/6 mouse | Charles River | C57BL/6NCtrl |
| Teklad Global 14% Protein Rodent Maintenance diet | Envirgo | 2014C |
| Mat1a−/− mouse | CIC bioGUNE animal facility | N/A |
| Mat1a+/+ mouse | CIC bioGUNE animal facility | N/A |
| Oligonucleotides | ||
| Gls Ambion® In vivo Pre-designed siRNA | Ambion (ThermoFischer) | #Cat4390771 ID:s66756 |
| Silencer™ Negative Control No. 1 siRNA | Ambion (ThermoFischer) | AM4635 |
| siRNA targeting sequence: Gls1: Fw 5′- GCAAUAGGAUAUUACUUAATT- 3′ Rv 5′- UUAAGUAAUAUCCUAUUGCAA- 3′ | Sigma-Aldrich | N/A |
| siRNA targeting sequence: Negative Control: Fw 5′- AAUUCUCCGAACGUGUCACGU[dT][dT]- 3′ Rv 5′- ACGUGACACGUUCGGAGAAUU [dT][dT]- 3′ | Sigma-Aldrich | N/A |
| Recombinant DNA | ||
| Software and Algorithms | ||
| ImageJ | NIH | https://imagej.nih.gov/ij/index.html RRID SCR_003070 |
| MassLynx MS | Waters Corp | http://www.waters.com/waters/es_ES/MassLynx-Mass-Spectrometry-Software-/nav.htm?cid=513164&locale=es_ES RRID: SCR_014271 |
| QuanLynx Application Manager | Waters Corp | #Cat720001677EN http://www.waters.com/waters/educationInstance.htm?eiid=10137743 |
| Prism 5 | Graphad Software | https://www.graphpad.com/ RRID: SCR_00278 |
| DDS Research sample size calculator | DSS Research | https://www.dssresearch.com/resources/calculators/sample-size-calculator-average/ |
| Other | ||
| 300SL Liquid Scintillation Counter | LabLogic | N/A |
| Acquity-SQD UPLC | Waters Corp | N/A |
| AXIO Imager A1 Manual | Carl Zeiss | N/A |
| AXIO Imager D1 Upright Fluorescence Microscope | Carl Zeiz | N/A |
| BD FACSCanto™ II | BD Biosciences | N/A |
| BEH Amide Collumn | Waters Corp | #Cat186004802 |
| BEH C18 Column | Waters Corp | #Cat186002350 |
| Cryostate | Leica Biossystems | CM 1850 UV |
| Medical X-Ray Film | Fujifilm | #Cat47410 19289 |
| Nitrocellulose transfer membrane | ThermoFischer | LC2009 |
| Omnican ® 100 | Braun | #Cat9151141 |
| Precellys Tissue Homogenizer | Bertin Instruments | N/A |
| Seahorse XF24-3 Extracellular Flux Analyzer | Seahorse Bioscience (Agilent technologies) | N/A |
| Selectra Junior® Spinlab 100 analyser | Vital Scientific | #Cat0200-013-01 |
| Spectramax M2 | bioNova | N/A |
| SpeedVac™ | Thermofischer | SPD131DDA |
| SYNAPT G2 HDMS TOF | Waters Corp | N/A |
| Thin-layer chromatography (TLC) silica sheets 20x20cm | Merck Millipore | #Cat1055530001 |
| Tissue Homogenizer | FasPrep | N/A |
| Type 50.4 Ti Fixed-Angle Titanium Rotor | Beckamn Coulter | #Cat377299 |
| ViiA 7 Real-Time PCR System | Applied Biosystems (ThermoFischer) | #Cat4453545 |
| Whatman paper 3MM Chr | GE Healthcare | #Cat3030-931 |
| XF24-3 fluxpak | Seahorse Bioscience (Agilent technologies) | #Cat102070-001 |
Highlights.
The high activity glutaminase isoform, GLS1, is augmented in NASH.
GLS1 inhibition reduces steatosis in NASH by increasing VLDL export.
GLS1 inhibition diminishes oxidative stress in pre-clinical models of NASH.
GLS1 targeting may be a valuable therapeutic approach in NASH.
Context and Significance.
Non-alcoholic steatohepatitis (NASH) is a global health problem and an important risk factor for cirrhosis, liver cancer and cardiovascular diseases. Currently, in spite of huge investments made by researchers and the pharmaceutical industry, there are still no approved therapies for the treatment of NASH. Herein, investigators of CIC bioGUNE in Spain and their colleagues have studied the role of the amino acid glutamine and its catabolism in NASH progression. They show that a liver enzyme, glutaminase 1, involved in glutamine metabolism is elevated in the liver biopsies of patients with NASH and in mouse models of the disease. Therapeutically, they reveal that GLS1 inhibition in pre-clinical models reduces both liver steatosis and oxidative stress without associated side-effects. Overall, GLS1 targeting could be a valuable therapeutic approach for the clinical management of NASH.
Acknowledgements
This work was supported by grants from NIH (US Department of Health and Human services)-R01AT001576 (S.C.L., J.M.M and M.L.M-C.), Gobierno Vasco-Departamento de Salud 2013111114 (to M.L.M.-C), ELKARTEK 2016, Departamento de Industria del Gobierno Vasco (to M.L.M-C), Ministerio de Ciencia, Innovación y Universidades MICINN: SAF2017-87301-R, SAF2017-88041-R and RTI2018-096759-1-100 integrado en el Plan Estatal de Investigación Cientifica y Técnica y Innovación, cofinanciado con Fondos FEDER (to M.L.M-C, J.M.M., and T.C.D respectively), BIOEF (Basque Foundation for Innovation and Health Research): EITB Maratoia BIO15/CA/014; Asociacion Española contra el Cáncer (T.C.D, P.F-T and M.L.M-C), Daniel Alagille award from EASL (to T.C.D), Fundación Científica de la Asociación Española Contra el Cancer (AECC Scientific Foundation) Rare Tumor Calls 2017 (to M.L.M), La Caixa Foundation Program (to M.L.M), Programma di Ricerca Regione-Università 2007-2009 and 2011-2012, Regione Emilia-Romagna (to E.V.), Ayudas para apoyar grupos de investigación del sistema Universitario Vasco IT971-16 (P.A.), MINECO SAF2015-64352-R (P.A.). Work produced with the support of a 2017 Leonardo Grant for Researchers and Cultural Creators, BBVA Foundation (M.V-R) and Gilead Sciences International Research Scholars Program in Liver Disease (to MVR).The authors would like to acknowledge Agios Pharmaceuticals for providing us the antibody against GLS1. Ciberehd_ISCIII_MINECO is funded by the Instituto de Salud Carlos III. We thank MINECO for the Severo Ochoa Excellence Accreditation to CIC bioGUNE (SEV-2016-0644).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
Dr. Mato consults for, advises for, and owns stock in Owl. He consults for and advises for Abbott. He consults for Galmed. Dr. Martinez-Chantar advises for Mitotherapeutix LLC. Dr. Alonso and Dr. Castro are OWL Metabolomics’ employees.
References
- Alberghina L, and Gaglio D (2014). Redox control of glutamine utilization in cancer. Cell Death Dis 5, e1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alonso C, Fernandez-Ramos D, Varela-Rey M, Martinez-Arranz I, Navasa N, Van Liempd SM, Lavin JL, Mayo R, Ilisso CP, de Juan VG, et al. (2017). Metabolomic Identification of Subtypes of Nonalcoholic Steatohepatitis. Gastroenterology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ascha MS, Hanouneh IA, Lopez R, Tamimi TA, Feldstein AF, and Zein NN (2010). The incidence and risk factors of hepatocellular carcinoma in patients with nonalcoholic steatohepatitis. Hepatology 51, 1972–1978. [DOI] [PubMed] [Google Scholar]
- Barr J, Caballeria J, Martinez-Arranz I, Dominguez-Diez A, Alonso C, Muntane J, Perez-Cormenzana M, Garcia-Monzon C, Mayo R, Martin-Duce A, et al. (2012). Obesity-dependent metabolic signatures associated with nonalcoholic fatty liver disease progression. J Proteome Res 11, 2521–2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunelli L, Caiola E, Marabese M, Broggini M, and Pastorelli R (2014). Capturing the metabolomic diversity of KRAS mutants in non-small-cell lung cancer cells. Oncotarget 5, 4722–4731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caballero F, Fernandez A, Matias N, Martinez L, Fucho R, Elena M, Caballeria J, Morales A, Fernandez-Checa JC, and García-Ruiz C (2010). Specific contribution of methionine and choline in nutritional nonalcoholic steatohepatitis: impact on mitochondrial S-adenosyl-L-methionine and glutathione. J Biol Chem 285, 18528–18536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canbay A, and Sowa JP (2019). L-Ornithine L-Aspartate (LOLA) as a Novel Approach for Therapy of Non-alcoholic Fatty Liver Disease. Drugs 79, 39–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cano A, Buque X, Martinez-Una M, Aurrekoetxea I, Menor A, Garcia-Rodriguez JL, Lu SC, Martinez-Chantar ML, Mato JM, Ochoa B, et al. (2011). Methionine adenosyltransferase 1A gene deletion disrupts hepatic very low-density lipoprotein assembly in mice. Hepatology 54, 1975–1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassago A, Ferreira AP, Ferreira IM, Fornezari C, Gomes ER, Greene KS, Pereira HM, Garratt RC, Dias SM, and Ambrosio AL (2012). Mitochondrial localization and structure-based phosphate activation mechanism of Glutaminase C with implications for cancer metabolism. Proc Natl Acad Sci U S A 109, 1092–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassidy S, and Syed BA (2016). Nonalcoholic steatohepatitis (NASH) drugs market. Nat Rev Drug Discov 15, 745–746. [DOI] [PubMed] [Google Scholar]
- Cefalu AB, Pirruccello JP, Noto D, Gabriel S, Valenti V, Gupta N, Spina R, Tarugi P, Kathiresan S, and Averna MR (2013). A novel APOB mutation identified by exome sequencing cosegregates with steatosis, liver cancer, and hypocholesterolemia. Arterioscler Thromb Vasc Biol 33, 2021–2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen NC, Yang F, Capecci LM, Gu Z, Schafer AI, Durante W, Yang XF, and Wang H (2010). Regulation of homocysteine metabolism and methylation in human and mouse tissues. FASEB J 24, 2804–2817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole LK, Vance JE, and Vance DE (2012). Phosphatidylcholine biosynthesis and lipoprotein metabolism. Biochim Biophys Acta 1821, 754–761. [DOI] [PubMed] [Google Scholar]
- Dasarathy S, Kasumov T, Edmison JM, Gruca LL, Bennett C, Duenas C, Marczewski S, McCullough AJ, Hanson RW, and Kalhan SC (2009). Glycine and urea kinetics in nonalcoholic steatohepatitis in human: effect of intralipid infusion. Am J Physiol Gastrointest Liver Physiol 257, G567–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Chiara F, Heeboll S, Marrone G, Montoliu C, Hamilton-Dutoit S, Ferrandez A, Andreola F, Rombouts K, Gronbaek H, Felipo V, et al. (2018). Urea cycle dysregulation in non-alcoholic fatty liver disease. J Hepatol 69, 905–915. [DOI] [PubMed] [Google Scholar]
- De Chiara F, Thomsen KL, Habtesion A, Jones H, Davies N, Gracia-Sancho J, Manicardi N, Hall A, Andreola F, Paish HL, et al. (2019). Ammonia Scavenging Prevents Progression of Fibrosis in Experimental Nonalcoholic Fatty Liver Disease. Hepatology. [DOI] [PubMed] [Google Scholar]
- DeLaBarre B, Gross S, Fang C, Gao Y, Jha A, Jiang F, Song JJ, Wei W, and Hurov JB (2011). Full-length human glutaminase in complex with an allosteric inhibitor. Biochemistry 50, 10764–10770. [DOI] [PubMed] [Google Scholar]
- Di Filippo M, Moulin P, Roy P, Samson-Bouma ME, Collardeau-Frachon S, Chebel-Dumont S, Peretti N, Dumortier J, Zoulim F, Fontanges T, et al. (2014). Homozygous MTTP and APOB mutations may lead to hepatic steatosis and fibrosis despite metabolic differences in congenital hypocholesterolemia. J Hepatol 61, 891–902. [DOI] [PubMed] [Google Scholar]
- Diaz-Herrero MM, del Campo JA, Carbonero-Aguilar P, Vega-Perez JM, Iglesias-Guerra F, Perinan I, Minano FJ, Bautista J, and Romero-Gomez M (2014). THDP17 decreases ammonia production through glutaminase inhibition. A new drug for hepatic encephalopathy therapy. PLoS One 9, e109787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du K, Hyun J, Premont RT, Choi SS, Michelotti GA, Swiderska-Syn M, Dalton GD, Thelen E, Rizi BS, Jung Y, et al. (2018). Hedgehog-YAP Signaling Pathway Regulates Glutaminolysis to Control Activation of Hepatic Stellate Cells. Gastroenterology 154, 1465–1479 e1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenberg S, and Olivecrona T (1979). Very low density lipoprotein. Fate of phospholipids, cholesterol, and apolipoprotein C during lipolysis in vitro. J Lipid Res 20, 614–623. [PubMed] [Google Scholar]
- Elgogary A, Xu Q, Poore B, Alt J, Zimmermann SC, Zhao L, Fu J, Chen B, Xia S, Liu Y, et al. (2016). Combination therapy with BPTES nanoparticles and metformin targets the metabolic heterogeneity of pancreatic cancer. Proc Natl Acad Sci U S A 113, E5328–5336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elizalde M, Urtasun R, Azkona M, Latasa MU, Goni S, Garcia-Irigoyen O, Uriarte I, Segura V, Collantes M, Di Scala M, et al. (2014). Splicing regulator SLU7 is essential for maintaining liver homeostasis. J Clin Invest 124, 2909–2920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Embade N, Fernandez-Ramos D, Varela-Rey M, Beraza N, Sini M, Gutierrez de Juan V, Woodhoo A, Martinez-Lopez N, Rodriguez-Iruretagoyena B, Bustamante FJ, et al. (2012). Murine double minute 2 regulates Hu antigen R stability in human liver and colon cancer through NEDDylation. Hepatology 55, 1237–1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faubert B, Vincent EE, Griss T, Samborska B, Izreig S, Svensson RU, Mamer OA, Avizonis D, Shackelford DB, Shaw RJ, et al. (2014). Loss of the tumor suppressor LKB1 promotes metabolic reprogramming of cancer cells via HIF-1alpha. Proc Natl Acad Sci U S A 111, 2554–2559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felig P, Wahren J, Hendler R, and Brundin T (1974). Splanchnic glucose and amino acid metabolism in obesity. J Clin Invest 53, 582–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frades I, Andreasson E, Mato JM, Alexandersson E, Matthiesen R, and Martinez-Chantar ML (2015). Integrative genomic signatures of hepatocellular carcinoma derived from nonalcoholic Fatty liver disease. PLoS One 10, e0124544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita K, Nozaki Y, Wada K, Yoneda M, Fujimoto Y, Fujitake M, Endo H, Takahashi H, Inamori M, Kobayashi N, et al. (2009). Dysfunctional very-low-density lipoprotein synthesis and release is a key factor in nonalcoholic steatohepatitis pathogenesis. Hepatology 50, 772–780. [DOI] [PubMed] [Google Scholar]
- Gao P, Tchernyshyov I, Chang TC, Lee YS, Kita K, Ochi T, Zeller KI, De Marzo AM, Van Eyk JE, Mendell JT, et al. (2009). c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 458, 762–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao X, van der Veen JN, Hermansson M, Ordonez M, Gomez-Munoz A, Vance DE, and Jacobs RL (2015). Decreased lipogenesis in white adipose tissue contributes to the resistance to high fat diet-induced obesity in phosphatidylethanolamine N-methyltransferase-deficient mice. Biochim Biophys Acta 1851, 152–162. [DOI] [PubMed] [Google Scholar]
- Gross MI, Demo SD, Dennison JB, Chen L, Chernov-Rogan T, Goyal B, Janes JR, Laidig GJ, Lewis ER, Li J, et al. (2014). Antitumor activity of the glutaminase inhibitor CB-839 in triple-negative breast cancer. Mol Cancer Ther 13, 890–901. [DOI] [PubMed] [Google Scholar]
- Gutierrez-de-Juan V, Lopez de Davalillo S, Fernandez-Ramos D, Barbier-Torres L, Zubiete-Franco I, Fernandez-Tussy P, Simon J, Lopitz-Otsoa F, de Las Heras J, Iruzubieta P, et al. (2017). A morphological method for ammonia detection in liver. PLoS One 12, e0173914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirschey MD, Shimazu T, Goetzman E, Jing E, Schwer B, Lombard DB, Grueter CA, Harris C, Biddinger S, Ilkayeva OR, et al. (2010). SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature 464, 121–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Q, Stalnecker C, Zhang C, McDermott LA, Iyer P, O’Neill J, Reimer S, Cerione RA, and Katt WP (2018). Characterization of the interactions of potent allosteric inhibitors with glutaminase C, a key enzyme in cancer cell glutamine metabolism. J Biol Chem 293, 3535–3545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyotylainen T, Jerby L, Petaja EM, Mattila I, Jantti S, Auvinen P, Gastaldelli A, Yki-Jarvinen H, Ruppin E, and Oresic M (2016). Genome-scale study reveals reduced metabolic adaptability in patients with non-alcoholic fatty liver disease. Nat Commun 7, 8994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iozzo P, Bucci M, Roivainen A, Nagren K, Jarvisalo MJ, Kiss J, Guiducci L, Fielding B, Naum AG, Borra R, et al. (2010). Fatty acid metabolism in the liver, measured by positron emission tomography, is increased in obese individuals. Gastroenterology 139, 846–856, 856, e841–846. [DOI] [PubMed] [Google Scholar]
- Iruarrizaga-Lejarreta M, Varela-Rey M, Fernandez-Ramos D, Martinez-Arranz I, Delgado TC, Simon J, Juan VG, delaCruz-Villar L, Azkargorta M, Lavin JL, et al. (2017). Role of Aramchol in steatohepatitis and fibrosis in mice. Hepatol Commun 1, 911–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jalan R, Wright G, Davies NA, and Hodges SJ (2007). L-Ornithine phenylacetate (OP): a novel treatment for hyperammonemia and hepatic encephalopathy. Med Hypotheses 69, 1064–1069. [DOI] [PubMed] [Google Scholar]
- Karpe F, Olivecrona T, Olivecrona G, Samra JS, Summers LK, Humphreys SM, and Frayn KN (1998). Lipoprotein lipase transport in plasma: role of muscle and adipose tissues in regulation of plasma lipoprotein lipase concentrations. J Lipid Res 39, 2387–2393. [PubMed] [Google Scholar]
- Koliaki C, Szendroedi J, Kaul K, Jelenik T, Nowotny P, Jankowiak F, Herder C, Carstensen M, Krausch M, Knoefel WT, et al. (2015). Adaptation of hepatic mitochondrial function in humans with non-alcoholic fatty liver is lost in steatohepatitis. Cell Metab 21, 739–746. [DOI] [PubMed] [Google Scholar]
- Kuge O, and Nishijima M (1997). Phosphatidylserine synthase I and II of mammalian cells. Biochim Biophys Acta 1348, 151–156. [DOI] [PubMed] [Google Scholar]
- Kuge O, Saito K, and Nishijima M (1997). Cloning of a Chinese hamster ovary (CHO) cDNA encoding phosphatidylserine synthase (PSS) II, overexpression of which suppresses the phosphatidylserine biosynthetic defect of a PSS I-lacking mutant of CHO-K1 cells. J Biol Chem 272, 19133–19139. [DOI] [PubMed] [Google Scholar]
- Leclercq IA, Lebrun VA, Starkel P, and Horsmans YJ (2007). Intrahepatic insulin resistance in a murine model of steatohepatitis: effect of PPARgamma agonist pioglitazone. Lab Invest 87, 56–65. [DOI] [PubMed] [Google Scholar]
- Li B, Cao Y, Meng G, Qian L, Xu T, Yan C, Luo O, Wang S, Wei J, Ding Y, et al. (2018). Targeting glutaminase 1 attenuates stemness properties in hepatocellular carcinoma by increasing reactive oxygen species and suppressing Wnt/beta-catenin pathway. EBioMedicine. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Wang L, Yan X, Liu Q, Yu C, Wei H, Li Y, Zhang X, He F, and Jiang Y (2011). A proton nuclear magnetic resonance metabonomics approach for biomarker discovery in nonalcoholic fatty liver disease. J Proteome Res 10, 2797–2806. [DOI] [PubMed] [Google Scholar]
- Maddocks OD, Labuschagne CF, Adams PD, and Vousden KH (2016). Serine Metabolism Supports the Methionine Cycle and DNA/RNA Methylation through De Novo ATP Synthesis in Cancer Cells. Mol Cell 61, 210–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maldonado EN, Delgado I, Furland NE, Buque X, Iglesias A, Aveldano MI, Zubiaga A, Fresnedo O, and Ochoa B (2014). The E2F2 transcription factor sustains hepatic glycerophospholipid homeostasis in mice. PLoS One 9, e112620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mates JM, Segura JA, Martin-Rufian M, Campos-Sandoval JA, Alonso FJ, and Marquez J (2013). Glutaminase isoenzymes as key regulators in metabolic and oxidative stress against cancer. Curr Mol Med 13, 514–534. [DOI] [PubMed] [Google Scholar]
- Mato JM, Martinez-Chantar ML, and Lu SC (2013). S-adenosylmethionine metabolism and liver disease. Ann Hepatol 12, 183–189. [PMC free article] [PubMed] [Google Scholar]
- Millar JS, Cromley DA, McCoy MG, Rader DJ, and Billheimer JT (2005). Determining hepatic triglyceride production in mice: comparison of poloxamer 407 with Triton WR-1339. J Lipid Res 46, 2023–2028. [DOI] [PubMed] [Google Scholar]
- Miller RA, Shi Y, Lu W, Pirman DA, Jatkar A, Blatnik M, Wu H, Cardenas C, Wan M, Foskett JK, et al. (2018). Targeting hepatic glutaminase activity to ameliorate hyperglycemia. Nat Med 24, 518–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noga AA, Zhao Y, and Vance DE (2002). An unexpected requirement for phosphatidylethanolamine N-methyltransferase in the secretion of very low density lipoproteins. J Biol Chem 277, 42358–42365. [DOI] [PubMed] [Google Scholar]
- Nossen JO, Rustan AC, Gloppestad SH, Malbakken S, and Drevon CA (1986). Eicosapentaenoic acid inhibits synthesis and secretion of triacylglycerols by cultured rat hepatocytes. Biochim Biophys Acta 879, 56–65. [DOI] [PubMed] [Google Scholar]
- Pitman JL, Bonnet DJ, Curtiss LK, and Gekakis N (2011). Reduced cholesterol and triglycerides in mice with a mutation in Mia2, a liver protein that localizes to ER exit sites. J Lipid Res 52, 1775–1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porteiro B, Fondevila MF, Delgado TC, Iglesias C, Imbernon M, Iruzubieta P, Crespo J, Zabala-Letona A, Ferno J, Gonzalez-Teran B, et al. (2017). Hepatic p63 regulates steatosis via IKKbeta/ER stress. Nat Commun 8, 15111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman A, Smith FP, Luc PT, and Woolley PV (1985). Phase I study and clinical pharmacology of 6-diazo-5-oxo-L-norleucine (DON). Invest New Drugs 3, 369–374. [DOI] [PubMed] [Google Scholar]
- Rahman SM, Qadri I, Janssen RC, and Friedman JE (2009). Fenofibrate and PBA prevent fatty acid-induced loss of adiponectin receptor and pAMPK in human hepatoma cells and in hepatitis C virus-induced steatosis. J Lipid Res 50, 2193–2202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizki G, Arnaboldi L, Gabrielli B, Yan J, Lee GS, Ng RK, Turner SM, Badger TM, Pitas RE, and Maher JJ (2006). Mice fed a lipogenic methionine-choline-deficient diet develop hypermetabolism coincident with hepatic suppression of SCD-1. J Lipid Res 47, 2280–2290. [DOI] [PubMed] [Google Scholar]
- Romestaing C, Piquet MA, Letexier D, Rey B, Mourier A, Servais S, Belouze M, Rouleau V, Dautresme M, Ollivier I, et al. (2008). Mitochondrial adaptations to steatohepatitis induced by a methionine- and choline-deficient diet. Am J Physiol Endocrinol Metab 294, E110–119. [DOI] [PubMed] [Google Scholar]
- Rosca MG, Vazquez EJ, Chen Q, Kerner J, Kern TS, and Hoppel CL (2012). Oxidation of fatty acids is the source of increased mitochondrial reactive oxygen species production in kidney cortical tubules in early diabetes. Diabetes 61, 2074–2083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz JI, and Ochoa B (1997). Quantification in the subnanomolar range of phospholipids and neutral lipids by monodimensional thin-layer chromatography and image analysis. J Lipid Res 38, 1482–1489. [PubMed] [Google Scholar]
- Sahai A, Pan X, Paul R, Malladi P, Kohli R, and Whitington PF (2006). Roles of phosphatidylinositol 3-kinase and osteopontin in steatosis and aminotransferase release by hepatocytes treated with methionine-choline-deficient medium. Am J Physiol Gastrointest Liver Physiol 291, G55–62. [DOI] [PubMed] [Google Scholar]
- Sappington DR, Siegel ER, Hiatt G, Desai A, Penney RB, Jamshidi-Parsian A, Griffin RJ, and Boysen G (2016). Glutamine drives glutathione synthesis and contributes to radiation sensitivity of A549 and H460 lung cancer cell lines. Biochim Biophys Acta 1860, 836–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satapati S, Kucejova B, Duarte JA, Fletcher JA, Reynolds L, Sunny NE, He T, Nair LA, Livingston KA, Fu X, et al. (2015). Mitochondrial metabolism mediates oxidative stress and inflammation in fatty liver. J Clin Invest 125, 4447–4462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sellmann C, Jin CJ, Degen C, De Bandt JP, and Bergheim I (2015). Oral Glutamine Supplementation Protects Female Mice from Nonalcoholic Steatohepatitis. J Nutr 145, 2280–2286. [DOI] [PubMed] [Google Scholar]
- Shukla K, Ferraris DV, Thomas AG, Stathis M, Duvall B, Delahanty G, Alt J, Rais R, Rojas C, Gao P, et al. (2012). Design, synthesis, and pharmacological evaluation of bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl)ethyl sulfide 3 (BPTES) analogs as glutaminase inhibitors. J Med Chem 55, 10551–10563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirtori CR, Pavanello C, and Bertolini S (2014). Microsomal transfer protein (MTP) inhibition-a novel approach to the treatment of homozygous hypercholesterolemia. Ann Med 46, 464–474. [DOI] [PubMed] [Google Scholar]
- Soderberg C, Stal P, Askling J, Glaumann H, Lindberg G, Marmur J, and Hultcrantz R (2010). Decreased survival of subjects with elevated liver function tests during a 28-year follow-up. Hepatology 51, 595–602. [DOI] [PubMed] [Google Scholar]
- Sunny NE, Parks EJ, Browning JD, and Burgess SC (2011). Excessive hepatic mitochondrial TCA cycle and gluconeogenesis in humans with nonalcoholic fatty liver disease. Cell Metab 14, 804–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thangavelu K, Pan CQ, Karlberg T, Balaji G, Uttamchandani M, Suresh V, Schuler H, Low BC, and Sivaraman J (2012). Structural basis for the allosteric inhibitory mechanism of human kidney-type glutaminase (KGA) and its regulation by Raf-Mek-Erk signaling in cancer cell metabolism. Proc Natl Acad Sci U S A 109, 7705–7710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas AG, Rojas C, Tanega C, Shen M, Simeonov A, Boxer MB, Auld DS, Ferraris DV, Tsukamoto T, and Slusher BS (2013). Kinetic characterization of ebselen, chelerythrine and apomorphine as glutaminase inhibitors. Biochem Biophys Res Commun 438, 243–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Wier B, Balk JM, Haenen GR, Giamouridis D, Bakker JA, Bast BC, den Hartog GJ, Koek GH, and Bast A (2013). Elevated citrate levels in non-alcoholic fatty liver disease: the potential of citrate to promote radical production. FEBS Lett 587, 2461–2466. [DOI] [PubMed] [Google Scholar]
- Wang JB, Erickson JW, Fuji R, Ramachandran S, Gao P, Dinavahi R, Wilson KF, Ambrosio AL, Dias SM, Dang CV, et al. (2010). Targeting mitochondrial glutaminase activity inhibits oncogenic transformation. Cancer Cell 18, 207–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf MJ, Adili A, Piotrowitz K, Abdullah Z, Boege Y, Stemmer K, Ringelhan M, Simonavicius N, Egger M, Wohlleber D, et al. (2014). Metabolic activation of intrahepatic CD8+ T cells and NKT cells causes nonalcoholic steatohepatitis and liver cancer via cross-talk with hepatocytes. Cancer Cell 26, 549–564. [DOI] [PubMed] [Google Scholar]
- Younossi ZM, Koenig AB, Abdelatif D, Fazel Y, Henry L, and Wymer M (2016). Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 64, 73–84. [DOI] [PubMed] [Google Scholar]
- Yu D, Shi X, Meng G, Chen J, Yan C, Jiang Y, Wei J, and Ding Y (2015). Kidney-type glutaminase (GLS1) is a biomarker for pathologic diagnosis and prognosis of hepatocellular carcinoma. Oncotarget 6, 7619–7631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan L, Sheng X, Clark LH, Zhang L, Guo H, Jones HM, Willson AK, Gehrig PA, Zhou C, and Bae-Jump VL (2016). Glutaminase inhibitor compound 968 inhibits cell proliferation and sensitizes paclitaxel in ovarian cancer. Am J Transl Res 8, 4265–4277. [PMC free article] [PubMed] [Google Scholar]
- Yuneva MO, Fan TW, Allen TD, Higashi RM, Ferraris DV, Tsukamoto T, Mates JM, Alonso FJ, Wang C, Seo Y, et al. (2012). The metabolic profile of tumors depends on both the responsible genetic lesion and tissue type. Cell Metab 15, 157–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Su B, Jacobs RL, Kennedy B, Francis GA, Waddington E, Brosnan JT, Vance JE, and Vance DE (2009). Lack of phosphatidylethanolamine N-methyltransferase alters plasma VLDL phospholipids and attenuates atherosclerosis in mice. Arterioscler Thromb Vasc Biol 29, 1349–1355. [DOI] [PubMed] [Google Scholar]
- Zubiete-Franco I, Fernandez-Tussy P, Barbier-Torres L, Simon J, Fernandez-Ramos D, Lopitz-Otsoa F, Gutierrez-de Juan V, de Davalillo SL, Duce AM, Iruzubieta P, et al. (2017). Deregulated neddylation in liver fibrosis. Hepatology 65, 694–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate any unique datasets or code.