

An innovative biomimetic polymer integrates zwitterionic non-fouling properties with immunomodulatory functionality.
Abstract
Superhydrophilic zwitterionic polymers are a class of nonfouling materials capable of effectively resisting any nonspecific interactions with biological systems. We designed here a functional zwitterionic polymer that achieves a trade-off between nonspecific interactions providing the nonfouling property and a specific interaction for bioactive functionality. Built from phosphoserine, an immune-signaling molecule in nature, this zwitterionic polymer exhibits both nonfouling and immunomodulatory properties. Its conjugation to uricase is shown to proactively eradicate all unwanted immune response, outperforming the zwitterionic polymers. On the other hand, this polymer could significantly prolong the half-life of protein drugs in vivo, overcoming the innate drawback of phosphoserine in inducing accelerated clearance. Our demonstration of a nonfouling zwitterionic material with built-in immunomodulatory functionality provides new insights into the fundamental design of biomaterials, as well as far-reaching implications for broad applications such as drug delivery, implants, and cell therapy.
INTRODUCTION
Zwitterions are molecules that contain equal numbers of cationic and anionic moieties while maintaining an overall neutral charge. This combination of oppositely charged groups in zwitterions grants them superior hydration capacity, prompting significant efforts in transforming zwitterions into zwitterionic polymers (1). Because of the superior hydrophilicity, polymers of zwitterions are able to effectively resist the nonspecific adsorption or “fouling” from biomolecules and microorganisms (1–3). Surfaces coated with zwitterionic polymers are “nonfouling” in complex media, inhibiting nonspecific protein adsorption in undiluted human serum to an ultralow level [<0.3 ng/cm2; (4)]. This unique capacity of zwitterionic polymers in minimizing nonspecific interactions is also highly appealing in many in vivo applications. For example, zwitterionic hydrogels have been shown to prevent the occurrence of foreign body reactions including capsule formation for at least 3 months in mice (5). Moreover, nanoparticles coated with zwitterionic polymers bear reduced interactions with biological systems and are able to escape the immune recognition, exhibiting “immune stealth” (6–9). Up to date, a number of zwitterionic polymers including poly(2-methacryloyloxyethyl phosphorylcholine) (MPC), poly(sulfobetaine), poly(carboxy betaine), and poly(trimethylamine N-oxide) have been developed (2, 3, 10), which are being extensively explored to improve the longevity and performance of medical devices, implants, and drug delivery systems. While all these efforts have revolved around the nonfouling property of bioinert zwitterionic polymers, functional zwitterionic materials are still scanty. The ability of specific molecular and biological interactions is known to render biomaterials with new functions and enhanced performance (11). Given the chemical and structural diversities of zwitterions (12), we envision that it is possible to develop a bioactive zwitterionic material by reaching a trade-off between the minimal nonspecific interaction, which is essential to nonfouling property, and the specific biointeraction that may result in bioactive functionality. To demonstrate this concept, we here designed a zwitterionic phosphoserine-mimetic polymer (ZPS; Fig. 1A) with built-in immunomodulatory functions beyond the conventional design of biologically inert zwitterionic materials.
Fig. 1. Illustration of PS-mimetic polymers.
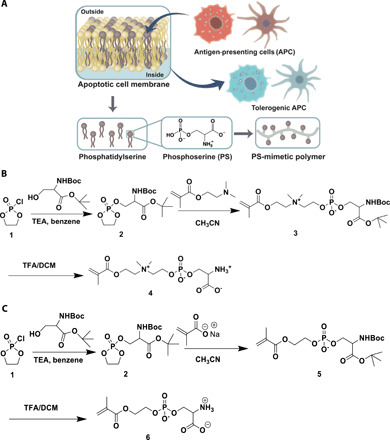
(A) Design of PS-mimetic polymers built from phosphoserine, an immunomodulatory molecule naturally occurring on the outside membranes of apoptotic cells. (B) Synthesis of the polymerizable ZPS monomer. (C) Synthesis of the polymerizable NZPS monomer.
There have been increasing interests in using biomaterials to actively regulate the immune system (13–16). Currently, unwanted activation of the immune system represents a serious threat to human health as it causes various diseases (e.g., autoimmune diseases, allergies, and chronic inflammatory diseases) and compromises the safety and efficacy of implants and biopharmaceuticals (17, 18). In particular, many protein drugs are immunogenic and can elicit immune responses that deprive patients of life-sustaining therapies and even cause lethal adverse effects (19). Clinical solutions to this challenge include the systemic bolus administration of immunosuppressants such as anti-inflammatory drugs, corticosteroids, and rapamycin. However, these treatments broadly suppress the immune system resulting in opportunistic pathogen infections. Biomaterial carriers have enabled codelivery of antigens and immunoregulators as a more specific strategy to drive immunosuppression (20, 21). For example, poly(lactide-co-glycolide) nanoparticles codelivering protein antigens and rapamycin to dendritic cells (DCs) efficiently induced antigen-specific tolerance toward the protein, which otherwise may generate immune responses (22–24). These biomaterial-aided immunosuppressants need to be preadministered or coadministered as adjunct therapies, increasing the complexity of treatment regimens as well as the burden to patients. In addition, although biomaterials delivering regulatory cues have demonstrated promise, the encapsulation, adsorption, or conjugation of immunoregulators to biomaterial vehicles is still a rather complicated process. Hence, a “drug-free” biomaterial built with the ability to directly regulate the immune response is highly preferred.
Phosphatidylserine, a natural lipid with phosphoserine (PS) as the head group, serves as an immunomodulatory signal under physiological conditions (25). During cell apoptosis, phosphatidylserine is externalized from the inner membrane to the outer membrane where it can serve as an “eat-me” signal to phagocytes, promoting the clearance of apoptotic cells by phagocytosis. Meanwhile, phosphatidylserine elicits an anti-inflammatory program in the engulfing phagocyte, induces immune tolerance to components of the apoptotic cell, and thus prevents aberrant immune response to self-antigens. Structure-binding studies have shown that the tolerogenic effect of phosphatidylserine mainly comes from its PS head group rather than its glycerol domain and fatty acid chains (26). The PS molecule is found to trigger the release of regulatory cytokines such as transforming growth factor–β (TGF-β) by directly or indirectly interacting with the PS receptor (PSR) on immune cells. Inspired by the naturally occurring immunological tolerance mediated by PS, we built a biofunctional zwitterionic polymer, ZPS, from its structure. On one hand, the specific interaction of ZPS with PSR enables it to induce immune tolerogenic effects, distinguishing ZPS from conventional bioinert zwitterionic materials such as MPC. On the other hand, the zwitterionic nature of ZPS allows it to exhibit nonfouling properties like MPC. Comparison of ZPS to a nonzwitterionic PS-mimetic polymer (NZPS) indicates that transforming PS into a zwitterionic form can subdue the innate vulnerability of PS to phagocytosis during blood circulation, thus avoiding the rapid clearance mediated by the mononuclear phagocyte system (MPS). Furthermore, to demonstrate the enhanced functionality and performance of ZPS over MPC and NZPS, we conjugated these polymers to uricase, a highly immunogenic enzyme, and examined their impacts on the immunological and pharmacokinetic (PK) profiles.
RESULTS AND DISCUSSION
Synthesis of zwitterionic and nonzwitterionic PS-mimetic monomers
The design of the ZPS monomers seeks to combine the immunomodulatory function of PS with the nonfouling property inherent to zwitterionic materials. To accomplish this, the overall synthesis of ZPS monomers involves the attachment of anionic PS to a cationic tertiary amine moiety and then to a methacrylate group, forming a polymerizable zwitterionic product (Fig. 1B). First, a five-membered cyclic phosphotriester ring attached with protected PS (dioxaphospholane ring) was generated in situ by having the hydroxyl group of protected serine to react with 2-chloro-2-oxo-1,3,2-dioxaphospholane. Then, the nucleophilic ring opening of this five-membered dioxaphopholane ring in nonprotic media with an amine nucleophile, N,N′-dimethylaminoethyl methacrylamide could generate protected ZPS monomer, which subsequently was purified using flash column chromatography. Last, ZPS monomer was collected after the deprotection step using trifluoroacetic acid. As a nonzwitterionic control, NZPS retaining the anionic property of PS was also synthesized in parallel. The synthesis of NZPS is similar to that of ZPS except for using a carboxylate anion (nucleophile) from sodium methacrylate instead to open the five-membered cyclic dioxaphopholane ring (Fig. 1C). The cleavage of the dioxaphospholane ring was readily achieved using highly reactive anionic reagents rather than neutral nucleophiles. During the synthesis of NZPS, 18-crown-6-ether was necessitated to increase the nucleophilicity of carboxylate anion by chelating with the positive sodium ion. It should be noted that ZPS with a neutral charge balance was observed to have better solubility in water than that of the negatively charged NZPS.
Resistance to nonspecific protein and cell adsorption
The adsorption of biomolecules and adhesion of cells to biomaterials may cause the loss of their functions and trigger a variety of iatrogenic complications such as foreign-body reactions, inflammation, and rapid clearance (10). Hence, biofouling resistance has been a critical property to prevent biomaterials from failure in complex biological environments. Superhydrophilic zwitterionic polymers are renowned for their excellent nonfouling properties as they can efficiently create hydration shells that repel the nonspecific adsorption of biomolecules or adhesion of cells (27). Here, we examined this characteristic in ZPS by testing its ability to resist the nonspecific adsorption of fibrinogen, a plasma protein abundant in the blood. Both ZPS and NZPS hydrogel disks (D = 5 mm) were prepared and exposed to a highly concentrated fibrinogen solution (10 mg/ml). Hydrogels made of MPC, a zwitterionic phosphocholine-derived polymer known with very good nonfouling property, and tissue culture polystyrene (TCPS) disks in a similar size were prepared and tested in parallel as negative and positive controls, respectively. After 2-hour incubation, the amount of adsorbed fibrinogen onto each disk sample was quantitatively analyzed via enzyme-linked immunosorbent assay (ELISA). Compared with the TCPS disk, ZPS, NZPS, and MPC hydrogels have displayed varied capacity in reducing fibrinogen adsorption (Fig. 2A). While NZPS reduced 40% fibrinogen adsorption, ZPS exhibited better nonfouling performance by reducing 85% fibrinogen adsorption, similar to that of MPC. This result indicates that like other zwitterionic materials, ZPS is also resistant to nonspecific protein adsorption due to enhanced hydration effect driven by zwitterions. Moreover, to evaluate the resistance of each polymer against the adhesion of immune cells, we seeded RAW 264.7 macrophages onto TCPS disks, ZPS, NZPS, and MPC hydrogels and analyzed the number of adhered cells after 1-day cell culture (fig. S5). Consistent with the result in protein adsorption, a large number of macrophages were also observed on TCPS disks (>300 cells per 1 mm2). Hydrogels made from two zwitterionic polymers, MPC and ZPS, both exhibited much better nonfouling property than NZPS hydrogel (~80 cells per 1 mm2). ZPS hydrogel (~13 cells per 1 mm2) showed slightly higher susceptibility to macrophage adhesion than MPC (~2 cells per 1 mm2). This result indicates that while ZPS could effectively resist nonspecific protein adsorption, it still maintains the ability of PS to specifically interact with immune cells, resulting in a potential trade-off of ZPS between nonspecific and specific interactions.
Fig. 2. Test of nonfouling property, immunomodulatory effect, and phagocytosis.
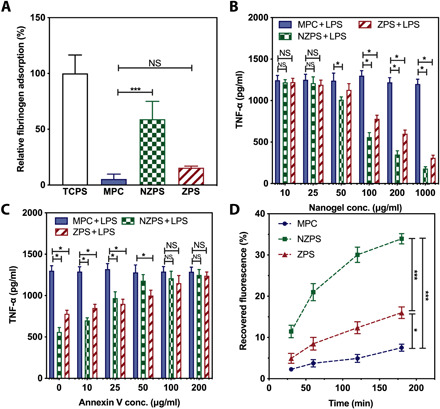
(A) Fibrinogen adsorbed onto TCPS, MPC, NZPS, and ZPS hydrogel surfaces measured by ELISA. (B) RAW 264.7 macrophages (105 per well) were treated with the MPC, NZPS, or ZPS nanogels at various concentrations (10, 25, 50, 100, 200, and 1000 μg/ml) for 18 hours followed by the stimulation of LPS (100 ng/ml) for 48 hours. The level of TNF-α secretion in the supernatant was measured by an ELISA kit. (C) The MPC, NZPS, or ZPS nanogels (100 μg/ml) were preincubated with an annexin V solution at various concentrations (0, 10, 25, 50, 100, and 200 μg/ml) for 6 hours. RAW 264.7 macrophages (105 per well) were then treated with these nanogels (100 μg/ml) for 18 hours followed by the stimulation of LPS (100 ng/ml) for 48 hours. The level of TNF-α secretion in the supernatant was measured by the ELISA kit. (D) RAW 264.7 macrophages (105 per well) were incubated with MPC, NZPS, or ZPS nanogels encapsulating FITC-BSA for 30, 60, 120, and 180 min, after which the cells were washed and lysed for the detection of recovered fluorescence. Statistical significance was determined using Student’s t test. NS, no significance. *P < 0.05 and ***P < 0.001. Data are represented as mean ± SEM.
Immunomodulatory effect of PS-mimetic polymers
During cell apoptosis, natural phosphatidylserines exposed on the outer membranes of apoptotic cells can prevent the occurrence of unexpected immune activation by directing antigen-presenting cells, such as macrophages and DCs that differentiate into the tolerogenic phenotype, and promoting the release of anti-inflammatory cytokines (28). To examine whether the PS-mimetic polymers can emulate the modulating effect of apoptotic cells and elicit anti-inflammatory responses in activated macrophages, we investigated the potential effect of ZPS, NZPS, and MPC on RAW 264.7 macrophages in the presence of lipopolysaccharide (LPS), an immunostimulatory molecule. For a fair comparison, ZPS, NZPS, and MPC were prepared into nanogels with uniform sizes (~40 nm) and surfaces (table S1). The zeta potential of anionic NZPS nanogel was negative (~−20 mv), while those of zwitterionic ZPS and MPC nanogels were close to neutral. RAW 264.7 macrophages (105 per well) were treated with nanogels composed of MPC, NZPS, or ZPS at various concentrations (10, 25, 50, 100, 200, and 1000 μg/ml) for 18 hours followed by the stimulation of LPS (100 ng/ml). The status of macrophage was evaluated by measuring the production and release of proinflammatory cytokines [tumor necrosis factor–α (TNF-α), interleukin-6 (IL-6), and CXCL-10] in the supernatant. Macrophages exposed to MPC nanogels were still activated by LPS and were directed toward proinflammatory phenotypes, as evidenced by the high level of proinflammatory cytokines (Fig. 2B and fig. S6, A and B). By contrast, NZPS and ZPS nanogels effectively mitigated the activation of macrophages in a dose-dependent manner, demonstrating the immunomodulatory function of PS-mimetic polymers.
To further confirm the role of PS-mediated pathway in the immunomodulation process, a blocking experiment was conducted using annexin V, a protein that specifically binds PS. The binding of annexin V is known to block the interaction of PS with PSR or bridging molecules and thus inhibits the immune tolerogenic effect of PS (29). Here, nanogels composed of MPC, NZPS or ZPS (100 μg/ml) were preincubated with annexin V solution at various concentrations (0, 10, 25, 50, 100, and 200 μg/ml) for 6 hours before being added to RAW 264.7 macrophages (105 per well). After 18-hour incubation with nanogels, macrophages were stimulated with LPS (100 ng/ml), followed by the detection of proinflammatory cytokines (TNF-α, IL-6, and CXCL-10) in the supernatant. As shown in Fig. 2C and fig. S6 (C and D), the status of macrophages exposed to MPC nanogels was not affected by annexin V, displaying a persistently high level of proinflammatory cytokines. On the other hand, the escalation in the concentration of annexin V led to an increased level of TNF-α in macrophages incubated with NZPS and ZPS nanogels, indicating the attenuated anti-inflammatory effects of PS-mimetic polymers. Such a blocking effect of annexin V confirms the essential role of PS-mediated immune signaling in enabling the immunomodulatory function of NZPS and ZPS. The interaction of PS-mimetic polymers with annexin V was reflected in the change of nanogel size. Results show that while the size of MPC nanogel was unaffected after the incubation with annexin V, the sizes of NZPS and ZPS nanogels both increased with the elevated concentration of annexin V, which suggests the specific adsorption of annexin V to their surfaces (fig. S7).
In addition to the immunoregulatory role, PS also serves as an eat-me signal since its binding to PSR or bridging molecules such as MFG-E8 and annexing A1 and A2 could trigger the engulfment by phagocytes and thus promote the clearance of dying cells (30–32). To interrogate the interaction of PS-mimetic polymers with the immune cells, we studied the phagocytosis of PS-mimetic polymers by macrophages. To quantify cell uptake, nanogels were loaded with fluorescein isothiocyanate–bovine serum albumin (FITC-BSA) for the purpose of fluorescent labeling. FITC-BSA could be stably retained inside the nanogels as its hydrodynamic size is larger than the pore size of nanogels. Then, these fluorescently labeled nanogels (MPC, NZPS, and ZPS) were respectively incubated with RAW 264.7 macrophages (105 per well) for 30, 60, 120, and 180 min. At each time point, cells were washed and lysed for the detection of recovered fluorescence (Fig. 2D). Consistent with cell adhesion test, MPC displayed a slow uptake due to its resistance to interactions with macrophages, while NZPS was quickly taken up by macrophages as NZPS well retains the immunomodulatory function of PS as an eat-me signal in facilitating the uptake by phagocytes. It should be noted that such a high vulnerability of NZPS to phagocytosis is an undesirable feature in biomedical applications that require longevity. In comparison, the uptake of ZPS by macrophages, although slightly quicker than MPC, was much delayed compared with NZPS, confirming that the zwitterionic property of ZPS decreased the binding affinity of its PS head groups to PSR. The reduced susceptibility of ZPS to phagocytosis is favorable to overcome the innate limitation of PS as an eat-me signal and is highly meaningful to the clinical translation of this naturally occurring immunomodulatory molecule.
Effect of PS-mimetic polymers on the immunogenicity and PK of proteins
In the past decades, advances in biotechnology have significantly boosted the discovery of proteins with high potency and selectivity, enriching the arsenal of drug candidates against diverse diseases (33). However, applying proteins to clinical use remains a challenging task due to their immunogenic nature and inadequate circulation time (34–36). To improve the immunological and PK profiles of protein drugs, a variety of strategies have been proposed, among which polymer conjugation is the most straightforward and widely used one (23, 37–39). Covalent binding of bulky hydrophilic polymers such as poly(ethylene glycol) (PEG) (40, 41) and zwitterionic polymers can physically shield proteins from immune recognition, thus alleviating their immunogenicity in a passive manner (42). Yet, many immunogenic proteins only contain limited accessible functional groups for conjugation, limiting the density of the polymer to fully cover protein surfaces that can be grafted onto protein surfaces (43). As a result, the incomplete cloak afforded by sparse polymers is oftentimes not able to eradicate the unwanted immune response. Typical examples include pegloticase, a PEGylated uricase approved by the U.S. Food and Drug Administration in 2010 for the treatment of refractory chronic gout. During the clinical use of pegloticase, more than 40% of patients developed high-titer anti-drug antibodies (ADA), resulting in the nonresponsiveness of treatment and even life-threatening adverse reactions (44–46).
Recognizing the shortcoming of conventional polymers, we envision that PS-mimetic polymers hold the potential to fully address the immunogenic issue as they may not only supply proteins with physical protections but also incorporate immunomodulatory cues into protein formulations, proactively preventing the immune response. To test this presumption, we covalently attached MPC, NZPS, and ZPS polymers to the surface of uricase using an in situ graft-from method (10), producing MPC-uricase, NZPS-uricase, and ZPS-uricase conjugates with similar hydrodynamic sizes (fig. S8 and table S2). The conjugation of polymers to proteins may reduce the activity of protein because of steric blockage of active sites. In the case of uricase, its bioactivity was maintained after polymer conjugation (>95%) because the substrate of this enzyme, uric acid, is small and can diffuse readily. The first step of protein-induced immune responses involves the uptake and processing by DCs, which will direct the subsequent differentiation of T cells and determine whether there will be an immunogenic or a tolerogenic response. Hence, we first evaluated the immunogenicity of each uricase sample by examining their impact on the status of DCs. As shown in Fig. 3, A and B, the exposure to native uricase induced a significant increase in the expression of surface molecules associated with DC maturation (CD40+ CD80+), and as a result, less than 50% DCs were still in purely immature status (CD40−CD80−) at the end of 72-hour incubation. By contrast, the conjugation of MPC, NZPS, and ZPS lowered the immunostimulatory effect of uricase on DCs at varied degrees, and higher percentages of immature DCs (>60%) were correspondingly retained. Furthermore, to understand the mechanism of polymer conjugation in immunogenicity mitigation, supernatants of culture media were collected for the measurement of immunomodulatory cytokines (TGF-β and IL-10) and immunostimulatory cytokine (IL-6). As typical immunomodulatory cytokines, both TGF-β and IL-10 can actively promote the differentiation of tolerogenic DCs and T regulatory (Treg) cells (47, 48). Compared with native uricase, MPC-uricase inhibited the secretion of IL-6 by DCs but did not affect the secretion of TGF-β and IL-10, indicating that MPC conjugation could alleviate the immunogenicity of uricase in a passive manner such as reducing protein endocytosis, interfering with protein proteolysis, and physically blocking major histocompatability complex II–epitope binding (Fig. 3C and fig. S9, A and B). In contrast, NZPS-uricase and ZPS-uricase up-regulated the secretion of TGF-β and IL-10 while effectively attenuating the secretion of IL-6. This result suggests that the relatively low tendency of NZPS-uricase and ZPS-uricase to provoke DC maturation might stem from their modulatory effect on immune cells. Such a proactive function distinguishes the conjugation of PS-mimetic polymers from conventional polymer conjugation techniques.
Fig. 3. In vitro immunogenicity of polymer-uricase conjugates.
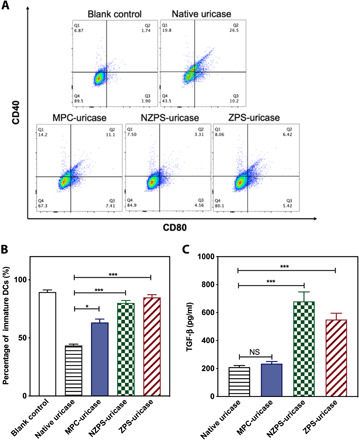
(A) DC 2.4 dendritic cells were incubated with native uricase, MPC-uricase, NZPS-uricase, or ZPS-uricase conjugates for 72 hours and stained for flow cytometry. (B) Summary of the percentage of DCs that maintained an immature status (CD40− CD80−). (C) The secretion of TGF-β into the supernatant was detected by an ELISA kit. Statistical significance was determined using Student’s t test. *P < 0.05 and ***P < 0.001. Data are represented as mean ± SEM.
To further demonstrate the benefit of NZPS and ZPS in improving immunological profiles of proteins, three intravenous injections of native uricase and polymer-uricase conjugates were respectively performed on mice at one dose per week. Mice were euthanized on the 21st day, and their sera were harvested for the detection of antibodies specific to uricase or uricase conjugates via ELISA test. Results indicate that while MPC-uricase displayed a reduced level of ADA formation [immunoglobulin M (IgM) titers, 1:400; IgG titers, 1:1600), much lower than that detected in the native uricase group (IgM titers, 1:1600; IgG titers, >1:25,600), the conjugation of MPC failed to completely prohibit the immune response (Fig. 4, A and B). In stark contrast, negligible levels of ADA were detected (IgM titers, <200; IgG titers, <200) in the mice treated with NZPS-uricase (Fig. 4C) or ZPS-uricase conjugates (Fig. 4D), demonstrating that the conjugation of PS-mimetic polymers with built-in immunomodulatory functions is more effective in eliminating the protein immunogenicity. Moreover, mouse spleens were harvested from each cohort for the extraction of splenocytes, which were cultured with each corresponding uricase sample. Consistent with the antibody test, an elevated secretion of IL-4, a marker of stimulated helper T cell proliferation, was detected in the cohorts treated with native uricase. In comparison, conjugation of three polymers could all attenuate the secretion of IL-4, while NZPS and ZPS could more effectively reduce the level of IL-4 than MPC (fig. S10). Furthermore, compared with native uricase and MPC-uricase, NZPS-uricase and ZPS-uricase significantly expanded the frequency of Treg cells, which play an essential role in modulating immune responses (Fig. 4, C and D). Hence, the conjugation of NZPS and ZPS polymers could tolerize immune systems to the underlying proteins and thus proactively prevent potential immune responses. The effect of these PS-mimetic polymers in inhibiting the protein immunogenicity is fundamentally distinct from that of inert polymers such as MPC and explains why NZPS-uricase and ZPS-uricase are more effective than MPC-uricase in eliminating the production of ADA.
Fig. 4. In vivo immunogenicity of polymer-uricase conjugates.
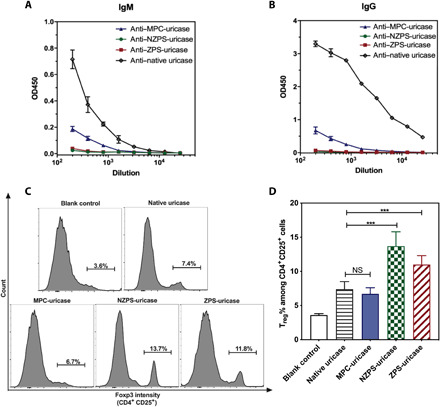
After three weekly administrations of uricase samples, mice were euthanized on the 21st day, and their sera were harvested for the detection of IgM (A) and IgG (B) specific to uricase or uricase conjugates via ELISA test. The mouse spleen was also harvested for the extraction of splenocytes, which were cultured in the presence of native uricase, ZPS-uricase, NZPS-uricase, or MPC-uricase conjugates for 72 hours and then stained for flow cytometry analysis (C). Summary of the percentage of Treg phenotype (Foxp3+) cells among CD4+ CD25+ splenocytes (D). Statistical significance was determined using Student’s t test.***P < 0.001. Data are represented as mean ± SEM. OD450, optical density at 450 nm.
For the tolerogenic effects of PS to be clinically relevant, attempts that directly use PS for immunogenicity mitigation have been previously made (49–52). However, PS as an eat-me signal can trigger rapid clearance by MPS, resulting in an inadequate retention time of drug formulations in vivo (31). For example, liposome formulations containing PS encountered severely reduced circulation half-lives (53). Given that the introduction of zwitterionic property in ZPS delayed the phagocytosis of ZPS by macrophages as compared with that of NZPS, we hypothesized that the adoption of ZPS might hurdle the innate drawback of PS in short circulation.
To study the PK profiles of uricase conjugated with PS-mimetic polymers, intravenous injections of native uricase, and uricase conjugated with MPC, NZPS or ZPS were performed on mice for three consecutive weeks, during which the rat serum was collected during the first and third week at various time points for the measurement of uricase concentration. The concentration-time kinetics of each uricase sample was fitted into the two-compartment model, based on which the PK parameters were derived (table S3). As shown in Fig. 5 (A and B), the first dose of MPC-uricase displayed an extended circulating behavior (t1/2β = 30.1 hours), nearly threefold longer than that of native uricase (t1/2β = 11.2 hours). However, the half-life of MPC-uricase in the blood dropped by 30% after three repetitive administrations. The shrunken PK profiles of MPC-uricase could be attributed to the accelerated blood clearance (ABC) driven by ADA. In contrast, the circulation time of NZPS-uricase had been consistent over the three administrations, without incurring the ABC effect due to the elimination of ADA. However, the half-lives of NZPS-uricase in the first dose and third dose were both limited (~15 hours), much shorter than that of MPC-uricase (Fig. 5C). This observation agrees with results in the phagocytosis study, which reveals that while NZPS retains the high activity of PS in immunomodulation, it also well emulates the function of PS as an eat-me signal, facilitating the rapid clearance by MPS. In other words, although NZPS conjugation could improve the immunological profiles of biological drugs, it would limit their PK profiles. Compared with MPC and NZPS, ZPS with dual functions in nonfouling and immunomodulation was demonstrated to not only get rid of the ABC effect but also persistently prolong the circulation time of uricase (first dose t1/2β = 27.9 hours, third dose t1/2β = 27.1 hours) (Fig. 5D). This result confirms that the susceptibility of PS to MPS clearance was attenuated in ZPS, while the immunomodulatory function of PS was well retained, suggesting ZPS as an effective tool to improve the safety and efficacy of biologic drugs.
Fig. 5. PK of polymer-uricase conjugates.
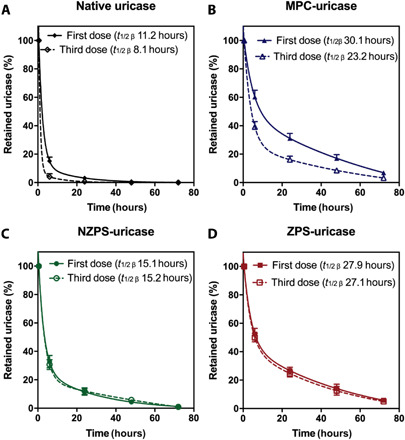
Circulation time of native uricase (A), MPC-uricase (B), NZPS-uricase (C), and ZPS-uricase (D) conjugates after the first and third IV injection in mice. Data are represented as mean ± SEM.
In previous studies, antigen-specific immunomodulatory nanoparticles have been reported to proactively inhibit the wanted immune responses (20). However, in the case of protein drugs, polymer conjugation is still necessitated for the purpose of PK enhancement. Furthermore, extra administrations of those immunosuppressants and polymer-conjugated protein therapeutics complicate the treatment regimen and increase the burden of patients. In this context, ZPS conjugation provides a simple yet effective tactic to simultaneously realize the twin incentives of immunogenicity elimination and circulation extension. As always, efficacy combined with simplicity is the key to successful clinical implementation on a large scale. The adoption of ZPS can accelerate the development of safe and efficacious biopharmaceuticals by saving the efforts in increasing accessible functional groups on proteins and truncating immunogenic epitopes via protein engineering. Moreover, we also envision ZPS as broadly useful for other applications that require immunomodulation such as medical devices, implants, and cell therapy.
CONCLUSIONS
Beyond the historically pursued conventional zwitterionic polymer with single nonfouling property, we successfully developed the first zwitterionic polymer integrated with immunomodulatory function, ZPS. Unlike zwitterionic MPC that is nonfouling but bioinert, ZPS is capable of actively suppressing undesired immune system activation. Distinct from NZPS that mimics the immunosuppressive function of PS but is nonzwitterionic, ZPS overcomes the innate drawback of PS in facilitating phagocytosis, displaying reduced vulnerability to phagocyte clearance. ZPS conjugated to uricase manifested to proactively eliminate the immunogenicity of uricase while significantly enhancing its PK profiles. To the best of our knowledge, ZPS represents the first polymer that simultaneously achieved nonfouling and immunomodulatory functions, which were generally considered to be distinct or opposite and has never been accomplished in one single material. This work sets new directions for the development of functional zwitterionic materials such as integrating bioactive properties into inert materials or reducing the nonspecific interaction of active biomolecules with complex media.
MATERIALS AND METHODS
Materials
Fibrinogen, FITC-BSA conjugate, uricase (Candida sp.), and all chemicals were purchased from Sigma-Aldrich unless otherwise noted and were used as received. Pierce 660 nm Protein Assay Reagent was purchased from Thermo Fisher Scientific. Goat anti-mouse IgM antibody and goat anti-mouse IgG antibody were purchased from Bethyl Laboratories Inc. TNF-α, IL-4, IL-6, IL-10, CXCL-10, and TGF-β Quantikine ELISA kits were purchased from R&D Systems. Anti–CD40-FITC, anti–CD80–phycoerythrin (PE), anti–CD4-PE, anti–CD25-FITC, and anti–Foxp3-Percep antibodies were purchased from BD Bioscience. PEG polymers with different molecule weights were purchased from BroadPharm. Mouse macrophages RAW 264.7 were purchased from American Type Culture Collection. Mouse dendritic cell line DC 2.4 was received from K. L. Rock (University of Massachusetts Medical Center, Worcester, MA) as a gift.
Synthesis of compound 3
N-Boc-O-tert-butyl-l-serine (N-Boc-Ser-OtBu; 1.0 g, 3.82 mmol) was dissolved in 30 ml of dry benzene, and the solution was cooled to 0°C. Next, triethylamine (0.62 ml, 4.6 mmol) was added followed by dropwise addition of 2-chloro-2-oxo-1,3,2-dioxaphospholane 1 (0.42 ml, 4.6 mmol) in 10 ml of dry benzene over a period of 30 min, and then the reaction contents were stirred at room temperature for another 3 hours. After completion of reaction, diethyl ether was poured into the reaction mixture, and the precipitated trimethylamine hydrochloride was filtered off. The filtrate was then concentrated under reduced pressure to give Compound 2 as an oil, which was used in next step without further purification. Compound 2 was redissolved in 30 ml of anhydrous acetonitrile, and 2-(dimethylamino)ethyl methacrylate (1.45 ml, 8.8 mmol) was added. The reaction mixture was then stirred at 55°C for 24 hours. The reaction contents were then concentrated under vacuum and purified by flash column chromatography to give compound 3 in 42% yield. 1H NMR (300 MHz, CDCl3) δ 6.18 (s, 1H), 5.67 (s, 1H), 4.63 to 4.54 (m, 2H), 4.35 to 4.14 (m, 3H), 4.10 to 3.99 (m, 2H), 3.83 to 3.76 (m, 2H), 3.40 to 3.27 (m, 2H), 2.82 (s, 6H), 1.97 (s, 3H), 1.46 (d, J = 11.7 Hz, 18H).
Synthesis of ZPS monomer (compound 4)
Compound 3 (0.84 g, 1.6 mmol) was dissolved in 5 ml of dichloromethane (DCM), and 30 ml of trifluoroacetic acid (TFA) was added. The reaction contents were stirred for 5 hours. After completion of the reaction, the reaction mixture was concentrated under vacuum to give a thick viscous liquid. The crude product was then crystallized with MeOH:diethyl ether (1:15) to give desired Compound 4 as white powder. 1H NMR (300 MHz, D2O) δ 6.04 (s, 1H), 5.63 (s, 1H), 4.44 to 4.35 (m, 2H), 4.31 to 4.11 (m, 2H), 3.95 to 3.76 (m, 3H), 3.67 to 3.60 (m, 2H), 3.46 to 3.38 (m, 2H), 2.83 (s, 6H), 1.79 (s, 3H).
Synthesis of compound 5
N-Boc-Ser-OtBu (1.0 g, 3.82 mmol) was dissolved in 30 ml of dry benzene, and the solution was cooled to 0°C. Next, triethylamine (0.62 ml, 4.6 mmol) was added followed by dropwise addition of 2-chloro-2-oxo-1,3,2-dioxaphospholane 1 (0.42 ml, 4.6 mmol) in 10 ml of dry benzene over a period of 30 min, and then the reaction contents were stirred at room temperature for another 3 hours. After completion of reaction, diethyl ether was poured into the reaction mixture, and the precipitated trimethylamine hydrochloride was filtered off. The filtrate was then concentrated under reduced pressure to give Compound 2 as an oil, which was used in next step without further purification. Compound 2 was redissolved in 30 ml of anhydrous acetonitrile, and sodium methacrylate (0.62 g, 5.7 mmol) along with 18-crown-6 ether (0.14 g, 0.53 mmol) was added to it. The reaction mixture was then stirred at 55°C for 72 hours. After the reaction, the reactions contents were filtered, concentrated under vacuum and purified by flash column chromatography to give compound 5 in 71% yield. 1H NMR (300 MHz, CDCl3) δ 6.15 (s, 1H), 5.57 (s, 1H), 4.41 to 4.27 (m, 2H), 4.24 to 4.04 (m, 4H), 3.86 to 3.78 (m, 1H), 1.95 (s, 3H), 1.46 (d, J = 5.4 Hz, 18H).
Synthesis of NZPS monomer (compound 6)
Compound 5 (1.2 g, 2.65 mmol) was dissolved in 5 ml of DCM, and 30 ml of TFA was added to it. The reaction contents were stirred for 5 hours. After completion of the reaction, the reaction mixture was concentrated under vacuum to give a thick viscous liquid. The crude product was then crystallized with MeOH:diethyl ether (1:20) to give desired Compound 6 as white powder. 1H NMR (300 MHz, D2O) δ 6.09 (s, 1H), 5.65 (s, 1H), 4.33 to 4.12 (m, 6H), 4.09 to 3.96 (m, 3H), 1.85 (s, 3H).
Preparation of hydrogels
ZPS hydrogel were fabricated by bulk photopolymerization with a hydrogel aqueous solution containing ZPS monomer (0.67 g of Milli-Q water, 330 mg), cross-linker N,N′ methylenebis(acrylamide) (MBA; 1 weight %, 3.3 mg), and photoinitiator 2-hydroxy-2-methylpropiophenone (0.33 mg). The hydrogel aqueous solution was placed between two glass slides separated by a 0.5-mm-thick polytetrafluoroethylene spacer and was then photopolymerized at room temperature for 30 min. After polymerization, hydrogels were removed from the casts and soaked in phosphate-buffered saline (PBS) for 3 days to remove unreacted chemicals and reach the fully hydrated hydrogel network. PBS was refreshed every 12 hours. Following the same protocol, MPC and NZPS hydrogels with the same cross-linking density were prepared.
Fibrinogen adsorption test
Biopsy punches were used to punch the hydrated MPC, NZPS, and ZPS hydrogels into 5-mm-diameter disks. Hydrogel disks were placed into a 24-well plate and incubated with 1 ml of fibrinogen in PBS buffer (1 mg/ml) for 1 hour, followed by five washes with pure PBS buffer. Hydrogel disks were then transferred to new wells and incubated with 1 ml of horseradish peroxidase (HRP)–conjugated anti-fibrinogen antibody (1 μg/ml) in PBS buffer for 1 hour. All hydrogel disks were then transferred to new wells after five washes with pure PBS buffer. Next, 1 ml of o-phenylenediamine (1 mg/ml) and 0.1 M citrate phosphate (pH 5.0) solution, containing 0.03% hydrogen peroxide was added. After 15-min incubation, the enzymatic reaction was stopped by adding an equal volume of 1 M HCl. The same procedure was conducted on of TCPS disks with the same surface area as the control. Absorbance value at 492 nm was recorded by a plate reader and was normalized to that TCPS sample. Average data were acquired from three specimens.
Cell adsorption test
RAW 264.7 macrophages were used for the cell adhesion test. The hydrogel samples were equilibrated in PBS (pH 7.4) buffer for 5 days by exchanging the buffer every day. The hydrogel samples were cut into a disk shape of 5 mm and sterilized under ultraviolet (UV) light (254 nm) for 60 min before the cell adhesion test. The macrophages were seeded on the hydrogel samples and TCPS at a density of 7.5 × 105 cells/ml and cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum for 24 hours in a 5% CO2 humidified atmosphere at 37°C. Afterward, the nonadherent macrophages were removed by rinsing with PBS buffer. Images of the adherent macrophages were obtained with a phase-contrast microscope (Nikon Eclipse TE2000-U) equipped with a digital camera using a 20× objective lens. Three replicates of each sample type including the control were analyzed, and three images were acquired for each sample on different areas.
Preparation of nanogels
Preparation of MPC, NZPS, and ZPS nanogels: sodium bis(2-ethylhexyl) sulfosuccinate (AOT; 237 mg) and poly(ethylene glycol) dodecyl ether (Brij 30, 459 mg) were added to a 20-ml glass vial to which a stir bar was added. The vial was sealed with a Teflon-lined septum cap and purged with dry nitrogen for 10 min. Nitrogen-deoxygenated hexane (10 ml) was then added to the vial under vigorous stirring. For the aqueous phase, ZPS monomers (20 mg) and cross-linker, MBA (2 mg), were dissolved in 200 μl of 10 mM Hepes buffer [140 mM NaCl and 2.5 mM CaCl2 (pH 7.4)]. Dry nitrogen was bubbled through the monomer solution for 2 min, after which the aqueous phase was slowly added to the organic continuous phase dropwise. The vial was sonicated to form a stable nanoemulsion. A 20% (w/v) solution of ammonium persulfate in deionized water (10 μl) was then added to the emulsion. After 5 min, polymerization was initiated by the addition of tetramethylethylenediamine (TEMED; 6 μl) and maintained at 4°C under rapid magnetic stirring. After the 2-hour reaction, the organic solvent was removed by rotary evaporator, and the nanogel was precipitated and washed with tetrahydrofuran (THF) three times. The nanogel was resuspended in 10 mM Hepes buffer [140 mM NaCl and 2.5 mM CaCl2 (pH 7.4)] and purified with 100-kDa molecular weight cutoff centrifugal filters to remove the unreacted monomer and cross-linker. Following the same protocol, MPC and NZPS hydrogels were prepared and purified. The size and zeta potential of nanogels were measured by Malvern Zetasizer. A similar protocol was followed for the preparation of fluorescent nanogels. Briefly, FITC-BSA (1 mg) was added into the aqueous solution of a monomer and a cross-linker in 200 μl of 10 mM Hepes buffer. Dry nitrogen was bubbled through the monomer solution for 2 min, after which the aqueous phase was slowly added to the organic continuous phase [AOT (237 mg) and Brij 30 (459 mg) in 10 ml of hexane] dropwise. The vial was sonicated to form a stable nanoemulsion. A 20% (w/v) solution of ammonium persulfate in deionized water (10 μl) was then added to the emulsion. After 5 min, polymerization was initiated by the addition of TEMED (6 μl) and maintained at 4°C under rapid magnetic stirring. After the 2-hour reaction, the organic solvent was removed by a rotary evaporator, and the nanogel was precipitated and washed with THF three times. The nanogel was resuspended in 10 mM Hepes buffer [140 mM NaCl and 2.5 mM CaCl2 (pH 7.4)] and purified with 100-kDa molecular weight cutoff centrifugal filters to remove the unloaded FITC-BSA and unreacted monomer/cross-linker. The encapsulation efficiency of FITC-BSA in each nanogel was determined by the loaded quantity of BSA protein using the Pierce 660 nm Protein Assay Kit and was around 50%.
Immunomodulatory effect test
RAW 264.7 cells were seeded (105 per well) and were exposed to MPC, NZPS, or ZPS nanogel solution at various concentrations (10, 25, 50, 100, 200, and 1000 μg/ml) for 18 hours. Then, these cells were stimulated by LPS solution (100 ng/ml) for another 48 hours, after which the cells were spun at 300g for 10 min. The level of TNF-α secretion in the supernatant was measured by a TNF-α Quantikine ELISA kit.
Annexin V–blocking experiment
Annexin V stock solution was prepared in 10 mM Hepes buffer [140 mM NaCl and 2.5 mM CaCl2 (pH 7.4)]. MPC, NZPS, or ZPS nanogel solutions (100 μg/ml) were preincubated with annexin V at various concentrations (0, 10, 25, 50, 100, and 200 μg/ml) for 6 hours and then concentrated using 10-kDa molecular weight cutoff centrifugal filters. Subsequently, RAW 264.7 macrophages (105 per well) were treated with these nanogel solutions (100 μg/ml) for 18 hours, followed by the stimulation of LPS (100 ng/ml) for 48 hours. The level of TNF-α secretion in the supernatant was measured by the TNF-α Quantikine ELISA kit. In a separate experiment, nanogels (100 μg/ml) were incubated with annexin V at various concentrations (0, 10, 25, 50, 100, and 200 μg/ml) for 30 min and then washed with the Hepes buffer using 100-kDa molecular weight cutoff centrifugal filters. The size of nanogels were measured by Malvern Zetasizer.
Phagocytosis assay
To visualize the phagocytosis of nanogels, fluorescent nanogels containing FITC-BSA were used. RAW 264.7 macrophages (105 per well) were incubated with MPC, NZPS, and ZPS nanogel encapsulating FITC-BSA (100 μg/ml) at 37°C for 30, 60, 120, and 180 min, after which the cells were washed three times with PBS buffer. Then, cells were lysed for the detection of fluorescence, and the percentage of recovered fluorescence was calculated as recovered fluorescence intensity/total fluorescent intensity at time 0.
Preparation of polymer-uricase conjugates
The uricase conjugates were prepared by grafting polymers from the surface of uricase. Briefly, uricase was first modified to introduce acryloyl group onto its surface. The reaction was performed by dissolving 2 mg uricase into 1 ml 50 mM Hepes buffer (pH 8.5), followed by adding 8 μl of N-acryloxysuccinimide (NAS) dimethyl sulfoxide solution (20 mg/ml) dropwise. The reaction was stirred at 4°C for 2 hours. The NAS-modified uricase was purified by 10-kDa molecular weight cutoff centrifugal filters.
AOT (237 mg) and Brij 30 (459 mg) were added to a 20-ml glass vial to which a stir bar was added. The vial was sealed with a Teflon-lined septum cap and purged with dry nitrogen for 10 min. Nitrogen-deoxygenated hexane (10 ml) was then added to the vial under vigorous stirring. For the aqueous phase, as-prepared NAS-modified uricase (2 mg) were mixed with ZPS monomers (60 mg) in 400 μl of 10 mM Hepes buffer [140 mM NaCl and 2.5 mM CaCl2 (pH 7.4)]. Dry nitrogen was bubbled through the monomer and protein solution for 2 min, after which the aqueous phase was slowly added to the organic continuous phase dropwise. The vial was sonicated to form a stable microemulsion. A 20% (w/v) solution of APS (15 μl) in Milli-Q water was then added to the emulsion. After 5 min, polymerization was initiated by the addition of TEMED (9 μl) and maintained at 4°C under rapid magnetic stirring. After the 2-hour reaction, the organic solvent was removed by rotary evaporator and the ZPS-uricase conjugate was precipitated and washed with THF three times. The ZPS-uricase conjugate was resuspended in PBS buffer and purified with high-resolution size exclusion chromatography (Sephacryl S-500 HR) to remove the free uricase. Last, the conjugates were washed and concentrated with Hepes buffer three times using a 100-kDa molecular weight cutoff centrifugal filter. Following the same protocol, MPC- and NZPS-uricase conjugates were prepared and purified.
All polymer-protein conjugates were processed in a 1260 Infinity binary high-performance liquid chromatography system equipped with a Waters Ultrahydrogel 1000 column (7.8 mm by 300 mm, 12-μm particle size), a UV detector (Agilent Technologies, Santa Clara, CA), a miniDAWN TREOS light scattering detector, and an Optilab T-rEX differential refractive index detector (Wyatt Technology, Santa Barbara, CA). The flow rate was set at 0.6 ml/min with the mobile-phase PBS (pH 7.4) with 0.02% sodium azide as a preservative.
In vitro immunogenicity study
DC 2.4 dendritic cells (105 per well) were incubated with PBS (blank control), native uricase, ZPS-uricase, NZPS-uricase, and MPC-uricase conjugates (2 mU/ml) for 72 hours. At the end of incubation, the cells were spun at 300g for 10 min, and the supernatant medium was collected for cytokine (IL-6, IL-10, and TGF-β) analysis by a cytokine Quantikine ELISA kit. The cells were harvested and washed twice with ice-cold sterile PBS. Cells were labeled with anti–CD40-FITC and anti–CD80-PE antibodies and analyzed using flow cytometry.
Animal studies
All animal experiments adhered to federal guidelines and were approved by the University of Washington Institutional Animal Care and Use Committee. Animals were randomized to treatment groups at the beginning of each study. A sample size of five animals per group was used. C57/BL6 mice (male; body weight, 20 to 30 g) were obtained from the Jackson laboratory (Seattle, WA).
For in vivo immunogenicity study, ZPS-uricase, NZPS-uricase, and MPC-uricase conjugates at a dose of 25 U/kg body weight were intravenously administered into the mice via retro-orbital injection. The administrations of uricase samples were repeated three times with 1 week as the time interval between each administration. At the end of 3 weeks (21st day), all the mice were euthanized, and their blood collected through cardiac puncture was handled for direct ELISA test. As the first step of direct ELISA test, 100-μl antigen solutions [uricase or polymer-uricase conjugates (10 μg/ml)] prepared in the coating buffer [0.1 M sodium carbonate buffer (pH 10.5)] were used to coat each well of 96-well plates. After overnight coating at 4°C overnight, the plates were washed five times using PBS buffer (pH 7.4) to remove the antigen solutions and then filled with blocking buffer [1% BSA solution in 0.1 M tris buffer (pH 8.0)] for 1-hour incubation at room temperature, subsequent to which the blocking buffer was removed. All wells were then washed by PBS buffer for another five times. Subsequently, serial dilutions of mouse sera in PBS buffer containing 1% BSA were added to the plates (100 μl per well) for 1-hour incubation at 37°C, subsequent to which the mouse sera were removed, and all wells were washed five times with PBS buffer. Next, goat anti-rat IgM or IgG (HRP-conjugated; Bethyl Laboratories) as the secondary antibody was added into each well for another 1-hour incubation at 37°C. Subsequently, all the wells were washed five times using PBS buffer before the addition of 100 μl per well HRP substrate 3,3′,5,5′-tetramethylbenzidine (Bethyl Laboratories). The plates were shaken for 15 min, and 100 μl of stop solution (0.2 M H2SO4) was added to each well. Absorbance at 450 (signal) and 570 nm (background) was recorded by a microplate reader. Mouse sera naïve to the administration of uricase samples were used as the negative control for all ELISA detections. Moreover, the mouse spleens were harvested on the 21st day for the isolation of splenocytes by 100-μm cell strainer (Fisherbrand). The mouse splenocytes from each group were cultured in a 12-well plate (106 per well) and restimulated with native uricase, MPC-uricase, NZPS-uricase, or ZPS-uricase (1 mg/ml). After 72 hours, the cell culture medium from each well was collected for the quantification of IL-4, while cells were stained with anti–CD4-PE, anti–CD25-FITC, and anti–Foxp3-Percep antibodies for analysis by flow cytometry.
For PK study, ZPS-uricase, NZPS-uricase, MPC-uricase conjugates at a dose of 25 U/kg body weight were intravenously administered into the mice via retro-orbital injection. The administrations of uricase samples were repeated three times with 1 week as the time interval between each administration. The mouse blood was collected at various time points (−1, 5 min, 6, 24, 48, and 72 hours) after the injection of the first dose and the third dose. The uricase concentration in plasma was estimated on the basis of the enzyme activity measured by Amplex Red Uric Acid/Uricase Assay Kit. To exclude the disturbance of mouse’s nature uricase and the potential dose-accumulating effect, uricase concentration in the rat sera at time point −1 min was subtracted as background in PK calculation. All PK parameters were calculated using PKSolver following the instructions.
Statistics
The Student’s t test was chosen to compare two small sets of quantitative data when data in each sample set were related, with *P < 0.05 being considered as statistically significant.
Supplementary Material
Acknowledgments
Funding: This work was supported by the Defense Threat Reduction Agency (HDTRA1-13-1-0044) and the University of Washington. Author contributions: B.L., Z.Y., and S.J. designed the experiment. B.L., Z.Y., P.J., H.-C.H., Y.H., X.L., and P.M. performed the experiment. B.L. and Y.H. drew the figures. B.L. wrote the manuscript. B.L. and S.J. edited the manuscript. Competing interests: B.L., P.J., and S.J. are inventors on a pending patent related to this work filed by University of Washington (PCT/US19/23921, 25 March 2019). All other authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/22/eaba0754/DC1
REFERENCES AND NOTES
- 1.Blackman L. D., Gunatillake P. A., Cass P., Locock K. E. S., An introduction to zwitterionic polymer behavior and applications in solution and at surfaces. Chem. Soc. Rev. 48, 757–770 (2019). [DOI] [PubMed] [Google Scholar]
- 2.Jiang S. Y., Cao Z. Q., Ultralow-fouling, functionalizable, and hydrolyzable zwitterionic materials and their derivatives for biological applications. Adv. Mater. 22, 920–932 (2010). [DOI] [PubMed] [Google Scholar]
- 3.Schlenoff J. B., Zwitteration: Coating surfaces with zwitterionic functionality to reduce nonspecific adsorption. Langmuir 30, 9625–9636 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ladd J., Zhang Z., Chen S., Hower J. C., Jiang S., Zwitterionic polymers exhibiting high resistance to nonspecific protein adsorption from human serum and plasma. Biomacromolecules 9, 1357–1361 (2008). [DOI] [PubMed] [Google Scholar]
- 5.Zhang L., Cao Z., Bai T., Carr L., Ella-Menye J.-R., Irvin C., Ratner B. D., Jiang S., Zwitterionic hydrogels implanted in mice resist the foreign-body reaction. Nat. Biotechnol. 31, 553–556 (2013). [DOI] [PubMed] [Google Scholar]
- 6.Li B., Yuan Z., Zhang P., Sinclair A., Jain P., Wu K., Tsao C., Xie J., Hung H.-C., Lin X., Bai T., Jiang S., Zwitterionic nanocages overcome the efficacy loss of biologic drugs. Adv. Mater. 30, e1705728 (2018). [DOI] [PubMed] [Google Scholar]
- 7.Zhang P., Liu E. J., Tsao C., Kasten S. A., Boeri M. V., Dao T. L., DeBus S. J., Cadieux C. L., Baker C. A., Otto T. C., Cerasoli D. M., Chen Y., Jain P., Sun F., Li W., Hung H.-C., Yuan Z., Ma J., Bigley A. N., Raushel F. M., Jiang S., Nanoscavenger provides long-term prophylactic protection against nerve agents in rodents. Sci. Transl. Med. 11, eaau7091 (2019). [DOI] [PubMed] [Google Scholar]
- 8.Liu S. J., Jiang S. Y., Zwitterionic polymer-protein conjugates reduce polymer-specific antibody response. Nano Today 11, 285–291 (2016). [Google Scholar]
- 9.Li B., Yuan Z., Hung H.-C., Ma J., Jain P., Tsao C., Xie J., Zhang P., Lin X., Wu K., Jiang S., Revealing the immunogenic risk of polymers. Angew. Chem. Int. Ed. Engl. 57, 13873–13876 (2018). [DOI] [PubMed] [Google Scholar]
- 10.Li B., Jain P., Ma J., Smith J. K., Yuan Z., Hung H.-C., He Y., Lin X., Wu K., Pfaendtner J., Jiang S., Trimethylamine N-oxide-derived zwitterionic polymers: A new class of ultralow fouling bioinspired materials. Sci. Adv. 5, eaax4659 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Green J. J., Elisseeff J. H., Mimicking biological functionality with polymers for biomedical applications. Nature 540, 386–394 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mi L., Jiang S. Y., Integrated antimicrobial and nonfouling zwitterionic polymers. Angew. Chem. Int. Ed. Engl. 53, 1746–1754 (2014). [DOI] [PubMed] [Google Scholar]
- 13.Watkins E. A., Hubbell J. A., Designing biofunctional immunotherapies. Nat. Rev. Mater. 4, 350–352 (2019). [Google Scholar]
- 14.Stabler C. L., Li Y., Stewart J. M., Keselowsky B. C., Engineering immunomodulatory biomaterials for type 1 diabetes. Nat. Rev. Mater. 4, 429–450 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gammon J. M., Jewell C. M., Engineering immune tolerance with biomaterials. Adv. Healthc. Mater. 8, e1801419 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sadtler K., Singh A., Wolf M. T., Wang X., Pardoll D. M., Elisseeff J. H., Design, clinical translation and immunological response of biomaterials in regenerative medicine. Nat. Rev. Mater. 1, 16040 (2016). [Google Scholar]
- 17.Luo X., Miller S. D., Shea L. D., Immune tolerance for autoimmune disease and cell transplantation. Annu. Rev. Biomed. Eng. 18, 181–205 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yanover C., Jain N., Pierce G., Howard T. E., Sauna Z. E., Pharmacogenetics and the immunogenicity of protein therapeutics. Nat. Biotechnol. 29, 870–873 (2011). [DOI] [PubMed] [Google Scholar]
- 19.Schellekens H., Bioequivalence and the immunogenicity of biopharmaceuticals. Nat. Rev. Drug Discov. 1, 457–462 (2002). [DOI] [PubMed] [Google Scholar]
- 20.Kishimoto T. K., Maldonado R. A., Nanoparticles for the induction of antigen-specific immunological tolerance. Front. Immunol. 9, 230 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li B., Yuan Z., Mullen P. M., Xie J., Jain P., Hung H.-C., Xu S., Zhang P., Lin X., Wu K., Jiang S., A chromatin-mimetic nanomedicine for therapeutic tolerance induction. ACS Nano 12, 12004–12014 (2018). [DOI] [PubMed] [Google Scholar]
- 22.Maldonado R. A., LaMothe R. A., Ferrari J. D., Zhang A.-H., Rossi R. J., Kolte P. N., Griset A. P., O’Neil C., Altreuter D. H., Browning E., Johnston L., Farokhzad O. C., Langer R., Scott D. W., von Andrian U. H., Kishimoto T. K., Polymeric synthetic nanoparticles for the induction of antigen-specific immunological tolerance. Proc. Natl. Acad. Sci. U.S.A. 112, E156–E165 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kishimoto T. K., Ferrari J. D., LaMothe R. A., Kolte P. N., Griset A. P., O'Neil C., Chan V., Browning E., Chalishazar A., Kuhlman W., Fu F.-N., Viseux N., Altreuter D. H., Johnston L., Maldonado R. A., Improving the efficacy and safety of biologic drugs with tolerogenic nanoparticles. Nat. Nanotechnol. 11, 890–899 (2016). [DOI] [PubMed] [Google Scholar]
- 24.Meliani A., Boisgerault F., Hardet R., Marmier S., Collaud F., Ronzitti G., Leborgne C., Costa Verdera H., Simon Sola M., Charles S., Vignaud A., van Wittenberghe L., Manni G., Christophe O., Fallarino F., Roy C., Michaud A., Ilyinskii P., Kishimoto T. K., Mingozzi F., Antigen-selective modulation of AAV immunogenicity with tolerogenic rapamycin nanoparticles enables successful vector re-administration. Nat. Commun. 9, 4098 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Birge R. B., Boeltz S., Kumar S., Carlson J., Wanderley J., Calianese D., Barcinski M., Brekken R. A., Huang X., Hutchins J. T., Freimark B., Empig C., Mercer J., Schroit A. J., Schett G., Herrmann M., Phosphatidylserine is a global immunosuppressive signal in efferocytosis, infectious disease, and cancer. Cell Death Differ. 23, 962–978 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tietjen G. T., Gong Z., Chen C.-H., Vargas E., Crooks J. E., Cao K. D., Heffern C. T. R., Henderson J. M., Meron M., Lin B., Roux B., Schlossman M. L., Steck T. L., Lee K. Y. C., Adams E. J., Molecular mechanism for differential recognition of membrane phosphatidylserine by the immune regulatory receptor Tim4. Proc. Natl. Acad. Sci. U.S.A. 111, E1463–E1472 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen S., Li L., Zhao C., Zheng J., Surface hydration: Principles and applications toward low-fouling/nonfouling biomaterials. Polymer 51, 5283–5293 (2010). [Google Scholar]
- 28.Fadok V. A., Bratton D. L., Frasch S. C., Warner M. L., Henson P. M., The role of phosphatidylserine in recognition of apoptotic cells by phagocytes. Cell Death Differ. 5, 551–562 (1998). [DOI] [PubMed] [Google Scholar]
- 29.Blankenberg F., Contag C., Hardy J., Annexin V blockade of the immunosuppressive effects of phosphatidylserine in 4T1 murine mammary tumors. J. Nucl. Med. 56, 1179 (2015). [Google Scholar]
- 30.van Genderen H. O., Kenis H., Hofstra L., Narula J., Reutelingsperger C. P. M., Extracellular annexin A5: Functions of phosphatidylserine-binding and two-dimensional crystallization. Biochim. Biophys. Acta 1783, 953–963 (2008). [DOI] [PubMed] [Google Scholar]
- 31.Segawa K., Nagata S., An apoptotic 'Eat Me' signal: Phosphatidylserine exposure. Trends Cell Biol. 25, 639–650 (2015). [DOI] [PubMed] [Google Scholar]
- 32.Hoffmann P. R., deCathelineau A. M., Ogden C. A., Leverrier Y., Bratton D. L., Daleke D. L., Ridley A. J., Fadok V. A., Henson P. M., Phosphatidylserine (PS) induces PS receptor-mediated macropinocytosis and promotes clearance of apoptotic cells. J. Cell Biol. 155, 649–660 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Aggarwal R. S., What's fueling the biotech engine-2012 to 2013. Nat. Biotechnol. 32, 32–39 (2014). [DOI] [PubMed] [Google Scholar]
- 34.Schellekens H., The immunogenicity of therapeutic proteins. Discov. Med. 9, 560–564 (2010). [PubMed] [Google Scholar]
- 35.Baker M. P., Reynolds H. M., Lumicisi B., Bryson C. J., Immunogenicity of protein therapeutics: The key causes, consequences and challenges. Self/nonself 1, 314–322 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dingman R., Balu-Iyer S. V., Immunogenicity of protein pharmaceuticals. J. Pharm. Sci. 108, 1637–1654 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.van Witteloostuijn S. B., Pedersen S. L., Jensen K. J., Half-life extension of biopharmaceuticals using chemical methods: Alternatives to PEGylation. ChemMedChem 11, 2474–2495 (2016). [DOI] [PubMed] [Google Scholar]
- 38.Zaman R., Islam R. A., Ibnat N., Othman I., Zaini A., Lee C. Y., Chowdhury E. H., Current strategies in extending half-lives of therapeutic proteins. J. Control. Release. 301, 176–189 (2019). [DOI] [PubMed] [Google Scholar]
- 39.Griswold K. E., Bailey-Kellogg C., Design and engineering of deimmunized biotherapeutics. Curr. Opin. Struct. Biol. 39, 79–88 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Harris J. M., Chess R. B., Effect of pegylation on pharmaceuticals. Nat. Rev. Drug Discov. 2, 214–221 (2003). [DOI] [PubMed] [Google Scholar]
- 41.Turecek P. L., Bossard M. J., Schoetens F., Ivens I. A., PEGylation of Biopharmaceuticals: A review of chemistry and nonclinical safety information of approved drugs. J. Pharm. Sci. 105, 460–475 (2016). [DOI] [PubMed] [Google Scholar]
- 42.Qi Y., Chilkoti A., Protein-polymer conjugation-moving beyond PEGylation. Curr. Opin. Chem. Biol. 28, 181–193 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yang Q., Lai S. K., Anti-PEG immunity: Emergence, characteristics, and unaddressed questions. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 7, 655–677 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ganson N. J., Kelly S. J., Scarlett E., Sundy J. S., Hershfield M. S., Control of hyperuricemia in subjects with refractory gout, and induction of antibody against poly(ethylene glycol) (PEG), in a phase I trial of subcutaneous PEGylated urate oxidase. Arthritis Res. Ther. 8, R12 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hershfield M. S., Ganson N. J., Kelly S. J., Scarlett E. L., Jaggers D. A., Sundy J. S., Induced and pre-existing anti-polyethylene glycol antibody in a trial of every 3-week dosing of pegloticase for refractory gout, including in organ transplant recipients. Arthritis Res. Ther. 16, R63 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sundy J. S., Baraf H. S., Yood R. A., Edwards N. L., Gutierrez-Urena S. R., Treadwell E. L., Vázquez-Mellado J., White W. B., Lipsky P. E., Horowitz Z., Huang W., Maroli A. N., Waltrip R. W. II, Hamburger S. A., Becker M. A., Efficacy and tolerability of pegloticase for the treatment of chronic gout in patients refractory to conventional treatment: Two randomized controlled trials. JAMA 306, 711–720 (2011). [DOI] [PubMed] [Google Scholar]
- 47.Johnston C. J. C., Smyth D. J., Dresser D. W., Maizels R. M., TGF-β in tolerance, development and regulation of immunity. Cell. Immunol. 299, 14–22 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dominguez-Villar M., Hafler D. A., Regulatory T cells in autoimmune disease. Nat. Immunol. 19, 665–673 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gaitonde P., Purohit V. S., Balu-Iyer S. V., Intravenous administration of Factor VIII-O-Phospho-L-Serine (OPLS) complex reduces immunogenicity and preserves pharmacokinetics of the therapeutic protein. Eur. J. Pharm. Sci. 66, 157–162 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Glassman F. Y., Balu-Iyer S. V., Subcutaneous administration of Lyso-phosphatidylserine nanoparticles induces immunological tolerance towards A mouse model. Int. J. Pharm. 548, 642–648 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schneider J. L., Dingman R. K., Balu-Iyer S. V., Lipidic nanoparticles comprising phosphatidylinositol mitigate immunogenicity and improve efficacy of recombinant human acid alpha-glucosidase in a murine model of pompe disease. J. Pharm. Sci. 107, 831–837 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ramani K., Miclea R. D., Purohit V. S., Mager D. E., Straubinger R. M., Balu-Iyer S. V., Phosphatidylserine containing liposomes reduce immunogenicity of recombinant human factor VIII (rFVIII) in a murine model of hemophilia A. J. Pharm. Sci. 97, 1386–1398 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ramani K., Purohit V., Miclea R., Gaitonde P., Straubinger R. M., Balu-Iyer S. V., Passive transfer of polyethylene glycol to liposomal-recombinant human FVIII enhances its efficacy in a murine model for hemophilia A. J. Pharm. Sci. 97, 3753–3764 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/22/eaba0754/DC1