

Abstract
Discovery strategies commonly focus on the identification of chemical libraries or natural products, but the modulation of endogenous ligands offers a much better therapeutic strategy due to their low adverse potential. Recently, we found that hexadecanamide (Hex) is present in hippocampal nuclei of normal mice as an endogenous ligand of PPARα. This study underlines the importance of Hex in inducing the expression of brain-derived neurotrophic factor (BDNF) from hippocampal neurons via PPARα. The level of Hex was lower in the hippocampi of 5XFAD mice as compared with that in non-Tg mice. Oral administration of Hex increased the level of this molecule in the hippocampus to stimulate BDNF and its downstream plasticity-associated molecules, promote synaptic functions in the hippocampus, and improve memory and learning in 5XFAD mice. However, oral Hex remained unable to stimulate hippocampal plasticity and improve cognitive behaviors in 5XFADPparα-null and 5XFADPparα-ΔHippo mice, indicating an essential role of hippocampal PPARα in Hex-mediated improvement in hippocampal functions. This is the first demonstration to our knowledge of protection of hippocampal functions by oral administration of a hippocampus-based drug, suggesting that Hex may be explored for therapeutic intervention in AD.
Keywords: Neuroscience
Keywords: Alzheimer’s disease
Oral administration of a hippocampus-based drug improves hippocampal functions in an animal model of Alzheimer’s disease.
Introduction
Alzheimer’s disease (AD) is an irreversible and a progressive form of dementia that slowly destroys cognitive functions; it is severe enough to interfere with activities of daily living. According to CDC, currently AD is officially ranked as the sixth leading cause of death in the United States (1). Accumulating evidences link AD pathology with reduced BDNF levels in the serum (2) and in the cognition-related structures, such the as hippocampus and frontal cortex (3). BDNF is a prominent neurotrophin that regulates many of the hippocampus-based biological processes that include maintenance and survival of hippocampal neurons (4), promoting hippocampal dendritic spine density and morphology (5), and enhancing hippocampal neurogenesis (6). Additionally, using BDNF-knockout mouse lines, researchers have implicated the involvement of BDNF in long-term potentiation (7, 8). Considering its essential role in providing trophic support to degenerating neurons and promoting postsynaptic functions, BDNF has, therefore, gained an enormous attention as a promising therapeutic target in countering AD pathology. However, current BDNF-based therapeutics are not that effective in delivering desirable relief to patients with AD. Therefore, enhancing the production of BDNF in vivo in the hippocampus using safer and effective approaches is presently an important area of research.
Recently, we found that hexadecanamide (Hex) is present in vivo in hippocampal nuclei of normal mice as an endogenous ligand of PPARα (9). Here, we demonstrated that Hex upregulated BDNF and stimulated synaptic functions in cultured hippocampal neurons and in vivo in the hippocampi of 5XFAD mice. In addition, we also noticed an improvement in hippocampus-based cognitive behaviors following oral administration of Hex in 5XFAD mice. While investigating the underlined mechanism, we found that Hex rendered these neuroprotective effects through hippocampal PPARα. These results highlight the protection of spatial learning and memory by a hippocampal drug in an animal model of AD.
Results
Hex upregulates the expression of BDNF in primary hippocampal neurons.
Since BDNF is known for its ability to modulate hippocampal plasticity, we wanted to explore the role of Hex in regulating the expression of hippocampal BDNF. Accordingly, we found marked upregulation of Bdnf mRNA in primary WT hippocampal neurons (Figure 1, A and B). In addition, immunoblot analyses (Figure 1C), densitometric analyses (Figure 1D), and immunofluorescence analyses (Figure 1E) further corroborated the finding that low-dose (2 μM) Hex significantly stimulated the level of BDNF in hippocampal neurons. Similar to BDNF, NT3 is also known to regulate synaptic plasticity through receptor tropomyosin-related kinase B (TrkB) (10). Accordingly, following Hex treatment, we also noticed a significant increase in mRNA expression of NT3 and TrkB in hippocampal neurons (Figure 1, A and B). However, levels of NT4 and p75NTR mRNAs remained unaltered following Hex treatment (Figure 1, A and B), suggesting the specificity of the effect. Consistent with mRNA analyses, we also observed a significant upregulation in NT3 (Figure 1, F–H) and TrkB proteins (Figure 1, I and J) in Hex-treated hippocampal neurons. Together, these results suggest that Hex is capable of stimulating hippocampal BDNF and related neurotrophic molecules.
Figure 1. Hex upregulates BDNF and other members of the neurotrophin family in the primary hippocampal neurons.

Primary hippocampal neurons derived from E18 C57BL6 mice pups were treated at 14 days in vitro (DIV) with increasing doses of Hex (μM) for 5 hours, followed by mRNA expression analyses of different members of the neurotrophin family (Bdnf, NT3, and NT4) along with their receptors (TrkB and P75NTR) using semiquantitative RT-PCR (A) and quantitative real-time PCR (B). Expression of GAPDH mRNA was used as loading control. Results are shown as mean ± SD, and 1-way ANOVA [Bdnf mRNA, F(2,6) = 17.990, P = 0.003; NT3 mRNA, F(2,6) = 18.058; NT4 mRNA, F(2,6) = 3.9702, P = 0.080; TrkB mRNA, F(2,6) = 27.393, P < 0.001; P75NTR mRNA, F(2,6) = 3.6634; P = 0.091] followed by Bonferroni’s multiple comparisons test, was used to assess significance of the mean; *P < 0.05, **P < 0.01 vs. control. Following dose-dependent treatment with Hex (μM) for 12 hours, the protein expression of BDNF (C), NT3 (F), and TrkB (I) was investigated in the primary hippocampal neurons by immunoblot analysis (see complete unedited blots in the supplemental material). Relative densities of BDNF (D), NT3 (G), and TrkB (J) protein expressions compared with β-actin were calculated by ImageJ. Results are shown as mean ± SD and 1-way ANOVA [BDNF protein, F(2,6) = 8.9312, P = 0.016; NT3 protein, F(2,6) = 52.019, P < 0.001; TrkB protein, F(2,6) = 8.7881, P = 0.02] followed by Bonferroni’s multiple comparisons test was used to assess significance of the mean; *P < 0.05, **P < 0.01 vs. control. BDNF and NT3 protein levels were also investigated by immunostaining Hex-treated primary hippocampal neurons with neuronal marker MAP2 (green) and BDNF (red) (E) or MAP2 (green) and NT3 (red) (H). Scale bars: 20 μm.
Hex upregulates the expression of BDNF and other plasticity-associated proteins in the 5XFAD mouse model of AD.
Characterizing drugs for improving synaptic plasticity is an important area of research. Recently, we delineated Hex, a carboxamide derived from the most abundant saturated fatty acid (palmitic acid), as an endogenous ligand of PPARα in the hippocampus of normal mouse brain (9). Therefore, we investigated the level of Hex in the hippocampi of six-month-old 5XFAD mice. Surprisingly, 5XFAD mice showed a markedly low endogenous level of Hex in their hippocampi when compared with that of age-matched non-Tg mice (Supplemental Figure 1; supplemental material available online with this article; https://doi.org/10.1172/jci.insight.136654DS1). Therefore, we examined whether oral administration of Hex could increase the level of Hex in the hippocampi of 5XFAD mice. Although in cultured hippocampal neurons, Hex showed efficacy at μM concentrations; for in vivo experiments, we used a dose of 5 mg/kg body weight/d of Hex for oral gavage. This is because fatty acid amides are metabolized by fatty acid amide hydrolases (FAAH) to free fatty acids (11) that undergo β-oxidation in mitochondria. Because, among all organs, the liver has the highest FAAH expression (12), after oral administration of Hex, a major part of it should be metabolized in the liver by hepatic FAAH, with a small part of Hex left for traveling to the hippocampus for the activation of hippocampal PPARα. Moreover, the brain is also rich in FAAH (13); therefore, we decided to treat mice via gavage daily. As evident from our gas chromatography–mass spectrometry (GC-MS) analyses, following 1 month of Hex treatment (5 mg/kg/d), the level of Hex markedly increased in the hippocampi of 5XFAD mice (Supplemental Figure 1). These results demonstrate that after oral supplementation, Hex is capable of entering into the CNS and adding on to its endogenous levels in the hippocampus.
Next, we investigated whether this increased endogenous level of Hex could counter any of the AD-associated pathology. Because BDNF restoration plays a pivotal role in attenuating behavioral deficits, preventing neuron loss, alleviating synaptic degeneration, and reducing neuronal abnormality in different AD mouse models (14, 15), we investigated whether oral administration of Hex could influence hippocampal BDNF and other plasticity-related molecules in 5XFAD mice. As evident from our custom qPCR-based microarray analysis summarized in a heatmap (Figure 2A) and Venn diagram (Figure 2B), Hex treatment caused a dramatic change, affecting approximately 60% of the total selected 69 plasticity-related genes. In addition to Bdnf gene, several other plasticity-associated genes (e.g., Gria1 and Grin2a [members of the ionotropic glutamate receptors family]; Adam10, Dlg4, and Adcy1 [genes encoding synaptic membrane-associated proteins]) were upregulated by Hex treatment. We validated some of these plasticity-associated genes, including Bdnf, by Western blot. Consistent with our custom qPCR-based microarray analysis, Hex treatment significantly increased the level of BDNF protein in the hippocampi of 5XFAD mice when compared with that in the vehicle-treated group (Figure 2, C and D). We revalidated this finding by double labeling hippocampal sections with BDNF and MAP2 (Figure 2E). The level of BDNF decreased in the hippocampi of 5XFAD mice as compared with that in non-Tg mice (Figure 2, E and F). However, Hex treatment significantly enhanced the level of hippocampal BDNF in 5XFAD mice (Figure 2, E and F). It has been reported that the level of postsynaptic density protein of 95 kDa (PSD95), involved in synaptic plasticity, is reduced in the hippocampi of 5XFAD mice (16) and patients with AD (17), probably due to aberrant plaque and tangle formation and neuroinflammation. It has been also shown that the BDNF/TrkB signaling pathway plays a key role in regulating the hippocampal PSD95 and various membrane proteins and signaling molecules present in the niche of postsynaptic density (18, 19). Hence, we analyzed whether Hex treatment could enhance the level of PSD95 in the hippocampi of 5XFAD mice. As evident from our immunoblot analyses (Figure 2C) and corresponding densitometric analyses (Figure 2G), oral administration of Hex significantly increased the level of PSD95 protein in the hippocampi of Hex-fed 5XFAD mice in comparison with vehicle-fed mice. To further characterize the role of Hex in the regulation of synaptic functions, we next investigated the expression of GLUR1 and NR2A, 2 proteins belonging to ionotropic glutamate receptor family that are long known to play important roles in postsynaptic calcium influx (20). We found that protein levels of both GluR1 and NR2A were significantly low in the hippocampi of vehicle-fed 5XFAD mice as compared with that in non-Tg mice (Figure 2, C, H, and I). However, oral treatment of Hex markedly enhanced the levels of GluR1 and NR2A in the hippocampi of 5XFAD mice (Figure 2, C, H, and I). Together, these results indicate that Hex may control hippocampal plasticity by regulating the hippocampal expression of some of the notable plasticity-associated molecules.
Figure 2. Hex upregulates BDNF and other hippocampal plasticity–associated proteins in the hippocampi of 5XFAD mice.

5XFAD mice (n = 6/group; 6–7 months old) were treated orally with Hex (5 mg/kg/d) or vehicle (0.1% methyl cellulose) for 1 month. Untreated non-Tg (NTG) mice (n = 6) were used as control. Following 1 month of Hex treatment, the mRNA expression of several plasticity-associated genes was analyzed by real-time PCR, followed by gene array analysis. GAPDH was used as an endogenous control. (A) Heatmap showing relative expression of differentially regulated plasticity genes in hippocampi of NTG or Hex-fed 5XFAD mice compared with that of vehicle-fed 5XFAD mice. (B) Venn diagram summarizing the numbers of differentially regulated plasticity genes. Relative expression greater than 2 was considered statistically significant. (C) Similarly, following 1 month of Hex treatment, protein levels in hippocampal homogenates of selected plasticity-associated molecules (BDNF, PSD95, GluR1, and NR2A) were analyzed by immunoblot analysis. β-Actin was used as a loading control. Uncropped Western blot images are shown in Supplemental Figure 6. Relative densities of BDNF (D), PSD95 (G), GLUR1 (H), and NR2A (I) immunoblots compared with that of β-actin were calculated by ImageJ. Results are shown as mean ± SEM. One-way ANOVA [BDNF protein, F(2,15) = 6.5000, P = 0.009; PSD95 protein, F(2,15) = 23.977, P < 0.001; GLUR1 protein, F(2,15) = 74.063, P < 0.001; NR2A protein, F(2,15) = 19.624, P < 0.001], followed by Bonferroni’s multiple comparisons test was used to assess significance of the mean; *P < 0.05 vs. vehicle-fed 5XFAD; **P < 0.01, ***P < 0.001 vs. vehicle-fed 5XFAD. Hippocampal sections were also double labeled for MAP2 (green) and BDNF (red) (E). Scale bars: 50 μm. (F) MFI of hippocampal BDNF was calculated using ImageJ. For quantification, 2 sections per mice (n = 6/group) were used. Results are shown as mean ± SEM. One-way ANOVA [BDNF MFI: F(2,33) = 115.77, P < 0.001] followed by Bonferroni’s multiple comparisons test was used to assess significance of the mean; **P < 0.01 vs. vehicle-fed 5XFAD.
Hex promotes hippocampal synaptic functions in 5XFAD mice.
It has been reported that cognitive decline positively correlates with reduced number of positive clusters for PSD95, an indicator of loss of actual synapses (21). Therefore, we next explored a possible association of Hex-mediated upregulation of PSD95 with altered spine morphology. Therefore, we immunolabeled the hippocampal slices harvested from mice across all groups with MAP2, a neuronal marker, and PSD95. As expected, the number of puncta positive for MAP2 and PSD95 was markedly decreased in 5XFAD mice compared with that in non-Tg mice (Figure 3, A and B). However, the observed reduction in puncta positive for MAP2 and PSD95 in hippocampi was significantly rescued in 5XFAD mice following Hex treatment (Figure 3, A and B). AMPA-mediated and NMDAR-mediated Ca2+ influx have been implicated in long-lasting forms of synaptic plasticity and higher cognitive functions (22, 23). Therefore, we also analyzed the effect of Hex on the ionotropic calcium influx through AMPA and NMDA receptors in hippocampal slices of 5XFAD mice. Consistent with the increased hippocampal expression of GluR1 and NR2A proteins (Figure 2, C, H, and I), oral administration of Hex upregulated AMPA- (Figure 3C), and NMDA-dependent (Figure 3D), calcium influx in hippocampal slices of 5XFAD mice as compared with that in vehicle-fed 5XFAD mice. In summary, Hex-mediated protective effects on postsynaptic marker clusters and calcium currents in the hippocampus suggest that Hex may ameliorate AD-associated cognitive defects by promoting these synaptic morphological and functional improvements.
Figure 3. Hex promotes synaptic functions in the hippocampi of 5XFAD mice.

(A) 5XFAD mice (n = 6/group; 6–7 months old) were treated orally with Hex (5 mg/kg/d) or vehicle (0.1% methyl cellulose) for 1 month. Untreated non-Tg (NTG) mice (n = 6) were used as control. Following 1 month of treatment, PSD95 protein levels were investigated by immunostaining hippocampal tissue harvested from hippocampi of NTG and vehicle fed- and Hex-fed 5XFAD mice with antibodies against neuronal marker MAP2 (green) and PSD95 (red). Scale bars: 50 μm. (B) Colocalization of PSD95 with MAP2 was analyzed by quantifying PSD95 and MAP2 immunoreactive puncta using ImageJ. For quantification, 2 sections per mice (n = 6/group) were used. Results are shown as mean ± SEM. One-way ANOVA [PSD95 puncta: F(2,33) = 39.712, P < 0.001], followed by Bonferroni’s multiple comparisons test, was used to assess significance of the mean; **P < 0.01 vs. vehicle-fed 5XFAD. (C) AMPA-dependent and (D) NMDA-dependent calcium currents measured in the hippocampal slices of NTG, vehicle-fed, and Hex-fed 5XFAD mice. The arrow indicates the application of AMPA and NMDA in the assay. To nullify the secondary involvement of AMPA receptor in NMDA-dependent calcium currents, hippocampal slices were treated with NMDA together with NASPM (Ca2+-permeable AMPA antagonist), followed by the recording of calcium influx. Similarly, AMPA-dependent calcium influx was measured in the presence of N20C (noncompetitive NMDA open-channel blocker). Calcium influx was monitored for 300 repeats in a PerkinElmer VICTOR X2 fluorimeter. Results represent the mean of 3 independent experiments.
Hex-mediated upregulation of hippocampal BDNF is dependent on PPARα.
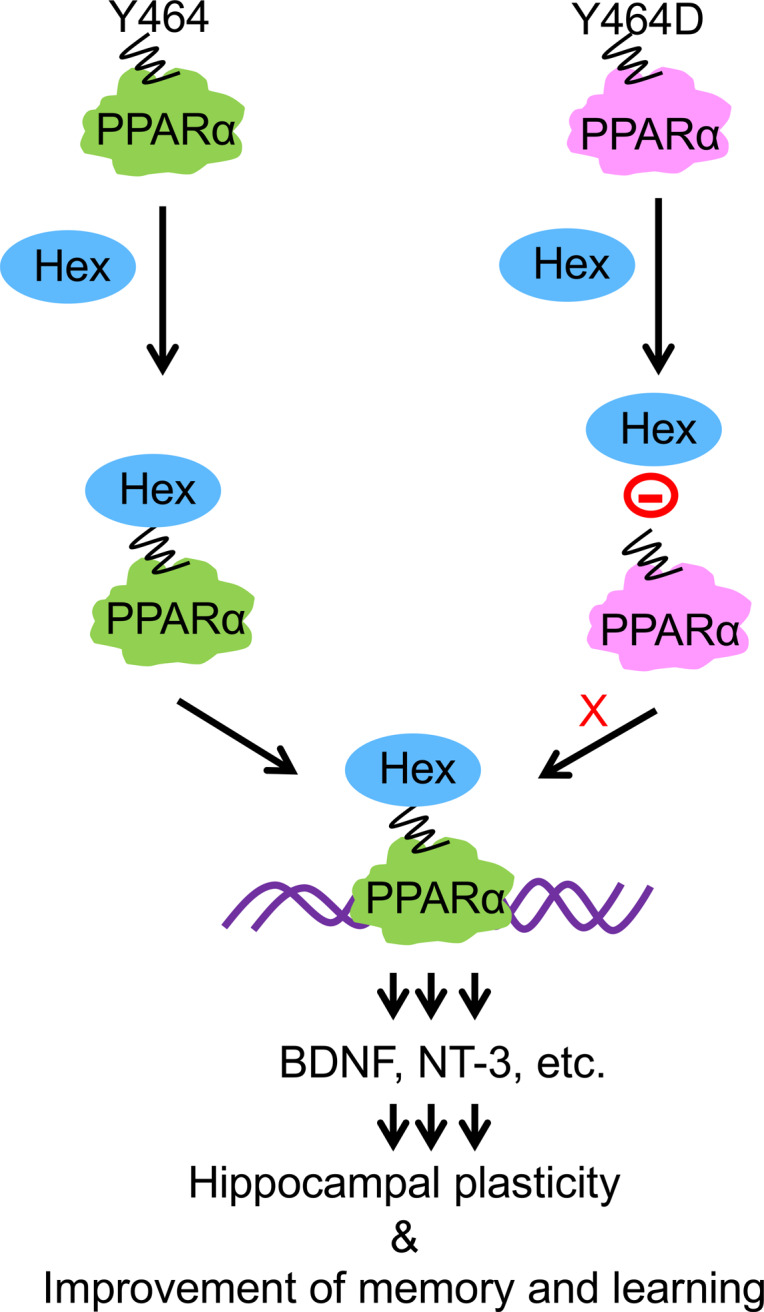
Next, we investigated mechanisms by which Hex upregulated BDNF in hippocampal neurons. BDNF is a well-established downstream target of cAMP response element–binding protein (CREB) (24), a transcription factor that plays an important role in promoting synaptic activity. Previously, we reported that PPARα directly binds to CREB promoter to promote the expression of CREB and CREB-dependent neurotrophic factors (25, 26). Therefore, we analyzed whether Hex required PPARα to stimulate the expression of BDNF in the hippocampal neurons. Interestingly, Hex stimulated the expression of Bdnf mRNA (Figure 4A) and protein (Figure 4, B and C) in WT, but not Pparα-null, hippocampal neurons. To further confirm a direct role for PPARα in the hippocampal BDNF upregulation, we overexpressed the lentiviral construct containing full-length PPARα (lenti-flPPARα) in Pparα-null hippocampal neurons, followed by stimulation with Hex and assessment of Bdnf mRNA (Figure 4D) and protein (Figure 4, E and F). Interestingly, transduction of lenti-flPPARα enhanced both mRNA and protein levels of BDNF in Hex-treated Pparα-null hippocampal neurons. Since HEX binds to Y464 of PPARα (9), we also transduced Pparα-null hippocampal neurons with mutated lenti-Y464D-PPARα. In contrast to lenti-flPPARα–transduced neurons, HEX remained unable to upregulate BDNF mRNA and protein in lenti-Y464D-PPARα–transduced Pparα-null hippocampal neurons (Figure 4, E and F). Taken together, these results suggest that HEX requires PPARα to upregulate BDNF in hippocampal neurons.
Figure 4. Hex upregulates BDNF in hippocampal neurons via PPARα.

(A) WT or Pparα-null hippocampal neurons were treated with Hex (2 μM) for 5 hours followed by Bdnf mRNA by real-time PCR. Results are mean ± SD of 3 independent experiments. Statistical analyses between groups were performed using 2-way ANOVA considering genotype [F(1,8) = 7.918, P = 0.02] and treatment [F(1,8) = 6.919, P = 0.03] as 2 independent variables. Interaction statistics between 2 independent variables was calculated as well [F(1,8) = 7.918, P = 0.02]. Bonferroni’s multiple comparisons test was applied to assess the significance of the mean; **P < 0.01 vs. control, nsP > 0.05 vs. control. (B) Cells were also treated with Hex for 12 hours and BDNF protein level was investigated by immunostaining. Scale bars: 20 μm. (C) MFI of hippocampal BDNF. Statistical analyses were performed using 2-way ANOVA considering genotype [F(1,32) = 44.55, P < 0.001] and treatment [F(1,32) = 19.53, P < 0.001] as 2 independent variables. Interaction statistics between 2 independent variables was calculated as well [F(1,32) = 23.54, P < 0.001]. Bonferroni’s multiple comparisons test was applied to assess the significance of the mean; ***P < 0.001 vs. control, nsP > 0.05 vs. control. (D) Pparα-null hippocampal neurons were transduced with either empty lentiviral vector (GFP) or with the lentiviral vector containing flPPARα or Y464DPPARα for 48 hours followed by stimulation with Hex. After 5 hours, cells were analyzed for Bdnf mRNA. Results are mean ± SD of 3 independent experiments. Statistical analyses of Bdnf mRNA expression analyses between groups were performed using 2-way ANOVA considering lentiviral treatment [F(2,12) = 9.318, P = 0.004] and Hex treatment [F(1,12) = 11.71, P = 0.005] as 2 independent variables. Interaction statistics between 2 independent variables was calculated [F(2,12) = 9.318, P = 0.004]. Bonferroni’s multiple comparisons test was applied to assess the significance of the mean; ***P < 0.001 vs. control, nsP > 0.05 vs. control. Similarly, BDNF protein was analyzed by immunofluorescence. (E) Representative images. Scale bars: 20 μm. (F) MFI of hippocampal BDNF. Statistical analyses of hippocampal BDNF between groups were performed using 2-way ANOVA considering lentiviral treatment [F(2,48) = 23.31, P < 0.001] and Hex treatment [F(1,48) = 20.53, P < 0.001] as 2 independent variables.
To further understand the functional significance of Hex-mediated upregulation of BDNF via PPARα, we examined the involvement of PPARα in Hex-mediated protection of hippocampal properties using 5XFAD and 5XFAD mice with global deletion of PPARα. 5XFADPparα-null mice were generated to study the role of PPARα in AD-related pathologies in mice (27, 28). As expected, following 1 month of Hex (5 mg/kg/d) treatment, we observed a significant increase in BDNF in the hippocampi of 5XFAD, but not that in 5XFADPparα-null, mice (Figure 5, A and B). The involvement of PPARα in Hex-mediated BDNF protein upregulation was again investigated by double labeling hippocampal sections with BDNF and MAP2 antibodies (Figure 5, C and D, and Supplemental Figure 2A). Hex-fed 5XFAD mice showed a significant increase in BDNF in the hippocampus as compared with that in vehicle-fed 5XFAD mice (Figure 5, C and D, and Supplemental Figure 2A). On the other hand, while analyzing BDNF expression in 5XFADPparα-null mice, we did not find any significant difference in BDNF between vehicle-fed 5XFADPparα-null mice and Hex-fed 5XFADPparα-null mice (Figure 5, C and D, and Supplemental Figure 2A). Together, these results suggest that PPARα plays an essential role in Hex-mediated induction of BDNF expression in the hippocampi of 5XFAD mice.
Figure 5. Hex upregulates BDNF and PSD95 in the hippocampi of 5XFAD mice via PPARα.

(A) 5XFAD and 5XFADPparα-null mice (n = 6/group; 6–7 months old) were treated orally with Hex (5 mg/kg/d) or vehicle (0.1% methyl cellulose) for 1 month followed by analysis of BDNF and PSD95 proteins by immunoblot. Uncropped Western blot images are shown in Supplemental Figure 6. (B) Relative density of BDNF and PSD95 protein expressions compared with β-actin was calculated by ImageJ. Results are shown as mean ± SEM. One-way ANOVA followed by Bonferroni’s multiple comparisons test was used to assess the significance of the mean; **P < 0.01 vs. vehicle-fed 5XFAD, nsP > 0.05 vs. vehicle-fed 5XFAD. (C) Hippocampal sections were double labeled with either BDNF (red) plus MAP2 (green) or PSD95 (red) plus MAP2 (green). Scale bar: 50 μm. Raw images are shown in Supplemental Figure 6. (D) MFI of hippocampal BDNF was quantified in 2 sections of each of 6 mice per group. One-way ANOVA [BDNF MFI: F(4,55) = 114.28, P < 0.001], followed by Bonferroni’s multiple comparisons test was used; **P < 0.01 vs. vehicle-fed 5XFAD, nsP > 0.05 vs. vehicle-fed 5XFAD. (E) Colocalization of PSD95 with MAP2 was analyzed by quantifying PSD95 and MAP2 immunoreactive puncta using ImageJ. For quantification, 2 sections per mice (n = 6 per group) was used. Results are shown as mean ± SEM. One-way ANOVA [PSD95 puncta: F(4,55) = 52.569, P < 0.001], followed by Bonferroni’s multiple comparisons test was used to assess the significance of the mean, **P < 0.01 vs. vehicle-fed 5XFAD, nsP > 0.05 vs. vehicle-fed 5XFAD.
Hex-mediated improved hippocampal synaptic functions is dependent on PPARα.
As shown above, following Hex treatment, we noticed a significant upregulation in PSD95 in the hippocampi of 5XFAD mice (Figure 5, A and B). However, Hex did not have any significant effect on hippocampal PSD95 expression in 5XFADPparα-null mice when compared with vehicle-fed 5XFAD mice (Figure 5, A and B). Consistently, as evident from increased PSD95- and MAP2-positive puncta, we also observed an increase in the colocalization of postsynaptic PSD95 and neuronal marker MAP2 in the hippocampi of Hex-fed 5XFAD mice as compared with that in vehicle-fed 5XFAD mice (Figure 5, C and E, and Supplemental Figure 2B). However, the effect of Hex on PSD95 appeared specific to PPARα, because we did not detect any significant differences in the number of PSD95- and MAP2-positive puncta in the hippocampus between vehicle-fed and Hex-fed 5XFADPparα-null mice (Figure 5, C and E, and Supplemental Figure 2B).
Since treatment with Hex, a hippocampal ligand of PPARα (9), elicited both AMPA-dependent and NMDA-dependent postsynaptic calcium currents in the hippocampi of 5XFAD mice (Figure 3, C and D), we next investigated whether global knockdown or hippocampus-specific deletion of PPARα interfered with Hex-induced postsynaptic calcium currents in 5XFAD mice. Therefore, using the Cre-lox mechanism, we first generated 5XFADPparα-ΔHippo mice, 5XFAD mice with deletion of PPARα in hippocampal neurons (Supplemental Figure 3). Next, we analyzed the effect of Hex treatment on the ionotropic calcium influx through NMDA and AMPA receptors in the hippocampal slices of 5XFAD mice with functional PPARα, 5XFADPparα-null mice with global knockdown of PPARα, and 5XFADPparα-ΔHippo mice with hippocampus-specific deletion of PPARα. Consistent with the increased hippocampal expression of BDNF and PSD95 proteins, Hex feeding upregulated AMPA-dependent (Figure 6A), and NMDA-dependent (Figure 6B), calcium influx, as measured in hippocampal slices of 5XFAD mice but neither 5XFADPparα-null nor 5XFADPparα-ΔHippo mice. Together, these results demonstrate that Hex requires hippocampal PPARα in controlling the expression of plasticity-associated molecules and their function in vivo in hippocampus.
Figure 6. Hex promotes calcium influx and ameliorates cognitive deficits in 5XFAD mice via hippocampal PPARα.

5XFAD, 5XFADPparα-null, and 5XFADPparα-ΔHippo mice (n = 6/group; 6–7 months old) were orally treated with Hex (5 mg/kg body weight) or vehicle (0.1% methyl cellulose) for 30 days. Age-matched non-Tg (NTG) mice without any treatment (n = 6) were used as controls. Following treatment, AMPA- (A) and NMDA-dependent (B) calcium currents were measured in the hippocampal slices of NTG, vehicle-fed or Hex-fed 5XFAD mice, vehicle-fed or Hex-fed 5XFADPparα-null mice, and vehicle-fed or Hex-fed 5XFADPparα-ΔHippo mice. Arrow indicates the application of AMPA and NMDA into the assay. Results represent 3 independent experiments. Following Hex treatment, hippocampus-dependent spatial behavior of mice across all groups was also analyzed using Barnes maze (BM). (C) Representative track plots summarizing the overall activity of mice on the apparatus recorded with a Noldus camera and visualized by Ethovision XT software. (D) Latency and (E) number of errors made by NTG, vehicle-fed or Hex-fed 5XFAD mice, vehicle-fed or Hex-fed 5XFADPparα-null mice, and vehicle-fed or Hex-fed 5XFADPparα-ΔHippo mice. Results are shown as mean ± SEM. One-way ANOVA [BM latency: F(6,35) = 23.28, P < 0.0001 and BM errors: F(6,35) = 16.002, P < 0.001], followed by Bonferroni’s multiple comparisons test, was used to assess the significance of the mean; ***P < 0.001 vs. vehicle-fed 5XFAD, nsP > 0.05 vs. vehicle-fed 5XFAD. Context-dependent hippocampal behavior was analyzed by T-maze test. (F) Number of positive turns and (G) number of errors of NTG, control and Hex-treated 5XFAD, 5XFADPparα-null and 5XFADPparα-ΔHippo mice on an appetitive T maze conditioning task were manually recorded. Results are shown as mean ± SEM. One-way ANOVA [TM positive turns: F(6,35) = 5.361, P = 0.0005 and TM errors: F(6,35) = 5.361, P = 0.0005], followed by Bonferroni’s multiple comparisons test, was used to assess the significance of the mean, *P < 0.05 vs. vehicle-fed 5XFAD, **P < 0.01 vs. vehicle-fed 5XFAD, nsP > 0.05 vs. vehicle-fed 5XFAD.
Hex protects hippocampus-based spatial and recognition memories via PPARα.
Finally, we investigated the effect of Hex at the therapeutic level. Therefore, we analyzed some of the hippocampus-dependent behaviors in the 5XFAD, 5XFADPparα-null, and 5XFADPparα-ΔHippo mice following Hex treatment. Hippocampus-based spatial learning and context-dependent behavior can be reliably monitored in the Barnes maze test and T-maze test, respectively (25, 27). As noted in our previous reports (26, 27, 29), 5XFAD mice displayed decreased spatial behaviors indicated by increased latency (Figure 6D) and errors (Figure 6E) as compared with age-matched non-Tg mice. However, Hex feeding significantly improved latency (Figure 6D) and decreased errors (Figure 6E) in 5XFAD mice. In contrast, Hex treatment remained unable to improve spatial learning in both 5XFADPparα-null and 5XFADPparα-ΔHippo mice, indicating an essential role for hippocampal PPARα in Hex-mediated improvement in hippocampus-dependent spatial memory (Figure 6, C and D). Consistently, our T-maze test also exhibited significant improvement in the performance of 5XFAD, as shown by increase in the number of positive turns (Figure 6F) and reduction of errors (Figure 6G), whereas 5XFADPparα-null mice and 5XFADPparα-ΔHippo mice did not display any improvement in T maze analyses (Figure 6, F and G), following Hex treatment, indicating a pivotal role of hippocampal PPARα in Hex-mediated improvement in spatial and contextual in 5XFAD mice. Furthermore, hippocampus is also critical for the processing of memory required for object recognition (30, 31). Therefore, we also investigated the effect of Hex on memory associated with recognition of an object by analyzing the performance of mice in the novel object recognition test (Supplemental Figure 4A). Interestingly, vehicle-fed 5XFAD mice were clearly impaired in their ability to distinguish the novel object, whereas 5XFAD mice treated with Hex behaved very similar to the non-Tg mice with increased preferences for the novel object (Supplemental Figure 4B). In contrast, Hex treatment was unable to improve the ability to distinguish the novel object in both 5XFADPparα-null mice and 5XFADPparα-ΔHippo mice (Supplemental Figure 4B). Together, these observations suggest that Hex requires hippocampal PPARα to improve the memory in 5XFAD mice. Because the decreased latency in either Barnes maze or T-maze test could be confounded with the increased locomotion of animals, we also monitored the locomotor activity of these animals (Supplemental Figure 5A). We did not observe any significant difference in total distance moved (Supplemental Figure 5B) and velocity (Supplemental Figure 5C) across all different groups of animals, nullifying the possibility of interference by increased locomotion in the hippocampus-dependent behaviors. Collectively, these results demonstrate that Hex upregulates BDNF and its downstream signaling pathways and improves hippocampus-based cognitive behaviors in a mouse model of AD via hippocampal PPARα.
Discussion
AD is the most common form of dementia, causing a progressive decline of cognitive functions (32). A growing body of evidences suggests that AD is associated with multimodal pathologies triggered by concurrent occurrence of various fundamental changes in the cellular signaling pathways (33). Among these AD-associated pathologies, one deleterious effect that has attracted much attention in AD research is the downregulation of hippocampus-based neuroprotective molecules, especially BDNF (34). BDNF is the most widely distributed neurotrophin in the CNS that plays a pivotal role in regulating synaptic plasticity and neuronal survival (4, 5, 35). It has been reported that BDNF polymorphisms and reduced BDNF expression in human brains are closely associated with the pathogenesis of AD (2, 3). Preclinical data are also available in the literature to provide valuable insight about the therapeutic potential of BDNF in the treatment for AD (36, 37). However, there has been difficulty in translating BDNF-based therapy to the clinic. One possible explanation for this is the inability of BDNF in its free form to penetrate the blood-brain barrier (38, 39). Therefore, it is necessary to find safe drugs that can stimulate the endogenous BDNF to its therapeutic levels, especially in the hippocampus (40).
Although Hex or palmitamide is a primary fatty acid amide derived from palmitic acid, the most common saturated fatty acid found in animals, plants, and microorganisms, its biological effects are poorly understood. In this study, we demonstrate for the first time to our knowledge that Hex is capable of upregulating neurotrophic factors in hippocampal neurons. In cultured mouse hippocampal neurons, Hex alone induced the expression of BDNF and NT3. This upregulation was specific, as Hex had no effect on NT4. While Hex induced the expression of TrkB, it remained unable to increase the level of p75NTR, again exhibiting the specificity. Because the loss of these neurotrophic factors has been implicated in the pathogenesis of various neurodegenerative diseases, our results provide a potentially important mechanism whereby Hex may ameliorate or delay neurodegeneration in AD.
While many drugs show therapeutic effects in cell culture models, very few exhibit efficacy in vivo in the brain. It is therefore remarkable that after oral feeding Hex entered into the hippocampus and exhibited a neurotrophic effect in the 5XFAD mouse model of AD. First, oral administration of Hex increased the level of BDNF and different plasticity-related molecules, including NMDA- and AMPA-regulated ionotropic receptors in the hippocampi of 5XFAD mice. Second, oral Hex upregulated Ca2+ influx in the hippocampi of 5XFAD mice through NMDA and AMPA receptors. Third, impaired Ca2+ influx through ionotropic glutamate receptors, including NMDA and AMPA receptors, is directly linked to the loss of learning and memory (41). Consistent with the increase in Ca2+ influx, Hex treatment also improved spatial learning and memory and short-term memory in 5XFAD mice. On the other hand, we did not notice any drug-related side effect (e.g., hair loss, weight loss, untoward infection, etc.) in any of the mice used during the course of the study. These results suggest that oral Hex may have therapeutic importance for the protection of hippocampal plasticity and memory in AD.
PPARα is a transcription factor that regulates fatty acid metabolism. Although liver, being the major fatty acid–metabolizing organ, is very rich in PPARα, we have seen that PPARα is constitutively expressed in hippocampal neurons (9, 25–27, 42). Interestingly, we found the presence of Hex in the nuclei of hippocampal neurons as an endogenous ligand of PPARα (9). A ligand-binding triad of S280, Y314, and Y464 is critical for the interaction of PPARα with endogenous ligands (9) and other ligands (26, 27, 29). We have delineated that Hex is mainly bound to Y464 of the PPARα ligand–binding domain (9). Consistently, here, we have shown that Hex is unable to upregulate BDNF in the absence of PPARα and that lentiviral overexpression of flPPARα, but not mutated Y464D-PPARα, restores Hex-mediated BDNF upregulation, highlighting an important role of Y464 of PPARα in neurotrophic function of Hex. Accordingly, Hex treatment also failed to upregulate hippocampal BDNF, stimulate hippocampal plasticity, and improve any of the hippocampus-based memories in 5XFAD mice with either whole-body PPARα knockdown or hippocampus-specific PPARα knockdown, indicating an essential role of hippocampal PPARα in rendering Hex-mediated neuroprotection against AD-associated pathologies.
Currently, there is no effective therapy for halting the progression of AD. Use of memantine and different antagonists of cholinesterase, such as Aricept, Exelon, Razadyne, Cognex, etc., has been the standard treatment for AD (43). However, they are often associated with a number of side effects and disappointing results. On the other hand, there are a number of advantages of Hex over these proposed anti-AD therapies. First, in contrast to available AD therapies, Hex is physiological and present in the hippocampus. Second, Hex is very economical. Third, being the simple amide of the most common saturated fatty acid, Hex is fairly nontoxic. It could be converted into palmitic acid by FAAH to be metabolized completely by mitochondrial β-oxidation. It is interesting because inhibitors of FAAH have been reported to support hippocampal plasticity and protect memory (13). Therefore, it is possible that FAAH antagonists help to stabilize fatty acid amides such as Hex in the hippocampus to ultimately protect/stimulate memory and learning via PPARα.
In summary, we have demonstrated that Hex increases hippocampal BDNF, stimulates hippocampal plasticity, and improves cognitive functions in a mouse model of AD via hippocampal PPARα. These results highlight a potentially novel neurotrophic and neuroprotective role of Hex and suggest that this hippocampal endogenous molecule may be explored for therapeutic intervention in AD.
Methods
For cell culture, semiquantitative RT-PCR, real-time PCR, immunoblotting analysis, immunofluorescence analysis, and behavioral analysis, an extended section is provided in Supplemental Methods.
Reagents and antibodies.
Antibodies, their applications, sources, and dilutions are listed in Supplemental Table 1. Cell culture materials (DMEMF/12, neurobasal, phenol red-free neurobasal, B27, L-glutamine, antibiotic/antimycotic) were purchased from Life Technologies. Hex (A5376) was purchased from MilliporeSigma. All molecular biology–grade chemicals were obtained from either MilliporeSigma or Bio-Rad. Alexa Fluor secondary antibodies used for immunofluorescence analyses were obtained from Jackson ImmunoResearch, and IR-Dye–labeled secondary antibodies used for immunoblotting analyses were from Li-Cor Biosciences.
Animals.
C57BL/6J mice, Pparα-null mice, and 5XFAD mice (44) were obtained from The Jackson Laboratory. Unless otherwise indicated, Pparα-null mice (45) were maintained as homozygous on a C57BL/6J background. 5XFADPparα-null mice, developed earlier in-house (42), were maintained Tg for the 5XFAD mutations and homozygous for the Pparα-null allele through genotyping using PCR on genomic DNA isolated from tail biopsies and detected by primers listed in Supplemental Table 2. PparαΔHippo mice that harbor conditional Pparα deletion in the hippocampal neuronal cells were generated by breeding Pparαflox mice (46) with B6.Cg-Tg (Camk2a-Cre) T29-1Stl/J mice expressing Cre recombinase under the guidance of the calcium/calmodulin-dependent protein kinase II α (Camk2a) promoter (obtained from The Jackson Laboratory). Genotyping of the Pparαflox locus and Camk2a-Cre Tg (Supplemental Figure 3) was performed using PCR on DNA from tail biopsies and detected by primers listed in Supplemental Table 2. Next, PparαΔHippo mice were crossed with 5XFAD mice to generate pups heterozygous for both the PparαΔHippo allele and the 5XFAD Tgs. These mice were then backcrossed with homozygous PparαΔHippo mice to generate approximately 25% 5XFADPparα-ΔHippo mice (Supplemental Figure 3). 5XFAD, 5XFADPparα-null, and 5XFADPparα-ΔHippo mice were aged 6 to 6.5 months at time of treatment and allowed to survive for another 4–6 weeks before they were sacrificed, and their brains were harvested for further analyses.
Organotypic calcium influx assay.
Calcium influx measurement in the mouse hippocampal slices was performed as described previously (26, 27). Briefly, 5XFAD mice, 5XFADPparα-null mice, 5XFADPparα-ΔHippo mice and age-matched non-Tg mice, were anesthetized, rapidly perfused with ice cold sterile PBS, and the whole brain was carefully removed from the cranium. Dorsoventral slices of the hippocampus were made at a thickness of 100 μm using an adult mouse brain slicer matrix with 1.0-mm coronal section slice intervals. The slices were placed in the glass tray filled with cutting solution (24.56 g sucrose, 0.9008 g dextrose, 0.0881 g ascorbate, 0.1650 g sodium pyruvate, and 0.2703 g myo-inositol in 500 mL distilled water) and continuously bubbled with 5% CO2 and 95% O2 gas mixture. The glass tray was kept ice cold during the slicing period. Slices were then carefully transferred into Fluo-4 dye–containing reaction buffer. The reaction buffer was made before the making of brain slices using 10 mL artificial CSF (119 mM NaCl, 26.2 mM NaHCO3, 2.5 mM KCl, 1 mM NaH2PO4, 1.3 mM MgCl2, 10 mM glucose, bubbled with 5% CO2 and 95% O2 followed by the addition of 2.5 mM CaCl2) added to 1 bottle of Fluo-4 dye (catalog F10471 , ThermoFisher) and 250 mM probenecid. Before transferring slices, a flat-bottom 96-well plate (BD Falcon, catalog 323519) was loaded with 50 μL reaction buffer per well, covered with aluminum foil, and kept in a dark place. One individual slice was placed in each well loaded with reaction buffer, and the plate was rewrapped with aluminum foil and kept at 37°C for 20 minutes. After that, fluorescence excitation and emission spectra were recorded in a Perkin-Elmer Victor X2 Luminescence spectrometer in the presence of NMDA (50 μM) and AMPA (50 μM). The recording was performed with 300 repeats at 0.1-ms intervals.
qPCR-based microarray analyses.
qPCR-based microarray using total RNA isolated from hippocampal tissues of non-Tg, vehicle-fed, and Hex-fed 5XFAD mice was performed as described previously (9, 47). Briefly, 5XFAD mice were either treated with vehicle (0.1% methyl cellulose) or with Hex (5 mg/kg/d) for 1 month orally. Age-matched non-Tg mice without any treatment were used as control. Following 1 month of treatment, mice across all groups were anesthetized, rapidly perfused with ice-cold sterile PBS, and decapitated. The whole brain was carefully removed from the cranium, and the hippocampus was harvested. Total RNA was isolated from hippocampal tissue using the QIAGEN RNA isolation kit following the manufacturer’s protocol. Next, cDNA was synthesized from isolated RNA as described previously (25, 27). Synthesized cDNA was then amplified using SYBR green technology in the ABI Prism 7700 sequence detection system and PCR primer sets of key genes listed in Supplemental Table 3. The resulting Ct value was normalized with the housekeeping gene GAPDH and analyzed using the matrix visualization and analysis platform Morpheus (https://software.broadinstitute.org/morpheus/). The expression of mRNAs with fold change >2 or <–2 was defined as differentially expressed genes.
GC-MS analysis.
The endogenous levels of Hex in the hippocampi of harvested brains from mice across all groups was performed using GC-MS as described previously (9, 26, 27). Briefly, 5XFAD mice were either treated with Hex (5 mg/kg/d) or vehicle (0.1% methyl cellulose). Following 1 month of treatment, mice were anesthetized, rapidly perfused with ice-cold sterile PBS, and decapitated. The whole brain was carefully removed from the cranium, and the hippocampus was harvested. Isolated hippocampus was homogenized in ice-cold nondetergent hypotonic buffer (10 mM HEPES [pH 7.9], 1.5 mM MgCl2, 10 mM KCl, 100 mM DTT, protease and phosphatase inhibitor cocktail). After 10 minutes of additional incubation in the hypotonic buffer, the hippocampal homogenate was centrifuged at 8000 g at 4°C for 10 minutes. Next, the pellet was homogenized in ice-cold extraction buffer (10 mM HEPES [pH 7.9], 1.5 mM MgCl2, 0.21 M NaCl, 0.2 mM EDTA, 25% [v/v] glycerol, 100 mM DTT, protease and phosphatase inhibitor cocktail), placed on a rotating shaker at 4°C for 1 hour, and then centrifuged at 18,000 g for 10 minutes. The supernatant was next transferred to a methanol/chloroform/water (4:3:1) mixture and then centrifuged at 18,000 g for 90 seconds. The organic phase was collected, evaporated in the speedvac (Centrivap Concentrator, Labconco), reconstituted with 30 μL chloroform, and analyzed by GC-MS.
DNA constructs and lentiviral transductions.
Generation of the pCMV6-AC-GFP lentiviral backbone–expressing TurboGFP (OriGene, PS100010) and flPPARα was described elsewhere (25). For biochemical experiments, 18 days in vitro Pparα-null hippocampal neurons were transduced with lentiviral particles at MOI 10 for 48 hours at 37°C. Viral integration was monitored by live GFP imaging.
Behavioral analysis.
Barnes maze, T-maze, novel object recognition, and open-field tests were performed as described previously (see the Supplemental Methods).
Statistics.
Statistical analysis was conducted using Graph Pad Prism software (version 8.2.0). Unless otherwise stated, 1-way or 2-way ANOVA was performed to determine the significance of differences between groups, followed by Bonferroni’s post hoc analyses for the significance of differences among multiple experimental groups. Data are expressed as mean ± SEM or mean ± SD. P values of less than 0.05 were considered statistically significant.
Study approval.
Mice were maintained and experiments conducted in accordance with NIH guidelines, and studies were approved (protocol 18-045) by the Rush University Medical Center Institutional Animal Care and Use Committee.
Author contributions
KP conceived the original idea, supervised the project, and edited the final version of the manuscript. DP, AR, and KP designed the study. DP, AR, MK, and SR performed the research. FJG contributed the new reagents/analytic tools. DP, AR, and KP analyzed the data. DP wrote the manuscript in consultation with AR and KP.
Supplementary Material
Acknowledgments
This study was supported by a grant from the NIH (AG050431), the Zenith Fellows Award (ZEN-17-438829) from the Alzheimer’s Association, and a merit award (1I01BX003033) from the US Department of Veterans Affairs to KP. Moreover, KP is the recipient of a Research Career Scientist Award (1IK6 BX004982) from the Department of Veterans Affairs.
Version 1. 04/21/2020
In-Press Preview
Version 2. 05/21/2020
Electronic publication
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Copyright: © 2020, American Society for Clinical Investigation.
Reference information: JCI Insight. 2020;5(10):e136654.https://doi.org/10.1172/jci.insight.136654.
Contributor Information
Avik Roy, Email: Avik_Roy@rush.edu.
Sumita Raha, Email: Sumita_Raha@rush.edu.
Madhuchhanda Kundu, Email: Madhuchhanda_Kundu@rush.edu.
Kalipada Pahan, Email: Kalipada_Pahan@rush.edu.
References
- 1.Melonie H. Deaths leading causes for 2017. Natl Vital Stat Rep. 2019;68(6):1–76. [PubMed] [Google Scholar]
- 2.Gezen-Ak D, et al. BDNF, TNFα, HSP90, CFH, and IL-10 serum levels in patients with early or late onset Alzheimer’s disease or mild cognitive impairment. J Alzheimers Dis. 2013;37(1):185–195. doi: 10.3233/JAD-130497. [DOI] [PubMed] [Google Scholar]
- 3.Rosas-Vidal LE, Do-Monte FH, Sotres-Bayon F, Quirk GJ. Hippocampal--prefrontal BDNF and memory for fear extinction. Neuropsychopharmacology. 2014;39(9):2161–2169. doi: 10.1038/npp.2014.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lipsky RH, Marini AM. Brain-derived neurotrophic factor in neuronal survival and behavior-related plasticity. Ann N Y Acad Sci. 2007;1122:130–143. doi: 10.1196/annals.1403.009. [DOI] [PubMed] [Google Scholar]
- 5.Kellner Y, Gödecke N, Dierkes T, Thieme N, Zagrebelsky M, Korte M. The BDNF effects on dendritic spines of mature hippocampal neurons depend on neuronal activity. Front Synaptic Neurosci. 2014;6:5. doi: 10.3389/fnsyn.2014.00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu PZ, Nusslock R. Exercise-mediated neurogenesis in the hippocampus via BDNF. Front Neurosci. 2018;12:52. doi: 10.3389/fnins.2018.00052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Korte M, Carroll P, Wolf E, Brem G, Thoenen H, Bonhoeffer T. Hippocampal long-term potentiation is impaired in mice lacking brain-derived neurotrophic factor. Proc Natl Acad Sci USA. 1995;92(19):8856–8860. doi: 10.1073/pnas.92.19.8856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Patterson SL, Abel T, Deuel TA, Martin KC, Rose JC, Kandel ER. Recombinant BDNF rescues deficits in basal synaptic transmission and hippocampal LTP in BDNF knockout mice. Neuron. 1996;16(6):1137–1145. doi: 10.1016/S0896-6273(00)80140-3. [DOI] [PubMed] [Google Scholar]
- 9.Roy A, et al. Identification and characterization of PPARα ligands in the hippocampus. Nat Chem Biol. 2016;12(12):1075–1083. doi: 10.1038/nchembio.2204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chao MV. Neurotrophins and their receptors: a convergence point for many signalling pathways. Nat Rev Neurosci. 2003;4(4):299–309. doi: 10.1038/nrn1078. [DOI] [PubMed] [Google Scholar]
- 11.Cravatt BF, Giang DK, Mayfield SP, Boger DL, Lerner RA, Gilula NB. Molecular characterization of an enzyme that degrades neuromodulatory fatty-acid amides. Nature. 1996;384(6604):83–87. doi: 10.1038/384083a0. [DOI] [PubMed] [Google Scholar]
- 12.Cravatt BF, et al. Supersensitivity to anandamide and enhanced endogenous cannabinoid signaling in mice lacking fatty acid amide hydrolase. Proc Natl Acad Sci USA. 2001;98(16):9371–9376. doi: 10.1073/pnas.161191698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Varvel SA, Wise LE, Niyuhire F, Cravatt BF, Lichtman AH. Inhibition of fatty-acid amide hydrolase accelerates acquisition and extinction rates in a spatial memory task. Neuropsychopharmacology. 2007;32(5):1032–1041. doi: 10.1038/sj.npp.1301224. [DOI] [PubMed] [Google Scholar]
- 14.Jiao SS, et al. Brain-derived neurotrophic factor protects against tau-related neurodegeneration of Alzheimer’s disease. Transl Psychiatry. 2016;6(10):e907. doi: 10.1038/tp.2016.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.de Pins B, et al. Conditional BDNF delivery from astrocytes rescues memory deficits, spine density, and synaptic properties in the 5xFAD mouse model of Alzheimer disease. J Neurosci. 2019;39(13):2441–2458. doi: 10.1523/JNEUROSCI.2121-18.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shao CY, Mirra SS, Sait HB, Sacktor TC, Sigurdsson EM. Postsynaptic degeneration as revealed by PSD-95 reduction occurs after advanced Aβ and tau pathology in transgenic mouse models of Alzheimer’s disease. Acta Neuropathol. 2011;122(3):285–292. doi: 10.1007/s00401-011-0843-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sultana R, Banks WA, Butterfield DA. Decreased levels of PSD95 and two associated proteins and increased levels of BCl2 and caspase 3 in hippocampus from subjects with amnestic mild cognitive impairment: Insights into their potential roles for loss of synapses and memory, accumulation of Abeta, and neurodegeneration in a prodromal stage of Alzheimer’s disease. J Neurosci Res. 2010;88(3):469–477. doi: 10.1002/jnr.22227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Béïque JC, Andrade R. PSD-95 regulates synaptic transmission and plasticity in rat cerebral cortex. J Physiol (Lond) 2003;546(Pt 3):859–867. doi: 10.1113/jphysiol.2002.031369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ehrlich I, Klein M, Rumpel S, Malinow R. PSD-95 is required for activity-driven synapse stabilization. Proc Natl Acad Sci USA. 2007;104(10):4176–4181. doi: 10.1073/pnas.0609307104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bollmann JH, Helmchen F, Borst JG, Sakmann B. Postsynaptic Ca2+ influx mediated by three different pathways during synaptic transmission at a calyx-type synapse. J Neurosci. 1998;18(24):10409–10419. doi: 10.1523/JNEUROSCI.18-24-10409.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.VanGuilder HD, et al. Hippocampal dysregulation of synaptic plasticity-associated proteins with age-related cognitive decline. Neurobiol Dis. 2011;43(1):201–212. doi: 10.1016/j.nbd.2011.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lau CG, Zukin RS. NMDA receptor trafficking in synaptic plasticity and neuropsychiatric disorders. Nat Rev Neurosci. 2007;8(6):413–426. doi: 10.1038/nrn2153. [DOI] [PubMed] [Google Scholar]
- 23.Chater TE, Goda Y. The role of AMPA receptors in postsynaptic mechanisms of synaptic plasticity. Front Cell Neurosci. 2014;8:401. doi: 10.3389/fncel.2014.00401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Palomer E, Carretero J, Benvegnù S, Dotti CG, Martin MG. Neuronal activity controls BDNF expression via Polycomb de-repression and CREB/CBP/JMJD3 activation in mature neurons. Nat Commun. 2016;7:11081. doi: 10.1038/ncomms11081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Roy A, et al. Regulation of cyclic AMP response element binding and hippocampal plasticity-related genes by peroxisome proliferator-activated receptor α. Cell Rep. 2013;4(4):724–737. doi: 10.1016/j.celrep.2013.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Roy A, et al. HMG-CoA reductase inhibitors bind to PPARα to upregulate neurotrophin expression in the brain and improve memory in mice. Cell Metab. 2015;22(2):253–265. doi: 10.1016/j.cmet.2015.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Patel D, et al. Aspirin binds to PPARα to stimulate hippocampal plasticity and protect memory. Proc Natl Acad Sci USA. 2018;115(31):E7408–E7417. doi: 10.1073/pnas.1802021115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chandra S, Jana M, Pahan K. Aspirin induces lysosomal biogenesis and attenuates amyloid plaque pathology in a mouse model of Alzheimer’s disease via PPARα. J Neurosci. 2018;38(30):6682–6699. doi: 10.1523/JNEUROSCI.0054-18.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chandra S, Roy A, Jana M, Pahan K. Cinnamic acid activates PPARα to stimulate lysosomal biogenesis and lower amyloid plaque pathology in an Alzheimer’s disease mouse model. Neurobiol Dis. 2019;124:379–395. doi: 10.1016/j.nbd.2018.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Barker GR, Warburton EC. When is the hippocampus involved in recognition memory? J Neurosci. 2011;31(29):10721–10731. doi: 10.1523/JNEUROSCI.6413-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bird CM. The role of the hippocampus in recognition memory. Cortex. 2017;93:155–165. doi: 10.1016/j.cortex.2017.05.016. [DOI] [PubMed] [Google Scholar]
- 32.Roy DS, Arons A, Mitchell TI, Pignatelli M, Ryan TJ, Tonegawa S. Memory retrieval by activating engram cells in mouse models of early Alzheimer’s disease. Nature. 2016;531(7595):508–512. doi: 10.1038/nature17172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Selkoe DJ. Alzheimer’s disease is a synaptic failure. Science. 2002;298(5594):789–791. doi: 10.1126/science.1074069. [DOI] [PubMed] [Google Scholar]
- 34.Garzon DJ, Fahnestock M. Oligomeric amyloid decreases basal levels of brain-derived neurotrophic factor (BDNF) mRNA via specific downregulation of BDNF transcripts IV and V in differentiated human neuroblastoma cells. J Neurosci. 2007;27(10):2628–2635. doi: 10.1523/JNEUROSCI.5053-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen HH, et al. Overexpression of brain-derived neurotrophic factor in the hippocampus protects against post-stroke depression. Neural Regen Res. 2015;10(9):1427–1432. doi: 10.4103/1673-5374.165510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nagahara AH, Tuszynski MH. Potential therapeutic uses of BDNF in neurological and psychiatric disorders. Nat Rev Drug Discov. 2011;10(3):209–219. doi: 10.1038/nrd3366. [DOI] [PubMed] [Google Scholar]
- 37.Wu CC, Lien CC, Hou WH, Chiang PM, Tsai KJ. Gain of BDNF function in engrafted neural stem cells promotes the therapeutic potential for Alzheimer’s disease. Sci Rep. 2016;6:27358. doi: 10.1038/srep27358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pardridge WM. Blood-brain barrier delivery. Drug Discov Today. 2007;12(1-2):54–61. doi: 10.1016/j.drudis.2006.10.013. [DOI] [PubMed] [Google Scholar]
- 39.Pardridge WM, Wu D, Sakane T. Combined use of carboxyl-directed protein pegylation and vector-mediated blood-brain barrier drug delivery system optimizes brain uptake of brain-derived neurotrophic factor following intravenous administration. Pharm Res. 1998;15(4):576–582. doi: 10.1023/A:1011981927620. [DOI] [PubMed] [Google Scholar]
- 40.Arriagada PV, Growdon JH, Hedley-Whyte ET, Hyman BT. Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer’s disease. Neurology. 1992;42(3 Pt 1):631–639. doi: 10.1212/wnl.42.3.631. [DOI] [PubMed] [Google Scholar]
- 41.Olney JW, Wozniak DF, Farber NB. Glumate receptor dysfunction and Alzheimer’s disease. Restor Neurol Neurosci. 1998;13(1-2):75–83. [PubMed] [Google Scholar]
- 42.Corbett GT, Gonzalez FJ, Pahan K. Activation of peroxisome proliferator-activated receptor α stimulates ADAM10-mediated proteolysis of APP. Proc Natl Acad Sci USA. 2015;112(27):8445–8450. doi: 10.1073/pnas.1504890112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Atri A. Effective pharmacological management of Alzheimer’s disease. Am J Manag Care. 2011;17 Suppl 13:S346–S355. [PubMed] [Google Scholar]
- 44.Oakley H, et al. Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: potential factors in amyloid plaque formation. J Neurosci. 2006;26(40):10129–10140. doi: 10.1523/JNEUROSCI.1202-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Peters JM, et al. Growth, adipose, brain, and skin alterations resulting from targeted disruption of the mouse peroxisome proliferator-activated receptor beta(delta) Mol Cell Biol. 2000;20(14):5119–5128. doi: 10.1128/MCB.20.14.5119-5128.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brocker CN, Yue J, Kim D, Qu A, Bonzo JA, Gonzalez FJ. Hepatocyte-specific PPARA expression exclusively promotes agonist-induced cell proliferation without influence from nonparenchymal cells. Am J Physiol Gastrointest Liver Physiol. 2017;312(3):G283–G299. doi: 10.1152/ajpgi.00205.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Roy A, Modi KK, Khasnavis S, Ghosh S, Watson R, Pahan K. Enhancement of morphological plasticity in hippocampal neurons by a physically modified saline via phosphatidylinositol-3 kinase. PLoS One. 2014;9(7):e101883. doi: 10.1371/journal.pone.0101883. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.