

Abstract
The application of Wolbachia in insect pest and vector control requires the establishment of genotypically stable host associations. The cytoplasmic incompatibility (CI) inducing Wolbachia strain wCer2 naturally occurs in the cherry fruit fly Rhagoletis cerasi as co-infection with other strains and was transferred to other fruit fly species by embryonic microinjections. We obtained wCer2 genome data from its native and three novel hosts, Drosophila simulans, Drosophila melanogaster, and Ceratitis capitata and assessed its genome stability, characteristics, and CI factor (cif) genes. De novo assembly was successful from Wolbachia cell-enriched singly infected D. simulans embryos, with minimal host and other bacterial genome traces. The low yield of Wolbachia sequence reads from total genomic extracts of one multiply infected R. cerasi pupa and one singly infected C. capitata adult limited de novo assemblies but was sufficient for comparative analyses. Across hosts wCer2 was stable in genome synteny and content. Polymorphic nucleotide sites were found in wCer2 of each host; however, only one nucleotide was different between R. cerasi and C. capitata, and none between replicated D. simulans lines. The wCer2 genome is highly similar to wAu (D. simulans), wMel (D. melanogaster), and wRec (Drosophila recens). In contrast to wMel and wRec (each with one cif gene pair) and wAu (without any cif genes), wCer2 has three pairs of Type I cif genes, and one Type V cifB gene without a cifA complement. This may explain previously reported CI patterns of wCer2, including incomplete rescue of its own CI modification in three novel host species.
Keywords: symbiosis, reproductive manipulation, prophage, microinjection, cifA, cifB, Rhagoletis cerasi
Introduction
Contrary to the common assumption that closely related taxa are unlikely to co-occur because of competition and natural selection (Hubbell 2001), closely related microorganisms can stably co-exist. An example is the maternally inherited bacterial endosymbiont Wolbachia (Alphaproteobacteria) that occurs in about half of all arthropod species (Weinert et al. 2015). It causes a multitude of host effects that contribute to its maintenance and spread in host populations (Teixeira et al. 2008; Werren et al. 2008; Hosokawa et al. 2010). Individuals of some host species harbor not just one, but two to five distinct Wolbachia strains (Arthofer et al. 2009; Morrow et al. 2014), and this constitutes a substantial challenge for genome sequencing, assembly, and attribution of host effects to any particular Wolbachia strain (Arthofer et al. 2011). The most prominent Wolbachia effects are host reproductive manipulations, with cytoplasmic incompatibility (CI) being the most common (Werren et al. 2008). CI is manifested as embryonic mortality in crosses of males infected with a CI-inducing Wolbachia strain that modifies sperm, and females not infected by the same, or a compatible strain that can rescue the sperm’s CI modification in the fertilized embryo. Wolbachia strains can also cause other host effects that are either beneficial, for example, protection of host from RNA viruses (Hedges et al. 2008; Teixeira et al. 2008) or deleterious, for example, pathology and shortened life-span (Min and Benzer 1997). Several Wolbachia genomes have been sequenced, mostly from host individuals, tissues, or tissue cultures infected with only one strain (Wu et al. 2004; Klasson et al. 2008; Klasson et al. 2009; Ellegaard et al. 2013; Metcalf et al. 2014; Sutton et al. 2014; Lindsey et al. 2016; Newton et al. 2016), but not from source material containing multiple Wolbachia strains.
The first fully sequenced and assembled Wolbachia genome was wMel (Wu et al. 2004), a strain naturally infecting Drosophila melanogaster (Hoffmann 1988; Riegler et al. 2005). Despite its relatively small genome (1.27 Mb), it contains an unexpectedly high number of mobile genetic elements, repetitive regions and the prophage regions WO-A and WO-B. Other Wolbachia genomes share these features, together with high rates of pseudogenisation and recombination (Klasson et al. 2008, 2009; Sutton et al. 2014; Lindsey et al. 2016). LePage et al. (2017) have recently demonstrated that the CI factor A (cifA) and B (cifB) genes, formerly WD0631 and WD0632 in the WO-B region of the wMel genome, encode proteins that mimic the modification and rescue traits of CI when expressed in transgenic D. melanogaster. Similar CI genes (classified as Type I), as well as more divergent orthologs (Types II–V) have been detected in other Wolbachia strains (LePage et al. 2017; Bing et al. 2020; Lindsey et al. 2018). Several Wolbachia genomes contain multiple copies of CI genes, such as the two pairs of CI deubiquitylase (cidA/B) and CI nuclease (cinA/B) genes in the strain wPip of Culex pipiens mosquitoes (Beckmann et al. 2017; Chen et al. 2019) and the phylogenetically distinct pairs of Cif genes in the recently sequenced genome of wIrr of Haematobia irritans hornflies (Madhav et al. 2020). Interestingly, CI levels appear to be higher when Wolbachia strains occur at high titer (Sinkins et al. 1995; Osborne et al. 2012; Martinez et al. 2014). A wMel-related strain, wMelPop, becomes virulent at high temperatures, resulting in premature host mortality (Min and Benzer 1997; Reynolds et al. 2003). This is linked to the presence of an extensive genomic amplification of a 20 kb octomom region encompassing eight genes from WD0507 to WD0514 (Chrostek et al. 2013). The wMel and wMelPop strains have been artificially transferred by embryonic microinjection, directly from their host D. melanogaster to other insect species, such as Drosophila simulans (Poinsot et al. 1998; McGraw et al. 2002), and wMelPop after tissue-culture adaptation to Aedes aegypti (McMeniman et al. 2008; Walker et al. 2011). In these novel hosts, CI and virulence are still observed, but the new intracellular environment produced different Wolbachia titers and levels of CI and virulence (McMeniman et al. 2008; Walker et al. 2011). Genome analyses of wMelPop prior to and after host transfers revealed some genomic changes after infection of cell culture but no changes after microinjection into the new host (Woolfit et al. 2013).
In contrast to wMel, the related Wolbachia supergroup A strain wAu, originally detected in D. simulans, does not cause CI (Hoffmann et al. 1996), does not have cif genes (LePage et al. 2017), yet has highly efficient maternal transmission in field populations (Hoffmann et al. 1996; Kriesner et al. 2013). It provides a fitness benefit to the host and this can lead to fixation in population cage experiments (Kriesner and Hoffmann 2018).
According to multi-locus sequence typing (MLST), the Wolbachia strain wCer2 (Riegler and Stauffer 2002) of the European cherry fruit fly Rhagoletis cerasi (Tephritidae) is phylogenetically very similar to both wAu and wMel (Arthofer et al. 2009). It is likely to be closer to wAu based on wsp sequence similarity (Charlat et al. 2004), whereas closer to wMel based on the analysis of variable number tandem repeats (Riegler et al. 2012). It has previously been suggested that wCer2 is responsible for the high levels of unidirectional CI detected between populations of R. cerasi across Europe (Riegler and Stauffer 2002) where it occurs in co-infections with other Wolbachia strains (Arthofer et al. 2009), with potential for recombination between strains and interactions of shared bacteriophages. Embryonic microinjection was performed to transfer wCer2 to D. simulans, thus isolating it from the other strains for phenotypic assessment in D. simulans where wCer2 caused fecundity costs, moderate levels of CI and, unexpectedly, incomplete rescue of its own CI modification (Riegler et al. 2004). Transfer to D. simulans enabled the comparison of its CI traits with the closely related strains wAu and wMel, and the more distant strain wRi of D. simulans within the same host species (Charlat et al. 2004). Later, wCer2 was also transferred to the Mediterranean fruit fly Ceratitis capitata (Zabalou et al. 2004) and from there to the olive fly Bactrocera oleae (Apostolaki et al. 2011), with the aim to establish and test its potential in the incompatible insect technique (IIT) for the two tephritid species as originally proposed for R. cerasi (Boller et al. 1976; Riegler and Stauffer 2002). In both new tephritid hosts, wCer2 caused complete CI, incomplete rescue of its own CI modification and other fitness costs, such as reduced fecundity and adult longevity (Zabalou et al. 2004, 2009; Apostolaki et al. 2011; Sarakatsanou et al. 2011).
Here, we obtained the genome sequence of wCer2. Individuals of its original host, R. cerasi, can carry up to five different strains, including a supergroup B strain and several closely related supergroup A strains (Arthofer et al. 2009, 2011) which we expected to interfere with genome assembly. Therefore, we used different genomic sources of wCer2: Its multiply infected natural host and the two novel hosts D. simulans and C. capitata that carried wCer2 as monoinfection. We aimed to 1) obtain reliable Wolbachia genomic data from a host that naturally carries multiple strains; 2) contrast success in Wolbachia genome assemblies by using singly versus multiply infected host samples; 3) assess genome stability in the context of experimental host switches by microinjection commonly used for the application of Wolbachia in pest and vector control; and 4) investigate the gene repertoire of wCer2, with a particular focus on the genes that have recently been identified as being involved in the modification and rescue functions of CI strains (Beckmann et al. 2017; LePage et al. 2017; Chen et al. 2019). This is of particular interest as wCer2 has repeatedly displayed incomplete rescue of its own CI modification after its transfer into three novel host species, D. simulans, C. capitata, and B. oleae. It could, therefore, be another example of a “suicide” Wolbachia strain that is not capable of fully rescuing its own modification (Werren 1997; Charlat et al. 2001; Zabalou et al. 2008).
Materials and Methods
Source of Wolbachia Strains
Sequences of wCer2 were obtained from four different host species: Its native host R. cerasi; and three novel hosts, D. simulans, C. capitata, and D. melanogaster into which wCer2 was transferred by embryonic microinjection using R. cerasi as donor (Riegler et al. 2004; Zabalou et al. 2004). The wCer2 donors were sampled from two Austrian R. cerasi populations 50 km apart from each other. However, only donor material for the C. capitata microinjection obtained from Stillfried was available for genome sequencing (fig. 1).
Fig. 1.

—Source populations for the wCer2 genome sequencing, together with the timeline of wCer2 microinjection experiments that resulted in the establishment of wCer2 monoinfections in Drosophila simulans, Drosophila melanogaster, and Ceratitis capitata by using its native host, multiply infected Rhagoletis cerasi as a donor.
In R. cerasi, wCer2 occurs in co-infections with up to four other strains, supergroup A strains wCer1 and wCer4, supergroup B strain wCer5, and a low prevalence supergroup A and B recombinant strain wCer3 (Arthofer et al. 2009). Only wCer1 appears fixed in all populations of R. cerasi (Riegler and Stauffer 2002; Arthofer et al. 2009). Artificial transfer of wCer2 to D. simulans (DsimRC) was performed in 2000 (Riegler et al. 2004). As Wolbachia donor for DsimRC, a population of R. cerasi was collected from honeysuckle (Lonicera xylosteum) in Schönbrunn, Vienna, Austria (RcerAV) in 1999. Based on Wolbachia-specific PCR this population harbored wCer1 and wCer2 (Riegler et al. 2004) and individuals may also have carried other strains at low titer. The microinjection experiment resulted in the establishment of six lines with wCer2. In some lines, wCer1 was detected in generation 1 (G1), but not in the following generations. In 2007, a follow-up study detected wCer2 in five tested lines at G140; four of these were also tested for wCer1 and, surprisingly, wCer1 was detected in RC20, RC33, and RC45, but not in RC50 (Schneider et al. 2013). In October 2011, ∼270 generations after injection, eggs from two of these isofemale D. simulans lines, RC45 and RC50 (hereafter DsimRC45 and DsimRC50) were collected for DNA extraction and whole genome sequencing. Furthermore, another microinjection experiment was performed in 2006 by using DsimRC50 as a wCer2 donor to infect a Wolbachia-free D. melanogaster w1118 line (D. Schneider and W.J. Miller, unpublished), resulting in a wCer2 infected D. melanogaster line 13A (hereafter DmelRC13A). In 2011, embryos of DmelRC13A were collected for DNA extraction and genome sequencing.
Wolbachia of R. cerasi was also used to establish infected lines of C. capitata by embryonic microinjections in 2002 (Zabalou et al. 2004). The Benakeion laboratory line of C. capitata was used as a recipient host, and as a donor for wCer2 another field population of R. cerasi was sourced from infested cherries in Stillfried, Austria (RcerAS), ∼50 km NE of the collection site of RcerAV, in 2001. Pupae were collected from the infested cherries in the laboratory. Some pupae of RcerAS were placed in pure ethanol and stored at –20 °C for later DNA extraction and genome sequencing. Other pupae were conditioned to break the pupal diapause, and donor embryos for microinjection were dissected from the ovaries of emerged females after maturation (Riegler et al. 2004). Based on Wolbachia-specific PCR, RcerAS harbored wCer1 and wCer2 (Riegler and Stauffer 2002) and possibly other Wolbachia strains at low titer. From this donor, the wCer2 infected C. capitata line WolMed 88.6 (hereafter Ccap88.6) was established, and this new host line had stable maternal transmission. For DNA extraction, we accessed adult Ccap88.6 flies provided by K. Bourtzis in 2004 (∼30 generations after injection).
DNA Extraction, Library Preparation, and Sequencing
Wolbachia DNA was extracted from embryos of three independently microinjected Drosophila lines, DsimRC45, DsimRC50, and DmelRC13A, using the protocol developed and described by Ellegaard et al. (2013). This involved for each line the enrichment of Wolbachia cells from 15 to 30 dechorionated embryos through selective centrifugation and filtration, thus isolating Wolbachia cells from host chromosomes and tissue. DNA from Wolbachia cells was amplified by multiple displacements using the Qiagen Repli-g Midi kit, then cleaned-up using the Qiagen DNA Mini kit, eluting in 50 µl of AE elution buffer. Prior to submission of DNA for next-generation sequencing, these samples were ethanol precipitated and resuspended in 40 µl nuclease-free water.
For RcerAS and Ccap88.6 no enrichment of Wolbachia was performed. Instead, total DNA was extracted from one RcerAS pupa and from the abdomen of one adult Ccap88.6 female. Prior to DNA extraction, the material was surface sterilized by immersion in 5% sodium hypochlorite for 1 min, followed by rinsing in triton-X and multiple washes of water. The QiaAmp DNA Mini kit was used according to the manufacturer’s instructions, including RNase treatment, with the exception that the final elution was with 50 µl of nuclease-free water. Genomic DNA quality was checked by gel electrophoresis. Whole genome amplification of 5–20 ng genomic DNA using the Qiagen Repli-G Midi kit was performed to generate the requisite amount of template for library preparation. The amplified DNA was cleaned again using the QiaAmp DNA kit and eluted in 50 µl nuclease-free water. DNA quality and yield were ascertained by gel electrophoresis, Nanodrop spectrophotometry and the Qubit double-stranded DNA quantification system. Libraries for each sample were prepared with TruSeq PCR-free (350 bp insert) library kit (Illumina), using 1 µg of input DNA, and the paired-end (2 × 125 bp) libraries were sequenced on the Illumina HiSeq 2500 platform.
Bioinformatics
Forward and reverse fastq files for each sequenced library were imported into CLC Genomics Workbench v10 (Qiagen) for quality control and further processing. Raw reads were trimmed by allowing no more than two ambiguous nucleotides, using the modified Mott trimming algorithm set to an error probability limit of 0.05, and for removing sequence matching TruSeq adapters. Finally, reads were filtered if <15 nucleotides remained in a read.
To check the Wolbachia strains present in the five libraries (two DsimRC lines, DmelRC13A, Ccap88.6, and RcerAS), the MLST (gatB, coxA, hcpA, fbpA, and ftsZ) and wsp profiles for the strains wCer1, wCer2, wCer4, and wCer5 (Arthofer et al. 2009) were downloaded from the Wolbachia MLST database (Baldo et al. 2006). To check for presence of the low prevalence strain wCer3 we used its wsp gene; its MLST profile is not available (Arthofer et al. 2009, 2011). Trimmed reads for each library were mapped at 100% similarity to all MLST sequences and the wsp genes of all strains, and only wCer2 reads were detected in all four sequenced libraries of the three novel hosts. This is in contrast to the detection of wCer1 and wCer4 in previous generations of DsimRC45 in 2011 (Schneider et al. 2013) and suggests the loss of a low titer infection or incomplete transmission. For RcerAS, the native host of wCer2, the sequencing reads mapped mostly to MLST sequences of wCer2 (high) but also of wCer1 (moderate) and wCer5 (low) at relative proportions 17:8:1 (based on the average coverage of reads mapped to the six marker genes). Furthermore, RcerAS did not contain any wsp sequence reads of wCer3. The sequenced libraries also contained the expected mitochondrial genome and nuclear sequences of each of the four host species.
Each of the five libraries (four with wCer2 as the only Wolbachia strain) was independently de novo assembled with trimmed paired reads using default parameters in CLC Genomics Workbench. The assembled sequences were then searched to identify Wolbachia genome contigs by 1) querying complete Wolbachia genomes (wAu GenBank: LK055284; wMel GenBank: AE017196; wRi GenBank: CP001391; wHa GenBank: NC_021089; wNo GenBank: NC_021084; and wPip GenBank: NC_010981) against all contigs in the assembly, and 2) using BlastN search of the NCBI nucleotide collection (nt) (downloaded July 2018) using an E-value cutoff of 1E–3 to identify any additional contigs that were not similar to the above six genomes. The total number and size of the Wolbachia contigs from each library was calculated, and this preliminary assessment revealed that DsimRC50 and DsimRC45 had a higher proportion of larger Wolbachia contigs than DmelRC13A, Ccap88.6, and RcerAS. Hence, DsimRC50, one of the four libraries to contain wCer2 as only Wolbachia strain, was chosen to assemble wCer2.
Several methods were employed and combined to produce the wCer2 draft genome. DsimRC50 contigs identified as Wolbachia sequence by either of the above methods were extracted and aligned against the wAu genome using Mauve (Darling et al. 2004). The contigs, which had been reordered to most closely match the gene order of the reference genome, were placed in a scaffold with gaps denoted by “N.” GapFiller (Boetzer and Pirovano 2012) was implemented to extend the sequence and close the gaps where possible. The scaffold was iteratively improved by mapping the trimmed paired reads onto the scaffold at a stringency of 0.95 over 80% of the read length, keeping only properly paired reads, and subjecting the resulting consensus sequence to another round of GapFiller analysis. This process was repeated several times until it was not possible to close the remaining gaps. The scaffold was then examined manually to identify remaining gaps and separated into component contigs. The process was repeated, with updated contigs realigned with alternative reference genomes, first wMel, then wRi, using Mauve and again applying GapFiller and read mapping to attempt to close the genome. The final wCer2 genome consensus sequence of DsimRC50 was mapped at a stringency of 0.99 over 95% of the read length, and comprised 11 contigs. The quality of the assembly was then verified using the reads from single-infected libraries DsimRC45 and Ccap88.6 by mapping at the same stringency.
Comparison of the wCer2 Genomes from the Five Different Libraries
The wCer2 genomes of DsimRC45, Ccap88.6, and RcerAS were generated by mapping the sequencing reads to the wCer2 genome assembly of DsimRC50. The data of DmelRC13A were not used because of poor coverage (0.09%, as shown in table 1). The sequencing reads were mapped to wCer2 at a stringency of 0.97 over 90% of the read length, and the new consensus sequence for each host was determined from properly paired reads, and verified by the reads mapped at a stringency of 0.99 over 95% of the read length.
Table 1.
Genome Reads Obtained from Five Different Genomic Host Libraries and Their Mapping Success to wCer2 and Mitochondrial Host Genomes
| Host Species | Line (Previous Name) | Donor (Host Plant) | Source (Isolation Strategy) | Wolbachia Strain | No. of Paired Reads after QC | No. Paired Reads Mapped to wCer2 | Percentage Mapped to wCer2 | Percentage Mapped to mtDNA |
|---|---|---|---|---|---|---|---|---|
| Drosophila simulans | DsimRC50 (RC50) | RcerAV (honeysuckle) | Embryos (purified Wolbachia cells) | wCer2 | 39,480,884 | 31,178,350 | 78.99% | 11.48% |
| 2,200,000 (sampled) | 1,736,722 | 78.94% | 11.47% | |||||
| DsimRC45 (RC45) | RcerAV (honeysuckle) | Embryos (purified Wolbachia cells) | wCer2 | 191,317,107 | 145,719,874 | 76.17% | 14.09% | |
| 2,200,000 (sampled) | 1,682,758 | 76.49% | 14.09% | |||||
| Drosophila melanogaster | DmelRC13A | DsimRC50 | Embryos (purified Wolbachia cells) | wCer2 | 110,991,840 | 100,278 | 0.09% | 91.44% |
| Ceratitis capitata | Ccap88.6 (WolMed 88.6) | RcerAS (cherries) | Single adult (abdomen) | wCer2 | 117,173,724 | 1,667,076 | 1.42% | 16.57% |
| Rhagoletis cerasi | RcerAS; Stillfried, Austria | Not applicable | Single pupa (whole individual) | wCer1; wCer2; wCer5 | 131,385,710 | 3,341,178 | 2.54% | 43.15% |
| 66,000,000 (sampled) | 1,681,128 | 2.55% | 43.16% |
Note.—Some libraries were subsampled to normalize the number of wCer2 reads for variant calling, based on the lowest number of wCer2 in Ccap88.6 (excluding the insufficient coverage of wCer2 in DmelRC13A). The percentage of mapping success is calculated accordingly.
To detect sequence variants in the wCer2 genome across the libraries, some libraries were first subsampled to normalize the number of wCer2 reads available for mapping. The full Ccap88.6 library was used, and contained 1,667,076 wCer2 reads out of a total of 117,173,724; however, both DsimRC libraries were sampled to 2.2 million reads each, and RcerAS was sampled to 66 million reads (table 1). DmelRC13A was excluded because the 100,278 wCer2 reads did not provide full coverage of the genome. These four libraries were separately mapped at a stringency of 0.97 over 97% of the read length to DsimRC50 wCer2, and employed basic variant detection in CLC genomics Workbench to find polymorphisms within the libraries, and to detect single nucleotide polymorphisms (SNPs) between libraries. Variants were identified when they were uniformly different from the reference sequence, or if they comprised two or more nucleotides at a given site at proportions that met the parameter thresholds. Here, variant detection parameters were set to minimum coverage of 40x and minimum frequency of 20%, to minimize inflation of variant calling due to sequence error or errors induced by the whole genome amplification step. Furthermore, in order to assess insertion site polymorphism of small repetitive elements (Wu et al. 2004; Riegler et al. 2005) the overall gene integrity and contig synteny was verified by a reliable coverage of the mapped reads within each contig.
Annotation and Comparative Genomics
The wCer2 genome was annotated with PROKKA v1.13.3 (Seemann 2014) using the annotation of wMel (NC_002978) as the first choice. Wolbachia genomes intended for comparison and phylogeny were also re-annotated with PROKKA—wMel, wAu, wRi, wHa, wNo, wPip, wRec (GenBank: NZ_JQAM01000001.1), wSuz (GenBank: NZ_CAOU02000000), and wVitA (GenBank: NZ_MUJM01000000). BUSCO (Benchmarking Universal Single-Copy Orthologs) (Simão et al. 2015) was utilized to check the completeness of the wCer2 draft genome against a standardized gene set for Proteobacteria, and compared with the nine reference genomes, that is, six complete and three scaffolded (wRec, wSuz, and wVitA) genomes. OrthoFinder was implemented with default parameters to detect orthologous genes from the coding sequences (CDS) identified by PROKKA for wCer2 and the nine reference genomes. Differences and commonalities between orthologs were visualized using the R package UpSetR (Conway et al. 2017).
A phylogeny was estimated from a filtered subset of the single-copy orthologs common to wCer2 and the nine reference genomes. Orthologs were excluded from analysis based on several parameters following Gerth and Bleidorn (2017). Recombining loci were identified using PhiPack by the pairwise homoplasy index (PHI) (Bruen et al. 2006), and excluded if recombination was indicated by P values <0.05. Recombination was also identified in gene trees that were nonmonophyletic for the supergroup A and B strains using the ape package in R. The remaining loci were examined for nucleotide substitution saturation using DAMBE v7.0.35 (Xia 2018) and excluded. Gene alignments were concatenated using FASconCAT (Kück and Meusemann 2010), and maximum likelihood trees estimated using IQ-TREE (Nguyen et al. 2015) with model selection by ModelFinder (Kalyaanamoorthy et al. 2017). For the Wolbachia phylogeny, a general time reversible base substitution model (GTR+F+R2), with empirically determined base frequencies and free rate model of heterogeneity with two categories was chosen. For the cifA tree, the TPM3+F+G4 model was used; for cifB, TIM3+F+G4 model was chosen.
Genome synteny was tested by aligning wCer2 with wMel, wAu and wRi in Progressive Mauve. Orthogroups of interest, including the cifA and cifB genes (and orthologous cid and cin genes), were identified by orthology to these genes in the wMel (supergroup A) and wNo and wPip (supergroup B) genomes. DNA sequences were codon-aligned in MEGA using Muscle. Protein domains were identified using HHPred, applying default parameters and using the SCOPe70 (v2.07), Pfam (v32.0), SMART (v6.0), and COG/KOG (v1.0) databases, and/or by alignment with the modules described in Lindsey et al. (2018). Furin cleavage sites were predicted using PiTou (Tian et al. 2012). Gene structures were drawn using IBS Data visualization. Prophage regions were identified using the web-based tool PHASTER (Zhou et al. 2011; Arndt et al. 2016). The wCer2 genome sequence of DsimRC50 was submitted to GenBank and the annotations (E3V96_00005–E3V96_06530) assigned by the NCBI Prokaryotic Genome Annotation Pipeline (Accession No. SOZK00000000) are used here.
Results
Sequence Data Quality and Assembly of the wCer2 Genome
Genomic data were obtained from three different developmental stages of fruit fly that were extracted and prepared for sequencing in two different ways. The D. simulans and D. melanogaster embryos underwent a Wolbachia cell-enrichment protocol prior to DNA extraction, amplification and library preparation, whereas DNA of the RcerAS pupa and Ccap88.6 female fly was extracted as total genomic DNA and amplified for library preparation. The two different approaches impacted the return of Wolbachia sequence read proportions in each library. Following contig assembly, identification of Wolbachia contigs, and read mapping, the data from DsimRC50 and DsimRC45 embryos contained substantially less bacterial and host contamination. The two samples had complete coverage of wCer2 with very high proportions of reads mapping to the Wolbachia genome (78.99% and 76.17% for DsimRC50 and DsimRC45, respectively; 11.48% and 14.09% of the remaining reads mapped to the mitochondrial genome). However, 91.44% of the DmelRC13A reads mapped to the mitochondrial genome of D. melanogaster, and only 0.09% mapped to the wCer2 genome, likely due to a low titer Wolbachia infection in the embryos, and therefore the wCer2 genome was incompletely covered in the data set of this fly line (table 1).
In contrast, the total genomic DNA extraction from the Ccap88.6 female fly and the RcerAS pupa without a Wolbachia cell-enrichment only yielded 1.42% and 2.54% reads mapped to the wCer2 genome, with the balance largely made up of host and gut bacterial DNA in Ccap88.6 (16.57% mitochondria and over 50% bacterial reads), and mostly host DNA including 43.15% mitochondrial reads in RCerAS (table 1).
De novo assemblies for DsimRC50 and DsimRC45 had higher N50 values than for DmelRC13A, Ccap88.6, and RcerAS, and contained a much higher proportion of Wolbachia contigs (table 2). DsimRC50 was chosen for the assembly of the wCer2 reference genome because the cumulative size of the 138 Wolbachia contigs was slightly larger than the 132 contigs from DsimRC45. After contigs were joined and most gaps were closed, the wCer2 draft genome of DsimRC50 comprised 11 contigs between 1,337 and 329,073 bp in length. It was not possible to join these contigs into a complete circular genome, but the cumulative length was 1,325,568 bp (table 3).
Table 2.
Key Parameters of the Genome Data Sets
| Sample | No. Contigs | N50 | Total nt | Average Size | Range | No. Wolbachia Contigs | Percentage Wolbachia Contigs (%) | Total nt | MLST |
||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| gatB | coxA | hcpA | fbpA | ftsZ | wsp | Strain | |||||||||
| DsimRC50 | 718 | 15,039 | 1,587,228 | 2,211 | 201–46,699 | 138 | 19.22 | 1,299,967 | 1*** | 1*** | 1*** | 1*** | 3*** | 23*** | wCer2 |
| DsimRC45 | 180 | 15,926 | 1,301,387 | 7,230 | 201–46,699 | 132 | 73.33 | 1,282,993 | 1*** | 1*** | 1*** | 1*** | 3*** | 23*** | wCer2 |
| DmelRC13A | 11,216 | 669 | 6,837,724 | 610 | 200–42,979 | 39 | 0.35 | 29,928 | 1* | 1* | 1* | 1* | 3* | 23* | wCer2 |
| Ccap88.6 | 279,253 | 2,936 | 367,056,147 | 1,314 | 200–565,329 | 323 | 0.12 | 1,881,617 | 1* | 1* | 1* | 1* | 3* | 23* | wCer2 |
| 1*** | 1*** | 1*** | 1*** | 3*** | 23*** | wCer2 | |||||||||
| 8** | 84** | 103** | 160** | 79** | 335** | wCer1 | |||||||||
| RcerAS | 580,642 | 1,144 | 428,039,057 | 737 | 200–38,978 | 1,625 | 0.28 | 2,596,908 | 101* | 85* | 40* | 4* | 22* | 116* | wCer5 |
Note.—The percentage of Wolbachia contigs of the total assembly is indicative of the level of host or other bacterial sequences in the sequencing libraries, with a high percentage obtained only for DsimRC50 and DsimRC45. The mapping coverage of the Wolbachia MLST genes and wsp (presented as MLST allele numbers) is shown (*** high, ** moderate, * low) to demonstrate the relative titer of sequencing reads for each of wCer1, wCer2, wCer3, wCer4, and wCer5 potentially harbored by Rhagoletis cerasi or its recipient hosts. Please note that no traces of wCer3 and wCer4 were found in any libraries.
NB * denotes relative titer of the Wolbachia strains in each library.
Table 3.
Genome Features of wCer2, Annotated with PROKKA
|
wCer2 Genome |
wAu |
wMel |
wRec |
wVitA |
wHa |
wRi |
wSuz |
wNo |
wPip |
||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Host | Drosophila simulans DsimRC50 | Drosophila simulans DsimRC45 | Ceratitis capitata Ccap88.6 | Rhagoletis cerasi RcerAS | Drosophila simulans | Drosophila melanogaster | Drosophila recens | Nasonia vitripennis | Drosophila simulans | Drosophila simulans | Drosophila suzukii | Drosophila simulans | Culex quinquefasciatus |
| Assembly | De novo | Resequenced | Resequenced | Resequenced | — | — | — | — | — | — | — | — | — |
| Genome size (bp) | 1,325,568 | 1,325,566 | 1,325,566 | 1,325,563 | 1,268,461 | 1,267,782 | 1,126,656 | 1,211,929 | 1,295,804 | 1,445,873 | 1,415,350 | 1,301,823 | 1,482,455 |
| Number of contigs | 11 | 11 | 11 | 11 | 1 | 1 | 43 | 142 | 1 | 1 | 110 | 1 | 1 |
| G+C content (%) | 35.18 | 35.18 | 35.18 | 35.18 | 35.22 | 35.23 | 35.17 | 34.9 | 35.09 | 35.16 | 35.21 | 34.01 | 34.19 |
| Predicted CDSs | 1,259 | 1,261 | 1,260 | 1,262 | 1,276 | 1,271 | 1,111 | 1,097 | 1,235 | 1,396 | 1,321 | 1,220 | 1,410 |
| Coding density (%) | 84.1 | 84.1 | 84.1 | 84.1 | 84.3 | 84 | 83.4 | 85.1 | 84.2 | 84.6 | 82.6 | 87 | 87.2 |
| Average gene size (bp) | 885 | 884 | 884 | 883 | 838 | 838 | 845 | 941 | 883 | 876 | 885 | 928 | 917 |
| Transfer RNAs | 34 | 34 | 34 | 34 | 34 | 34 | 34 | 34 | 34 | 34 | 34 | 34 | 34 |
| Ribosomal RNAs | 1 of each | 1 of each | 1 of each | 1 of each | 1 of each | 1 of each | 1 of each | 1 of each | 1 of each | 1 of each | 1 of each | 1 of each | 1 of each |
| Pseudogenes | 33 | 33 | 33 | 33 | 53 | 39 | 32 | 17 | 36 | 64 | 32 | 19 | 42 |
Read coverage of the genome was uneven within contigs of all sequencing data sets, even when only perfectly aligned paired reads were mapped (supplementary fig. 1, Supplementary Material online). However, this uneven coverage is typical of the amplification protocol (Ellegaard et al. 2013).
Genome Variability across the Four Different Genomic Libraries
The wCer2 genomes of the DsimRC45, Ccap88.6 and RcerAS libraries were constructed by mapping their reads against the DsimRC50 wCer2 genome (table 3). The DmelRC13A assembly was incomplete (average coverage was <5×), likely due to a low Wolbachia titer in this line, and not included in the analysis because many sections of the wCer2 genome were not covered. The genomes were then aligned and searched for variability. No insertion site polymorphism for small repetitive elements was detected. Each detected SNP was verified from the mapped reads; however, some of these sites were polymorphic within each library and represented by >20% of the reads (therefore unlikely to be due to a sequencing error, although could be caused by incorporation of an error during the whole genome amplification prior to library preparation). These polymorphisms suggest that wCer2 is a population of closely related genome variants, which is also supported by previously conducted Sanger sequencing analyses of PCR amplicons (Schneider et al. 2013). Variant detection (fig. 2 and supplementary tables 1 and 2, Supplementary Material online) identified 11, 18, and 10 polymorphic sites in DsimRC50, DsimRC45, and Ccap88.6, respectively. In contrast RcerAS had 1,248 variant sites (inclusive of the sites found in the other three libraries); however, this high number may be due to the presence of wCer1 and wCer5 that may be similar enough to sections of wCer2 for their reads to map to the wCer2 sequence at the stringency level selected here. Therefore, these variant sites must be regarded with caution.
Fig. 2.

—Variant nucleotides detected across four libraries (see supplementary table 1, Supplementary Material online). The horizontal axis represents nucleotide position numbers in the concatenated DsimRC50 genome sequence. The red bars represent the number of variants in each successive 10 kb region starting from position 1. The label for each panel denotes the total number of variants, with numbers above and below the label denoting the limits of the scale applied to each library. The top four panels show variants called within the DsimRC50, DsimRC45, Ccap88.6, and RCerAS libraries if they differed from the reference wCer2 consensus sequence of DsimRC50, or if they were polymorphic at a minimum frequency of 20% for the minor allele. DmelRC13A was not included in this analysis due to its low Wolbachia coverage preventing extensive genome analyses.
RcerAS was the Wolbachia donor population for Ccap88.6, and the Wolbachia genome sequences of these two hosts were compared. In RcerAS, five of the 1,248 sites had a fixed difference compared with DsimRC50 at >80% of reads. All other sites were polymorphic (i.e., at least two nucleotides at a given site with a minimum of 20% coverage frequency for each) but had a variant that was the same as DsimRC50 (supplementary tables 1 and 2, Supplementary Material online). The five variable nonreference nucleotide sites were manually checked in the RcerAS and Ccap88.6 mappings: Three were polymorphic but at a 5–20% frequency for the minor allele; the fourth at position 105,539 in contig 8 (position 1,170,698 in supplementary tables 1 and 2, Supplementary Material online) was the nonreference allele in both libraries at 99.44% and 100% frequency, respectively; and the fifth site (position 17,064 in contig 6; position 993,305 in supplementary table 1, Supplementary Material online) was not polymorphic in either library, but showed the reference nucleotide (G) in Ccap88.6 and the alternative (A) in RcerAS. This single difference did not alter the amino acid sequence of the gene involved, but is the only unequivocal difference between the RcerAS and Ccap88.6 libraries. We cannot exclude that the reference nucleotide for this site polymorphism was present in the donor population (as we only sequenced one individual).
The two DsimRC libraries were of fly lines established as isofemale lines from different microinjected individuals of the same microinjection experiment with RcerAV as Wolbachia donor. RcerAV was not available for genome sequencing here. Nevertheless, DsimRC50 had 7 of its 11 polymorphic sites shared with RcerAS (a field population 50 km distant from RcerAV), similarly DsimRC45 had 6 of 18 shared polymorphic sites with RcerAS, but the two nonreference variant sites in RcerAS (positions 1,170,698 and 993,305) were both reference nucleotides in DsimRC50 and DsimRC45. A direct comparison of DsimRC50 and DsimRC45 found no unique variants; each of the variant sites was polymorphic for the DsimRC50 reference nucleotide and an alternative nucleotide.
Comparative Analysis of wCer2 with Other Wolbachia Genomes
The wCer2 DsimRC50 genome and the nine published reference genomes were annotated with PROKKA to facilitate a comparison of gene content that was identified under the same parameters. The PROKKA annotation identified 1,259 CDS, 34 tRNAs, and three rRNAs in wCer2 of DsimRC50 (table 3). The CDSs were submitted to the BUSCO pipeline, which assessed the presence of proteins based on their similarity to the set of 221 universal single-copy orthologs for Proteobacteria. The wCer2 assembly is likely a complete representation of the genome, even though it had 36 missing genes from the BUSCO set. This is because it had similar gene content when compared with the complete genomes of wMel, wAu, wRi, wHa, wNo and wPip, wRec, wSuz, and wVitA (supplementary table 3, Supplementary Material online), and was missing the same 36 genes that are missing in wSuz. In comparison, other complete Wolbachia genomes had between 32 and 37 genes missing from the expected core set in Proteobacteria.
OrthoFinder identified 1,258 orthogroups (orthologous sequences shared by a minimum of two of the analyzed genomes); of these, 790 orthogroups were contained in all ten genomes, 77 orthogroups were shared solely between the two supergroup B genomes wPip and wNo, and 48 were common to the eight supergroup A genomes (supplementary fig. 2, Supplementary Material online). Out of all orthogroups, 728 were single gene copy clusters, and these were assessed for recombination: Each orthogroup nucleotide CDS was aligned and 268 loci were found to be recombinant using the PHI, with an additional four loci recombinant between supergroups A and B, as exhibited by polyphyletic gene trees. Therefore, 272 of the 728 single-copy orthologs (37%) produced signals of recombination events, including three of the five MLST marker genes: gatB, coxA, and fbpA.
For phylogenetic analysis, the recombinant genes were excluded, and an additional three loci were discarded for nucleotide substitution saturation. The remaining 453 loci were concatenated, and the maximum likelihood phylogeny estimated (fig. 3). Using an alignment of 375,108 sites, including 43,430 parsimony informative sites, wCer2 exhibited high genetic similarity to wAu, wMel, and wRec (pairwise nucleotide distance of wCer2 with wAu 0.07%, wMel 0.09%, and wRec 0.13%), and the four were placed in a well-supported clade in which wCer2 clustered with wAu, and wMel clustered with wRec. The relationship between wCer2, wAu, and wMel, based on phylogenetic analyses of wsp, also suggests a closer relationship of wCer2 with wAu than with wMel (Charlat et al. 2004), whereas according to the VNTR-141 locus wCer2 and wMel appear more similar (Riegler et al. 2012).
Fig. 3.

—Maximum likelihood phylogeny of 453 single-copy orthologous protein coding genes from eight supergroup A and two supergroup B strains. Codon-based nucleotide alignments were concatenated, producing an alignment of 375,105 sites. The general time reversible base substitution model (GTR+F+R2) was used to estimate the phylogenetic tree, with support by 1,000 bootstrap repetitions. Only bootstrap values >50% are shown. The scale bar represents the number of substitutions per site.
Whole genome alignment with Progressive Mauve provided the level of synteny between wCer2 and the two most closely related genomes wMel and wAu, and the more diverged wRi (wCer2 pairwise nucleotide distance 1.2%). Several rearrangements, including inversions, and large indels, had occurred between wCer2 and its two close relatives wAu and wMel (supplementary fig. 3, Supplementary Material online), but fewer than between wCer2 and wRi. The region between loci WD0400 and WD0535 in wMel is inverted in variant genotypes of wMel (such as wMelPop and wMelCS; Riegler et al. 2005) but is in synteny between wAu and wCer2. This region includes the octomom of wMel (WD0506–514), which is absent from wCer2. Although orthologs to WD0511 and WD0514 were found in separate parts of wCer2, genome alignment of wCer2, wAu, wRec, and wMel showed a contiguous sequence in wCer2 joining homologs of WD0505 directly to WD0519, representing an indel of 23,772 bp. Similarly, neither wAu nor wRec contained the octomom region, but both included a sequence homologous to WD0506, and were contiguous from WD0506 to WD0519 (supplementary fig. 4, Supplementary Material online).
PHASTER identified three prophage regions in the wCer2 genome, two were intact (with scores of 100 and 150) and one considered questionable (score 70). The latter, a ∼27-kb region (called WOCer2-C) encompassing CDSs E3V96_2955–E3V96_3090 in wCer2 contig 3 (28 CDSs, positions 48,421–76,885 in supplementary fig. 5, Supplementary Material online), was most similar to WOVitA1 (GenBank HQ906662) from Nasonia vitripennis, and was adjacent to one pair of cif genes (E3V96_2935–E3V96_2940) which are similar to the cif gene pair in wVitA (wVitA_RS00555/wVitA_RS00550). However, this entire 27 kb region bears little homology to any region of wMel and wAu. The two intact prophage regions were both in contig 4 and included a region 18.4 kb in length spanning E3V96_3160–E3V96_3275 (WOCer2-A: 24 CDSs, positions 8,214–26,511 bp in supplementary fig. 6, Supplementary Material online) and another region of 57.8 kb from E3V96_3445 to E3V96_3720 (WOCer2-B: 56 CDSs, positions 77,941–135,287 bp in supplementary fig. 7, Supplementary Material online). These two regions are homologous to the prophage regions previously identified in wMel, WO-A (WD0259–WD0294) and WO-B/P2-like (WD0565–WD0644). WO-A is complete in wCer2 (E3V96_3135–E3V96_3300) and contains the 18.4 kb prophage region detected using PHASTER, but is slightly different from wMel (0.93%) and wAu (0.84%). The WO-B region differs in structure from both wMel and wAu; however, the two sections orthologous to WD0565–WD0605 and WD0606–WD0644 are in synteny. These CDSs in wCer2 constitute E3V96_3310–E3V96_3685, are 86,883 bp and partially overlap the 57.8 kb prophage region. Combining results of PHASTER and homology searches, the identified prophage regions span ∼170 kb in total, which constitutes ∼13% of the wCer2 genome.
The wCer2 genome contains orthologs for the cifA (WD0631) and cifB (WD0632). Of the nine reference genomes, only wAu did not possess orthologous sequences for these genes. All cifA and cifB genes were aligned and maximum likelihood trees for both CI genes were constructed using IQ-TREE (fig. 4). For cifA, the alignment included 701 parsimony informative sites and for cifB the alignment contained 2,004 parsimony informative sites. Protein domains were identified, and within cif gene types, these conserved domains were generally present (fig. 4).
Fig. 4.

—Maximum likelihood phylogenies for the cifA and cifB genes, and their protein structures. Phylogenetic gene trees were constructed from codon alignments of the wCer2 cif genes plus reference sequences of the Types I–V orthologs obtained from GenBank. The archetypal cif genes are named according to the current GenBank annotation for consistency with other reference sequences: Here, wMel_RS02835 = WD0631 = cifA; wMel_RS06940 = WD0632 = cifB. The general domain structure of each cif gene type is shown, including the CIFA domains catalase (red); DUF3243 (light green); STE (light blue); Ulp1 deubiquitinase module in CIFB (dark blue); and the PDDEXK nuclease domains (yellow). The scale bars represent the number of substitutions per site.
In wCer2, two contiguous wCer2 CDS (E3V96_3425 and E3V96_3430) matched the Type I cifA and cifB genes with >99% similarity and identical length to the wMel cif genes, and were located in a region corresponding to the wMel WO-B prophage region (fig. 5). In addition to this pair, a second pair of adjacent genes (E3V96_2935 and E3V96_2940) was identified in wCer2 that most closely matched the pair of Type I cif genes from wVitA. These wCer2 and wVitA cifB genes have an extended open reading frame (an additional 362 amino acids) that shows homology to herpes simplex virus tegument sequence, that is not present in other cifB ORFs (Lindsey et al. 2018). A third copy of cifA was also Type I (E3V96_6520) but more similar to wPip and wHa sequences. While Orthofinder did not identify the adjacent ORF as a cifB ortholog (E3V96_6515), BLAST search of the entire contig containing this cifA elicited top hits to cidA and cidB from wPip, which are orthologous to the cif genes. This third cif gene pair was located in a small contig (6,316 bp) with a BLAST search hit to a degenerate prophage region in wSol of the fig wasp Ceratosolen solmsi (GenBank KC955252). Further investigation revealed the putative cifB gene of wCer2 was truncated at the end of the contig. This contig could not be reliably extended to complete the gene using any of the libraries containing wCer2 reads. Furthermore, mapping of the DsimRC50 reads to the wPip genome which harbored a close sequence match to this cifB gene did not highlight any reads from wCer2 that mapped to the 3′ end of the wPip cidB gene. The truncated gene was assessed with the other cifA and cifB for expected protein domains, and the Ulp1 protease domain that was present at the 3′ end in all other Type I cifB genes was absent (fig. 5). Nevertheless, this gene was included in the phylogeny of the cif genes. A sole cifB gene from wCer2 (E3V96_3720) was detected that was not coupled with a cifA gene and was more similar to the recently identified Type V cifB gene of wStriCN of Laodelphax striatellus (Bing et al. 2020). Like the wPip Type IV cinB gene which can recapitulate CI, the Type V cifB genes contained both PDDEXK nuclease domains, but also had an extended ORF (of over 2,600 amino acids in wCer2) that included an ankyrin and latrotoxin domain (fig. 5). The ankyrin/latrotoxin domains have also been found in the eukaryotic association module of prophage WOCauB3 derived from Ephestia kuehniella (Bordenstein and Bordenstein 2016); however, this wCer2 gene has a top BLAST hit to a long 4,513 amino acid CDS in wCauA from Carposina sasakii. In spider venom, latrotoxin proteins undergo posttranslational cleavage at furin recognition sites, which is thought to activate the toxin (Graudins et al. 2012). Five possible furin cleavage sites were identified in the E3V96_3720 protein sequence (fig. 5B), including a cleavage site at amino acid position 790 that would leave a protein of a similar size to CINB, with both PDDEXK nuclease domains intact. A BLAST search of the surrounding sequence was performed in order to detect a pseudogenised or divergent copy of cifA, but no sequence was found with similarity to known cifA genes. This putative cifB gene is within the intact 57.8 kb prophage region identified by PHASTER which shares some homology to WO-B of wMel (supplementary fig. 7, Supplementary Material online).
Fig. 5.

—Diagram of the predicted CIF proteins of wCer2. (A) Three contiguous pairs of CIFA and CIFB genes; (B) the putative unpaired CIFB gene with an extended ORF containing ankyrin and latrotoxin domains, and sites for furin cleavage. Protein domains shown are catalase (Cat) in red; DUF3243 (DUF) in light green; STE in light blue; Ulp1 deubiquitinase module (Ulp) in dark blue; PDDEXK (PDD) nuclease domains in yellow; herpes tegument (Herp) in orange; Ankyrin in purple; and Latrotoxin (Lat) in dark green. The cifB wCer2_E3V96_6515 sits at the end of a contig, and not terminated at a stop codon; the truncated protein sequence is denoted by *.
Discussion
We have used a multi-faceted approach to establish the genome sequence of the Wolbachia supergroup A strain wCer2 native to the European cherry fruit fly R. cerasi, a host species naturally infected by up to five strains, including other supergroup A strains. In order to confidently assemble the wCer2 genome, we obtained genomic DNA from two novel host species that only contained wCer2 and had received this strain by microinjection in independent experiments. Across the different host species, we have demonstrated synteny across large contigs and genome stability in terms of gene content and sequence across the donor and recipient hosts. We have identified three pairs of cif genes (all Type I), and an unpaired fourth cifB gene (Type V) without a cifA. One cif gene pair had high similarity to the wMel cif gene pair, and sits within WOCer2-B a prophage region that is homologous to wMel WO-B. However, there is no certainty that the other two cif gene pairs and the unpaired cifB gene are fully functional; two cifB genes have extended ORFs, and one of the paired cifB gene is truncated and lacks an important protein domain. Yet from previous host transfer experiments it is known that wCer2 is not capable of fully rescuing its own modification in three novel host species, D. simulans (Riegler et al. 2004), C. capitata (Zabalou et al. 2004), and B. oleae (Apostolaki et al. 2011). Therefore, wCer2 could be a “suicide” Wolbachia strain (Werren 1997; Charlat et al. 2001) when present as a single infection in a host species. In its original host, R. cerasi, it always occurs as coinfection with other strains (Riegler and Stauffer 2002; Arthofer et al. 2009) where rescue of CI appears complete in some crossing combinations (Boller and Bush 1974).
Genome Stability
The wCer2 genome was not closed, but each of the libraries showed consistent read mapping coverage, thus demonstrating synteny within each of the 11 contiguous sequences across its different hosts. In general, Wolbachia experiences high levels of recombination (Baldo et al. 2006), but no recombination was detected within the contigs of the wCer2 strain. However, the finding of nucleotide polymorphism was anticipated within hosts (Schneider et al. 2013), and this was demonstrated in all libraries. We found that within all host populations wCer2 was polymorphic but this polymorphism was shared across all donor and recipient hosts, whereas polymorphism was higher in the donor than the recipient hosts. According to mapping with MLST marker genes, the novel host lines did not contain any reads that represented other Wolbachia strains, such as wCer1, that had been detected in the donor flies, or in some cases in early generations of the recipient lines postinjection (Riegler et al. 2004; Schneider et al. 2013). The absence of these additional strains of a multiply infected donor host in novel hosts demonstrated that strains can easily be lost after microinjection due to random bottleneck effects and/or selective processes that Wolbachia is exposed to during the establishment of infected lines after microinjection.
Multiple cif genes
The presence of three pairs of cif genes of Type I within the wCer2 genome and an additional cifB gene without a cifA complement, is in stark contrast to the phylogenetically similar genomes which have either no cif genes, such as wAu, or a single contiguous pair, such as wMel and wRec. Like wCer2, other sequenced Wolbachia strains also harbor multiple cif gene pairs, including supergroup A strains wRi (Types I and II) and wVitA (Types I and III), and the supergroup B strain wPip (Types I and IV) (Lindsey et al. 2018) and wStriCN (Type V) (Bing et al. 2020).
wCer2 has a Type I cif gene pair that may be functional because of >99% amino acid sequence identity, identical length and comparable location within the WO-B prophage region to the wMel cifA and cifB genes that recapitulate CI in transgenic D. melanogaster lines (LePage et al. 2017). In D. melanogaster, both cifA and cifB are required to induce CI, and cifA rescues CI (Shropshire and Bordenstein 2019). Strains wRec, wHa, wRi, and wSuz also possess similar cif genes. The Type I cifB genes (and cidB ortholog in wPip) encode deubiquitylating enzymes that include a Ulp1 protease domain (Beckmann et al. 2017) and two other conserved protein modules (Lindsey et al. 2018). Other Type I cif genes that possess these canonical modules, but are more diverged from the wMel type, are found in the wCer2 genome and other CI-inducing strains, such as wVitA and wPip. However, E3V96_6515 has a shortened CDS that lacks the Ulp1 protease domain, and the extended open reading frame in E3V96_2940, which is also found in the wVitA cifB gene (Lindsey et al. 2018), increases the size of the protein by ∼30%. It is not yet known how severely this affects its functionality (fig. 5). wVitA has an additional Type III cifB gene, which may compensate for the extended cifB gene if indeed it is rendered nonfunctional.
Paralogous cif genes classified as Types II–V differ from Type I by the absence of the Ulp1 protease domain (LePage et al. 2017; Lindsey et al. 2018); however, they are hypothesized to act through a nuclease domain. The two very divergent cif paralogues in wPip (Type I cidA/B and Type IV cinA/B) can both emulate the modification-rescue function of CI in transgenic Saccharomyces cerevisiae and D. melanogaster, and the inactivation of the Ulp1 protease in cidB and the nuclease domain in cinB removes the modification function (Beckmann et al. 2017; Chen et al. 2019). Furthermore, the interaction specificity between cognate and noncognate pairs suggests in this system, cidA and cinA do not reciprocally rescue the modification induced by cidB or cinB (Beckmann et al. 2017). It appears the two modes of CI induction are not compatible. In wCer2, we found a Type V cifB gene, with low (~35%) amino acid similarity to the archetypal Type IV cinB, but containing the two PDDEXK nuclease domains as well as a furin cleavage site that possibly separates this part of the peptide from the extensive latrotoxin at the C-terminal end. However, the presence of a potentially functional Type V cifB gene, combined with the previously established CI crossing types where wCer2 was unable to completely rescue its own CI in the novel hosts D. simulans (Riegler et al. 2004), C. capitata (Zabalou et al. 2004), and B. oleae (Apostolaki et al. 2011), accords several possibilities. Firstly, the additional CIFB protein without the CIFA may induce weak CI or lethality by toxic levels of expression (Beckmann et al. 2017); secondly, the dual activity of this diverged cifB gene with one of the other expressed cifA genes, as described in the Two-By One model of CI (Shropshire and Bordenstein 2019) may induce weak CI, or conversely, only partial rescue is achieved by the Type I CIFA proteins. In either case, a severe fertility cost was inflicted on infected females in infected control crosses of new host species. Interestingly, the recently published wIrr genome also contains a cifB gene without a corresponding cifA complement (Madhav et al. 2020).
Previous studies have tested the bidirectional compatibility of different Wolbachia strains, for the purpose of studying the mod/resc pair relationship (Mercot et al. 1995; Poinsot et al. 1998; Charlat et al. 2004; Zabalou et al. 2008; Cattel et al. 2018), and to assess the potential of microinjected Wolbachia strains in IIT (Cattel et al. 2018). Many of these strains have been transferred by microinjection into D. simulans, so various strains and studies can be compared (fig. 6). Models for the CI mod/resc phenotypes have been proposed (Bossan et al. 2011) that account for many of the empirical results provided by bidirectional CI experiments, but since then more complete Wolbachia genomes have been obtained and CI genes identified. Variants of cifB in strains of wPip have been shown to cause variation in the modification phenotype in C. pipiens (Bonneau et al. 2019). Examining the genomic signals in the context of phenotypic outcomes of bidirectional crosses may shed light on the functionality and compatibility of the different types of cif genes.
Fig. 6.
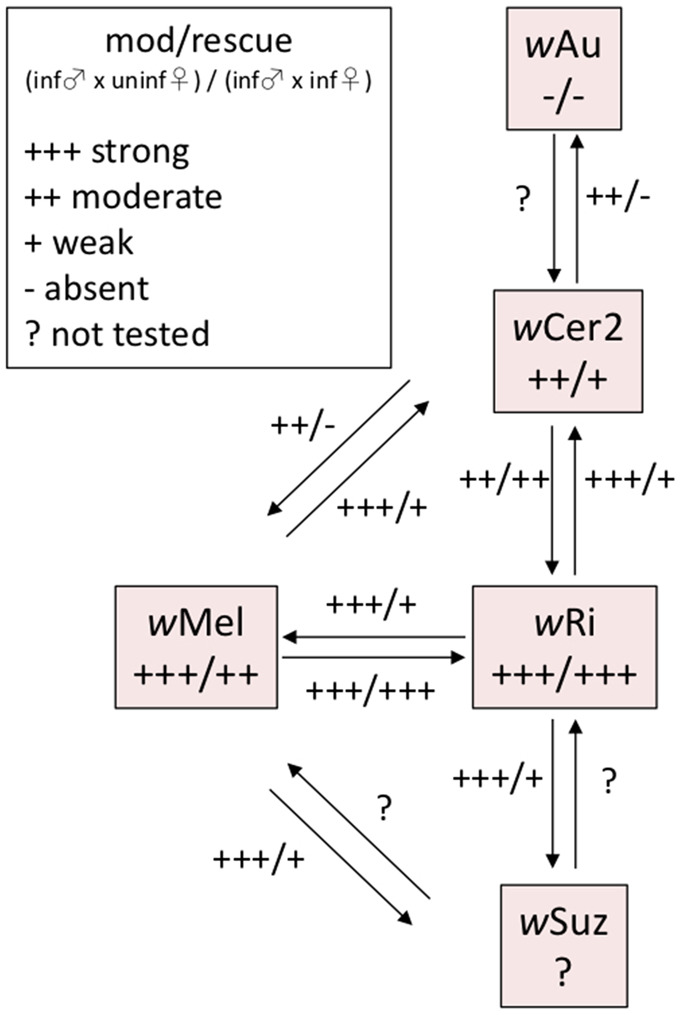
—Diagram of cytoplasmic incompatibility types of Wolbachia strains with different cif gene repertoires. Reciprocal crossing experiments were performed in Drosophila simulans and demonstrated differences in modification (mod) and rescue (resc) capacity of Wolbachia strains. Plus and minus symbols within boxes represent CI induction and rescue characteristics of individual Wolbachia strains; plus and minus symbols along arrows represent CI induction and rescue characteristics of strains when tested in reciprocal crosses of differently infected individuals.
Population Dynamics
An interaction of multiple functional cif genes within a strain’s genome may have implications for the spread of this strain into host populations. It could also influence the population dynamics of multiple infections in hosts and lead to more complex compatibility types (Bonneau et al. 2019). The transfer of wCer2 into the novel hosts D. simulans, C. capitata, and B. oleae has resulted in incomplete rescue of CI and relatively high host fitness costs. It appears that in its original, multiply infected host R. cerasi, the CI induced by wCer2 is rescued (Boller and Bush 1974). However, this is not fully conclusive and needs further investigation together with the assessment of any fitness costs in R. cerasi where wCer2 is maintained in high titer and high prevalence across large parts of the distribution of R. cerasi where it is currently invading (Riegler and Stauffer 2002; Schuler et al. 2016; Bakovic et al. 2018). Although R. cerasi harbors up to five strains of Wolbachia including wCer2, only wCer1 is fixed in all populations. Future genomic analyses and comparisons of wCer1 and the other Wolbachia strains co-infecting R. cerasi will reveal more about the interactions of these strains within populations (fig. 6).
Evolution of the wCer2 Genome
The genome of wCer2 is very similar to the genomes of wAu, wMel, and wRec, with relatively few SNPs but substantial differences due to intergenic rearrangements. The close phylogenetic relationship of the four strains detected by MLST analysis (Baldo et al. 2006; Morrow et al. 2014) was confirmed using 453 single-copy orthologous genes. Few single copy genes differentiated these four strains. Of the four genomes, wAu does not cause CI and possesses no copies of the cifA and cifB genes, but wMel, wRec, and wCer2 all contain contiguous cifA and cifB genes within the WO-B prophage, which are nearly identical across the three strains (Type I). The single cifB gene (E3V96_3720) of wCer2 adjacent to the WO-B prophage region (Type V) and the incomplete pair homologous to the cidA/B genes of wPip (Type I) were not orthologous to any genes in wMel and wAu. Furthermore, the contiguous cif pair E3V96_2935 and E3V96_2940 was found associated with a degenerate 27 kb prophage region that is homologous to WOVitA1 and not found in wMel or wAu. This additional pair of cif genes corresponds to the expansion of prophage regions in wCer2 because cif genes are generally found in the eukaryotic association modules of the prophage (Bordenstein and Bordenstein 2016).
Conclusion
Here, we report the genome sequence of the CI-inducing strain wCer2 and compare it to genomes of other supergroup A Wolbachia strains that have been characterized for their host effects and phylogenetic relationships. The advantage of utilizing single Wolbachia-infected hosts or tissue cultures as source material cannot be overstated. An assembly of the wCer2 genome from total genomic material of its native host that also harbored wCer1 and wCer5 would have been impossible because of the sequence similarity of wCer2 to wCer1, the evidence for recombination, and the expanded complement of CI genes found in wCer2. Therefore, we needed access to hosts that were singly infected with wCer2. For this we accessed wCer2 infected D. simulans and C. capitata. We then used comparative genomics to substantiate this strain’s phylogenetic placement within the Wolbachia, to characterize its diversity of cif genes in the context of compatibility with other strains, and found the expansion of prophage-related cif genes as a potential factor in the idiosyncratic expression of CI in wCer2.
Supplementary Material
Acknowledgments
We thank the reviewers for constructive feedback. This research was supported by the Australian Government through the Australian Research Council (ARC) Industrial Transformation Training Centre (ITTC) Fruit Fly Biosecurity Innovation (IC150100026) to M.R. and by the Austrian Science Fund FWF through the research grant P28255-B22 to W.J.M.
Data deposition: All data are available in public repositories SRA/GenBank/DDBJ/EMBL or in Supplementary Material Online. The wCer2 genome of DsimRC50 was submitted to NCBI GenBank under BioProject No. PRJNA528516; Acc. No. SOZK00000000. Raw reads (for DsimRC50 and DsimRC45) and wCer2 read alignments (for Ccap88.6 and RcerAS) were submitted to NCBI SRA (sequence read archive), also under BioProject No. PRJNA528516.AQ4
Literature Cited
- Apostolaki A, et al. 2011. Transinfection of the olive fruit fly Bactrocera oleae with Wolbachia: towards a symbiont-based population control strategy. J Appl Entomol. 135(7):546–553. [Google Scholar]
- Arndt D, et al. 2016. PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res. 44(W1):W16–W21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arthofer W, et al. 2009. Hidden Wolbachia diversity in field populations of the European cherry fruit fly, Rhagoletis cerasi (Diptera, Tephritidae). Mol Ecol. 18(18):3816–3830. [DOI] [PubMed] [Google Scholar]
- Arthofer W, et al. 2011. Allele intersection analysis: a novel tool for multi locus sequence assignment in multiply infected hosts. PLoS One 6(7):e22198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakovic V, Schebeck M, Telschow A, Stauffer C, Schuler H.. 2018. Spatial spread of Wolbachia in Rhagoletis cerasi populations. Biol Lett. 14(5):20180161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldo L, et al. 2006. Multilocus sequence typing system for the endosymbiont Wolbachia pipientis. Appl Environ Microbiol. 72(11):7098–7110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckmann JF, Ronau JA, Hochstrasser M.. 2017. A Wolbachia deubiquitylating enzyme induces cytoplasmic incompatibility. Nat Microbiol. 2(5):17007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bing XL, Zhao DS, Sun JT, Zhang KJ, Hong XY.. 2020. Genomic analysis of Wolbachia from Laodelphax striatellus (Delphacidae, Hemiptera) reveals insights into its “Jekyll and Hyde” mode of infection pattern. Genome Biol Evol. 12(2):3818–3831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boetzer M, Pirovano W.. 2012. Toward almost closed genomes with GapFiller. Genome Biol. 13(6):R56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boller EF, Bush GL.. 1974. Evidence for genetic variation in populations of the European cherry fruit fly, Rhagoletis cerasi (Diptera: Tephritidae) based on physiological parameters and hybridization experiments. Entomol Exp Appl. 17(2):279–293. [Google Scholar]
- Boller EF, Russ K, Vallo V, Bush GL.. 1976. Incompatible races of European cherry fruit fly, Rhagoletis cerasi (Diptera: Tephritidae), their origin and potential use in biological control. Entomol Exp Appl. 20(3):237–247. [Google Scholar]
- Bonneau M, et al. 2019. Variation in Wolbachia cidB gene, but not cidA, is associated with cytoplasmic incompatibility mod phenotype diversity in Culex pipiens. Mol Ecol. 28(21):4725–4736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bordenstein SR, Bordenstein SR.. 2016. Eukaryotic association module in phage WO genomes from Wolbachia Nat Commun. 7:13155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bossan B, Koehncke A, Hammerstein P.. 2011. A new model and method for understanding Wolbachia-induced cytoplasmic incompatibility. PLoS One 6(5):e19757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruen TC, Philippe H, Bryant D.. 2006. A simple and robust statistical test for detecting the presence of recombination. Genetics 172(4):2665–2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cattel J, et al. 2018. Back and forth Wolbachia transfers reveal efficient strains to control spotted wing drosophila populations. J Appl Ecol. 55(5):2408–2418. [Google Scholar]
- Charlat S, Calmet C, Merçot H.. 2001. On the mod resc model and the evolution of Wolbachia compatibility types. Genetics 159(4):1415–1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlat S, et al. 2004. Incipient evolution of Wolbachia compatibility types. Evolution 58(9):1901–1908. [DOI] [PubMed] [Google Scholar]
- Chen H, Ronau JA, Beckmann JF, Hochstrasser M.. 2019. A Wolbachia nuclease and its binding partner provide a distinct mechanism for cytoplasmic incompatibility. Proc Natl Acad Sci USA. 116(44):22314–22321. [DOI] [PMC free article] [PubMed]
- Chrostek E, et al. 2013. Wolbachia variants induce differential protection to viruses in Drosophila melanogaster: a phenotypic and phylogenomic analysis. PLoS Genet. 9(12):e1003896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conway JR, Lex A, Gehlenborg N.. 2017. UpSetR: an R package for the visualization of intersecting sets and their properties. Bioinformatics 33(18):2938–2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darling AC, Mau B, Blattner FR, Perna NT.. 2004. Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 14(7):1394–1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellegaard KM, Klasson L, Näslund K, Bourtzis K, Andersson SGE.. 2013. Comparative genomics of Wolbachia and the bacterial species concept. PLoS Genet. 9(4):e1003381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerth M, Bleidorn C.. 2017. Comparative genomics provides a timeframe for Wolbachia evolution and exposes a recent biotin synthesis operon transfer. Nature Microbiol. 2:16241. [DOI] [PubMed]
- Graudins A, et al. 2012. Cloning and activity of a novel α-latrotoxin from red-back spider venom. Biochem Pharmacol. 83(1):170–183. [DOI] [PubMed] [Google Scholar]
- Hedges LM, Brownlie JC, O’Neill SL, Johnson KN.. 2008. Wolbachia and virus protection in insects. Science 322(5902):702–702. [DOI] [PubMed] [Google Scholar]
- Hoffmann AA. 1988. Partial cytoplasmic incompatibility between two Australian populations of Drosophila melanogaster. Entomol Exp Appl. 48(1):61–67. [Google Scholar]
- Hoffmann AA, Clancy D, Duncan J.. 1996. Naturally-occurring Wolbachia infection in Drosophila simulans that does not cause cytoplasmic incompatibility. Heredity 76(1):1–8. [DOI] [PubMed] [Google Scholar]
- Hosokawa T, Koga R, Kikuchi Y, Meng XY, Fukatsu T.. 2010. Wolbachia as a bacteriocyte-associated nutritional mutualist. Proc Natl Acad Sci USA. 107(2):769–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbell SP. 2001. The unified neutral theory of biodiversity and biogeography. Princeton (NJ): Princeton University Press. [Google Scholar]
- Kalyaanamoorthy S, Minh BQ, Wong TKF, von Haeseler A, Jermiin LS.. 2017. ModelFinder: fast model selection for accurate phylogenetic estimates. Nat Methods. 14(6):587–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klasson L, et al. 2008. Genome evolution of Wolbachia strain wPip from the Culex pipiens group. Mol Biol Evol. 25(9):1877–1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klasson L, et al. 2009. The mosaic genome structure of the Wolbachia wRi strain infecting Drosophila simulans. Proc Natl Acad Sci USA. 106(14):5725–5730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriesner P, Hoffmann AA.. 2018. Rapid spread of a Wolbachia infection that does not affect host reproduction in Drosophila simulans cage populations. Evolution 72(7):1475–1487. [DOI] [PubMed] [Google Scholar]
- Kriesner P, Hoffmann AA, Lee SF, Turelli M, Weeks AR.. 2013. Rapid sequential spread of two Wolbachia variants in Drosophila simulans. PLoS Pathog. 9(9):e1003607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kück P, Meusemann K.. 2010. FASconCAT: convenient handling of data matrices. Mol Phylogenet Evol. 56(3):1115–1118. [DOI] [PubMed] [Google Scholar]
- LePage DP, et al. 2017. Prophage WO genes recapitulate and enhance Wolbachia-induced cytoplasmic incompatibility. Nature 543(7644):243–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsey ARI, et al. 2018. Evolutionary genetics of cytoplasmic incompatibility genes cifA and cifB in prophage WO of Wolbachia. Genome Biol Evol. 10(2):434–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsey ARI, Werren JH, Richards S, Stouthamer R.. 2016. Comparative genomics of a parthenogenesis-inducing Wolbachia symbiont. G3 6:2113–2123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madhav M, Parry R, Morgan JAT, James P, Asgari S.. 2020. Wolbachia endosymbiont of the horn fly Haematobia irritans irritans: a supergroup A strain with multiple horizontally acquired cytoplasmic incompatibility genes. Appl Environ Microbiol. 86(6):e02589–19. [DOI] [PMC free article] [PubMed]
- Martinez J, et al. 2014. Symbionts commonly provide broad spectrum resistance to viruses in insects: a comparative analysis of Wolbachia strains. PLoS Pathog. 10(9):e1004369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGraw EA, Merritt DJ, Droller JN, O’Neill SL.. 2002. Wolbachia density and virulence attenuation after transfer into a novel host. Proc Natl Acad Sci USA. 99(5):2918–2923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMeniman CJ, et al. 2008. Host adaptation of a Wolbachia strain after long-term serial passage in mosquito cell lines. Appl Environ Microbiol. 74(22):6963–6969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercot H, Llorente B, Jacques M, Atlan A, Montchamp-Moreau C.. 1995. Variability within the Seychelles cytoplasmic incompatibility system in Drosophila simulans. Genetics 141:1015–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metcalf JA, Jo M, Bordenstein SR, Jaenike J, Bordenstein SR.. 2014. Recent genome reduction of Wolbachia in Drosophila recens targets phage WO and narrows candidates for reproductive parasitism. PeerJ 2:e529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min KT, Benzer S.. 1997. Wolbachia, normally a symbiont of Drosophila, can be virulent, causing degeneration and early death. Proc Natl Acad Sci USA. 94(20):10792–10796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrow JL, Frommer M, Shearman DCA, Riegler M.. 2014. Tropical tephritid fruit fly community with high incidence of shared Wolbachia strains as platform for horizontal transmission of symbionts. Environ Microbiol. 16(12):3622–3637. [DOI] [PubMed] [Google Scholar]
- Newton IL, et al. 2016. Comparative genomics of two closely related Wolbachia with different reproductive effects on hosts. Genome Biol Evol. 8(5):1526–1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen L-T, Schmidt HA, von Haeseler A, Minh BQ.. 2015. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 32(1):268–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborne SE, Iturbe-Ormaetxe I, Brownlie JC, O'Neill SL, Johnson KN.. 2012. Antiviral protection and the importance of Wolbachia density and tissue tropism in Drosophila simulans. Appl Environ Microbiol. 78(19):6922–6929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poinsot D, Bourtzis K, Markakis G, Savakis C, Merçot H.. 1998. Wolbachia transfer from Drosophila melanogaster into D. simulans: host effect and cytoplasmic incompatibility relationships. Genetics 150(1):227–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds KT, Thomson LJ, Hoffmann AA.. 2003. The effects of host age, host nuclear background and temperature on phenotypic effects of the virulent Wolbachia strain popcorn in Drosophila melanogaster. Genetics 164(3):1027–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riegler M, Charlat S, Stauffer C, Mercot H.. 2004. Wolbachia transfer from Rhagoletis cerasi to Drosophila simulans: investigating the outcomes of host–symbiont coevolution. Appl Environ Microbiol. 70(1):273–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riegler M, Iturbe-Ormaetxe I, Woolfit M, Miller WJ, O’Neill SL.. 2012. Tandem repeat markers as novel diagnostic tools for high resolution fingerprinting of Wolbachia. BMC Microbiol. 12(Suppl 1):S12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riegler M, Sidhu M, Miller WJ, O’Neill SL.. 2005. Evidence for a global Wolbachia replacement in Drosophila melanogaster. Curr Biol. 15(15):1428–1433. [DOI] [PubMed] [Google Scholar]
- Riegler M, Stauffer C.. 2002. Wolbachia infections and superinfections in cytoplasmically incompatible populations of the European cherry fruit fly Rhagoletis cerasi (Diptera, Tephritidae). Mol Ecol. 11(11):2425–2434. [DOI] [PubMed] [Google Scholar]
- Sarakatsanou A, Diamantidis AD, Papanastasiou SA, Bourtzis K, Papadopoulos NT.. 2011. Effects of Wolbachia on fitness of the Mediterranean fruit fly (Diptera: Tephritidae). J Appl Entomol. 135(7):554–563. [Google Scholar]
- Schneider DI, et al. 2013. Uncovering Wolbachia diversity upon artificial host transfer. PLoS One 8(12):e82402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuler H, et al. 2016. The hitchhiker’s guide to Europe: the infection dynamics of an ongoing Wolbachia invasion and mitochondrial selective sweep in Rhagoletis cerasi. Mol Ecol. 25(7):1595–1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seemann T. 2014. Prokka: rapid prokaryotic genome annotation. Bioinformatics 30(14):2068–2069. [DOI] [PubMed] [Google Scholar]
- Shropshire JD, Bordenstein SR.. 2019. Two-By-One model of cytoplasmic incompatibility: synthetic recapitulation by transgenic expression of cifA and cifB in Drosophila. PLoS Genet. 15(6):e1008221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simão FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM.. 2015. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31(19):3210–3212. [DOI] [PubMed] [Google Scholar]
- Sinkins SP, Braig HR, O’neill SL.. 1995. Wolbachia pipientis: bacterial density and unidirectional cytoplasmic incompatibility between infected populations of Aedes albopictus. Exp Parasitol. 81(3):284–291. [DOI] [PubMed] [Google Scholar]
- Sutton ER, Harris SR, Parkhill J, Sinkins SP.. 2014. Comparative genome analysis of Wolbachia strain wAu. BMC Genomics 15(1):928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teixeira L, Ferreira Á, Ashburner M.. 2008. The bacterial symbiont Wolbachia induces resistance to RNA viral infections in Drosophila melanogaster. PLoS Biol. 6(12):e1000002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian S, Huajun W, Wu J.. 2012. Computational prediction of furin cleavage sites by a hybrid method and understanding mechanism underlying diseases. Sci Rep. 2(1):261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker T, et al. 2011. The wMel Wolbachia strain blocks dengue and invades caged Aedes aegypti populations. Nature 476(7361):450–453. [DOI] [PubMed] [Google Scholar]
- Weinert LA, Araujo-Jnr EV, Ahmed MZ, Welch JJ.. 2015. The incidence of bacterial endosymbionts in terrestrial arthropods. Proc R Soc B. 282(1807):20150249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werren JH. 1997. Biology of Wolbachia. Annu Rev Entomol. 42(1):587–609. [DOI] [PubMed] [Google Scholar]
- Werren JH, Baldo L, Clark ME.. 2008. Wolbachia: master manipulators of invertebrate biology. Nat Rev Microbiol. 6(10):741–751. [DOI] [PubMed] [Google Scholar]
- Woolfit M, et al. 2013. Genomic evolution of the pathogenic Wolbachia strain, wMelPop. Genome Biol Evol. 5(11):2189–2204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu M, et al. 2004. Phylogenomics of the reproductive parasite Wolbachia pipientis wMel: a streamlined genome overrun by mobile genetic elements. PLoS Biol. 2(3):E69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia X. 2018. DAMBE7: new and improved tools for data analysis in molecular biology and evolution. Mol Biol Evol. 35(6):1550–1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zabalou S, et al. 2004. Wolbachia-induced cytoplasmic incompatibility as a means for insect pest population control. Proc Natl Acad Sci USA. 101(42):15042–15045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zabalou S, et al. 2008. Multiple rescue factors within a Wolbachia strain. Genetics 178(4):2145–2160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zabalou S, et al. 2009. Incompatible insect technique: incompatible males from a Ceratitis capitata genetic sexing strain. Entomol Exp Appl. 132(3):232–240. [Google Scholar]
- Zhou Y, Liang Y, Lynch KH, Dennis JJ, Wishart DS.. 2011. PHAST: a fast phage search tool. Nucleic Acids Res. 39(Suppl):W347–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.