

Massive single-colony sequencing revealed that mutation rates varied between different bacilli from the same patient.
Abstract
Mycobacterium tuberculosis (Mtb) infection results in a spectrum of clinical and histopathologic manifestations. It has been proposed that the environmental and immune pressures associated with different contexts of infection have different consequences for the associated bacterial populations, affecting drug susceptibility and the emergence of resistance. However, there is little concrete evidence for this model. We prospectively collected sputum samples from 18 newly diagnosed and treatment-naïve patients with tuberculosis and sequenced 795 colony-derived Mtb isolates. Mutant accumulation rates varied considerably between different bacilli isolated from the same individual, and where high rates of mutation were observed, the mutational spectrum was consistent with reactive oxygen species–induced mutagenesis. Elevated bacterial mutation rates were identified in isolates from HIV-negative but not HIV-positive individuals, suggesting that they were immune-driven. These results support the model that mutagenesis of Mtb in vivo is modulated by the host environment, which could drive the emergence of variants associated with drug resistance in a host-dependent manner.
INTRODUCTION
Infection with Mycobacterium tuberculosis (Mtb) causes a spectrum of clinical outcomes, from latent infection to active disease. During latent infection, Mtb can survive in the infected host for decades, creating a reservoir that continuously fuels the global tuberculosis (TB) epidemic (1). Control of the TB epidemic has been complicated by the emergence of high-level antibiotic resistance, which occurs through de novo chromosomal mutations (2). We understand little about the rates and drivers of mutation in Mtb within the infected host where the bacterial population is thought to face a range of different environments and immunologic stresses in different disease states, as reflected by different histopathologic manifestations of infection, including granuloma formation and less-organized areas of inflammation. It is postulated that host immune stressors drive the bacterial population toward a nonreplicative state. Thus, replication-associated mutations are predicted to accumulate at very low rates (3–5). However, a study in the macaque TB model found that Mtb accumulated mutations at roughly the same rate during both active disease and early latent infection as during rapid growth in vitro (5). This mutant accumulation rate is remarkably similar to the molecular clock estimated from the study of human Mtb isolates, suggesting that there are additional drivers of mutation in vivo (6).
Whole-genome sequencing has been used to track the evolution of Mtb bacilli within the host during antibiotic treatment and demonstrate how drug-resistant mutations arise and become fixed in the population (7–10). When a patient develops active TB, they are estimated to harbor 1010 to 1012 bacilli (11), and such a large population should contain numerous genetic mutations that could be used to study the mutagenesis of Mtb in vivo (8, 10, 12). Because antibiotic treatment can reduce Mtb population and diversity very quickly, such mutational records should be preserved only in treatment-naïve patients. By applying whole-genome sequencing to large numbers of individual colonies recovered from 18 patients, we characterized the genetic diversity of Mtb populations at the onset of TB disease and reconstructed the within-host evolution of Mtb. This approach provided sufficient resolution to characterize the mutational signature of Mtb at the individual colony level, which should represent single bacilli in vivo. Our data indicate that mutagenesis of Mtb in vivo is modulated by the host environment, suggesting that the risk of de novo drug resistance varies in a host-dependent fashion.
RESULTS
Sampling Mtb population before antibiotics treatment
Between 1 February 2017 and 31 December 2018, we recruited 18 new, smear-positive patients with TB who had not taken any anti-TB drugs before the diagnosis. All of the isolates were pan-susceptible except for one isolate that was resistant only to isoniazid (table S1). We collected three to five sputum samples from the new patients with TB on the day of diagnosis, which allowed us to deeply sample the Mtb populations in their pulmonary lesions that communicated with the airways. The sampling size of the Mtb population was estimated to be between 25,000 and 150,000 bacilli per patient, according to the conversion index between smear microscopic scores and bacterial load (13, 14). The sputum sample from each patient was digested, dispersed, serially diluted, and then cultured on Löwenstein-Jensen (L-J) solid medium (Fig. 1A). The remainder of the undiluted sputum was cultured separately to represent the whole population of bacilli present. For each patient, we randomly picked ~50 well-separated colonies for whole-genome sequencing (Fig. 1A). In addition, we scraped all the colonies from the whole population plates of nine patients for deep whole-genome sequencing (Fig. 1A).
Fig. 1. Schematic diagram of the sampling approach and genetic diversity of the Mtb population within the host.
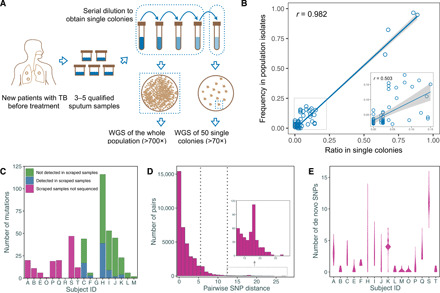
(A) We repeatedly collected three to five sputum samples from new and treatment-naïve patients with TB. All sputum samples from a single individual were mixed together and processed with a standard procedure and then serially diluted. The target dilutions were spread onto five plates, while the remaining undiluted samples were mixed together and spread onto two plates. WGS, whole-genome sequencing. (B) The correlation between the frequencies of single-nucleotide polymorphisms (SNPs) in scraped samples and the relative single-colony samples was tested by Pearson’s correlation coefficient (r); the small inset shows an enlargement of the dashed box on the left. (C) A bar plot showing the numbers of de novo SNPs that were detected in colony samples in different patients with the number of SNPs that were not detected in the corresponding scraped whole population samples highlighted. (D) A histogram showing the distribution of pairwise SNP distance between any two single colonies from the same patient with the two dashed lines indicating the two commonly used SNP thresholds for defining transmission clusters. (E) A violin plot showing the distribution of numbers of de novo SNPs in single colonies from each patient.
Sequencing single colonies can delineate the Mtb population structure
In total, we obtained whole-genome sequence data for 795 single colonies from 18 patients (average sequencing depth is 119.4) and nine scraped whole populations (average sequencing depth is 726.3). Sequencing reads were aligned to the reconstructed ancestral genome of the Mtb complex to detect single-nucleotide polymorphisms (SNPs) (15). The SNPs shared by all colonies from a given patient were defined as “inherited SNPs,” indicating phylogenetic SNPs that were fixed before the infection. The study then focused on the SNPs that were present in only a proportion of colonies that were termed “de novo SNPs” to indicate their de novo accumulation during infection. To verify that sequencing of ~50 colonies would represent the Mtb population structure in the original samples, we compared the frequency of SNPs detected in the deep-sequenced scraped whole population samples with their ratio in the single colonies. We detected 107 SNPs with a frequency above 1.5% in the nine scraped samples, of which 67 (62.6%) could be detected with a similar frequency in the corresponding single colonies (Pearson’s correlation coefficient: 0.982; Fig. 1B and fig. S1). The SNPs with higher frequencies showed better correlation, while the SNPs with frequencies between 1.5 and 15% presented considerable variation between the scraped population samples and the individual colonies (Pearson’s correlation coefficient: 0.503; Fig. 1B). In addition, we detected 224 SNPs that were present in only one or two single colonies but were not detected in the scraped samples, indicating the increased sensitivity of single-colony sequencing for capturing low-frequency mutations in the population.
Mtb diversity at the onset of TB disease
There was a broad range in the total number of de novo mutations found in the bacterial populations isolated from each patient (0 to 116 SNPs; Fig. 1C), with the average number of de novo mutations varying from 0 to 11.3 SNPs (fig. S2A). However, the SNP distance between any two colonies from the same patient was much lower than between any two strains from different patients (fig. S2, B and C). Mapping of both the fixed and unfixed SNPs from each patient to our phylogenomic database for the identification of multiple evolutionary paths (16) did not suggest any mixed infections in our samples. Of the 18 Mtb strains, 17 were lineage 2 strains and one belonged to lineage 4 (table S1).
Because the SNP distance between different bacilli within a single individual is an important reference for establishing the SNP threshold for epidemiologically defining transmission clusters (17), we calculated the pairwise SNP distance between any two single colonies from each patient (Fig. 1D). For 91.0% of the pairs, the difference was ≤5 SNPs, and for 98.8% of the pairs, the difference was ≤12 SNPs, indicating that these two widely used thresholds (5 or 12 SNPs) encompass most of the genetic distance between different bacilli within a patient. However, in 7 of 18 patients, there were colony pairs that differed by more than 5 SNPs and 3 of 18 patients had colony pairs that differed by more than 12 SNPs, indicating that these larger SNP distances can occur within an individual and thus could occasionally be found between isolates from cases linked by direct transmission.
Two patterns of Mtb population growth
We next used the minimum evolution method to reconstruct phylogenetic trees for the single colonies from each patient, setting the inferred ancestral genome as the root (Fig. 2). We found that the in vivo populations of Mtb separated into two different patterns: “starlike expansion” and “stepwise growth”. Ten patients were characterized by the starlike expansion model (Fig. 2A). For these cases, only a few colonies showed de novo SNPs, while the majority maintained the ancestral genome (Fig. 2A). This pattern suggests a bacterial population derived from a recent expansion, with a starting population that was genetically homogeneous. This observation was consistent with a model in which a small number of Mtb bacilli established the infection, proliferated, and caused TB disease relatively rapidly (18).
Fig. 2. Phylogenetic trees of Mtb populations from different patients.
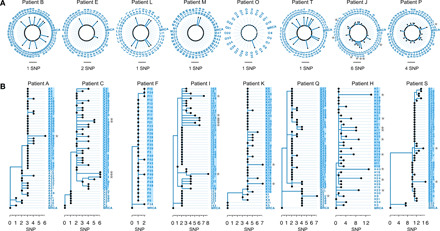
All trees are rooted to the inferred ancestral genome and all the “inherited SNPs” were excluded before the phylogenetic reconstruction. The length of solid lines represents the number of de novo SNPs. (A) “Starlike expansion” trees for these patients are shown in a circle format. Trees for patients G and R were not shown because no de novo SNPs were detected. (B) “Stepwise growth” trees for these patients are shown in rectangular format. Gray stars indicate those colonies with excessive de novo SNPs and the taxa names with blue backgrounds highlight the recently expanded populations.
By contrast, eight patients presented a pattern of stepwise growth, typically suggesting two or three stages (Fig. 2B): (i) growth after the initial infection with divergence into subpopulations containing different SNP markers, (ii) a limited number of subpopulations achieving a second wave of growth with further genetic differentiation of the within-host Mtb population, and (iii) recent expansion of one or a very few of the subpopulations. It appeared that 68.7 to 96.0% of the colonies were the result of the expansion at the third stage. These inferences rely on the assumption that the initial infection was established by a single or a few genetically homogeneous bacilli of Mtb, as appeared to be typical in the cases with starlike expansions. However, the possibility of initial infections with several different, closely related bacilli cannot be conclusively ruled out. In either scenario, the patterns suggested that a large proportion of the Mtb population at the time of clinically detectable TB disease derived from a recently expanded subpopulation.
Mutation rate varies between bacilli within a single individual
There was a variation in the number of de novo SNPs that accumulated in different colonies from the same patient (Fig. 1E). For example, for patients J and P in the starlike group, and patients A, C, I, K, Q, H, and S in the stepwise group, a few colonies (marked by gray stars) accumulated 4 to 14 de novo SNPs, while most colonies maintained the ancestral genotypes (Fig. 2). Because the in vivo mutation rate of Mtb is approximately 0.24 to 0.5 SNPs per genome per year (17, 19–21), this disparity suggested that the mutation rate in vivo might not be uniform. To test this, we simulated Mtb population growth in silico using a growth model with a constant mutation rate (Wright-Fisher model) (22). The simulated data showed that the distribution of numbers of de novo SNPs appeared as a very convergent Poisson distribution with only one peak (Fig. 3). In contrast, the observed data for patients H, I, J, and S showed multiple peaks in the distribution of the number of de novo SNPs. A comparison of the 0.9 quantiles in the observed and simulated data shows that H, I, J, and S had several colonies with more de novo SNPs than were predicted (Fig. 3), suggesting a deviation from a constant mutation rate model. An alternative explanation might be positive selection that preferentially selected beneficial mutations. To test this possibility, we compared the proportion of nonsynonymous to synonymous mutations (pNS, similar to dN/dS; see details in Materials and Methods) in the de novo SNPs from the four patients whose colonies had the most SNPs (H, I, J, and S) against those of the other patients. We found that the average pNS value for the four patients with high SNP colonies was 0.64 (H, 0.67; I, 0.56; J, 0.83; S, 0.46), while it was 0.79 for the other patients. This indicates a purifying selection in all patients and argues against positive selection as an explanation for the high SNP colonies.
Fig. 3. Comparison of the distribution of de novo SNP numbers between simulated and observed populations.
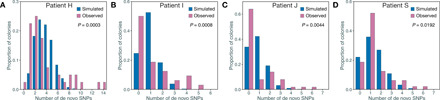
(A to D) The comparisons of the Mtb populations from patients H, I, J, and S, respectively. The height of histograms shows the proportions of colonies with the relative number of de novo SNPs. The P values indicate the hypergeometric test for 0.9-quantile SNP numbers for the simulated and observed populations.
Enhanced mutation rate likely induced by reactive oxygen species
The four patients, H, I, J, and S, whose colonies suggested non-constant mutation rates also had larger numbers of total de novo SNPs (Fig. 1C), and the majority of the SNPs were C>T and G>A, the base changes associated with oxidative damage (Fig. 4A) (23). Although oxidative damage mutations are the major source of genetic changes during in vivo growth (6, 23), the ratio of these mutations in patients H, I, J, and S was significantly higher than the average level for all patients (80.6% versus 67.1%, P < 0.0001) and even higher than that of the fixed SNPs in a collection of 8399 genomes from global isolates (80.6% versus 47.2%, P < 0.0001; fig. S3). The most notable case was patient H, where 101 of the 116 (87.1%) total de novo SNPs were due to the base changes associated with oxidative damage. We further created histograms of mutation-type compositions for each colony and mapped them to the phylogenetic tree for each patient (Fig. 4B and fig. S4). For the 40 colonies from patient H, 27 had only 0 to 3 de novo SNPs, while 13 colonies had 4 to 14 de novo SNPs. The extra SNPs in these 13 colonies were exclusively mutations associated with oxidative damage—C-T and double CC-TT mutations (Fig. 4B)—which are thought to indicate exposure to reactive oxygen species (ROS) (24, 25). We also observed a similar pattern with I, J, and S (Fig. 4B). Moreover, the colonies with longer mutation branches from patients A, K, Q, and P can also be explained by C-T and CC-TT mutations (fig. S4).
Fig. 4. Oxidative damage mutations are the source of excessive de novo SNPs.
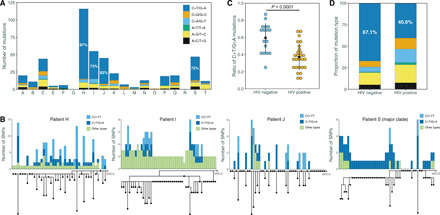
(A) The proportions of different mutation types in the samples from patients H, I, J, and S showed a deviation from the average level. (B) Integration of phylogenetic tree and mutation composition histograms for patients H, I, J, and S. For patient S, only the major clade (the 48 colonies) is shown here. Different colors refer to different mutation types. The gray dot “MRCA” represents the reconstructed ancestor of each Mtb population. For patients H, J, and S, the MRCA strains were detected in the single colonies. (C) A comparison of C-T mutation ratio between samples from HIV-negative and HIV-positive hosts (P < 0.0001 by t test), with each dot representing one patient. (D) The bar plot shows the ratio of C-T mutations in all samples from HIV-negative and HIV-positive hosts, respectively.
The elevated mutation rate was host dependent
This elevated mutation rate could be explained either by an increased susceptibility of these particular Mtb strains to oxidative damage or by increased bacterial exposure to oxidative stress in a subset of the patients. If some of the strains had increased ROS susceptibility, all of the colonies derived from these Mtb strains should exhibit similar patterns of mutations and comparable numbers of SNPs, but this was not what was observed; different colonies from the same strain showed a wide variation in de novo SNPs (Fig. 4B). In addition, strains that gave rise to colonies with higher mutation rates should have more C-T and CC-TT inherited mutations than the other strains, but there was no difference in the ratio of C-T and CC-TT between the two groups for the inherited mutations (fig. S5). Last, the four strains whose colonies had more mutations (H, I, J, and S) were not clustered in the phylogenetic tree, showing that the increased mutation rate was not a feature of a particular phylogenetic clade (fig. S6); besides, we found no homoplastic mutation between any two of these four strains. Together, these results suggest that increased bacterial susceptibility to ROS seemed to be not a plausible explanation for the higher mutation rates in a subset of colonies.
As the production and regulation of ROS and reactive nitrogen species are important components of the host immune response to pathogen infections, we hypothesized that the variation in the bacterial mutation rate could be host dependent. We compared the ratio of C-T mutations in Mtb samples collected from HIV-negative hosts (all 18 patients) in this study with the ratio from HIV-positive hosts in a previous study of 2587 Mtb single colonies from 42 HIV-positive hosts (12). This comparison showed that Mtb strains from HIV-negative hosts accumulated more C-T mutations than those from HIV-positive hosts (P < 0.0001, Fig. 4, C and D), and the difference remained when we restricted this comparison to lineage 2 strains from the two groups (P = 0.0005; fig. S7). Furthermore, the double CC-TT mutations were not observed in the Mtb colonies from HIV-positive hosts, and in these hosts, the phylogenetic trees of the de novo SNPs did not show colonies with “long branches” (fig. S8). These analyses thus suggest that the signature of ROS-associated mutations could be driven by immune pressure.
DISCUSSION
This work analyzed the population structure and mutation signatures of Mtb by genome sequencing colonies isolated directly from clinical specimens, thereby allowing us to make inferences about the growth and evolution of the bacterial population during infection. The data present at least four clear findings: (i) distinct levels of bacterial diversity were observed in the Mtb populations from different patients; (ii) the pairwise SNP distance between any two colonies within a host could occasionally exceed the SNP thresholds used to define a transmission cluster; (iii) the number of de novo SNPs varied significantly between different bacilli within the same host, suggesting that the mutation rate varies between different bacteria within a host; and (iv) the elevated mutation rate in some colonies carries the signature of ROS-induced mutagenesis.
The estimated mutation rate of Mtb, about 0.5 mutations in every 10,000 genomes copied (6), is low compared to most other bacteria, yet the high rates of acquired resistance in clinical strains raise the question of whether the mutation rate within the host could somehow be higher (4). The rate calculated from an in vivo macaque infection model was very similar to that inferred in vitro through fluctuation experiments, despite the differences in bacterial replication rates in vitro and in vivo (3, 5), and deep whole-genome sequencing of serial sputum samples from patients with TB taken during treatment revealed a high degree of genetic diversity with tens of unfixed SNPs (8, 26). In the current study, we found an increased rate of ROS-associated mutations that could perhaps explain the high rate of acquired resistance during clinical treatment. Those patients in whom Mtb is subjected to high levels of ROS mutagenesis should carry larger pools of mutations and, thus, perhaps have a higher risk for developing drug resistance. Further studies are warranted to understand why some patients induce higher levels of ROS mutagenesis, how to identify them, and how to reduce their risk of developing of drug resistance.
Unexpectedly, only 4 of the 18 patients had bacterial populations wset of the colonies from each of these four patients. Because the excess mutations were predominantly the C-T changes, the results suggest that ROS stress can vary between hosts and also that ROS heterogeneity exists in different microenvironments within the same host. This model is consistent with the growing knowledge of granuloma heterogeneity, where each granuloma represents a microenvironment that can be independently influenced by the local immune response (27, 28). This inference was further supported by the lower ratio of C-T mutations in strains of HIV-positive patients, implying that the host immune status plays a role in the frequency of oxidative damage mutations.
Because unfixed SNPs were detected in 39% of the single colonies, we scanned them for CC-TT mutations and, as expected, found them in the genomes of colonies isolated from the patients with an elevated mutation rate: H (9), I (11), J (14), and S (3). Unexpectedly, we also found CC-TT mutations in colonies from patients without elevated mutation rates: 26 in colonies from E, 2 from F, and 3 from O (fig. S9A). This suggests that the rates of ROS-induced mutations could have been underestimated in our study because of the limitations of our sampling size and a mixture of different colonies.
We considered positive selection as an alternative explanation for the high numbers of SNPs found in some colonies but discarded this explanation for the following reasons. First, pNS analysis suggested that a negative selection was operating on the bacterial population of strains H, I, J, and S, which was consistent with previous findings that within-host selective pressure on Mtb is governed by negative selection (8, 29, 30). Second, in the colonies with large numbers of SNPs, the mutation type of excessive SNPs was almost exclusively C-T or CC-TT. It seems unlikely that a particular mutation type would be favored by positive selection, which should be based only on the functional impact of the mutations on the genes without a preference for a specific type of mutation. Third, if a positive selection was operating, we should expect to see some homoplasy or gene enrichment in the different colonies with large numbers of SNPs isolated from a single patient. However, we found neither homoplastic mutations nor gene enrichment across the colonies from each of the four strains that had high SNP colonies.
Our analysis suggested that increased bacterial susceptibility to ROS might not be a plausible explanation for the higher mutation rates in a subset of colonies, but we cannot completely rule out its role in the phenomenon we observed. Heterogeneity in the strains’ susceptibility to ROS could act together with host environmental stress and perhaps help explain why colonies with high numbers of SNPs were isolated only from some patients. Although we found no homoplastic mutations between any two of these four strains (H, I, J, and S) or mutations on the same gene but in different codons, it is still possible that mutations in different genes could have similar functional effects in increasing the susceptibility to ROS stress. The absence of C-T or CC-TT mutational signature in the inherited mutations was a strong argument, but it cannot exclude the possibility that these four strains only acquired an increased “ROS susceptibility” very recently, for which they would not have accumulated many C-T or CC-TT in the inherited mutations.
The biological process of Mtb infection is difficult to study in humans, but PET-CT (positron emission tomography–computed tomography) tracking of the dynamic course of infection in cynomolgus macaques revealed that different lesions in the same patient followed diverse trajectories. While some granulomas were sterilized in both active and latent cases, others grew and ultimately determined the clinical outcome of infection (27). Typically, only one or a few granulomas that are unable to contain the bacteria are probably responsible for bacterial dissemination and the onset of active disease (12, 27, 31). Our results are consistent with these observations. We found that the burden of TB disease in humans often appeared the result of the recent expansion of a subpopulation of the bacilli present in the individual. Together, these findings suggest a “turning point” theory for the progression of TB disease, in which the deterioration of a particular lesion leads to clinical disease. Hence, finding biomarkers that can predict such a transition or detect it early would allow preventive interventions that might prevent the development of clinical TB disease.
For those patients with more mutational diversity, the probability was higher that the genomes of any two selected colonies differed by more than 5 SNPs, and pairs that differed by more than 12 SNPs were not rare (fig. S9, B and C). This finding implies that the dominant subpopulation cultured and sequenced from one patient could differ by >12 SNPs from a variant that infects a second patient. In the second host, this transmitted bacillus could again mutate and generate a more distant variant that dominates when the second patient’s sputum is cultured and sequenced. The commonly used SNP thresholds of ≤5 SNP differences for direct transmission and ≤ 12 SNPs for transmission clustered strains have been supported by epidemiologic data (17, 32), but our results suggest that strains with slightly more SNP differences might occasionally belong to the same transmission chain and deserve to be included when conducting epidemiological investigations.
The extent to which the sputum bacillary population is representative of the total mycobacterial population within an individual is unknown. An ideal and complete characterization of the Mtb diversity within a host would require sampling from each lesion, which is not feasible in patients with TB. The bacilli in sputum mainly reflect Mtb population in lesions that are open and connected to the airway, so bacilli in lesions that were not open or connected to airways may not have been sampled and therefore not represented in our data. However, expectorated tubercle bacilli are thought to originate from sites of extensive bacterial growth that are the major contributors to active disease (33, 34), so we assume that we sampled at least some of the most clinically relevant sites. In addition, different sputum samples collected on the same day could vary in the Mtb growth sites from which they derive, so to avoid bias, we repeatedly sampled from the same patient and pooled the specimens.
In conclusion, this study provides a baseline characterization of Mtb population diversity within the host during active TB disease, and the two patterns of population growth extend our understanding of Mtb proliferation in vivo. We show that the mutation rate of Mtb in vivo appears to vary, presumably related to the host environment. The possibility that the mutation rate may increase under certain in vivo conditions could help to understand how drug resistance evolves.
MATERIALS AND METHODS
Study cohort
The patients were enrolled in the Shanghai Public Health Clinical Center (a designated hospital for TB), in Shanghai, China, and the study was approved by the ethics committee of this clinical center. The recruitment criteria for the patients with TB were as follows: (i) no previous history of TB disease, (ii) no previous anti-TB antibiotics, (iii) positive sputum smear for bacilli on microscopy with a score >1+, (iv) a qualified sputum volume at least 5 ml, and (v) drug-susceptible Mtb isolates. Initially, 20 patients were enrolled, but samples from two patients were excluded because one contained Mycobacterium kansasii and the other was contaminated with fungus, leaving 18 patient specimens for analysis (table S1). In the isolate from patient F, an inhA −15 C-T promoter mutation was found and drug sensitivity testing reported isoniazid resistance after whole-genome sequencing was performed. Because this patient had not previously taken any anti-TB drugs, the strain was included. The methods of this study were carried out in accordance with the approved guidelines, and written informed consent was obtained from the patients before the study.
Sample collection and processing
Patients suspected to have TB were first screened according to recruitment criteria 1 and 2. Routine smear microscopy was performed on the sputum samples, and the remaining sediment was temporarily stored at 4°C. Sputum quality was monitored and specimens with purulent sputum were preferentially frozen. The clinical center routinely performed GeneXpert MTB/RIF tests and culture-based drug susceptibility testing for isoniazid, rifampicin, ethambutol, and pyrazinamide. GeneXpert MTB/RIF results were reported on the same day as diagnosis. When a sputum sample had a microscopic score >1+ and was rifampicin sensitive by GeneXpert, a further three to five sputum samples were collected from these patients before the treatment was started. All sputum samples from each patient were combined to reach a total of at least 5 ml for each patient. The combined sputum samples were subjected to digestion with 1:1 of 2% NaOH-NaCl and left standing for 15 min. Phosphate-buffered saline (PBS; pH 6.8) was added to a final volume of 50 ml and the samples were then centrifuged at 3000g for 15 min. The supernatant was discarded and the sediment was suspended in 1 ml of PBS solution. We then performed serial dilutions for each of the samples, and the dilution index was estimated based on the sputum smear results. For each target dilution, eight L-J medium plates were spread to obtain an estimated 50 colonies on each plate. The remainder of the original, undiluted sputum specimens from each patient was centrifuged and spread onto two L-J medium plates. From most patient isolates, 50 well-separated colonies were selected from different plates, but only 10 colonies were obtained from sample G. Each selected colony was spread onto a fresh L-J plate for a short amplification culture of 1 to 2 weeks and then collected for DNA extraction. For nine patients, all of the colonies from the two plates spread with the undiluted sputa were scraped into a single tube for DNA extraction and designated scraped, whole population samples.
Illumina sequencing and SNP calling
Genomic DNA from both the single-colony isolates and scraped whole population samples was extracted with the cetyltrimethylammonium bromide lysozyme method (35). A 300–base pair fragment length library was constructed for each DNA sample and paired-end–sequenced on an Illumina HiSeq 2500 instrument. A previously validated pipeline was used for SNP calling (35). Fixed mutations with a frequency of ≥90% and at least 10 supporting reads were identified. Whole-genome sequence data of 8399 Mtb isolates from previous studies were downloaded and subjected to SNP calling to identify the ratio of C-T/G-A mutations in each isolate (35).
Filter for unfixed SNPs
A previously validated pipeline was used to filter out false positives and detect unfixed SNPs (8). We considered only unfixed SNPs whose frequencies were estimated to be ≥1.5% in whole population samples. Because the false positives shared similar patterns in the strains with close genetic backgrounds, we further used repetitive unfixed SNPs called from Shanghai Mtb isolates that had been previously sampled and sequenced as a background filter (32). Ideally, we should not find unfixed mutations in single-colony samples if each colony derived from a single bacillus (8). However, we detected ≥1 unfixed SNP with allele frequencies above 10% but below 90% in 39.0% (310 of 795) of the single-colony samples. Because these unfixed SNPs survived our filters for false positives, we further examined their prevalence. We found that some unfixed SNPs detected in one colony from a patient were fixed SNPs (≥90%) in other colonies from the same patient but absent in colonies from other patients, suggesting that these unfixed SNPs were present because the colony had grown from more than one original, isolated bacillus or that colonies from separate bacilli grew into one another. Given these possibilities, we included the unfixed SNPs with frequencies above 50%, which represent the major clones for the subsequent analysis.
Phylogenetic reconstruction
For phylogenetic reconstructions, all SNP locations for each isolate were combined into a nonredundant consensus list and recalled with the mpileup2cns function of VarScan (version 2.3.9) (36). Nucleotide positions with missing calls in more than 5% of the isolates were removed. An alignment of the remaining polymorphic positions from all strains was used for phylogeny reconstruction with MEGA 6.0 (37). To make the branch lengths represent the number of de novo SNPs, we used the minimum evolution method to infer the phylogenetic trees under the “No. of differences” model with both transitions and transversions included. The bootstrap method was used with 500 replications for each test. Phylogeny trees were visualized in FigTree (version 1.4.3) (http://tree.bio.ed.ac.uk/software/figtree/).
Proportion of nonsynonymous to synonymous mutations
We wished to determine whether the excessive mutations in some colonies of the four patients, H, I, J, and S, were due to positive selection, which would accelerate the mutation frequency and fixation in the population. We therefore used pNS to evaluate the selective pressure in these patients’ samples and compared the values to those of the remaining patients (8). The basic principle of the pNS method is similar to that of dN/dS, but dN/dS is used to compare two real sequences that exist in nature, while pNS can be applied to concatenated sequences of all the mutations in the genomes. A codon substitution matrix was generated using a base substitution model that takes into account the proportion of guanine and cytosine in the genome (percentage GC content, 0.656). Briefly, for each variant codon, we used a custom Python script to simulate 50,000 individual introductions of a single mutation into the codon and scored the outcomes as either synonymous or nonsynonymous. We considered the average number of nonsynonymous outcomes of the simulations as an estimate of the probability that a mutation in the given codon would be nonsynonymous. The formula used to calculate the pNS was described previously (8).
Simulating Mtb growth
We generated Mtb growth with continuously accumulating neutral mutations under a constant growth rate in silico, starting with a single bacillus cell and ending when the population size reached 108. The mutation rate that we chose here was 2.01 × 10−10 mutations per site per generation (6). Because our analysis excluded the mutations in PPE/PE-PGRS family genes and other mobile sequences, which accounts for 8.9% of the whole genome, the genome size was set as 4.41 × 106 × 91.1% = 4.02 × 106. Then, the mutation rate (μ) used was 0.0008 per genome per generation. Mtb bacilli in exponential growth are represented by
where Nt is the number of bacilli in generation t with Nt = 1 and a is a constant number. If the average number of de novo mutations in the TB lineage of a patient is k, the number of generations from the first bacilli cell to 108 is k/μ. Therefore, the parameter .
The Wright-Fisher model (22) was used to simulate discrete TB growth. In generation t, the expected number of bacilli is Nt = e0.0147t/k. The bacilli in generation t were randomly sampled as progenies of cells from generation t − 1. A newborn cell had a probability μ of accumulating a de novo mutation in each cell division. The workflow of the simulation is displayed in fig. S10.
Test the heavy-tailed distribution
To test whether the de novo mutation number distribution of an observed TB lineage is a heavy-tailed distribution, we sorted the mutation number of observed data and simulated data from lowest to highest separately and used a hypergeometric test to compare the 0.9-quantile mutation number of the observed data and simulated data.
Ethics statement
This study was approved by the Research Ethics Review Committee of the Shanghai Public Health Clinical Center, Fudan University.
Supplementary Material
Acknowledgments
We thank L.-D. Lyu (School of Basic Medical Sciences, Fudan University) and C. Yang (School of Public Health, Yale University) for the fruitful discussions. Funding: This work was supported by the National Natural Science Foundation of China (91631301 and 81661128043 to Q.G., 81701975 to Q.L., and 31771416 to X.L.), the National Science and Technology Major Project of China (2017ZX10201302 to Q.G. and 2018ZX10714002-001-005 to Z.Z.), the Sanming Project of Medicine in Shenzhen (SZSM201611030 to Q.G.), European Research Council 638553-TB-ACCELERATE (to I.C.), the Key Research Program of the Chinese Academy of Sciences (KFZD-SW-220-1 to X.L.), and the CAS Light of West China Program (to X.L.). Y.F. is supported in part by NIH R01HG009524. Support was also received from NIH awards P01 AI132130 and AI142793 to S.M.F.. Author contributions: Q.L. and Q.G. designed and implemented the study. Q.G. and S.M.F. supervised this work. Y.-X.F. provided important suggestions and consultants during the design of this study. M.W., Y.Lu, and F.L. recruited patients and collected sputum samples. J.W., J.S., X.Q., Z.Z., and H.W. performed the culture experiments to obtain single colonies. Q.L., M.G., and M.G.L. analyzed the sequencing reads and performed the genetic analysis. Y.Li and X.L. designed and conducted the mathematic simulation of population growth of Mtb. Q.J. performed statistical analysis. Q.L., I.C., H.E.T., S.M.F., and Q.G. drafted the manuscript. All authors critically reviewed and approved the final version of the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors. Sequencing reads have been submitted to the European Nucleotide Archive (EMBL-EBI) under study accession PRJEB34582 and PRJEB34609. The analysis scripts used in this study are available online at GitHub (https://github.com/StopTB/Single_Colony_Project).
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/22/eaba4901/DC1
REFERENCES AND NOTES
- 1.Gengenbacher M., Kaufmann S. H., Mycobacterium tuberculosis: Success through dormancy. FEMS Microbiol. Rev. 36, 514–532 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dookie N., Rambaran S., Padayatchi N., Mahomed S., Naidoo K., Evolution of drug resistance in Mycobacterium tuberculosis: A review on the molecular determinants of resistance and implications for personalized care. J. Antimicrob. Chemother. 73, 1138–1151 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kempker R. R., Kipiani M., Mirtskhulava V., Tukvadze N., Magee M. J., Blumberg H. M., Acquired drug resistance in Mycobacterium tuberculosis and poor outcomes among patients with multidrug-resistant tuberculosis. Emerg. Infect. Dis. 21, 992–1001 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McGrath M., Gey van Pittius N. C., van Helden P. D., Warren R. M., Warner D. F., Mutation rate and the emergence of drug resistance in Mycobacterium tuberculosis. J. Antimicrob. Chemother. 69, 292–302 (2014). [DOI] [PubMed] [Google Scholar]
- 5.Ford C. B., Shah R. R., Maeda M. K., Gagneux S., Murray M. B., Cohen T., Johnston J. C., Gardy J., Lipsitch M., Fortune S. M., Mycobacterium tuberculosis mutation rate estimates from different lineages predict substantial differences in the emergence of drug-resistant tuberculosis. Nat. Genet. 45, 784–790 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ford C. B., Lin P. L., Chase M. R., Shah R. R., Iartchouk O., Galagan J., Mohaideen N., Ioerger T. R., Sacchettini J. C., Lipsitch M., Flynn J. L., Fortune S. M., Use of whole genome sequencing to estimate the mutation rate of Mycobacterium tuberculosis during latent infection. Nat. Genet. 43, 482–486 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Eldholm V., Norheim G., von der Lippe B., Kinander W., Dahle U. R., Caugant D. A., Mannsåker T., Mengshoel A. T., Dyrhol-Riise A. M., Balloux F., Evolution of extensively drug-resistant Mycobacterium tuberculosis from a susceptible ancestor in a single patient. Genome Biol. 15, 490 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Trauner A., Liu Q., Via L. E., Liu X., Ruan X., Liang L., Shi H., Chen Y., Wang Z., Liang R., Zhang W., Wei W., Gao J., Sun G., Brites D., England K., Zhang G., Gagneux S., Barry C. E. III, Gao Q., The within-host population dynamics of Mycobacterium tuberculosis vary with treatment efficacy. Genome Biol. 18, 71 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Didelot X., Walker A. S., Peto T. E., Crook D. W., Wilson D. J., Within-host evolution of bacterial pathogens. Nat. Rev. Microbiol. 14, 150–162 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu Q., Via L. E., Luo T., Liang L., Liu X., Wu S., Shen Q., Wei W., Ruan X., Yuan X., Zhang G., Barry III C. E., Gao Q., Within patient microevolution of Mycobacterium tuberculosis correlates with heterogeneous responses to treatment. Sci. Rep. 5, 17507 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Colijn C., Cohen T., Ganesh A., Murray M., Spontaneous emergence of multiple drug resistance in tuberculosis before and during therapy. PLOS ONE 6, e18327 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lieberman T. D., Wilson D., Misra R., Xiong L. L., Moodley P., Cohen T., Kishony R., Genomic diversity in autopsy samples reveals within-host dissemination of HIV-associated Mycobacterium tuberculosis. Nat. Med. 22, 1470–1474 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hobby G. L., Holman A. P., Iseman M. D., Jones J. M., Enumeration of tubercle bacilli in sputum of patients with pulmonary tuberculosis. Antimicrob. Agents Chemother. 4, 94–104 (1973). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Singhal R., Myneedu V. P., Microscopy as a diagnostic tool in pulmonary tuberculosis. Int. J. Mycobacteriol. 4, 1–6 (2015). [DOI] [PubMed] [Google Scholar]
- 15.Comas I., Coscolla M., Luo T., Borrell S., Holt K. E., Kato-Maeda M., Parkhill J., Malla B., Berg S., Thwaites G., Yeboah-Manu D., Bothamley G., Mei J., Wei L., Bentley S., Harris S. R., Niemann S., Diel R., Aseffa A., Gao Q., Young D., Gagneux S., Out-of-Africa migration and Neolithic coexpansion of Mycobacterium tuberculosis with modern humans. Nat. Genet. 45, 1176–1182 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gan M., Liu Q., Yang C., Gao Q., Luo T., Deep whole-genome sequencing to detect mixed infection of Mycobacterium tuberculosis. PLOS ONE 11, e0159029 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Walker T. M., Ip C. L. C., Harrell R. H., Evans J. T., Kapatai G., Dedicoat M. J., Eyre D. W., Wilson D. J., Hawkey P. M., Crook D. W., Parkhill J., Harris D., Walker A. S., Bowden R., Monk P., Smith E. G., Peto T. E., Whole-genome sequencing to delineate Mycobacterium tuberculosis outbreaks: A retrospective observational study. Lancet Infect. Dis. 13, 137–146 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Martin C. J., Cadena A. M., Leung V. W., Lin P. L., Maiello P., Hicks N., Chase M. R., Flynn J. L., Fortune S. M., Digitally barcoding Mycobacterium tuberculosis reveals in vivo infection dynamics in the macaque model of tuberculosis. MBio 8, e00312–17 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Folkvardsen D. B., Norman A., Andersen Å. B., Michael Rasmussen E., Jelsbak L., Lillebaek T., Genomic epidemiology of a major Mycobacterium tuberculosis outbreak: Retrospective cohort study in a low-incidence setting using sparse time-series sampling. J Infect Dis 216, 366–374 (2017). [DOI] [PubMed] [Google Scholar]
- 20.Roetzer A., Diel R., Kohl T. A., Rückert C., Nübel U., Blom J., Wirth T., Jaenicke S., Schuback S., Rüsch-Gerdes S., Supply P., Kalinowski J., Niemann S., Whole genome sequencing versus traditional genotyping for investigation of a Mycobacterium tuberculosis outbreak: A longitudinal molecular epidemiological study. PLOS Med. 10, e1001387 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xu Y., Cancino-Munoz I., Torres-Puente M., Villamayor L. M., Borrás R., Borrás-Máñez M., Bosque M., Camarena J. J., Colomer-Roig E., Colomina J., Escribano I., Esparcia-Rodríguez O., Gil-Brusola A., Gimeno C., Gimeno-Gascón A., Gomila-Sard B., González-Granda D., Gonzalo-Jiménez N., Guna-Serrano M. R., López-Hontangas J. L., Martín-González C., Moreno-Muñoz R., Navarro D., Navarro M., Orta N., Pérez E., Prat J., Rodríguez J. C., Ruiz-García M. M., Vanaclocha H., Colijn C., Comas I., High-resolution mapping of tuberculosis transmission: Whole genome sequencing and phylogenetic modelling of a cohort from Valencia Region, Spain. PLOS Med. 16, e1002961 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.D. Hartl, A. Clark, Principles of Population Genetics, Fourth Edition. (Sinauer Associates, Inc. Publishers, ed. 4, 2007). [Google Scholar]
- 23.Kreutzer D. A., Essigmann J. M., Oxidized, deaminated cytosines are a source of C → T transitions in vivo. Proc. Natl. Acad. Sci. U.S.A. 95, 3578–3582 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hutchinson F., Induction of tandem-base change mutations. Mutat. Res. 309, 11–15 (1994). [DOI] [PubMed] [Google Scholar]
- 25.Reid T. M., Loeb L. A., Tandem double CC→TT mutations are produced by reactive oxygen species. Proc. Natl. Acad. Sci. U.S.A. 90, 3904–3907 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sun G., Luo T., Yang C., Dong X., Li J., Zhu Y., Zheng H., Tian W., Wang S., Barry C. E. III, Mei J., Gao Q., Dynamic population changes in Mycobacterium tuberculosis during acquisition and fixation of drug resistance in patients. J Infect Dis 206, 1724–1733 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lin P. L., Ford C. B., Coleman M. T., Myers A. J., Gawande R., Ioerger T., Sacchettini J., Fortune S. M., Flynn J. L., Sterilization of granulomas is common in active and latent tuberculosis despite within-host variability in bacterial killing. Nat. Med. 20, 75–79 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cadena A. M., Fortune S. M., Flynn J. L., Heterogeneity in tuberculosis. Nat. Rev. Immunol. 17, 691–702 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hershberg R., Lipatov M., Small P. M., Sheffer H., Niemann S., Homolka S., Roach J. C., Kremer K., Petrov D. A., Feldman M. W., Gagneux S., High functional diversity in Mycobacterium tuberculosis driven by genetic drift and human demography. PLOS Biol. 6, e311 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pepperell C. S., Casto A. M., Kitchen A., Granka J. M., Cornejo O. E., Holmes E. C., Birren B., Galagan J., Feldman M. W., The role of selection in shaping diversity of natural M. tuberculosis populations. PLOS Pathog. 9, e1003543 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Coleman M. T., Maiello P., Tomko J., Frye L. J., Fillmore D., Janssen C., Klein E., Lin P. L., Early changes by 18Fluorodeoxyglucose positron emission tomography coregistered with computed tomography predict outcome after Mycobacterium tuberculosis infection in cynomolgus macaques. Infect. Immun. 82, 2400–2404 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang C., Luo T., Shen X., Wu J., Gan M., Xu P., Wu Z., Lin S., Tian J., Liu Q., Yuan Z., Mei J., DeRiemer K., Gao Q., Transmission of multidrug-resistant Mycobacterium tuberculosis in Shanghai, China: A retrospective observational study using whole-genome sequencing and epidemiological investigation. Lancet Infect. Dis. 17, 275–284 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.G. Canetti, The Tubercle Bacillus in the Pulmonary Lesion of Man: Histobacteriology and its Bearing on the Therapy of Pulmonary Tuberculosis (Springer Publishing Company, 1955). [Google Scholar]
- 34.Young D. B., Duncan K., Prospects for new interventions in the treatment and prevention of mycobacterial disease. Annu. Rev. Microbiol. 49, 641–673 (1995). [DOI] [PubMed] [Google Scholar]
- 35.Liu Q., Ma A., Wei L., Pang Y., Wu B., Luo T., Zhou Y., Zheng H. X., Jiang Q., Gan M., Zuo T., Liu M., Yang C., Jin L., Comas I., Gagneux S., Zhao Y., Pepperell C. S., Gao Q., China’s tuberculosis epidemic stems from historical expansion of four strains of Mycobacterium tuberculosis. Nat. Ecol. Evol. 2, 1982–1992 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Koboldt D. C., Zhang Q., Larson D. E., Shen D., McLellan M. D., Lin L., Miller C. A., Mardis E. R., Ding L., Wilson R. K., VarScan 2: Somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 22, 568–576 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tamura K., Stecher G., Peterson D., Filipski A., Kumar S., MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/22/eaba4901/DC1