

Formation of neutrophil extracellular traps worsens cerebrovascular outcomes after traumatic brain injury.
Abstract
Traumatic brain injury (TBI) is a major cause of mortality and morbidity. Preventative measures reduce injury incidence and/or severity, yet one-third of hospitalized patients with TBI die from secondary pathological processes that develop during supervised care. Neutrophils, which orchestrate innate immune responses, worsen TBI outcomes via undefined mechanisms. We hypothesized that formation of neutrophil extracellular traps (NETs), a purported mechanism of microbial trapping, exacerbates acute neurological injury after TBI. NET formation coincided with cerebral hypoperfusion and tissue hypoxia after experimental TBI, while elevated circulating NETs correlated with reduced serum deoxyribonuclease-1 (DNase-I) activity in patients with TBI. Functionally, Toll-like receptor 4 (TLR4) and the downstream kinase peptidylarginine deiminase 4 (PAD4) mediated NET formation and cerebrovascular dysfunction after TBI. Last, recombinant human DNase-I degraded NETs and improved neurological function. Thus, therapeutically targeting NETs may provide a mechanistically innovative approach to improve TBI outcomes without the associated risks of global neutrophil depletion.
INTRODUCTION
Traumatic brain injury (TBI) is a major public health issue, killing or debilitating more individuals than breast cancer, AIDS, multiple sclerosis, and spinal cord injury combined. In contrast to primary injuries that occur at the time of impact, secondary injuries develop while patients are under supervised medical care. The fixed volume of the skull imposes a space constraint such that any imbalance in the movement of blood or fluid into and out of the brain may contribute toward elevated intracranial pressure (ICP), cerebral hypoperfusion, inadequate tissue oxygenation, and neurological deterioration. In particular, the venous system, which contains 70 to 80% of total cerebral circulatory volume, regulates cerebrovascular resistance (1); thus, reduced venous outflow increases capillary blood volume, promotes hydrostatic edema, and elevates ICP to exacerbate venous congestion and restrict cerebral outflow. Although surgical decompression and osmotherapy remain mainstays in the clinical management of cerebral edema, these limited approaches do not proactively inhibit the molecular cascades that promote brain swelling.
Mechanical trauma induces widespread cellular necrosis, which temporally correlates with innate immune activation after TBI. Surgical excision of necrotic brain tissue during the acute injury phase reduces elevated ICP and improves 6-month mortality rates in patients with severe TBI (2), suggesting a deleterious, yet poorly understood, effect of early immune activation. We and others reported that elevated plasma or cerebrospinal fluid (CSF) levels of the damage-associated molecular pattern molecule high-mobility group box protein-1 (HMGB1) independently predicted mortality and unfavorable outcomes in patients with severe TBI undergoing surgical ICP management, while the immediate, passive release of HMGB1 from necrotic neurons increased leukocyte activation and promoted cerebral edema via a Toll-like receptor 4 (TLR4)–dependent mechanism after experimental TBI (3–5). Similarly, TLR4 activation was associated with poor TBI outcomes in patients (6), whereas global genetic TLR4 inhibition reduced neuroinflammation and attenuated the development of cerebral edema after preclinical TBI (4). We reported that VGX-1027, a selective TLR4 antagonist, maximally suppressed cerebral edema when administered between 0.5 to 4 hours after TBI, mirroring peak central nervous system (CNS) innate immune activation (4); however, the molecular and cellular mechanisms linking TLR4 activation with edema development, elevated ICP, and poor neurological function remain poorly defined.
Neutrophils are short-lived, circulating cells that rapidly mobilize to sites of infection or injury as a first line of host defense. The blood-brain barrier limits the entry of neutrophils into the noninjured CNS; however, disruptions within the blood-brain barrier permit neutrophil infiltration following injury. Infiltrating neutrophils roll along cerebral arterioles and venules within 5 min of human closed head injury, with adherence limited to postcapillary venules in hypoperfused brain regions (7). Neutrophils subsequently fill the subarachnoid and subdural spaces within 4 hours after injury and exhibit a fourfold increase in circulating levels, with infiltration into the cortical and subcortical parenchyma observed over the ensuing days (8, 9). Functionally, neutrophil activation induced microcirculatory dysfunction, cerebral hypoperfusion, and edema formation in experimental models of TBI (10, 11). Moreover, thiopental, a barbiturate used to manage refractory ICP, was associated with induced neutropenia in patients with TBI (12). Despite the deleterious effect of acute neutrophil activation, the potential benefits of global neutrophil depletion strategies are outweighed by a heightened infection risk in critically ill patients.
Neutrophils engulf pathogens into phagosomes, which fuse with cytoplasmic granules containing antimicrobial enzymes; however, this mechanism of host protection is unlikely to explain the deleterious effects of neutrophils during sterile injuries. More recently, neutrophils were shown to extrude a meshwork of chromatin fibers studded with globular domains made of granule proteins to produce cloud-like neutrophil extracellular traps (NETs) that aid in pathogen trapping/killing. Of note, activation of TLR4 increased histone hypercitrullination, a posttranslational modification that initiates NET formation in human and mouse neutrophils ex vivo, via a mechanism involving the enzyme peptidylarginine deiminase 4 (PAD4) (13–15). Histone hypercitrullination and NET production were undetectable in global PAD4−/− mice, yet these mice did not exhibit increased vulnerability to severe polymicrobial sepsis (16). As the regulatory mechanisms and functional roles for NETs remain largely unexplored after neurological injury, we explored whether NETs promote cerebrovascular dysfunction following acute brain injury. The primary objective of this study was to test the hypothesis that NETs exacerbate neurological deterioration after TBI. A secondary objective was to determine whether activation of TLR4 initiates NET formation after TBI.
RESULTS
NETs localize within hypoperfused brain tissue after controlled cortical impact in mice
A precipitous reduction in cerebral blood flow (CBF) preceded persistent cerebral hypoperfusion (fig. S1, A and B), tissue hypoxia (fig. S1C), and reduced brain-tissue oxygenation (fig. S1D) after experimental TBI. Using laser speckle contrast imaging (LSCI), cerebral hypoperfusion was observed within 30 min of injury, remaining suppressed for over 3 days after injury (fig. S1, A and B). This reduction in perfusion was mirrored by tissue hypoxia, as assessed by the histological detection of pimonidazole adducts (fig. S1C), and by a reduction in pO2 (fig. S1D) throughout the pericontusional cortex after TBI, as compared to sham-injured mice. Within hypoperfused brain tissue, we observed that increased expression of leukocyte adhesion molecules, including intercellular adhesion molecule–1 (ICAM-1), vascular cell adhesion molecule–1 (VCAM-1), E-selectin, and P-selectin (fig. S2A), spatially overlapped with neutrophil infiltration into the hypoxic brain tissue (fig. S2B). In parallel, use of a discovery-based array revealed that the elevated expression of leukocyte chemoattractant molecules [e.g., chemokine (C-X-C) ligand 10 (CXCL10), CXCL1, CCL3, and CXCL2] (fig. S3, A and B) spatially and temporally overlapped with neutrophil infiltration into regions of tissue hypoxia after TBI.
We next sought to characterize the CNS-infiltrated neutrophils after brain injury. Using scanning electron microscopy, we observed extracellular NET-like structures adjacent to leukocytes, extravasated erythrocytes, and necrotic/degenerating cells within the traumatized parenchyma (Fig. 1A). The presence of NETs was restricted to contusional and pericontusional brain regions, as these structures were not present in sham-operated control brain tissue or in anatomical sites distant from the contusion after TBI. Further work using transmission electron microscopy revealed that citrullinated histone H3 (Cit-H3), a marker of NETs, colocalized within an infiltrated neutrophil in the pericontusional cortex, while the granule enzyme neutrophil elastase (NE) exhibited a thread-like appearance, consistent with the structure of NETs, in perivascular regions after TBI (Fig. 1, B and C). Moreover, NE was localized around the pericontusional vasculature after TBI (Fig. 1, D and E).
Fig. 1. Generation of NETs following experimental TBI.
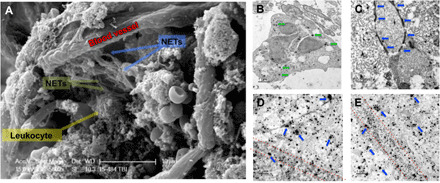
Mixed-sex adult CD-1 mice were subjected to TBI, and pericontusional tissue was prepared for electron microscopy at 24 hours after injury. (A) Representative scanning electron micrograph showing NET-like structures adjacent to an injured blood vessel in the pericontusional cortex after TBI. Data are representative of n = 5 mice. Scale bar, 10 μm. (B) Immunogold labeling showing the extranuclear presence of citrullinated histone H3 (Cit-H3; green arrows), a marker of early-stage NET generation, in a pericontusional neutrophil at 24 hours after TBI. Scale bar, 1 μm. (C) Immunogold labeling of the neutrophil granule enzyme, neutrophil elastase (NE), showing a thread-like localization (blue arrows). Scale bar, 0.5 μm. (D and E) Dual immunogold labeling of the vascular marker, laminin (small spots), and NE (large spots, blue arrows). Red dotted lines demarcate the location of blood vessels. Note the extravascular and clustered appearance of NE, indicative of NET formation. Scale bar, 1 μm.
NET formation correlates with elevated ICP and worse neurological function in patients with TBI
An inverse association was observed between the activity of serum deoxyribonuclease-1 (DNase-I), an endogenous NET-degrading enzyme, and levels of circulating NETs in patients with TBI undergoing CSF diversion due to elevated ICP (Fig. 2, A to C). A significant inverse correlation was observed between serum DNase levels and ICP in patients with severe TBI (R = −0.7850; P = 0.0036) (Fig. 2D). Similarly, Glasgow Coma Scale (GCS) score, a clinically reliable measure of neurological function, strongly correlated with serum DNase (R = 0.8450; P = 0.001) (Fig. 2E), raising the unexplored possibility that NETs contribute to the development of cerebral edema.
Fig. 2. Increased NET formation in patients with severe neurotrauma.
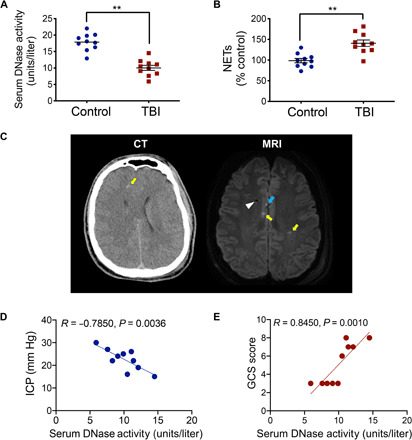
(A) Serum DNase-I activity and (B) myeloperoxidase (MPO)–DNA binding, a sensitive measure of NET formation, were quantified by EIA in blood collected from control patients (n = 10) or patients with severe TBI undergoing CSF diversion due to elevated ICP (n = 10). Data are presented as means ± SEM and analyzed using a Student’s t test (**P < 0.01 versus control). (C) Axial computed tomography (CT) scan without contrast and magnetic resonance imaging (MRI) axial diffusion-weighted image from a representative patient (19-year-old male, GCS = 7) used in blood collection. Note the effacement of the gray-white delineation and partial effacement of the right lateral ventricle, consistent with diffuse cerebral edema on the CT scan. Arrow indicates a hyperdense area at the gray-white junction in the right frontal lobe, consistent with diffuse axonal injury/tissue tear hemorrhage at the gray-white junctions. On the MRI image, yellow arrows indicate hyperintense areas, consistent with diffuse axonal injury. The more anterior right-sided hyperintensity is cortical and represents a microcontusion with associated diffuse axonal injury (see blue arrow). The hypointense area coincides with the tract of the ventricular catheter that was placed for both CSF diversion and ICP monitoring (white arrowhead). Correlation analysis between patient serum DNase activity and (D) ICP or (E) Glasgow Coma Scale (GCS) score. Pearson’s correlation coefficient (R) and P value are shown as insets.
Activation of TLR4 promotes NET formation after experimental TBI
We next sought to elucidate the mechanism whereby TBI induces NETs. Both circulating and CNS-infiltrated neutrophils exhibited elevated TLR4 expression after TBI (Fig. 3A). C3H/HeJ mice, which lack functional TLR4, displayed less NET formation (Fig. 3B and fig. S4), exhibited improvements in cerebral perfusion, and displayed less edema development after TBI, as compared to wild-type C3H/OuJ mice (Fig. 3C). Adoptive transfer of isolated C3H/OuJ neutrophils to either C3H/OuJ [wild type (WT) > WT] or C3H/HeJ [WT > mutant (MUT)] mice increased NET formation after TBI (Fig. 3D). In contrast, administration of C3H/HeJ neutrophils to either C3H/OuJ (MUT > WT) or C3H/HeJ (MUT > MUT) mice resulted in less NET formation (Fig. 3D). In keeping with these findings, WT > WT and WT > MUT mice exhibited reduced CBF and elevated edema after TBI, whereas improved CBF and attenuated edema were observed in MUT > MUT and MUT > WT mice (Fig. 3, E and F). While demonstrating a direct role for TLR4 activation in NET formation, serum DNase-I was also reduced in C3H/OuJ mice after TBI (fig. S5), similar to that observed in TBI patient serum (Fig. 2A); however, this effect was lost in C3H/HeJ mice (fig. S5), suggesting that TLR4 enhances NET production while simultaneously limiting NET degradation.
Fig. 3. TLR4 activation promotes NETs and neurovascular injury after TBI.
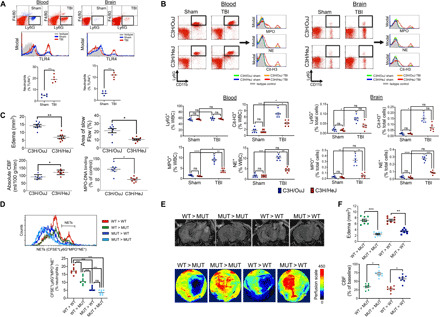
(A) Increased TLR4 expression in neutrophils from blood or pericontusional brain tissue at 24 hours after sham (blue)/TBI (red). Scatterplots show % TLR4+ neutrophils from n = 5 mice per group. (B) Extracellular expression of MPO, NE, and Cit-H3 in CD11b+Ly6G+ neutrophils derived from blood or brain from C3H/OuJ (blue) or C3H/HeJ (red) mice after sham/TBI. Representative flow cytometry scatterplots are provided along with quantified data. Brain panels are depicted as % total brain cells, and blood panels are shown as % leukocytes [% white blood cell (WBC)]. (C) Cerebral perfusion and cerebral edema were quantified by MRI in C3H/OuJ or C3H/HeJ mice at 24 hours after TBI. Cerebral hypoperfusion, cerebral edema, and the region of slow flow are attenuated in C3H/HeJ mice, indicative of improved cerebrovascular function following TLR4 inhibition. MPO-DNA binding, a quantitative marker of NET formation, was measured in blood from mice immediately following the final imaging session. (D) Carboxyfluorescein diacetate succinimidyl ester (CFSE)–labeled wild-type (WT; C3H/OuJ) or mutant (MUT; C3H/HeJ) neutrophils were administered to WT or MUT mice at the time of TBI. NET formation was elevated in WT > WT and WT > MUT mice, as assessed by flow cytometry, as compared to MUT > MUT and MUT > WT mice. (E and F) Representative images showing changes in cerebral edema (top) and CBF (bottom), as assessed by MRI and LSCI, respectively. For all panels, data are means ± SEM from n = 8 mice per group from two independent experiments. Data were analyzed using a Student’s t test or one-way analysis of variance (ANOVA) followed by Tukey’s post hoc test (*P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001; ns, not statistically significant).
TLR4 activation increased histone hypercitrullination, and NET formation via a PAD4-dependent mechanism in isolated human neutrophils and circulating levels of Cit-H3, a product of PAD4 activity, is an acute-phase biomarker of injury severity after lipopolysaccharide-induced shock. Following TBI, PAD4 colocalized in activated TLR4-expressing neutrophils, exhibiting increased extracellular myeloperoxidase (MPO) and NE expression (Fig. 4A). Functionally, C3H/HeJ mice displayed reduced extracellular Cit-H3 and MPO expression in neutrophils (Fig. 3B), while Cl-amidine, a selective, irreversible, and cell-permeable PAD4 inhibitor, reduced ex vivo NET formation after exposure to the TLR4 agonist, lipopolysaccharide (fig. S6). Similarly, administration of Cl-amidine dose-dependently reduced CNS neutrophil infiltration, attenuated NET production after TBI (Fig. 4B), reduced cerebral edema (Fig. 4C), improved CBF (Fig. 4C), and ameliorated neurological deficits after TBI (Fig. 4D).
Fig. 4. PAD4 promotes NET formation and exacerbates neurological injury after TBI.
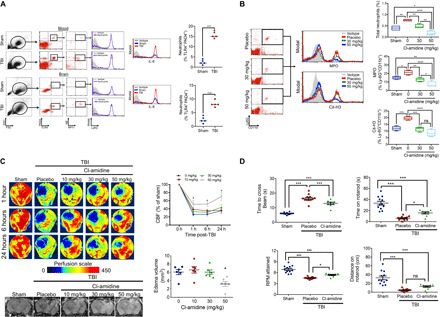
(A) Blood and pericontusional brain tissue were collected at 24 hours after sham/TBI, and a population of TLR4+PAD4+ neutrophils were selected by flow cytometry. This population was further gated for the extracellular expression of NE and MPO and for intracellular expression of interleukin-8 (IL-8; nine- and eightfold increase in blood and brain, respectively, after TBI), an activated neutrophil marker and autocrine inducer of NETs. The % TLR4+PAD+ neutrophils expressing IL-8 and extracellular NE/MPO are shown. (B) Placebo or Cl-amidine (30 to 50 mg/kg i.p.) was administered at 10 min after sham/TBI, and pericontusional brain tissue was collected for flow cytometry at 24 hours after injury. Representative histograms and quantified data indicate that Cl-amidine reduces total neutrophil infiltration and attenuates both extracellular MPO and Cit-H3 expression in Ly6G+CD11b+ neutrophils. (C) Placebo or Cl-amidine (10 to 50 mg/kg i.p.) was administered at 10 min after sham/TBI. Representative LSCI (top) and MRI (bottom) images show a reduction in cerebral edema (24 hours after injury) and improved cerebral perfusion (1 to 24 hours after injury) following Cl-amidine (50 mg/kg) treatment. Data are means ± SEM from n = 8 mice per group. (D) Administration of Cl-amidine (50 mg/kg) at 10 min after TBI reduced the time to cross a narrow beam and improved both revolutions per minute (RPM) attained and time spent on the rotarod, as compared to placebo-treated mice after TBI. Data are means ± SEM from n = 10 to 12 mice per group. For all panels, data were analyzed by one-way ANOVA followed by Tukey’s post hoc test (*P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001; ns, not statistically significant). SSC, side scatter; FSC, forward scatter.
Degradation of NETs improves cerebrovascular function in mice after TBI
TLR4 inhibitors exhibited a narrow therapeutic window to improve outcomes after experimental TBI, suggesting a limited translational value for targeting NET formation after TBI. Instead, therapies that accelerate the degradation of NETs may provide a wider opportunity for clinical intervention. The presence of a chromatin backbone renders NETs highly susceptible to digestion by DNase-1 (17), an enzyme that was suppressed after both experimental (Fig. 2A) and clinical TBI (fig. S5). Thus, we explored whether restoration of serum DNase-I level may balance circulating NETs and improve TBI outcomes. Intravenous administration of recombinant human DNase-I (rhDNase) degraded both circulating and CNS-infiltrated NETs, as assessed by the extracellular localization of NE and MPO in Ly6G+TLR4+ neutrophils (Fig. 5, A and B). In parallel, rhDNase reduced the development of edema, as assessed by magnetic resonance imaging (MRI), and improved cerebral perfusion, as determined by MRI and LSCI (Fig. 5, C and D). These improvements in NET degradation and cerebrovascular function resulted in enhanced acute (fig. S7) and chronic neurobehavioral outcomes after TBI, including better motor, cognitive, and psychiatric function at 2 months after injury (Fig. 5E).
Fig. 5. Degradation of NETs using rhDNase improves neurological outcomes after TBI.
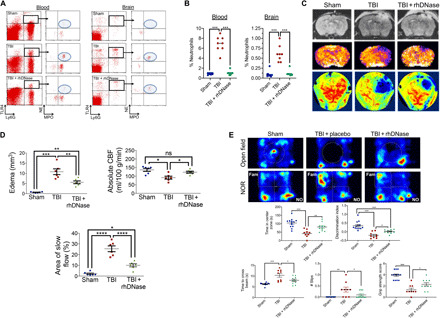
(A) Administration of rhDNase (5 mg/kg i.v.) at 1 hour after TBI reduced the extracellular expression of MPO and NE on Ly6G+TLR4+ neutrophils in both blood and brain tissue after TBI. (B) Quantification of data from (A). Scatterplots are representative of n = 8 mice per group and depict the % TLR4+ neutrophils exhibiting extracellular expression of NE and MPO. (C) Administration of rhDNase (5 mg/kg i.v.) at 1 hour after TBI reduced cerebral edema by MRI (top) and improved cerebral perfusion by MRI (middle) and LSCI (bottom) at 24 hours after injury. Representative images are provided from n = 8 mice per group. (D) Quantification of edema, absolute CBF, and area of slow flow/reduced reflow from (C). Data were analyzed using a one-way ANOVA followed by Tukey’s post hoc test. (*P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001; ns, not statistically significant). (E) Administration of rhDNase (5 mg/kg i.v.) at 1 hour after TBI improved chronic neurological function, as assessed at 2 months after TBI, as compared to placebo-treated mice. Top panels depict representative heat maps obtained in the open-field test, which demonstrated an increased time spent within the center zone (a measure of decreased anxiety) after rhDNase treatment following TBI (see middle panel, left). Representative heat maps obtained following the NOR test show that rhDNase increased the time spent exploring a novel object. The increased discrimination index (middle, right) suggests that rhDNase improves recognition memory at 8 weeks after TBI. Bottom panels demonstrate improvements in chronic, posttraumatic motor function following rhDNase treatment. Administration of rhDNase at 1 hour after TBI reduced the time required to traverse a narrow beam and decreased the number of slips, indicative of better motor function and coordination (bottom left and middle). Treatment with rhDNase also improved grip strength after TBI, as compared to placebo-treated mice, suggestive of better neuromuscular function. Data are means ± SEM from n = 9 to 13 mice per group and were analyzed by one-way ANOVA followed by Tukey’s post hoc test (*P < 0.05, **P < 0.01, and ***P < 0.001).
DISCUSSION
Elevated ICP, a frequent and life-threatening neurological complication, is directly associated with poor patient outcomes after acute brain injuries. Cerebral edema is a major contributor toward elevated ICP; however, the mechanisms underlying the development of brain swelling remain poorly defined, contributing toward a lack of efficacious therapeutic approaches. Global neutrophil depletion limited microcirculatory dysfunction, cerebral hypoperfusion, edema formation, and tissue loss after experimental TBI, yet the translational value of these broadly targeted approaches is limited by the development of associated side effects (10, 18). In this study, we identified NETs as mechanistically distinct mediators of cerebral hypoperfusion and edema formation after TBI. Given the lack of effective treatment options to improve patient outcomes, NETs may provide a previously unrecognized target to limit the deleterious consequences while simultaneously maintaining the beneficial aspects of neutrophils after acute brain injury. In addition to the therapeutic implications, our data implicate NETs as potential prognostic biomarkers to prospectively identify “at risk” patients requiring neuromonitoring and aggressive intervention.
The mechanisms whereby NETs worsen cerebrovascular outcomes remain undefined. Suppression of serum DNase-I activity correlated with the increased presence of circulating NETs in patients with severe neurotrauma requiring CSF diversion due to elevated ICP, while serum DNase activity was reduced for up to 1 week after surgical trauma (19). Thrombus formation is a major source of morbidity and mortality in hospitalized neurosurgical patients; thus, it is notable that reduced DNase-I activity similarly correlated with impaired NET degradation in patients with acute thrombotic microangiopathies (20). Host DNases prevent noncanonical intravascular occlusion by degrading circulating NETs during sterile neutrophilia and septicemia, while NETs purportedly provide a scaffold to propagate thrombogenesis (21, 22). In line with these observations, Cit-H3, a biomarker of NETs, was present within thrombi retrieved from patients with acute ischemic stroke and was independently associated with mortality (23). As cerebral microthrombi occluded up to 70% of postcapillary venules within 2 hours after experimental TBI (24), NETs may promote microthrombus formation to restrict cerebral venous outflow and increase venous congestion after TBI. The resultant accumulation of blood within brain capillaries may, in turn, increase hydrostatic edema, elevate ICP, and compress the venous system to further restrict cerebral outflow. Consistent with these possibilities, deficits in venous outflow exacerbated the development of early fatal cerebral edema in patients with ischemic stroke, whereas reduced cerebral venous flow velocity was predictive of elevated ICP and poor outcomes in patients with subarachnoid hemorrhage (25). Hence, restoration of serum DNase-I may provide a viable therapeutic strategy to reduce or eliminate NETs, which may explain the observed improvements in acute cerebrovascular function after TBI.
Neutrophils, which mediate host protection against pathogens, may promote repair or injury, depending on timing, context, and microenvironmental cues. The selective expression of leukocyte adhesion molecules within postcapillary venules provides a mechanism to localize neutrophils and NETs within the pericontusional cortex after TBI (26). Increased expression of P-selectin, E-selectin, and VCAM-1 was associated with leukocyte-endothelial adhesion, whereas ICAM-1 increased neurological demise after experimental and clinical TBI (26). Notably, CXCL10 enhanced neutrophil infiltration after lung injury (27), CCL3 and CXCL1 increased CNS neutrophil trafficking after brain abscess or epilepsy (28), and CXCL2, a powerful neutrophil chemoattractant, increased NET formation in cystic fibrosis (29). Thus, increased expression of leukocyte chemoattractants and adhesion molecules may localize circulating neutrophils within cerebral venules of pericontusional brain tissue after TBI.
We reported that HMGB1 enhanced leukocyte activation to initiate edema development via a TLR4-dependent mechanism after experimental TBI (4). Similarly, plasma and CSF levels of HMGB1 independently predicted unfavorable outcomes in patients with severe TBI receiving extraventricular drainage for elevated ICP, while TLR4 activation clinically correlated with negative TBI outcomes (6); however, the precise mechanisms whereby TLR4 activation worsened TBI outcomes remain undefined. The efficacy of pharmacological TLR4 inhibitors was restricted to the first several hours after experimental TBI, temporally paralleling neutrophil infiltration into the CNS (4). In line with the findings reported herein, HMGB1 induced pro-inflammatory gene expression in human neutrophils, increased histone hypercitrullination and NET formation via a TLR4-dependent mechanism ex vivo (30), and facilitated both NET formation and venous thrombosis in mice (31). In line with these data, we found that PAD4, which induced histone hypercitrullination and increased NET formation in isolated blood neutrophils, during necrotizing fasciitis and after deep vein thrombosis (13, 32, 33), mediated the effects of TLR4 activation after TBI. In addition, Cl-amidine, a selective, first-generation PAD inhibitor, reduced NET formation while simultaneously improving cerebrovascular function after TBI. Thus, the immediate, passive release of HMGB1 from necrotic neurons may initiate a TLR4-dependent molecular signaling cascade in neutrophils, culminating in NET formation and the development of cerebral edema.
Our translational studies support the clinical repurposing of rhDNase (Dornase Alfa, Pulmozyme), a U.S. Food and Drug Administration–approved drug with an outstanding long-term clinical safety record in pediatric and adult populations, to reduce life-threatening edema and elevated ICP after TBI. Despite the high clinical potential of rhDNase, our study has several limitations. Extracellular DNA is not normally present within the circulation, supporting the rationale for injury-specific therapeutic targeting; however, we cannot rule out the possibility that degradation of “off-target” (e.g., non-NETs) sources of extracellular DNA could mediate the observed improvements after TBI. For example, elevated levels of mitochrondrial DNA (mtDNA) are present within the blood/CSF after TBI (34). mtDNA induces immune responses, including NET formation in neutrophils from patients with trauma, via a mechanism requiring TLR9 (35); however, TLR9 expression was inversely correlated with edema formation after TBI (36). While a reduction in NETs after rhDNase administration could potentially heighten the risk of infection in critically ill patients, PAD4−/− mice did not exhibit increased vulnerability to severe polymicrobial sepsis, despite an inability to produce NETs (16). Moreover, no clinical evidence suggests that rhDNase adversely affects the host response to infection in patients, further supporting the conclusion that NETs may be dispensable for pathogen defense. Nonetheless, given the skyrocketing costs of therapeutic development, rhDNase may represent a readily available, cost-effective, low-risk/high-reward candidate to degrade NETs and improve acute cerebrovascular function after neurological injury. Although unexplored in this study, we anticipate that rhDNase may exhibit an extended therapeutic window with equivalent efficacy in both sexes and across all ages. Our future experimental and clinical studies will further define the molecular mechanisms whereby rhDNase limits cerebral edema and improves cerebrovascular function in addition to addressing these important translational questions.
MATERIALS AND METHODS
Experimental design
Mice were used as the primary research subject in controlled laboratory experiments. Primary study endpoints included NET formation, edema, CBS, and neurobehavioral outcomes. Blood derived from patients with severe neurotrauma was used to extend the translational relevance of our preclinical modeling studies. We used a double-blinded study design whereby mice were assigned a unique subject number and then randomized to treatment groups in a predetermined manner by a blinded study coordinator separate from the surgeon. Power analyses were conducted a priori to determine sample sizes using α = 0.05 and β = 0.10. Blinded investigators performed all data acquisition of outcome measures (e.g., MRI, behavior, and flow cytometry). Following final data acquisition, mice were decoded and final analyses were performed. No experimental subjects were removed from the study, and all data were included in the final analysis.
Controlled cortical impact
The Institutional Animal Care and Use Committee at Augusta University approved all animal studies, in compliance with National Institutes of Health guidelines. Adult mixed-sex CD-1 (Charles River, Wilmington, MA), C3H/OuJ (WT; the Jackson laboratories, stock no. 000635), or C3H/HeJ (TLR4 mutant; the Jackson laboratories, stock no. 000659) mice were subjected to a sham injury or controlled cortical impact. Briefly, mice were anesthetized using 2% isoflurane, placed in a stereotaxic frame, and a craniotomy was made in the right parietal bone midway between bregma and lambda with the medial edge 1 mm lateral to the midline, leaving the dura intact. Mice were impacted at 3 m/s with an 85-ms dwell time and 3.0-mm depression using a 3-mm-diameter convex tip (PinPoint PCI3000 Precision Cortical Impactor, Hatteras Instruments, Cary, NC). Sham-operated mice underwent the identical surgical procedures but were not impacted. The skin incision was closed and mice were allowed to recover in a clean, warm cage. Body temperature was maintained at 37°C using a small-animal temperature controller throughout all procedures (Kopf Instruments, Tujunga, CA, USA). Food and water were provided ad libitum. For drug administration studies, rhDNase was administered via the tail vein, and Cl-amidine (Cayman Chemical; >95% purity) was administered via an intraperitoneal injection.
Laser speckle contrast imaging
LSCI was performed, as reported by our laboratory (37). Perfusion images were acquired via a midline incision in anesthetized mice using PeriCam high-resolution LSCI (PSI System, Perimed) with a 70-mW built-in laser diode for illumination and 1388 × 1038 pixels charge-coupled device (CCD) camera installed 10 cm above the skull (speed, 19 Hz; and exposure time, 6 ms). Acquired images were analyzed for dynamic changes in CBF using PIMSoft. As both anesthesia and injury affect cerebral perfusion within the contralateral hemisphere, the absolute value from the ipsilateral side was normalized to sham and calculated as percent change. Body temperature was maintained at 37° ± 0.5°C throughout the experiment.
Magnetic resonance imaging
Mice were anesthetized with isoflurane (3% for induction and 1.5% for maintenance in a 2:1 mixture of N2:O2) and imaged using a horizontal 7-T BioSpec MRI spectrometer (Bruker instruments, Bellerica, MA) equipped with a 12-cm self-shielded gradient set (45 G/cm max). Radio frequency pulses were applied using a standard transmit/receive volume coil (72-mm inner diameter) actively decoupled from the two-channel Bruker quadrature receiver coil positioned over the centerline of the skull. Stereotaxic ear bars were used to minimize movement during the imaging procedure. Mouse temperature was maintained at 37° ± 0.5°C using a pad heated by a recirculating water bath. After positioning using a triplanar fast low angle shot (FLASH) sequence, MR studies were performed using T1-, T2-, and T2*-weighted MRI scans. Edema was quantified using a T2W map, while cerebral perfusion was quantified using flow sensitive alternating inversion recovery (FAIR)–rapid acquisition with relaxation enhancement (RARE) sequences that were acquired concurrently with edema measurements. Perfusion map values of a region of interest in injured and noninjured brain regions were used for normalization using Paravision 5.1 software.
Preparative and analytic flow cytometry
At designated time points, blood (200 μl) was collected by cardiac puncture and placed into ice-cold heparinized tubes. Mice were perfused with ice-cold phosphate-buffered saline (PBS), and whole brains were carefully harvested. A 3-mm coronal brain section centered on the contusion was prepared using an acrylic brain matrix. Freshly harvested brain tissue (0.2 g) was sieved through a 100 μM cell strainer, followed by centrifugation (252g, 10 min) to prepare single-cell suspensions. Cells were incubated with conjugated antibodies against the following markers to detect the extracellular presence of TLR4 (BioLegend, catalog no. 145409), CD11b (BioLegend, catalog no. 101212), F4/80 (BioLegend, catalog no. 123107), Ly6G (BioLegend, catalog no. 127608), NE (Bioss, catalog no. bs-6982R), MPO (Invitrogen, catalog no. PA5-16672), and Cit-H3 (Cell Signaling Technology, catalog no. 9715). Following a wash, cells were fixed and permeabilized using a Fixation/Permeabilization Concentrate (Affymetrix eBioscience) and then incubated with antibodies to detect the intracellular labeling of PAD4 (BioLegend, catalog no. 684202) or interleukin-8 (Affymetrix eBioscience, catalog no. BMS136FI). After a final wash, cells were analyzed using a FACSCalibur flow cytometer and CellQuest software, as routine to our laboratory (38). Isotype-matched controls were analyzed to set the appropriate gates for each sample. For each marker, samples were analyzed in duplicate. To minimize false-positive events, the number of double-positive events detected with the isotype controls was subtracted from the number of double-positive cells stained with corresponding antibodies (not isotype control), respectively. Viable cells were visibly differentiated from debris by gating on live cells with high forward scatter (FSC) and positivity for specific antibodies. Single stains were performed for compensation controls, controls to check for fluorescence spread, and isotype controls were used to determine the level of nonspecific binding. Cells expressing a specific marker were reported as a percentage of the number of gated events.
Arrays
MPO, which is contained within intracellular neutrophil granules under basal conditions, is released into the extracellular space after infection/injury to associate with chromatin during NET formation. Hence, the interaction between MPO and chromatin is a highly sensitive and specific method to quantify NETs in tissue and fluids (39). Toward this end, NETs were quantified in blood and brain tissue using a high-sensitivity capture enzyme-linked immunosorbent assay (ELISA) based on the association between MPO and DNA, as detailed (40). Briefly, an anti-MPO capture monoclonal antibody (1:500; Millipore) was coated in a 96-well enzyme linked immunoassay (EIA) plate. Brain tissue lysate or human plasma was incubated with a peroxidase-labeled anti-DNA monoclonal antibody (1:25 Cell Death Detection ELISAPLUS; Roche). Following addition of peroxidase substrate, absorbance at 405 nm was measured to quantify MPO-DNA interactions. DNase-I activity was measured in patient blood samples using a human DNase-I ELISA kit (LifeSpan Biosciences), per manufacturer’s recommended protocol. Protein lysates from pericontusional brain tissue or anatomically matched brain tissue from sham-injured mice was quantified by bicinchoninic acid (BCA) protein assay and then analyzed for chemokine expression using a Proteome Profiler Mouse Cytokine Array Kit, Panel A (#ARY006, R&D Systems, Minneapolis, MN), according to manufacturer’s recommended protocol.
Patient specimens
All studies were approved by the Institutional Review Board at Augusta University. Blood was obtained from consecutive adult severe TBI (GCS of 3T-8) or patients with normal pressure hydrocephalus (NPH) requiring CSF diversion as a part of routine medical care in the Department of Neurosurgery at the Medical College of Georgia. Specimens were collected without regard for age, race, gender, or socioeconomic status. Patients with NPH served as a control population as these patients present with edema in the absence of a traumatic event. All samples were de-identified and coded by the attending physician before transport to the laboratory.
Quantitative RT-PCR
Total RNA was isolated using an SV RNA isolation kit, and quantitative reverse transcription polymerase chain reaction (qRT-PCR) was performed on a Bio-Rad iCycler. Primers used were as follows: E-selectin, 5′-aggtccctgtcatgcttctg-3′ (forward) and 5′-gctgctggtgatcctcttgt-3′ (reverse); P-selectin, 5′-atgaaggtctccaccactgc-3′ (forward) and 5′-cccaggtctctttggagtca-3′ (reverse); ICAM-1, 5′-gtcctcaggtattggctgga-3′ (forward) and 5′-gggtcagcacagatctcctt-3′ (reverse); VCAM-1, 5′-gctgggattcacctcaagaa-3′ (forward) and 5′-tggggacaccttttagcatc-3′ (reverse); CXCL10, 5′-aagtgctgccgtcattttct-3′ (forward) and 5′-gtggcaatgatctcaacacg-3′ (reverse); CXCL1, 5′-gctgggattcacctcaagaa-3′ (forward) and 5′-tggggacaccttttagcatc-3′ (reverse); CCL2, 5′-aggtccctgtcatgcttctg-3′ (forward) and 5′-gctgctggtgatcctcttgt-3′ (reverse); CCL12, 5′-gtcctcaggtattggctgga-3′ (forward) and 5′-gggtcagcacagatctcctt-3′ (reverse); CCL3, 5′-atgaaggtctccaccactgc-3′ (forward) and 5′-cccaggtctctttggagtca-3′ (reverse); and CXCL2, 5′-agtgaactgcgctgtcaatg-3′ (forward) and 5′-ttcagggtcaaggcaaactt-3′ (reverse). Product specificity was confirmed by melting curve analysis. Gene expression levels were quantified, and data were normalized to RPS3 or 18S, housekeeping genes that were unaffected by TBI. Data are expressed as fold change versus placebo treatment.
Western blotting
Whole-cell protein lysates were prepared from the plasma of C3H/OuJ or C3H/HeJ mice at 24 hours after sham/TBI. Protein concentrations were quantified using a BCA protein assay kit. Thirty micrograms of protein was resolved on a 4 to 20% SDS–polyacrylamide gel and transferred onto a polyvinylidene difluoride membrane. Blots were incubated overnight at 4°C in a primary antibody (1:200 anti-mouse DNase I, clone B-4; Santa Cruz Biotechnology, sc-376207) in 5% nonfat dry milk followed by a 1-hour incubation with a donkey anti-mouse Alexa Fluor 680–tagged secondary antibody (1:3000) at room temperature. Total protein loading was visualized by Ponceau S staining. Blots were visualized using a Li-Cor Odyssey near-infrared imaging system, and densitometry analysis was performed using Quantity One software (Bio-Rad, Foster City, CA).
Electron microscopy
For scanning electron microscopy, tissue was fixed overnight in 4% paraformaldehyde, 2% glutaraldehyde in 0.1 M sodium cacodylate (NaCac) buffer, pH 7.4, postfixed in 2% osmium tetroxide in NaCac buffer, dehydrated with a graded ethanol series (25 to 100%), and critical point dried from CO2. The dried tissue was mounted on aluminum stubs with carbon adhesive tabs and sputter coated for 6 min with gold-palladium (Anatech Hummer 6.2, Union City, CA). Tissue was observed and imaged in a FEI XL30 scanning electron microscope at 10 kV. For transmission electron microscopy, tissue was fixed in 4% paraformaldehyde and 0.2% glutaraldehyde in 0.1 M NaCac buffer and processed through a graded series of ethanol incubations. The tissue was processed in a 95% ethanol:LR resin (1:1) for 1 hour, followed by pure LR White resin overnight at 4°C, and then embedded in pure LR White resin and allowed to polymerize overnight in a 60°C oven. Thin (75-nm-thick) sections were cut on a Leica UC6 ultramicrotome and collected on 200-mesh nickel grids. Sections were etched in 2% hydrogen peroxide in PBS for 20 min and then washed in PBS. Before immunogold staining, sections were blocked in 0.4% bovine serum albumin for 2 to 4 hours followed by incubation with anti-mouse primary antibodies at a 1:50 dilution overnight at 4°C. Sections were then rinsed in PBS three times for 10 min per wash, followed by incubation with anti-mouse–specific secondary antibody conjugated to streptavidin nanogold (1:1000 in 0.4% bovine serum albumin in PBS; Nanoprobes Inc.) for 2 hours at room temperature. Last, sections were washed three times in PBS and twice in distilled water, followed by an HQ Silver enhancement for 12 min. Last, sections were washed with distilled water and stained with 2% uranyl acetate. Dried grids were observed using a JEM 1230 transmission electron microscope (JEOL USA Inc., Peabody, MA) at 110 kV and imaged with an UltraScan 4000 CCD camera and a First Light digital camera controller (Gatan Inc., Pleasanton, CA).
Adoptive transfer
Total splenocytes from C3H/OuJ or C3H/HeJ mice were collected and enriched, and Ly6G+ neutrophils were consecutively sorted three times by magnetic bead isolation (Miltenyi Biotec) to achieve >95% purity. Purified neutrophils were labeled with 5 μM carboxyfluorescein diacetate succinimidyl ester (CFSE; Molecular Probes, Eugene, OR), a green fluorescent cell staining dye, and resuspended in sterile PBS. A total of 5 × 106 cells per mouse were injected via the tail vein immediately after sham/TBI. Brains were collected 24 hours after TBI, and NET formation was analyzed in CFSE+ neutrophils by flow cytometry.
Ex vivo neutrophil treatment
Naïve neutrophils were isolated from pooled spleens and sorted using a Beckman Coulter Mo-Flo XDP cell sorter. Cells were gated on the basis of forward and side scatter properties and of marker combinations (CD11b+Ly6G+) to select cells of interest. This highly purified fraction of neutrophils was washed twice and resuspended in Dulbecco’s modified Eagle medium (DMEM), containing high glucose and 10% ultralow endotoxin fetal bovine serum without antibiotics. Neutrophils (60,000 cells per chamber) were plated on poly-l-lysine–coated four-chambered culture slides. At 3 hours after plating, adherent cells were washed and pretreated for 3 hours with Cl-amidine (50 μM in a serum-free DMEM), followed by a 3-hour exposure to lipopolysaccharide (100 ng/ml). At the end of the incubation, cells were washed and fixed with 4% paraformaldehyde overnight at 4°C. Fixed neutrophils were washed three times and blocked with 2% donkey serum in 0.3% Triton X-100 in PBS for 0.5 hours. Cells were washed three times with 0.1% Tween-20 in PBS and then incubated with primary antibodies [rat anti-mouse neutrophil 7/4 (Abcam no. ab53457) 1:200 or goat anti-mouse MPO (R&D System no. AF3667) 1:200] overnight at 4°C followed by incubation with secondary donkey anti-rat immunoglobulin G (IgG) tagged with AF594 1:400 or donkey anti-goat IgG tagged with AF488 1:400 (the Jackson laboratories, Burlingame, CA) for 1 hour at room temperature. Last, cells were washed and mounted with VectaMount Permanent Mounting Media. Fluorescent images were captured by Zeiss LSM 780 upright microscope integrated with Zen2.1 software.
Immunohistochemistry
Hypoxyprobe-1, a pimonidazole compound, was administered via tail vein at 3 hours after sham or TBI. At 1.5 hours after injection, anesthetized mice were perfused with saline, followed by fixation with 4% paraformaldehyde in 0.1 M phosphate buffer (pH 7.4). Brains were postfixed overnight in paraformaldehyde followed by cryoprotection with 30% sucrose (pH 7.4) until brains permeated. Serial coronal sections (12 μm) were prepared using a cryostat microtome and directly mounted onto glass slides. For comparisons, sections from sham-operated mice were prepared from tissue located directly below the craniectomy (e.g., same anatomical brain region). Sections were incubated at room temperature with 5% normal donkey serum in PBS containing 0.4% Triton X-100 for 1 hour, followed by incubation with anti-pimonidazole mouse IgG monoclonal antibody (1:200) or rat anti-mouse neutrophil 7/4 (1:100) overnight at 4°C. Sections were then washed and incubated with the appropriate Alexa Fluor–tagged secondary antibodies. Omission of primary antibody served as a negative control. Slides were imaged on a Keyence BZ-X700 microscope.
Cytospin
Freshly harvested brain tissue was sieved through a 100-μm cell strainer, followed by centrifugation (252g, 10 min) to prepare single-cell suspensions. Neutrophils (CD11b+, Ly6G+, and Gr-1+) were sorted from single-cell suspension by magnetic bead isolation (Miltenyi Biotec), as previously described (38). Neutrophils were suspended in PBS at a concentration of 20,000 cells/300 μl, loaded with the aid of a cytology funnel, and centrifuged at 55g for 5 min using a Shandon Cytospin 4 Cytocentrifuge to adhere cells onto glass microscope slides. Slides were dried and fixed with 4% paraformaldehyde for 5 min. Last, slides were dried and kept at 4°C until used for immunostaining with the following: anti-NE antibody (NovusBio USA, catalog no. 950334), anti–Cit-H3 antibody (NovusBio USA, catalog no. NB2-36468PE), anti-MPO antibody (Abcam, catalog no. ab90812), or 4′,6-diamidine-2′-phenylindole dihydrochloride (Sigma-Aldrich, catalog no. 10236276001). Immunofluorescence was determined using a Zeiss LSM510 Meta confocal laser microscope. Cellular colocalization was determined in Z-stack mode using 63× oil immersion Neofluor objective (numerical aperture, 1.3) with the image size set at 512 × 512 pixels. Z-stacks (20 optical slices) were collected at optimal pinhole diameter at 12-bit pixel depth and converted into three-dimensional projection images using LSM510 Meta imaging software.
Neurobehavioral outcomes
Rotarod
Motor coordination was assessed using an accelerating rotarod task. Briefly, mice were trained for three consecutive days before injury and reassessed at day 3 after TBI. Mice were placed on an accelerating rotarod cylinder that was slowly increased from 4 to 40 rpm over a 5-min trial. The length of time balance was maintained while walking on top of the drum was recorded. A trial ended once the mouse fell off or gripped the cylinder for two consecutive revolutions. Motor coordination was further determined by measuring the time required to traverse a stationary 1-m narrow beam (6-mm width). Each mouse was tested three times. The average time to traverse the beam and number of paw slips were recorded.
Hanging wire test
Grip strength was assessed by placing mice on an apparatus consisting of a 50-cm string pulled between two vertical supports. Mice were evaluated as follows: 0, falls off; 1, hangs onto string by two forepaws; 2, same as for 1 but attempts to climb on string; 3, hangs onto string by two forepaws plus one or both hind limbs; 4, hangs onto string by forepaws with tail wrapped around string; and 5, escapes. The average reading of three successive trials was taken for each animal.
Open-field test
Mice were tested in a square box (40 cm by 40 cm by 40 cm) for 10 min, and activity was digitally recorded. Distance traveled, mean velocity, and time spent in the center zone were analyzed with Ethovision XT video-tracking software (Noldus Information Technology, Asheville, NC).
NOR test
Cognitive function was determined using a two-object novel object recognition (NOR) task, which assesses recognition memory. Mice were placed in an enclosed box with two identical objects that were placed within a 10 cm circle, located a set distance apart. Mice were then removed from the environment for a set amount of time, and one of the two previously used (familiar) objects was replaced with a novel object that was different from the familiar object in shape, texture, and appearance. The ability of the mouse to discriminate between the familiar and novel object was quantified as a discrimination index, DI = (Tn − Tf)/(Tn + Tf), where Tn is the time spent by the mouse with the novel object, and Tf indicates the time spent with the familiar object. All behavioral analyses were done by blinded investigators.
Statistical analysis
All data were analyzed using GraphPad Prism 8 software. Multigroup comparisons were made using a one-way analysis of variance (ANOVA), with adjustments for multiple comparisons. Data were further analyzed by Tukey’s post hoc test. Two group comparisons were analyzed by Student’s t test. Associations between serum DNase, ICP, and GCS score were evaluated using Pearson correlation coefficients. Results are expressed as mean ± SEM. A P < 0.05 was considered to be statistically significant.
Supplementary Material
Acknowledgments
We thank L. Perry and B. Marshall for technical assistance with electron microscopy and J. Pihkala for assistance with flow cytometry. Funding: Financial support for this project was provided by grants from the National Institutes of Neurological Diseases and Stroke (NS065172 and NS097825 to K.M.D. and NS110378 to K.M.D., B.B., and A.S.A.), the National Institutes of Child Health and Development (HD094606 to K.V.), and the American Heart Association (GRNT33700286 to K.M.D.). Author contributions: K.V., M.B., N.H., B.B., and K.M.D. conceived the project, designed all studies, and drafted the manuscript. K.V., M.B., M.H.R., and A.S.A. performed imaging studies and associated data analysis. M.B., K.V., M.B.K., N.H., M.F.W., and D.C.H. performed CBF measurements and analysis. A.W., C.B., T.S., and J.R.V. provided patient specimens. A.M., H.K., and B.B. performed flow cytometry studies. K.V., M.B., K.A., A.K., and A.W. performed behavioral and biochemical assessments. K.M.D. oversaw the research team and ensured compliance with all regulatory units. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/22/eaax8847/DC1
REFERENCES AND NOTES
- 1.Kulik T., Kusano Y., Aronhime S., Sandler A. L., Winn H. R., Regulation of cerebral vasculature in normal and ischemic brain. Neuropharmacology 55, 281–288 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kawamata T., Katayama Y., Surgical management of early massive edema caused by cerebral contusion in head trauma patients. Acta Neurochir. Suppl. 96, 3–6 (2006). [DOI] [PubMed] [Google Scholar]
- 3.King M. D., Laird M. D., Ramesh S. S., Youssef P., Shakir B., Vender J. R., Alleyne C. H., Dhandapani K. M., Elucidating novel mechanisms of brain injury following subarachnoid hemorrhage: An emerging role for neuroproteomics. Neurosurg. Focus. 28, E10 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Laird M. D., Shields J. S., Sukumari-Ramesh S., Kimbler D. E., Fessler R. D., Shakir B., Youssef P., Yanasak N., Vender J. R., Dhandapani K. M., High mobility group box protein-1 promotes cerebral edema after traumatic brain injury via activation of Toll-like receptor 4. Glia 62, 26–38 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang K. Y., Yu G. F., Zhang Z. Y., Huang Q., Dong X. Q., Plasma high-mobility group box 1 levels and prediction of outcome in patients with traumatic brain injury. Clin. Chim. Acta 413, 1737–1741 (2012). [DOI] [PubMed] [Google Scholar]
- 6.Zhang D., Li H., Li T., Zhou M., Hao S., Yan H., Yu Z., Li W., Li K., Hang C., TLR4 inhibitor resatorvid provides neuroprotection in experimental traumatic brain injury: Implication in the treatment of human brain injury. Neurochem. Int. 75, 11–18 (2014). [DOI] [PubMed] [Google Scholar]
- 7.Hallenbeck J. M., Dutka A. J., Tanishima T., Kochanek P. M., Kumaroo K. K., Thompson C. B., Obrenovitch T. P., Contreras T. J., Polymorphonuclear leukocyte accumulation in brain regions with low blood flow during the early postischemic period. Stroke 17, 246–253 (1986). [DOI] [PubMed] [Google Scholar]
- 8.Clark R. S., Schiding J. K., Kaczorowski S. L., Marion D. W., Kochanek P. M., Neutrophil accumulation after traumatic brain injury in rats: Comparison of weight drop and controlled cortical impact models. J. Neurotrauma 11, 499–506 (1994). [DOI] [PubMed] [Google Scholar]
- 9.Liao Y., Liu P., Guo F., Zhang Z. Y., Zhang Z., Oxidative burst of circulating neutrophils following traumatic brain injury in human. PLOS ONE 8, e68963 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kenne E., Erlandsson A., Lindbom L., Hillered L., Clausen F., Neutrophil depletion reduces edema formation and tissue loss following traumatic brain injury in mice. J. Neuroinflammation 9, 17 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Soares H. D., Hicks R. R., Smith D., McIntosh T. K., Inflammatory leukocytic recruitment and diffuse neuronal degeneration are separate pathological processes resulting from traumatic brain injury. J. Neurosci. 15, 8223–8233 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Frenette A. J., Perreault M. M., Lam S., Williamson D. R., Thiopental-induced neutropenia in two patients with severe head trauma. Pharmacotherapy 27, 464–471 (2007). [DOI] [PubMed] [Google Scholar]
- 13.Martinod K., Demers M., Fuchs T. A., Wong S. L., Brill A., Gallant M., Hu J., Wang Y., Wagner D. D., Neutrophil histone modification by peptidylarginine deiminase 4 is critical for deep vein thrombosis in mice. Proc. Natl. Acad. Sci. U.S.A. 110, 8674–8679 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mastronardi F. G., Wood D. D., Mei J., Raijmakers R., Tseveleki V., Dosch H. M., Probert L., Casaccia-Bonnefil P., Moscarello M. A., Increased citrullination of histone H3 in multiple sclerosis brain and animal models of demyelination: A role for tumor necrosis factor-induced peptidylarginine deiminase 4 translocation. J. Neurosci. 26, 11387–11396 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Neeli I., Khan S. N., Radic M., Histone deimination as a response to inflammatory stimuli in neutrophils. J. Immunol. 180, 1895–1902 (2008). [DOI] [PubMed] [Google Scholar]
- 16.Martinod K., Fuchs T. A., Zitomersky N. L., Wong S. L., Demers M., Gallant M., Wang Y., Wagner D. D., PAD4-deficiency does not affect bacteremia in polymicrobial sepsis and ameliorates endotoxemic shock. Blood 125, 1948–1956 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.von Kockritz-Blickwede M., Chow O. A., Nizet V., Fetal calf serum contains heat-stable nucleases that degrade neutrophil extracellular traps. Blood 114, 5245–5246 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Uhl M. W., Biagas K. V., Grundl P. D., Barmada M. A., Schiding J. K., Nemoto E. M., Kochanek P. M., Effects of neutropenia on edema, histology, and cerebral blood flow after traumatic brain injury in rats. J. Neurotrauma 11, 303–315 (1994). [DOI] [PubMed] [Google Scholar]
- 19.Lundqvist E. H., Sjövall K., Eneroth P. H., Influences of diet and surgical trauma on serum alkaline DNase activity levels. Clin. Chim. Acta 205, 43–49 (1992). [DOI] [PubMed] [Google Scholar]
- 20.Jimenez-Alcazar M., Napirei M., Panda R., Köhler E. C., Kremer Hovinga J. A., Mannherz H. G., Peine S., Renné T., Lämmle B., Fuchs T. A., Impaired DNase1-mediated degradation of neutrophil extracellular traps is associated with acute thrombotic microangiopathies. J. Thromb. Haemost. 13, 732–742 (2015). [DOI] [PubMed] [Google Scholar]
- 21.Fuchs T. A., Brill A., Duerschmied D., Schatzberg D., Monestier M., Myers D. D. Jr., Wrobleski S. K., Wakefield T. W., Hartwig J. H., Wagner D. D., Extracellular DNA traps promote thrombosis. Proc. Natl. Acad. Sci. U.S.A. 107, 15880–15885 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jimenez-Alcazar M., Rangaswamy C., Panda R., Bitterling J., Simsek Y. J., Long A. T., Bilyy R., Krenn V., Renné C., Renné T., Kluge S., Panzer U., Mizuta R., Mannherz H. G., Kitamura D., Herrmann M., Napirei M., Fuchs T. A., Host DNases prevent vascular occlusion by neutrophil extracellular traps. Science 358, 1202–1206 (2017). [DOI] [PubMed] [Google Scholar]
- 23.Valles J., Lago A., Santos M. T., Latorre A. M., Tembl J. I., Salom J. B., Nieves C., Moscardó A., Neutrophil extracellular traps are increased in patients with acute ischemic stroke: Prognostic significance. Thromb. Haemost. 117, 1919–1929 (2017). [DOI] [PubMed] [Google Scholar]
- 24.Schwarzmaier S. M., Kim S. W., Trabold R., Plesnila N., Temporal profile of thrombogenesis in the cerebral microcirculation after traumatic brain injury in mice. J. Neurotrauma 27, 121–130 (2010). [DOI] [PubMed] [Google Scholar]
- 25.Niesen W. D., Rosenkranz M., Schummer W., Weiller C., Sliwka U., Cerebral venous flow velocity predicts poor outcome in subarachnoid hemorrhage. Stroke 35, 1873–1878 (2004). [DOI] [PubMed] [Google Scholar]
- 26.Carlos T. M., Clark R. S., Franicola-Higgins D., Schiding J. K., Kochanek P. M., Expression of endothelial adhesion molecules and recruitment of neutrophils after traumatic brain injury in rats. J. Leukoc. Biol. 61, 279–285 (1997). [DOI] [PubMed] [Google Scholar]
- 27.Ichikawa A., Kuba K., Morita M., Chida S., Tezuka H., Hara H., Sasaki T., Ohteki T., Ranieri V. M., dos Santos C. C., Kawaoka Y., Akira S., Luster A. D., Lu B., Penninger J. M., Uhlig S., Slutsky A. S., Imai Y., CXCL10-CXCR3 enhances the development of neutrophil-mediated fulminant lung injury of viral and nonviral origin. Am. J. Respir. Crit. Care Med. 187, 65–77 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kielian T., Barry B., Hickey W. F., CXC chemokine receptor-2 ligands are required for neutrophil-mediated host defense in experimental brain abscesses. J. Immunol. 166, 4634–4643 (2001). [DOI] [PubMed] [Google Scholar]
- 29.Marcos V., Zhou Z., Yildirim A. O., Bohla A., Hector A., Vitkov L., Wiedenbauer E. M., Krautgartner W. D., Stoiber W., Belohradsky B. H., Rieber N., Kormann M., Koller B., Roscher A., Roos D., Griese M., Eickelberg O., Döring G., Mall M. A., Hartl D., CXCR2 mediates NADPH oxidase-independent neutrophil extracellular trap formation in cystic fibrosis airway inflammation. Nat. Med. 16, 1018–1023 (2010). [DOI] [PubMed] [Google Scholar]
- 30.Tadie J. M., Bae H. B., Jiang S., Park D. W., Bell C. P., Yang H., Pittet J. F., Tracey K., Thannickal V. J., Abraham E., Zmijewski J. W., HMGB1 promotes neutrophil extracellular trap formation through interactions with Toll-like receptor 4. Am. J. Physiol. Lung Cell. Mol. Physiol. 304, L342–L349 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stark K., Philippi V., Stockhausen S., Busse J., Antonelli A., Miller M., Schubert I., Hoseinpour P., Chandraratne S., von Brühl M. L., Gaertner F., Lorenz M., Agresti A., Coletti R., Antoine D. J., Heermann R., Jung K., Reese S., Laitinen I., Schwaiger M., Walch A., Sperandio M., Nawroth P. P., Reinhardt C., Jäckel S., Bianchi M. E., Massberg S., Disulfide HMGB1 derived from platelets coordinates venous thrombosis in mice. Blood 128, 2435–2449 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li P., Li M., Lindberg M. R., Kennett M. J., Xiong N., Wang Y., PAD4 is essential for antibacterial innate immunity mediated by neutrophil extracellular traps. J. Exp. Med. 207, 1853–1862 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang Y., Li M., Stadler S., Correll S., Li P., Wang D., Hayama R., Leonelli L., Han H., Grigoryev S. A., Allis C. D., Coonrod S. A., Histone hypercitrullination mediates chromatin decondensation and neutrophil extracellular trap formation. J. Cell Biol. 184, 205–213 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Walko T. D. III, Bola R. A., Hong J. D., Au A. K., Bell M. J., Kochanek P. M., Clark R. S., Aneja R. K., Cerebrospinal fluid mitochondrial DNA: A novel DAMP in pediatric traumatic brain injury. Shock 41, 499–503 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang Q., Raoof M., Chen Y., Sumi Y., Sursal T., Junger W., Brohi K., Itagaki K., Hauser C. J., Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature 464, 104–107 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hua F., Wang J., Ishrat T., Wei W., Atif F., Sayeed I., Stein D. G., Genomic profile of Toll-like receptor pathways in traumatically brain-injured mice: Effect of exogenous progesterone. J. Neuroinflammation 8, 42 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hoda M. N., Siddiqui S., Herberg S., Periyasamy-Thandavan S., Bhatia K., Hafez S. S., Johnson M. H., Hill W. D., Ergul A., Fagan S. C., Hess D. C., Remote ischemic perconditioning is effective alone and in combination with intravenous tissue-type plasminogen activator in murine model of embolic stroke. Stroke 43, 2794–2799 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Braun M., Vaibhav K., Saad N., Fatima S., Brann D. W., Vender J. R., Wang L. P., Hoda M. N., Baban B., Dhandapani K. M., Activation of myeloid TLR4 mediates T lymphocyte polarization after traumatic brain injury. J. Immunol. 198, 3615–3626 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Caudrillier A., Kessenbrock K., Gilliss B. M., Nguyen J. X., Marques M. B., Monestier M., Toy P., Werb Z., Looney M. R., Platelets induce neutrophil extracellular traps in transfusion-related acute lung injury. J. Clin. Invest. 122, 2661–2671 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kessenbrock K., Krumbholz M., Schönermarck U., Back W., Gross W. L., Werb Z., Gröne H. J., Brinkmann V., Jenne D. E., Netting neutrophils in autoimmune small-vessel vasculitis. Nat. Med. 15, 623–625 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/22/eaax8847/DC1