

Cationic nanoparticles ameliorate severe sepsis progression through scavenging cell-free DNA.
Abstract
Severe sepsis represents a common, expensive, and deadly health care issue with limited therapeutic options. Gaining insights into the inflammatory dysregulation that causes sepsis would help develop new therapeutic strategies against severe sepsis. In this study, we identified the crucial role of cell-free DNA (cfDNA) in the regulation of the Toll-like receptor 9–mediated proinflammatory pathway in severe sepsis progression. Hypothesizing that removing cfDNA would be beneficial for sepsis treatment, we used polyethylenimine (PEI) and synthesized PEI-functionalized, biodegradable mesoporous silica nanoparticles with different charge densities as cfDNA scavengers. These nucleic acid–binding nanoparticles (NABNs) showed superior performance compared with their nucleic acid–binding polymer counterparts on inhibition of cfDNA-induced inflammation and subsequent multiple organ injury caused by severe sepsis. Furthermore, NABNs exhibited enhanced accumulation and retention in the inflamed cecum, along with a more desirable in vivo safety profile. Together, our results revealed a key contribution of cfDNA in severe sepsis and shed a light on the development of NABN-based therapeutics for sepsis therapy, which currently remains intractable.
INTRODUCTION
Sepsis is a leading cause of death in the intensive care unit of hospital (1). Despite substantial efforts to advance its medical care, sepsis remains a great challenge worldwide and causes more than 8 million deaths annually (2). Most clinical trials have seen little success because of the complexity of this disease (3). Current start-of-the-art approaches for treating severe sepsis and septic shock are mainly supportive treatments, which need intensive patient care and only give limited efficacy (4). Pathologically, sepsis is a deleterious and nonresolving systemic inflammatory response syndrome (SIRS), induced by infection, ultimately evolving to acute organ failure through irreversible damage on endothelial, epithelial, and immune cells (5, 6). Mechanistically, inflammatory dysregulation is typically initiated and driven by the excessive activation of Toll-like receptors (TLRs), which recognize pathogen-associated molecular patterns (PAMPs) or damage-associated molecular patterns, such as bacterial toxins, proteins, and nucleic acids (7, 8). Among those circulating danger signals, cell-free DNAs (cfDNAs), including pathogen-derived CpG, damaged cell-released mitochondrial DNA, and nuclear DNA, as well as neutrophil extracellular traps, have been demonstrated to not only represent prognostic, predictive biomarkers of sepsis but also contribute to the magnitude and duration of inflammatory response through TLR activation in immune cells (9, 10). In this context, neutralization of cfDNA may modulate the overwhelming immune response and ameliorate organ injury caused by severe sepsis.
Because cfDNAs comprise different structures, sequences, or modifications, it is difficult to specifically neutralize these cfDNAs through complementary sequences or structural interactions (11). However, they may be bound by cationic polymers used widely for gene delivery (12). We have demonstrated that nucleic acid–binding polymers (NABPs) can scavenge proinflammatory nucleic acids to modulate inflammation at the injured site (13, 14). We have previously found that third generation polyamidoamine dendrimer (PAMAM-G3), a classic NABP, could prevent TLR activation in the target immune cells through exogenous scavenging of proinflammatory nucleic acids and nucleic acid–protein complexes and was beneficial for treating acute liver failure (13), lupus (15), cancer metastasis (16), and influenza infection (17), although PAMAM-G3 also showed notable toxic effects in vivo (18–20). Compared with soluble NABPs, nucleic acid–binding nanoparticles (NABNs) may produce different scavenging effects due to their distinctive transport properties. We have shown in a murine rheumatoid arthritis model that cationic nanoparticles can outperform the soluble NABP counterpart, owing to their preferential accumulation in the inflamed joints and in the endosomal compartment where TLR9 resides (21).
Here, we first examined the effect of PAMAM-G3 on both the cecal ligation and puncture (CLP)–induced severe sepsis model and the CpG-induced fatal SIRS model. We confirmed that cfDNA played a crucial role in the TLR-mediated proinflammatory activation of macrophages in the peritoneal space, where the dysregulation of immune response and pathogenesis of severe sepsis occur, and that cfDNA scavenging could help alleviate the symptom and achieve a therapeutic effect. We then synthesized two biodegradable mesoporous silica nanoparticles functionalized with polyethylenimine (MSN-PEI) with controllable charge density. We compared the scavenging capabilities and anti-inflammatory effects between PEI and MSN-PEI with different charge densities in vitro and in vivo. We also investigated the tissue biodistribution and intracellular trafficking of these scavengers to help interpret the results and to determine their safety profile for defining the translation potential. Collectively, the findings demonstrate that NABNs with higher charge density can better ameliorate septic injury in the CLP model and suggest the potential of using these NABNs as promising cfDNA scavengers for treating intractable, severe sepsis.
RESULTS
Therapeutic potential of PAMAM-G3 in severe sepsis
PAMAM-G3 not only exhibited high binding affinity with calf thymus DNA (ct-DNA) (fig. S1A) but also inhibited CpG-mediated TLR9 activation in engineered human embryonic kidney (HEK)–TLR9 reporter cells (fig. S1B) and reduced the proinflammatory cytokine tumor necrosis factor–α (TNF-α) secreted by RAW 264.7 macrophages (fig. S1C). The serum cfDNA level in septic patients was significantly higher than that in healthy volunteers (Fig. 1A). The septic serum would activate TLR9 (Fig. 1B), and addition of PAMAM-G3 blocked the TLR9 activation. Furthermore, treating macrophages with PAMAM-G3 attenuated the septic serum–induced production of TNF-α (Fig. 1C). These findings indicate that the cfDNA in septic serum induced TLR9 activation and proinflammatory cytokine secretion, while this pathological phenotype could be abrogated by PAMAM-G3.
Fig. 1. PAMAM-G3 reduces the cfDNA-driven proinflammatory response in vitro and in CLP-induced severe sepsis.
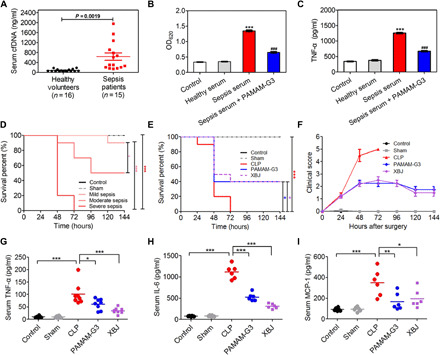
(A) Serum levels of cfDNA in healthy volunteers (n = 16) and patients with sepsis (n = 15). Data are expressed as the means ± SEM, and differences were assessed with Student’s t test. (B) Activation of HEK-TLR9 reporter cells by either healthy human serum or sepsis patient serum in the absence or presence of PAMAM-G3 (10 μg/ml) for 24 hours. The corresponding embryonic alkaline phosphatase (SEAP) activity in supernatants from each group was determined with a QUANTI-Blue assay with optical density at 620 nm (OD620). (C) RAW 264.7 macrophages were stimulated with sepsis patient serum in the absence or presence of PAMAM-G3 (10 μg/ml) for 24 hours. Supernatants were assayed for TNF-α via enzyme-linked immunosorbent assay (ELISA). In (B) and (C), differences were assessed via one-way analysis of variance (ANOVA) with Tukey’s multiple comparison tests (***P < 0.001, compared with healthy serum; ###P < 0.05, compared with sepsis serum). The data are expressed as the means ± SEM. (D) The indicated BALB/c mice were subjected to CLP of different grades. Survival was monitored for 144 hours (n = 10 mice per group; *P < 0.05 and ***P < 0.001, Kaplan-Meier survival analysis). (E) High-grade CLP was performed on BALB/c mice, followed by intraperitoneal injection of PAMAM-G3 or Xuebijing (XBJ) (20 mg/kg) 12 hours before and 1 and 12 hours after surgery. Survival was monitored for 144 hours (n = 10 mice per group; *P < 0.05 and ***P < 0.001, Kaplan-Meier survival analysis). (F) Mice were monitored for 144 hours after CLP for clinical scoring. The clinical scoring of sepsis was defined according to a range from 0 (no symptoms) to 5 (loss of self-righting reflex). The data are expressed as the means ± SEM. (G to I) High-grade CLP was performed on BALB/c mice, followed by treatment as described in (E). The levels of the proinflammatory cytokines (G) TNF-α, (H) interleukin-6 (IL-6), and (I) monocyte chemoattractant protein-1 (MCP-1) were measured in the blood 24 hours after CLP. Differences were assessed via one-way ANOVA with Tukey’s multiple comparison tests (n = 6 to 8 mice per group; *P < 0.05, **P < 0.01, and ***P < 0.001). The data are expressed as the means ± SEM.
The CLP model, which triggers polymicrobial peritonitis and ultimately leads to sepsis, is one of the gold standards in studying sepsis. It shares similar characteristics on a multitude of TLR activation relevant to the clinical sepsis (22, 23). We developed the CLP septic model with different grades by varying the distance of cecum ligation in wild-type BALB/c mice (Fig. 1D). The highest clinical score (fig. S2A) and the highest serum and peritoneal cfDNA levels (fig. S2, B and C) were designated as the severe grade. This severe model was chosen to demonstrate the protective potential of PAMAM-G3. All CLP-induced animals without any treatments died within 72 hours. In contrast, repetitive intraperitoneal administration of PAMAM-G3 (20 mg/kg) in a protective manner (at 12 hours before CLP and 1 and 12 hours after CLP) produced notable protection (40% surviving rate) against the lethal condition (Fig. 1E), which triggered death starting at 24 hours after CLP induction (Fig. 1F). The survival prolongation conferred by PAMAM-G3 corresponded with a significant reduction in the proinflammatory cytokines, including TNF-α (Fig. 1G), interleukin-6 (IL-6; Fig. 1H), and monocyte chemoattractant protein-1 (MCP-1; Fig. 1I), in serum. In addition, PAMAM-G3 ameliorated lung, kidney, liver, and heart tissue injury (tissue destruction, necrosis, and leukocyte infiltration; fig. S3). Consistently, biochemical analyses on liver [alanine aminotransferase (ALT), aspartate aminotransferase (AST), and bilirubin (TBIL)], kidney [blood urea nitrogen (BUN) and creatinine (CRE)], and heart [creatine kinase (CK)] functions confirmed the protective effects of PAMAM-G3 in preventing the failure of these organs (fig. S4). PAMAM-G3 exhibited an efficacy similar to that provided by Xuebijing (XBJ), the drug that the Chinese State Food and Drug Administration approved for sepsis treatment (24, 25). These data suggest that PAMAM-G3 is able to reduce septic death and ameliorate multiple organ injury in a clinically relevant severe sepsis model.
Scavenging of cfDNA protects mice against septic death through TLR9
After demonstrating the protective effects of PAMAM-G3 in the CLP-induced severe sepsis model, we checked its effect in a CpG-induced fatal SIRS model (26). Intraperitoneal injection of CpG in d-galactosamine–sensitized mice induces proinflammatory cytokine storm and septic lethal shock through the direct TLR9 activation, which conserves the characteristics of severe sepsis-induced SIRS in clinics. Mice were intraperitoneally treated with PAMAM-G3 or XBJ at 30 min after CpG challenge. All the mice died within 48 hours after CpG injection, while PAMAM-G3 remarkably reduced mortality by 90% (fig. S5A). In addition to the improvement on survival, we also found significant reduction in other inflammatory markers, such as fewer TLR9+ peritoneal cells (fig. S5B) and reduced serum TNF-α (fig. S5C) and IL-6 (fig. S5D) levels. These findings clearly verified that the protective effect of PAMAM-G3 and its downstream anti-inflammatory responses in CpG-induced SIRS model were from cfDNA scavenging.
Next, we sought to decipher the role of cfDNA in the progression of sepsis. As expected, PAMAM-G3 significantly lowered the elevation of both serum (Fig. 2A) and peritoneal (Fig. 2B) cfDNA level. The increased secretion of proinflammatory TNF-α, IL-6, and MCP-1 in peritoneal lavage fluid (PLF) of septic mice (fig. S6, A to C) also indicated the proinflammatory condition in the infective microenvironment. Correspondingly, the amount of TLR9+ peritoneal cells in the PAMAM-G3–treated septic mice was remarkably lower than that in the untreated CLP mice (Fig. 2C). Since macrophage is an important player in the immunopathogenesis of sepsis, we asked whether cfDNA-mediated TLR9 activation was pivotal for M1 macrophage polarization, thereby resulting in proinflammatory cytokine release during sepsis development. We harvested peritoneal macrophages (fig. S6D) and identified that a higher portion of them (6.3%) were CD11c+ (M1-polarized macrophages; Fig. 2D) in the septic mice, while the PAMAM-G3 treatment reduced the population of those cells (2.5%). In addition, the M1-polarized markers, TNF-α (Fig. 2E) and inducible nitric oxide synthase (iNOS) (Fig. 2F), which were up-regulated in CLP-challenged peritoneal macrophages, were significantly repressed by PAMAM-G3, and the expression of the M2-polarized marker, Arg-1, was partly restored (Fig. 2G). As the TLR9-MyD88 (myeloid differentiation primary-response protein 88)–nuclear factor κB (NF-κB) axis is the classic pathway involved in cfDNA-triggered TLR9 activation, we further investigated whether cfDNA scavenging affected the expression of the components in this pathway, including TLR9, MyD88, and NF-κB phosphorylated p65 (p-p65) (fig. S6E). The expression of TLR9 (Fig. 2H) and MyD88 (Fig. 2I) was markedly up-regulated in the peritoneal macrophages of model mice, and the level of p-p65 (Fig. 2J) increased substantially as well. In contrast, treatment with PAMAM-G3 significantly limited the activation of TLR9-MyD88–NF-κB signaling pathway. These data supported our hypothesis and suggested that PAMAM-G3 reversed the M1 polarization of peritoneal macrophages through TLR9-MyD88–NF-κB signaling pathway during severe sepsis progression.
Fig. 2. PAMAM-G3 reverses M1 polarization of peritoneal macrophages through the TLR9-MyD88–NF-κB signaling pathway during severe sepsis.
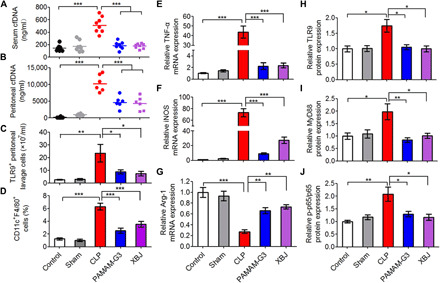
High-grade CLP was performed on BALB/c mice, followed by intraperitoneal injection of PAMAM-G3 or XBJ (20 mg/kg) 12 hours before and 1 and 12 hours after surgery. (A) Serum and (B) peritoneal cfDNA levels were analyzed after 24 hours after CLP. (C) The number of TLR9+ cells and (D) the percentage of M1-polarized macrophages (CD11c+F4/80+) were assessed in PLF by flow cytometry 8 hours after CLP. Differences were assessed via one-way ANOVA with Tukey’s multiple comparison tests (n = 6 to 8 mice per group; *P < 0.05, **P < 0.01, and ***P < 0.001). The data are expressed as the means ± SEM. (E to J) Peritoneal macrophages were collected 8 hours after CLP, and mRNA was extracted, converted to complementary DNA, and analyzed via real-time polymerase chain reaction (PCR) for (E) TNF-α, (F) iNOS, and (G) Arg-1 gene expression. The data are expressed as fold change relative to the saline-treated normal group and normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) gene expression. In parallel, macrophages were lysed in radioimmunoprecipitation assay (RIPA) buffer before analysis of (H) TLR9, (I) MyD88, and (J) p-p65 protein expression via Western blotting. The data are expressed as fold change relative to the control group and normalized to GAPDH or p65 protein expression. Differences were assessed via one-way ANOVA with Tukey’s multiple comparison tests (n = 5 mice per group; *P < 0.05, **P < 0.01, and ***P < 0.001). The data are expressed as the means ± SEM (n = 3 independent experiments in triplicate).
Charge density affects the efficacy of MSN-PEI on cfDNA-driven inflammation
After seeing the therapeutic potential of cfDNA scavenging for treating severe sepsis, we hypothesized that NABNs, rather than NABPs, might achieve more efficient and safer cfDNA scavenging in severe sepsis due to favorable accumulation in inflamed tissue (Fig. 3A). We and other groups have previously demonstrated that large-pore MSN is a versatile carrier for delivering biologics, such as nucleic acids and proteins, due to its large surface area and ease for functionalization (27–29). We therefore synthesized a large-pore, disulfide-bridged MSN (150 nm in diameter with 7-nm pores) as a model for developing NABNs (fig. S7, A to C). Its glutathione (GSH)–responsive degradation capability (Fig. 3, B to D) with low cytotoxicity (fig. S7D) also motivated us to select this biomaterial. To evaluate the surface charge density effect, we designed three NABNs with different charge densities through functionalizing the MSNs with or without PEIs [number-average molecular weight (Mn), 25 kDa or 800 Da]. After the conjugation, PEI mass content and surface area were characterized (fig. S7, E to H) and calculated (table S1) to determine the charge density of each MSN-PEI, with the charge density defined as the average molar nitrogen atoms per unit area of the nanoparticle surface. Although MSN-PEI 25K and MSN-PEI 800 have similar size, zeta potential (table S1), and degradable behavior (fig. S8), the former held a stronger charge density (represents as molar nitrogen atoms per surface area) than the latter one (10.85 versus 8.55 μM N/m2), as corroborated by a higher DNA binding capacity described below. In addition, the cationic MSNs (MSN-NH2) with the lowest charge density (0.64 μM N/m2) were used for further comparison.
Fig. 3. PEI-functionalized biodegradable MSNs block the proinflammatory response in vitro.
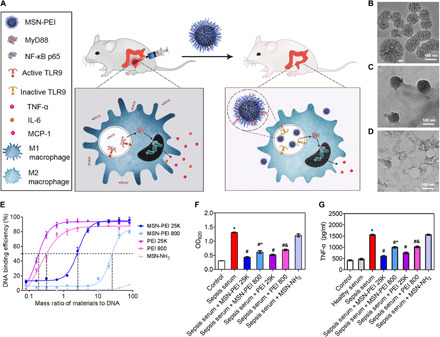
(A) The mechanism by which NABNs scavenge cfDNA to inhibit the proinflammatory response and treat severe sepsis. NABNs with a cationic corona are formed by conjugating PEI of different molecular weights onto the surface of biodegradable MSNs. After intraperitoneal injection into the CLP-induced severe sepsis models, NABNs exhibited favorable accumulation and retention in the inflamed cecum, protecting mice against death by attenuating proinflammatory effects and multiple organ injury through scavenging of cfDNA. (B to D) Transmission electron microscopy images of MSNs after incubation in buffer containing 5 × 10−3 M GSH for (B) 0 days, (C) 1 day, and (D) 3 days. Scale bar, 10 nm (B, inset). (E) ct-DNA binding efficiency of NABNs or NABPs with different mass ratios at 37°C. The data are expressed as the means ± SEM (n = 3 independent experiments in triplicate). (F) Activation of HEK-TLR9 reporter cells by either healthy human sera or sepsis patient sera in the absence or presence of NABNs or NABPs (10 μg/ml) for 24 hours. The corresponding SEAP activity in supernatants from each group was determined with a QUANTI-Blue assay at OD620. (G) RAW 264.7 macrophages were stimulated with sepsis patient sera in the absence or presence of NABNs or NABPs (10 μg/ml) for 24 hours. Supernatants were assayed for TNF-α via ELISA. In (F) and (G), differences were assessed via one-way ANOVA with Tukey’s multiple comparison tests (*P < 0.05, compared with healthy serum; #P < 0.05, compared with sepsis serum; ^P < 0.05, compared with sepsis serum + MSN-PEI 25K; &P < 0.05, compared with sepsis serum + PEI 25K). The data are expressed as the means ± SEM (n = 3 independent experiments in triplicate).
Both types of MSN-PEI, but not MSN-NH2, complexed with cfDNA in a dose-depending manner (fig. S9A) and formed stable scavenger-DNA complexes (fig. S9B). Notably, MSN-PEI 25K had median effective dose in binding compared with MSN-PEI 800 but lower than its PEI counterpart (Fig. 3E). Using the HEK-TLR9 reporter cells, we found that all NABNs and NABPs significantly inhibited both CpG-induced (fig. S9, C and D) and sepsis serum–induced (Fig. 3F) TLR9 activation under the same dosage. MSN-PEI 25K exhibited comparable TLR9 inhibition with PEI 25K on a mass-based comparison, although the MSN-PEI 25K has a lower N content. Among all the nanoparticles, the NABN with the higher charge density was more efficient in blocking the TLR9 activation. We saw similar trends in TNF-α secretion from those danger signals–activated macrophages (Fig. 3G and fig. S9E). As a result, we could confirm that charge density played an important role in NABN-mediated cfDNA scavenging and inflammation inhibition.
MSN-PEI attenuates proinflammatory effects and multiple organ injury in severe sepsis
Repetitive intraperitoneal administration of both NABNs conferred protection against CLP-induced death, which was consistent with transient clinical scores from the 24- to 48-hour time points (Fig. 4, A and B). The MSN-PEI 25K group outperformed the MSN-PEI 800 group in agreement with the in vitro results. Neither PEI 25K nor PEI 800, the soluble cationic counterparts, could improve the survival of CLP-induced mice compared with the control group. Consistent with the decreased mortality, the serum (Fig. 4C) and peritoneal (Fig. 4D) cfDNA levels and number of TLR9+ peritoneal cells (Fig. 4E) in the MSN-PEI 25K–treated mice were significantly lower than those in other groups (fig. S10, A to C). Similarly, MSN-PEI 25K administration significantly reduced CLP-induced TNF-α (Fig. 4F), IL-6 (Fig. 4G), and MCP-1 (Fig. 4H) release in both the serum and peritoneal fluid (fig. S10, D to I). The serum cfDNA and proinflammatory levels of the mice treated with its counterpart (PEI 25K) were somewhat higher, suggesting toxicity, although the peritoneal cfDNA level was decreased. In addition, MSN-PEI 25K resulted in less septic death, accompanied with reduced cfDNA levels, and lower number of TLR9+ cells and M1-polarized macrophages in PLF through inhibition of the TLR9-MyD88–NF-κB pathway, when compared with PAMAM-G3 (fig. S11).
Fig. 4. MSN-PEI 25K protects mice against CLP-induced severe sepsis.
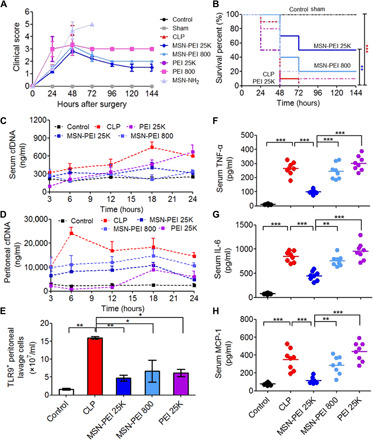
High-grade CLP was performed on BALB/c mice, followed by intraperitoneal injection of MSN-PEI 25K, MSN-PEI 800, PEI 25K, PEI 800, or MSN-NH2 (20 mg/kg) 12 hours before and 1 and 12 hours after surgery. (A) Mice were monitored for 144 hours after CLP for clinical scoring. The data are expressed as the means ± SEM. (B) Survival was monitored for 144 hours (n = 10 mice per group; **P < 0.01, ***P < 0.001, Kaplan-Meier survival analysis). (C) Serum and (D) peritoneal cfDNA levels were measured at 3, 6, 12, 18, and 24 hours after CLP. (E) The number of TLR9+ cells was assessed in PLF by flow cytometry 8 hours after CLP. (F to H) 24 hours after CLP, levels of the proinflammatory cytokines (F) TNF-α, (G) IL-6, and (H) MCP-1 in serum were measured. (E to H) Differences were assessed via one-way ANOVA with Tukey’s multiple comparison tests (n = 6 to 8 mice per group; *P < 0.05, **P < 0.01, and ***P < 0.001). The data are expressed as the means ± SEM.
Since multiple organ failure is the main consequence of severe sepsis (30), we carried out histopathological and biochemical analyses for the mice treated with NABPs at 24 hours after CLP induction. In the septic mice, hematoxylin and eosin (H&E)–stained sections revealed severe inflammatory cell infiltration in the lung, kidney, heart, liver, and spleen, showing typical multiple organ injuries, e.g., interalveolar septum thickened in the lung, tubular epithelial cell swelling in the kidney, and inflammatory cell infiltration in the liver and heart (Fig. 5A and fig. S12A). In agreement with the survival data, administration of MSN-PEI 25K significantly attenuated the multiple organ injury of the liver (Fig. 5B), kidney (Fig. 5E), and other organs, thereby improving the injury score (fig. S12, B to E). Consistent with these changes in organ histology, MSN-PEI 25K markedly reversed the increased serum ALT (Fig. 5C), AST (Fig. 5D), BUN (Fig. 5F), and CRE (Fig. 5G), as well as CK and TBIL levels (fig. S13). Similarly, the PEI 25K group held comparable score for each organ and significantly increased the level of serum biochemical indicators. These in vivo data agreed with the in vitro results that NABNs were better than NABPs and the NABN with the higher charge density was more effective.
Fig. 5. MSN-PEI 25K attenuates multiple organ injury in CLP-induced severe sepsis.

High-grade CLP was performed on BALB/c mice, followed by treatment as described in Fig. 4. (A) Twenty-four hours after CLP, the lung, kidney, heart, liver, and spleen tissues were collected, stained with H&E, and analyzed. Scale bars, 20 μm. Multiple organ injury representations, including interalveolar septum thickened in the lung, tubular epithelial cell swelling in the kidney, inflammatory cell infiltration in the liver and heart, and nuclear debris from dying cells in the spleen, are marked with arrows. The corresponding (B) liver and (E) kidney injury scores were determined and calculated according to the established criteria. In parallel, the blood serum biochemistry parameters (C) ALT, (D) AST, (F) BUN, and (G) CRE were measured. Differences were assessed via one-way ANOVA with Tukey’s multiple comparison tests (n = 6 mice per group; *P < 0.05, **P < 0.01, and ***P < 0.001). The data are expressed as the means ± SEM.
MSN-PEI exhibits higher accumulation in inflamed tissue with negligible toxicity
To better understand the underlying mechanisms responsible for the different therapeutic effects between NABNs and NABPs, we first examined the cellular internalization of the scavengers. With CpG labeled with Cy5.5, the PEI or MSN-PEI was labeled with fluorescein isothiocyanate (FITC). Cy5.5-labeled CpG alone mostly appeared in the cytosol of macrophages, whereas addition of PEI or MSN-PEI formed a punctate pattern in the lysosomes/endosomes (Fig. 6A and fig. S14A). Both NABNs of PEI 800 and PEI 25K exhibited higher cellular retention rate than their NABP counterparts (fig. S14B). The MSN-PEI 25K more efficiently interacted with the CpG, as reflected in a lowered cellular retention of CpG compared with MSN-PEI 800 (fig. S14C). We next studied the biodistribution of the Cy7-labeled NABNs and NABPs in abdominal organs through ex vivo near-infrared fluorescence (NIRF) imaging. In the sham mice, the NABNs and NABPs were mainly observed in the liver, with little difference of retention in the cecum (Fig. 6B and fig. S15). However, in septic mice, both NABNs and NABPs preferentially localized in in the cecum, with NABNs showing longer retention in the colonic tissue than NABPs (Fig. 6C and fig. S15). MSN-PEI 25K exhibited higher retention in the colonic tissue than MSN-PEI 800 (Fig. 6D). Correspondingly, its counterpart PEI 25K had much higher accumulation in the liver and kidney. This differential accumulation was confirmed by quantitative measurement of Si in various tissues, reinforcing the NIRF imaging results that higher charge density correlated with higher accumulation in the inflamed site (Fig. 6E).
Fig. 6. MSN-PEI 25K shows inflammatory-specific accumulation and retention with negligible toxicity in vivo.
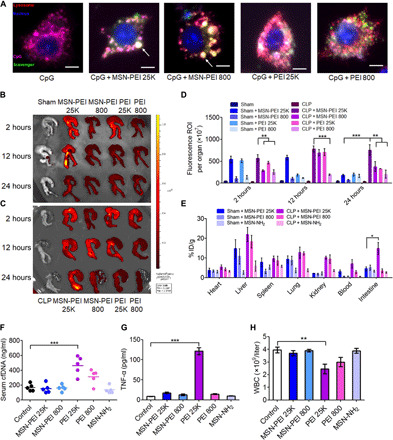
(A) Merged images show intracellular localization of cationic materials and CpG in RAW 264.7 cells after 6 hours of incubation. Scale bars, 5 μm. The large white spots in NABNs-treated cells are marked with an arrow. (B to D) Ex vivo NIRF images of cecum from (B) sham and (C) CLP mice 2, 12, and 24 hours after intraperitoneal injection of Cy7-labeled NABNs or NABPs (20 mg/kg). (D) Semiquantitative analysis of ex vivo fluorescence images of the cecum in (B) and (C). Differences were assessed via one-way ANOVA with Tukey’s multiple comparison tests (n = 3 mice per group; **P < 0.01 and ***P < 0.001). The data are expressed as the means ± SEM. ROI, region of interest. (E) Quantification analysis of Si content in the lung, kidney, heart, liver, spleen, and cecum of sham or CLP mice via inductively coupled plasma optical emission spectrometry (ICP-OES) after intraperitoneal injection of MSN-PEI 25K, MSN-PEI 800, or MSN-NH2 (20 mg/kg) 12 hours before surgery and 1 and 12 hours after surgery. The data are expressed as the means ± SEM (n = 5 mice per group). % ID/g, percentage of the injected dose per gram of tissue. (F to H) Normal mice were intraperitoneally injected three times with MSN-PEI 25K, MSN-PEI 800, PEI 25K, PEI 800, or MSN-NH2 (20 mg/kg) at 0, 13, and 24 hours. (F) cfDNA and (G) TNF-α levels and (H) the white blood cell (WBC) count were measured in the blood 24 hours after the last administration. Differences were assessed via one-way ANOVA with Tukey’s multiple comparison tests (n = 5 to 6 mice per group; *P < 0.05,**P < 0.01, and ***P < 0.001). The data are expressed as the means ± SEM.
As toxicity is a major concern in clinical application of cationic materials (31), we found that MSN-PEI 25K showed reduced toxicity compared with the soluble PEI 25K in macrophages [with a median inhibitory concentration (IC50) of 61.45 μg/ml versus 22.98 μg/ml for PEI], and as expected, the one with higher charge density was more toxic in both RAW 264.7 and human umbilical cord endothelial cells (fig. S16). Subsequently, we systematically determined the safety profile of NABNs and NABPs in the healthy mice. In line with the repetitive administration procedure, we evaluated the acute toxicity of these scavengers with thrice-repeated dosing within 24 hours (table S2). All of the PEI-treated mice (both 25K and 800) died at the dose of 40 mg/kg. In contrast, all of the mice in both MSN-PEI 25K and MSN-PEI 800 groups behaved normally under the same dosage, showing no mortality at 320 mg/kg but suffering a 50 and 20% mortality, respectively, at the dose of 640 mg/kg. We further probed the possible mechanism of NABP-induced toxicity and observed that PEI 25K increased the levels of biochemical parameters in comparison with those of the control group (fig. S17). Correspondingly, markedly higher levels of cfDNAs and proinflammatory cytokines, abnormal number of blood cells, and damaged tissue of the liver, kidney, and lung in the PEI 25K group were also observed (Fig. 6, F to H, and figs. S18 to S20), implying that the toxicity originated from the systematic proinflammatory response, resulting in thrombosis. Both MSN-PEI 25K and MSN-PEI 800 did not significantly change the level of any serum biochemical indicators, blood cell number, or proinflammatory cytokine level, consistent with negligible pathological changes in the major organs described earlier.
DISCUSSION
Sepsis, severe sepsis, and septic shock represent increasingly severe systemic inflammation (4). Among them, severe sepsis is the most lethal, associated with hypotension, hypoperfusion or organ dysfunction, and high mortality (4, 32). As severe sepsis involves multiple inflammatory cytokines and other pathological conditions, therapeutic strategies simply targeting cytokines, coagulation factors, or virulence factors only offer limited benefit (5). The complexity of the inflammatory response in sepsis represents a significant challenge for the development of effective therapies. Recent evidences have shown that poorly regulated activation of TLRs triggers imbalance of innate immune system, thereby leading to elaborate proinflammatory cytokine storm during SIRS (33, 34). As a result, targeting the interactions between danger signals and TLR activation may be a fruitful direction for sepsis therapy. Clinically, septic patients often have elevated circulating cfDNA released from pathogens or/and infected host cells (10). Similar in our hands, we found the levels of circulating cfDNA increased in both septic mice and patients. We found that there is a different time-dependent manner of serum and peritoneal cfDNA levels in CLP mice. We attributed this phenomenon to the fact that (i) since the early infection and inflammation happened in cecum, the peak of peritoneal cfDNA at 6 hours after CLP was mainly from the bacteria-released CpG; (ii) the released CpG might enter the blood, combined with the later organ damage causing a peak of serum cfDNA at 18 hours after CLP. In both cases, the decrease in cfDNA level in 24 hours after CLP might be explained by the metabolic clearance of this cfDNA by deoxyribonuclease (DNase) or other phagocytes. Although the cfDNA level decreased at 24 hours after CLP, their level was still much higher than the normal group. It is worth noting that the higher cfDNA level in our severe septic model was strongly associated with mortality. Although underlying mechanisms have yet to be fully elucidated, scavenging cfDNA or blocking abnormal cfDNA-sensing pathways appears fruitful in ameliorating systemic inflammation in severe sepsis.
To validate the scavenging concept, we first used PAMAM-G3, a widely studied NABP that has previously been demonstrated to neutralize proinflammatory TLR9 agonists, such as CpG (13), to establish that a cfDNA scavenger could play a beneficial role in sepsis by modulating the cytokine storm and prevent multiple organ failure. Our observation was also aligned with the study reporting that degradation of cfDNA by DNase treatment induced a protective effect in the same septic model (35). Furthermore, we observed that MCP-1, an important chemoattractant for macrophage migration toward dead cells (36), was up-regulated markedly in the peritoneal fluid of septic mice, indicating an important role of macrophage during sepsis. Together with the increase of TLR9+ peritoneal cells, both in vitro and in vivo results revealed that cfDNA promoted the proinflammatory phenotype of macrophages, at least partially, via the TLR9-MyD88–NF-κB signaling pathway. This speculation comes from the finding that PAMAM-G3 could elicit anti-inflammatory effects not only at the site of infection but also at other tissues, including the lung, liver, kidney, and heart, through the blocking of M1 macrophage polarization. Similar to the protective effect in CLP model, PAMAM-G3 significantly improved the survival, reduced TLR9+ cell number, and attenuated proinflammatory cytokines in the CpG-induced fatal model. PAMAM-G3 exhibited comparable efficiency with a clinically approved antisepsis drug XBJ at the standard dose in the lethal CLP model (25). In light of this, we believe that cfDNA instigated the macrophage-mediated proinflammatory response through the TLR9-MyD88–NF-κB signaling pathway, and cfDNA scavengers can intervene at this signaling pathway for severe sepsis.
Despite its wide use in drug delivery and positive outcome in treating severe sepsis, the significant toxicity of PAMAM-G3 would hinder its translation (18); the batch-to-batch variability of the commercially available PAMAM-G3 observed in our hands is also challenging. PEI may be equally problematic in toxicity, although its synthesis is straightforward and reproducible. Nevertheless, we chose PEI in this proof-of-concept study because these stable PEI-functionalized MSNs have been widely used for gene delivery without showing significant toxicity in vivo (29, 37). In gene delivery, the surface charge plays a role on the transfection efficacy (37), which is also a factor determining cfDNA scavenging and the downstream therapeutic efficacy. To study this effect, we chose two PEIs with different molecular weights (25 kDa and 800 Da) for MSN functionalization. The MSN was chosen for its high surface area, and the disulfide-bridged MSN would facilitate eventual translation (38, 39). Although MSN-PEI 25K was comparable with its counterpart, PEI 25K, in blocking CpG or septic serum–triggered TLR9 activation and proinflammatory effect in vitro, MSN-PEI 25K exhibited better performance in the septic mice model with improved survival, decreased cfDNA level, and lower proinflammatory cytokine level, along with diminished organ failure. Unexpectedly, regardless of the reduction of cfDNA affected by the soluble PEI 25K in the intraperitoneal space, the serum cfDNA level was elevated, which exacerbated proinflammation and organ damage. This phenomenon might be attributed to the fact that PEI 25K could distribute into the blood and cause unwanted toxicity to release cfDNA, resulting in periodically increased serum cfDNA level. This finding suggests that serum cfDNA is a better biomarker than peritoneal cfDNA level in predicting therapeutic outcomes. Clinical observations have supported the correlation between cfDNA and organ dysfunction (40); our findings suggest that not only circulating cfDNA but also peritoneal cfDNA are relevant with multiple organ failure in the CLP-induced mice. The higher efficacy of nanoparticulate NABNs compared with the soluble NABPs is consistent with our previous findings that cationic nanoparticles outperformed the soluble polycations in scavenging cfDNA for rheumatoid arthritis treatment (21).
The structure-property relationship of cationic polymer in gene transfection and toxicity has been extensively investigated (41–43). Increase in molecular weight and charge density of PEI not only yielded higher nucleic acid complexation capacity and transfection efficiency but also increased toxicity (44). Here, we have demonstrated that the NABNs with a higher charge density exhibited stronger nucleic acid–binding affinity, thereby being more effective on attenuating cfDNA-mediated proinflammation in vitro and rescuing the septic animals from lethal inflammation in vivo. On the other hand, although PEI 25K worked well in vitro, it failed to prevent septic death in vivo. This may be attributed to its increased toxicity, resulting in exacerbating lethal inflammation rather than alleviating proinflammatory response.
To understand why NABNs would outperform NABPs, our intracellular trafficking and tissue distribution studies suggest that the nanoparticulate scavengers were more favorably accumulated in the site of action, when compared with its soluble counterparts. The former would have the nanoparticles sequestered into the endolysosomal compartment, where TLR9 resides, more efficiently, as evidenced by the higher intensity and persistence of the nanoparticulate scavenger signal. The charge density effect came into play in more efficiently binding the CpG, probably in the extracellular space. We believe that some scavenger-DNA complexes have localized on cell membrane rather than cytoplasm or endosome; the cell membrane–attached complexes were able to enter the cells with increasing incubation time, resulting in the increased fluorescence intensity of both scavengers and CpG. In tissue biodistribution, the cationic nanoparticles could reach the inflamed sites due to the leaky microvasculature, and once reached, there would be retained for a longer period of time. Hence, the nanoparticle would enjoy an enhanced permeation and retention effect similar to that observed in a murine tumor microenvironment (45, 46). All these extra- and intracellular transport factors contribute to the success of the nanoparticulate scavengers. In addition, they also have better safety profiles. We found that MSN-PEI 25K and PEI 25K were more cytotoxic than MSN-PEI 800 and PEI 800, respectively. Our finding supports the previous reports regarding the correlation between nanoparticle’s cytotoxicity and surface charge density (37, 47); the IC50 of MSN-PEI 25K was higher than that of PEI 25K. The reduced toxicity of NABNs benefits the fact that PEI was covalently attached to the surface of MSNs rather than encapsulated, thus avoiding undesired release of PEI. The MSN-PEI 25K exhibited a good safety profile for intraperitoneal administration, even at a high dose of 640 mg/kg. However, PEI 25K–exposed mice showed elevated serum tissue enzymes, proinflammatory immune cells, and cytokines, as well as pathological phenotypes in the liver, kidney, heart, and lung.
There are two other nanotherapeutic approaches targeting sepsis in the literature (48, 49). Soh et al. (48) doped ceria nanoparticles with zirconia to form 2-nm Ce0.7Zr0.3O2 ceria-zirconia nanoparticles (7CZ) with strong reactive oxygen species (ROS) scavenging capacity because the Zr4+ ions improve the conversion of Ce4+ to Ce3+ oxidation state, thereby scavenging the O2− and OH species more effectively. Administration of 7CZ through intraperitoneal route at a dose of 2 mg/kg improved the survival rate to ~50% (2.5-fold compared with control) in a CLP model that we would characterize as between moderate and severe. Although the upstream scavenging mechanisms of our approach appears to differ from the aforementioned study, PAMP-specific versus ROS-dominant, these two approaches may share common downstream molecular pathways in the intraperitoneal macrophages, as suggested by the comparable performance in survival prolongation. Another nanotherapeutic approach (49) relies on targeting the sialic acid–binding immunoglobulin-like lectin-E (Siglec-E) receptor, which plays an important role in down-regulating inflammatory mediators via inhibition of neutrophil infiltration and/or TLR pathways. When 150-nm PLGA [poly(lactic-co-glycolic acid] nanoparticles decorated with a Siglec ligand were intraperitoneally administered at a dose of 2 mg per mouse (~80 to 100 mg/kg) on a daily basis (total of nine injections) to a CLP model that we characterized as less severe, a 100% survival could be achieved. Our study cannot match this impressive performance, although we used a lower dose of 20 mg/kg of three injections each and in a more severe sepsis model. The underlying mechanisms of the therapeutic outcome also appear different, although both studies saw a reduction in TNF-α and IL-6 levels in vitro and in vivo. It would be tempting to investigate whether a combination of these nanoparticulate targeting strategies could achieve a synergistic effect to tackle the complicated and challenging septic conditions.
In summary, we have revealed a role of cfDNA in stimulating the TLR9-mediated proinflammatory response and provided a new target for severe sepsis management. Activation of the TLR9-MyD88–NF-κB pathway in peritoneal macrophage by cfDNA is crucial and may contribute to the pathogenesis and progression of severe sepsis. We showed that PAMAM-G3, a classic NABP, could reduce septic death and ameliorate multiple organ injury in the severe sepsis model. By balancing efficacy and toxicity of cfDNA scavengers for severe sepsis therapy, we have developed NABNs consisting of PEI-functionalized biodegradable MSNs with favorable accumulation and retention behavior in inflammatory tissue, resulting in improved survival and reduced multiple organ injury compared with the soluble NABP counterparts. The NABNs with higher charge density gave stronger inhibition on cfDNA-driven proinflammation in vitro and showed better therapeutic efficacy in vivo. Together, our findings suggest a new nanoparticulate scavenging approach to treating severe sepsis and shed light on the development of safe and effective NABNs for lethal inflammatory disorders.
MATERIALS AND METHODS
Synthesis of MSN-PEI
PEI-conjugated MSNs were fabricated according to our previously reported protocol (29). Briefly, 4.0 g of cetyltrimethylammonium tosylate, 1.6 g of triethanolamine, and 200 ml of deionized water were stirred at 80°C for 1 hour. Then, a solution containing 3.2 g of tetraethyl orthosilicate and 2.4 g of bis[3-(triethoxysilyl)propyl]tetrasulfide was added dropwise to the surfactant solution. The resulting mixture was stirred at 80°C for another 4 hours at 1000 rpm. The reaction mixture was centrifuged to collect the products, and then the products were washed three times with ethanol and refluxed in an ethanol solution of ammonium nitrate (1%, w/v) for 12 hours. The disulfide-bridged MSNs were collected, washed, and dried under vacuum. Next, the MSNs (1.0 g) were suspended in 250 ml of toluene under 1 hour of sonication, and (3-glycidyloxypropyl)trimethoxysilane (1.5 ml) was added; the mixture was then refluxed at 80°C for 24 hours. The epoxysilane-functionalized MSNs were separated, collected, washed, and dried under vacuum. Last, epoxysilane-functionalized MSNs (500 mg) were suspended in 250 ml of PEI 25K or PEI 800 solution (1 mg/ml) under 1 hour of sonication. The reaction was performed at room temperature for 24 hours. PEI-functionalized MSNs were collected, washed, and redispersed in water for further use.
Patient samples
Serum samples from 15 patients with sepsis and 16 healthy volunteers were obtained from the Second Affiliated Hospital of Jilin University. Collection of samples was performed with the approval of the Ethics Committee at the Second Affiliated Hospital of Jilin University. Sepsis was confirmed by the Third International Consensus Definitions for sepsis and septic shock (Sepsis-3) (50).
DNA binding assay
The efficiency of NABNs or NABPs binding with ct-DNA was determined on the basis of our previously reported protocol with minor modification. Briefly, 25 μl of ct-DNA (10 μg/ml) and 25 μl of PicoGreen were mixed with 50 μl of Milli-Q water in wells of a 96-well plate. The mixture was shaken for 30 min in the dark to form DNA-dye complex. Then, 100 μl of NABNs or NABPs at different concentrations was added. After incubation at 37°C for 1 hour, the fluorescence intensity at 520 nm was measured with a multiwell plate reader (BioTek, Winooski, VT) via excitation at 490 nm.
In vitro TLR9 activation assay
HEK-Blue TLR9 reporter cell lines were obtained from InvivoGen (San Diego, CA) and cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin cocktail. Then, CpG 1826 (1 μg/ml) or 5 μl of human sera was incubated with HEK-Blue TLR9 reporter cells at a density of 5 × 104 per well in a 96-well plate for 30 min, and NABPs or NABNs (10 μg/ml) were added in a final volume of 200 μl. After incubation for 24 hours, the supernatants were collected and incubated with QUANTI-Blue (InvivoGen, San Diego, CA). The corresponding embryonic alkaline phosphatase (SEAP) activity in each well was determined by measuring the optical density at 620 nm (OD620) using a multiwall plate reader.
In vitro anti-inflammatory assays
RAW 264.7 macrophages were purchased from American Type Culture Collection (Manassas, VA) and maintained in DMEM supplemented with 10% FBS, 1 mM sodium pyruvate, and 1% penicillin-streptomycin cocktail. Then, CpG 1826 (1 μg/ml) or 5 μl of human sera were incubated with 2 × 104 per well in a 96-well plate for 30 min, and NABPs or NABNs (10 μg/ml) were added in a final volume of 200 μl. After incubation for 24 hours, the supernatants were collected, and the TNF-α level was measured by enzyme-linked immunosorbent assay (ELISA) kit (Proteintech, Rosemont, USA).
Animals
All animal experiments were performed in accordance with the procedures and protocols approved by the Ethics Committee for the Use of Experimental Animals of Jilin University (Changchun, China). Six- to eight-week-old male BALB/c mice were purchased from Changchun Yisi Laboratory Animal Technology. All of the mice were maintained in the animal facility of the College of Basic Medical Sciences, Jilin University. Before experiments, all of the mice were acclimatized for at least 1 week.
CLP-induced sepsis model and treatment
Sepsis was developed in mice using a CLP procedure as previously described (23). Briefly, mice were anesthetized with ketamine [80 to 100 mg/kg, intraperitoneally (i.p.)] and xylazine (10 to 12.5 mg/kg, i.p.) after a 12-hour fasting. The abdominal cavity was opened with a midline incision in layers, and the cecum was gently exteriorized and then ligated with 4-0 silk without causing intestinal obstruction. Then, the cecal stump was punctured with a 21-gauge needle twice. The laparotomy site was closed with the 4-0 silk, and each of the mice was subcutaneously injected with resuscitative prewarmed sterile saline. In the sham group, only the abdominal laparotomy procedure was performed. After CLP, the mice were monitored for 144 hours to record the survival rate and clinical scores according to a previously established method (49). Mice were scored every 12 hours using the following criteria: score 0, no symptoms; score 1, piloerection and huddling; score 2, piloerection, diarrhea, and huddling; score 3, lack of interest in surroundings and severe diarrhea; score 4, decreased movement and listless appearance; and score 5, loss of self-righting reflex. Mice were humanely euthanized when they exhibited a score of 5. The mice were divided into three groups and subjected to different grades of CLP: (i) mild sepsis, 10% of the cecum was ligated and punctured with a 21-gauge needle twice; (ii) moderate sepsis, 50% of the cecum was ligated and punctured with a 21-gauge needle twice; and (iii) severe sepsis, 100% of the cecum was ligated and punctured with a 21-gauge needle twice.
In the severe sepsis model, NABNs or NABPs (20 mg/kg, i.p.), XBJ (4 ml/kg) was administered 12 hours before CLP and 1 and 12 hours after CLP. Mice (n = 8 to 10) were monitored for survival rate and clinical score as described above. Twenty-four hours after CLP, serum was collected for analysis of cfDNA, TNF-α, IL-6, and MCP-1 levels and tissue enzyme levels. Peritoneal fluids were collected simultaneously for detection of cfDNA, TNF-α, IL-6, and MCP-1 levels and to determine the number of TLR9+ cells and macrophages. The lung, kidney, liver, spleen, and heart were collected and fixed with 4% paraformaldehyde for histopathological examination.
Extraction and quantification of cfDNA
Extraction of cfDNA from serum or peritoneal fluid was performed with a DNeasy Blood & Tissue Kit (QIAGEN, Germany). The concentration of cfDNA was measured using the Quant-iT PicoGreen double-stranded DNA Assay Kit (Thermo Fisher Scientific, Waltham, MA) according to the manufacturer’s instructions.
Flow cytometric analysis
To quantitatively determine the number of TLR9+ immune cells in peritoneal fluid, the peritoneal fluid was collected. The collected cells were washed twice and resuspended in staining buffer and then calculated. Then, the cells were fixed and permeabilized by adding Fixation/Permeabilization solution (BD Biosciences, USA). Then, the cells were incubated with CD289 (TLR9) monoclonal antibody at 4°C for 30 min. After washing, 1 × 104 cells in each mouse were analyzed by fluorescence-activated cell sorting (FACS) (BD Accuri C6).
Peritoneal macrophage isolation
Peritoneal macrophages were isolated as described previously (51). Briefly, peritoneal lavages were performed with 1 ml of phosphate-buffered saline. Cells were suspended in RPMI 1640 containing 10% fetal bovine serum (FBS) and incubated in an atmosphere of 5% CO2 at 37°C for 90 min. The nonadherent cells were removed, and the adherent cells were further cultured.
Quantitative real-time polymerase chain reaction assay
Peritoneal macrophages were isolated as described previously. TRIzol reagent was used to extract the total RNA, and 1 μg of the total RNA was reverse-transcribed using TransScript II Reverse Transcriptase (TransGen Biotech, Beijing, China). Quantitative polymerase chain reaction (PCR) was then performed using FastStart Universal SYBR Green Master mix (Roche Diagnostics Ltd., Lewes, UK). The amplified transcripts were quantified using the comparative Ct method.
Western blotting assay
NF-κB p65 (phospho S536), NF-κB p65, MyD88, and TLR9 were detected via Western blot analysis. After isolation of peritoneal macrophages, the cells were lysed with the protease inhibitors supplemented radioimmunoprecipitation assay (RIPA) lysis buffer and centrifuged. Then, the cell lysate was collected and mixed with 5× loading buffer. The samples were heated at the temperature of 100°C for 5 min; an equal volume of the supernatant was loaded onto 10% polyacrylamide gels. Then, the separated proteins were transferred and probed with primary antibodies against NF-κB p65 (phospho S536), NF-κB p65, MyD88, TLR9, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH), followed by incubating with the secondary antibody. The bands were analyzed with an enhanced chemiluminesence Western blotting detection system (Tanon 4200), and quantitative analysis was performed with Quantity One software.
Cellular colocalization and retention of cationic materials and CpG
RAW 264.7 cells were seeded into 24-well culture plates and reach full attachment. Then, the cells were treated with Cy5.5-labeled CpG 1826 (1 μg/ml). After 30 min of incubation, cells were exposed to FITC-labeled NABNs or NABPs (10 μg/ml) for another 6 hours and then stained with 4′,6-diamidino-2-phenylindole and LysoTracker Red DNA-99 to observe the fluorescence via fluorescence microscopy. In parallel, after Cy5.5-CpG and FITC-NABNs or FITC-NABPs treatment for 3 hours, the fresh medium was added into each well, and cells were incubated for another 3, 6, 12, 18, and 24 hours. The relative fluorescence intensity of CpG and scavengers in RAW 264.7 cells at each time point was determined by FACS.
In vivo fluorescence imaging
The mice were divided into 10 groups: sham group, MSN-PEI 25K–treated sham group, MSN-PEI 800–treated sham group, PEI 25K–treated sham group, PEI 800–treated sham group, CLP group, MSN-PEI 25K–treated CLP group, MSN-PEI 800–treated CLP group, PEI 25K–treated CLP group, and PEI 800–treated CLP group. At 1 hour after surgery, the cationic materials were intraperitoneally injected into the mice at a dose of 20 mg/kg. At 2, 12, and 24 hours after surgery, mice were euthanized, and the images of the heart, lung, liver, spleen, kidney, and intestines were collected using an NIR imaging system (IVIS Spectrum, Caliper Life Sciences).
Biodistribution of NABNs
The mice were randomly divided into eight groups, including the sham group, MSN-NH2–treated sham group, MSN-PEI 25K–treated sham group, MSN-PEI 800–treated sham group, CLP group, MSN-NH2–treated CLP group, MSN-PEI 25K–treated CLP group, and MSN-PEI 800–treated CLP group. At 12 hours before CLP and 1 and 12 hours after CLP procedure, NABNs (20 mg/kg) were intraperitoneally injected into the mice. Twenty-four hours after surgery, the main organs (intestines, spleen, heart, liver, lung, and kidney) and blood were disrupted with concentrated nitric acid. The concentrations of Si ion in the solutions were determined by inductively coupled plasma optical emission spectrometry (ICP-OES), and the contents in each organ were calculated through normalization of control group. Standards were prepared and measured along with the samples to calculate the percentage of the injected dose per gram of tissue (% ID/g).
Statistical analysis
Statistical analyses were performed using GraphPad Prism software 7 (La Jolla, CA), and the results were expressed as means ± SEM. The differences between groups were assessed by either Student’s t test (for simple two-sample comparison) or one-way analysis of variance (ANOVA) with Tukey’s post hoc test (for multiple comparison). The Kaplan-Meier method was used to compare differences in survival rates.
Supplementary Material
Acknowledgments
We are grateful to C. Yang, H. Li, X. Zheng, B. Huang, Z. Tu, Y. Zhong, F. Chen, H. Huang, X. Meng, C. Guo, and P. Wang for expert technical assistance. Funding: This study was supported by the National Natural Science Foundation of China (81503122, 51873208, and 51520105004), the NIH (AR073935), and the Science and Technology Development projects of Jilin Province (20170623062TC and 20180201025YY). Author contributions: J.D., MD.S., YW.Z., D.S., HZ.Y., Y.-H.L., HZ.H., LZ.C., XY.L., F.L., C.-W.C and Y.Z. contributed to the collection of experimental data. J.D., MD.S., YW.Z., D.S., HZ.Y., MQ.L., M.Z., HY.T. and XS.C. analyzed the data. J.D., MD.S., YW.Z., D.S., K.W.L., and L.Ch. contributed to writing and revising the paper. D.S., K.W.L., and L.Ch. supervised the research. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/22/eaay7148/DC1
REFERENCES AND NOTES
- 1.Rhee C., Dantes R., Epstein L., Murphy D. J., Seymour C. W., Iwashyna T. J., Kadri S. S., Angus D. C., Danner R. L., Fiore A. E., Jernigan J. A., Martin G. S., Septimus E., Warren D. K., Karcz A., Chan C., Menchaca J. T., Wang R., Gruber S., Klompas M.; CDC Prevention Epicenter Program , Incidence and trends of sepsis in US hospitals using clinical vs claims data, 2009-2014. JAMA 318, 1241–1249 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dellinger R. P., Levy M. M., Rhodes A., Annane D., Gerlach H., Opal S. M., Sevransky J. E., Sprung C. L., Douglas I. S., Jaeschke R., Osborn T. M., Nunnally M. E., Townsend S. R., Reinhart K., Kleinpell R. M., Angus D. C., Deutschman C. S., Machado F. R., Rubenfeld G. D., Webb S., Beale R. J., Vincent J.-L., Moreno R.; The Surviving Sepsis Campaign Guidelines Committee including The Pediatric Subgroup , Surviving Sepsis Campaign: International guidelines for management of severe sepsis and septic shock, 2012. Intensive Care Med. 39, 165–228 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arnold C., News feature: The quest to solve sepsis. Proc. Natl. Acad. Sci. U.S.A. 115, 3988–3991 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Angus D. C., van der Poll T., Severe sepsis and septic shock. N. Engl. J .Med. 369, 840–851 (2013). [DOI] [PubMed] [Google Scholar]
- 5.Hotchkiss R. S., Karl I. E., The pathophysiology and treatment of sepsis. N. Engl. J .Med. 348, 138–150 (2003). [DOI] [PubMed] [Google Scholar]
- 6.Kaukonen K.-M., Bailey M., Pilcher D., Cooper D. J., Bellomo R., Systemic inflammatory response syndrome criteria in defining severe sepsis. N. Engl. J. Med. 372, 1629–1638 (2015). [DOI] [PubMed] [Google Scholar]
- 7.Bosmann M., Ward P. A., The inflammatory response in sepsis. Trends Immunol. 34, 129–136 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.van der Poll T., van de Veerdonk F. L., Scicluna B. P., Netea M. G., The immunopathology of sepsis and potential therapeutic targets. Nat. Rev. Immunol. 17, 407–420 (2017). [DOI] [PubMed] [Google Scholar]
- 9.Saukkonen K., Lakkisto P., Pettila V., Varpula M., Karlsson S., Ruokonen E., Pulkki K.; Finnsepsis Study Group , Cell-free plasma DNA as a predictor of outcome in severe sepsis and septic shock. Clin. Chem. 54, 1000–1007 (2008). [DOI] [PubMed] [Google Scholar]
- 10.Dwivedi D. J., Toltl L. J., Swystun L. L., Pogue J., Liaw K. L., Weitz J. I., Cook D. J., Fox-Robichaud A. E., Liaw P. C.; Canadian Critical Care Translational Biology Group , Prognostic utility and characterization of cell-free DNA in patients with severe sepsis. Crit. Care 16, R151 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kilpinen H., Waszak S. M., Gschwind A. R., Raghav S. K., Witwicki R. M., Orioli A., Migliavacca E., Wiederkehr M., Gutierrez-Arcelus M., Panousis N. I., Yurovsky A., Lappalainen T., Romano-Palumbo L., Planchon A., Bielser D., Bryois J., Padioleau I., Udin G., Thurnheer S., Hacker D., Core L. J., Lis J. T., Hernandez N., Reymond A., Deplancke B., Dermitzakis E. T., Coordinated effects of sequence variation on DNA binding, chromatin structure, and transcription. Science 342, 744–747 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lachelt U., Wagner E., Nucleic acid therapeutics using polyplexes: A journey of 50 years (and beyond). Chem. Rev. 115, 11043–11078 (2015). [DOI] [PubMed] [Google Scholar]
- 13.Lee J., Sohn J. W., Zhang Y., Leong K. W., Pisetsky D., Sullenger B. A., Nucleic acid-binding polymers as anti-inflammatory agents. Proc. Natl. Acad. Sci. U.S.A. 108, 14055–14060 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pisetsky D. S., Lee J., Leong K. W., Sullenger B. A., Nucleic acid-binding polymers as anti-inflammatory agents: Reducing the danger of nuclear attack. Expert Rev. Clin. Immunol. 8, 1–3 (2012). [DOI] [PubMed] [Google Scholar]
- 15.Stearns N. A., Lee J., Leong K. W., Sullenger B. A., Pisetsky D. S., The inhibition of anti-DNA binding to DNA by nucleic acid binding polymers. PLOS ONE 7, e40862 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Naqvi I., Gunaratne R., McDade J. E., Moreno A., Rempel R. E., Rouse D. C., Herrera S. G., Pisetsky D. S., Lee J., White R. R., Sullenger B. A., Polymer-mediated inhibition of pro-invasive nucleic acid DAMPs and microvesicles limits pancreatic cancer metastasis. Mol. Ther. 26, 1020–1031 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Holl E. K., Shumansky K. L., Borst L. B., Burnette A. D., Sample C. J., Ramsburg E. A., Sullenger B. A., Scavenging nucleic acid debris to combat autoimmunity and infectious disease. Proc. Natl. Acad. Sci. U.S.A. 113, 9728–9733 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Roberts J. C., Bhalgat M. K., Zera R. T., Preliminary biological evaluation of polyamidoamine (PAMAM) StarburstTM dendrimers. J. Biomed. Mater. Res. 30, 53–65 (1996). [DOI] [PubMed] [Google Scholar]
- 19.Jones C. F., Campbell R. A., Brooks A. E., Assemi S., Tadjiki S., Thiagarajan G., Mulcock C., Weyrich A. S., Brooks B. D., Ghandehari H., Grainger D. W., Cationic PAMAM dendrimers aggressively initiate blood clot formation. ACS Nano 6, 9900–9910 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jones C. F., Campbell R. A., Franks Z., Gibson C. C., Thiagarajan G., Vieira-de-Abreu A., Sukavaneshvar S., Mohammad S. F., Li D. Y., Ghandehari H., Weyrich A. S., Brooks B. D., Grainger D. W., Cationic PAMAM dendrimers disrupt key platelet functions. Mol. Pharm. 9, 1599–1611 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liang H., Peng B., Dong C., Liu L., Mao J., Wei S., Wang X., Xu H., Shen J., Mao H.-Q., Gao X., Leong K. W., Chen Y., Cationic nanoparticle as an inhibitor of cell-free DNA-induced inflammation. Nat. Commun. 9, 4291 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Buras J. A., Holzmann B., Sitkovsky M., Animal models of sepsis: Setting the stage. Nat. Rev. Drug Discov. 4, 854–865 (2005). [DOI] [PubMed] [Google Scholar]
- 23.Rittirsch D., Huber-Lang M. S., Flierl M. A., Ward P. A., Immunodesign of experimental sepsis by cecal ligation and puncture. Nat. Protoc. 4, 31–36 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shi H., Hong Y., Qian J., Cai X., Chen S., Xuebijing in the treatment of patients with sepsis. Am. J. Emerg. Med. 35, 285–291 (2017). [DOI] [PubMed] [Google Scholar]
- 25.Wang Q., Wu X., Tong X., Zhang Z., Xu B., Zhou W., Xuebijing ameliorates sepsis-induced lung injury by downregulating HMGB1 and RAGE expressions in mice. Evid.-Based Complementary Altern. Med. 2015, 860259 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Meng X., Sun W., Ren Y., Xiao Y., Zhao P., Lu W., Hua L., Wang L., Wang L., Yu Y., Protective role of surface Toll-like receptor 9 expressing neutrophils in local inflammation during systemic inflammatory response syndrome in mice. Mol. Immunol. 90, 74–86 (2017). [DOI] [PubMed] [Google Scholar]
- 27.Knežević N. Z., Durand J.-O., Large pore mesoporous silica nanomaterials for application in delivery of biomolecules. Nanoscale 7, 2199–2209 (2015). [DOI] [PubMed] [Google Scholar]
- 28.Shao D., Li M., Wang Z., Zheng X., Lao Y.-H., Chang Z., Zhang F., Lu M., Yue J., Hu H., Yan H., Chen L., Dong W.-f., Leong K. W., Bioinspired diselenide-bridged mesoporous silica nanoparticles for dual-responsive protein delivery. Adv. Mater. 30, 1801198 (2018). [DOI] [PubMed] [Google Scholar]
- 29.Pinese C., Lin J., Milbreta U., Li M., Wang Y., Leong K. W., Chew S. Y., Sustained delivery of siRNA/mesoporous silica nanoparticle complexes from nanofiber scaffolds for long-term gene silencing. Acta Biomater. 76, 164–177 (2018). [DOI] [PubMed] [Google Scholar]
- 30.Graetz T. J., Hotchkiss R. S., Sepsis: Preventing organ failure in sepsis—The search continues. Nat. Rev. Nephrol. 13, 5–6 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nel A., Xia T., Meng H., Wang X., Lin S., Ji Z., Zhang H., Nanomaterial toxicity testing in the 21st century: Use of a predictive toxicological approach and high-throughput screening. Acc. Chem. Res. 46, 607–621 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stoller J., Halpin L., Weis M., Aplin B., Qu W., Georgescu C., Nazzal M., Epidemiology of severe sepsis: 2008-2012. J. Crit. Care 31, 58–62 (2016). [DOI] [PubMed] [Google Scholar]
- 33.Zhang Q., Raoof M., Chen Y., Sumi Y., Sursal T., Junger W., Brohi K., Itagaki K., Hauser C. J., Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 464, 104–107 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kang J.-W., Kim S.-J., Cho H.-I., Lee S.-M., DAMPs activating innate immune responses in sepsis. Ageing Res. Rev. 24, 54–65 (2015). [DOI] [PubMed] [Google Scholar]
- 35.McDonald B., Urrutia R., Yipp B. G., Jenne C. N., Kubes P., Intravascular neutrophil extracellular traps capture bacteria from the bloodstream during sepsis. Cell Host Microbe 12, 324–333 (2012). [DOI] [PubMed] [Google Scholar]
- 36.Yadav A., Saini V., Arora S., MCP-1: Chemoattractant with a role beyond immunity: A review. Clin. Chim. Acta 411, 1570–1579 (2010). [DOI] [PubMed] [Google Scholar]
- 37.Xia T., Kovochich M., Liong M., Meng H., Kabehie S., George S., Zink J. I., Nel A. E., Polyethyleneimine coating enhances the cellular uptake of mesoporous silica nanoparticles and allows safe delivery of siRNA and DNA constructs. ACS Nano 3, 3273–3286 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Croissant J. G., Fatieiev Y., Khashab N. M., Degradability and clearance of silicon, organosilica, silsesquioxane, silica mixed oxide, and mesoporous silica nanoparticles. Adv. Mater. 29, 1604634 (2017). [DOI] [PubMed] [Google Scholar]
- 39.Du X., Kleitz F., Li X., Huang H., Zhang X., Qiao S.-Z., Disulfide-bridged organosilica frameworks: Designed, synthesis, redox-triggered biodegradation, and nanobiomedical applications. Adv. Funct. Mater. 28, 1707325 (2018). [Google Scholar]
- 40.Timmermans K., Kox M., Scheffer G. J., Pickkers P., Plasma nuclear and mitochondrial DNA levels, and markers of inflammation, shock, and organ damage in patients with septic shock. Shock 45, 607–612 (2016). [DOI] [PubMed] [Google Scholar]
- 41.Mastrobattista E., Hennink W. E., Polymers for gene delivery: Charged for success. Nat. Mater. 11, 10–12 (2011). [DOI] [PubMed] [Google Scholar]
- 42.Ren Y., Jiang X., Pan D., Mao H.-Q., Charge density and molecular weight of polyphosphoramidate gene carrier are key parameters influencing its DNA compaction ability and transfection efficiency. Biomacromolecules 11, 3432–3439 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Grigsby C. L., Leong K. W., Balancing protection and release of DNA: Tools to address a bottleneck of non-viral gene delivery. J. R. Soc. Interface 7, ( suppl 1), S67–S82 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fischer D., Bieber T., Li Y., Elsässer H.-P., Kissel T., A novel non-viral vector for DNA delivery based on low molecular weight, branched polyethylenimine: Effect of molecular weight on transfection efficiency and cytotoxicity. Pharm. Res. 16, 1273–1279 (1999). [DOI] [PubMed] [Google Scholar]
- 45.Crielaard B. J., Lammers T., Schiffelers R. M., Storm G., Drug targeting systems for inflammatory disease: One for all, all for one. J. Control. Release 161, 225–234 (2012). [DOI] [PubMed] [Google Scholar]
- 46.Nehoff H., Parayath N. N., Domanovitch L., Taurin S., Greish K., Nanomedicine for drug targeting: Strategies beyond the enhanced permeability and retention effect. Int. J. Nanomedicine 9, 2539–2555 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Soenen S. J., Rivera-Gil P., Montenegro J.-M., Parak W. J., De Smedt S. C., Braeckmans K., Cellular toxicity of inorganic nanoparticles: Common aspects and guidelines for improved nanotoxicity evaluation. Nano Today 6, 446–465 (2011). [Google Scholar]
- 48.Soh M., Kang D.-W., Jeong H.-G., Kim D., Kim D. Y., Yang W., Song C., Baik S., Choi I.-Y., Ki S. K., Kwon H. J., Kim T., Kim C. K., Lee S.-H., Hyeon T., Ceria-Zirconia nanoparticles as an enhanced multi-antioxidant for sepsis treatment. Angew. Chem. Int. Ed. 56, 11399–11403 (2017). [DOI] [PubMed] [Google Scholar]
- 49.Spence S., Greene M. K., Fay F., Hams E., Saunders S. P., Hamid U., Fitzgerald M., Beck J., Bains B. K., Smyth P., Themistou E., Small D. M., Schmid D., O'Kane C. M., Fitzgerald D. C., Abdelghany S. M., Johnston J. A., Fallon P. G., Burrows J. F., McAuley D. F., Kissenpfennig A., Scott C. J., Targeting Siglecs with a sialic acid-decorated nanoparticle abrogates inflammation. Sci. Transl. Med. 7, 303ra140 (2015). [DOI] [PubMed] [Google Scholar]
- 50.Seymour C. W., Liu V. X., Iwashyna T. J., Brunkhorst F. M., Rea T. D., Scherag A., Rubenfeld G., Kahn J. M., Shankar-Hari M., Singer M., Deutschman C. S., Escobar G. J., Angus D. C., Assessment of clinical criteria for sepsis: For the third international consensus definitions for sepsis and septic shock (sepsis-3). JAMA 315, 762–774 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Huang X., Venet F., Wang Y. L., Lepape A., Yuan Z., Chen Y., Swan R., Kherouf H., Monneret G., Chung C. S., Ayala A., PD-1 expression by macrophages plays a pathologic role in altering microbial clearance and the innate inflammatory response to sepsis. Proc. Natl. Acad. Sci. U.S.A. 106, 6303–6308 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li H., Wang S., Zhan B., He W., Chu L., Qiu D., Li N., Wan Y., Zhang H., Chen X., Fang Q., Shen J., Yang X., Therapeutic effect of Schistosoma japonicum cystatin on bacterial sepsis in mice. Parasit. Vectors 10, 222 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stewart S., Winters G. L., Fishbein M. C., Tazelaar H. D., Kobashigawa J., Abrams J., Andersen C. B., Angelini A., Berry G. J., Burke M. M., Demetris A. J., Hammond E., Itescu S., Marboe C. C., McManus B., Reed E. F., Reinsmoen N. L., Rodriguez E. R., Rose A. G., Rose M., Suciu-Focia N., Zeevi A., Billingham M. E., Revision of the 1990 working formulation for the standardization of nomenclature in the diagnosis of heart rejection. J. Heart Lung Transplant. 24, 1710–1720 (2005). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/22/eaay7148/DC1