

Abstract
The essential role of membrane associated guanylate kinase 2 (MAGI2) in podocytes is indicated by the phenotypes of severe glomerulosclerosis of both MAGI2 knockout mice and in patients with congenital nephrotic syndrome (CNS) caused by mutations in MAGI2. Here, we show that MAGI2 forms a complex with the Rap1 guanine nucleotide exchange factor, RapGEF2, and that this complex is lost when expressing MAGI2 CNS variants. Co-expression of RapGEF2 with wild-type MAGI2, but not MAGI2 CNS variants, enhanced activation of the small GTPase Rap1, a central signaling node in podocytes. In mice, podocyte-specific RapGEF2 deletion resulted in spontaneous glomerulosclerosis, with qualitative glomerular features comparable to MAGI2 knockout mice. Knockdown of RapGEF2 or MAGI2 in human podocytes caused similar reductions in levels of Rap1 activation and Rap1-mediated downstream signaling. Furthermore, human podocytes expressing MAGI2 CNS variants show severe abnormalities of cellular morphology and dramatic loss of actin cytoskeletal organization, features completely rescued by pharmacological activation of Rap1 via a non-MAGI2 dependent upstream pathway. Finally, immunostaining of kidney sections from patients with congenital nephrotic syndrome and MAGI2 mutations showed reduced podocyte Rap1-mediated signaling. Thus, MAGI2-RapGEF2-Rap1 signaling is essential for normal podocyte function. Hence, disruption of this pathway is an important cause of the renal phenotype induced by MAGI2 CNS mutations.
Keywords: focal segmental glomerulosclerosis, nephrotic syndrome, podocyte
MAGI2 belongs to the MAGUK family of scaffolding proteins. It is composed of 5 PDZ domains, 2 WW domains, an Src homology (SH3) domain, and a catalytically inactive guanylate kinase domain. These multiple protein-protein interacting domains predict dual functions as a molecular scaffold and signaling hub. The MAGI subfamily has 3 members, designated MAGI1, MAGI2, and MAGI3, which share identical protein domains and similar molecular structure. In the kidney, MAGI2 is exclusively expressed in podocytes,1 the terminally differentiated epithelial cells that line the outer aspect of the glomerular basement membrane and regulate the filtration of blood to form urine. Within the podocyte, MAGI2 is specifically expressed at the slit diaphragm,1,2 a specialized cell-cell junction that bridges adjacent interdigitating podocyte foot processes. All forms of human proteinuric kidney disease, regardless of specific etiology, result in podocyte injury with loss of normal foot process architecture, a pathologic process known as effacement.
Several recent individual MAGI2 knockout (KO) mouse models all demonstrate the gene’s essential roles in podocyte development and function.3–6 Two global KO mouse models died within the first 24 hours after birth from anuric renal failure with absence of slit diaphragm formation.3,5 In the third global KO model4 and in a podocyte-specific KO model,6 severe focal segmental and global glomerulosclerosis developed with renal failure and early lethality. In humans as well, convincing disease-causing mutations in MAGI2 give rise to CNS.7,8 Most cases present with severe nephrotic syndrome in the first few months of life. Whereas these findings establish the critical role of MAGI2 in proper podocyte function in mice and humans, the underlying mechanisms are poorly understood.
MAGI proteins previously have been implicated as upstream activators of the small GTPase Rap1,9,10 which regulates multiple essential downstream signaling pathways in podocytes, including activation of β1 integrin.11 In fact, severe glomerulosclerosis develops in mice with reduced podocyte Rap1 expression, and they die early of renal failure by 8 weeks of age.11 Rap1 cycles between an active guanosine triphosphate (GTP)–bound form and an inactive guanosine diphosphate–bound form. The balance between active and inactive Rap1 is tightly regulated by numerous upstream factors including guanine nucleotide exchange factors (GEFs), which turn on signaling by catalyzing the exchange of guanosine diphosphate to GTP, and GTPase activating proteins, which terminate signaling by hydrolyzing GTP to guanosine diphosphate.12 Rap1-specific GEFs include RapGEF1, RapGEF2 (PDZ-GEF1), RapGEF3 (EPAC1), RapGEF4 (EPAC2), RapGEF5, and RapGEF6 (PDZ-GEF2). It has been shown that RapGEF2 forms a complex with MAGI proteins,10 has enriched expression in the mammalian glomerulus,13 and is important in maintenance of the glomerular filtration barrier in zebra fish.14 With this background, we aimed to investigate the RapGEF2-MAGI2 protein complex as a potential upstream regulator of podocyte Rap1 activation.
RESULTS
MAGI2 variants that cause congenital nephrotic syndrome fail to form a complex with RapGEF2 and are unable to induce Rap1 activation
Two homozygous truncating point mutations in the human MAGI2 gene (pGly39* and pTyr746*) have been identified as directly causative of CNS7 (comparison of domain organizations, Figure 1a). Both affected patients presented with severe CNS early in childhood.7 Reciprocal coimmunoprecipitation experiments performed between FLAG-RapGEF2 and full-length green fluorescent protein (GFP)–MAGI2 show a robust protein complex formation (Figure 1b). However, this protein complex is completely lost when substituting either of the GFP-tagged MAGI2 disease-causing variants. GFP-Toca, used as a negative control, also failed to form a complex with FLAG-RapGEF2.
Figure 1 |. Membrane-associated guanylate kinase 2 (MAGI2), but not variants that cause congenital nephrotic syndrome (CNS), form a complex with RapGEF2.
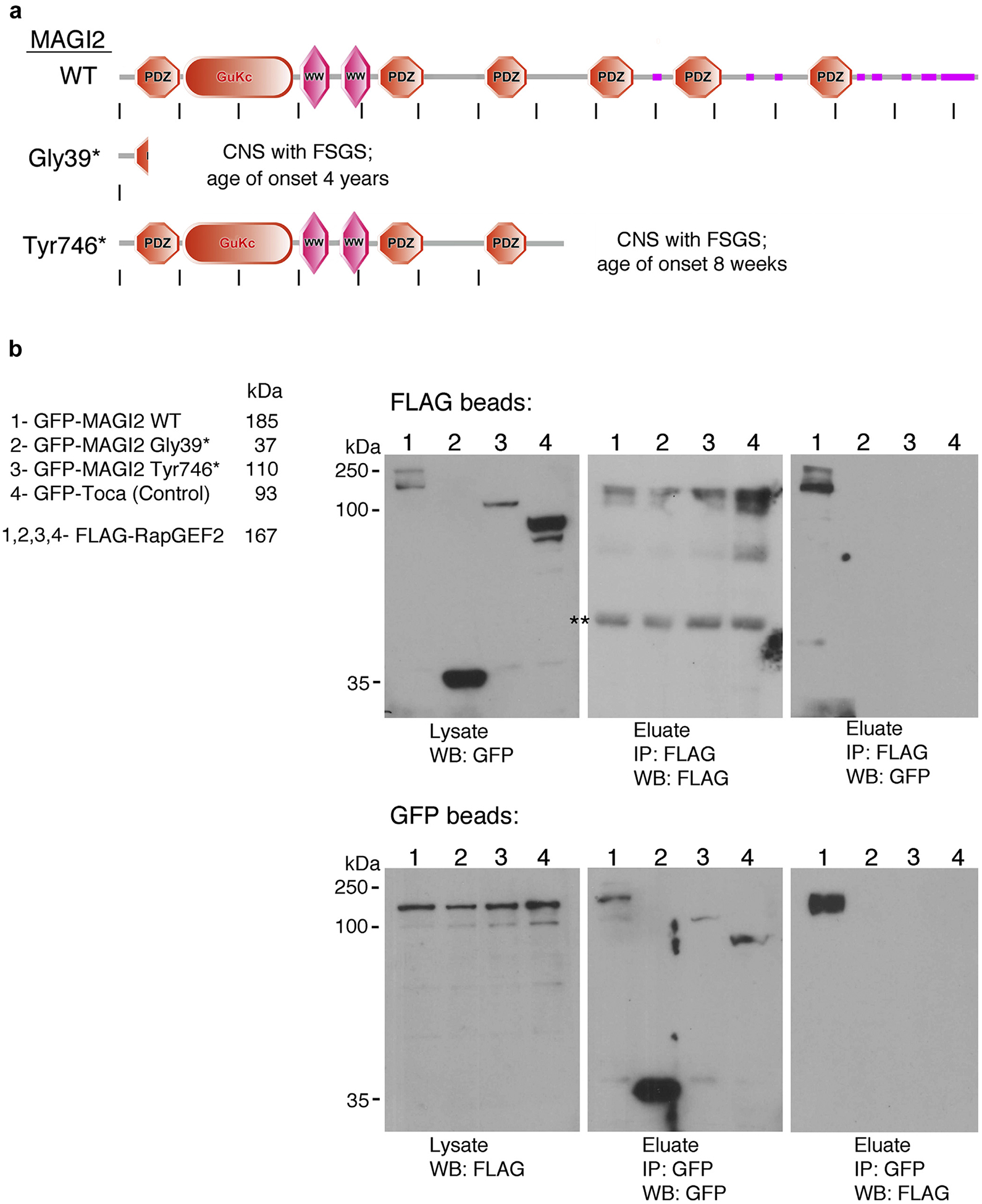
(a) Schematic representation of protein domain organization of wild-type MAGI2 and 2 of its disease-causing variants, Gly39* and Tyr746*. Each variant carries a homozygous truncating point mutation. Image was generated using the Simple Modular Architecture Research Tool (SMART) web resource and then modified. (b) Reciprocal co-immunoprecipitations between FLAG-tagged RapGEF2 and green fluorescent protein (GFP)–tagged MAGI2 were performed using antibodies to their tags. FLAG-RapGEF2 and the indicated GFP-tagged MAGI2 protein lysates were mixed at equal concentration and incubated overnight with FLAG (upper blot) or GFP (lower blot) antibodies bound to agarose. A robust protein complex is present between RapGEF2 and wild-type MAGI2 but is completely lost for MAGI2 mutant variants or for the negative control, GFP-Toca. Predicted molecular weights for each protein are as follows: GFP-MAGI2 WT, 185 kDa; GFP-MAGI2 Gly39*, 37 kDa; GFP-MAGI2 Tyr746*, 110 kDa; GFP-Toca, 93 kDa; and FLAG-RapGEF2, 167 kDa. FSGS, focal segmental glomerulosclerosis. **Nonspecific band.
To look for a functional consequence to the loss of this protein complex, we tested the ability of each to induce Rap1 activation (Figure 2a). We utilized calcium switch as a means to induce cellular Rap1 activation using a previously established protocol.9,10 Confluent transfected 293T cells were exposed briefly to calcium chelation to disrupt intercellular contacts (ethyleneglycol-bis-(β-aminoethylether)-N,N,N’,N’-tetraacetic acid [EGTA] washout), after which full calcium-containing medium was restored (calcium switch). Consistent with published data, we found a general increase in levels of Rap1 activation in all transfectants after calcium switch (Figure 2a). However, this effect was dramatically enhanced in 293T cells expressing both RapGEF2 and full-length MAGI2 but was not present in cells expressing both RapGEF2 and either of the MAGI2 mutant variants. Previous work also had implicated MAGI1 as a regulator of podocyte Rap1 activation.10 However, using an identical calcium switch system, the combined expression of MAGI1 and RapGEF2 did not augment Rap1 activation beyond baseline (Figure 2b). Overall, these findings demonstrate that the protein complex of MAGI2, but not disease-causing MAGI2 variants, with RapGEF2 functions as a potent stimulus for Rap1 activation.
Figure 2 |. Combined expression of RapGEF2 and membrane-associated guanylate kinase 2 (MAGI2), but not disease-causing MAGI2 variants, induce Rap1 activation.
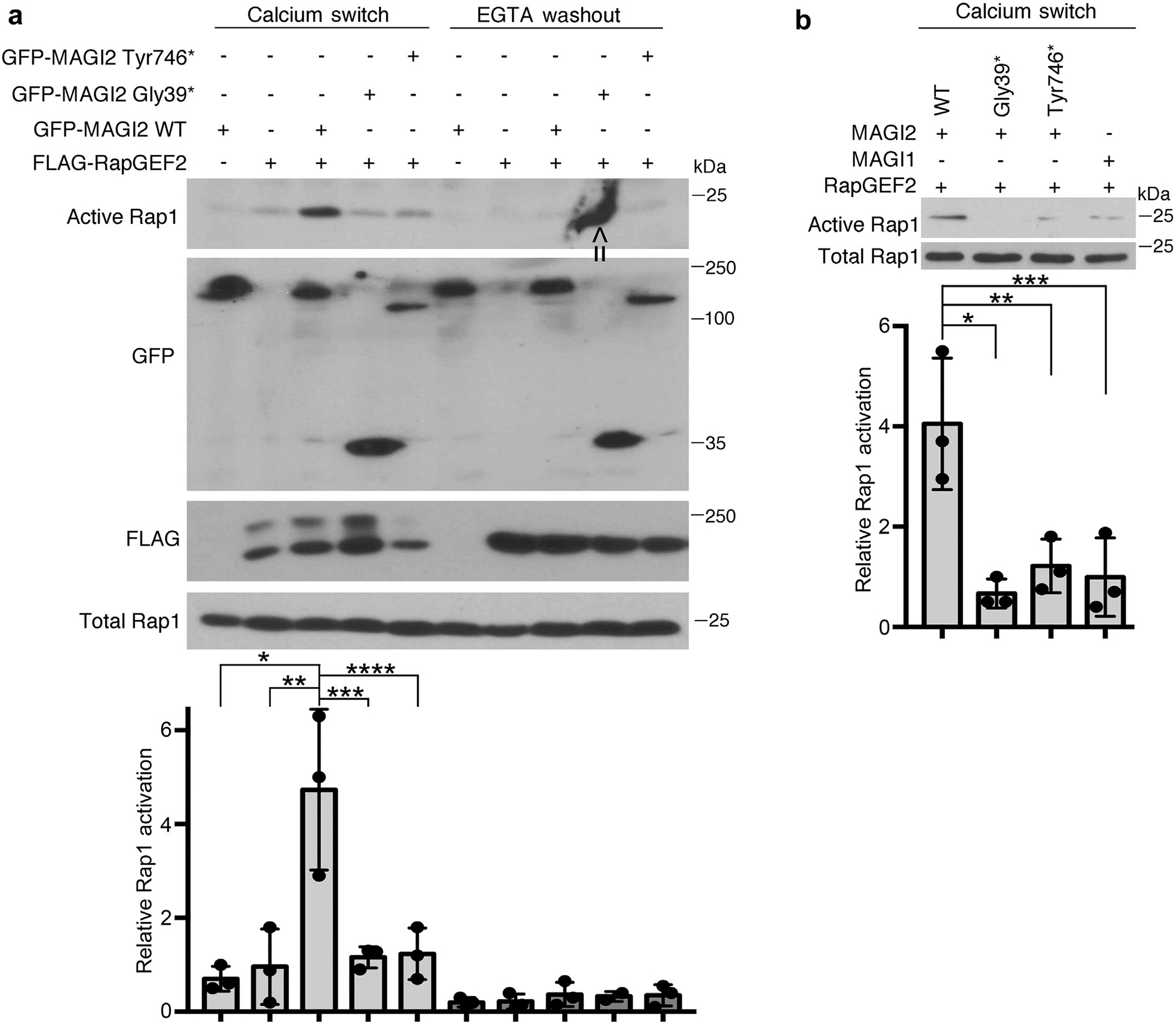
(a) Confluent transfected 293T cells were exposed briefly to calcium chelation followed by restoration of full calcium-containing medium (calcium switch). Cellular lysates were collected both before and after the calcium switch and assayed for levels of active Rap1. Whereas there was a generalized increase in Rap1 activation in all samples after calcium switch, this response was dramatically augmented in cells that expressed both RapGEF2 and MAGI2, but not in cells expressing RapGEF2 and disease-causing MAGI2 alleles. 293T cells were transfected with the following expression plasmids: lanes 1 and 6: green fluorescent protein (GFP)–MAGI2 wild type (WT); lanes 2 and 7: FLAG-RapGEF2; lanes 3 and 8: GFP-MAGI2 WT and FLAG-RapGEF2; lanes 4 and 9: GFP-MAGI2 Gly39* and FLAG-RapGEF2; and lanes 5 and 10: GFP-MAGI2 Tyr746* and FLAG-RapGEF2. Lower panel shows the Western blot densitometric quantification comparing relative levels of Rap1 activation between groups. The ratio of active to total Rap1 was calculated for 3 individual experiments and then normalized. *P < 0.025, **P < 0.035, ***P < 0.03, ****P < 0.04. Arrow indicates nonspecific blotting artifact over the ninth lane, which was excluded from densitometric analysis. (b) Levels of Rap1 activation also were not augmented in cells expressing RapGEF2 and MAGI1. 293T cells were transfected with the following plasmids: lane 1: GFP-MAGI2 WT and FLAG-RapGEF2; lane 2: GFP-MAGI2 Gly39* and FLAG-RapGEF2; lane 3: GFP-MAGI2 Tyr 746* and RapGEF2; and lane 4: GFP-MAGI1 and FLAG-RapGEF2. Densitometric quantificationof active to total Rap1 was calculated for 3 separate experiments and then normalized. *P < 0.02, **P < 0.025, ***P < 0.03. EGTA, ethyleneglycol-bis-(b-aminoethylether)-N,N,N’,N’-tetraacetic acid.
Podocyte-specific RapGEF2 KO mice develop focal segmental glomerulosclerosis (FSGS) with comparable qualitative glomerular features compared with MAGI2 KO mice
We next performed detailed immunofluorescence studies to localize the RapGEF2 protein within wild-type kidney (Figure 3a). Unlike MAGI2, RapGEF2 is robustly expressed in both the glomerular and tubular compartments. Within the glomerulus, it is strongly expressed in the podocyte cytoplasm, where it co-localizes with the podocyte cytoplasmic marker nestin.
Figure 3 |. Glomerular RapGEF2 expression is lost in podocyte-specific RapGEF2 knockout (KO) mice.
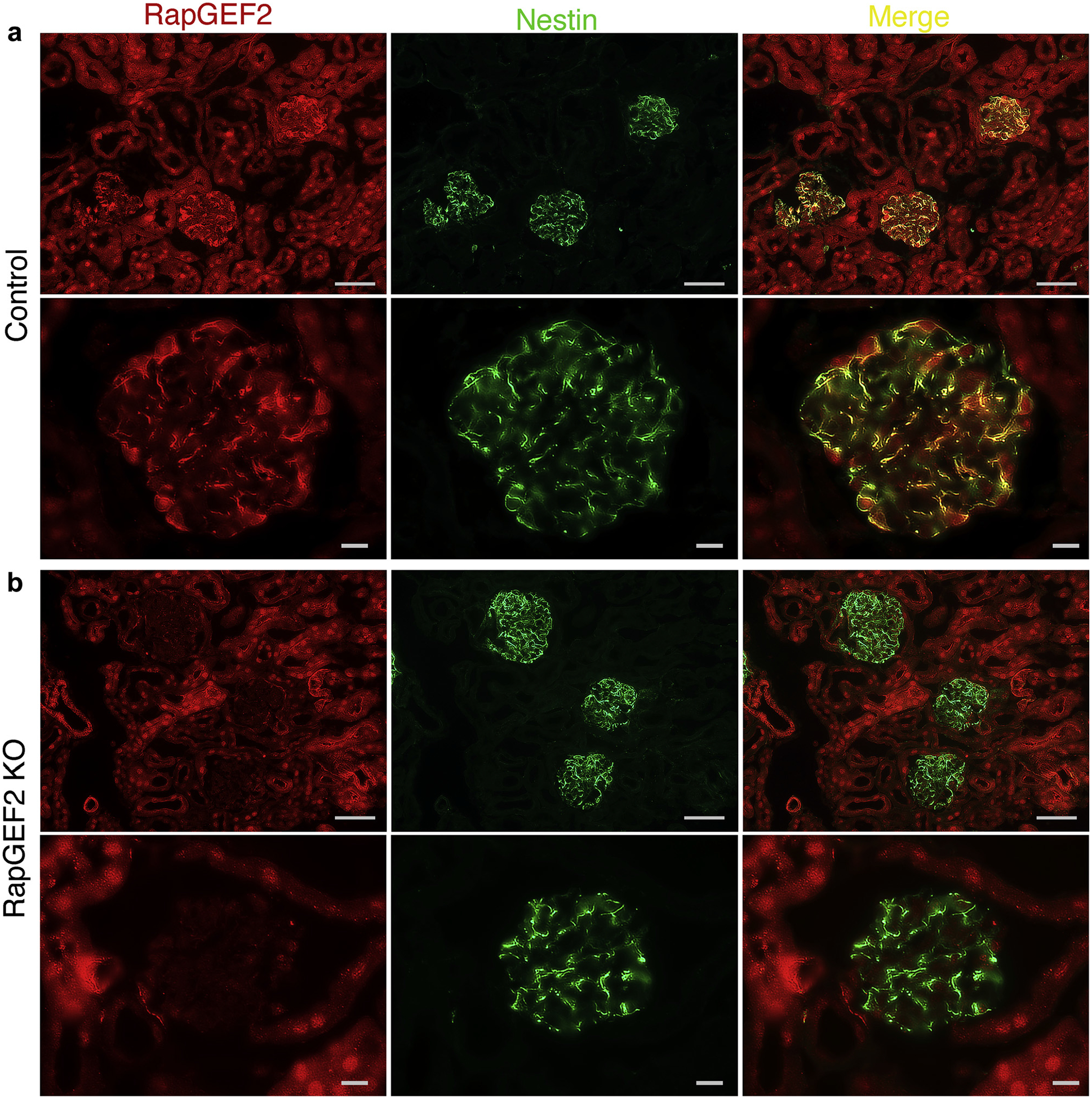
(a) Immunostaining for RapGEF2 on kidney sections of control mice demonstrates widespread expression in both tubules and podocytes. Its podocyte expression is mainly cytoplasmic, where it co-localizes with the podocyte cytoplasmic marker nestin. Bar = low power, 50 μm; high power, 10 μm. (b) In podocyte-specific RapGEF2 KO mice, glomerular RapGEF2 expression is absent but tubular expression is preserved. Bar = low power, 50 μm; high power, 10 μm.
We hypothesized that if RapGEF2 function were to be mediated by its ability to form a complex with MAGI2, then RapGEF2 KO mice, similar to MAGI2-deficient mice, would demonstrate severe podocyte dysfunction. Because global RapGEF2 KO mice are early embryonic lethal,15 we bred a conditional floxed RapGEF2 KO mouse line15 to a transgenic mouse line that expresses cre recombinase under the podocyte-specific promoter podocin.16 This approach allowed the generation of podocyte-specific KO of RapGEF2. RapGEF2 immunostaining verifies loss of glomerular RapGEF2 expression but preservation of tubular RapGEF2 expression in these mice (Figure 3b).
Overt proteinuria develops in podocyte-specific RapGEF2 KO mice between 6 and 16 weeks of age (Figure 4a) with progressive glomerulosclerosis and severe proteinuria by 20 weeks of age (Figure 4b). The renal phenotype is comparable to that described for podocyte-specific MAGI2 KO6 and global MAGI2 KO mice,4 although the onset of proteinuria for the RapGEF2 model is later and glomerulosclerosis scores overall are less severe (Figure 4c). Renal histologic analysis reveals similar qualitative glomerular features between RapGEF2 and MAGI2 models that include focal segmental and global glomerulosclerosis, mesangial expansion, podocyte loss, and glomerular epithelial cell proliferation with pseudocrescent formation (Figure 4d). Electron microscopic features are similar between the 2 models: diffuse severe foot process effacement with basal actin matting, simplification of overall podocyte cellular shape, loss of normal primary and secondary process architecture, and cytoplasmic vacuolization (Figure 4e). Podocyte loss is a major driving force for development of glomerulosclerosis.17 To quantify podocyte depletion in our model, we calculated the number of Wilms tumor 1 (WT1)-positive cells per glomerular tuft area (Figure 4f). Consistent with the severity of their pathologic findings, podocyte loss was greater in MAGI2 KO mice than in RapGEF2 KO mice, but both models showed substantially fewer podocytes compared with control mice.
Figure 4 |. odocyte-specific RapGEF2 knockout (KO) mice develop focal segmental glomerulosclerosis (FSGS) with comparable qualitative glomerular features compared with membrane-associated guanylate kinase 2 (MAGI2) KO mice.
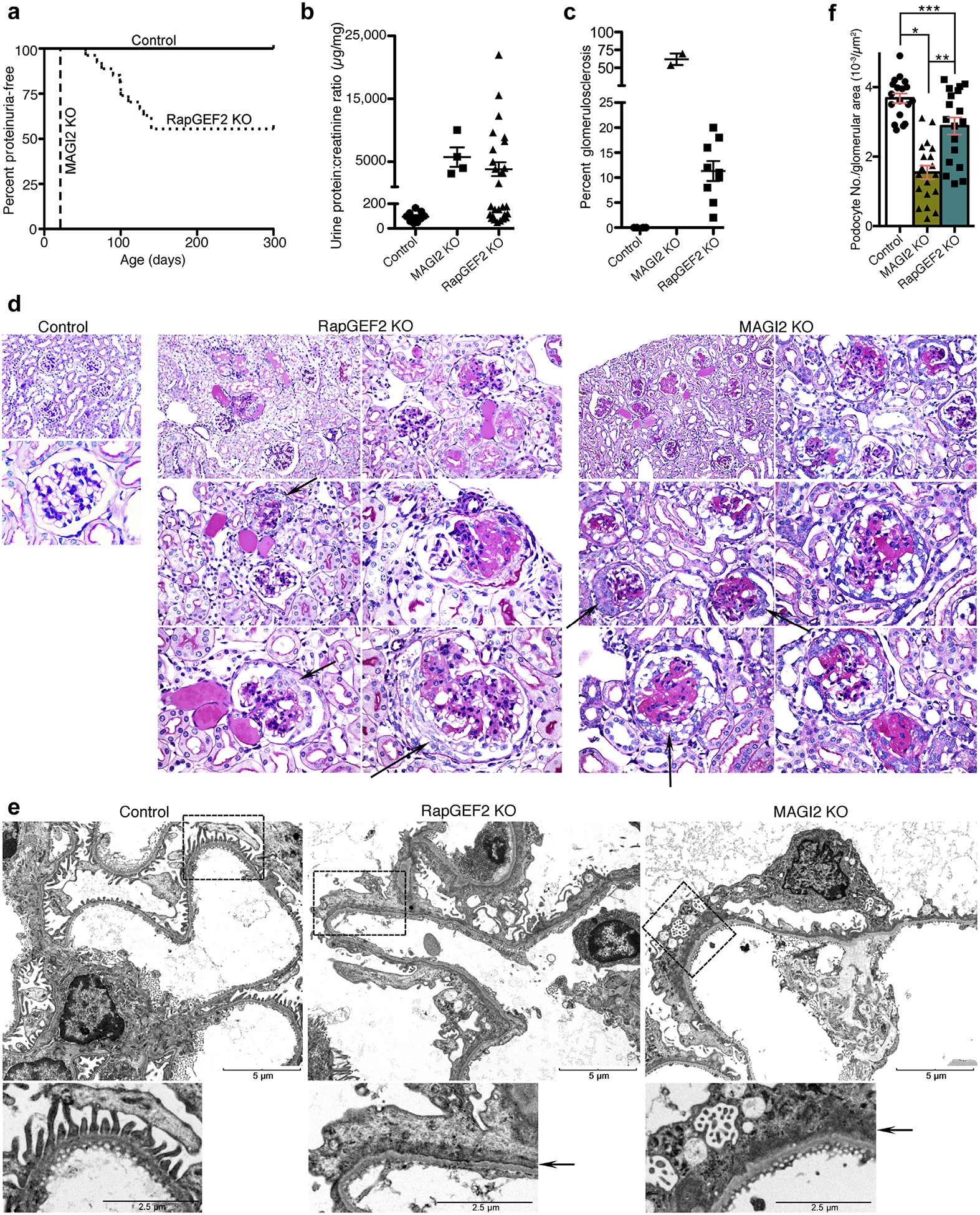
(a) Survival curve showing age of onset of proteinuria. Urine protein:creatinine ratios were measured weekly with the onset of proteinuria defined by a protein:creatinine ratio of greater than 400 mg/mg for 2 consecutive weeks. (b) Calculated protein:creatinine ratios for RapGEF2 KO mice (n = 12, at age 20 weeks or at the time they were killed) and global MAGI2 KO mice (n = 4, age 5 weeks). Severe proteinuria develops in both models. (c) For each mouse with proteinuria, the percentage of glomeruli demonstrating glomerulosclerosis on periodic acid–Schiff staining was calculated. (d) Comparative pathologic analysis of proteinuric RapGEF2 KO mice (age 12 weeks) with MAGI2 KO mice (age 5 weeks). Both models demonstrate focal segmental and global glomerulosclerosis, mesangial expansion, and parietal cell proliferation with pseudocrescent formation. Original magnification for control: upper, ×200; lower, ×600. Original magnification for RapGEF2 and MAGI2 KO: upper left, ×200; upper right and middle left, ×400; middle right and lower left and right, ×600. Arrow shows parietal cell proliferation with pseudocrescent. (e) Electron microscopy reveals comparable qualitative features between proteinuric podocyte-specific RapGEF2 KO and MAGI2 KO mice. Both show diffuse foot process effacement, actin cytoskeletalmatting parallel to the glomerular basement membrane, and simplification and loss of primary and secondary processes. Bar = low power, 5 μm; high power, 2.5 mm. Arrow shows actin cytoskeletal matting present in both models. (f) Podocyte loss was evident in both MAGI2 and proteinuric RapGEF2 KO mice. The number of Wilms tumor 1 (WT1)–positive cells per glomerular tuft area was determined for glomeruli where the macula densa was clearly evident; n = 3 mice per group, with 18 total glomeruli analyzed for each group (*P < 0.0001, **P < 0.0002, ***P < 0.008).
RapGEF2 or MAGI2 knockdown in podocytes similarly diminish Rap1-GTP activation and signaling
We hypothesized that podocyte dysfunction in both the RapGEF2 and MAGI2 KO mouse models was caused by similar reductions in podocyte Rap1 activation and downstream Rap1 signaling pathway activation. To investigate this possibility, we transduced a short hairpin (sh)RNA expression plasmid that targeted either RapGEF2 or MAGI2 RNA into a conditionally immortalized human podocyte cell line.18 To eliminate the possibility of nonspecific off-target effects, we utilized 2 different hairpins per gene, each with a distinct RNA target sequence. In this way, we generated 2 distinct RapGEF2 knockdown and 2 distinct MAGI2 knockdown podocyte cell lines. By Western blotting, we confirmed severely reduced RapGEF2 or MAGI2 protein expression in each of the knockdown podocyte cell lines compared with control podocytes that were transduced with a scrambled shRNA expression plasmid (Figure 5a).
Figure 5 |. RapGEF2 or membrane-associated guanylate kinase 2 (MAGI2) knockdown in podocytes similarly diminish Rap1 activation and Rap1-mediated downstream signaling.
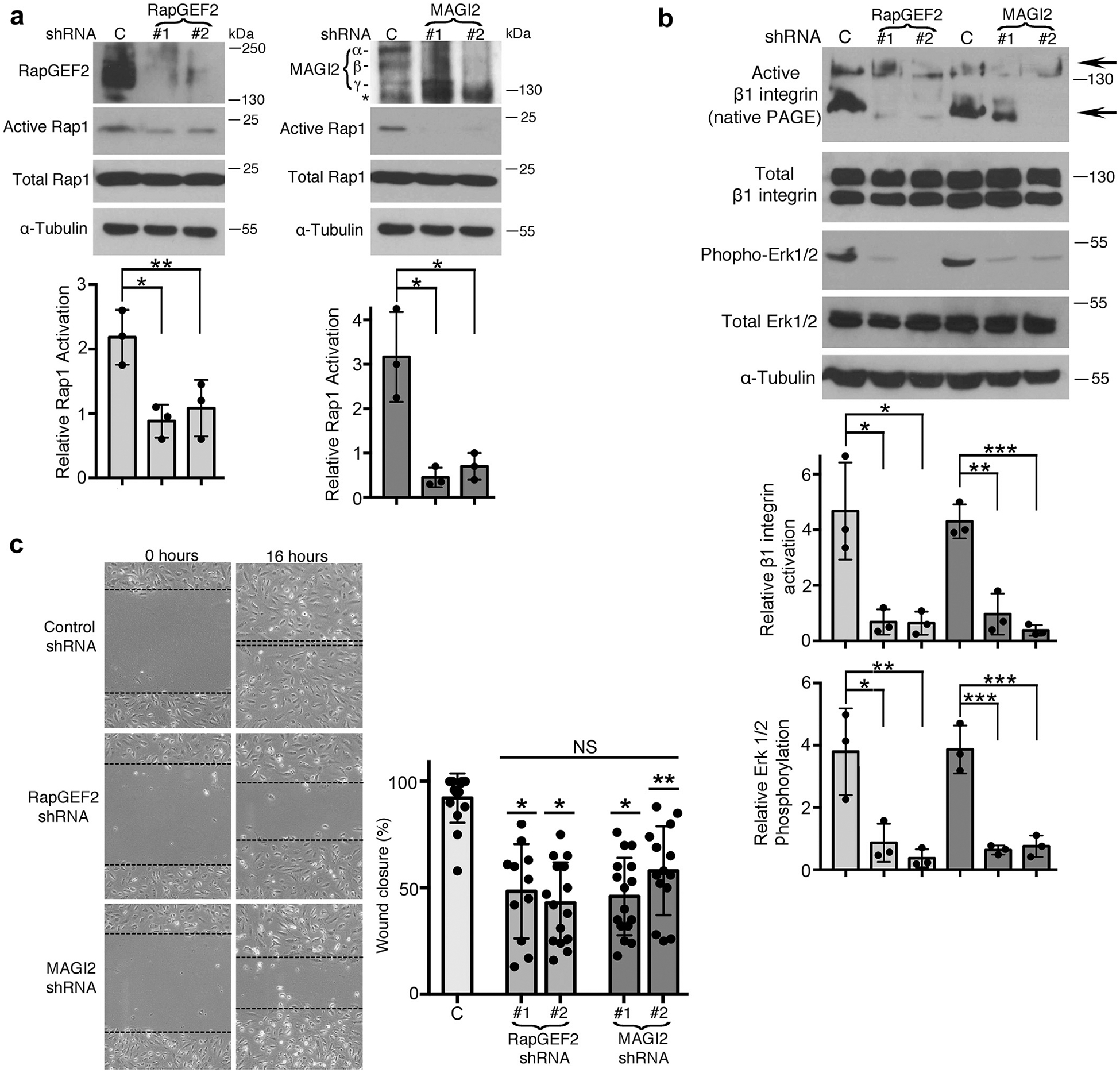
(a) Human cultured podocytes transduced with short hairpin (sh)RNA expression plasmid targeting RapGEF2 or MAGI2 mRNA demonstrated diminished RapGEF2 or MAGI2 protein expression, respectively, compared with podocytes expressing a scrambled shRNA. Two distinct shRNA plasmids were used for both RapGEF2 and for MAGI2, each with a unique mRNA target sequence. Levels of Rap1 activation induced by calcium switch were dramatically reduced in all 4 knockdown cell lines compared with control cells. Ratio of active to total Rap1 was calculated for 3 individual experiments and then normalized (for control cells vs. RapGEF2 #1 and #2, *P < 0.03, **P < 0.04; for control cells vs. MAGI2 #1 and #2, *P < 0.01 for both). α, β, and γ indicate major MAGI2 splice isoforms. The asterisk represents a nonspecific band. (b) Western blotting of podocytes transduced as indicated was performed after calcium switch. Levels of β1 integrin activation and Erk1/2 phosphorylation (both major Rap1 downstream signals) weresubstantially diminished. Densitometric ratios of active to total β1 integrin were calculated for 3 individual experiments and then normalized. *P < 0.02, **P < 0.01, ***P < 0.001. Quantification combined the intensities of the 2 active bands for β1 integrin (shown by arrows; the upper 130 kDa band represents its fully mature glycosylated form, and the lower 110 kDa band represents its precursor form) and compared it with the combined intensities of the mature and immature forms of total β1 integrin. Densitometric quantificationfor phospho to total Erk1/2 is also shown. *P < 0.03, **P < 0.02, ***P < 0.01. (c) RapGEF2 and MAGI2 knockdown cells migratedmore slowly than control cells. The percent wound closure was quantified at fixed locations along the scratch. *P < 0.0001, **P < 0.002 (each vs. control subjects). NS, not significant; PAGE, polyacrylamide gel electrophoresis.
In the setting of calcium switch, RapGEF2 and MAGI2 knockdown podocytes showed similar reductions in the level of Rap1 activation (Figure 5a), as well as dramatically reduced levels of essential Rap1-mediated downstream signals including b1 integrin activation and Erk phosphorylation (Figure 5b). To detect levels of activated β1 integrin, native polyacrylamide gel electrophoresis (nonreducing and nondenaturing sample buffer) was used; under these conditions, the monoclonal antibody 12G10 specifically detects the activated form of β1 integrin. Next, we studied cellular migration rates, which are substantially reduced in podocytes that have diminished cellular Rap1 activation,11 a reliable phenotype observed in podocyte-specific knockdown of other genes known to cause nephrotic syndrome.7,19,20 Again, RapGEF2 and MAGI2 knockdown podocyte cell lines behaved similarly, each showing dramatic reductions in migratory rates (Figure 5c). Diminished cellular Rap1 activation also was previously reported to enhance podocyte susceptibility to apoptosis.11 With use of Annexin V staining, we found comparable high rates of podocyte apoptosis under basal condition for both RapGEF2 and MAGI2 knockdown podocytes compared with control cells (Figure 6a and b). Taken together, these data demonstrate that loss of either RapGEF2 or MAGI2 in podocytes causes disruption to Rap1 activation and downstream signaling.
Figure 6 |. RapGEF2 or membrane-associated guanylate kinase 2 (MAGI2) knockdown in podocytes results in similarly high rates of apoptosis.
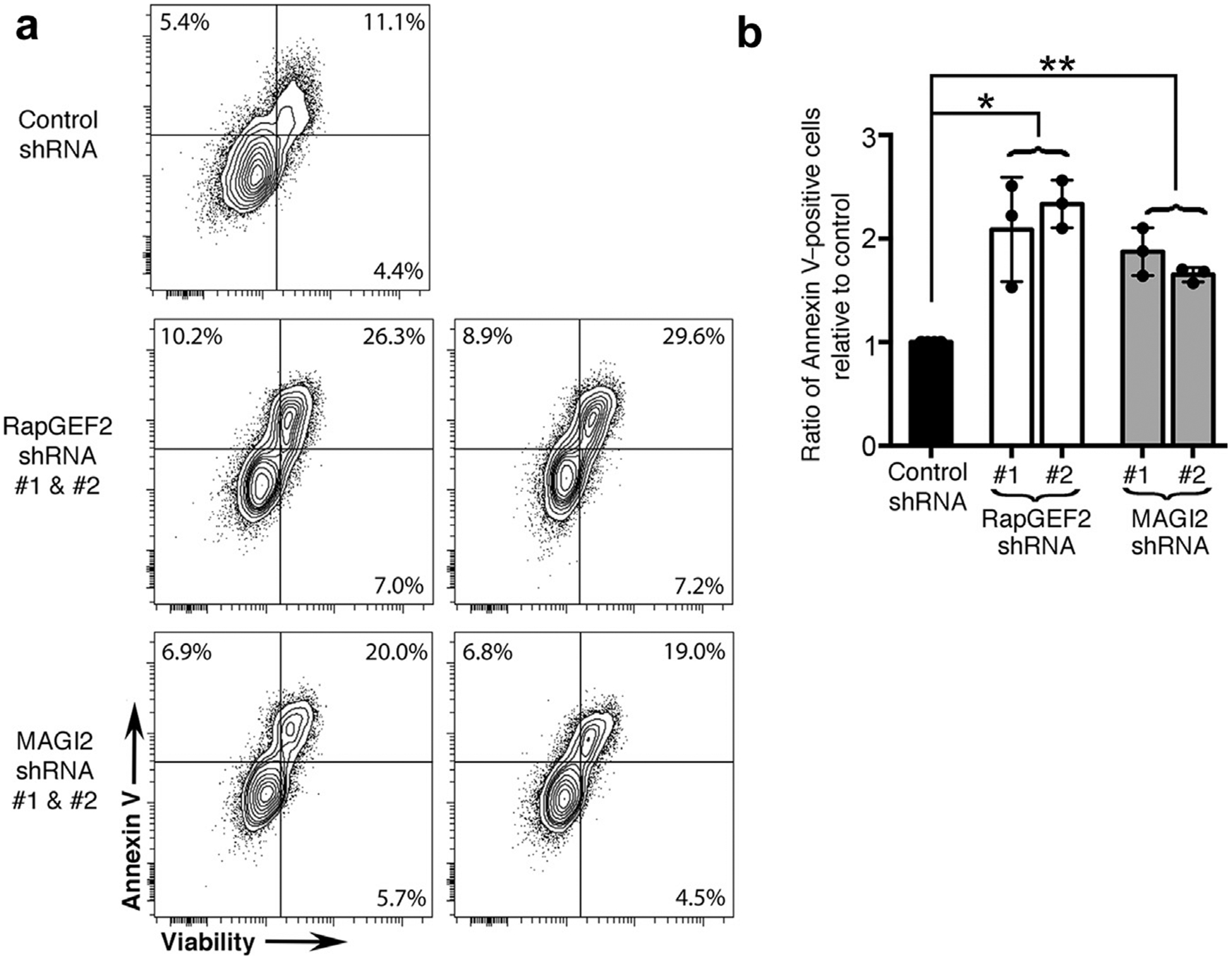
(a) Differentiated RapGEF2 and MAGI2 knockdown podocytes were cultured for 48 hours under basal conditions, after which adherent and nonadherent cells were collected, combined, and then assayed for Annexin V and 7-AAD by fluorescence-activated cell sorting analysis. Both RAPGEF2 and MAGI2 knockdown podocytes show enhanced levels of apoptosis compared with control subjects. Experiments were repeated a total of 3 times using newly grown cells each time. (b) The ratio of Annexin V–positive cells relative to control subjects was calculated for each experiment, and results were averaged. *P < 0.0005 (RapGEF2 vs. control), **P < 0.0002 (MAGI2 vs. control), no significant difference between RapGEF2 and MAGI2. shRNA, short hairpin RNA.
Human podocytes that express MAGI2 CNS-causing variants are phenotypically rescued by pharmacologic activation of Rap1
Here we generated individual human podocyte cell lines that express FLAG-tagged wild-type MAGI2 and each of the FLAG-tagged CNS MAGI2 human mutant variants (pGly39* and pTyr746*). This step was accomplished by transduction of a lentiviral expression plasmid into human podocytes under permissive conditions (vector backbone pSF-Lenti), followed by selection with puromycin. Robust expression of wild-type and mutant versions of MAGI2 in transduced podocytes is shown by immunofluorescence using an anti-FLAG antibody compared with control vector–alone transduced podocytes (Figure 7a). Interestingly, whereas wild-type MAGI2 strongly localized to sites of intercellular contact, both of the MAG2 mutant proteins were completely mislocalized, showing robust accumulation in cytoplasmic vesicles and almost no expression at intercellular contacts. Control podocytes lacked any immunoreactivity.
Figure 7 |. Podocytes expressing membrane-associated guanylate kinase 2 (MAGI2) congenital nephrotic syndrome (CNS) variants are rescued by pharmacologic activation of Rap1.
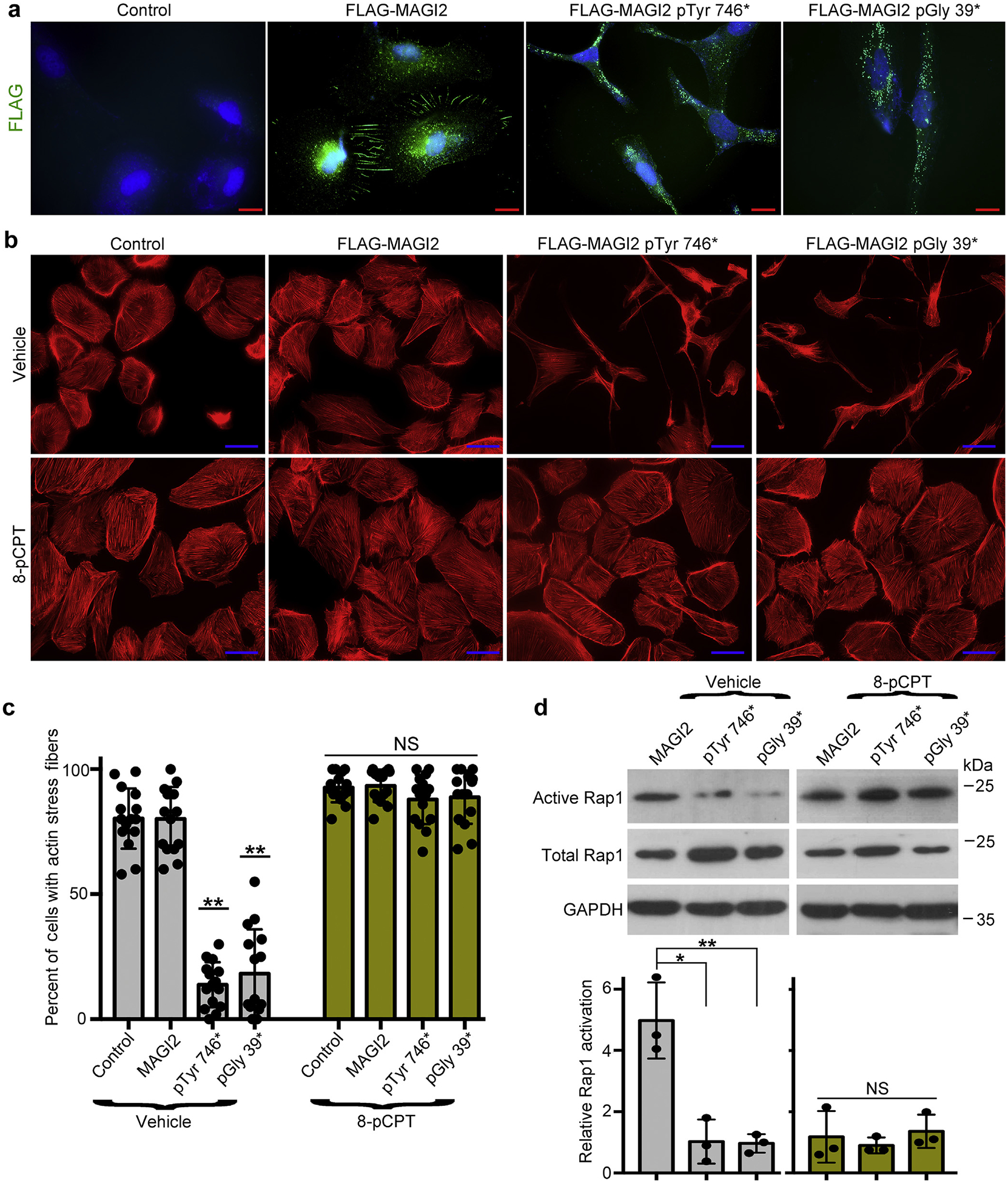
(a) Human podocytes were transduced with lentiviral expression plasmidscarrying full-length FLAG-MAGI2, FLAG-MAGI2 pTyr746*, or FLAG-MAGI2 pGly 39* or with lentiviral vector alone (control). Each podocyte line was fully differentiated, and then immunofluorescence was performed using an anti-FLAG antibody. Wild-type FLAG-MAGI2 robustly accumulated at sites of intercellular contacts. In both of the CNS-causing MAGI2 variants, however, FLAG-MAGI2 was dramatically mislocalized, demonstrating punctate protein accumulation throughout the cytoplasm. Control vector–transduced podocytes showed no immunoreactivity. Bar = 20 μm. (b) Each podocyte cell line was differentiated for 7 days either with or without the potent Rap1 activator 8-pCPT-2′-O-Me-cAMP (8-pCPT) and then stained for actin using phalloidin. Whereas vector-alone and wild-type MAGI2-transduced podocyte lines each demonstrated a normal structure and an intricate actin cytoskeleton, podocytes that expressed either of the human MAGI2 disease-causing variants demonstrated severe abnormalities of the cellular structure and a dramatic loss of actin filament organization (top row). However, when cultured in the presence of 8-pCPT, the MAGI2 mutant phenotype was completely rescued with the normalization of the cell shape and restoration of a robust actin cytoskeletal structure (lower row). Bar = 50 μm. (c) Quantification of actin staining is shown. Three blinded observers each assessed 500 phalloidin-stained cells for each podocyte line (a total of 1500 cells were assessed per line). Cells were considered positive when actin filaments were robust throughout the entire cell. Each individual dot represents the percentage of positive cells per 100 cells analyzed. **P < 0.0001 (each vs. control or MAGI2). NS, not significant. (d) Podocyte lines were differentiated with either vehicle or 8-pCPT, and protein then was harvested for active Rap1 pull-down assays or Western blotting. Levels of active Rap1 were substantially reduced in podocytes expressing mutant MAGI2 variants, but in the presence of 8-pCPT they were restored to levels similar to podocytes expressing wild-type MAGI2. Densitometric quantification ofactive to total Rap1 was calculated for 3 separate experiments and then normalized. *P < 0.02, **P < 0.01. NS, not significant. GADPH, glyceraldehyde-3-phosphate dehydrogenase.
Differentiated podocytes that expressed either of the human MAGI2 disease-causing variants demonstrated severe abnormalities of cellular structure, including an elongated cellular shape and a disorganized actin cytoskeleton (Figure 7b and c). Vector-alone and wild-type MAGI2 transduced podocyte lines each demonstrated normal structure and intricate actin filament organization. Podocytes expressing either of the MAGI2 disease-causing mutations show diminished basal Rap1 activation compared with podocytes expressing wild-type MAGI2 (Figure 7d). To show that inactivation of the MAGI2-RapGEF2-Rap1 signal is the cause of podocyte dysfunction induced by disease-causing MAGI2 mutations, rescue experiments were performed using pharmacologic activation of Rap1 via a non–MAGI2-dependent upstream pathway. Individual podocyte cell lines were split from the same flask prior to moving to 37 °C and then were maintained under nonpermissive conditions for 7 days, either with or without the potent Rap1-activating drug 8-pCPT-2′-O-Me-cAMP (10 μM).21 In the presence of the drug, levels of activated Rap1 were dramatically increased in the mutant MAGI2-expressing podocyte cell lines to levels similar to wild-type MAGI2-expressing cells (Figure 7d). Impressively, mutant MAGI2-transduced podocytes were completely rescued by pharmacologic Rap1 activation, including normalization of cellular shape with restoration of a robust and well-organized actin cytoskeletal structure (Figure 7b and c). These findings suggest that inactivation of RapGEF2-Rap1 signaling is an important cause of the renal phenotype induced by MAGI2 CNS mutations.
Immunostaining of kidney sections from patients with CNS who have MAGI2 mutations is consistent with reduced glomerular Rap1GTPase-mediated signaling
To corroborate our findings in humans, we performed immunohistochemistry on kidney sections from patients who had CNS as a result of mutations in MAGI2.8 Because direct staining for activated Rap1 is not feasible, we investigated activation of the Rap1-mediated downstream signal Erk1/2 (Figure 8a and b). Although phosphorylated Erk1/2 is highly enriched in podocytes of control kidney, it is essentially undetectable in podocytes from patients with CNS caused by MAGI2 mutations. Levels of Erk1/2 phosphorylation were only moderately reduced in podocytes from patients who had idiopathic FSGS compared with control subjects, suggesting that such dramatic loss of podocyte Erk1/2 phosphorylation cannot be generalized to all forms of podocyte injury. Importantly, total Erk1/2 was ubiquitously expressed in all samples, including substantially in podocytes, suggesting that podocyte loss is not the main explanation for our observed reductions in phosphorylated Erk1/2 staining. Overall, these findings are consistent with reduced levels of podocyte Rap1 activation in patients with CNS who harbor mutations in MAGI2.
Figure 8 |. Immunohistochemistry of kidney from patients with congenital nephrotic syndrome (CNS) caused by membrane-associated guanylate kinase 2 (MAGI2) mutations is consistent with reduced podocyte Rap1 signaling.
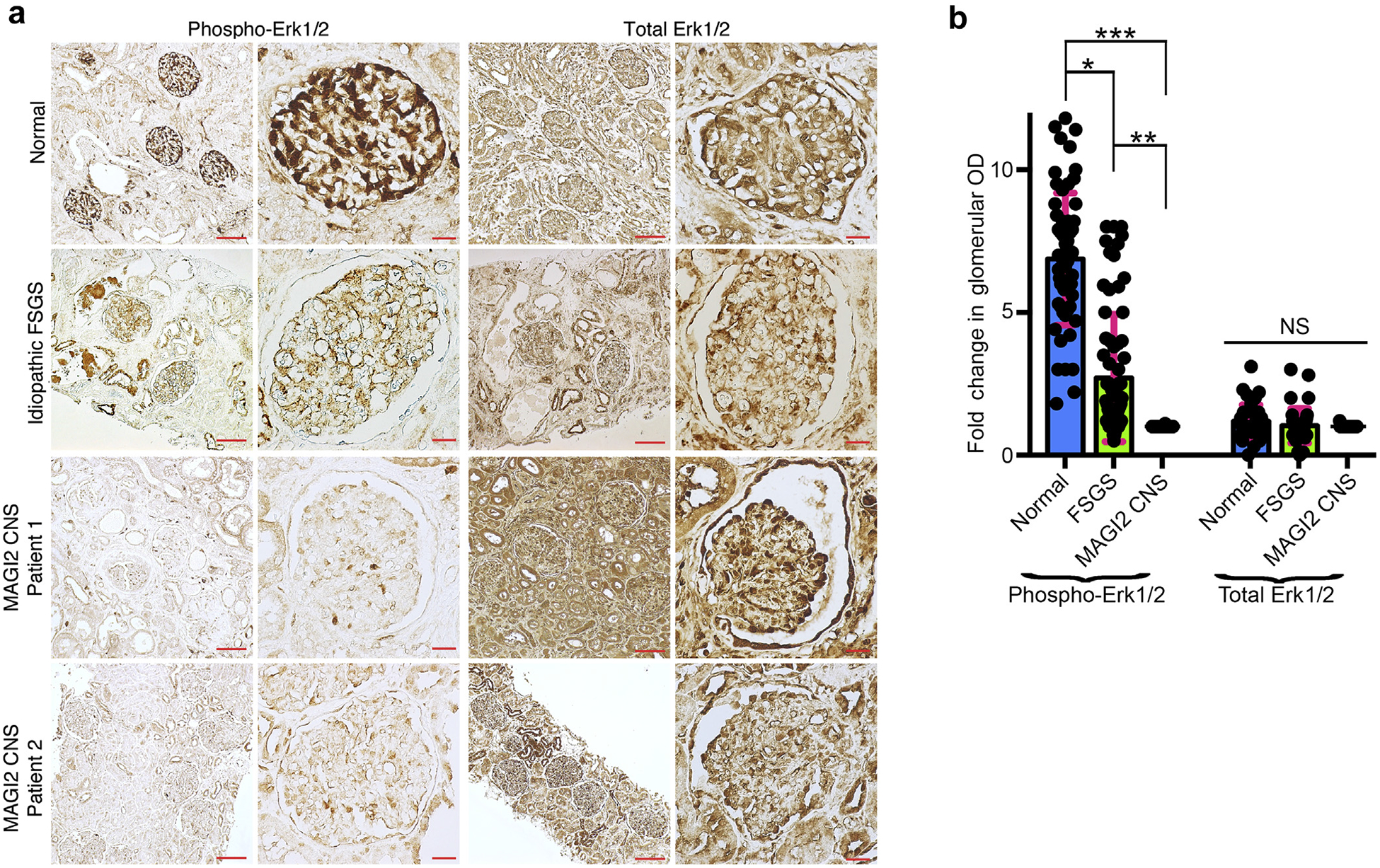
(a) Immunostaining for the Rap1 downstream effector phospho-Erk1/2 was performed on paraffin-embedded human kidney sections. Representative images are shown. Bar = low power, 100 μm; high power, 20 μm. (b) Quantification of glomerular staining intensity was calculated and is shown as a fold change relative to MAGI2 CNS. Podocyte phospho-Erk1/2 staining is severely reduced in patients with MAGI2 CNS comparedwith control subjects but is only intermediately reduced in patients with idiopathic focal segmental glomerulosclerosis (FSGS). The intensity of total Erk1/2 staining is identical between the groups. Total number of glomeruli analyzed: n = 60 control (20 glomeruli from each of 3 individual patients), n = 72 idiopathic FSGS (12 glomeruli from each of 6 individual patients), n = 50 MAGI2 CNS (2 individual patients, 32 glomeruli from nephrectomy sample, 18 glomeruli from biopsy sample; *P < 0.001, **P < 0.002, ***P < 0.0001; NS, not significant).
DISCUSSION
In the current work, we propose a novel mechanism of podocyte injury in patients with CNS caused by mutations in MAGI2. In normal podocytes under physiologic conditions, MAGI2 forms a complex with RapGEF2, and this complex is required to sustain adequate levels of Rap1 activation and Rap1-mediated downstream signaling. In patients with MAGI2 mutations, however, MAGI2 and RapGEF2 are unable to form a complex, resulting in the inability to activate Rap1. Over time, the resulting inadequate levels of podocyte Rap1 activation, as well as Rap1-mediated downstream signaling, induce podocyte injury and mediate FSGS. We conclude that under the basal condition, the MAGI2-RapGEF2 protein complex is a critical upstream signal for Rap1 activation in podocytes.
The FSGS phenotypes of global and podocyte-specific MAGI2 KO mice4,6 are more severe than for podocyte-specific RapGEF2 KO mice. This difference is reflected in a more completely penetrant phenotype, earlier age of onset of proteinuria, more severe pathologic findings, including higher glomerulosclerosis, and earlier lethality. There are several potential explanations for this finding. First, it is likely that MAGI2 has important functions beyond its protein complex with RapGEF2. MAGI2 protein contains multiple protein interacting domains (PDZ and WW) and mediates numerous protein-protein interactions essential for proper podocyte function.2,22,23 For example, the intricate cytoplasmic protein complex at the slit diaphragm that is mediated by nephrin also includes MAGI2.2,24 Two of the 3 described global MAGI2 knockout mouse models have demonstrated alterations in nephrin expression and localization, suggesting that MAGI2 may have a role in nephrin stability and/or signaling.4,5 Another proposed function of MAGI2, via a protein complex that includes dendrin, is as regulator of Hippo signaling,3,25 which recently has been shown to be critical to podocyte function.26 In podocyte-specific MAGI2 knockout mice, loss of MAGI2 induced dendrin translocation to the nucleus, alterations in the Hippo regulator YAP, and podocyte apoptosis.6 Beyond these known protein complexes that include nephrin and dendrin, MAGI2 may mediate other yet uncharacterized protein interactions governing podocyte maintenance. A second potential explanation for the milder phenotype of RapGEF2 KO mice is redundancy in the network of proteins that regulate Rap1 function. RapGEF2 loss of function, under the appropriate conditions, might be compensated by upregulation of other Rap1 activators, including other Rap1-specific GEFs. A suitable candidate activator is RapGEF6, which is structurally similar to RapGEF2.27 Ongoing studies will look at whether RapGEF6 is expressed in podocytes, and if so, whether it also can activate Rap1 by forming a protein complex with MAGI2.
We acknowledge that because several upstream signaling cascades can induce Erk1/2 phosphorylation, measuring its staining intensity as a secondary readout for levels of Rap1 activation has limitations. However, our available starting material from patients with MAGI2 CNS was limited to paraffin-embedded slides only. Because there is no technically feasible way to stain directly for levels of Rap1 activation on such sections, our best option was to assay for a major downstream Rap1-mediated signal as a surrogate readout. We chose to use immunostaining for Erk1/2 phosphorylation because it is a major downstream target of Rap1 that can be detected reliably on paraffin-embedded material by a well-characterized commercially available antibody and is also relatively podocyte specific in kidney. To minimize the possibility of a secondary effect, we analyzed levels of podocyte Erk1/2 phosphorylation in multiple cases of idiopathic FSGS and compared them with MAGI2 CNS. Levels of podocyte Erk1/2 phosphorylation were uniformly lower in MAGI2 CNS compared with idiopathic FSGS, suggesting that the effect is specific and could not be generalized to all forms of podocyte injury. Overall, these findings are consistent with our observations in MAGI2 knockdown cultured podocytes, where it was technically possible to demonstrate that diminished Erk1/2 phosphorylation was downstream of reductions in Rap1 activation.
MAGI2 expression is one of the most dramatically downregulated of all podocyte-expressed genes in human proteinuric kidney diseases, including diabetic nephropathy and idiopathic FSGS5 (nephroseq database). Levels of podocyte-activated Rap1 are likewise reduced in the same conditions.11 Further studies will be required to determine whether reduced expression of MAGI2 is a cause of diminished podocyte Rap1 in these diseases. What is clear, however, is that the activation state of podocyte Rap1 is tightly regulated both under the basal condition and in the setting of podocyte injury. Importantly, several of the regulatory proteins located upstream of Rap1, including MAGI2 but not RapGEF2, are mainly podocyte specific. An innovative therapeutic approach would be to pharmacologically target such podocyte-specific regulatory proteins. This targeted approach would increase levels of activated Rap1 specifically in podocytes and thereby avoid potentially undesirable effects in other cell types.
METHODS
Mice
Podocin-cre transgenic mice,16 floxed RapGEF2 conditional KO mice,15 and global MAGI2 KO mice4 were previously described and genotyped according to established protocols.
Human kidney samples
We analyzed paraffin-embedded kidney sections (one biopsy, one nephrectomy) from 2 patients with CNS caused by MAGI2 mutations.8 Paraffin-embedded de-identified human kidney sections from archived human biopsies were provided by Vivette D’Agati with appropriate Institutional Review Board approval (5 separate cases). All biopsies were clinically indicated and carried a confirmed pathologic diagnosis of idiopathic FSGS. Paraffin-embedded normal human kidney sections were obtained from nephrectomy samples from the Department of Pathology at Icahn School of Medicine. In all cases, only remaining tissue not required for diagnostic purposes was used.
Antibodies
The following antibodies were used: Rap1 (16120, ThermoFisher Scientific, Waltham, MA), total Erk1/2 (137F5, Cell Signaling Technology, Danvers, MA), phospho-Erk1/2 ([Thr202/Tyr204] D13.14.4E, Cell Signaling Technology), activated β1 integrin (clone 12G10, Millipore, Burlington, MA), total β1 integrin (clone 18, BD Biosciences, Franklin Lakes, NJ), MAGI2 (M2441, Sigma Aldrich, St. Louis, MO), RapGEF2 (clone 1E8, WB, Sigma Aldrich), RapGEF2 (LS-B12608, IF, Lifespan Biosciences, Seattle, WA), WT1 (CAN-R9 (IHC)-56–2, Abcam, Cambridge, United Kingdom), nestin (556309, BD Biosciences), FLAG (M2, Sigma Aldrich), and GFP (632377, Clontech, Mountain View, CA).
Plasmids
Expression plasmids for GFP-MAGI2 wild type, GFP-MAGI2 pGly39*, and GFP-MAGI2 pTry746* were previously described.7 The expression plasmids for FLAG-RapGEF228 and myc-MAGI19 have been described previously. The lentiviral expression plasmid also encoding puromycin resistance, pSF-Lenti, was purchased from MilliporeSigma (Burlington, MA). FLAG-MAGI2 wild type, FLAG-MAGI2 pGly39*, and FLAG-MAGI2 pTyr746* were synthesized and cloned into pSF-Lenti by GenScript Biotech Corporation, Piscataway, NJ.
Histopathology and immunohistochemistry
All kidneys were perfused in vivo with 4% paraformaldehyde. For histologic analysis, kidneys were left in paraformaldehyde overnight and then embedded in paraffin. Sections were cut at 2 μm and stained with periodic acid–Schiff. For immunofluorescence, kidneys were fixed in paraformaldehyde for 4 hours, transferred to 18% sucrose overnight, and then flash frozen in optimal cutting temperature medium. For electron microscopy, samples were cut into 1-mm cubes and fixed overnight in 2.5% glutaraldehyde before being embedded in epoxy using standard techniques (JEOL 1011 electron microscope, JEOL Inc., Peabody, MA).
Measurement of proteinuria
Urine albumin concentration was measured using an enzyme-linked immunosorbent assay kit (Bethyl Laboratories, Montgomery, TX) per the manufacturer’s protocol. Urine creatinine concentrations were quantified by calorimetric assay (Cayman Chemical Co., Ann Arbor, MI) according to the manufacturer’s directions.
Podocyte cell culture
A conditionally immortalized human podocyte cell line was propagated and differentiated as described previously.18
Lentiviral infection and production
All shRNA plasmids were purchased from OriGene, Rockville, MD, already cloned into the pGFP-C-shLenti backbone: RapGEF2 shRNA plasmids (TL316875), MAGI2 shRNA plasmids (TL311600), and scrambled shRNA control (TR30021). Hairpin sequences specific for human RapGEF2 are as follows: CAATGTCAGTGAGGCGA GAACTCTGTGCT (#1) and TGGTCAGTCTCAAGATGACAGCA TAGTAG (#2). Hairpin sequences specific for human MAGI2 are as follows: GAACCTGAGCCATACAGAAGTAGTGGATA (#1) and TCTACTTCATTGACCATAACACAAAGACA (#2). All lentiviral preparation and infections were performed as previously described.29 Infections were done at the permissive temperature in conditionally immortalized human podocytes, and then stable cell lines were established by selection with puromycin at a concentration of 1 to 2 ug/ml. Knockdown and control transduced cell lines were grown at 37 °C for at least 1 week prior to use in experiments.
Reciprocal co-immunoprecipitations
Plates of 293T cells were individually transfected with FLAG-RapGEF2, GFP-MAGI2 wild type, GFP-MAGI2 pGly39*, and GFP-MAGI2 pTry746*. Reciprocal co-immunoprecipitations were performed using antibodies to each protein’s tag (either GFP-agarose [Allele Biotechnology, San Diego, CA]) or anti-FLAG M2 affinity gel (Sigma Aldrich). Total cellular lysates were harvested in radioimmunoprecipitation assay buffer, combined at equal concentrations, and then incubated overnight with either the GFP-agarose or FLAG gel. Beads were washed with radioimmunoprecipitation assay buffer × 3 and then boiled. Western blotting was performed on resulting eluates and on input lysates.
Rap1-GTP pull-downs
Relative levels of active Rap1 were assessed by pull-downs using a GST-tagged fusion protein, corresponding to amino acids 788–884 of the human RalGDS-RAP-binding domain bound to glutathioneagarose. Experiments were performed using the Active Rap1 Pull-down and Detection Kit (16120, ThermoFisher Scientific).
Calcium switch
Confluent cells were serum-deprived overnight in medium containing 1% serum. To chelate extracellular calcium, cells were exposed to 1 mM EGTA (293T cells) or 2 mM EGTA (podocytes) for 2 minutes (293T cells) or 10 minutes (podocytes). Cells were washed twice with PBS and then returned to full serum calcium-containing medium for the indicated time.
Apoptosis Assay
Staining was performed with Annexin V APC (eBioscience, San Diego, CA) and 7-AAD (eBioscience). Samples were collected using a FACSCanto II flow cytometer (BD Biosciences, San Jose, CA) and analyzed using Cytobank (Santa Clara, CA).
Migration Assays
Differentiated podocytes were plated to complete confluence on collagen type I–coated 6-well plates. A scratch was created using a 200-ml sterile pipette, and loosely adherent cells were removed by washing. At specific time intervals, images were obtained at several fixed locations along the scratch. Percent wound closure was calculated using the ImageJ processing program.
Quantification of immunostaining
Immunostained images with a final magnitude of 400× were obtained. The level of immunostaining was measured using ImageJ software. First, acquired images were converted to 8-bit grayscale. Glomerular regions were selected for measurement of area and integrated intensity. Background intensity was measured by selecting 3 distinct areas that showed no staining. The corrected optical density was calculated as previously described.30
Calculation of podocyte number
The number of WT1-positive cells per glomerular tuft was determined only for glomeruli where the macula densa could be clearly visualized, which ensured that all glomeruli analyzed were from a similar level of sectioning. Glomerular area was calculated using ImageJ software.
Pharmacologic Rap1 Activation
Podocytes were split into 2 flasks at time of thermoshift to 37 °C and allowed to differentiate for 1 day. At this point, we added the potent Rap1 activating drug, 8-pCPT-2′-O-Me-cAMP (from Sigma Aldrich) at a concentration of 10 μM to 1 flask and vehicle only to the other. Cells were allowed to continue to differentiate under these conditions for an additional 6 days prior to performing experiments. The drug 8-pCPT-2′-O-Me-cAMP activates Rap1 through EPAC,21 a pathway independent of MAGI2-mediated Rap1 activation.
Statistics
All experiments were repeated at least 3 or more times. Bar graphs represent combined results from all experiments. All data are represented as mean ± SEM. Statistical significance was determined by utilizing the 2-tailed Student t test. A P value of less than 0.05 was considered significant. Prism software (GraphPad, San Diego, CA) was used for comparison between groups.
Study Approval
All mouse studies were approved by the Institutional Animal Care and Use Committee at Icahn School of Medicine at Mount Sinai, New York, New York, USA, in accordance with National Institutes of Health guidelines.
Translation Statement.
All forms of proteinuric renal disease are characterized by injury to specialized glomerular epithelial cells called podocytes. CNS typically is caused by an autosomalrecessive mutation to a critical podocyte gene and presents clinically as severe nephrotic syndrome in early childhood. In the current work, we elucidate the mechanism by which mutations in MAGI2, recently identified as a causal gene of CNS, produce podocyte injury. We find that in normal podocytes, MAGI2 forms a complex with the Rap1 guanine nucleotide exchange factor, RapGEF2, to sustain activation of the small GTPase Rap1, which we previously showed to be essential for proper podocyte function. In children with CNS caused by mutations in MAGI2, however, the MAGI2-RapGEF2 complex is lost, which results in an inability to activate podocyte Rap1 and essential Rap1-induced downstream signaling. A novel therapeutic approach would be to pharmacologically target Rap1 regulatory proteins, in particular those that are mainly podocyte specific, such as MAGI2, in order to augment podocyte Rap1 activation in the setting of injury.
ACKNOWLEDGMENTS
This work was supported by National Institute of Diabetes and Digestive and Kidney Diseases of the National Institutes of Health, grant No. R01 DK104712 (to LK), and the National Natural Science Foundation of China project (81803921).
Footnotes
DISCLOSURE
All the authors declared no competing interests.
REFERENCES
- 1.Ihara K, Nishimura T, Fukuda T, et al. Generation of Venus reporter knock-in mice revealed MAGI-2 expression patterns in adult mice. Gene Expr Patterns. 2012;12:95–101. [DOI] [PubMed] [Google Scholar]
- 2.Lehtonen S, Ryan JJ, Kudlicka K, et al. Cell junction-associated proteins IQGAP1, MAGI-2, CASK, spectrins, and alpha-actinin are components of the nephrin multiprotein complex. Proc Natl Acad Sci U S A. 2005;102: 9814–9819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ihara K, Asanuma K, Fukuda T, et al. MAGI-2 is critical for the formation and maintenance of the glomerular filtration barrier in mouse kidney. Am J Pathol. 2014;184:2699–2708. [DOI] [PubMed] [Google Scholar]
- 4.Balbas MD, Burgess MR, Murali R, et al. MAGI-2 scaffold protein is critical for kidney barrier function. Proc Natl Acad Sci U S A. 2014;111:14876–14881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lefebvre J, Clarkson M, Massa F, et al. Alternatively spliced isoforms of WT1 control podocyte-specific gene expression. Kidney Int. 2015;88:321–331. [DOI] [PubMed] [Google Scholar]
- 6.Shirata N, Ihara KI, Yamamoto-Nonaka K, et al. Glomerulosclerosis induced by deficiency of membrane-associated guanylatekinase inverted 2 in kidney podocytes. J Am Soc Nephrol. 2017;28:2654–2669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ashraf S, Kudo H, Rao J, et al. Mutations in six nephrosis genes delineate a pathogenic pathway amenable to treatment. Nat Commun. 2018;9: 1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bierzynska A, Soderquest K, Dean P, et al. MAGI2 mutations cause congenital nephrotic syndrome. J Am Soc Nephrol. 2017;28:1614–1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ni J, Bao S, Johnson RI, et al. MAGI-1 Interacts with nephrin to maintain slit diaphragm structure through enhanced Rap1 activation in podocytes. J Biol Chem. 2016;291:24406–24417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sakurai A, Fukuhara S, Yamagishi A, et al. MAGI-1 is required for Rap1 activation upon cell-cell contact and for enhancement of vascular endothelial cadherin-mediated cell adhesion. Mol Biol Cell. 2006;17:966–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Potla U, Ni J, Vadaparampil J, et al. Podocyte-specific RAP1GAP expression contributes to focal segmental glomerulosclerosis-associated glomerular injury. J Clin Invest. 2014;124:1757–1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kooistra MR, Dube N, Bos JL. Rap1: a key regulator in cell-cell junction formation. J Cell Sci. 2007;120(pt 1):17–22. [DOI] [PubMed] [Google Scholar]
- 13.Takemoto M, He L, Norlin J, et al. Large-scale identification of genes implicated in kidney glomerulus development and function. EMBO J. 2006;25:1160–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ebarasi L, He L, Hultenby K, et al. A reverse genetic screen in the zebrafish identifies crb2b as a regulator of the glomerular filtration barrier. Dev Biol. 2009;334:1–9. [DOI] [PubMed] [Google Scholar]
- 15.Satyanarayana A, Gudmundsson KO, Chen X, et al. RapGEF2 is essential for embryonic hematopoiesis but dispensable for adult hematopoiesis. Blood. 2010;116:2921–2931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moeller MJ, Sanden SK, Soofi A, et al. Podocyte-specific expression of cre recombinase in transgenic mice. Genesis. 2003;35:39–42. [DOI] [PubMed] [Google Scholar]
- 17.Wharram BL, Goyal M, Wiggins JE, et al. Podocyte depletion causes glomerulosclerosis: diphtheria toxin-induced podocyte depletion in rats expressing human diphtheria toxin receptor transgene. J Am Soc Nephrol. 2005;16:2941–2952. [DOI] [PubMed] [Google Scholar]
- 18.Saleem MA, O’Hare MJ, Reiser J, et al. A conditionally immortalized human podocyte cell line demonstrating nephrin and podocin expression. J Am Soc Nephrol. 2002;13:630–638. [DOI] [PubMed] [Google Scholar]
- 19.Rao J, Ashraf S, Tan W, et al. Advillin acts upstream of phospholipase C 1 in steroid-resistant nephrotic syndrome. J Clin Invest. 2017;127:4257–4269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gee HY, Saisawat P, Ashraf S, et al. ARHGDIA mutations cause nephrotic syndrome via defective RHO GTPase signaling. J Clin Invest. 2013;123: 3243–3253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Parnell E, Palmer TM, Yarwood SJ. The future of EPAC-targeted therapies: agonism versus antagonism. Trends Pharmacol Sci. 2015;36:203–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kawata A, Iida J, Ikeda M, et al. CIN85 is localized at synapses and forms a complex with S-SCAM via dendrin. J Biochem. 2006;139:931–939. [DOI] [PubMed] [Google Scholar]
- 23.Nagashima S, Kodaka M, Iwasa H, Hata Y. MAGI2/S-SCAM outside brain. J Biochem. 2015;157:177–184. [DOI] [PubMed] [Google Scholar]
- 24.Hirabayashi S, Mori H, Kansaku A, et al. MAGI-1 is a component of the glomerular slit diaphragm that is tightly associated with nephrin. Lab Invest. 2005;85:1528–1543. [DOI] [PubMed] [Google Scholar]
- 25.Meliambro K, Wong JS, Ray J, et al. The Hippo pathway regulator KIBRA promotes podocyte injury by inhibiting YAP signaling and disrupting actin cytoskeletal dynamics. J Biol Chem. 2017;292:21137–21148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schwartzman M, Reginensi A, Wong JS, et al. Podocyte-specific deletion of yes-associated protein causes FSGS and progressive renal failure. J Am Soc Nephrol. 2016;27:216–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dube N, Kooistra MR, Pannekoek WJ, et al. The RapGEF PDZ-GEF2 is required for maturation of cell-cell junctions. Cell Signal. 2008;20:1608–1615. [DOI] [PubMed] [Google Scholar]
- 28.Ye T, Ip JP, Fu AK, Ip NY. Cdk5-mediated phosphorylation of RapGEF2 controls neuronal migration in the developing cerebral cortex. Nat Commun. 2014;5:4826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Husain M, Gusella GL, Klotman ME, et al. HIV-1 Nef induces proliferation and anchorage-independent growth in podocytes. J Am Soc Nephrol. 2002;13:1806–1815. [DOI] [PubMed] [Google Scholar]
- 30.Zhong F, Chen H, Xie Y, et al. Protein S protects against podocyte injury in diabetic nephropathy. J Am Soc Nephrol. 2018;29:1397–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]