

The reversal of 2-cell–like state in mESCs involves an intermediate state and is driven by nonsense-mediated mRNA decay of Dux.
Abstract
In mouse embryonic stem cell (ESC), a small cell population displays totipotent features by expressing a set of genes that are transiently active in 2-cell–stage embryos. These 2-cell–like (2C-like) cells spontaneously transit back into the pluripotent state. We previously dissected the transcriptional dynamics of the transition from pluripotency to the totipotent 2C-like state and identified factors that modulate the process. However, how 2C-like cells transit back into the pluripotent state remains largely unknown. In this study, we analyzed the transcriptional dynamics from the 2C-like state to pluripotent ESCs and identified an intermediate state. The intermediate state characterized by two-wave step up-regulation of pluripotent genes is different from the one observed during the 2C-like entry transition. Nonsense-mediated Dux mRNA decay plays an important role in the 2C-like state exit. Thus, our study not only provides a transcriptional roadmap for 2C-like–to–pluripotent state transition but also reveals a key molecular event driving the transition.
INTRODUCTION
During preimplantation development in mice, the quiescent zygotic genome is activated at the late 1-cell and 2-cell stages through a process known as zygotic genome activation (ZGA) (1). ZGA coincides with the gain of totipotency, the ability of a cell to generate embryonic and extraembryonic cell types of an organism (2) and transient expression of a group of genes (e.g., Zscan4 gene cluster) and repeats (e.g., murine endogenous retrovirus with leucine tRNA primer (MERVL) repeats) (3, 4). In serum/leukemia inhibitory factor (LIF) culture conditions, a small population of embryonic stem cells (ESCs) (<1%) exhibit several features of 2-cell embryos (4, 5), including expression of 2-cell embryo–specific genes and repeats (e.g., Zscan4 genes and MERVL repeats) (4), increased histone mobility (6), dispersed chromocenters (7), and the capacity to contribute to both embryonic and extraembryonic tissues (4). Using a MERVL promoter–driven reporter, 2-cell–like (2C-like) cells can be easily isolated from ESC culture. Compared to other totipotent cell models (8–10), 2C-like cells exhibit a unique transcriptome similar to that of the 2-cell embryos, making it a good model for understanding ZGA and totipotency (2, 11).
The 2C-like state is transient and can spontaneously reverse to the pluripotent state under mouse ESC (mESC) culture conditions (4). Multiple factors, including Tet proteins (12), components of noncanonical PRC1 (Polycomb repressive complex 1) (13), and miR-34a (14), have been shown to mediate the pluripotent–to–2C-like state transition. However, the rare occurrence of the 2C-like transition event in the mESC culture makes it technically challenging to study how 2C-like cells transit back to the pluripotent state.
The transcription factor Dux is necessary and sufficient to drive the ESC to 2C-like cell transition (15–17). Dux-induced 2C-like cells highly resemble the spontaneous 2C-like cells and are able to exit 2C-like state spontaneously (18). Taking advantage of this property, we recently characterized the transcriptional dynamics of pluripotent–to–2C-like state transition and showed that the transition involves a two-step reprogramming process, a first step characterized by the down-regulation of pluripotent genes and a second step characterized by the expression of 2-cell embryo–specific transcripts (18). We also identified Dnmt1 and Myc as negative regulators of the pluripotent–to–2C-like transition (18).
In this study, we characterized the transcriptional dynamics of the reverse process. We found that during the reverse transition, pluripotent genes are activated in two waves, distinct from a direct reversal of the pluripotent–to–2C-like transition process, indicating that the entry and exit of 2C-like state follow distinct transcriptional paths. In addition, our results indicated that nonsense-mediated Dux mRNA decay (NMD) plays an important role in driving the reversal process.
RESULTS
Establishment of a cell model to study reversal of Dux-induced 2C-like transition
To study the transition between the pluripotent and 2C-like state, we previously constructed a reporter ES cell line containing MERVL promoter–driving tdTomato transgene in which activation of the reporter gene serves as an indicator of 2C-like state (4) and doxycycline-inducible Dux transgene (Fig. 1A) (18). To study the reversal process, we first sorted 2C-like cells (D1 2C+) after 24 hours of Dux induction. Then, the purified 2C-like cells were cultured for an additional 24 hours to allow them to exit the 2C-like state (Fig. 1B). Last, the cells were sorted into tdTomato-negative (D2 2C−) and tdTomato-positive (D2 2C+) cell populations for RNA sequencing (RNA-seq) analysis (Fig. 1B and fig. S1, A and B).
Fig. 1. D2 2C− cells exhibit intermediate transcriptome between 2C-like and pluripotent cells.
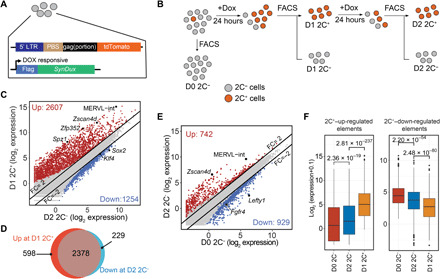
(A) Schematic representation of the reporter constructs. Tdtomato is under the MERVL promoter control. synDux refers to codon-optimized exogenous Dux. PBS, primer binding site. LTR, long terminal repeats. (B) Diagrammatic representation of the different cell populations used for RNA sequencing (RNA-seq). (C) Scatterplot comparing the mean expression profiles between D2 2C− and D1 2C+ (2C-like cells) cell population. Criteria: FC > 2 and FDR < 0.001. Genes/repeats up-regulated in 2C-like cells are shown in red, and elements up-regulated in D2 2C− are shown in blue. (D) Venn diagram showing the overlap between the up-regulated genes during ESC–to–2C-like transition (red) and down-regulated genes during the exit from 2C-like state (blue). (E) Scatterplot comparing the mean expression profile between spontaneous D2 2C− and D0 2C− (ESCs). Criteria: FC > 2 and FDR < 0.001. Genes and repeats up-regulated in D2 2C− cells are indicated in red; Genes and repeats up-regulated in D0 2C− are indicated in blue. (F) Box plot showing the log2 expression level of 2C+–up-regulated transcripts and 2C+–down-regulated transcripts in each of the D0 2C−, D2 2C−, and D1 2C+ cell populations. P values were calculated by two-tailed Mann-Whitney U test. All data were derived from two biological RNA-seq repeats.
Compared to that of the starting cells (D1 2C+), the transcriptome of D1 2C+ cells and that of D2 2C+ cells are almost identical (Pearson correlation = 0.96). Only 276 and 49 genes were activated or repressed [fold change (FC) > 2, false discovery rate (FDR) < 0.001; fig. S1C and table S1], respectively, indicating that the D2 2C+ cells remain in 2C-like state. Although the transcriptome of D2 2C+ cells and that of D1 2C+ cells are almost identical, D2 2C+ cells exhibit Gadd45b/g up-regulation (fig. S1C), p53/ATM serine/threonine kinase signaling enrichment, and metabolic alteration (fig. S1D), indicating that prolonged maintenance in the 2C-like state may activate cell death pathway.
In contrast, D2 2C− cells exhibit substantial transcriptional alterations compared to that of D1 2C+ cells with 1254 and 2607 genes and repeats, respectively, up- or down-regulated (FC > 2, FDR < 0.001; Fig. 1C and table S1). The down-regulated genes and repeats include many known 2-cell embryo–specific transcripts such as MERVL repeats, Zscan4 genes, Spz1, and Zfp352 (Fig. 1C), while the up-regulated genes and repeats include pluripotent genes, such as Sox2 and Klf4, and are enriched in pluripotency and signaling pathways critical for pluripotency (Fig. 1C and fig. S1E). In addition, ~80% of 2C+–up-regulated transcripts are repressed in D2 2C− cells (Fig. 1D). Collectively, these results confirm that D2 2C− cells exit from the 2C-like state and thus the system can be used for studying the reverse process of 2C-like transition.
D2 2C− cells exhibit intermediate transcriptome between pluripotent and 2C-like cells
Because transcriptome indicates that D2 2C− cells have exited from the 2C-like state (Fig. 1, C and D), we next asked whether these cells return to the original pluripotent state. To this end, we compared the transcriptome between D2 2C− cells and the original pluripotent cells (D0 2C−). We found that, intriguingly, D2 2C− cells did not completely reverse back to the pluripotent state based on transcriptome analysis. Compared to the pluripotent ESCs (D0 2C−), D2 2C− cells exhibited distinct transcriptome (Fig. 1E and table S1), including up-regulation of 2-cell embryo–specific transposable elements, Zscan4d and MERVL-int, and down-regulation of some pluripotency-related genes, including Lefty1 and Fgfr4, suggesting that these cells exhibit a transcriptome between pluripotent and 2C-like state. We found that D2 2C− cells exhibit an intermediate expression level of 2C+–up-regulated or –down-regulated genes and repeats between pluripotent and 2C-like cells (Fig. 1F).
To determine the extent to which the intermediate transcriptome of D2 2 C− cells is different from that in the pluripotent ESC and 2C-like cells, we compared it to the transcriptomes of D0 2C− (pluripotent cells) and D1 2C+ (2C-like cells). We identified that 4702 transcripts exhibit different dynamics between the three datasets. The majority of them (~80%), referred to as “group 1,” show consistent expression between D2 2C− cells and ESCs (D0 2C−) (table S2), indicating that these genes have been successfully reversed to the pluripotent state in D2 2C− cells (Fig. 2, A and B). The down-regulated group 1 genes and repeats, including Zscan4 and MERVL, are enriched for assembly of the RNA polymerase II complex, while the up-regulated group 1 genes include pluripotent gene Sox2 and are enriched in mESC pluripotency pathways (Fig. 2, A and C), indicating that the pluripotency network is at least partially restored in D2 2C− cells. Another group of genes and repeats (group 2) maintains a similar expression level between D2 2C− and D1 2C+ cells (Fig. 2, A and B, and table S2), indicating that expression of these genes has not been completely restored to the pluripotent level. The down-regulated group 2 genes during the reversal process include Itga3 and Skp1a and are enriched for actin cytoskeleton signaling and ubiquitination pathway, while the up-regulated group 2 genes include Fgfr4 and Mapk12 and are enriched for FGF and MAPK signaling (Fig. 2, A and C). This indicated that the D2 2C− cells exhibit distinct molecular and cellular features compared to the pluripotent cells.
Fig. 2. Transcriptional dynamics during 2C-like state–to–pluripotent state transition.
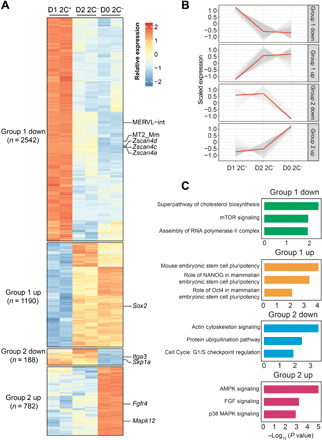
(A) Heatmap showing the relative expression level of group 1 and group 2 genes from two biologically independent samples of D1 2C+, D2 2C−, and D0 2C− cell population. The number of genes in each category is indicated on the left side of the heatmap. (B) The expression dynamics of the transcripts in group 1 and group 2 genes. Each transcript expression is shown as a gray line; the red line represents the mean expression. (C) Bar plot showing the −log10 (P value) of the pathways enriched in each category of genes (right, right-tailed Fisher’s exact test). All data were derived from two biological RNA-seq repeats.
There are two possible ways to explain the intermediate transcriptome of the D2 2C− cells. One is that an intermediate cell population emerges during the reversal of 2C-like state and the D2 2C− cells represent a mixed cell population consisting of intermediate and pluripotent cells. Alternatively, D2 2C− cells are a homogeneous cell population with altered transcriptome.
Single-cell RNA-seq analysis indicates that the D2 2C− cells are a mixed cell population
To determine which of the above two possibilities are true, we performed single-cell RNA-seq (scRNA-seq) at different time points of the reverse transition process (Fig. 3A). We isolated Dux-induced 2C-like (D1 2C+) cells and continue culturing them for 24 hours to initiate the spontaneous reversal process. In total, we obtained 1304 and 994 single cells with >800 transcripts at 0- and 24-hour time points, respectively (Fig. 3A, fig. S2A, and table S3; see Materials and Methods).
Fig. 3. scRNA-seq reveals an intermediate state during the 2C-like–to–pluripotent state transition.
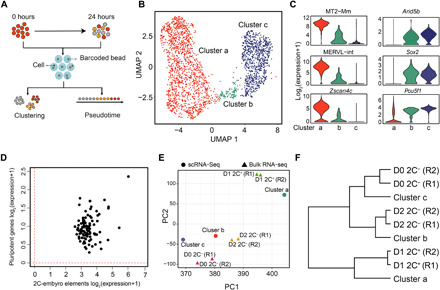
(A) The workflow of the experimental and data analysis process. A total of 1304 and 994 cells with >800 transcripts were obtained at 0 and 24 hours after the exit from 2C-like state, respectively. (B) Uniform Manifold Approximation and Projection (UMAP) of the sequenced cells. Cluster a (1397 cells), cluster b (101 cells), and cluster c (800 cells) are indicated. (C) Violin plot showing the log2 expression level of representative 2-cell-embryo and pluripotency marker genes in each cluster. (D) Scatterplot showing the log2 mean expression of 2-cell-embryo transcripts (x axis) and pluripotency transcripts (y axis) in cluster 2 cells. The dashed red lines indicate the threshold for expression. (E) Principal components analysis projection of the single-cell clusters and the fluorescence-activated cell sorting (FACS) isolated cell populations based on their transcriptional profiles. R1 and R2 indicate two replicates. (F) Hierarchical clustering (complete linkage) of the relationship between cell clusters derived from scRNA-seq and FACS-isolated bulk RNA-seq cell populations based on their transcriptome profiles. R1 and R2 indicate two replicates.
As the exit of 2C-like state is an unsynchronized process (Fig. 1B), we pooled all the cells from the two time points for clustering analysis. The analysis revealed three major cell clusters with distinct transcriptional profiles (Fig. 3, B and C, and fig. S2B). Cluster a (1397 cells) shows characteristics of 2C-like cells with the expression of Zscan4c and MERVL (Fig. 3C and fig. S2B) and a low-level expression of Sox2 and Arid5b. Cluster c (800 cells) shows ESC features with high expression of pluripotency genes such as Sox2, Pou5f1, and Arid5b but almost no expression of MERVL and Zscan4c (Fig. 3C and fig. S2B). Cluster b (101 cells), on the other hand, shows intermediate expression of both pluripotency genes (e.g., as Sox2, Pou5f1, and Arid5b) and 2-cell embryo elements (e.g., MERVL and Zscan4c) and thus may represent an intermediate cell population (Fig. 3C and fig. S2B). We further confirmed that cells in cluster b are in an intermediate state as each cell in this cluster simultaneously expresses pluripotent genes and 2-cell embryo transcripts (2C+–up-regulated ZGA genes and repeats) (Fig. 3D).
To further confirm that the cells we analyzed belong to different cell clusters, we performed clustering and principal components analysis (PCA) with both scRNA-seq and bulk RNA-seq samples. The transcriptional profiles of the three cell clusters derived from scRNA-seq respectively correlate with D1 2C+, D2 2C−, and D0 2C− cell populations (Fig. 3, E and F), supporting that they represent 2C-like, intermediate, and pluripotent cells. While scRNA-seq reveals that the 2C− cells obtained at 24 hours (cluster b and c cells) mainly consist of pluripotent cells (cluster c) (fig. S2A), the bulk transcriptomic profile of these cells (D2 2C− transcriptome) is more similar to that of cluster b cells (Fig. 3, E and F), indicating that cluster b cells contribute to the intermediate transcriptome of D2 2C− cells. For example, we found that several group 2 genes (Fig. 2A), such as Camk1d, Trim 28, and Utp3, are expressed at a similar level between cluster b and cluster a cells, suggesting that cluster b cells are the major contributors of the expression of these genes in D2 2C− cells (fig. S2C). In contrast, the expression of many group 1 genes, such as Sox2 and Zfp352, is altered in both cluster b and cluster c cells compared to cluster a cells (fig. S2C), which is consistent with the fact that their expression is restored to the pluripotent state in D2 2C− cells.
Previous studies indicate that 2C-like cells restore to the pluripotent state after 3-day culture (19), indicating that the intermediate cluster we identified eventually transits back to the pluripotent state. To confirm that this is indeed the case, we isolated D2 2C− cells, which include the cluster b cells, and cultured them for an additional 2 days (D4 2C− cells). We found that while D2 2C− cells activated the 2-cell embryo–specific transcripts, such as Zscan4d, Spz1, MERVL-int, and Zfp352 compared to pluripotent cells (fig. S2D, left), the majority of these transcripts are expressed at a similar level between D4 2C− cells and ESCs (fig. S2D, right). In addition, PCA shows that the transcriptome of D4 2C− cells are more similar to that of ESCs compared to that of D2 2C− cells (fig. S2E). Collectively, these results suggest that cluster b cells transit back into the pluripotent state after further culture.
In conclusion, by performing scRNA-seq analysis, we demonstrate the existence of an intermediate state during the transition from the 2C-like state to the pluripotent state, suggesting that the D2 2C− cells consisted of a mixture of intermediate and pluripotent cells.
The two intermediate states of forward and reverse transition between pluripotent and 2C-like states are distinct
The identification of an intermediate-state cell population suggests that the reversal of the 2C-like state is also a stepwise process. To dissect the transcriptional dynamics of this process, we performed a pseudotime analysis (Fig. 4A). By coloring cells along the pseudotime (gray: beginning of the pseudotime, red: end of the pseudotime), we noticed that cluster a cells (2C-like cells) are mainly located at the beginning of the projected timeline (Fig. 4A); cluster b cells (intermediate) are mainly located in the middle of the timeline, while cluster c cells (pluripotent) are at the end of the timeline (Fig. 4A). The pseudotime also captures the expression dynamics of key pluripotent and totipotent genes, with the totipotency marker genes (e.g., Zscan4c and MT2-Mm) gradually decreasing and the pluripotency genes (e.g., Sox2 and Arid5b) suddenly up-regulating at the intermediate state (fig. S3A).
Fig. 4. The intermediate states of forward and reverse transition are distinct.
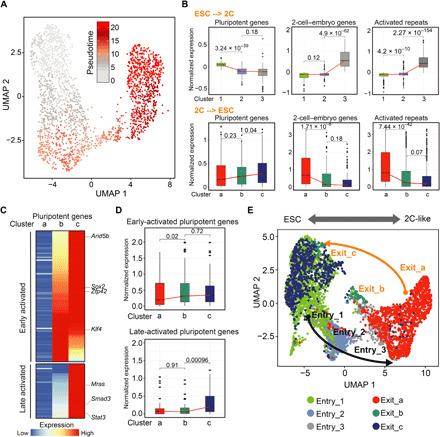
(A) UMAP plot showing the pseudotime progression. Each cell is colored according to its position in the pseudotime. (B) Box plot showing the expression of 2C+–down-regulated pluripotent genes (n = 135 genes), 2-cell-embryo genes (2C+–up-regulated ZGA genes, n = 349 genes), and 2C+-activated repeats (n = 501 repeats) during the entry (top) and exit from the 2C-like state (bottom). (C) Heatmap showing the expression of 2C+–down-regulated pluripotent genes in each cluster. (D) Box plot showing the expression of early-activated and late-activated pluripotent genes during the exit from the 2C-like state. (E) UMAP projection of cells during entry and exit from the 2C-like state. P values (shown as numbers in the figures) are calculated by two-tailed Mann-Whitney U test.
The similarity between the transcriptional dynamics of totipotent and pluripotent genes during preimplantation development and the reverse of 2C-like state (fig. S3A) (20) prompted us to ask whether reversal of the 2C-like state recapitulates the transcriptional dynamics of preimplantation development. A comparative analysis of transcriptome revealed that the 2C-like cluster (cluster a) is similar to 2-cell embryo; the pluripotent cluster (cluster c) is similar to the inner cell mass, while the intermediate cluster (cluster b) is most similar to the 4-cell/8-cell embryos (fig. S3B). Furthermore, hierarchical clustering analysis revealed that the intermediate cells are clustered with 4-cell/8-cell embryos, while clusters a and c are grouped with 2-cell embryos and inner cell mass, respectively (fig. S3C). These data support that the reversal of the 2C-like state at least partly recapitulates the transcriptional dynamics of preimplantation development.
We have recently demonstrated that transition from the pluripotent state to the 2C-like state follows a two-step transcriptional dynamics: (i) down-regulation of pluripotency genes and (ii) activation of 2-cell embryo–specific genes and repeats (18) (Fig. 4B, top). To determine whether the 2C-like state–to–pluripotency state transition simply follows a reverse transcriptional dynamics, we analyze the expression dynamics of pluripotent genes, 2-cell embryo genes, and activated repeats during the reversal process and found that activated 2-cell embryo genes and repeats are majorly silenced from the 2C-like state to the intermediate state (Fig. 4B, bottom; fig. S3D; and table S4), which follows a direct opposite dynamics of what we observed during the entry of the 2C-like state. In contrast, pluripotent genes are gradually up-regulated during the entire reversal process (Fig. 4B, bottom). Further analysis of the pluripotent gene activation revealed a two-step process (Fig. 4, C and D, and fig. S3E). Early-activated pluripotent genes that are up-regulated in the first step (2C-like state to intermediate state) mainly consist of crucial pluripotency factors including Sox2, Esrrb, Arid5b, and Zfp42 that are involved in embryonic development and stem cell maintenance (Fig. 4C, fig. S3F, and table S4), indicating that establishment of a pluripotent network is initiated at the beginning of the reversal process, while the late-activated pluripotent genes that are up-regulated during the second step (intermediate state to pluripotent state) include many signaling-related genes (such as Mras, Stat3, and Smad3) that are involved in cellular signaling, actin cross-link, and cell junction regulation (Fig. 4C, fig. S3F, and table S4).
The two-step up-regulation of pluripotent genes during the exit of the 2C-like state is not a direct reversal of the pluripotent to 2C-like state, where down-regulation of pluripotent genes takes place at the early stage during the transition (Fig. 4B). This indicates that 2C-like cells may exit the 2C-like state following a distinct path from the path for the entry of 2C-like state. An integrative analysis of scRNA-seq datasets from the 2C-like entry process (18) and the exit process indicated that the entry and exit of the 2C-like state follow different transcriptional paths (Fig. 4E). Together, our scRNA-seq analyses revealed the distinct features of the transcriptional dynamics for the reversal of the 2C-like state and indicated that 2C-like cells go through a different intermediate state during the reverse transition.
NMD mediates Dux mRNA degradation and contributes to the 2C-like–to–pluripotent state transition
Dux plays an important role in the pluripotent–to–2C-like state transition (15–17). The reverse transition from 2C-like to pluripotent state may be through down-regulation of Dux or a Dux-independent pathway. Transcriptomic data analysis revealed that the Dux mRNA level is significantly down-regulated in D2 2C− cells but maintained at a high level in D2 2C+ cells (Fig. 5A), suggesting that down-regulation of Dux may contribute to the reverse transition from the 2C-like state to the pluripotent state.
Fig. 5. Smg7 degrades Dux mRNA and contributes to the 2C-like–to–pluripotent state transition.
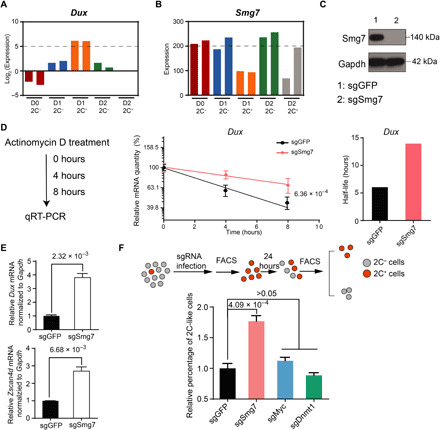
(A and B) Bar graph showing the expression levels of Dux (A) and Smg7 (B) in different cell populations. (C) Western blot confirming efficient Smg7 knockdown. (D) Measurement of Dux half-life. The half-life of Dux is calculated from the Dux degradation curve upon actinomycin D treatment. P value (shown as number) is calculated using a standard t test. (E) Relative Dux and Zscan4d mRNA levels normalized to Gapdh in Dux-activated mESCs. (F) Workflow for measuring the percentage of 2C-like state cell population (top) and the relative percentage of 2C-like cells under different genetic manipulation (bottom). P values are shown as numbers in the figure. Data presented in (D) to (F) are mean ± SD, n = 3. By unpaired t test, two-tailed, two-sample unequal variance unless otherwise indicated.
The rapid decrease of Dux mRNA during the exit of the 2C-like state suggests that an active RNA degradation mechanism may be involved in degrading Dux mRNA during the process. A previous study has shown that nonsense-mediated mRNA decay (NMD) is involved in degrading DUX4, a Dux homolog in humans, by recognizing the second 3′ untranslated region (3′UTR) intron of DUX4 in facioscapulohumeral muscular dystrophy (21). Although Dux lacks the 3′UTR intron, Dux mRNA transcripts contain a relatively long 3′UTR [~700 base pairs (bp)] (3), which is one of the signatures that can be targeted by NMD (22, 23). In addition, we noticed that one core component of the NMD pathway (24), Smg7, negatively correlates with 2C-like transition (Fig. 5B) and Smg7 expression is up-regulated in cells that exit the 2C-like state but remains low in cells that stay in the 2C-like state (fig. S4A), suggesting that Smg7 is likely involved in the 2C-like transition dynamics. These results suggest that Dux may be targeted by NMD for rapid degradation.
To test this hypothesis, we generated Smg7-deficient mESCs using single guide RNA targeting Smg7 and confirmed its depletion by Western blot analysis (Fig. 5C). We found that the mRNA half-life of Dux is significantly increased upon Smg7 depletion, while the mRNA half-life of Zscan4d, a downstream target of Dux, is not significantly altered (Fig. 5D and fig. S4B), confirming that Dux is degraded by NMD. In addition, upon Dux derepression, the mRNA levels of Dux and Zscan4d are significantly increased in Smg7-deficient cells compared to that of control cells (Fig. 5E). The general expression level of 2C+–up-regulated transcripts is further increased upon Smg7 depletion (fig. S4C and table S5). These results support the notion that Dux mRNA is degraded by Smg7-mediated NMD. Most of the mRNA biogenesis genes (Kyoto Encyclopedia of Genes and Genomes, BR:mmu03019) are not altered upon Smg7 perturbation (fig. S4D), indicating that Smg7 directly modulates Dux stability through NMD. We found that zfp36l2, which destabilizes mRNA through binding to the AU-rich element (ARE) of the 3′UTR (25), is decreased upon Smg7 perturbation (table S5). However, Dux mRNA does not contain ARE at its 3′UTR (26), and thus, it is unlikely that Smg7 destabilizes Dux mRNA through Zfp36l2. Last, perturbation of Upf1, another core factor in NMD, similarly stabilizes Dux mRNA (fig. S4, E and F). Together, our results support the notion that Smg7 destabilizes Dux mRNA directly through NMD.
Next, we asked whether NMD modulates the 2C-like state–to–pluripotent state transition. To this end, we sorted out spontaneous 2C-like cells (without exogenous Dux expression) from control and Smg7-depleted cells and examined the 2C-like state–to–pluripotent state transition rate. Compared to control cells, Smg7 depletion resulted in a higher percentage of cells maintaining their 2C-like state after 24-hour culture, indicating that the 2C-like–to–pluripotent state transition is inhibited by Smg7 depletion (Fig. 5F). In contrast, depletion of Dnmt1 and Myc, which mediates 2C-like transition independent of Dux expression (18), did not significantly alter the reversal of the 2C-like state (Fig. 5F). In addition, Upf1 perturbation similarly impedes the reversal of the 2C-like state (fig. S4G). Together, our results support the idea that NMD degrades Dux mRNA and modulates 2C-like state maintenance and that Dux degradation contributes to the reversal of the 2C-like state.
DISCUSSION
The 2C-like state is transient and reversible in mESCs. We previously characterized the transcriptional dynamics during the ESC–to–2C-like cell transition and identified factors regulating this process (18). However, how mESCs exit the 2C-like state to transit back to the pluripotent state and what factors are involved in this process are not clear. In this study, we performed bulk and scRNA-seq to dissect the molecular roadmap during the 2C-like–to–pluripotent state transition. Our analysis revealed that the transition process involves an intermediate state with a two-stage up-regulation of pluripotent genes, which is distinct from the intermediate state during the pluripotent–to–2C-like state transition. However, the lack of a suitable cell surface marker prevented the isolation and characterization of the intermediate cell state. A detailed functional characterization of this intermediate state cell population awaits the development of tools for isolation of the cell population.
Although Dux is a key regulator for the pluripotent–to–2C-like state transition in mESCs (15–17), the function of Dux in the reversal process and what factors drive the reversal transition are important and unanswered questions. We found that during the reversal of the 2C-like state, Dux mRNA is rapidly degraded and NMD at least partly contributes to its degradation. Transcriptome analysis suggests that NMD will likely modulate Dux degradation without affecting other pathways involved in RNA surveillance and degradation (table S5). However, we could not rule out the possibility that NMD may indirectly destabilize Dux through uncharacterized factors or via modulating other RNA-degradation–related factors at the translational or posttranslational level. NMD contributes to the reversal process as depletion of an NMD factor, Smg7 or Upf1, increased the cell population that remains in the 2C-like state. Together, our study demonstrates that the degradation of Dux mRNA, particularly through NMD-mediated mRNA clearance, drives the reversal of the 2C-like state. Additional machinery may exist in mESCs to mediate Dux degradation at the mRNA level and protein level to ensure the efficient reversal of the 2C-like state in the mESC culture.
During mouse preimplantation development, embryos are able to efficiently eliminate Dux transcripts in the late 2-cell embryo to gradually establish pluripotency. A failure in timely Dux elimination may lead to embryonic developmental arrest (20, 27, 28). By exploiting the reversal of the 2C-like state, we revealed that the reverse process coincides with a rapid decrease of Dux mRNA and activation of pluripotent genes, which partially recapitulates the transcriptional dynamics of preimplantation development. Our analysis indicates that the reversal of the 2C-like state can serve as an in vitro model for understanding preimplantation development. In addition, we revealed that NMD-mediated mRNA degradation might be a previously unidentified mechanism contributing to the timely Dux clearance in early embryos. The 2C-like transition system exhibits potential discrepancy compared to the in vivo embryonic development (27, 29, 30). Thus, future studies with the development of new reagents and tools are needed to examine the role of NMD in Dux clearance in preimplantation embryos.
In conclusion, this study, together with our previous study (18), provides a comprehensive transcriptional roadmap in the forward and reversal transition between the pluripotent and 2C-like states in mESCs. The studies also revealed factors involved in the transitions. Furthermore, the studies suggest that the reversal of 2C-like transition can serve as an in vitro model to understand the molecular events, such as the rapid clearance of Dux mRNA, during preimplantation development.
MATERIALS AND METHODS
ESC culture and establishment of cell lines with inducible Dux expression
The ES-E14 cells were cultured on plates coated with 0.1% gelatin. The medium for ES-E14 cells is standard LIF/serum medium containing 15% fetal bovine serum (FBS) (Sigma-Aldrich, catalog no. F6178), mouse LIF (1000 U/ml) (MilliporeSigma, catalog no. ESG1107), 0.1 mM nonessential amino acids (Gibco, catalog no. 11140), 0.055 mM β-mercaptoethanol (Gibco, catalog no. 21985023), 2 mM GlutaMAX (Gibco, catalog no. 35050), 1 mM sodium pyruvate (Gibco, catalog no. 11360), and penicillin-streptomycin (100 U/ml) (Gibco, catalog no. 15140). The transcriptome of our mESCs is close to the transcriptome of a published ES-E14 (Pearson correlation, r = 0.92) (31). The MERV-L-LTR-tdTomato reporter constructs were a gift from the laboratory of Samuel L. Pfaff (4) and were linearized and transfected into mESCs. Colonies containing tdTomato-positive cells were isolated. Codon-optimized Dux (table S5) was synthesized by Integrated DNA Technologies (IDT) and inserted into pCW57-MCS1-P2A-MCS2 (Neo) (Addgene 89180). Lentivirus infection was used to knock in Dux genes into ESC with 2C::tdTomato Reporter. Single clones were picked for further analysis.
Fluorescence-activated cell sorting
Flow cytometry was performed via BD FACSCanto II, and cell sorting was performed using the BD FACSAria II cell sorter. mESCs were suspended in PBS with 5% FBS for fluorescence-activated cell sorting (FACS). Data and image were processed using FlowJo software. The gating strategy is presented in fig. S1A. The cell viability and apoptotic index of D2 2C− cells isolated via FACS are 99.91 and 1.11%, respectively. The cell viability and apoptotic index of D2 2C+ cells isolated via FACS are 98.44 and 20.2%, respectively.
RNA isolation, quantitative polymerase chain reaction, and bulk RNA-seq
Cellular RNA was isolated using Qiagen Allprep RNA/DNA Mini Kit (Qiagen, catalog no. 80204). Complementary DNA was prepared by using the SuperScript III First-Strand Synthesis System (Thermo Fisher Scientific, catalog no. 18080051) and quantitative reverse transcription polymerase chain reaction was performed by using the Fast SYBR Green Master Mix (Thermo Fisher Scientific, catalog no. 4385612). Relative quantification was performed using the comparative cycle threshold (CT) method normalized to Gapdh. Bulk RNA-seq sample was generated by the NEBNext Ultra Directional RNA Library Prep Kit for Illumina (NEB, catalog no. E7420S).
mRNA half-life calculation
Actinomycin D (4 μg/ml, Sigma-Aldrich, A1410-10MG) was used to prevent transcription. The quantitative polymerase chain reaction results were used for linear fitting to the log-transformed solution log N(t) = −λt + log N(0) of the exponential decay function dN = −λN, where N is the amount of a specific mRNA, λ is the decay rate constant, and t is the time elapsed after the initial time point. Half-life of an mRNA was calculated using the equation h = log(2)/λ, where h is the half-life.
Western blotting
Cellular protein was isolated using M-PER Mammalian Protein Extraction Reagent (Thermo Fisher Scientific, catalog no. 78501) with protease inhibitor (Roche, catalog no. 4693159001). Western blotting was performed with 4 to 12% bis-tris gradient gel (Invitrogen, NP0322BOX) with the following antibodies: Smg7 (1:1000, Bethyl Laboratories, A302-170), Upf1 (1:1000, Bethyl Laboratories, A300-036), Gapdh (1:10,000, Ambion, AM4300, 6C5), goat anti-mouse immunoglobulin G (IgG) (H + L) secondary antibody, horseradish peroxidase (HRP) (1:10,000, Invitrogen, 31430), and goat anti-rabbit IgG (H + L) secondary antibody, HRP (1:10,000, Invitrogen, 31460).
10× scRNA-seq
10× scRNA-seq was performed as previously described (32). 2C-like cells were sorted and cultured for 24 hours before harvested for scRNA-seq.
Genomic annotation files preparation
The gene transfer format (GTF) file corresponding to the mm10 Ensembl GRCm38.85 transcriptome was downloaded from the Ensemble database. Sequences of the synthetic-Dux and TdTomato were added to the mm10 genomic annotation file. The GTF files were converted to refFlat format using the UCSC gtfToGenePred tool and then the refFlat file was converted to a format compatible with the droplet sequencing (Drop-seq) tool using a custom script.
Repeat pseudo-genome preparation
As repeat elements tend to have multiple highly similar copies, it is difficult to accurately align and estimate their expression. Hence, we create a repeat pseudo-genome. We use a slightly modified version of the RepEnrich software (33). Briefly, for each repetitive element subfamily, a pseudo-chromosome is created by concatenating all genomic instances of that subfamily along with their flanking 15-bp genomics sequences and a 200-bp spacer sequence (a sequence of Ns). The pseudo-genome was then indexed using Spliced Transcripts Alignment to a Reference (STAR) (34) and the corresponding GTF and refFlat files were created using custom scripts and each pseudo-chromosome was considered as one gene.
Sequencing alignment for coding genes
Raw reads were first trimmed using Trimmomatic (v.0.36). Illumina sequence adaptors were removed, the leading and tailing low-quality base pairs (less than 3) were trimmed, and a 4-bp sliding window was used to scan the reads and was trimmed when the window mean quality dropped below 15. Only reads having at least 50 bp were kept. The resulting reads were mapped to the mm10 genome using STAR (v.2.5.2b) (34) with the following parameters: --outSAMtype BAM SortedByCoordinate --outSAMunmapped Within --outFilterType BySJout --outSAMattributes NH HI AS NM MD --outFilterMultimapNmax 20 --outFilterMismatchNmax 999 --quantMode TranscriptomeSAM GeneCounts. The gene expression count files generated by STAR were then used for estimating gene expression.
Sequencing alignment for repeats
Multi-mapped reads and reads mapping to intronic or intergenic regions were extracted and then mapped to the repeat pseudo-genome. First, the TagReadWithGeneExon command of the Drop-seq tools (35) was used to tag the reads into UTR, coding, intergenic, and intronic reads using the bam tag “XF”. Multi-mapped reads and intergenic and intronic reads were extracted and mapped to the repeat pseudo-genome using STAR. The STAR read counts were used as an estimate of repeats expression.
Bulk RNA-seq normalization
For each sample, the genes and repeats expression matrices were merged together and then the “Trimmed Mean of M-values” (TMM) normalization method (36) from the R/Bioconductor package and edgeR package was used to calculate the normalized expression (37, 38).
Differential gene expression analysis of bulk RNA-seq data
The R/Bioconductor edgeR package (37, 38) was used to detect the differentially expressed genes between the different samples using the generalized linear model–based method. Genes showing more than twofold expression change and an FDR < 0.0001 were considered as differentially expressed.
Single-cell raw gene expression generation
Raw reads were preprocessed using the cellranger software (v.13.1) (39). The “cellranger mkfastq” command was used to demultiplex the different samples, and the “cellranger count” command was used to generate the gene-per-cell expression matrices for each sample by aligning the reads to the mm10 genome and quantifying expression of the Ensembl genes (GRCm38.93).
Single-cell raw repeats expression generation
To quantify the expression of repeats, we used the same pseudo-genome reference created for RNA-seq analysis. First, the “cellranger mkref” command was used to create a custom 10× pseudo-genome reference. Next, all the reads that do not map to known gene (reads not having the GX or the AN tag) were extracted using a custom python script. Then, the “cellranger count” command was used to generate the repeat-per-cell expression matrices for each sample.
Identification of valid cells, clustering, and dimension reduction
As the 2C-like cell transcriptome is dominated by repeats expression and the ESC transcriptome is dominated by protein-coding gene expression, cellranger will fail to detect valid cells when quantifying the gene expression of 2C-like cells and also fail to detect valid cells when quantifying the repeats expression for ESCs. To distinguish between valid cells and empty droplets, we used the emptyDrops function from the DropletUtils R/Bioconductor package (40, 41) with an FDR cutoff of 0.01. A total of 2972 and 1112 valid cells were detected at 0 and 12 hours, respectively. Valid cells were then filtered to select cells with >800 transcripts to obtain a final total of 1347 and 996 cells at 0 and 12 hours, respectively. The scran package was then used to normalize the counts. First, the quickCluster method with parameter min.size = 50 was used to cluster similar cells based on their rank correlations, then the size factors were calculated using the computeSumFactors function, and, last, the counts were normalized with the normalize function of the scater package. The Seurat package v.3 was then used for clustering analysis. Uniform Manifold Approximation and Projection (UMAP) plots were generated using the uwot package.
Integration of 2C-like entry and exit scRNA-seq data
To combine the 2C-like cell scRNA-seq entry data (Drop-seq) and exit data (10×), we used the FindIntegrationAnchors method available in the Seurat package to “anchor” datasets together (dims = 1:30). The anchors were then passed through the IntegrateData function to generate a batch-corrected expression matrix. The integrative UMAP plot was then generated using the uwot package.
Pseudotime analysis
Monocle v.3 was used for the pseudotime analysis. First, the UMAP plot obtained from the uwot package was passed to the Monocle object. Next, the learn_graph function was called to fit the principal pseudotime graph. The pseudotime ordering was then generated using the cell at the top of the umap2 axis as root.
Quantification and statistical analysis
Data are expressed as mean ± SD. Statistical significance was determined using Student’s t test (two-tailed) or nonparametric Mann-Whitney U test for datasets with nonnormal distribution, as indicated in the corresponding figure legends. For the t test, Welch’s correction was used for unequal variance. Boxes in all box plots extend from the 25th to 75th percentiles, with a line at the median. Whiskers show minimum and maximum values. Statistical tests were performed using Prism7 (GraphPad Software) or R. Pearson correlation was calculated using the cor function in R. P values for mRNA degradation curves are calculated using a standard t test on the interaction term between time and the experiment type in the regression model of the degradation curves. Bulk RNA-seq was performed in two biological replicates with high correlation. All other experiments were performed in at least three biological replicates and were repeated at least twice independently with similar results. Differences were considered statistically significant at P ≤ 0.05. The n values are specified in the figure legends.
Comparison between scRNA-seq and preimplantation embryo data
The raw RNA-seq preimplantation data were downloaded from GSE71434 (42) and then mapped to the mm10 using STAR. The gene expression level was quantified using RNA-Seq by Expectation-Maximization RSEM and then TMM-normalized. The differentially expressed genes between the different stages were detected using edgeR (FC > 3 and P <0.001). To compare with scRNA-seq expression, we first averaged the expression of each gene in each scRNA-seq cluster. The common genes between the variable scRNA-seq genes and the differential bulk RNA-seq genes were used to correct for batch effect using the scBatchCpp function from the scBatch package (43).
Supplementary Material
Acknowledgments
We thank R. Chen for help in scRNA-seq library construction, S. L. Pfaff for providing the MERVL-tdTomato reporter, F. Lu and X. Wu for establishing the reporter cell line, and Z. Chen for help in some experiments. This project was supported by NIH (R01HD092465) and HHMI. Y.Z. is an investigator of the Howard Hughes Medical Institute. Author contributions: Y.Z. conceived the project. X.F. performed the experiments. M.N.D. performed data analysis. X.F., M.N.D., and Y.Z. interpreted the data and wrote the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: Bulk RNA-seq and single-cell RNA-seq for the reversal of 2C-like transition reported in this paper have been deposited to NCBI Gene Expression Omnibus (GEO), GEO: GSE133234. All other RNA-seq samples used in the analysis are available on NCBI GEO, GEO: GSE121459. All other data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/22/eaay5181/DC1
REFERENCES AND NOTES
- 1.Lee M. T., Bonneau A. R., Giraldez A. J., Zygotic genome activation during the maternal-to-zygotic transition. Annu. Rev. Cell Dev. Biol. 30, 581–613 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lu F., Zhang Y., Cell totipotency: Molecular features, induction, and maintenance. Natl. Sci. Rev. 2, 217–225 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Falco G., Lee S.-L., Stanghellini I., Bassey U. C., Hamatani T., Ko M. S. H., Zscan4: A novel gene expressed exclusively in late 2-cell embryos and embryonic stem cells. Dev. Biol. 307, 539–550 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Macfarlan T. S., Gifford W. D., Driscoll S., Lettieri K., Rowe H. M., Bonanomi D., Firth A., Singer O., Trono D., Pfaff S. L., Embryonic stem cell potency fluctuates with endogenous retrovirus activity. Nature 487, 57–63 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li M., Izpisua Belmonte J. C., Deconstructing the pluripotency gene regulatory network. Nat. Cell Biol. 20, 382–392 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bošković A., Eid A., Pontabry J., Ishiuchi T., Spiegelhalter C., Ram E. V. R., Meshorer E., Torres-Padilla M. E., Higher chromatin mobility supports totipotency and precedes pluripotency in vivo. Genes Dev. 28, 1042–1047 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ishiuchi T., Enriquez-Gasca R., Mizutani E., Bošković A., Ziegler-Birling C., Rodriguez-Terrones D., Wakayama T., Vaquerizas J. M., Torres-Padilla M.-E., Early embryonic-like cells are induced by downregulating replication-dependent chromatin assembly. Nat. Struct. Mol. Biol. 22, 662–671 (2015). [DOI] [PubMed] [Google Scholar]
- 8.Yang Y., Liu B., Xu J., Wang J., Wu J., Shi C., Xu Y., Dong J., Wang C., Lai W., Zhu J., Xiong L., Zhu D., Li X., Yang W., Yamauchi T., Sugawara A., Li Z., Sun F., Li X., Li C., He A., Du Y., Wang T., Zhao C., Li H., Chi X., Zhang H., Liu Y., Li C., Duo S., Yin M., Shen H., Belmonte J. C. I., Deng H., Derivation of pluripotent stem cells with in vivo embryonic and extraembryonic potency. Cell 169, 243–257.e25 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yang J., Ryan D. J., Wang W., Tsang J. C. H., Lan G., Masaki H., Gao X., Antunes L., Yu Y., Zhu Z., Wang J., Kolodziejczyk A. A., Campos L. S., Wang C., Yang F., Zhong Z., Fu B., Eckersley-Maslin M. A., Woods M., Tanaka Y., Chen X., Wilkinson A. C., Bussell J., White J., Ramirez-Solis R., Reik W., Göttgens B., Teichmann S. A., Tam P. P. L., Nakauchi H., Zou X., Lu L., Liu P., Establishment of mouse expanded potential stem cells. Nature 550, 393–397 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bao S., Tang W. W. C., Wu B., Kim S., Li J., Li L., Kobayashi T., Lee C., Chen Y., Wei M., Li S., Dietmann S., Tang F., Li X., Surani M. A., Derivation of hypermethylated pluripotent embryonic stem cells with high potency. Cell Res. 28, 22–34 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Baker C. L., Pera M. F., Capturing totipotent stem cells. Cell Stem Cell 22, 25–34 (2018). [DOI] [PubMed] [Google Scholar]
- 12.Lu F., Liu Y., Jiang L., Yamaguchi S., Zhang Y., Role of Tet proteins in enhancer activity and telomere elongation. Genes Dev. 28, 2103–2119 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rodriguez-Terrones D., Gaume X., Ishiuchi T., Weiss A., Kopp A., Kruse K., Penning A., Vaquerizas J. M., Brino L., Torres-Padilla M.-E., A molecular roadmap for the emergence of early-embryonic-like cells in culture. Nat. Genet. 50, 106–119 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Choi Y. J., Lin C.-P., Risso D., Chen S., Kim T. A., Tan M. H., Li J. B., Wu Y., Chen C., Xuan Z., Macfarlan T., Peng W., Lloyd K. C. K., Kim S. Y., Speed T. P., He L., Deficiency of microRNA miR-34a expands cell fate potential in pluripotent stem cells. Science 355, eaag1927 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Whiddon J. L., Langford A. T., Wong C.-J., Zhong J. W., Tapscott S. J., Conservation and innovation in the DUX4-family gene network. Nat. Genet. 49, 935–940 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.De Iaco A., Planet E., Coluccio A., Verp S., Duc J., Trono D., DUX-family transcription factors regulate zygotic genome activation in placental mammals. Nat. Genet. 49, 941–945 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hendrickson P. G., Doráis J. A., Grow E. J., Whiddon J. L., Lim J. W., Wike C. L., Weaver B. D., Pflueger C., Emery B. R., Wilcox A. L., Nix D. A., Peterson C. M., Tapscott S. J., Carrell D. T., Cairns B. R., Conserved roles of mouse DUX and human DUX4 in activating cleavage-stage genes and MERVL/HERVL retrotransposons. Nat. Genet. 49, 925–934 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fu X., Wu X., Djekidel M. N., Zhang Y., Myc and Dnmt1 impede the pluripotent to totipotent state transition in embryonic stem cells. Nat. Cell Biol. 21, 835–844 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eckersley-Maslin M. A., Svensson V., Krueger C., Stubbs T. M., Giehr P., Krueger F., Miragaia R. J., Kyriakopoulos C., Berrens R. V., Milagre I., Walter J., Teichmann S. A., Reik W., MERVL/Zscan4 network activation results in transient genome-wide DNA demethylation of mESCs. Cell Rep. 17, 179–192 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xue Z., Huang K., Cai C., Cai L., Jiang C. Y., Feng Y., Liu Z., Zeng Q., Cheng L., Sun Y. E., Liu J.-y., Horvath S., Fan G., Genetic programs in human and mouse early embryos revealed by single-cell RNA sequencing. Nature 500, 593–597 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Feng Q., Snider L., Jagannathan S., Tawil R., van der Maarel S. M., Tapscott S. J., Bradley R. K., A feedback loop between nonsense-mediated decay and the retrogene DUX4 in facioscapulohumeral muscular dystrophy. eLife 4, e04996 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kebaara B. W., Atkin A. L., Long 3'-UTRs target wild-type mRNAs for nonsense-mediated mRNA decay in Saccharomyces cerevisiae. Nucleic Acids Res. 37, 2771–2778 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hogg J. R., Goff S. P., Upf1 senses 3'UTR length to potentiate mRNA decay. Cell 143, 379–389 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Unterholzner L., Izaurralde E., SMG7 acts as a molecular link between mRNA surveillance and mRNA decay. Mol. Cell 16, 587–596 (2004). [DOI] [PubMed] [Google Scholar]
- 25.Hudson B. P., Martinez-Yamout M. A., Dyson H. J., Wright P. E., Recognition of the mRNA AU-rich element by the zinc finger domain of TIS11d. Nat. Struct. Mol. Biol. 11, 257–264 (2004). [DOI] [PubMed] [Google Scholar]
- 26.Fallmann J., Sedlyarov V., Tanzer A., Kovarik P., Hofacker I. L., AREsite2: an enhanced database for the comprehensive investigation of AU/GU/U-rich elements. Nucleic Acids Res. 44, D90-5 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guo M., Zhang Y., Zhou J., Bi Y., Xu J., Xu C., Kou X., Zhao Y., Li Y., Tu Z., Liu K., Lin J., Yang P., Gao S., Wang Y., Precise temporal regulation of Dux is important for embryo development. Cell Res. 29, 956–959 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Percharde M., Lin C. J., Yin Y., Guan J., Peixoto G. A., Bulut-Karslioglu A., Biechele S., Huang B., Shen X., Ramalho-Santos M., A LINE1-Nucleolin partnership regulates early development and ESC identity. Cell 174, 391–405.e19 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen Z., Zhang Y., Loss of DUX causes minor defects in zygotic genome activation and is compatible with mouse development. Nat. Genet. 51, 947–951 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.De Iaco A., Verp S., Offner S., Grun D., Trono D., DUX is a non-essential synchronizer of zygotic genome activation. Development 147, dev177725 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.ENCODE Project Consortium , An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bhattacherjee A., Djekidel M. N., Chen R., Chen W., Tuesta W., Zhang Y., Cell type-specific transcriptional programs in mouse prefrontal cortex during adolescence and addiction. Nat. Commun. 10, 4169 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Criscione S. W., Zhang Y., Thompson W., Sedivy J. M., Neretti N., Transcriptional landscape of repetitive elements in normal and cancer human cells. BMC Genomics 15, 583 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dobin A., Davis C. A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T. R., STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Macosko E. Z., Basu A., Satija R., Nemesh J., Shekhar K., Goldman M., Tirosh I., Bialas A. R., Kamitaki N., Martersteck E. M., Trombetta J. J., Weitz D. A., Sanes J. R., Shalek A. K., Regev A., McCarroll S. A., Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell 161, 1202–1214 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Robinson M. D., Oshlack A., A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 11, R25 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Robinson M. D., McCarthy D. J., Smyth G. K., edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McCarthy D. J., Chen Y., Smyth G. K., Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res. 40, 4288–4297 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zheng G. X. Y., Terry J. M., Belgrader P., Ryvkin P., Bent Z. W., Wilson R., Ziraldo S. B., Wheeler T. D., McDermott G. P., Zhu J., Gregory M. T., Shuga J., Montesclaros L., Underwood J. G., Masquelier D. A., Nishimura S. Y., Schnall-Levin M., Wyatt P. W., Hindson C. M., Bharadwaj R., Wong A., Ness K. D., Beppu L. W., Deeg H. J., McFarland C., Loeb K. R., Valente W. J., Ericson N. G., Stevens E. A., Radich J. P., Mikkelsen T. S., Hindson B. J., Bielas J. H., Massively parallel digital transcriptional profiling of single cells. Nat. Commun. 8, 14049 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Griffiths J. A., Richard A. C., Bach K., Lun A. T. L., Marioni J. C., Detection and removal of barcode swapping in single-cell RNA-seq data. Nat. Commun. 9, 2667 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lun A. T. L., Riesenfeld S., Andrews T., Dao T. P., Gomes T.; participants in the 1st Human Cell Atlas Jamboree, Marioni J. C., EmptyDrops: Distinguishing cells from empty droplets in droplet-based single-cell RNA sequencing data. Genome Biol. 20, 63–63 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang B., Zheng H., Huang B., Li W., Xiang Y., Peng X., Ming J., Wu X., Zhang Y., Xu Q., Liu W., Kou X., Zhao Y., He W., Li C., Chen B., Li Y., Wang Q., Ma J., Yin Q., Kee K., Meng A., Gao S., Xu F., Na J., Xie W., Allelic reprogramming of the histone modification H3K4me3 in early mammalian development. Nature 537, 553–557 (2016). [DOI] [PubMed] [Google Scholar]
- 43.Fei T., Zhang T., Shi W., Yu T., Mitigating the adverse impact of batch effects in sample pattern detection. Bioinformatics 34, 2634–2641 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/22/eaay5181/DC1