

Abstract
Brain interstitial pH (pHbrain) alterations play an important role in the mechanisms of neuronal injury in neonatal hypoxic-ischemic encephalopathy (HIE) induced by perinatal asphyxia. The newborn pig is an established large animal model to study HIE, however, only limited information on pHbrain alterations is available in this species and it is restricted to experimental perinatal asphyxia (PA) and the immediate reventilation. Therefore, we sought to determine pHbrain over the first 24h of HIE development in piglets. Anaesthetized, ventilated newborn pigs (n = 16) were instrumented to control major physiological parameters. pHbrain was determined in the parietal cortex using a pH-selective microelectrode. PA was induced by ventilation with a gas mixture containing 6%O2-20%CO2 for 20 min, followed by reventilation with air for 24h, then the brains were processed for histopathology assessment. The core temperature was maintained unchanged during PA (38.4±0.1 vs 38.3±0.1°C, at baseline versus the end of PA, respectively; mean±SEM). In the arterial blood, PA resulted in severe hypoxia (PaO2: 65±4 vs 23±1*mmHg, *p<0.05) as well as acidosis (pHa: 7.53±0.03 vs 6.79±0.02*) that is consistent with the observed hypercapnia (PaCO2: 37±3 vs 160±6*mmHg) and lactacidemia (1.6±0.3 vs 10.3±0.7*mmol/L). Meanwhile, pHbrain decreased progressively from 7.21±0.03 to 5.94±0.11*. Reventilation restored pHa, blood gases and metabolites within 4 hours except for PaCO2 that remained slightly elevated. pHbrain returned to 7.0 in 29.4±5.5 min and then recovered to its baseline level without showing secondary alterations during the 24 h observation period. Neuropathological assessment also confirmed neuronal injury. In conclusion, in spite of the severe acidosis and alterations in blood gases during experimental PA, pHbrain recovered rapidly and notably, there was no post-asphyxia hypocapnia that is commonly observed in many HIE babies. Thus, the neuronal injury in our piglet model is not associated with abnormal pHbrain or low PaCO2 over the first 24 h after PA.
Introduction
Perinatal asphyxia (PA), defined as O2 deprivation around the time of delivery, is one of the primary causes of neonatal morbidity and mortality worldwide, affecting ~4 million neonates annually [1]. The interruption of the placental or pulmonary gas exchange induces immediate metabolic changes (hypoxemia, hypercapnia and mixed acidosis) that trigger cardiovascular responses in favour of maintaining O2 delivery to the myocardium and the brain (i.e. centralized circulation). When these compensatory mechanisms are exhausted, critical tissue hypoxia/ischemia will occur, and the subsequent metabolic crisis will lead to hypoxic-ischemic encephalopathy (HIE) in the survivors [2].
Despite the partial efficacy of therapeutic hypothermia to mitigate the adverse outcome of PA/HIE [3,4], preclinical animal models are still required to study the precise pathophysiology of HIE development, and also to test putative neuroprotective approaches [5]. Similar to the heterogeneity of the aetiology, severity, and duration of the human PA/HIE syndrome, there is also considerable heterogeneity in the preclinical models, both in the species, the prenatal/postnatal age of the animals, the methodology to induce PA/HIE, and the length of the post-asphyxial observation period. These differences do not allow easy comparison and translation of the accumulated data. The newborn pig has long been identified and accepted as a general preclinical model to study the term human neonate [6,7]. There are a number of important similarities between the two species in brain structure, metabolism, development [8,9], and in cerebrovascular physiology [10] that make piglets adequate for translational PA/HIE research. We have recently published a newborn piglet PA/HIE model [11] that produced major hallmarks of human PA/HIE and was able to elicit significant neuronal injury, but did not include manipulations that occur never or very rarely in humans such as bilateral carotid artery occlusion or transient hemorrhagic hypotension.
In the present study, we set out to investigate the course of brain interstitial pH (pHbrain) changes during PA and the subacute phase of HIE development in our piglet model. In HIE patients using magnetic resonance spectroscopy (MRS) techniques at < 2 weeks of age, brain intracellular pH (pHi) levels were found to be rather elevated [12]. This alkalosis persisted for months and correlated with adverse neurological outcome, and the described pH alteration may be one of the features of the so-called secondary energy failure [13,14]. However, changes in pHi may not truthfully reflect pHbrain alterations as transmembrane pH gradients are subject to change during HIE development, and furthermore, intracellular and extracellular pH have different targets via which they may change brain activity and energy consumption [15]. There are very limited data on pHbrain changes elicited by PA/HIE from piglets [16], and these studies did not follow the course of pHbrain beyond 4 hours after the completion of the hypoxic-ischemic stress. We now report quantitative cerebrocortical pHbrain data in our well-characterized piglet PA/HIE model both during the PA and the first 24 hours of HIE development.
Materials and methods
All experimental procedures involving animals were approved in a three-step process. First, the detailed experimental plan was carefully reviewed and approved by the Institutional Animal Care and Use Committee of the University of Szeged (IACUC, in Hungarian: SZTE Munkahelyi Állatjóléti Bizottság). Second, the approval of the IACUC-endorsed experimental plan was requested from the National Ethical Committee on Animal Experiments (in Hungarian: Állatkísérletes Tudományos Etikai Tanács, ÁTET). Third, the National Food Chain Safety and Animal Health Directorate of Csongrád county, Hungary on behalf of the Hungarian Government issued the permit based on the ÁTET recommendation (permit nr: XIV./1414/2015). All animal experiments complied with (1) the guidelines of the Scientific Committee of Animal Experimentation of the Hungarian Academy of Sciences (updated Law and Regulations on Animal Protection: 40/2013. (II. 14.) Gov. of Hungary), (2) the EU Directive 2010/ 63/EU on animal protection used for scientific research, and (3) with the ARRIVE guidelines. Furthermore, we state that all procedures in the present study were performed on anesthetized animals, also that all animals were anesthetized and treated with analgetics along with intensive monitoring of vital signs throughout the observation period (24 h) without expected or observed mortality. At the end of the experiments the animals were euthanized with an overdose of pentobarbital sodium (300 mg, Release; Wirtschaftsgenossenschaft deutscher Tierärzte eG, Garbsen, Germany).
Newborn (≤1 day old) male Landrace pigs (body weight: 1.5–2.5 kg, n = 16) were obtained from a local company (Pigmark Ltd., Co., H-6728, Rózsamajor út 13., Szeged, Hungary) and delivered to the laboratory on the morning of the experiments. Anaesthesia was induced by sodium thiopental (45 mg/kg ip; Sandoz, Kundl, Austria). Piglets were intubated via tracheotomy and artificially ventilated by a pressure-controlled small animal respirator with warmed, humidified medical air (21% O2, balance N2) that could be optionally supplemented with O2. Respiratory settings (fraction of inspired O2: 0.21–0.25; respiratory rate (RR): 30–35 1/min, peak inspiratory pressure: 120–135 mmH2O) were adjusted to maintain blood gas values and O2 saturation in the physiologic range. The right femoral vein was catheterized under aseptic conditions to maintain anaesthesia/analgesia with a bolus injection of morphine (100 μg/kg; Teva, Petach Tikva, Israel) and midazolam (250 μg/kg; Torrex Pharma, Vienna, Austria), then with continuous infusion of morphine (10 μg/kg/h), midazolam (250 μg/kg/h) and fluids (5% glucose, 0.45% NaCl 3–5 ml/kg/h). A second catheter was placed into the right carotid artery for continuous monitoring of mean arterial blood pressure (MABP) and heart rate (HR). This artery was chosen as ligation of the femoral artery would have resulted in critical ischemia of the hindlimb over the 24h reventilation period (personal observations), in contrast, unilateral carotid artery occlusion has been shown not to affect cerebral blood flow [17]. Rectal temperature was measured continuously and kept in the physiologic range (38.5±0.5°C) with a servo-controlled water circulation heating-cooling pads (Blanketrol III., Cincinnati Sub-Zero, Cincinnati, Ohio, USA). O2 saturation, MABP and HR were continuously monitored using a Hewlett-Packard M1094 monitor (Palo Alto, California, USA) and recorded online (MecifView, Arlington, Mass., USA). These parameters were recorded at baseline, during PA and and then for 10 minutes at the beginning of each reventilation hour. Arterial blood samples (~300 μl/sample) were analysed with a blood analysis system (EPOC Blood Analysis, Epocal Inc., Ottawa Canada) at baseline, at the end of asphyxia; then at selected intervals up to 20 hours to determine arterial blood pH, gas tensions, base excess, central oxygen saturation, hemoglobin, bicarbonate, glucose and lactate concentrations. Prophylactic antibiotics were given iv.: penicillin (50 mg/kg/12 h, Teva, Petah Tikva, Israel) and gentamicin (2.5 mg/kg/12 h, Sanofi, Paris, France). The urinary bladder was tapped by suprapubic puncture at 12 hour after asphyxia.
pHbrain measurements
Measurements were performed inside a self-made Faraday cage with 4 Hz sampling rate. pH-selective microelectrodes (external tip diameter: 50 μm) were obtained from Unisense (Aarhus, Denmark), whereas glass reference microelectrodes (external tip diameter: ~20 μm and filled with 150 mM NaCl; resistance: ~4-5x1010 Ohm) were self-made and used with Ag/AgCl wire electrodes. The electrodes were mounted on stereotaxic manipulators for calibration in 3 different warmed (38°C) buffer solutions (pH: 6.10, 7.10, and 8.10, respectively) before each experiment. The piglet head was fixed in a stereotaxic frame and after retracting the scalp, two small circular craniotomies (Ø≅5 mm) were made over the fronto-parietal cortex, and the dura mater was gently removed. The tips of the pH and reference microelectrodes were installed ~1–2 mm deep into the exposed cortex, and a Ag/AgCl ground electrode was placed under the scalp. The electrode signals were recorded, digitized and stored either using a custom-built differential electrometer (>1014 Ohm input impedance; 16 Hz low pass cut-off), a 16-bit analog-to-digital converter (National Instruments, Austin, TX) and WinEDR software (Dr. John Dempster, University of Strathclyde, UK), or using a Microsensor Multimeter and SensorTrace Logger software (Unisense, Aarhus, Denmark). Evaluation of the recordings was performed offline: by applying linear regression analysis, the signals from the calibration solutions were fitted with a curve and the data were converted to pH values using linear interpolation [15,18,19]. As the technique allows stable continuous pHbrain measurements reliably only for 3–4 hours, in different animals, different time windows were chosen to be assessed (baseline, PA and the first 4 hours of reventilation (n = 6), 8th-14th hours (n = 8) and 20th-24th hours (n = 3) of reventilation as presented in the Results).
Induction of asphyxia
After surgery, a 1h recovery period allowed stabilization of the monitored physiological parameters prior to obtaining their baseline values. PA was induced by switching ventilation from medical air to a hypoxic-hypercapnic gas mixture (6% O2, 20% CO2, balance N2) for 20 minutes, simultaneously reducing the RR to 15 1/min and stopping the fluid/glucose administration. Piglets (n = 13) were reventilated (RR: 30 1/min) with medical air for the remaining time of the experiment.
In three additional animals, before inducing PA, the effect of graded normoxic hypercapnia on pHbrain was evaluated by 5% step-wise increases in inhaled CO2 from 0% to 20%, for 7 to 8 min each. After the graded hypercapnia, normocapnia was restored for 30 min. These animals were euthanized with an overdose of pentobarbital sodium (300 mg, Release; Wirtschaftsgenossenschaft deutscher Tierärzte eG, Garbsen, Germany) at 2 hours after PA.
Neuropathology
The objective of the neuropathology examination was to test if the asphyxia-induced neuronal injury was similar to what we reported previously using this PA/HIE model at 24 hours after asphyxia [11]. Accordingly, out of the 13 piglets exposed to PA, only those animals which were maintained for 24 hours (n = 8) were included. The brains of the eight anesthetized animals were perfused with cold (4°C) physiological saline through the catheterized common carotid arteries 24 hours after the end of asphyxia. The brains were gently removed and immersion-fixed in 4°C, 4% paraformaldehyde solution before further processing. Paraffin embedded, 4 μm sections were stained with haematoxylin-eosin, and neuropathology was assessed by light microscopic evaluation (Leica Microsystems, Wetzlar, Germany). Damaged neurons were identified using the major hallmarks of dark eosinophilic cytosol, as well as pyknotic or disrupted nuclei. The degree of cerebrocortical neuronal damage in the frontal, parietal, temporal, and occipital cortices was determined adapting a previously published scoring system [11,20]. Briefly, the pattern of neuronal injury (none < scattered < grouped < panlaminar) was determined in 20–20 non-overlapping fields of vision under 20x magnification in each assessed cortical region. Then, scores (0–9) were given to each region based on the frequency (% of 20 examined fields) of the most severe pattern of injury observed. The neuronal damage in the putamen, thalamus and the hippocampal CA1 regions was assessed with cell counting in non-overlapping areas (in 5-5-3 fields of vision respectively; under 200x magnification) as in [11,21]. Neuronal injury in these regions was expressed as the percentage of damaged neurons.
Statistical analysis
Results were analysed offline and plotted using SigmaPlot (v12.0, Systat Software Inc., San Jose, CA, USA) or a MATLAB environment (Mathworks Inc., Natick, MA, USA). Neuropathology scores are expressed as median, 25–75 and 5–95 percentiles. All other data are expressed as mean±SEM. Normality was tested with the Shapiro-Wilk test. The correlation between PaCO2 and pHbrain data were calculated with MATLAB’s polynomial curve fitting. Parametric data were compared with one-way repeated measure of analysis of variance (RM ANOVA) followed by the Student-Newman-Keuls post hoc test. Level of significance (p) was set at 0.05.
Results
pHbrain changes during PA and HIE development
Induction of PA elicited a reduction in pHbrain that continued without levelling off over the 20 min insult, during which pHbrain dropped from the baseline value of 7.21±0.03 to 5.94±0.11 by the end of asphyxia (n = 6; Fig 1A). Upon reventilation, pHbrain was restored to 7.0 in 29.4±5.5 minutes with subsequent stabilization at a level that was virtually indistinguishable from the original baseline. Thereafter, from 2 h onwards pHbrain remained slightly below baseline (on average, by 0.10±0.02 pH units) without showing any marked alterations at any observed time point within the 24-hour follow-up period (Fig 1B).
Fig 1. pHbrain changes during PA and the reventilation period.

(A) pHbrain changes (grey lines–individual tracings, bold black line–mean) are plotted 10 min prior the onset asphyxia, during asphyxia, and the first hour of reventilation. pHbrain fell progressively during asphyxia showing severe cerebrocortical acidosis, by the end of the insult the pH drop exceeded 1.0 pH unit virtually in all asphyxiated animals. Reventilation quickly restored pHbrain to baseline levels. (B) pHbrain alterations upon PA and over the 24-hour period after PA: after recovery from the PA-induced severe acidosis, no further significant pHbrain alterations were detected at the selected time intervals (n = 6 at PA and between 1–4 hours; n = 8 between 8–14 hours and n = 3 between 20–24 hours; respectively). B: baseline, A: at the end of 20 minute PA. Panel B data points are presented as mean±SEM. *p<0.05 vs. baseline.
During the 20 min asphyxia period, O2 saturation fell to below 30% and HR increased from about 140 1/min to nearly 200 1/min within the first 2 min, after which they showed no major changes, whereas the response in MABP was biphasic with a transient rise to about 90 mmHg followed by a slower decrease to below baseline (Fig 2). A rapid recovery of O2 saturation to above 80% was seen during the first 2–3 min of reventilation, paralleled by transient increases in HR and MABP to above 240 1/min and 80 mmHg, respectively, after which the signals gradually recovered towards their normal values.
Fig 2. Hemodynamic and oxygenation changes during PA.
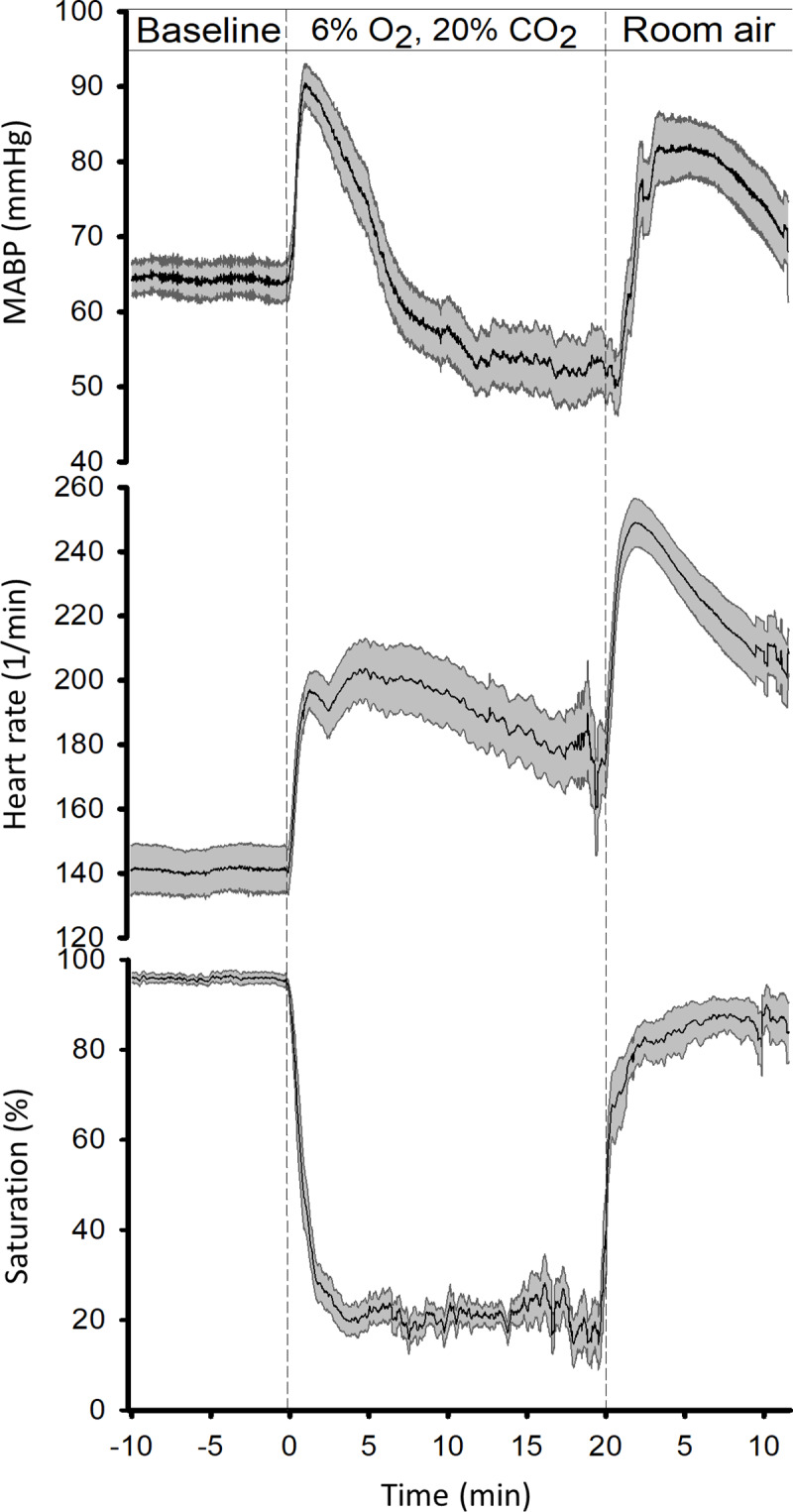
In all panels, solid lines indicate the mean and the grey shaded area the SEM. (n = 13). After obtaining baselines, PA was initiated resulting first in the transient elevation of mean arterial blood pressure (MABP, top panel) and heart rate (HR, middle panel) accompanying the rapid fall in blood oxygen saturation (pulsoxymetry, bottom panel). While blood oxygen saturation remained ~20% throughout the asphyxia, after the initial peak, MABP fell continuously below baseline, whereas the drop in HR was more moderate and HR remained elevated. Reventilation with air quickly restored oxygen saturation and induced a second transient elevation in both MABP and HR.
Longer-term monitoring of the physiological parameters reflected well the expected effects of PA and reventilation. Blood hemoglobin concentration (baseline: 8.0±0.3 g/dl; time course not illustrated), core body temperature (controlled by heating, see Methods), O2 saturation, MABP and HR were within the normal range at baseline before PA and the values were not significantly different from baseline throughout the survival (Fig 3A–3D). In addition to the pulsoxymetry data, arterial blood gas analysis at the end of PA also confirmed the development of central haemoglobin desaturation (from 94±4 to 13±4%) along with severe acidosis, hypoxia and hypercapnia (Fig 3E–3H). Indeed, the fall in the arterial blood pH (pHa) from 7.53±0.03 to 6.79±0.02 was substantial and paralleled by a rise in PaCO2 to 160±6 mmHg, however, pHa remained more than 0.8 pH unit higher than pHbrain. Blood glucose and lactate levels were also profoundly raised (Fig 3I and 3J) [22], indicating the metabolic response to PA. The large drop in base excess by 17.4±1.5 mmol/L and reduction in bicarbonate concentration (Fig 3K and 3L) together with the low pHa (6.79) at the end of PA indicate that asphyxia developed much beyond the key clinical criteria of severe BA in human neonates (pH <7.0 and base deficit ≥12 to 16 mmol/L [3,4]). Reventilation quickly restored normoxia in arterial blood, but PaCO2 levels remained slightly elevated, although the change was statistically significant only at 4 hours. Base excess was already normalized by this time point, and pHa returned to 7.39±0.02, normal for piglets [23]. In a similar fashion, blood glucose and lactate levels also returned to baseline by 4 hours, although both were still significantly elevated at 1 hour after PA.
Fig 3. Physiological parameters and blood chemistry changes during PA and the subsequent 24h reventilation period.

Core temperature of the animals (n = 13) was maintained in the physiologic range (38.5±0.5°C) during the whole experiment (A). Blood oxygen saturation by pulsoxymetry (B) and mean arterial blood pressure (MABP, (C)) returned to baseline levels soon after asphyxia, however, the heart rate remained moderately elevated (D). Arterial blood gas analysis revealed that asphyxia resulted in severe acidosis (E), hypoxemia (F), hypercapnia (G), and central (arterial blood) desaturation (H). Plasma glucose (I) and lactate levels (J) were markedly elevated along with large drops in base excess (K) and significant reductions in blood bicarbonate concentrations (L). Reventilation restored most of the deranged parameters by 4 hours, and they were not significantly different from baseline levels afterwards, except for pH that was restored to the normal values [23] and not to the slightly alkalotic baseline. B: baseline, A: at the end of 20 minute PA. Bars and whiskers represent mean±SEM, *p<0.05 vs. baseline values.
Neuropathology analysis confirmed the development of HIE by revealing medium/severe neuronal damage in all examined neocortical regions (Fig 4A and 4C), and also in the hippocampal CA1 region, the thalamus and the putamen (Fig 4B). These findings are in accordance with previously reported neuronal injury using the same PA stress [11].
Fig 4. Neuronal injury evaluated at 24 hours after PA.

(A) Representative photomicrographs showing injured red neurons at 24-hours of after asphyxia in the CA1 hippocampal region, the putamen, and the thalamus (scale bar: 100μm) (B) Cell counting revealed moderate neuronal damage in these regions (n = 8, mean±SEM). (C) Asphyxia also induced moderate/severe neocortical damage shown by medium/high neuropathology scores (lines, boxes, and whiskers represent the median, the 25th-75th, and the 5th-95th percentiles, respectively).
pHbrain changes during graded normoxic hypercapnia
These experiments were performed to evaluate the contribution of the respiratory component to the pHbrain changes recorded during PA. Step-wise, 5% increases in inhaled CO2 (Fig 5A) resulted in proportional step-wise reductions in pHbrain (Fig 5B and 5C). Arterial blood gas analysis revealed similar graded reductions in pHa, thus the difference between pHbrain and pHa remained unchanged during hypercapnia (Fig 5B). Maximal changes were observed during the inhalation of 20% CO2 when pHa dropped from 7.52±0.06 to 6.98±0.02, whereas pHbrain from 7.32±0.01 to 6.77±0.02. The pHbrain and pHa changes were fully reversed by restoration of normocapnia.
Fig 5. Cerebral acidosis induced by graded normoxic hypercapnia.

(A) pHbrain changes during graded normoxic hypercapnia induced by ventilation with 5–20% CO2 followed by restoration of normocapnia (grey lines–individual tracings, bold black line–mean). (B) Simultaneous reductions in pHa and pHbrain during graded normoxic hypercapnia were in accordance with the elevations in PaCO2, the values were (mmHg) 5%: 59.6±7.8, 10%: 84.7±9.0, 15%: 107.0±6.7, 20%: 120.3±8.2, (mean±SEM; n = 3). Corresponding PaO2 values were (mmHg) 5%: 75.5±12.8, 10%: 78.2±11.3, 15%: 85.5±8.2, 20%: 84.5±9.8, (mean±SEM; n = 3). (C) Linear regression shows close correlation between PaCO2 and pHbrain (R2 = 0.8546) values obtained during graded hypercapnia (5–20% CO2).
Discussion
In this study we present in vivo real-time recorded pHbrain values during PA and the subacute phase of HIE in a translational piglet PA/HIE model. The major findings of the present study are the following: (1) our experimental model elicited PA reflecting its major hallmarks and triggered neuronal injury corresponding to moderate/severe HIE revealed by neuropathology; (2) cerebrocortical pHbrain dropped drastically in response to PA, the acidosis exceeding one pH unit in the brain compared to the arterial blood; (3) reventilation/reoxygenation allowed the restoration of pHbrain to baseline levels, then pHbrain remained stable around baseline levels without major shifts over the 24h observation period.
Every PA/HIE model using postnatal animals inevitably carries the limitation that PA is induced after and not before/during the cardiorespiratory adaptation to extrauterine life. However, we believe that in our study this disadvantage has been minimized by two factors. First, we used truly newborn piglets, thus their vulnerability to PA was least likely to have considerably changed by postnatal development. Second, we elicited PA that resulted in severe enough arterial pH and blood gas alterations that are known to signal human HIE development. This statement is justified by a clinical study that reported significant differences in the degree of acidosis (pHa: 6.75±0.18 vs 6.90±0.18) and hypercapnia (PaCO2: 141±37 vs PaCO2: 94±22 mmHg) between asphyxiated babies presenting vs not presenting with HIE [24]. In our model the corresponding values were pHa: 6.79±0.02 and PaCO2: 160±6 mmHg, closely matching the data from human HIE patient group. Interestingly, virtually identical values from umbilical artery blood samples (pHa: 6.69±0.04 and PaCO2: 156±4 mmHg) were obtained in piglets undergoing spontaneous PA during delivery also indicating the translational value of the piglet as a PA/HIE model species [25]. We have developed and used this PA/HIE protocol to test the putative neuroprotective effect of molecular hydrogen [11]. We report virtually identical neocortical, hippocampal, and subcortical neuronal injury in the present study compared to those in [11]. Based on the similar neuropathology findings, we can assume that the surgical manipulations associated with pHbrain measurements did not affect HIE development in this study, lending support to the translational value of the present pHbrain findings.
The importance of pHbrain determining neurological outcome following hypoxic-ischemic stress has long been acknowledged [26]. However, quantitative data on pHbrain changes during/after PA in piglet PA/HIE models are very scarce in the literature [16,27,28]. Bender et al. [16] assessed pHbrain using a similar technique in 1–3 days old piglets. PA was induced also with a hypoxic-hypercapnic gas mixture (5–8%O2-7%CO2) for 30 min. At the end of PA, the measured pHbrain was 6.26±0.14, which is about 0.3 pH unit higher than in our study and likely at least part due to the much lower PaCO2 values (61±1 vs. 160±6 mmHg [16] vs present study, respectively). After PA, the reventilation commenced with 100% O2 ventilation, in addition, sodium bicarbonate (2 mEq/kg, iv) was infused for rapid correction of arterial pH. Both of these interventions are likely to decrease the translational value of the study by Bender et al. [16] as they are not included in current guidelines of neonatal care, and they may have affected the recovery of pHbrain that was completed in 90 min. Moreover, there was no significant neuronal injury compared to sham operated animals, except experiments where PA was combined with hemorrhagic hypotension, suggesting that the applied PA as such was not severe enough to elicit HIE. Corbett et al. [27] used MRS in 8±3 day old piglets to determine pHi during and after ischemia but not genuine PA. Incomplete cerebral ischemia was elicited by combining bilateral carotid artery occlusion and hemorrhagic hypotension for 25 min followed by 90 min of reperfusion [28]. Severe acidosis developed during the ischemia that was dependent on blood glucose levels during the stress. Brain pHi dropped below 5.6 in fed piglets that responded to ischemia with hyperglycemia (9.4-15 mmol/L), whereas in fasted piglets responding with hypoglycaemia (1.4-2.6 mmol/L) the nadir at the end of ischemia was mere pHi≅6.6. Upon reperfusion, pHi was again normalized within the 90 min observation period.
From the above discussed studies it is clear that higher levels of hypercapnia, cerebral blood flow, and blood glucose all promote the development of cerebral acidosis during PA. Increases in PaCO2 to 140–160 mmHg observed both in human and piglet PA will alone reduce pHi to 6.5–6.6 under normoxic conditions [29]. Also in the present study, we provide evidence that under normoxic conditions the inhalation of 20% CO2 alone (PaCO2:120 mmHg) results in a pHbrain drop to 6.8. Using linear regression (Fig 4C), we then calculated that the developing hypercapnia (at PaCO2: 160 mmHg) alone results in a pHbrain drop to 6.50 in our PA model, in full agreement with previous results. Further acidification during PA will be dominantly determined by the increases in the rate of anaerobic glycolysis fuelling the subsequent lactic acid production. The rate of glycolysis is limited by glucose delivery to the hypoxic brain, as its reduction by either reducing cerebral blood flow [16] or blood glucose levels [28] attenuated the development of acidosis. We have previously shown that in our PA model significant cerebral ischemia does not develop [11] as in most animals the drop in MABP during the 20 min PA (Fig 4) does not reach the lower limit of blood flow autoregulation [30]. Admittedly, PA in our model has not been long enough for the development of hypotension and bradycardia commonly seen in severely asphyxiated infants, however, it can nicely represent the PA phase before the cardiovascular adaptation mechanisms are exhausted and likely the most pronounced pHbrain alterations occur. In summary, our current study provides new compelling experimental evidence that not only cerebral ischemia but bona fide PA combining clinically relevant levels of hypoxia, hypercapnia and hyperglycemia is sufficient to elicit cerebral acidosis that is severe enough (pHbrain <6.0) to strongly affect neuronal viability [31]. The developing ≅0.8 pH unit difference between pHbrain and pHa signals a >6-fold H+ gradient across the blood-brain barrier that is in compliance with previous findings that the blood-brain barrier is mature in newborn pigs and not severely compromised by PA [32].
Alterations in pHbrain may play important pathophysiological roles not only during acute PA, but also after reventilation/reoxygenation. Previously, pHbrain was reported to be stable in piglets after restoration from PA, but it was not followed up beyond four hours of recovery [16]. In contrast, our current study extended the post-asphyxia observation period to 24 hours. We found no major secondary alterations in pHbrain in this piglet PA/HIE model during the 24-hour period after PA. Our present findings are in compliance with previous pHi data obtained with MRS in a piglet HIE model using hypoxic-ischemic stress instead of PA [33]. In this study, pHi remained at baseline levels after restoration from the ischemic stress for 48 hours, furthermore, the development of secondary energy failure was not reflected in pHi alterations in this time period. In a human MRS study, pHi was also reported to be normal (7.13±0.05) in asphyxiated normocapnic newborns during the first day of life [34], in accordance with our present study. Importantly, brain alkalosis developed during the following days, and then persisted for weeks, even months [13,34]. However, we would like to point out that our PA/HIE piglet model represents the subgroup of asphyxiated babies that require intubation and respiratory support, but not those who breathe spontaneously. While the mechanically ventilated babies stay normocapnic, the spontaneously breathing babies often hyperventilate and may develop hypocapnia [24]. This response can reflect a relative hyperventilation, secondary to the induction of hypoxic hypometabolism resulting in reduced CO2 production [35]. Reduction in PaCO2 due to hyperventilation tends to elevate pHbrain and this notion is of special interest as hypocapnia has been identified as an independent risk factor for adverse neurological outcome [36,37]. In piglets, moderate hypocapnia was sufficient to elicit a reduction in cerebral perfusion with a simultaneous increase in pH and lactate levels even in the absence of asphyxia [38]. Indeed, our findings indirectly suggest that maintenance of normocapnia/ slight hypercapnia may prevent secondary pHbrain alterations. In a recent work aimed to establish a translationally valid small-animal model of birth asphyxia in rats and guinea pigs, higher PaCO2 levels during simulated birth asphyxia and gradual restoration of normocapnia afterwards were found to elicit beneficial effects on cerebral metabolic acidosis, oxygen and lactate levels [39]. Clearly, further studies are warranted to explore these effects in the piglet model. Our current study had some additional limitations. We collected pHbrain data from the neocortex using only one level of asphyxia, leaving other brain regions and levels of stress unexplored.
Conclusions
Our translational piglet PA/HIE model reproduces all major hallmarks of birth asphyxia and elicits significant neuronal injury without employing carotid artery occlusions and/or hemorrhagic hypotension. In this model, pHbrain drops below 6.0, ≅0.8 pH unit lower than pHa during PA, establishing a pathogenetic role of severe acidosis in neuronal injury. However, secondary pHbrain alterations after restoration of baseline levels were not observed during the 24-hour subacute period, perhaps due to the prevention of secondary hypocapnia by controlled mechanical ventilation.
Data Availability
All experimental data are available at Open Science Framework (osf.io): DOI 10.17605/OSF.IO/MUTGA.
Funding Statement
JN,GR,VTSZ,VV,VK,FD; 2.0 1.3 2017 1,2.1 NKP 2017 00002; Hungarian Brain Research Program JN,GR,VTSZ,VV,VK,FD; EFOP-3.6.1-16-2016-00014; EU-funded Hungarian grant EFOP JN,GR,VTSZ,VV,VK,FD; GINOP 2.3.2. 15 2016 00034; GINOP The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Lawn JE, Manandhar A, Haws RA, Darmstadt GL. Reducing one million child deaths from birth asphyxia—A survey of health systems gaps and priorities. Heal Res Policy Syst. 2007;5: 1–10. 10.1186/1478-4505-5-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Volpe JJ. Neonatal encephalopathy: An inadequate term for hypoxic-ischemic encephalopathy. Ann Neurol. 2012;72: 156–166. 10.1002/ana.23647 [DOI] [PubMed] [Google Scholar]
- 3.Azzopardi D V., Strohm B, Edwards AD, Dyet L, Halliday HL, Juszczak E, et al. Moderate hypothermia to treat perinatal asphyxial encephalopathy. N Engl J Med. 2009;361: 1349–1358. 10.1056/NEJMoa0900854 [DOI] [PubMed] [Google Scholar]
- 4.Thoresen M. Who should we cool after perinatal asphyxia? Semin Fetal Neonatal Med. 2015;20: 66–71. 10.1016/j.siny.2015.01.002 [DOI] [PubMed] [Google Scholar]
- 5.Yager JY, Ashwal S. Animal Models of Perinatal Hypoxic-Ischemic Brain Damage. Pediatr Neurol. 2009;40: 156–167. 10.1016/j.pediatrneurol.2008.10.025 [DOI] [PubMed] [Google Scholar]
- 6.Bustad LK, McClellan RO. Swine in Biomedical Research. Science. 1966;152: 1526–1530. 10.1126/science.152.3728.1526 [DOI] [PubMed] [Google Scholar]
- 7.Book SA, Bustad LK. The Fetal and Neonatal Pig in Biomedical Research. Am J Anim Sci. 1974;38: 997–1002. [DOI] [PubMed] [Google Scholar]
- 8.Conrad MS, Johnson RW. The Domestic Piglet: An Important Model for Investigating the Neurodevelopmental Consequences of Early Life Insults. Annu Rev Anim Biosci. 2015;3: 245–264. 10.1146/annurev-animal-022114-111049 [DOI] [PubMed] [Google Scholar]
- 9.Dobbing J, Sands J. Comparative aspects of the brain growth spurt. Early Hum Dev. 1979;311: 79–83. 10.1016/0378-3782(79)90022-7 [DOI] [PubMed] [Google Scholar]
- 10.Busija DW. Cerebral Circulation of the Fetus and Newborn In: Bevan RD, Bevan JA, editors. The Human Brain Circulation. Humana Press; 1994. pp. 259–269. [Google Scholar]
- 11.Nemeth J, Toth-Szuki V, Varga V, Kovacs V, Remzso G, Domoki F. Molecular hydrogen affords neuroprotection in a translational piglet model of hypoxic-ischemic encephalopathy. J Physiol Pharmacol. 2016;67: 677–689. [PubMed] [Google Scholar]
- 12.Azzopardi D, Wyatt JS, Cady EB, Delpy DT, Baudin J, Stewart AL, et al. Prognosis of Newborn Infants with Hypoxic-Ischemic Brain Injury Assessed by Phosphorus Magnetic Resonance Spectroscopy. Pediatr Res. 1989;25: 445–451. 10.1203/00006450-198905000-00004 [DOI] [PubMed] [Google Scholar]
- 13.Robertson NJ, Cox IJ, Cowan FM, Counsell SJ, Azzopardi D, Edwards AD. Cerebral Intracellular Lactic Alkalosis Persisting Months after Neonatal Encephalopathy Measured by Magnetic Resonance Spectroscopy. Pediatr Res. 1999;46: 287–296. [DOI] [PubMed] [Google Scholar]
- 14.Robertson NJ, Cowan FM, Cox IJ, Edwards AD. Brain alkaline intracellular pH after neonatal encephalopathy. Ann Neurol. 2002;52: 732–742. 10.1002/ana.10365 [DOI] [PubMed] [Google Scholar]
- 15.Helmy MM, Tolner EA, Vanhatalo S, Voipio J, Kaila K. Brain alkalosis causes birth asphyxia seizures, suggesting therapeutic strategy. Ann Neurol. 2011;69: 493–500. 10.1002/ana.22223 [DOI] [PubMed] [Google Scholar]
- 16.Bender TM, Johnston JA, Manepalli AN, Mink RB. Association between brain tissue pH and brain injury during asphyxia in piglets. Resuscitation. 2003;59: 243–254. 10.1016/S0300-9572(03)00207-7 [DOI] [PubMed] [Google Scholar]
- 17.Laptook AR, Stonestreet BS, Oh W. The effect of carotid artery ligation on brain blood flow in newborn piglets. Brain Res. 1983;276: 51–54. 10.1016/0006-8993(83)90547-4 [DOI] [PubMed] [Google Scholar]
- 18.Voipio J, Kaila K. Interstitial PCO2 and pH in rat hippocampal slices measured by means of a novel fast CO2/H+-sensitive microelectrode based on a PVC-gelled membrane. Pflügers Arch Eur J Physiol. 1993;423: 193–201. 10.1007/BF00374394 [DOI] [PubMed] [Google Scholar]
- 19.Voipio J, Pasternack M, MacLeod K. Ion-sensitive microelectrodes. 2nd editio. Microelectrode Techniques–The Plymouth Workshop Handbook 2nd editio Cambridge: The Company of Biologists Limited; 1994. pp. 275–316. [Google Scholar]
- 20.Foster KA, Colditz PB, Lingwood BE, Burke C, Dunster KR, Roberts MS. An improved survival model of hypoxia/ischaemia in the piglet suitable for neuroprotection studies. Brain Res. 2001;919: 122–131. 10.1016/S0006-8993(01)03011-6 [DOI] [PubMed] [Google Scholar]
- 21.Oláh O, Tóth-Szki V, Temesvári P, Bari F, Domoki F. Delayed neurovascular dysfunction is alleviated by hydrogen in asphyxiated newborn pigs. Neonatology. 2013;104: 79–86. 10.1159/000348445 [DOI] [PubMed] [Google Scholar]
- 22.Randall GCB. Studies on the effect of acute asphyxia on the fetal pig in utero. Biol Neonate. 1979;36: 63–69. 10.1159/000241208 [DOI] [PubMed] [Google Scholar]
- 23.Randall GCB. pH Values and Blood-Gas Tensions in the Normal Piglet during the First 48 Hours of Life. Biol Neonate. 1972;20: 68–73. 10.1159/000240447 [DOI] [PubMed] [Google Scholar]
- 24.Engle WD, Laptook AR, Perlman JM. Acute changes in arterial carbon dioxide tension and acid-base status and early neurologic characteristics in term infants following perinatal asphyxia. Resuscitation. 1999;42: 11–17. [DOI] [PubMed] [Google Scholar]
- 25.Randall GC. The relationship of arterial blood pH and pCO2 to the viability of the newborn piglet. Can J Comp Med. 1971;35: 141–146. [PMC free article] [PubMed] [Google Scholar]
- 26.Siesjö BK. Acid-Base Homeostasis in the Brain: Physiology, Chemistry, and Neurochemical Pathology. Prog Brain Res. 1985;63: 121–154. 10.1016/S0079-6123(08)61980-9 [DOI] [PubMed] [Google Scholar]
- 27.Corbett RJT, Laptook AR, Nunnally RL, Hassan A, Jackson J. Intracellular pH, Lactate, and Energy Metabolism in Neonatal Brain During Partial Ischemia Measured In Vivo by 31P and 1H Nuclear Magnetic Resonance Spectroscopy. J Neurochem. 1988;51: 1501–1509. 10.1111/j.1471-4159.1988.tb01118.x [DOI] [PubMed] [Google Scholar]
- 28.Laptook AR, Corbett RJT, Nunnally RL. Effect of Plasma Glucose Concentration on Cerebral Metabolism During Partial Ischemia in Neonatal Piglets. Stroke. 1990;21: 435–440. 10.1159/000448585 [DOI] [PubMed] [Google Scholar]
- 29.Corbett RJT, Laptook AR, Hassan A, Nunnally RL. Quantitation of acidosis in neonatal brain tissue using the 31P NMR resonance peak of phosphoethanolamine. Magn Reson Med. 1988;6: 99–106. 10.1002/mrm.1910060112 [DOI] [PubMed] [Google Scholar]
- 30.Laptook A, Stonestreet BS, Oh W. Autoregulation of brain blood flow in the newborn piglet: Regional differences in flow reduction during hypotension. Early Hum Dev. 1982;6: 99–107. 10.1016/0378-3782(82)90063-9 [DOI] [PubMed] [Google Scholar]
- 31.Li P-A, Shamloo M, Katsura K, Smith M-L, Siesjö BK. Critical values for plasma glucose in aggravating ischaemic brain damage: correlation to extracellular pH. Neurobiol Dis. 1995;2: 97–108. 10.1006/nbdi.1995.0010 [DOI] [PubMed] [Google Scholar]
- 32.Stonestreet BS, Burgess GH, Cserr HF. Blood-brain barrier integrity and brain water and electrolytes during hypoxia/hypercapnia and hypotension in newborn piglets. Brain Res. 1992;590: 263–270. 10.1016/0006-8993(92)91104-M [DOI] [PubMed] [Google Scholar]
- 33.Lorek A, Takei Y, Cady EB, Wyatt JS, Penrice J, Edwards AD, et al. Delayed (“Secondary”) Cerebral Energy Failure after Acute Hypoxia-Ischemia in the Newborn Piglet: Continuous 48-Hour Studies by Phosphorus Magnetic Resonance Spectroscopy. Pediatr Res. 1994;36: 699–706. 10.1203/00006450-199412000-00003 [DOI] [PubMed] [Google Scholar]
- 34.Hope PL, Cady EB, Tofts PS, Hamilton PA, Costello AMDL, Delpy DT, et al. Cerebral Energy Metabolism Studied With Phosphorus NMR Spectroscopy In Normal And Birth-Asphyxiated Infants. Lancet. 1984;324: 366–370. [DOI] [PubMed] [Google Scholar]
- 35.Mortola JP. How newborn mammals cope with hypoxia. Respiration Physiology. 1999. pp. 95–103. 10.1016/S0034-5687(99)00038-9 [DOI] [PubMed] [Google Scholar]
- 36.Lingappan K, Kaiser JR, Srinivasan C, Gunn AJ. Relationship between PCO2 and unfavorable outcome in infants with moderate-to-severe hypoxic ischemic encephalopathy. Pediatr Res. 2016;80: 204–208. 10.1038/pr.2016.62 [DOI] [PubMed] [Google Scholar]
- 37.Pappas A, Shankaran S, Laptook AR, Langer JC, Bara R, Ehrenkranz RA, et al. Hypocarbia and adverse outcome in neonatal hypoxic-ischemic encephalopathy. J Pediatr. 2011;158: 752–758. 10.1016/j.jpeds.2010.10.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ringer SK, Clausen NG, Spielmann N, Weiss M. Effects of moderate and severe hypocapnia on intracerebral perfusion and brain tissue oxygenation in piglets. Paediatr Anaesth. 2019;29: 1114–1121. 10.1111/pan.13736 [DOI] [PubMed] [Google Scholar]
- 39.Pospelov AS, Puskarjov M, Kaila K, Voipio J. Endogenous brain‐sparing responses in brain pH and P O2 in a rodent model of birth asphyxia. Acta Physiol (Oxf). 2020; e13467 10.1111/apha.13467 [DOI] [PubMed] [Google Scholar]