

Abstract
Cortical layer 1 (L1) interneurons have been proposed as a hub for attentional modulation of underlying cortex, but the transformations that this circuit implements are not known. We combined genetically targeted voltage imaging with optogenetic activation and silencing to study the mechanisms underlying sensory processing in mouse barrel cortex L1. Whisker stimuli evoked precisely timed single spikes in L1 interneurons, followed by strong lateral inhibition. A mild aversive stimulus activated cholinergic inputs and evoked a bimodal distribution of spiking responses in L1. A simple conductance-based model that only contained lateral inhibition within L1 recapitulated the sensory responses and the winner-takes-all cholinergic responses, and correctly predicted that the network would function as a spatial and temporal high-pass filter for excitatory inputs. Our results demonstrate that all-optical electrophysiology can reveal basic principles of neural circuit function in vivo and suggest an intuitive picture for how L1 transforms sensory and modulatory inputs.
Keywords: Layer 1, barrel cortex, lateral inhibition, acetylcholine, optical electrophysiology, E/I balance
Graphical Abstract
In brief
By simultaneously combining genetically targeted voltage imaging with optogenetic modulation of neuronal activity, Fan et al. demonstrate that all-optical electrophysiology in awake animals can be a powerful tool for revealing hidden principles of neural circuit function.
Introduction
The brain receives myriad sensory inputs. It must distinguish the relevant from the irrelevant. An input can merit attention either through its intrinsic properties (novelty, salience) or through learned associations. The sparse interneurons of neocortical Layer 1 (L1) have been hypothesized as a hub for integrating these factors and modulating the underlying cortex to gate sensory processing (Jiang et al., 2013; Letzkus et al., 2011; Takesian et al., 2018). L1 interneurons receive thalamic (Cruikshank et al., 2012; Takesian et al., 2018; Zhu, 2004), cortico-cortical (Lee et al., 2013; Palmer et al., 2012), and neuromodulatory (cholinergic (Letzkus et al., 2011; Poorthuis et al., 2018; Takesian et al., 2018), serotonergic (Lee et al., 2010; Poorthuis et al., 2018) and adrenergic (Lam and Sherman, 2019)) inputs. The primary outputs of L1 appear to be via inhibition of deeper-lying interneurons and thereby dis-inhibition of pyramidal neurons (Jiang et al., 2013; Lee et al., 2013; Letzkus et al., 2011; Takesian et al., 2018), and via inhibition of apical dendrites of deeper layer pyramidal cells (Abs et al., 2018; Lee et al., 2015; Takesian et al., 2018).
Electrophysiological, genetic, and anatomical studies have identified four sub-types of interneurons within L1 (Schuman et al., 2018). These cells fall into two broad classes (Jiang et al., 2015). Laterally projecting neurogliaform cells primarily synapse within L1 and show late-spiking non-adapting firing pattern in acute slices (Cadwell et al., 2016; Chu et al., 2003; Cruikshank et al., 2012; Schuman et al., 2018). Downward projecting single bouquet-like cells primarily dis-inhibit underlying pyramidal neurons and show a non-late spiking adapting firing pattern (Jiang et al., 2013; Lee et al., 2013; Letzkus et al., 2011; Takesian et al., 2018). It is not known how these properties combine in awake animals to transform L1 inputs to outputs. Indeed, the function of L1 has been called a “crowning mystery” (Schuman et al., 2018).
L1 is unique among cortical layers for having purely inhibitory short-range connectivity. In mutually inhibitory networks, competition between concurrently activated units can lead to winner-takes-all or point-attractor dynamics (Koyama et al., 2016). This motif has been proposed as a general means to implement multiple forced-choice calculations, i.e. in which small disparities in excitatory inputs are amplified to select one among multiple possible firing modes (Machens et al., 2005). Considering the proposed role of L1 in gating sensory processing—where multiple inputs compete for attention—it is interesting to ask what kinds of dynamics arise in this circuit in vivo.
Electrophysiological studies in L1 in vivo have been challenging due to the sparseness of neuronal cell bodies. While a few whole-cell patch clamp recordings have been performed in anesthetized rats (Egger et al., 2015; Jiang et al., 2013; Zhu, 2004), technical difficulties prevented similar acquisitions in awake animals. Recent advances in genetically encoded voltage indicators (GEVIs) enabled voltage imaging with single-neuron, single-spike resolution in vivo in near-surface neurons (Abdelfattah et al., 2019; Adam et al., 2019; Piatkevich et al., 2019; Villette et al., 2019). Paired expression of near infrared GEVIs based on Archaerhodopsin 3 (Arch) (Kralj et al., 2012) with blue-excited channelrhodopsin actuators enabled optical measurements of excitability in vivo (Optopatch) (Adam et al., 2019; Lou et al., 2016) and of synaptic transmission in primary culture and acute slices (Fan et al., 2018). These advances open the possibility for optical explorations of L1 circuit function in awake animals.
Using a holographic structured illumination imaging system and a recently developed soma-localized GEVI derived from Arch, SomArchon (Piatkevich et al., 2019), we probed the circuit function of L1 interneurons in barrel cortex of awake mice. We developed an all-optical technique to resolve the separate contributions of excitatory and inhibitory synaptic inputs to membrane potential. Experiments with patterned optogenetic activation and silencing, paired with voltage imaging, revealed that strong lateral inhibition within the L1 microcircuit plays a dominant role in setting the network responses to sensory and neuromodulatory inputs. Mutual inhibition led to high-pass temporal and spatial filtering of excitatory inputs, while under strong cholinergic drive the circuit showed winner-takes-all attractor dynamics.
A simple conductance-based model that incorporates the known electrophysiology and connectivity of L1 interneurons captured the main features of our sensory and neuromodulatory data and predicted responses to several types of inputs which we tested experimentally. The model also predicted that the sensitivity of the L1 microcircuit to thalamic inputs should show an inverted-U dependence on cholinergic drive, a prediction which can be tested in future experiments.
Results
In vivo voltage imaging with holographic patterned illumination
We analyzed the sources of signal and noise in optical voltage recordings with the goal to achieve millivolt-sensitivity recordings of subthreshold dynamics in vivo. Voltage signals in tissue arise solely from the neuronal membrane. In epifluorescence images, the soma perimeter appeared brighter than the center, a geometrical projection effect from viewing membranes edgeon. We thus reasoned that incident photons would most efficiently produce signal if targeted to the soma perimeter. Confocal-like excitation combined with spatially filtered emission also minimized optical crosstalk from out-of-focus cells.
We built a holographic structured illumination system, similar to Ref. (Lutz et al., 2008), to achieve this precisely targeted illumination with red (λ = 639 nm) light for excitation of SomArchon (Fig. 1A, Table S1, Methods and Methods S1). SomArchon fluorescence from all holographically targeted spots was recorded simultaneously on a scientific CMOS camera. All recordings were performed at a 1 kHz frame rate. Spatial filters were applied digitally in postprocessing to separate signal from background (Methods). A digital micromirror device (DMD) patterned blue illumination for targeted optogenetic stimulation (Fig. 1A, Table S1, Methods S1).
Figure 1. All-optical electrophysiology in vivo.

(A) Optical system for holographic structured illumination voltage imaging (red) and micromirror-patterned optogenetic stimulation (blue). Details in Methods. (B) Comparison of wide-field epifluorescence, soma-targeted, and membrane-targeted holographic illumination of the same field of view containing a SomArchon-expressing cortical neuron. Arrow indicates location of cell in wide-field image. Scale bar 20 μm. (C) Left: Signal-to-background ratio for wide-field and membrane-targeted imaging modalities (n = 8 cells). Right: SNR of action potentials with soma-wide and membrane-targeted holographic illumination (n = 10 cells). (D) Voltage imaging in cortical neurons at different depths with different promoters in awake mice. Grey bands indicate simultaneously recorded cells. Right: magnified views. Traces corrected for photobleaching but not otherwise filtered. (E) SNR of action potentials at different depths (error bars: mean ± s.d.; n = 7 – 22 cells per depth). (F) Spike-triggered autocorrelogram showing refractory period (n = 27 cells). (G) Simultaneous optogenetic stimulation and voltage imaging in L1 interneurons in awake mice expressing Optopatch4. Groups of 1 – 3 cells were stimulated with patterned blue light (1.8 to 21 mW/mm2). Bottom: Spike raster (n = 23 cells, 3 mice). (H) Top: Three targeted cells, with red and blue illumination overlaid. Scale bar 20 μm. Bottom: Fluorescence traces from the three cells in response to blue illumination. (I) Simultaneous optogenetic inhibition and voltage imaging (i-Optopatch). Cells co-expressed stGtACR2 and SomArchon. Groups of 1 – 3 cells were inhibited with patterned blue light (1.8 to 21 mW/mm2). Bottom: Spike raster (n = 14 cells, 2 mice). (J) Magnified view showing hyperpolarization and silencing upon blue light onset. (K) Left: Mean fluorescence response to blue illumination (1.8 mW/mm2). Right: Blue light stimulation significantly decreased the spontaneous firing rate (n = 14 cells, 2 mice, 1.8 mW/mm2).
We characterized the performance of the imaging system in cortical L1 and superficial L2/3 in vivo (Methods). Under wide-field red illumination, SomArchon-expressing neurons were not visible due to high background from scattered and out-of-focus light (Fig. 1B). Illumination targeted to the somas revealed individual cells (Fig. 1B, Methods S1). Holographic membrane focal illumination significantly improved the signal-to-noise ratio of optically detected spikes (spike amplitude:baseline noise in a 1 kHz bandwidth 11.4 ± 1.4 membrane focal vs. 7.5 ± 1.3 whole soma, mean ± s.e.m., paired measurements in n = 10 cells, matched laser power per cell between illumination patterns; Fig. 1C).
A challenge with 1-photon voltage imaging is to ensure that the fluorescence ascribed to each cell is not contaminated by crosstalk from other simultaneously imaged cells or from out-of-focus background. To characterize crosstalk between in-focus cells, we quantified the fluorescence signal (spike amplitude) in concentric rings centered on individual holographically illuminated cells (Fig. S1). At 6 μm from the cell boundary, signal amplitude decayed to 7 ± 6% of the on-cell signal (mean ± s.d., n = 4 cells), and was undetectable (< 1% of on-cell signal) at 20 μm from the boundary.
To characterize out-of-focus crosstalk, we recorded from individual cells as a function of optical defocus. With membrane focal illumination, fluorescence amplitude decayed by 50% over 15 μm deeper defocus, but more slowly with shallower defocus, whereas with whole-soma illumination this fluorescence decay was ~two-fold slower (Fig. S1).
We used activity-based image segmentation to remove residual out-of-focus crosstalk. A penalized matrix decomposition algorithm separated true voltage signals from crosstalk based on the different spatial profiles of these two signal sources (Buchanan et al., 2018). Application of the algorithm to simulated data with realistic noise (Adam et al., 2019), and to composite movies where optical crosstalk was inserted ‘by hand’ confirmed that the extracted signals were highly robust to crosstalk (Fig. S1, Methods).
Finally, we used simultaneous patch clamp and fluorescence measurements in acute slices to assess the precision of the optical voltage measurements. Optical and electrical recordings showed close correspondence, both for spikes and subthreshold events (Fig. S1).
The holographic optical system enabled high-resolution recordings in several genetically defined cell types across L1 and superficial L2/3 in awake head-fixed mice (Fig. 1D). 5HT3AR-Cre transgenic mice expressed primarily in L1 interneurons (Lee et al., 2010; Takesian et al., 2018); somatostatin (SST)-Cre transgenic mice expressed in interneurons in deeper layers (Neske et al., 2015); and CKII(0.4)-Cre virus drove dense pan-neuronal expression in wild-type mice. In all three populations, two-photon fluorescence images of an appended eGFP tag showed membrane-localized and somatically restricted expression (Fig. S1).
Action potentials were recorded with SNR 12 ± 4 (mean ± s.d., n = 16 cells) throughout L1 at depths between 20 and 150 μm at a mean laser power of 3 mW/cell. In SST-Cre mice optical recordings were made in superficial L2/3 (100 – 230 μm depth) with SNR 9 ± 4 (mean ± s.d., n = 6 cells). Fig. 1D shows a recording at depth of ~230 μm with SNR 7. SNR was approximately two-fold greater for near-surface neurons (< 100 μm depth, 14 ± 4, mean ± s.d., n = 22 neurons) than at 150 – 200 μm (8 ± 3, mean ± s.d., n = 7 neurons, Fig. 1E). We imaged multiple neurons simultaneously to a depth of 150 μm (Fig. 1D).
For an SNR of 7 and a spike-detection threshold set at 4σ above the baseline noise, the expected false-positive rate is < 0.3 spikes per 10 s recording, and the expected false-negative rate (missed spikes) is < 0.14% of true spikes (Methods). We used the refractory period after true spikes to test the false-positive rate. A spike-triggered autocorrelogram showed a probability < 10−3 of two spikes occurring within 5 ms of each other (n = 27 cells, Fig. 1F), confirming the physiological origin of the detected spikes.
Simultaneous optogenetic perturbations and voltage imaging: Optopatch and i-Optopatch
Next, we sought to combine optogenetic perturbations (activation and silencing) with simultaneous voltage imaging. For optogenetic activation, we paired SomArchon with a blue light-activated soma-localized channelrhodopsin, SomCheRiff (Adam et al., 2019; Fan et al., 2018; Hochbaum et al., 2014). This channelrhodopsin has previously been shown to have negligible activation at the 639 nm wavelength used to excite Arch-derived GEVIs (Hochbaum et al., 2014). For co-expression in vivo, we made a Cre-dependent bicistronic AAV construct which we called Optopatch4.
In awake head-fixed 5-HT3AR-Cre mice expressing Optopatch4, single-cell targeted optogenetic stimulation through a cortical window (I = 1.8 - 21 mW/mm2) evoked sustained spiking which was clearly resolved via holographically targeted voltage imaging (Fig. 1G, H). Measurements using laterally offset spots showed little crosstalk of stimulation to surrounding cells, which had a mean spacing of ~60 μm (Meyer et al., 2013) (Fig. S2).
Optogenetic silencing can be a powerful tool to determine the roles of specific neural populations in network dynamics, but optogenetic silencing has not previously been paired with voltage imaging. The action spectrum of a soma-localized blue-shifted chloride channel, stGtACR2 (Mahn et al., 2018) suggested it could be spectrally orthogonal to SomArchon. In human embryonic kidney (HEK293) cells expressing stGtACR2, blue light (I = 0.2 – 1 mW/mm2) evoked large inhibitory photocurrents (~0.5 nA), but red light at intensities used for voltage imaging (635 nm, 0.1 – 10 W/mm2) did not induce observable photocurrents, nor did it impair blue light-mediated channel gating (Fig. S3). Thus SomArchon and stGtACR2 constitute a spectrally orthogonal pair appropriate for inhibitory Optopatch (i-Optopatch).
In head-fixed awake mice expressing i-Optopatch in L1 (Methods), targeted optogenetic stimuli (I = 1.8 - 21 mW/mm2) inhibited spontaneous spiking (spontaneous spike rate 3.2 ± 0.7 Hz vs. 0.4 ± 0.3 Hz with blue light at I = 1.8 mW/mm2, p = 3 × 10−3, two-sided paired-sample t-test, Fig. 1I, K). Optogenetic stimulation also hyperpolarized resting membrane potential by - 13.5 ± 4% of spike height (mean ± s.e.m., n = 14 neurons, I = 1.8 mW/mm2, 2 mice, Fig. 1J, K). These results establish i-Optopatch as a tool for assessing how targeted optogenetic silencing affects network dynamics.
Whisker stimulation evoked excitation followed by inhibition in L1
We used this suite of tools to dissect the circuit function of L1 during sensory processing (Fig. 2A, B). We first characterized the intrinsic excitability properties of L1 neurons. In acute slices from 5HT3AR-Cre mice expressing Optopatch4, single-cell L1-targeted optogenetic stimuli evoked characteristic firing patterns, including bursting adapting and late-spiking non-adapting phenotypes, as previously reported (Chu et al., 2003), though not all neurons had a clear classification (Fig. S4). Neurons recorded in vivo sequentially under isoflurane anesthesia and then wakefulness showed characteristic firing patterns preserved between the two brain states (Fig. S4). The in vivo recordings did not clearly resolve into distinct sub-classes based on firing patterns, consistent with prior results (Cadwell et al., 2016; Chu et al., 2003). We therefore treated all 5-HT3AR-positive neurons measured in vivo as a single population.
Figure 2. Sensory-evoked responses in L1 neurons in vivo.

(A) 5-HT3AR-positive interneurons in barrel cortex L1 receive thalamocortical (sensory) and neuromodulatory inputs. (B) Simultaneous sensory stimulation and voltage imaging in L1. (C) Fluorescence transients in single L1 interneurons evoked by whisker stimuli (20 ms deflections at 0.5 Hz) in isoflurane anesthetized mice. Left, top: example fluorescence traces. Horizontal stripes indicate simultaneously recorded cells. Bottom: spike raster (n = 18 neurons, 3 mice). Right: Fluorescence waveforms from the boxed region at left. (D) Same as C but in awake mice. (E) Top: Distribution of delays between stimulus onset and peak of evoked spike (n = 24 cells, 135 trials). Bottom: Distribution of relative delays in sensory-evoked action potential peaks between simultaneously recorded pairs of cells (n = 14 pairs, 66 trials). (F) Spike-triggered average waveform of spontaneous (top, n = 17 neurons) and whisker stimulus-evoked (bottom, n = 24 neurons) action potentials. (G) Same as F but in an awake mouse. Spontaneous: n = 22 neurons. Evoked: n = 21 neurons, 3 mice. Stimulus-evoked spikes had smaller after-spike hyperpolarization under anesthesia than under wakefulness (anesthetized: 11 ± 2% spike height, n = 24 neurons, 3 mice vs. awake: 17 ± 1% of spike height, n = 21 neurons, 3 mice, p = 0.02, two-tailed t-test), consistent with a more depolarized resting potential under wakefulness (Constantinople and Bruno, 2011).(H) Sensory stimulation induced a period of reduced spontaneous activity in awake mice.
Next, we used voltage imaging to characterize the sensory-evoked responses in L1 interneurons. Barrel fields corresponding to individual whiskers (B2, C2, D2) were identified by intrinsic imaging (Fig. S5, Methods). In mice anesthetized with isoflurane and in awake mice, brief stimuli to individual whiskers (~1 mm deflection, ~8 mm from the base, 20 ms duration, repeated at 0.5 Hz, Methods) elicited excitatory post-synaptic potentials (EPSPs) and often single spikes in L1 neurons in the corresponding barrel fields (Fig. 2C, D). Whisker stimuli were substantially more effective in eliciting spikes in anesthetized mice than in awake mice (anesthetized: 135 spikes from 153 trials, n = 24 cells vs. awake: 73 spikes from 126 trials, n = 24 cells), consistent with prior studies in other cortical layers (Haider et al., 2013).
The delay from stimulus onset to spike peak was 16 ± 2 ms in anesthetized mice (mean ± s.d., n = 135 events, 24 neurons, 3 mice, Fig. 2E) and 16 ± 3 ms in awake mice (mean ± s.d., n = 73 events, 21 neurons, 3 mice, Fig. S5). Similar delay and jitter were previously reported in L4 pyramidal and fast-spiking neurons, both of which receive direct thalamic inputs (Gabernet et al., 2005). To assess how much of the jitter reflected variation in input timing vs. variation in the intrinsic responses of individual L1 neurons, we then compared the relative delay in spike time between simultaneously recorded pairs of neurons in anesthetized mice. The mean relative delay was 0.9 ms (n = 14 pairs, 66 trials in which both cells spiked, Fig. 2E). Remarkably, despite the small delay, the order of firing was preserved in all pairs in all trials, to within our 1 ms time resolution, i.e. if cell A fired before cell B in a single trial, then cell A fired before cell B in all trials. These observations indicate that abrupt sensory inputs drove millisecond-precision spike sequences in L1.
We observed striking differences in the subthreshold dynamics between spontaneous vs. whisker-evoked spikes, in both anesthetized and awake animals. Spike-triggered average (STA) waveforms of spontaneous spikes rode atop a baseline depolarization that both preceded and followed the spike, whereas whisker-evoked spikes arose abruptly and were followed by a period of hyperpolarization (Fig. 2F, G). Stimulus-triggered average waveforms of whisker deflection trials that did not induce spikes also showed a depolarization followed by a hyperpolarization (Fig. S5). Together, these results implied that whisker stimulus evoked synaptic excitation followed by synaptic inhibition.
Consistent with this hypothesis, we found that in awake animals, whisker stimulation evoked single spikes and then transiently suppressed spontaneous L1 activity (before stimulus firing rate: 3.6 ± 0.85 Hz; 30 to 105 ms post-stimulus: 1.2 ± 0.65 Hz, mean ± s.e.m., n = 24 cells p = 0.004, Kruskal-Wallis test, Fig. 2H). Sensory-evoked network suppression was particularly apparent when the sensory stimulus fortuitously arrived during a burst of spontaneous activity (Fig. 2H).
Sensory-evoked network suppression also affected subsequent sensory-evoked responses. When a whisker was subjected to a series of 20 ms deflections at 10 Hz in an anesthetized mouse, the response to the second stimulus was undetectable, while the mean responses to the third and fourth stimuli were ~75% of the response to the first stimulus (Fig. S5). While this observation might be explained by effects upstream of L1, it is also consistent with rapid sensory-evoked inhibition onto L1.
Optical dissection of sensory-evoked excitation and inhibition
We developed an all-optical technique to resolve the distinct contributions of excitatory and inhibitory synaptic inputs to sensory-evoked responses. Rapid inhibition is mediated by GABAA receptors, ligand-gated chloride channels with a reversal potential of ~−70 mV. L1 interneurons in anesthetized rats have been reported to rest at −65 to −70 mV (Zhu, 2004), suggesting that inhibitory inputs should have only small effects on membrane potential at rest. Borrowing from well-established patch clamp protocols (Segal and Barker, 1984), we reasoned that optogenetic depolarization would increase the driving force for inward chloride current, and thereby amplify the impact of GABAA receptor activation on the inhibitory postsynaptic potential (IPSP) (Fig. 3A,B).
Figure 3. Optical dissection of excitation and inhibition in L1 interneurons in awake mice.

(A) Whisker stimuli and single cell-targeted optogenetic stimuli were paired in 5HT3AR-Cre mice expressing Optopatch4. (B) Conductance-based model of subthreshold membrane potential. This simple model only contained passive conductances, with gating by light (Channelrhodopsin, ChR), glutamate (AMPAR), and GABA (GABAR). A leak conductance set the resting potential. (C) Three recordings from a single neuron showing response to (top) whisker stimulus, (middle) targeted optogenetic stimulus, and (bottom) simultaneous optogenetic and whisker stimuli. Arrows show whisker stimulus-evoked inhibition. (D) Mean spike rate evoked by whisker stimuli atop different levels of targeted optogenetic depolarization. In the absence of optogenetic stimulation, whisker stimuli evoked precisely timed single spikes. In the presence of optogenetic stimulation, whisker stimuli suppressed spiking. The suppression decreased in amplitude and duration as the strength of the optogenetic stimulus increased, a consequence of ChR shunting. Shading represents s.e.m. from n = 27 neurons, 4 mice. (E) Mean whisker stimulus-evoked subthreshold waveforms at different levels of optogenetic drive. Spikes were digitally removed prior to averaging (Methods). (F) Simulated membrane voltage waveforms under different levels of optogenetic drive, using the model shown in B. Excitation was assumed to lead inhibition by 2 ms (Methods). (G) Comparison of simulated (red) and measured (points) PSP amplitude as a function of optogenetic stimulus strength. (H) Repetitive measurements of whisker stimulus-evoked responses in anesthetized mice, with and without baseline optogenetic stimulation. Top: example recordings. Bottom: spike raster (n = 21 neurons, 3 mice). (I) Stimulus-triggered mean fluorescence responses without (black) and with (red) baseline optogenetic stimulation. Traces were aligned vertically to their peak. Inset: Magnified view showing dynamics near the peak.
In both awake and anesthetized mice, whisker stimuli in the absence of optogenetic stimulation evoked clear spikes or EPSPs in L1 interneurons, as in prior experiments (Fig. 3C, S5). Optogenetic stimuli targeted to single cells (500 ms duration, 1.8 to 21 mW/mm2, repeated at 1 Hz) reliably evoked stimulus intensity-dependent spiking. Remarkably, whisker stimuli applied during single-cell optogenetic stimulation hyperpolarized membrane potential and suppressed spiking (Fig. 3C, D, E, S5). We quantified the sensory-evoked subthreshold waveforms by digitally removing spikes (Methods) and calculating a stimulus-triggered average at different optogenetic stimulus strengths (Fig. 3E, S5). In both awake and anesthetized brain states, whisker stimuli had opposite effects in the absence vs. presence of baseline optogenetic depolarization, illustrating dramatic non-additivity of sensory and optogenetic inputs to the same neuron. This observation mimicked the prior observation that sensory stimuli evoked single spikes in quiescent neurons but suppressed ongoing bursts in active neurons (Fig. 2H).
We developed a simple biophysical model to test our interpretation that single cell-targeted optogenetic stimulation amplified the effect of network inhibition. We assumed a transient excitatory synaptic input followed shortly by a transient inhibitory input. With only passive conductances (leak, channelrhodopsin, AMPA receptor and GABAA receptor), the model captured the main features of the subthreshold dynamics (Fig. 3F, G, S5, Methods), including several subtleties. In the anesthetized state, as the strength of the optogenetic drive increased, the sensory-evoked IPSP amplitude first increased—as explained above—but then decreased (IPSP amplitude 29 ± 5% of spike height at Iblue = 5.8 mW/mm2, vs. 16 ± 4% of spike height at Iblue = 21 mW/mm2, mean ± s.e.m., n = 15 neurons, 3 mice, p = 0.001, two-sided paired-sample t-test, Fig. S5). The model revealed that this decrease was due to shunting of the membrane potential toward the CheRiff reversal potential (~0 mV) at high CheRiff conductance (Methods). This simple model thus connected the complex context-dependent whisker-evoked responses in L1 interneurons to basic membrane biophysics.
To constrain the possible sources of network inhibition, we next studied the relative timing of excitatory and inhibitory inputs. We delivered whisker stimuli alternately with and without baseline weak optogenetic stimulation targeted to single neurons (5.8 mW/mm2, Fig. 3H). In trials that evoked spikes, stimulus-triggered average waveforms +/− blue light overlapped for the first 2 ms after onset of whisker-evoked depolarization. Thereafter, the waveform in the presence of optogenetic stimulation fell below the waveform in the absence, signaling the onset of inhibition (Fig. 3I, inset). This finding implied a ~2 ms delay between onset of excitation and inhibition, suggesting at most a difference of one synapse in the respective paths (Gabernet et al., 2005). Slower inhibitory signals (e.g. from GABAB receptors or polysynaptic mechanisms) may also have contributed to inhibition at later times.
Center/surround optogenetic stimulation reveals lateral inhibition in L1
We sought to identify the source of the sensory-evoked inhibition. Patch clamp measurements in acute slices identified inhibitory connections between L1 interneurons (Chu et al., 2003; Cruikshank et al., 2012; Schuman et al., 2018). We hypothesized that whisker stimulation evoked near-synchronous spiking in L1, which then led to network inhibition via lateral connections within the L1 population. To test whether L1 activation was sufficient to evoke lateral network inhibition we performed an all-optical measurement of functional connectivity in vivo (Fig. 4A).
Figure 4. Center/surround optogenetic stimulation reveals lateral inhibition in barrel cortex L1.

(A) Simple model of L1 circuit with lateral inhibition. Tonic optogenetic depolarization increased the driving force for inhibitory currents in the central neuron. Pulsed optogenetic stimulation of the surrounding neurons (blue) evoked lateral inhibition, revealed by voltage imaging (red). (B) Configuration of stimulation and imaging spots to probe lateral inhibition in L1 of 5-HT3AR-Cre mice. (C) Epifluorescence images showing the illumination patterns in vivo. (D) Fluorescence waveforms from the central neurons under center/surround optogenetic stimulation. (E) Spike raster (n = 25 neurons, 3 mice). (F) Mean spike rate during central stimulation, before and after surround stimulation, n = 25 neurons, 3 mice. Shading represents s.e.m. (G) Mean subthreshold voltage in an optogenetically depolarized central neuron during, before and after surround stimulation. The initial voltage increase was due to scattered light from the surround stimulus (Fig. S2). Shading represents s.e.m.
We expressed Optopatch4 in 5-HT3AR-Cre mice and targeted voltage imaging to 1-3 L1 interneurons in the center of the field of view. We then defined two optogenetic stimulus patterns. The first pattern comprised small disks targeted individually to the central neurons, with sustained optogenetic depolarization (500 ms, 25 mW/mm2) to increase the driving force for inhibitory currents. The second pattern comprised an annulus (inner diameter ~200 μm, outer diameter of ~400 μm, Fig. 4B, C, Methods), surrounding the central neurons. Midway through the stimulation of the central neurons, a brief flash (20 ms, 25 mW/mm2) was applied to the annulus to evoke synchronized spiking of the surrounding cells.
Optogenetic stimulation of the central neurons evoked robust spiking (41 ± 6 Hz, n = 25 neurons, 3 mice, mean ± s.e.m.). Stimulation of the surrounding neurons transiently suppressed this spiking (spike rate 12 ± 4 Hz in the 25 ms following the annular flash, p = 4 × 10−4, two-sided paired-sample t-test, Fig. 4D, E, F). The mean fluorescence waveform following the annular flash showed robust hyperpolarization of the central neurons (−27 ± 3% of spike height, Fig. 4G). (Control experiments without the central optogenetic stimulus revealed that the initial depolarization after the annular flash was an artifact from light scatter, Fig. S2). The spike patterns and subthreshold hyperpolarization dynamics in these experiments closely resembled the corresponding data for a sensory stimulus (Fig. 3E, F, I). We observed similar results in neuronderived neurotrophic factor (Ndnf)-Cre mice (Poorthuis et al., 2018; Tasic et al., 2016) which drove Optopatch4 expression selectively in neurogliaform and canopy cells whose axons project laterally within L1 (Schuman et al., 2018) (Fig. S6). Together, these results established that synchronous activation of laterally projecting L1 neurons was sufficient to elicit rapid lateral inhibition within the L1 microcircuit.
Patterned optogenetic inhibition reveals that sensory stimulation evokes lateral inhibition
The existence of a lateral inhibitory connection within L1 did not prove that this connection was responsible for the observed sensory-evoked inhibition. To address this question, we tested how optogenetic inhibition of neurons in an annular region modulated sensory-evoked responses in a centrally located neuron (Fig. 5A, B).
Figure 5. Optogenetic silencing reveals that L1 network activity is necessary for sensory-evoked lateral inhibition.

(A,B) Sensory-evoked responses in centrally located neurons were recorded (red) either without or with optogenetic silencing of surrounding L1 neurons (blue). (C, D) Targeted gene expression. Cre-on SomArchon-EGFP expressed in all 5-HT3AR-Cre+ neurons. Cre-on-flp-off stGtACR2-CFP and low-titer Flpo virus were combined, so stGtACR2-CFP expressed in Cre+/Flp− neurons. (D) Left: schematic of gene expression patterns. Colors indicate constructs in (C). Right: Composite two-photon fluorescence image of GFP fluorescence from SomArchon-EGFP and CFP fluorescence from stGtACR2-CFP. Voltage imaging was performed only in neurons that did not express stGtACR2. (E) Paired recordings in awake mice of sensory-evoked responses from individual centrally located neurons, either without (red) or with (blue) inhibition of surrounding neurons. Asterisks indicate recordings where sensory stimuli evoked delayed spiking when lateral inhibition was suppressed. (F) Whisker stimulus-triggered average membrane potential without (red) or with (blue) surround inhibition (n = 13 neurons, 2 mice, shading represents s.e.m.).
We expressed Cre-on-SomArchon-EGFP and Cre-on-flp-off-stGtACR2-CFP in 5-HT3AR-Cre mice (Fig. 5C, Methods). To prevent scattered blue light from inducing spurious hyperpolarization of the measured cells, we used low-titer Flpo virus to turn off expression of stGtACR2 in a sparse subset of L1 neurons. We used 2-photon imaging of the appended fluorescent tags (GFP and CFP) to identify neurons that expressed SomArchon and not stGtACR2 (Fig. 5D). These cells were targeted for voltage imaging in awake mice. We then illuminated a surrounding annulus with blue light (inner diameter ~200 μm, outer diameter ~400 μm) to inhibit the surrounding cells while delivering whisker stimuli.
Whisker stimuli in the absence of surround optogenetic inhibition evoked excitation followed by inhibition (−12 ± 4% of spike height, mean ± s.e.m., n = 13 neurons, 2 mice, Fig. 5E,F), recapitulating earlier experiments performed with a different reporter construct (Fig. 2G). The same cells were then recorded during whisker stimulation with surround optogenetic inhibition (500 ms, 5.8 mW/mm2, Fig. 5E,F right). The post-stimulus hyperpolarization was replaced by a post-stimulus depolarization (+16 ± 5% of spike height, Fig. 5F). Some cells that showed prompt single spikes in response to whisker stimulation alone showed delayed and sustained spiking when lateral inhibition was suppressed. Together, these results established that sensory stimulation evoked lateral inhibition between L1 interneurons and that this lateral inhibition contributed to precise timing of whisker stimulus-evoked spikes.
Cholinergic activation induces diverse neuronal responses
Cholinergic modulation of L1 interneurons can enhance cortical L2/3 responses to sensory inputs (Letzkus et al., 2011). As a first step toward understanding the integration of modulatory and sensory inputs in L1, we studied the response of barrel cortex L1 neurons to a stimulus designed to activate cholinergic inputs (Fig. 6A, B). An air puff to the face excites cholinergic neurons in basal forebrain (Hangya et al., 2015), and these neurons are known to innervate cortical L1 (Eggermann et al., 2014; Letzkus et al., 2011; Mechawar et al., 2000).
Figure 6. Cholinergic inputs drive heterogeneous excitatory responses in L1 interneurons.

(A) Neuromodulatory inputs excite L1 interneurons. L1 interneuron activation leads to mutual inhibition. (B) Experimental configuration. Optopatch4 measurements were performed in barrel cortex L1 interneurons of awake 5-HT3AR-Cre mice while a mild air puff was applied to the ipsilateral eye. (C) Air puffs evoked an increase in spike rate which was blocked by an a4 nAChR blocker, DHβE. Top: paired measurements on the same cells before and after drug administration. Bottom: spike raster before and after drug. (D) Quantification of effects of air puff before and after DHβE. (E) Mean subthreshold responses to air puff before and after administration of DHβE. (F) DHβE significantly reduced the amplitude of the subthreshold response to air puff. (G) Top: Fluorescence recordings from single cells showing responses to air puff stimulation in the absence and presence of baseline optogenetic stimulation. Bottom: Spike raster (n = 21 neurons, 4 mice). (H) Mean spike rate during air puff stimulation without and with baseline optogenetic depolarization (n = 21 neurons, 4 mice). (I) Mean subthreshold response to air puff without and with baseline optogenetic stimulation. Spikes were digitally removed before averaging.
We imaged L1 neurons in awake mice while delivering a mild air puff (100 ms duration, ~5 psi) to the ipsilateral eye (to avoid incidental whisker stimulation, Fig. 6B). The air puff induced a significant increase in mean spike rate (from 2 ± 1 Hz to 8 ± 4 Hz, 50 to 200 ms after stimulus onset, p = 0.048, n = 15 neurons, 3 mice, paired two-sample t-test) and evoked a mean depolarization that grew over ~100 ms and lasted for ~1600 ms (Fig. 6C).
The a4 nicotinic acetylcholine receptor is highly expressed in L1 interneurons (Arroyo et al., 2012; Takesian et al., 2018), so we hypothesized that this receptor mediated the neuromodulatory response. We made paired recordings of the same L1 interneurons before and after systemic administration of the a4 nAChR blocker dihydro-β-erythroidine hydrobromide (DHβE, 1.5 mg/kg i.p., Fig. 6C). The AD50 for this drug in mice has been reported to range from 0.45 to 1.7 mg/kg (Damaj et al., 1995), implying partial block at the dose used (higher doses are toxic (Damaj et al., 1995)). DHβE did not significantly affect the spontaneous spike rate (before drug: 2 ± 1 Hz, after drug 3 ± 3 Hz, p = 0.53, two-sided paired-sample t-test, n = 15 neurons, 3 mice, Fig. 6D), but the drug abolished the air puff-induced increase in spike rate (3 ± 3 Hz before air puff to 3 ± 2 Hz after air-puff, p = 0.79; Fig. 6D), and significantly decreased the air puff-induced subthreshold depolarization from 22 ± 4% of spike height to 10 ± 3% (p = 0.02, two-sided paired-sample t-test, Fig. 6E,F). Thus a4-mediated cholinergic excitation was largely responsible for the air puff-induced responses, though we cannot rule out other contributions to these responses.
The single-cell traces showed striking variability, so we explored in detail the responses of individual cells. In 15 of 21 L1 interneurons, the air puff evoked a clear depolarization. In 6 of these neurons the air puff evoked one or more spikes and in 3 of these neurons, the air puff evoked a barrage of firing that lasted ~1 s, strikingly different from the precisely timed single spikes evoked by whisker stimulation (Fig. 6G). In pairs of simultaneously recorded neurons, we often observed distinct air puff-induced responses in the two cells, indicating that the variability in response was intrinsic to the L1 circuit and was not due to trial-to-trial variation (Fig. S7).
To explore how cholinergic activation affected synaptic inputs, we paired the air puff with single cell-targeted optogenetic depolarization. When the air puff was applied in the middle of an optogenetic stimulus (500 ms, 5.8 mW/mm2), there was no significant change in mean spike rate (Fig. 6H), though some individual neurons showed dramatic increases or decreases in spike rate (see e.g. Fig. 6G, second and fourth traces). During baseline optogenetic stimulation, the air puff on average depolarized the membrane potential (Fig. 6I), opposite to the hyperpolarizing transient evoked by a whisker stimulus under comparable conditions (e.g. Fig. 3C). However, individual neurons sometimes showed strong air puff-induced hyperpolarization (Fig. S7). There are many possible mechanisms underlying the heterogeneous air puff responses. Below we suggest a simple mechanism by which this heterogeneity could arise in a mutually inhibitory network.
Network model of L1 dynamics
Due to the strong mutual inhibition between L1 neurons, one cannot intuitively predict the network responses to excitatory inputs. We developed a simple numerical model of the L1 microcircuit that explained much of our data and predicted network responses to some new inputs (Fig. 7A, Methods S2). The model contained elongated neurogliaform cells (eNGCs) with mutually inhibitory connections, and downward-projecting single-bouquet cells (SBCs) that received inhibitory inputs from eNGC neurons but did not feed back into the simulated network. Both cell types received thalamic and neuromodulatory inputs (Cruikshank et al., 2012).
Figure 7. Numerical simulations capture input-output properties of L1 microcircuit.

(A) Model of L1 function. Thalamic, cholinergic, and optogenetic inputs are modulated by lateral inhibition. (B) Conductance-based models for two subtypes of L1 interneurons mimic the intrinsic firing patterns. (C) Simulation of L1 network with network-wide thalamic excitation and single-cell targeted optogenetic stimulation. The traces show two simulations in which the optogenetic stimulation and voltage measurement were targeted either to an eNGC cell or to an SBC-like cell. Compare to Figs. 3C, S5. (D) Simulation of L1 microcircuit response to patterned optogenetic stimulation. Localized optogenetic stimulation induced sustained firing while global optogenetic stimulation induced firing in all neurons only at the onset of the stimulation. (E) Population-average firing rates in response to local or global stimulation. (F) Population-average membrane potential in response to local or global stimulation. (G-I) Experimental test of the predictions in D – F with patterned optogenetic stimulation. For the local stimulation, spot sizes were 30 μm diameter. For global stimulation, spot sizes were 400 μm in diameter. (J) Simulated responses to cholinergic inputs showing winner-takes-all dynamics. (K) Experimental responses to air puffs (same data as in Fig. 6G). The air puff responses closely matched the simulated winner-takes-all dynamics.
Conductance-based point-neuron simulations approximately recapitulated the firing properties of the two major cell classes in L1 (Fig. 7B, Methods, Table S2) (Wang and Buzsáki, 1996). We used published anatomical and patch clamp data to set the soma positions, synaptic weights, and thalamocortical input strength (Chu et al., 2003; Jiang et al., 2015; Schuman et al., 2018) (Methods S2, Table S2). Our simulations comprised 51 neurons (34 eNGCs, 17 SBCs), approximately matching the number of interneurons above a single whisker barrel (Meyer et al., 2013).
We simulated the L1 network with thalamic excitation and patterned optogenetic stimulation. Simulated thalamic inputs alone evoked precisely timed single spikes followed by a period of hyperpolarization and suppressed spontaneous activity (Fig. S7), recapitulating the results of Fig. 2. Simulations containing single-cell targeted optogenetic stimulation amplified the effect of network inhibition in the targeted cell, closely matching the experimental results in Fig. 3 (Fig. 7C). We also reproduced the center/surround excitation experiments from Fig. 4 (Fig. S7).
Simulations predicted that localized excitation would evoke sustained spiking, while wide-area excitation would lead to strong firing rate adaptation due to lateral inhibition (Fig. 7D, E, F). Experiments in anesthetized mice confirmed that optogenetic stimulation of individual cells led to sustained spiking (Fig. 7G, H, I). Optogenetic stimulation of a 400 μm wide spot elicited a prompt spike in nearly all neurons (17 of 20 cells), followed by a complete suppression of spiking in nearly all neurons (18 of 20 cells) during the subsequent 20 ms (Fig. 7G, H). Most neurons remained silent during the subsequent ~100 ms, though a few (4 of 20 cells) resumed tonic spiking (Fig. 7G). The sub-threshold potentials also showed a clear difference between localized and distributed optogenetic excitation (Fig. 7I). Localized excitation led to a sustained depolarization, whereas distributed excitation led to a peak in potential followed by a ~100 ms inhibition (Fig. 7I).
These results demonstrate the input-output transformations of L1. The circuit acts as a temporal high-pass filter for spatially distributed inputs. This filtering manifests in the precisely timed single spikes from whisker stimulation. The circuit also acts as a spatial high-pass filter for temporally sustained inputs, i.e. spatially localized inputs can evoke sustained local firing, but spatially distributed inputs cannot.
We modeled cholinergic input as activation of an excitatory conductance in all L1 interneurons, with rise time of 300 ms and duration ~1 s. To our surprise, sustained spatially distributed excitation of all cells led to winner-takes-all spiking patterns above a critical excitation strength. Some cells spiked a little faster than their neighbors, suppressing spiking of the neighbors and amplifying the disparity in spike rates. For cholinergic inputs above the critical strength, the simulated single-cell firing traces (Fig. 7J) fell into two classes (sustained spiking vs. transient depolarization or single spikes), closely resembling the bimodal single-cell cholinergic responses we observed in response to an air puff (Fig. 7K). A similar bimodal distribution of cholinergic responses had previously been noted in L1 interneurons in auditory cortex in response to a tail shock (Letzkus et al., 2011), but had not been explained. We emphasize that the bimodal responses in the model are an emergent property of a homogeneous mutually inhibitory network; not the result of hard-wired single-cell properties or network connections. These simulation results suggest that strong cholinergic input to L1 can temporarily lock the circuit into a winner-takes-all firing pattern.
Discussion
By combining soma-localized voltage indicators with holographic structured illumination microscopy we achieved high-sensitivity genetically targeted recordings of membrane voltage in cortical L1 and superficial L2/3 in awake mice. We introduced optogenetic techniques for resolving excitatory from inhibitory contributions to subthreshold voltage and for mapping functional connectivity in vivo. We developed tools for simultaneous optogenetic inhibition and voltage imaging (i-Optopatch) to reveal the roles of specific neural populations in network function.
We applied these tools to study the input-output properties of L1. Whisker deflections evoked precisely timed single spikes in L1 interneurons, followed by periods of hyperpolarization, reduced spontaneous activity and reduced response to sensory inputs. Optogenetic depolarization of individual cells revealed that whisker stimuli evoked excitation, followed by strong network inhibition. Optogenetic stimulation of neighboring L1 neurons evoked similar patterns of network inhibition. Optogenetic silencing of neighboring L1 neurons abolished sensory-evoked network inhibition. Together these experiments showed that lateral inhibition within L1 plays a critical role in determining the network response to sensory inputs. Our results in L1 are reminiscent of similar results showing sensory-evoked concurrent excitation and inhibition in pyramidal cells in L4 (Gabernet et al., 2005; Wehr and Zador, 2003) and in L2/3 (Petersen et al., 2003; Sachdev et al., 2004).
Our results further showed that neuromodulatory excitation drove tonic spiking in a subset of cells, and drove single spikes or purely subthreshold depolarizations in other cells. While the origin of this heterogeneity is not known, simulations suggested that this heterogeneity could arise in a homogeneous mutually inhibitory network via winner-takes-all dynamics. However, alternate explanations are not ruled out. The heterogeneous cholinergic responses may reflect variations in acetylcholine receptor expression between L1 neuronal subtypes (Schuman et al., 2018), or specific features of the wiring. A definitive test will require population-level voltage imaging in defined subtypes of L1 interneurons, combined with precisely controlled cholinergic activation. This type of experiment is likely to be technically feasible in the near future.
The convergence of sensory and cholinergic inputs onto L1 interneurons raises the question of how these modalities interact. Our model predicts that weak neuromodulatory excitation sensitizes the network to thalamic inputs by bringing all cells are closer to threshold; but strong neuromodulatory excitation transiently locks the network into a symmetry-broken winner-takes-all state comprising strongly excited and strongly inhibited neurons. In this state, responses to thalamic inputs are suppressed. Thus the sensitivity of the L1 network to sensory inputs is predicted to show an inverted-U dependence on cholinergic drive.
Considering the importance of L1 in modulating sensory processing, we speculate that the L1 microcircuit may contribute to the inverted-U dependence of performance on arousal observed in many sensory processing tasks (McGinley et al., 2015), often called the Yerkes-Dodson law (Yerkes and Dodson, 1908). We further speculate that rapid firing in a subset of L1 interneurons under strong cholinergic drive may be important for L1-mediated plasticity (Abs et al., 2018; Doron et al., 2019). A clear goal for future work will be to test these predictions by studying the interaction of sensory and modulatory inputs in each subclass of L1 interneurons.
Future applications of the tools described here will benefit from voltage imaging deeper in the brain. Due to light scattering, the present 1-photon imaging approach faces a tradeoff between the density of signal sources and the imaging depth, with a maximum cortical depth for sparse cells of ~230 mm. Several approaches to circumventing this tradeoff are conceivable, such as adaptive optics (Ji, 2017), graded-index (GRIN) lens endoscopes (Jung et al., 2004) or prism-based imaging approaches (Andermann et al., 2013). Efforts are also underway to develop two-photon voltage imaging in vivo (Villette et al., 2019), though improvements in molecular indicators and optics are likely necessary. Two-photon targeted optogenetic stimulation has recently been paired with calcium imaging in vivo (Emiliani et al., 2015; Forli et al., 2018; Mardinly et al., 2018; Marshel et al., 2019; Packer et al., 2015; Rickgauer et al., 2014). Combination of two-photon stimulation with voltage imaging may enable fine-grained circuit dissection in vivo.
The all-optical tools introduced here could be used to dissect circuit mechanisms in many contexts. For instance, interactions between excitation and inhibition are critical in network models of reinforcement learning (Schultz et al., 1997) and predictive coding (Rao and Ballard, 1999) but many aspects of these models have not been tested. The ability to silence or activate defined sub-populations while imaging voltage can reveal otherwise hidden inhibition, and elucidate the necessity and sufficiency of specific connections for network function.
STAR Methods
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Adam Cohen (cohen@chemistry.harvard.edu). Plasmids generated in this study and their sequences are available from Addgene.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines
HEK293T cell line were bought from ATCC and incubated in DMEM supplemented with 10% FBS and penicillin/streptomycin in a 37 °C incubator under 5% CO2.
Mice
All procedures involving animals were in accordance with the National Institutes of Health Guide for the care and use of laboratory animals and were approved by the Institutional Animal Care and Use Committee at Harvard University.
For slice experiments, P16-P28 5HT3AR-Cre+/− mice were used.
For in vivo experiments, 35-60-day-old wild-type C57BL/6, 5HT3AR-Cre+/−, SST-Cre+/−, and NDNF-Cre+/− mice were used. All mice were housed in standard conditions (reverse 12-hour light/dark cycles, with water and food ad libitum). Mice of both sexes were used.
METHOD DETAILS
Design of Optopatch4
Optopatch4 construct (SomArchon-eGFP-P2A-somCheRiff) was cloned into an AAV vector with Cre-dependent expression driven by the hSyn promoter.
LZF1735 pAAV_hSyn-DiO-SomArchon-eGFP-P2A-somCheRiff (Optopatch4, Addgene #126512)
For expression of SomArchon alone we used: pAAV_CAG-FLEX-SomArchon-eGFP (Addgene #126943)
High-titer AAV2/9 virus with Optopatch4 (1.74×1013 GC/mL, LZF1735) was obtained from the Janelia Farm Vector Core. High-titer AAV2 virus with SomArchon (6.30×1012 GC/mL) was obtained from the UNC Vector Core. High-titer AAV9 virus with CKII(0.4)-Cre (2.8× 1013 GC/mL) was obtained from UPenn Vector Core.
Design of i-Optopatch
i-Optopatch constructs include:
LZF1826 pAAV_hSyn-DiO-SomArchon-eGFP-P2A-stGTACR2 (Addgene #135412)
LZF1827 pAAV_hSyn-Flp-off-Cre-on-stGtACR2-CFP (Addgene #135413)
pAAV_hSyn1-SIO-stGtACR2-FusionRed was a gift from Ofer Yizhar (Addgene plasmid # 105677). stGtACR2 cDNA segments were generated from the template of pAAV_hSyn1-SIO-stGtACR2-FusionRed (Addgene #105677).
High-titer AAV2/9 virus with LZF1826 (2.30×1013 GC/mL) and LZF1827 (2.50×1013 GC/mL) were obtained from the Janelia Farm Vector Core. pAAV-EF1a-Flpo was a gift from Karl Deisseroth (Addgene plasmid #55637). High-titer AAV5 virus with EF1a-Flpo (2.6×1012 GC/mL) was obtained from the UNC Vector Core.
Optical system for holographically targeted voltage imaging and patterned optogenetic stimulation
The optical system combined a red laser (λ = 639 nm) path for holographic targeted illumination voltage imaging, a blue laser (λ = 488 nm) path for micromirror-patterned optogenetic stimulation, a two-photon (2P) path for structural imaging, and a wide-field epifluorescence imaging path.
Red laser path.
A red laser (CNI Inc., MRL-FN-639, λ = 639 nm, 700 mW single transverse mode) was coupled into the setup via a photonic crystal polarization maintaining fiber (NKT Photonics, LMA-PM-15). The fiber output was collimated with an f = 100 mm focal length lens (Thorlabs, AC254-100-A-ML) to form a beam with approximately ~10 mm diameter. The polarization of the beam was set with a zero-order half-wave plate. The beam was directed onto a holographic reflection-mode liquid crystal spatial light modulator (SLM, Meadowlark 1920SLM VIS) with a resolution of 1920 x 1152 pixels. Zero-order diffraction was blocked by a home-made anti-pinhole comprised of a dot of solder on a glass slide, placed in a plane conjugate to the sample image plane. The SLM was re-imaged onto the back-focal plane of the objective via a series of relay optics. The objective lens was a 25× water immersion objective, numerical aperture 1.05 (Olympus XLPLN25XWMP2). A mechanical shutter blocked the red laser between data acquisitions. A series of OD filters were placed after the red laser for modulating intensity.
In the first generation of the setup, we used a variable focal length camera lens (Sigma macro 18-200 mm) to control the magnification of the SLM at the back focal plane of the objective. Demagnifying the SLM decreased the effective numerical aperture of the illumination at the sample, leading to bigger spots in the sample, but also to a larger region that could be targeted with red light. In the second-generation system, we used a fixed lens after the SLM to minimize aberrations. All relay lenses are specified in Table S1.
The SLM device was controlled by custom software. A user specified a set of lines for the SLM to target by drawing on a wide-field epifluorescence image or a 2P fluorescence image. These lines were discretized into a set of spots. The SLM phased pattern was calculated using the Gerchberg-Saxton algorithm.
Red laser intensity was ~ 3 mW per cell for in vivo imaging, ~ 1 mW per cell for acute slice imaging.
Blue laser path.
A blue laser (Cobolt, 06-01 series, λ = 488 nm, 60 mW) was modulated in intensity via an acoustooptic tunable filter (AOTF; Gooch and Housego TF525-250-6-3-GH18A). The beam was focused into a singlemode optical fiber. The output was collimated with an f = 60 mm focal length lens (Thorlabs, AC254-060-A-ML) to form a beam with approximately a ~17 mm diameter. The beam was then sent to a digital micromirror device with a resolution of 1024 x 768 pixels (DMD, Vialux, V-7001 VIS). The patterned blue beam was combined with the patterned red beam via a dichroic mirror. The DMD was re-imaged onto the sample at a magnification such that one DMD pixel corresponded to 0.62 μm in the sample plane. The DMD optical system enabled patterned blue light stimulation across a field of view of ~450 x ~520 μm.
The DMD was controlled by custom software. For excitability measurement, a pixel bitmap was preloaded onto and projected from the DMD. For lateral inhibition experiments, pixel bitmaps were loaded into the on-board RAM and digital clock pulses triggered the DMD to sequence through the pre-defined set of exposure patterns.
Wide-field fluorescence imaging path.
The image was relayed from the objective to the camera via a series of three lenses. The final image formation step was performed by a 4x objective (Olympus XLFLUOR 4X/340) serving the role of the tube lens. Fluorescence was collected on a scientific CMOS camera (Hamamatsu ORCA-Flash 4.0). The final magnification of the optical system was 16.7, corresponding to 0.39 μm in the sample plane per camera pixel.
Fluorescence from the sample was separated from the blue and red excitation beams via a dichroic mirror (Di03-R405/488/561/635-t3-40x55). An emission filter (Semrock 635 nm long-pass, BLP01-635R-25) further separated SomArchon fluorescence from scattered excitation light. An IR-blocking emission filter (Semrock, FF01-842/SP-25) was placed for blocking scattered infrared excitation light.
All movies are acquired at 1 kHz. To image at 1 kHz, the camera region of interest (ROI) was restricted to typically 200 rows, centered on the image-sensor midline.
The imaging system was designed for a magnification lower than the nominal 25x of the objective for two reasons. First, lower magnification increased the number of neurons that could be imaged simultaneously onto the limited detector area accessible at 1 kHz. Second, by concentrating sample photons onto as few camera pixels as possible, we sought to minimize the contribution from camera electronic noise, so that all signals would be in the shot noise-limited regime.
Two-photon imaging path.
Light from a femtosecond tunable pulsed infrared laser (Spectra Physics DeepSee) was sent to a pair of galvo mirrors (Cambridge Technologies 6215H). The galvos were re-imaged onto the back focal plane of the objective via an optimized scan lens (Thorlabs, SL50-CLS2) and tube lens (Thorlabs, TL200-CLS2). The visible (blue and red) and near-infrared beams were combined using a 785 nm long-pass dichroic mirror (Semrock, Di03-R785-t3-40x55). GFP fluorescence was directed to the 2P detection path via a removable 550 nm long-pass dichroic. Scattered excitation light was blocked by a 550 nm long-pass emission filter, an IR-blocking emission filter (BSP01-785R-25) and a band-pass emission filter (FF03-525/50-25). A pair of lenses (focal lengths 75 mm and 16 mm) re-imaged the back-aperture of the objective onto a photomultiplier tube (Hamamatsu, H11706P-40). The output of the photomultiplier was amplified and low pass filtered through an amplifier unit (Hamamatsu C7319) and then digitized.
For i-Optopatch experiments, GFP fluorescence was excited by 950 nm and selected by a band-pass emission filter (Chroma, ET525/36m). CFP fluorescence was excited by 900 nm and selected by a band-pass emission filter (FF01-475/28-25).
Control software.
The entire setup was controlled by custom software written in LabView. Interfacing was via a National Instruments DAQ (NI PCIe-6363).
The software contained routines for registration of the DMD, SLM, 2P microscope coordinates to the camera via affine transformations. The camera served as the global reference coordinate system.
Experimental protocols were specified by a set of images (to the SLM and the DMD), output waveforms (to the galvos, the AOTF, the shutters, the update clock on the DMD, the piezo whisker stimulator, and the air puff controller), and analog input streams (from the PMT, the camera exposure clock, and a patch clamp electrophysiology setup not used in the present work).
The Hamamatsu camera uses an internal 100 kHz clock to synchronize image row readout. We found that when the camera exposure times were triggered by the DAQ in synchronous mode, the camera rounded the exposure time to the nearest 10 μs, leading to 1% jitter in exposure time for 1 kHz imaging. To address this noise source, we used a custom firmware upgrade to access the 100 kHz camera clock. This clock became the master clock for the DAQ system, and all analog and digital input/output functions were synchronized to the camera clock.
Imaging in acute slices
All procedures involving animals were in accordance with the National Institutes of Health Guide for the care and use of laboratory animals and were approved by the Institutional Animal Care and Use Committee at Harvard University.
Virus injection for acute slice measurements.
Virus comprising AAV2/9 hSyn-Dio-SomArchon-eGFP-P2A-somCheRiff (1.74×1013 GC/mL) was diluted in PBS and injected at a final titer of ~2×1012 GC/mL.
5HT3AR-Cre+/− mice were crossed with wild-type C57BL/6 mice. Pups were cryo-anesthetized at P0-P2 and immobilized dorsal side up under a stereotaxic microscope. Injections were made using home-pulled micropipettes (Sutter P1000 pipette puller), mounted in a microinjection pump (World Precision Instruments Nanoliter 2010) controlled by a microsyringe pump controller (World Precision Instruments Micro4). The micropipette was positioned using a stereotaxic instrument (Stoelting Digital Mouse Stereotaxic Instrument). Pups were injected in the left hemisphere, 1 mm lateral and 1.2 mm anterior to lambda. Starting at a depth of 0.3 mm beneath the surface of the skull, virus injections (40 nL, 1 nL/s) were performed at 0.1 mm increments as the pipette was withdrawn. Pups were placed back in their home cage once they were awake.
Genotyping.
Genotyping for 5HT3AR-Cre, SST-Cre and NDNF-Cre was performed with the PCR primer pairs: Cre 5': 5' TAT CTC ACG TAC TGA CGG TG 3' and Cre 3': 5' AGA CTA ATC GCC ATC TTC CAG C 3' to yield a 500 bp band from Cre.
Acute slice preparation.
Acute brain slices were prepared from P16–P28 5HT3AR-Cre+/− mice. The mice were anesthetized by isoflurane and then perfused with carbogen (95% O2, 5% CO2)-saturated ice-cold slicing solution with the following composition (in mM): 110 choline chloride, 2.5 KCl, 1.25 NaH2PO4, 25 NaHCO3, 25 glucose, 0.5 CaCl2, 7 MgCl2, 11.6 Na-ascorbate, and 3.1 Na-pyruvate. Mice were then decapitated and the brains were rapidly coronally sliced with 300 μm thickness on a vibratome (Leica VT 1200S).
Slices were incubated for 45 min at 34 °C in a carbogenated artificial CSF (ACSF) with the following composition (in mM): 127 NaCl, 2.5 KCl, 1.25 NaH2PO4, 25 NaHCO3, 25 glucose, 2 CaCl2, and 1 MgCl2. The osmolarity of all solutions was adjusted to 300–310 mOsm and the pH was maintained at 7.3 under constant bubbling with carbogen.
Imaging acute slices.
Measurements were conducted in ACSF at 23 °C under ambient atmosphere. The slice was immobilized in a slice recording chamber using a slice anchor (Warner Instruments, SHD-40/2). ACSF, perfused with carbogen, was flowed through the chamber at a rate of 2 mL/minute.
Cranial windows and virus injections
Cranial window surgery and virus injection for imaging barrel cortex L1.
For Optopatch experiments, virus comprising AAV2/9 hSyn-Dio-SomArchon-eGFP-P2A-somCheRiff (1.74×1013 GC/mL) was diluted in PBS and injected at a final titer of ~3×1012 GC/mL.
For i-Optopatch experiments (Fig. 1I-K and Fig. 5), virus comprised AAV2/9 hSyn-Flp-off-Cre-on-stGtACR2-CFP (final concentration ~1×1012 GC/mL) mixed with AAV2 CAG-FLEX-SomArchon-eGFP (final concentration ~6.3×1011 GC/mL) and AAV5 EF1a-Flpo (final concentration ~2×1011 GC/mL).
The procedure for surgery and imaging in barrel cortex L1 followed the protocol from Andermann (Goldey et al., 2014). 35-60-day-old heterozygous 5HT3AR-Cre mice (male and female) were deeply anesthetized with 2% isoflurane and maintained with ~1% isoflurane throughout the surgery. Eyes were kept moist using ophthalmic eye ointment. Body temperature was continuously monitored and maintained at 37 °C using a heating pad (WPI, ATC2000). The skull was exposed and thoroughly dried and a 3 mm round craniotomy (3.3 – 3.4 mm lateral, 1.6 mm caudal of bregma) was opened using a biopsy punch (Miltex). Virus was then injected in 4 - 8 locations in the center of the craniotomy. Starting at a depth of 0.2 mm beneath the surface of the dura, virus injections (60 nL, 30 - 60 nL/min) were performed at 0.1 mm increments as the pipette was withdrawn. Brain surface was kept moist with saline throughout the injection.
A window was prepared prior to the surgery and comprised two 3 mm round #1 cover glasses and one 5 mm round #1 cover glass (Harvard apparatus) cured together with UV curable adhesive (Norland Products, NOA 81). Following the virus injection, the window was then placed covering the barrel cortex and cemented to the skull with dental cement (C&B metabond, Parkell, No. 242-3200). After the window cured, a titanium headplate (similar to the design in Ref. (Goldey et al., 2014)) was glued around the window and any exposed skull was covered with dental cement. Animals were returned to their home cage for recovery and treated for 3 days with Carprofen (5 mg/kg) and Buprenorphine (0.1 mg/kg) twice a day. To avoid damage to the implant, mice were housed in separate cages.
Cranial window surgery and virus injection for L1-3 imaging (Fig. 1D).
The procedure for surgery and imaging in visual cortical L1-3 followed the same protocol as above, except that 35-60-day-old wild-type C57BL/6 mice or heterozygous SST-Cre mice (male and female) were used. The coordinates for the 3 mm round craniotomy were 2.4 mm lateral and 2.7 mm caudal of bregma. Virus was then injected in 4 - 8 locations in the center of the craniotomy. Starting at a depth of 0.3 mm beneath the surface of the dura, virus injections (60 nL, 30 - 60 nL/min) were performed at 0.2 mm increments as the pipette was withdrawn.
For wild-type mice, injected virus comprised AAV2 CAG-FLEX-SomArchon-eGFP (final titer ~0.5×1012 GC/mL) mixed with CKII(0.4)-Cre virus (UPenn vector core, final titer ~1×10n GC/mL).
For SST-Cre mice, injected virus comprised AAV2 CAG-FLEX-SomArchon-eGFP (final titer ~0.5×1012 GC/mL).
Tracking, whisker stimulation, intrinsic imaging and in vivo voltage imaging
For general behavior monitoring during acquisitions we recorded video of the mouse’s face. An IR LED light (850 nm) was placed in front of the animal. A PointGrey camera (GS3-U3-51S5M-C, Mono Grasshopper3 USB 3.0 Camera) with a Fuji lens (Fuji Photo Optical 1:1.4/25 Fujinon-TV Camera Lens) and an IR-passing optical filter (Thorlabs, FB850-40) was placed on the side to track the animal’s face (whisker motion and eye blinks) during data acquisitions.
Whisker stimulation.
An individual whisker was selected for stimulation (typically B2, C2 or D2). Other whiskers were trimmed to prevent direct contact with the glass pipette. The whisker was inserted into a glass pipette glued to a piezoelectric actuator. The actuator was connected to an amplifier (Krohn-Hite 7602M) and controlled by the DAQ. A spike was classified as ‘evoked’ if it occurred within 30 ms of onset of whisker stimulus.
Intrinsic imaging.
Intrinsic imaging was performed on the same setup as described above. A 4x objective (Olympus XLFLUOR 4X/340) was used to image the entire 3 mm cranial window. The whisker stimulation was 10 Hz for 4 s with a 16 s interstimulus interval. A red LED (625 nm) illuminated the window surface from the side. Reflected light was imaged onto the camera at 10 Hz frame rate. A decrease in reflectance from the brain indicated the barrel, which could be localized relative to the blood vessel pattern as visualized with 488 nm illumination.
Imaging anesthetized animals.
Imaging started 3 weeks post-surgery. Mice were lightly anesthetized (0.7–1% isoflurane), head-fixed under the upright microscope using the titanium head plate and held in a body tube. Eyes were kept moist using ophthalmic eye ointment. Body temperature was continuously monitored and maintained at 37 °C using a heating pad (WPI, ATC2000). A typical imaging session lasted 1–2 hours, and then animals quickly recovered and returned to their home cage. Recordings targeting L1 neurons in vivo were performed at a depth < 150 μm in both anesthetized and awake animals. Recordings typically targeted 1 – 4 neurons simultaneously.
Habituation and imaging awake animals.
Habituation started 2 weeks post-surgery. Each animal was acclimated to the head restraint in a body tube for at least 3 days before starting the imaging sessions. For imaging in awake animals, a 3D-printed paw blocker was placed in front of the forepaws to prevent them from pushing away the glass pipette for whisker stimulation.
Air puff.
The timing of the air puff was controlled by a solenoid valve (WPI). The strength of the air puff was set ~ 5 psi and controlled by a pressure regulator (Festo, pressure regulator LRP-1/4-4). The air puff was delivered through a blunt needle at ~ 5 mm from the eye ipsilateral to the brain hemisphere used for voltage imaging (to avoid spurious whisker stimulation arriving in the imaged barrels). Air pressure and needle position were adjusted to achieve an air puff strength just strong enough to evoke an eye blink response and increase the pupil diameter.
DHβE administration.
To modulate cholinergic signaling, dihydro-β-erythroidine hydrobromide (DHβE, Tocris; 2349) was diluted in saline. The drug was administered systemically (1.5 mg/kg i.p.). Optopatch and air puff measurements were performed before, and then 30 min. after drug administration, on the same sets of cells. The experiment was performed on each animal twice, on successive days.
Center/surround optogenetic stimulation
For lateral inhibition experiments, we defined two optogenetic stimulus patterns. “Central masks” covered individually the cell bodies of 1 – 3 neurons at the center of the field of view. We calculated the geometrical centers of these masks individually. The mean of these centers was set as the coordinates for the center for the surrounding “annulus mask”.
The annulus inner radius was set to be ~100 –m from the most non-centered central mask. This distance was selected to minimize the impact of scattered light from the annulus mask. The outer radius of the annulus mask was set as the largest value at which the annulus would be contained within the FOV. Typical outer radii were ~200 μm.
The image sequence was composed of three composite masks: (1) central masks only; (2) central mask and annulus mask together; (3) annulus mask only. These masks were preloaded into the on-board RAM and digital clock pulses triggered the DMD to sequence through the pre-defined set of exposure patterns.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data were analyzed with homemade code written in MATLAB.
Corrections for photobleaching and motion artifacts.
Movies were first corrected for motion using the NoRMCorre algorithm (Pnevmatikakis and Giovannucci, 2017). Movies were then corrected for photobleaching by dividing the movie by an exponential fit of the mean fluorescence.
Image segmentation and waveform extraction.
We divided the movie into sub-movies comprising single cells and performed activity-based image segmentation separately in each sub-movie. Whereas correlations often arose between subthreshold voltages and out-of-focus background, we assumed that spiking was not correlated with background, and furthermore that the spatial footprint associated with spiking would be the same as for true subthreshold dynamics. For segmentation purposes, we first removed subthreshold signals via a 100 Hz high-pass filter. Movies were then segmented semi-automatically using one of two activity-based segmentation algorithms. In one approach, principal components analysis was followed by time-domain independent components analysis (PCA/ICA) (Mukamel et al., 2009). The spatial masks from PCA/ICA were then applied to the original movies without high-pass filtering to extract fluorescence traces.
In a second approach, high-pass filtered movies were segmented via penalized matrix decomposition nonnegative matrix factorization algorithm (PMD-NMF) (Buchanan et al., 2018). The spatial masks were applied to the original movies without high-pass filtering to extract initial fluorescence traces. An estimate of the background signal was calculated from pixels not on the targeted cells. Signals from the cellular footprints were calculated with the constraint that the background spatial profile should be maximally smooth. Both approaches to image segmentation gave similar results, a consequence of the sparsity of L1 interneurons and the optical sectioning from tightly focused holographic excitation.
Validation of the PMD-NMF signal extraction algorithm.
The algorithm has been extensively tested on simulated data comprising realistic shot-noise limited signals from individual cells, correlations in sub-threshold voltages between cells, and correlated out-of-focus background (See Extended Data Fig. 6 of Ref (Adam et al., 2019)). To test the algorithm with data from cortical L1 interneurons, we took independent single-cell recordings and extracted the fluorescence dynamics from each. We made composite movies by adding two movies together, with ~50% overlap between cell bodies. We extracted the signals from the composite movies, and compared to the ground truth data extracted from the individual movies. This test is more stringent than any real-world data because in-focus cells never physically overlapped in real data.
Removing scattering background for lateral inhibition measurements.
Flashes of blue light delivered in an annular pattern induced tissue autofluorescence which bled into the voltage recordings from centrally located neurons. To correct for this crosstalk, background fluorescence from a region surrounding the central imaged cells was subtracted from the fluorescence of the recorded cells.
Spike finding and scaling of fluorescence recordings.
A simple threshold-and-maximum procedure was applied for spike detection. Fluorescence traces were first high-pass filtered, and initial threshold was set at 4 times the noise level. This threshold was then manually adjusted.
All fluorescence signals were normalized to spike height for spike-triggered average or stimulation-triggered average.
Spike detection: false-positive and false-negative rates.
For an SNR of 7 and a spike-detection threshold set at 4σ about the baseline noise, the false-positive rate is calculated as the probability that samples from a Gaussian distribution lie more than 4σ above the mean. This probability is (1-p4)/2*1 kHz*10 s, where p4 = 0.999937. The false-negative rate is calculated as the probability that spike height values fall more than 3σ below the mean. This probability is (1-p3)/2, where p3 = 0.9973.
Spike removal for calculation of subthreshold waveforms.
Spikes were digitally removed and replaced with linear interpolations of the surrounding data. Spike width was estimated by viewing individual fluorescence recordings. Linear interpolations were performed between data-points 1 ms beyond the edges of the spike.
Statistics
All error ranges represent standard error of the mean, unless otherwise specified. For the same neurons before and after drug administration, in anesthetized and awake states, with and without surrounding inhibition, and under local and global optogenetic stimulation, paired sample t-test was used. For two-sample comparisons of a single variable, student’s t-test was used. In cases where the underlying distributions were non-Gaussian, the Kruskal-Wallis test was used. Probabilities of the null hypothesis p < 0.05 were judged to be statistically significant.
Biophysical model of subthreshold membrane potential
The evolution of subthreshold membrane potential in the presence of synaptic inputs and optogenetic stimulation (Fig. 3) was simulated with a passive single compartment model using the following equation:
| (1) |
where Cm is the membrane capacitance, ge, gi, gl and gChR are the conductance of excitatory, inhibitory, leak and channelrhodopsin channels, and Ee, Ei, El and EChR are the respective reversal potentials. The time course of conductance upon excitatory or inhibitory synaptic input was simulated using an alpha function:
| (2) |
Here gsyn is the strength of the synaptic input, t0 is the time of the synaptic input, t is a time-constant of synaptic input, and gbaseline reflects the tonic level of synaptic input excluding the event of interest.
In principle, the values of gbaseline for the inhibitory and excitatory synaptic inputs could be wrapped into the definitions of gl and El. Doing so would not affect the solutions to Eq. 1. We chose to keep the baseline synaptic conductances as separate parameters to facilitate explorations of the model under different brain states (e.g. anesthesia vs. wakefulness). In this approach, gl and El reflect cell-autonomous leak conductances (e.g. Kir channels), assumed to be independent of brain state, while gbaseline captures the effect of network-dependent inputs.
To simulate lateral inhibition in L1, we assumed inhibition lagged excitation by 2 ms. Other parameters are listed below:
| Parameter | Value |
|---|---|
| ge syn | 1.5 nS |
| ge baseline | 0.1 nS |
| gi syn | 5 nS |
| gi baseline | 0.1 nS |
| gl | 3.33 nS |
| gChR | 0 – 10 nS |
| Ee | −5 mV |
| Ei | −70 mV |
| El | −70 mV |
| EChR | 0 mV |
| Cm | 150 pF |
| τ | 1 ms |
Equation 1 was numerically integrated using Euler’s method.
The model above can be solved analytically for the steady-state voltage by setting , which yields:
| (3) |
The PSP amplitudes are obtained by calculating the difference in steady-state voltage, ΔVPSP, after vs. before the sensory perturbations to ge and gi, assuming that all other parameters do not vary during the synaptic event. If one assumes that ge and gi are both zero before the synaptic event (i.e. by absorbing the pre-stimulus values of ge and gi into the definition of gl and El), then one finds:
| (4) |
Since only differences in voltage appear in Eq. 4, one can arbitrarily choose to set one of the voltages to zero, and measure all other voltages relative to this reference. For convenience, we set El to zero for the fitting. Eq. 4 is linear in gChR in the numerator, and quadratic in gChR in the denominator, suggesting 5 fitting parameters to specify the function ΔVIPSP(gChR). However, the proportionality between ΔV and ΔF is not a priori known. Also the proportionality between gChR and I488 is not a priori known.
To estimate the waveforms of the post-synaptic potentials, we first normalized all fluorescence traces by spike height. We then calculated the mean fluorescence over 10 ms before the whisker stimulus, and the mean fluorescence over 10 ms starting 30 ms after the whisker stimulus. The difference between these values was taken as the amplitude of the post-synaptic potential. Equation 4 was fitted to the data using the nonlinear least-squares method in Matlab.
Analytical approximation.
To gain an intuition for the responses, one can make a simple estimate of the amplitude of the IPSP by assuming that ge = 0 at all times, that gi = 0 before the synaptic input, and that the resting potential is the same as the inhibitory reversal potential, i.e. El ≈ Ei. One then obtains:
| (5) |
Within the blue light intensity range used in our experiments, the CheRiff conductance is well approximated by a linear function of the blue intensity, though the proportionality factor depends on the (unknown) CheRiff expression level and attenuation of the blue light by scattering.
Eq. 5 shows that for small gChR the amplitude ΔVIPSP is linear in gChR, while for large gChR the quadratic term in the denominator dominates and ΔVIPSP decreases inversely with gChR. The value of gChR that gives the largest amplitude IPSP is , or for weak inhibition, . If gl is large, then the shunting from gChR is suppressed. The membrane time constant is approximately . The inverse relation between τ and gChR is consistent with our observation of faster recovery at stronger stimulus strength.
Numerical model of L1 dynamics
Single-cell properties.
We considered two broad classes of L1 interneurons: laterally projecting neurogliaform cells (eNGC) and downward projecting single bouquet-like cells (SBC). To capture the firing properties of SBC, we used a modified form of the conductance-based model for hippocampal interneurons in Ref (Wang and Buzsáki, 1996) (Table S2). The voltage gated conductances responsible for the late-spiking pattern of eNGC have not been identified (Overstreet-Wadiche and McBain, 2015). We included an A-type inactivating potassium conductance to capture the firing pattern (Bard Ermentrout and Terman, 2010). We then connected the two neuronal subtypes with distance dependent inhibitory synapses. ENGC cells synapsed onto each other and onto SBC cells. SBC cells did not synapse within L1. Simulations comprised 51 L1 interneurons (34 eNGC, 17 SBC) corresponding approximately to a single barrel (Table S2).
We judged more detailed channel-based biophysical models to have too many unknown parameters. Simpler spike rate-based models did not capture the details of the spike timing or subthreshold potentials, which we judged important for L1 circuit function.
Each SBC neuron was described by a single compartment and simulated with the following equation:
| (6) |
| (7) |
| (8) |
| (9) |
where Cm = 1 μF/cm2; and I(t) is the injected current; the conductance of leak current, gleak = 0.23 mS/cm2 so that the passive time constant τ = 4.3 ms; Eleak = −66.8 mV; gNa = 35 mS/cm2; ENa = 55 mV; φ = 5; gk = 9 mS/cm2; Ek = − 90 mV. The quantity ϵ(t) = N(0, σ2) represents a Gaussian distributed white noise current with σ = A (δt)1/2, where A ~ 1 pA cm−2 ms−1/2 and δt = 0.02 ms (or in some cases 0.05 ms) is the integration time-step. The equations were integrated using the Euler method.
The dynamics of eNGC neurons followed similar equations with the following difference: we included an A-type inactivating potassium conductance to capture the firing pattern. This channel contributed a current:
where gA = 10 mS/cm2; EA = −75 mV.
To introduce firing rate adaptation in SBC cells, we included a slow outward potassium current. This channel contributed a current:
where gsk2 = 10 mS/cm2; Esk2 = −70 mV.
For the figure in Fig. S7, in order to produce high spontaneous firing, we set the resting potential higher at −55 mV for both eNGCs and SBCs.
Channelrhodopsin activation.
The channelrhodopsin CheRiff was modeled as an excitatory conductance with reversal potential 0 mV and conductance proportional to blue light illumination intensity. Channel gating kinetics were assumed to be instantaneous. Patterns of blue light were targeted to one or more cells and modulated in time during the simulation.
Synaptic properties: Inhibition.
Examination of patch clamp recordings of inhibitory post-synaptic potentials (IPSPs) in acute slices showed a rapid onset followed by a slow recovery. The recovery was not well captured by a single exponential. To approximate these dynamics we used the following function:
τ1 = 5 ms, τ2 = 30 ms. We found that the qualitative network dynamics were insensitive to variations in the functional form or time constants. One may think of the two terms as representing GABAA and GABAB receptors respectively, though a more accurate implementation of a GABAB-mediated hyperpolarization would use the K+ reversal potential (−90 mV) rather than the Cl− reversal potential (−70 mV).
To calibrate the value of for eNGC → eNGC and eNGC → SBC synapses, we set up a model circuit with one eNGC cell synapsing onto an eNGC cell and an SBC cell. We adjusted a simulated channelrhodopsin conductance (EChR = 0 mV) to depolarize the downstream cells to −55 mV. We then triggered the upstream cell to spike. We adjusted to induce IPSP amplitudes of −1.5 to −2 mV, to match the patch clamp data. The model did not contain short term inhibitory synaptic plasticity.
The IPSP amplitudes recorded via patch clamp were assumed to represent the strongest possible IPSPs. The IPSP strength between each pair of neurons in the circuit was modulated by a Gaussian function of separation, with a length-scale set by the sum of sizes of the presynaptic axonal arbor and the postsynaptic dendritic arbor (Jiang et al., 2015).
Synaptic properties: Excitation.
We modeled thalamocortical excitation in L1 using the function:
We calibrated the amplitude of and τ by simulating thalamic inputs to eNGC cells and SBC-like cells and matching to literature data which showed that whisker-evoked EPSP amplitudes in vivo were 3 – 7 mV. We assumed that the timecourse of thalamic excitation to all neurons was identical. Excitatory synaptic strengths were randomized by 20-30% between cells to prevent numerical degeneracies. The model did not contain short-term excitatory synaptic plasticity.
Synaptic properties: Neuromodulation.
The greatest uncertainty in the model surrounded the time-course and strength of neuromodulatory inputs. We assumed that neuromodulatory inputs activated an excitatory conductance with reversal potential 0 mV. We assumed that the strength and timecourse of neuromodulatory action was the same on the eNGC and SBC-like neurons. If the coupling of neuromodulatory input to the SBC-like neurons was sufficiently strong, then these neurons could be driven to spike by neuromodulatory inputs, even in the absence of thalamic inputs.
Omissions from the model.
Many possibly relevant features were omitted from the model. These include: gap junction connections between eNGC neurons (Chu et al., 2003), activation of GABAB receptors or of muscarinic acetylcholine receptors (Brombas et al., 2014), and possible feedback inhibition from deeper layer Martinotti cells (Abs et al., 2018). We did not consider a finer classification of L1 interneurons into sub-types. Our model also did not include cortico-cortical inputs. We did not study or simulate the effects of L1 interneuron activation on apical dendrites of deeper layer pyramidal cells. Activation of 5HT3A ionotropic serotonin receptors is expected to have similar electrophysiological effects to activation of nicotinic acetylcholine receptors, though the distribution of these two receptor-types in the different L1 interneuron sub-classes may be different.
DATA AND CODE AVAILABILITY
Data, code and custom software are available upon request.
Supplementary Material
Fig. S1. Crosstalk-free measurement of subthreshold voltage dynamics in tissue, Related to Figure 1. Minimization of optical crosstalk between cells was achieved through a combination of hardware, software, and protein engineering approaches. (A) Left: Mean fluorescence was calculated in annular regions centered on single neurons subjected to holographic membrane-targeted illumination. Right: Fluorescence traces from the regions with corresponding colors show rapid decay of signal amplitude with distance. (B) To quantify fluorescence originating from the targeted neuron, spikes were found in the neuron trace in (A) and a spike-triggered average of fluorescence was calculated in each annular region. Spike amplitude was then calculated as a function of distance from the center of the targeted neuron. Error bars represent mean ± s.d., n = 4 cells. (C) Quantification of relative total fluorescence as a function of optical defocus. A region of interest mask was defined on the in-focus image, and then applied to images taken at a series of defocus values. These plots have not been corrected for tissue autofluorescence, so some of the fluorescence at negative defocus could be independent of the neuron. (D) Composite movies were composed from distinct single-cell recordings. Two separately acquired movies were first analyzed to extract the single-cell fluorescence traces (‘input signals’). The two movies were then added together such that the cell centers were 15 μm apart. (E) The composite movies were then analyzed via PMD-NMF and the results (‘output signals’) were compared to the input signals via cross-correlation analysis. (F) Mean cross-correlograms of six composite movies processed as described. The PMD-NMF algorithm accurately reproduced the correlational structure of the inputs even when the cells overlapped in the composite movie. Shading represents s.e.m. (G) Simultaneous fluorescence and patch clamp recordings from neurons expressing SomArchon in acute brain slices. Top: Fluorescence changes recorded during subthreshold excitatory post-synaptic potentials. Bottom: Example simultaneous fluorescence and patch clamp recording showing correspondence of optical and electrical traces. (H) Two-photon fluorescence images of GFP fluorescence from Cre-dependent SomArchon-EGFP in vivo. Soma localization of reporter and actuator minimized crosstalk between cells. 5-HT3AR-Cre and SST-Cre labeling was via transgenic mice, while CKII(0.4)-Cre labeling was via an AAV Cre virus co-injected with the SomArchon. Expression was predominantly localized to cell bodies and proximal dendrites.
{kind=link}
Fig. S2. Patterned 1-photon optogenetic stimulation in vivo, Related to Figures 1, 3-7. (A) Two epifluorescence images of the same field of view. Left: Fluorescence in the GFP channel showing micromirror-patterned blue illumination targeted to the soma of a neuron expressing SomArchon-EGFP. Right: the same illumination pattern was shifted 30 μm to the bottom right (white arrow). Contrast in both images is the same. The shifted illumination spot evoked little EGFP fluorescence from the cell body, suggesting little scatter of blue light onto the cell body. (B) Mean fluorescence as a function of distance from the center of an illuminated disk focused onto a cell-free region of the brain tissue (error bars: ± s.d., n = 7 regions). The observed signal combines scatter of excitation light and scatter of emission, whereas spurious off-target channelrhodopsin activation would only come from scatter of excitation. (C) Optically recorded spiking of neurons during soma-targeted vs. shifted illumination. (D) Comparison of spike rates between on-cell (37.4 ± 11.2 Hz), laterally shifted (10.3 ± 3.9 Hz), and no (2.9 ± 2.3 Hz) blue illumination (mean ± s.e.m., p = 0.022, two-sided paired-sample t-test, blue light intensity 13 mW/mm2). Detailed anatomical studies have shown a mean spacing between L1 neurons of ~60 μm (Meyer et al., 2013), implying that targeted 1-photon optogenetic activation in L1 preferentially activated single cells or small clusters. (E) Light scatter contributed a depolarizing transient when a neuron was surrounded by a ring stimulus. Mean fluorescence response of a central neuron during a 20 ms annular stimulus to surrounding neurons (25 mW/mm2). This experiment is the same as in Fig. 4G, except that the central neuron was not subjected to direct optogenetic stimulation. The experiment shown here and in Fig. 4G were performed on the same set of neurons in interleaved trials +/− central optogenetic depolarization (n = 25 neurons, 3 mice). The time-course of subthreshold depolarization matches the expectation from direct stimulation of the central neuron via scattered blue light from the surrounding annulus. In approximately half of trials the scattered blue light evoked a spike in the central neuron.
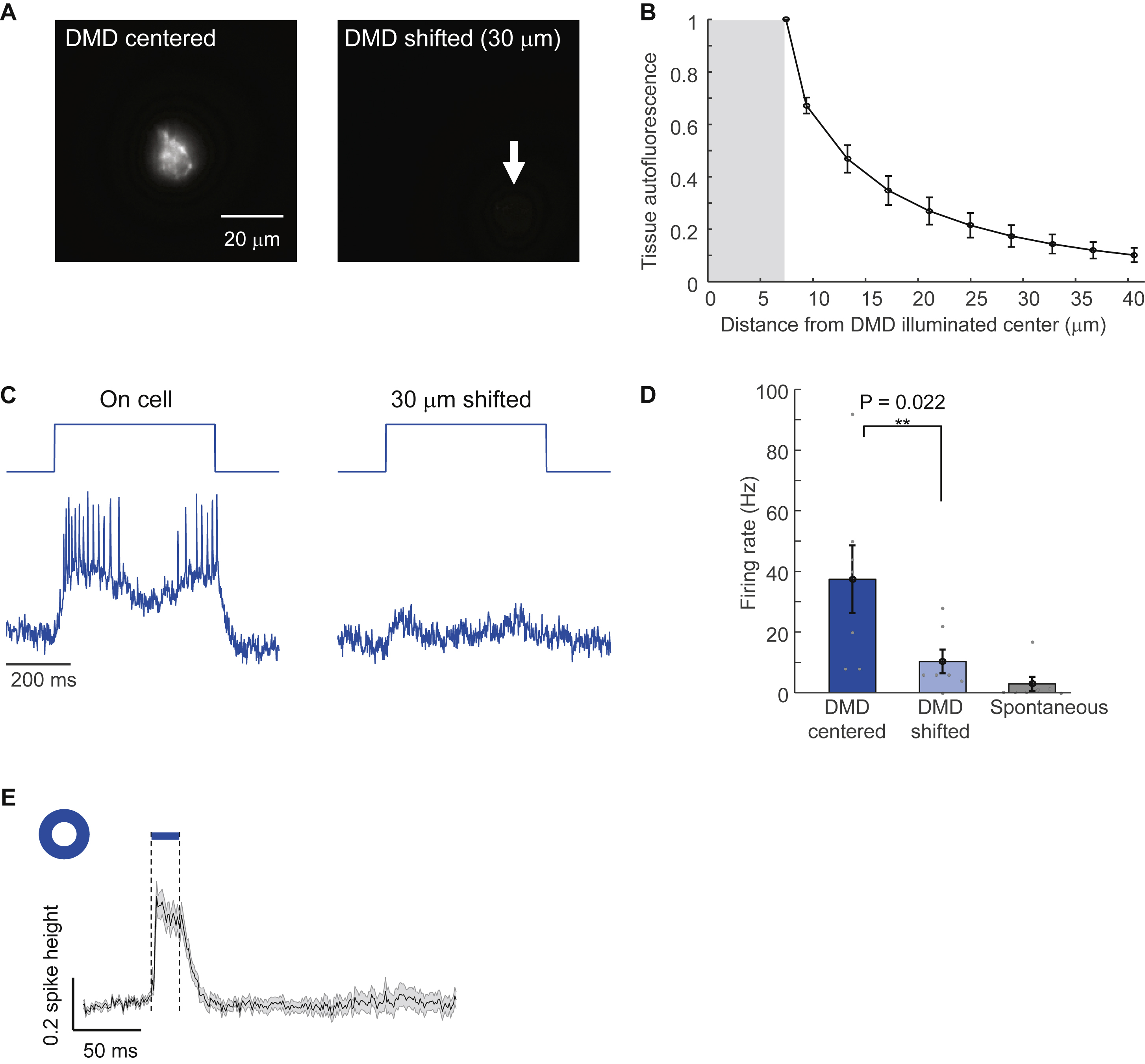{kind=link}
Fig. S3. Characterization of stGtACR2 for combined optogenetic silencing and voltage imaging, Related to Figure 1. stGtACR2 was expressed in HEK293 cells and photocurrents were recorded by manual patch clamp. The membrane potential was clamped at −15 mV. (A) Photocurrent of GtACR2 under blue illumination (500 ms pulses, 20 −100 mW/cm2). (B) Photocurrent of GtACR2 under red illumination (500 ms pulses, 1000-300-100-10 W/cm2). Top: same current scale as in (A), Bottom: expanded current scale. (C) Comparison of mean photocurrent for blue only, simultaneous red and blue, and red only illumination. Intensities were 80 mW/cm2 for blue and 1000 W/cm2 for red (mean ± s.d., n = 3 cells).
{kind=link}
Fig. S4. Optopatch excitability measurement of cortical L1 neurons in acute slices, under anesthesia and wakefulness, Related to Figure 1. (A) In cortical L1 neurons from a 5-HT3AR-Cre mouse expressing Optopatch4, the SomArchon fluorescence reported action potentials with high SNR. Blue: Individual neurons were illuminated with steps of blue light (500 ms duration, 0.2 to 2.1 mW/mm2, repeated at 1 Hz), followed by a ramp of blue light. Red: fluorescence of SomArchon during optogenetic stimulation, recorded at 1 kHz. The signal-to-noise ratio (spike:baseline noise) was 21 ± 1 in a 1 kHz bandwidth (n = 15 cells, 1 mW red laser per cell, mean ± s.e.m.). Some spike waveforms have been labeled as “Late spiking” (LS) or “Bursting” (B) in correspondence with established L1 firing phenotypes. Not all cells clearly fell into one of these classes. (B) Recordings of L1 neurons in an isoflurane anesthetized 5-HT3AR-Cre mouse expressing Optopatch4. Individual L1 neurons in vivo were illuminated with steps of blue light (500 ms duration, 1.8 to 21 mW/mm2, repeated at 1 Hz), followed by a ramp of blue light. Voltage was recorded at 1 kHz via holographic focused excitation of SomArchon fluorescence. Neuron coordinates were recorded relative to blood vessel landmarks. Anesthesia was then ended. After the animal awoke the measurements were repeated on the same set of cells. Each row represents repeated recordings of the same cell (n = 23 neurons from 3 mice). (C) Spike raster for the complete data-set recorded under anesthesia and wakefulness. (D) Mean spike rate during each stimulation epoch. Error bars represent s.e.m.. Awake data are from the same dataset as in Fig. 1G.
{kind=link}
Fig. S5. Sensory and optogenetic responses in vivo, Related to Figures 2, 3. (A) In an anesthetized mouse, the surface of the brain was imaged via reflected 640 nm light. A single whisker was periodically stimulated (10 Hz, 4 s, followed by 16 s rest) for 5 min. Images were acquired at 10 Hz. The mean of the image acquired during the stimulated epochs was subtracted from the mean of the image acquired during the rest epochs. A dark spot highlighted the active barrel. A reference image taken with back-scattered blue light identified the blood vessel landmarks around the active barrel. (B) Histogram of delays between stimulus onset and action potential peak in awake mice (complement of Fig. 2F). (C) Stimulus-triggered mean responses to stimuli that failed to evoke spikes. In both anesthetized and awake mice, these stimuli evoked an EPSP followed by an IPSP, indicating a synaptic as opposed to cell-autonomous origin of the post-stimulus hyperpolarization. Data from n = 9 neurons (anesthetized) and n = 16 neurons (awake), 3 mice in both cases. (D) Responses in anesthetized mice to four whisker stimuli at 10 Hz. Top: single-trial recordings. Bottom: mean response (n = 7 neurons). After the first stimulus, sensory-evoked responses were suppressed. Sensory-evoked responses recovered concurrent with recovery of the sub-threshold potential, suggesting that the suppression was at least partly due to L1 network inhibition. These experiments did not assess whether upstream mechanisms (e.g. short-term plasticity at thalamocortical synapses) also contributed to the suppressed response. (E) Three recordings from a single neuron in an anesthetized mouse showing response to (top) whisker stimulus, (middle) optogenetic stimulus, and (bottom) simultaneous optogenetic and whisker stimuli. Arrows show whisker stimulus-evoked inhibition. (F) Mean spike rate evoked by whisker stimuli atop different levels of optogenetic stimulus. In the absence of optogenetic stimulation, whisker stimuli evoked single spikes. In the presence of optogenetic stimulation, whisker stimuli suppressed spiking. The suppression decreased in amplitude and duration as the strength of the optogenetic stimulus increased. Shading represents s.e.m. from n = 15 neurons, 3 mice. (G) Mean whisker stimulus-evoked subthreshold waveforms at different levels of optogenetic drive. Spikes were digitally removed prior to averaging (Methods). (H) Comparison of IPSP amplitude as a function of optogenetic stimulus strength with numerical simulation from a simple conductance-based model.
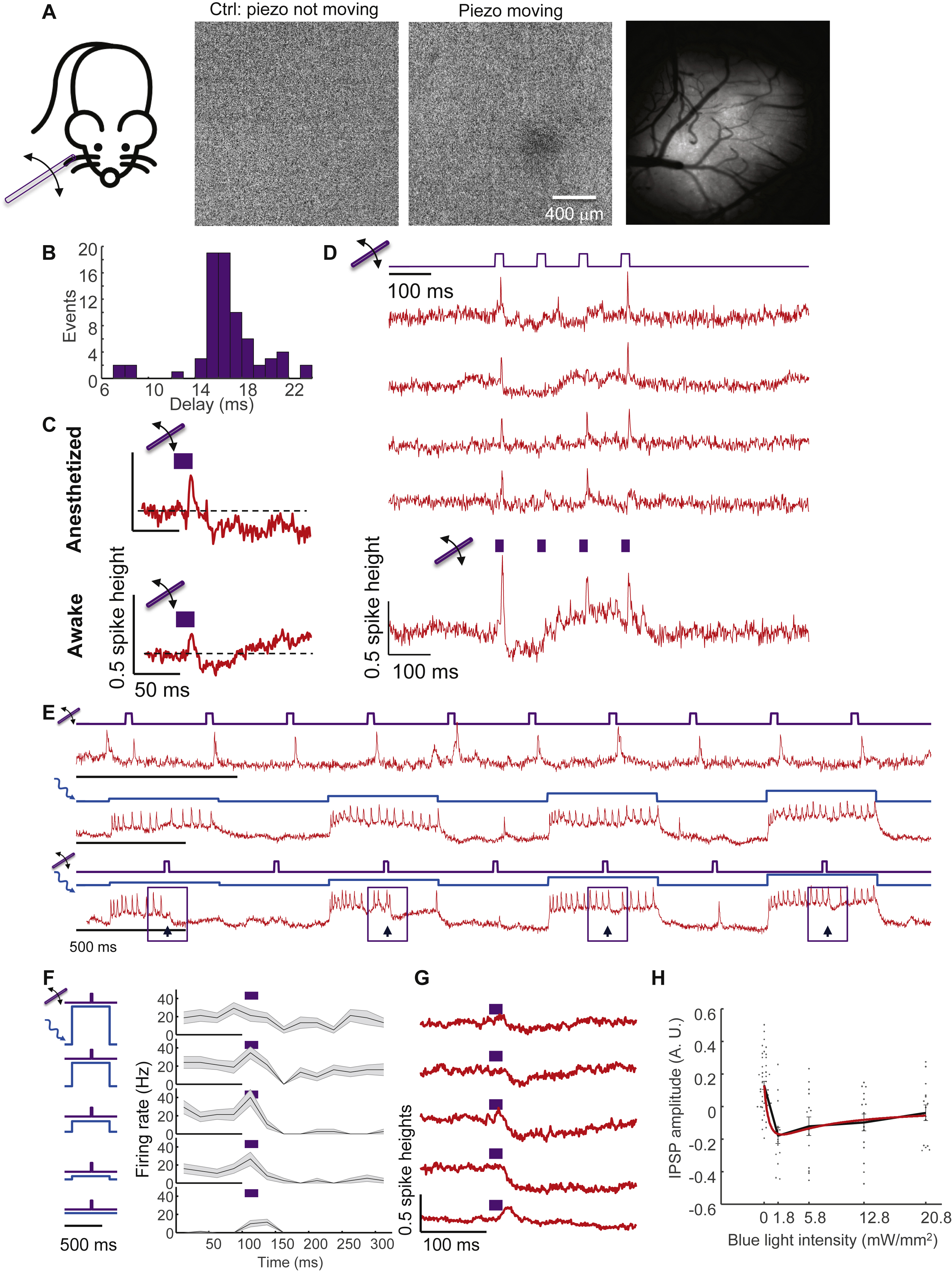{kind=link}
Figure S6. Center/surround optogenetic stimulation reveals lateral inhibition in NDNF- positive neurons, Related to Figure 4. This experiment is the same as in Fig. 4, with the replacement of 5-HT3AR-Cre by NDNF-Cre driver mice. (A) Simple model of L1 circuit with lateral inhibition. (B) Experiment to probe lateral inhibition in L1. NDNF-Cre mice expressed Optopatch4 in barrel cortex. Optogenetic stimuli were delivered separately to central and surrounding neurons. Voltage imaging was performed only in central neurons. Experiments were performed in anesthetized mice. (C) Epifluorescence images showing the illumination patterns in vivo. (D) Fluorescence waveforms from the central neurons under center/surround optogenetic stimulation. Central stimulation depolarized the targeted neurons and evoked spiking. Surround stimulation hyperpolarized the targeted neurons and suppressed spiking. (E) Spike raster showing responses from n = 19 neurons, 3 mice. (F) Mean spike rate during central stimulation, before and after surround stimulation. Surround stimulation caused spike rate to drop from 5.9 ± 1.9 Hz to 0 ± 0 Hz, n = 19 neurons, 3 mice (p = 0.007, two-sided paired-sample t-test). Shading represents s.e.m. (G) Mean subthreshold voltage during central stimulation, before and after surround stimulation. Surround stimulation caused inhibition in the central neuron. The initial spike in membrane voltage in the central neuron was due to scattered light from the surround which drove direct CheRiff activation. Shading represents s.e.m..
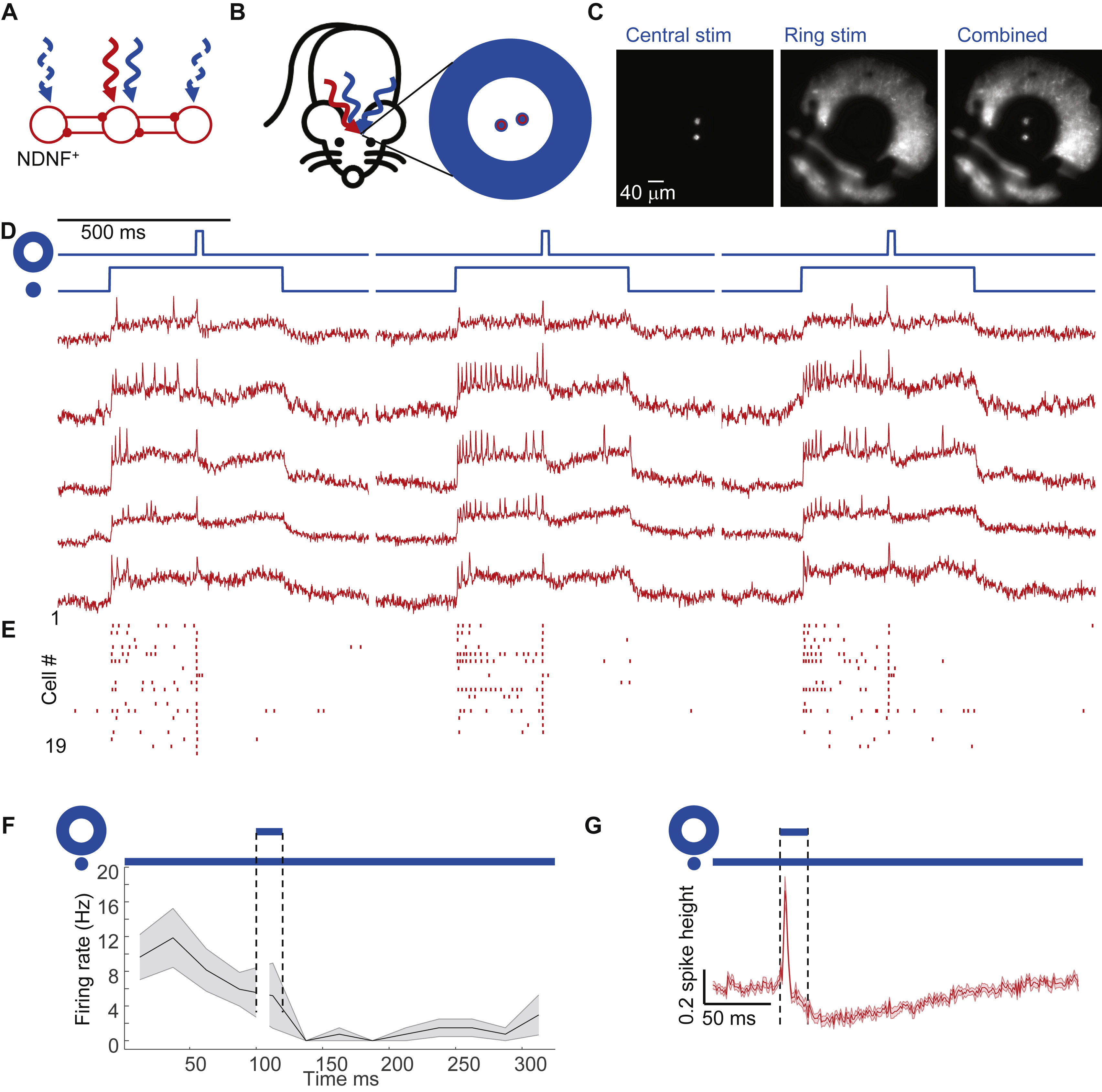{kind=link}
Fig. S7. Data and simulation of thalamic, optogenetic and cholinergic responses in L1 interneurons, Related to Figures 6, 7. (A) Simulated membrane voltage of single cells embedded in an L1 network during thalamocortical excitation. Here input noise and tonic depolarization were adjusted to achieve a spontaneous spike rate that matched experiment. (B) Stimulus-triggered average membrane potential with simulated thalamic inputs shows depolarization followed by a period of hyperpolarization. (C) Simulated thalamic inputs induced a period of reduced spontaneous activity. Firing rate quantified in 75 ms before the thalamic inputs (t1) were significantly higher than after the thalamic inputs (t2) (3.4 ± 1.1 Hz vs. 0.5 ± 0.4 Hz, n = 51 cells, p = 0.02, Kruskal-Wallis test, mean ± s.e.m.). (D) Top: Left: Geometry of annular optogenetic stimulation in a simulated L1 barrel. Cells were randomly positioned in a region 300 μm on an edge, 150 μm deep. A single SBC-like cell was manually defined to reside at the center of the region. The central neuron was subjected to tonic optogenetic depolarization. Neurons at radii r > 100 μm from the center were subjected to pulsed optogenetic stimulation. Right: Surround stimulation hyperpolarized the targeted neurons and suppressed spiking. Bottom: The same as top except that a central eNGC cell was subjected to tonic optogenetic depolarization. Compare to Fig. 4D. (E) Top: Air puff-induced responses in pairs of simultaneously recorded neurons, shown with shared background shading. Air puffs induced distinct concurrent responses, e.g. cell 3 showed a barrage of sustained firing while cell 4 did not spike; yet both cells had similar optogenetic excitability. Bottom: Air puff on top of blue light single-cell targeted optogenetic stimulation occasionally induced hyperpolarization (arrows). (F) Simulation of an L1 network response to a graded cholinergic input. The cholinergic input activated an excitatory conductance in all neurons. Under strong input, a few eNGC neurons become tonically active, suppressing spiking of the rest of the network. The plots show a randomly selected subset of the 51 neurons in the circuit. In the simulations, tonic activity was always in the eNGC cells, never in the SBC cells. (G) Numerical simulation showing spontaneous symmetry breaking in spike rate under strong cholinergic drive. L1 networks of 51 neurons were simulated with steady state cholinergic drive. At each value of gACh, 200 simulations were run with randomly reconfigured networks. For each simulation the value of gACh was held constant for 5 s and the spike rate was determined for each cell. The plot shows a histogram of the steady-state spike rate of all simulated neurons. The red overlay is a guide to the eye showing the bifurcation in firing rate at a critical stimulus threshold. Under strong stimulation approximately 10% of cells fired tonically while 90% were silent.
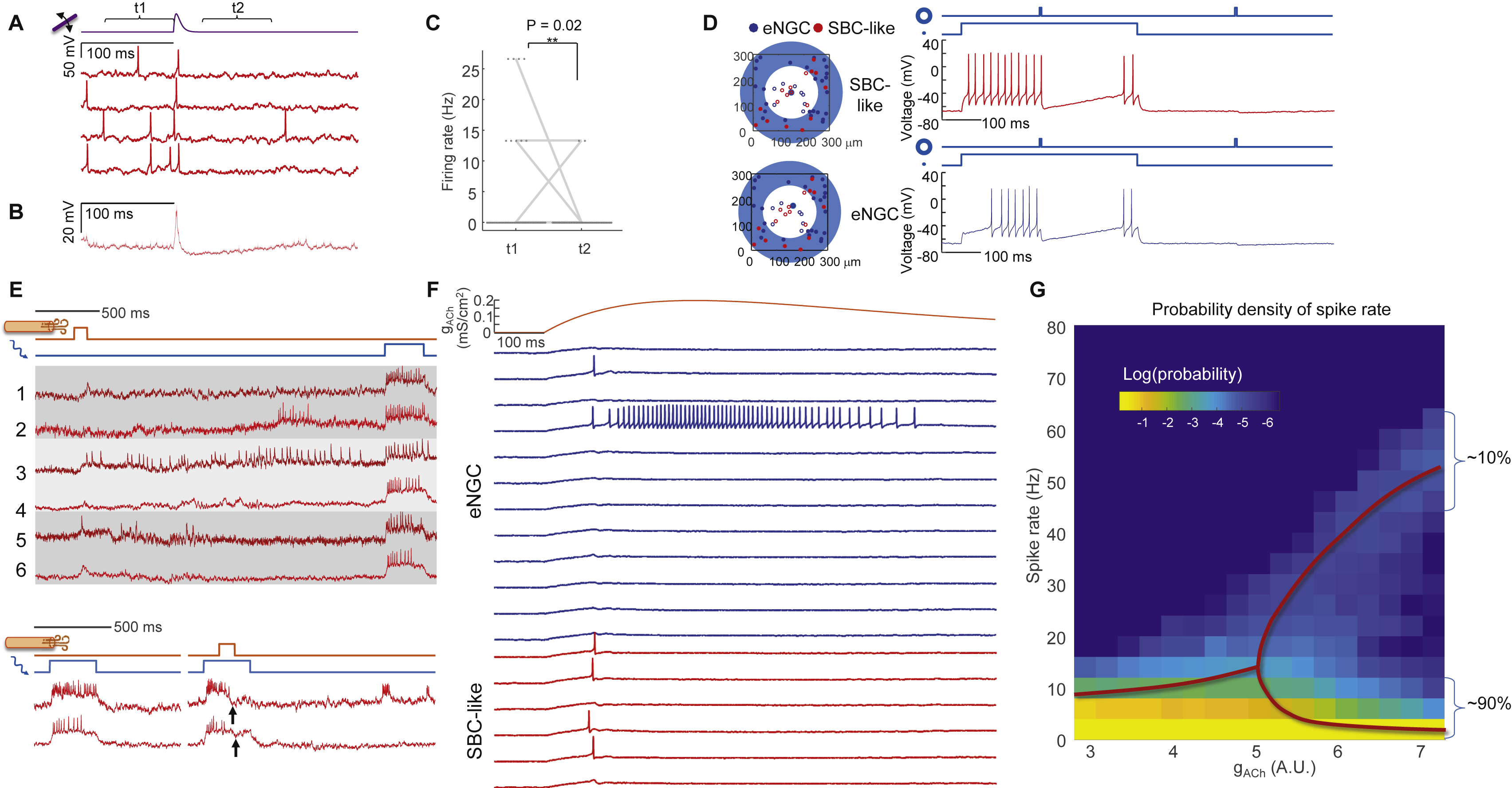{kind=link}
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and Virus Strains | ||
| LZF1735 pAAV_hSyn-DiO-SomArchon-eGFP-P2A-somCheRiff (Optopatch4) | Janelia Farm Vector Core | N/A |
| pAAV_CAG-FLEX-SomArchon-eGFP | UNC Vector Core | N/A |
| CKII(0.4)-Cre | UNC Vector Core | N/A |
| LZF1826 pAAV hSyn-DiO-SomArchon-eGFP-P2A-stGTACR2 | Janelia Farm Vector Core | N/A |
| LZF 1827 pAAV_hSyn-Flp-off-Cre-on-stGtACR2-CFP | Janelia Farm Vector Core | N/A |
| pAAV-EF1a-Flpo | UNC Vector Core | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| dihydro-β-erythroidine hydrobromide (DHβE) | Tocris | cat# 2349 |
| TransIT-293 | Mirus | cat# MIR2704 |
| glass capillaries | World precision Instrument | cat# TW120-3 |
| Experimental Models: Cell Lines | ||
| Hek293T | ATCC | CRL-11268 |
| Experimental Models: Organisms/Strains | ||
| 5HT3AR-Cre transgenic mice | Takao Hensch Lab | N/A |
| C57BL/6 wild-type mice | Charles River | N/A |
| SST-Cre transgenic mice | Jackson Lab | Stock #013044 |
| NDNF-Cre transgenic mice | Jackson Lab | Stock #028536 |
| Recombinant DNA | ||
| LZF1735 pAAV_hSyn-DiO-SomArchon-eGFP-P2A-somCheRiff (Optopatch4) | This work | Addgene 126512 |
| LZF1826 pAAV hSyn-DiO-SomArchon-eGFP-P2A-stGTACR2 | This work | Addgene 135412 |
| LZF1827 pAAV_hSyn-Flp-off-Cre-on-stGtACR2-CFP | This work | Addgene 135413 |
| pAAV_hSyn1-SIO-stGtACR2-FusionRed | Ofer Yizhar Lab | Addgene 105677 |
| UB061 FCK-stGtACR2-fusionRed (Lenti-vector) | This Work | Addgene 135765 |
| Software and Algorithms | ||
| Matlab R2015b | Mathworks | https://www.mathworks.com/products/matlab.html |
| Labview | National Instrument | N/A |
| Other | ||
| Holographic structured illumination microscope | This work | N/A |
Highlights.
All-optical electrophysiology reveals synaptic excitation and inhibition, in vivo.
Whisker stimuli evoke concurrent excitation and inhibition in L1 interneurons.
Cholinergic inputs evoke winner-takes-all spiking in L1 interneurons.
Lateral inhibition within L1 governs whisker and cholinergic responses.
Acknowledgments:
We thank B.L. Sabatini, C.D. Harvey, N. Uchida, R. Wilson and G. Yellen for advice and discussion; G. Vargish and T. Hensch for the 5HT3AR-Cre mouse line; S. Begum, K. Williams, A. Preecha and H. Dahche for technical assistance; B. Ermentrout, S. Baccus and E. Marder for advice on network simulations; M. Gomez-Ramirez, C. Moore and F. Wang for advice on whisker stimulation; H. Pi for advice on air puff; B. Gmeiner for advice on optics; Y. Adam for advice on mouse surgeries.
Funding: This work was supported by the Howard Hughes Medical Institute. KEE was supported by award Number T32GM007753 from the National Institute of General Medical Science. AET was supported by NIDCD award R21DC016991 and the Nancy Lurie Marks Family Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests: A.E.C. is a founder of Q-State Biosciences and is on the scientific advisory board. A.E.C., L.Z.F and S.K. have filed a patent on the imaging system.
Data and materials availability: Plasmids are available on Addgene. Data, code, and materials used in the analysis are available upon reasonable request to A.E.C..
References:
- Abdelfattah AS, Kawashima T, Singh A, Novak O, Liu H, Shuai Y, Huang Y-C, Campagnola L, Seeman SC, Yu J, et al. (2019). Bright and photostable chemigenetic indicators for extended in vivo voltage imaging. Science. 365, 699–704. [DOI] [PubMed] [Google Scholar]
- Abs E, Poorthuis RB, Apelblat D, Muhammad K, Pardi MB, Enke L, Kushinsky D, Pu DL, Eizinger MF, Conzelmann KK, et al. (2018). Learning-Related Plasticity in Dendrite-Targeting Layer 1 Interneurons. Neuron 100, 684–699.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adam Y, Kim JJ, Lou S, Zhao Y, Xie ME, Brinks D, Wu H, Mostajo-Radji MA, Kheifets S, Parot V, et al. (2019). Voltage imaging and optogenetics reveal behaviour-dependent changes in hippocampal dynamics. Nature 569, 413–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andermann ML, Gilfoy NB, Goldey GJ, Sachdev RNS, Wölfel M, McCormick DA, Reid RC, and Levene MJ (2013). Chronic Cellular Imaging of Entire Cortical Columns in Awake Mice Using Microprisms. Neuron 80, 900–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arroyo S, Aziz D, Bennett C, Hestrin S, and Brown SP (2012). Prolonged Disynaptic Inhibition in the Cortex Mediated by Slow, Non-7 Nicotinic Excitation of a Specific Subset of Cortical Interneurons. J. Neurosci 32, 3859–3864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bard Ermentrout G, and Terman DH (2010). Mathematical Foundations of Neuroscience. Vol. 35 Springer Science & Business Media, 2010. [Google Scholar]
- Brombas A, Fletcher LN, and Williams SR (2014). Activity-Dependent Modulation of Layer 1 Inhibitory Neocortical Circuits by Acetylcholine. J. Neurosci 34, 1932–1941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchanan EK, Kinsella I, Zhou D, Zhu R, Zhou P, Gerhard F, Ferrante J, Ma Y, Kim S, Shaik M, et al. (2018). Penalized matrix decomposition for denoising, compression, and improved demixing of functional imaging data. BioRxiv 334706. [Google Scholar]
- Cadwell CR, Palasantza A, Jiang X, Berens P, Deng Q, Yilmaz M, Reimer J, Shen S, Bethge M, Tolias KF, et al. (2016). Electrophysiological, transcriptomic and morphologic profiling of single neurons using Patch-seq. Nat. Biotechnol 34, 199–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu Z, Galarreta M, and Hestrin S (2003). Synaptic Interactions of Late-Spiking Neocortical Neurons in Layer 1. J. Neurosci 23, 96–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Constantinople CM, and Bruno RM (2011). Effects and mechanisms of wakefulness on local cortical networks. Neuron 69, 1061–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruikshank SJ, Ahmed OJ, Stevens TR, Patrick SL, Amalia N, Elmaleh M, and Connors BW (2012). Thalamic control of layer 1 circuits in prefrontal cortex. J. Neurosci 32, 17813–17823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damaj MI, Welch SP, and Martin BR (1995). In vivo pharmacological effects of dihydro-β-erythroidine, a nicotinic antagonist, in mice. Psychopharmacology (Berl). 117, 67–73. [DOI] [PubMed] [Google Scholar]
- Doron G, Shin JN, Takahashi N, Bocklisch C, Skenderi S, Drúke M, Mont L. de, Toumazo M, von Heimendahl M, Brecht M, et al. (2019). Perirhinal input to neocortical layer 1 controls learning. BioRxiv 713883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egger R, Schmitt AC, Wallace DJ, Sakmann B, Oberlaender M, and Kerr JND (2015). Robustness of sensory-evoked excitation is increased by inhibitory inputs to distal apical tuft dendrites. Proc. Natl. Acad. Sci 112, 14072–14077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggermann E, Kremer Y, Crochet S, and Petersen CCH (2014). Cholinergic Signals in Mouse Barrel Cortex during Active Whisker Sensing. Cell Rep. 9, 1654–1660. [DOI] [PubMed] [Google Scholar]
- Emiliani V, Cohen AE, Deisseroth K, and Hausser M (2015). All-Optical Interrogation of Neural Circuits. J. Neurosci 35, 13917–13926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan LZ, Nehme R, Adam Y, Jung ES, Wu H, Eggan K, Arnold DB, and Cohen AE (2018). All-optical synaptic electrophysiology probes mechanism of ketamine-induced disinhibition. Nat. Methods 15, 823–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forli A, Vecchia D, Binini N, Succol F, Bovetti S, Moretti C, Nespoli F, Mahn M, Baker CA, Bolton MM, et al. (2018). Two-Photon Bidirectional Control and Imaging of Neuronal Excitability with High Spatial Resolution In Vivo. Cell Rep. 22, 3087–3098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabernet L, Jadhav SP, Feldman DE, Carandini M, and Scanziani M (2005). Somatosensory integration controlled by dynamic thalamocortical feed-forward inhibition. Neuron 48, 315–327. [DOI] [PubMed] [Google Scholar]
- Goldey GJ, Roumis DK, Glickfeld LL, Kerlin AM, Reid RC, Bonin V, Schafer DP, and Andermann ML (2014). Removable cranial windows for long-term imaging in awake mice. Nat. Protoc 9, 2515–2538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haider B, Hausser M, and Carandini M (2013). Inhibition dominates sensory responses in the awake cortex. Nature 493, 97–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hangya B, Ranade SP, Lorenc M, and Kepecs A (2015). Central Cholinergic Neurons Are Rapidly Recruited by Reinforcement Feedback. Cell 162, 1155–1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochbaum DR, Zhao Y, Farhi SL, Klapoetke N, Werley CA, Kapoor V, Zou P, Kralj JM, MacLaurin D, Smedemark-Margulies N, et al. (2014). All-optical electrophysiology in mammalian neurons using engineered microbial rhodopsins. Nat. Methods 11, 825–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji N (2017). Adaptive optical fluorescence microscopy. Nat. Methods 14, 374–380. [DOI] [PubMed] [Google Scholar]
- Jiang X, Wang G, Lee AJ, Stornetta RL, and Zhu JJ (2013). The organization of two new cortical interneuronal circuits. Nat. Neurosci 16, 210–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X, Shen S, Cadwell CR, Berens P, Sinz F, Ecker AS, Patel S, and Tolias AS (2015). Principles of connectivity among morphologically defined cell types in adult neocortex. Science. 350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung JC, Mehta AD, Aksay E, Stepnoski R, and Schnitzer MJ (2004). In vivo mammalian brain imaging using one- and two-photon fluorescence microendoscopy. J. Neurophysiol 92, 3121–3133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koyama M, Minale F, Shum J, Nishimura N, Schaffer CB, and Fetcho JR (2016). A circuit motif in the zebrafish hindbrain for a two alternative behavioral choice to turn left or right. Elife 5, e16808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kralj JM, Douglass AD, Hochbaum DR, Maclaurin D, and Cohen AE (2012). Optical recording of action potentials in mammalian neurons using a microbial rhodopsin. Nat. Methods 9, 90–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam YW, and Sherman SM (2019). Convergent synaptic inputs to layer 1 cells of mouse cortex. Eur. J. Neurosci 49, 1388–1399. [DOI] [PubMed] [Google Scholar]
- Lee AJ, Wang G, Jiang X, Johnson SM, Hoang ET, Lanté F, Stornetta RL, Beenhakker MP, Shen Y, and Julius Zhu J (2015). Canonical Organization of Layer 1 Neuron-Led Cortical Inhibitory and Disinhibitory Interneuronal Circuits. Cereb. Cortex 25, 2114–2126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Hjerling-Leffler J, Zagha E, Fishell G, and Rudy B (2010). The Largest Group of Superficial Neocortical GABAergic Interneurons Expresses Ionotropic Serotonin Receptors. J. Neurosci 30, 16796–16808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Kruglikov I, Huang ZJ, Fishell G, and Rudy B (2013). A disinhibitory circuit mediates motor integration in the somatosensory cortex. Nat. Neurosci 16, 1662–1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letzkus JJ, Wolff SBE, Meyer EMM, Tovote P, Courtin J, Herry C, and Luthi A (2011). A disinhibitory microcircuit for associative fear learning in the auditory cortex. Nature 480, 331–335. [DOI] [PubMed] [Google Scholar]
- Lou S, Adam Y, Weinstein EN, Williams E, Williams K, Parot V, Kavokine N, Liberles S, Madisen L, Zeng H, et al. (2016). Genetically Targeted All-Optical Electrophysiology with a Transgenic Cre-Dependent Optopatch Mouse. J. Neurosci 36, 11059–11073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutz C, Otis TS, DeSars V, Charpak S, DiGregorio DA, and Emiliani V (2008). Holographic photolysis of caged neurotransmitters. Nat. Methods 5, 821–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machens CK, Romo R, and Brody CD (2005). Flexible control of mutual inhibition: A neural model of two-interval discrimination. Science. 307, 1121–1124. [DOI] [PubMed] [Google Scholar]
- Mahn M, Gibor L, Patil P, Cohen-Kashi Malina K, Oring S, Printz Y, Levy R, Lampl I, and Yizhar O (2018). High-efficiency optogenetic silencing with soma-targeted anionconducting channelrhodopsins. Nat. Commun 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mardinly AR, Oldenburg IA, Pégard NC, Sridharan S, Lyall EH, Chesnov K, Brohawn SG, Waller L, and Adesnik H (2018). Precise multimodal optical control of neural ensemble activity. Nat. Neurosci 21, 881–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshel JH, Kim YS, Machado TA, Quirin S, Benson B, Kadmon J, Raja C, Chibukhchyan A, Ramakrishnan C, Inoue M, et al. (2019). Cortical layer–specific critical dynamics triggering perception. Science. 365, eaaw5202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGinley MJ, David SV, and McCormick DA (2015). Cortical Membrane Potential Signature of Optimal States for Sensory Signal Detection. Neuron 87, 179–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mechawar N, Cozzari C, and Descarries L (2000). Cholinergic innervation in adult rat cerebral cortex: A quantitative immunocytochemical description. J. Comp. Neurol 428, 305–318. [DOI] [PubMed] [Google Scholar]
- Meyer HS, Egger R, Guest JM, Foerster R, Reissl S, and Oberlaender M (2013). Cellular organization of cortical barrel columns is whisker-specific. Proc. Natl. Acad. Sci 110, 19113–19118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukamel EA, Nimmerjahn A, and Schnitzer MJ (2009). Automated Analysis of Cellular Signals from Large-Scale Calcium Imaging Data. Neuron 63, 747–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neske GT, Patrick SL, and Connors BW (2015). Contributions of diverse excitatory and inhibitory neurons to recurrent network activity in cerebral cortex. J. Neurosci 35, 1089–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overstreet-Wadiche L, and McBain CJ (2015). Neurogliaform cells in cortical circuits. Nat. Rev. Neurosci 16, 458–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Packer AM, Russell LE, Dalgleish HWP, and Hausser M (2015). Simultaneous all-optical manipulation and recording of neural circuit activity with cellular resolution in vivo. Nat. Methods 12, 140–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer LM, Schulz JM, Murphy SC, Ledergerber D, Murayama M, and Larkum ME (2012). The cellular basis of GABAB-mediated interhemispheric inhibition. Science. 335, 989–993. [DOI] [PubMed] [Google Scholar]
- Petersen CCH, Hahn TTG, Mehta M, Grinvald A, and Sakmann B (2003). Interaction of sensory responses with spontaneous depolarization in layer 2/3 barrel cortex. Proc. Natl. Acad. Sci. U. S. A 100, 13638–13643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piatkevich KD, Bensussen S, Tseng H. an, Shroff SN, Lopez-Huerta VG, Park D, Jung EE, Shemesh OA, Straub C, Gritton HJ, et al. (2019). Population imaging of neural activity in awake behaving mice. Nature 574, 413–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pnevmatikakis EA, and Giovannucci A (2017). NoRMCorre: An online algorithm for piecewise rigid motion correction of calcium imaging data. J. Neurosci. Methods 291, 83–94. [DOI] [PubMed] [Google Scholar]
- Poorthuis RB, Muhammad K, Wang M, Verhoog MB, Junek S, Wrana A, Mansvelder HD, and Letzkus JJ (2018). Rapid Neuromodulation of Layer 1 Interneurons in Human Neocortex. Cell Rep. 23, 951–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao RPN, and Ballard DH (1999). Predictive coding in the visual cortex: A functional interpretation of some extra-classical receptive-field effects. Nat. Neurosci 2, 79–87. [DOI] [PubMed] [Google Scholar]
- Rickgauer JP, Deisseroth K, and Tank DW (2014). Simultaneous cellular-resolution optical perturbation and imaging of place cell firing fields. Nat. Neurosci 17, 1816–1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sachdev RNS, Ebner FF, and Wilson CJ (2004). Effect of subthreshold up and down states on the whisker-evoked response in somatosensory cortex. J. Neurophysiol 92, 3511–3521. [DOI] [PubMed] [Google Scholar]
- Schultz W, Dayan P, and Montague PR (1997). A neural substrate of prediction and reward. Science. 275, 1593–1599. [DOI] [PubMed] [Google Scholar]
- Schuman B, Fishell GJ, Machold RP, Hashikawa Y, Fuzik J, and Rudy B (2018). Four Unique Interneuron Populations Reside in Neocortical Layer 1. J. Neurosci 39, 125–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal M, and Barker JL (1984). Rat hippocampal neurons in culture: voltage-clamp analysis of inhibitory synaptic connections. J. Neurophysiol 52, 469–487. [DOI] [PubMed] [Google Scholar]
- Takesian AE, Bogart LJ, Lichtman JW, and Hensch TK (2018). Inhibitory circuit gating of auditory critical-period plasticity. Nat. Neurosci 21, 218–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasic B, Menon V, Nguyen TN, Kim TK, Jarsky T, Yao Z, Levi B, Gray LT, Sorensen SA, Dolbeare T, et al. (2016). Adult mouse cortical cell taxonomy revealed by single cell transcriptomics. Nat. Neurosci 19, 335–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villette V, Chavarha M, Dimov IK, Bradley J, Pradhan L, Mathieu B, Evans SW, Chamberland S, Shi D, Yang R, et al. (2019). Ultrafast Two-Photon Imaging of a High-Gain Voltage Indicator in Awake Behaving Mice. Cell 179, 1590–1608.e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XJ, and Buzsáki G (1996). Gamma oscillation by synaptic inhibition in a hippocampal interneuronal network model. J. Neurosci 16, 6402–6413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wehr M, and Zador AM (2003). Balanced inhibition underlies tuning and sharpens spike timing in auditory cortex. Nature 426, 442–446. [DOI] [PubMed] [Google Scholar]
- Yerkes RM, and Dodson JD (1908). The relation of strength of stimulus to rapidity of habitformation. J. Comp. Neurol. Psychol 18, 459–482. [Google Scholar]
- Zhu Y (2004). Rapid Arrival and Integration of Ascending Sensory Information in Layer 1 Nonpyramidal Neurons and Tuft Dendrites of Layer 5 Pyramidal Neurons of the Neocortex. J. Neurosci 24, 1272–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Crosstalk-free measurement of subthreshold voltage dynamics in tissue, Related to Figure 1. Minimization of optical crosstalk between cells was achieved through a combination of hardware, software, and protein engineering approaches. (A) Left: Mean fluorescence was calculated in annular regions centered on single neurons subjected to holographic membrane-targeted illumination. Right: Fluorescence traces from the regions with corresponding colors show rapid decay of signal amplitude with distance. (B) To quantify fluorescence originating from the targeted neuron, spikes were found in the neuron trace in (A) and a spike-triggered average of fluorescence was calculated in each annular region. Spike amplitude was then calculated as a function of distance from the center of the targeted neuron. Error bars represent mean ± s.d., n = 4 cells. (C) Quantification of relative total fluorescence as a function of optical defocus. A region of interest mask was defined on the in-focus image, and then applied to images taken at a series of defocus values. These plots have not been corrected for tissue autofluorescence, so some of the fluorescence at negative defocus could be independent of the neuron. (D) Composite movies were composed from distinct single-cell recordings. Two separately acquired movies were first analyzed to extract the single-cell fluorescence traces (‘input signals’). The two movies were then added together such that the cell centers were 15 μm apart. (E) The composite movies were then analyzed via PMD-NMF and the results (‘output signals’) were compared to the input signals via cross-correlation analysis. (F) Mean cross-correlograms of six composite movies processed as described. The PMD-NMF algorithm accurately reproduced the correlational structure of the inputs even when the cells overlapped in the composite movie. Shading represents s.e.m. (G) Simultaneous fluorescence and patch clamp recordings from neurons expressing SomArchon in acute brain slices. Top: Fluorescence changes recorded during subthreshold excitatory post-synaptic potentials. Bottom: Example simultaneous fluorescence and patch clamp recording showing correspondence of optical and electrical traces. (H) Two-photon fluorescence images of GFP fluorescence from Cre-dependent SomArchon-EGFP in vivo. Soma localization of reporter and actuator minimized crosstalk between cells. 5-HT3AR-Cre and SST-Cre labeling was via transgenic mice, while CKII(0.4)-Cre labeling was via an AAV Cre virus co-injected with the SomArchon. Expression was predominantly localized to cell bodies and proximal dendrites.
Fig. S2. Patterned 1-photon optogenetic stimulation in vivo, Related to Figures 1, 3-7. (A) Two epifluorescence images of the same field of view. Left: Fluorescence in the GFP channel showing micromirror-patterned blue illumination targeted to the soma of a neuron expressing SomArchon-EGFP. Right: the same illumination pattern was shifted 30 μm to the bottom right (white arrow). Contrast in both images is the same. The shifted illumination spot evoked little EGFP fluorescence from the cell body, suggesting little scatter of blue light onto the cell body. (B) Mean fluorescence as a function of distance from the center of an illuminated disk focused onto a cell-free region of the brain tissue (error bars: ± s.d., n = 7 regions). The observed signal combines scatter of excitation light and scatter of emission, whereas spurious off-target channelrhodopsin activation would only come from scatter of excitation. (C) Optically recorded spiking of neurons during soma-targeted vs. shifted illumination. (D) Comparison of spike rates between on-cell (37.4 ± 11.2 Hz), laterally shifted (10.3 ± 3.9 Hz), and no (2.9 ± 2.3 Hz) blue illumination (mean ± s.e.m., p = 0.022, two-sided paired-sample t-test, blue light intensity 13 mW/mm2). Detailed anatomical studies have shown a mean spacing between L1 neurons of ~60 μm (Meyer et al., 2013), implying that targeted 1-photon optogenetic activation in L1 preferentially activated single cells or small clusters. (E) Light scatter contributed a depolarizing transient when a neuron was surrounded by a ring stimulus. Mean fluorescence response of a central neuron during a 20 ms annular stimulus to surrounding neurons (25 mW/mm2). This experiment is the same as in Fig. 4G, except that the central neuron was not subjected to direct optogenetic stimulation. The experiment shown here and in Fig. 4G were performed on the same set of neurons in interleaved trials +/− central optogenetic depolarization (n = 25 neurons, 3 mice). The time-course of subthreshold depolarization matches the expectation from direct stimulation of the central neuron via scattered blue light from the surrounding annulus. In approximately half of trials the scattered blue light evoked a spike in the central neuron.
Fig. S3. Characterization of stGtACR2 for combined optogenetic silencing and voltage imaging, Related to Figure 1. stGtACR2 was expressed in HEK293 cells and photocurrents were recorded by manual patch clamp. The membrane potential was clamped at −15 mV. (A) Photocurrent of GtACR2 under blue illumination (500 ms pulses, 20 −100 mW/cm2). (B) Photocurrent of GtACR2 under red illumination (500 ms pulses, 1000-300-100-10 W/cm2). Top: same current scale as in (A), Bottom: expanded current scale. (C) Comparison of mean photocurrent for blue only, simultaneous red and blue, and red only illumination. Intensities were 80 mW/cm2 for blue and 1000 W/cm2 for red (mean ± s.d., n = 3 cells).
Fig. S4. Optopatch excitability measurement of cortical L1 neurons in acute slices, under anesthesia and wakefulness, Related to Figure 1. (A) In cortical L1 neurons from a 5-HT3AR-Cre mouse expressing Optopatch4, the SomArchon fluorescence reported action potentials with high SNR. Blue: Individual neurons were illuminated with steps of blue light (500 ms duration, 0.2 to 2.1 mW/mm2, repeated at 1 Hz), followed by a ramp of blue light. Red: fluorescence of SomArchon during optogenetic stimulation, recorded at 1 kHz. The signal-to-noise ratio (spike:baseline noise) was 21 ± 1 in a 1 kHz bandwidth (n = 15 cells, 1 mW red laser per cell, mean ± s.e.m.). Some spike waveforms have been labeled as “Late spiking” (LS) or “Bursting” (B) in correspondence with established L1 firing phenotypes. Not all cells clearly fell into one of these classes. (B) Recordings of L1 neurons in an isoflurane anesthetized 5-HT3AR-Cre mouse expressing Optopatch4. Individual L1 neurons in vivo were illuminated with steps of blue light (500 ms duration, 1.8 to 21 mW/mm2, repeated at 1 Hz), followed by a ramp of blue light. Voltage was recorded at 1 kHz via holographic focused excitation of SomArchon fluorescence. Neuron coordinates were recorded relative to blood vessel landmarks. Anesthesia was then ended. After the animal awoke the measurements were repeated on the same set of cells. Each row represents repeated recordings of the same cell (n = 23 neurons from 3 mice). (C) Spike raster for the complete data-set recorded under anesthesia and wakefulness. (D) Mean spike rate during each stimulation epoch. Error bars represent s.e.m.. Awake data are from the same dataset as in Fig. 1G.
Fig. S5. Sensory and optogenetic responses in vivo, Related to Figures 2, 3. (A) In an anesthetized mouse, the surface of the brain was imaged via reflected 640 nm light. A single whisker was periodically stimulated (10 Hz, 4 s, followed by 16 s rest) for 5 min. Images were acquired at 10 Hz. The mean of the image acquired during the stimulated epochs was subtracted from the mean of the image acquired during the rest epochs. A dark spot highlighted the active barrel. A reference image taken with back-scattered blue light identified the blood vessel landmarks around the active barrel. (B) Histogram of delays between stimulus onset and action potential peak in awake mice (complement of Fig. 2F). (C) Stimulus-triggered mean responses to stimuli that failed to evoke spikes. In both anesthetized and awake mice, these stimuli evoked an EPSP followed by an IPSP, indicating a synaptic as opposed to cell-autonomous origin of the post-stimulus hyperpolarization. Data from n = 9 neurons (anesthetized) and n = 16 neurons (awake), 3 mice in both cases. (D) Responses in anesthetized mice to four whisker stimuli at 10 Hz. Top: single-trial recordings. Bottom: mean response (n = 7 neurons). After the first stimulus, sensory-evoked responses were suppressed. Sensory-evoked responses recovered concurrent with recovery of the sub-threshold potential, suggesting that the suppression was at least partly due to L1 network inhibition. These experiments did not assess whether upstream mechanisms (e.g. short-term plasticity at thalamocortical synapses) also contributed to the suppressed response. (E) Three recordings from a single neuron in an anesthetized mouse showing response to (top) whisker stimulus, (middle) optogenetic stimulus, and (bottom) simultaneous optogenetic and whisker stimuli. Arrows show whisker stimulus-evoked inhibition. (F) Mean spike rate evoked by whisker stimuli atop different levels of optogenetic stimulus. In the absence of optogenetic stimulation, whisker stimuli evoked single spikes. In the presence of optogenetic stimulation, whisker stimuli suppressed spiking. The suppression decreased in amplitude and duration as the strength of the optogenetic stimulus increased. Shading represents s.e.m. from n = 15 neurons, 3 mice. (G) Mean whisker stimulus-evoked subthreshold waveforms at different levels of optogenetic drive. Spikes were digitally removed prior to averaging (Methods). (H) Comparison of IPSP amplitude as a function of optogenetic stimulus strength with numerical simulation from a simple conductance-based model.
Figure S6. Center/surround optogenetic stimulation reveals lateral inhibition in NDNF- positive neurons, Related to Figure 4. This experiment is the same as in Fig. 4, with the replacement of 5-HT3AR-Cre by NDNF-Cre driver mice. (A) Simple model of L1 circuit with lateral inhibition. (B) Experiment to probe lateral inhibition in L1. NDNF-Cre mice expressed Optopatch4 in barrel cortex. Optogenetic stimuli were delivered separately to central and surrounding neurons. Voltage imaging was performed only in central neurons. Experiments were performed in anesthetized mice. (C) Epifluorescence images showing the illumination patterns in vivo. (D) Fluorescence waveforms from the central neurons under center/surround optogenetic stimulation. Central stimulation depolarized the targeted neurons and evoked spiking. Surround stimulation hyperpolarized the targeted neurons and suppressed spiking. (E) Spike raster showing responses from n = 19 neurons, 3 mice. (F) Mean spike rate during central stimulation, before and after surround stimulation. Surround stimulation caused spike rate to drop from 5.9 ± 1.9 Hz to 0 ± 0 Hz, n = 19 neurons, 3 mice (p = 0.007, two-sided paired-sample t-test). Shading represents s.e.m. (G) Mean subthreshold voltage during central stimulation, before and after surround stimulation. Surround stimulation caused inhibition in the central neuron. The initial spike in membrane voltage in the central neuron was due to scattered light from the surround which drove direct CheRiff activation. Shading represents s.e.m..
Fig. S7. Data and simulation of thalamic, optogenetic and cholinergic responses in L1 interneurons, Related to Figures 6, 7. (A) Simulated membrane voltage of single cells embedded in an L1 network during thalamocortical excitation. Here input noise and tonic depolarization were adjusted to achieve a spontaneous spike rate that matched experiment. (B) Stimulus-triggered average membrane potential with simulated thalamic inputs shows depolarization followed by a period of hyperpolarization. (C) Simulated thalamic inputs induced a period of reduced spontaneous activity. Firing rate quantified in 75 ms before the thalamic inputs (t1) were significantly higher than after the thalamic inputs (t2) (3.4 ± 1.1 Hz vs. 0.5 ± 0.4 Hz, n = 51 cells, p = 0.02, Kruskal-Wallis test, mean ± s.e.m.). (D) Top: Left: Geometry of annular optogenetic stimulation in a simulated L1 barrel. Cells were randomly positioned in a region 300 μm on an edge, 150 μm deep. A single SBC-like cell was manually defined to reside at the center of the region. The central neuron was subjected to tonic optogenetic depolarization. Neurons at radii r > 100 μm from the center were subjected to pulsed optogenetic stimulation. Right: Surround stimulation hyperpolarized the targeted neurons and suppressed spiking. Bottom: The same as top except that a central eNGC cell was subjected to tonic optogenetic depolarization. Compare to Fig. 4D. (E) Top: Air puff-induced responses in pairs of simultaneously recorded neurons, shown with shared background shading. Air puffs induced distinct concurrent responses, e.g. cell 3 showed a barrage of sustained firing while cell 4 did not spike; yet both cells had similar optogenetic excitability. Bottom: Air puff on top of blue light single-cell targeted optogenetic stimulation occasionally induced hyperpolarization (arrows). (F) Simulation of an L1 network response to a graded cholinergic input. The cholinergic input activated an excitatory conductance in all neurons. Under strong input, a few eNGC neurons become tonically active, suppressing spiking of the rest of the network. The plots show a randomly selected subset of the 51 neurons in the circuit. In the simulations, tonic activity was always in the eNGC cells, never in the SBC cells. (G) Numerical simulation showing spontaneous symmetry breaking in spike rate under strong cholinergic drive. L1 networks of 51 neurons were simulated with steady state cholinergic drive. At each value of gACh, 200 simulations were run with randomly reconfigured networks. For each simulation the value of gACh was held constant for 5 s and the spike rate was determined for each cell. The plot shows a histogram of the steady-state spike rate of all simulated neurons. The red overlay is a guide to the eye showing the bifurcation in firing rate at a critical stimulus threshold. Under strong stimulation approximately 10% of cells fired tonically while 90% were silent.
Data Availability Statement
Data, code and custom software are available upon request.