

Oncoprotein SND1 induces ER-associated degradation for nascent MHC-I heavy chain, leading to impaired CD8+ T cell response in tumor.
Abstract
SND1 is highly expressed in various cancers. Here, we identify oncoprotein SND1 as a previously unidentified endoplasmic reticulum (ER) membrane–associated protein. The amino-terminal peptide of SND1 predominantly associates with SEC61A, which anchors on ER membrane. The SN domain of SND1 catches and guides the nascent synthesized heavy chain (HC) of MHC-I to ER-associated degradation (ERAD), hindering the normal assembly of MHC-I in the ER lumen. In mice model bearing tumors, especially in transgenic OT-I mice, deletion of SND1 promotes the presentation of MHC-I in both B16F10 and MC38 cells, and the infiltration of CD8+ T cells is notably increased in tumor tissue. It was further confirmed that SND1 impaired tumor antigen presentation to cytotoxic CD8+ T cells both in vivo and in vitro. These findings reveal SND1 as a novel ER-associated protein facilitating immune evasion of tumor cells through redirecting HC to ERAD pathway that consequently interrupts antigen presentation.
INTRODUCTION
Exploring the strategies for tumor immunotherapy is highly dependent on the discovery of molecular mechanisms of tumor immune escape. Tumor cells can escape immune response through loss of antigenicity and/or immunogenicity or by coordinating a suppressive immune microenvironment. Therefore, distinct therapeutic strategies may be required, depending on the mechanisms. Tumor immunotherapy strategies mediated by T cells rely on the functional competence of multiple immunological elements. For example, therapeutic monoclonal antibodies designed to disrupt inhibitory signals received by T cells through the Cytotoxic T lymphocyte-associated antigen-4 (CTLA-4) and Programmed cell death protein 1 (PD-1) have been demonstrating long-term survival benefits for some patients with metastatic melanoma (1, 2). However, not all tumors appear to respond effectively. The heterogeneity of cancer suggests the necessity to explore the additional immunoregulatory mechanisms. Defect in the surface expression of major histocompatibility complex class I (MHC-I) molecules is one of the most important reasons for tumor immune escape due to decreased recognition by CD8+ T cells, which has been found in approximately 20 to 60% of common solid cancers, including melanoma and lung, breast, renal, prostate, and bladder cancers, (3, 4). The molecular mechanisms underlying these changes vary according to the tumor type. These alterations can be genetic or regulatory at the transcriptional or posttranscriptional level (5–11).
Staphylococcal nuclease and tudor domain containing 1 (SND1) is a newly identified oncoprotein that is highly expressed in almost all the detected different tumor cells (12–14). SND1 was first identified as a transcriptional coactivator for Epstein-Barr virus nuclear antigen 2. It is a ubiquitously expressed and highly conserved protein in mammals and plays important physiological roles in a variety of cellular processes (15). It comprises a tandem repeat of four staphylococcal nuclease (SN)–like domains (referred to as SN domains) at the N terminus and a fusion of a Tudor domain with a partial SN domain at the C terminus (referred to as TSN domain) (16). Our current studies and others’ studies have demonstrated that SND1 regulates the differentiation and migration of different tumor cells via variant signal pathways at the cellular level (17–19). For example, SND1 expression is up-regulated in breast cancer tissues, and it associates with transforming growth factor–β signaling pathway to promote epithelial-mesenchymal transition in breast cancer (20). SND1 regulates the cadherin switch for epithelial-mesenchymal transition in ovary SKOV3 cells (17). However, the fundamental impact of SND1 on the tumorigenesis in vivo is largely unknown. In the present study, we demonstrate that in tumor cells, SND1 is a novel endoplasmic reticulum (ER)–associated protein “hijacking” the nascent heavy chain (HC) of MHC-I to ER-associated degradation (ERAD) process. With reduced expression of MHC-I on tumor cell membrane, it would be easy for tumor cells to orchestrate a cancer-favored immune microenvironment and escape immune response.
RESULTS
SND1 physically associates and colocalizes with the HC of MHC-I
To investigate the fundamental role of oncoprotein SND1 in tumor proliferation, we performed affinity purification and mass spectrometry to identify SND1-associated proteins from cellular extracts of HeLa cells with stable expression of SND1-FLAG. As shown in Fig. 1A, a group of ER-related proteins were coprecipitated with SND1, including human leukocyte antigen–A (HLA-A; the HC of human MHC-I), Valosin-containing protein (VCP), SEC61 translocon subunit alpha (SEC61A), and ribosomal protein L7a (RPL7A). As MHC-I molecule plays essential roles in antigen presentation, we therefore focused on investigating the relationship between SND1 and HLA-A. Coimmunoprecipitation (Co-IP) assay was performed to verify the association of SND1 and HLA-A in HeLa cells. As shown in Fig. 1B, the endogenous HLA-A was efficiently associated with ectopically overexpressed SND1-FLAG. In addition, anti-HC10 antibody that could specifically recognize immature (unfolded/partially folded) conformation of HLA-A, HLA-B, and HLA-C was used to detect the in vivo physical association of endogenous SND1 and HLA-A. The endogenous SND1 was efficiently coimmunoprecipitated with anti-HC10 antibody (Fig. 1C) and vice versa. HLA-A or HLA-B was coimmunoprecipitated with anti-SND1 antibody (Fig. 1D). Consistently, the Duolink assay [Fig. 1E, red dots indicate the proximity ligation assay (PLA) probe signal] and immunofluorescence assay (Fig. 1F) further confirmed the cellular colocalization of SND1 and HLA-A. We then mapped the interaction domain between SND1 and HLA-A by glutathione S-transferase (GST) pull-down assay. The bacterially produced GST-fusion protein containing full-length SND1 (GST-SND1), SN domain (GST-SN), TSN domain (GST-TSN) (as indicated in Fig. 1G), or GST alone was purified using glutathione agarose beads and used to incubate with HLA-A in vitro–translated from rabbit reticulocytes. As shown in Fig. 1G, the full length of SND1 or SN domain, but not TSN domain, efficiently associated with HLA-A–FLAG. Likewise, the GST-fusion proteins containing full-length HLA-A (GST–HLA-A), A1 domain (GST–HLA-A–A1), A2 domain (GST–HLA-A–A2), A3 domain (GST–HLA-A–A3), C domain (GST–HLA-A–C) (as indicated in Fig. 1H), or GST alone were used to incubate with recombinant histidine-tagged SND1 (His-SND1) purified from Escherichia coli. As shown in Fig. 1H, the full length of HLA-A, domain A1, domain A3, or domain C, but not domain A2, efficiently associated with His-SND1. To further consolidate the molecular interface required for the interaction between SND1 and HLA-A, FLAG-tagged different domain deletion mutants of SND1 were generated. Immunoprecipitation analysis in HeLa SND1-KO (knockout) cells demonstrated that the SN3 region of SND1 was required for the interaction of HLA-A (Fig. 1I). These data prompted us to interrogate the three-dimensional conformation for the complex of SND1 and HLA-A. Because the complex structure was not determined experimentally, we performed docking and molecular dynamics simulation to predict their associated conformation (21–23). The resulting structure (Fig. 1J) showed that the interacting interface was located between the SN3 region of SND1 and domains A1 and A3 of HLA-A. The electrostatic interaction between the basic and acidic amino acids on SN3 region and A3 domain might play an important role in the association process. The key residues on the interface included K484, K496, K490, K401, K450, and R384 in SN3 domain and E232, E229, and D227 in A3 domain. The interaction between K401-E232, K496-E229, and K496-D227 were identified to be vital for the association of SND1 with HLA-A. These results indicated that SND1 could physically interact with the immature form of HC in partially folded/unfolded conformation, which raises the question about the functional association of SND1 with “nascent” HC of MHC-I.
Fig. 1. SND1 is physically associated and colocalized with MHC-I HC.
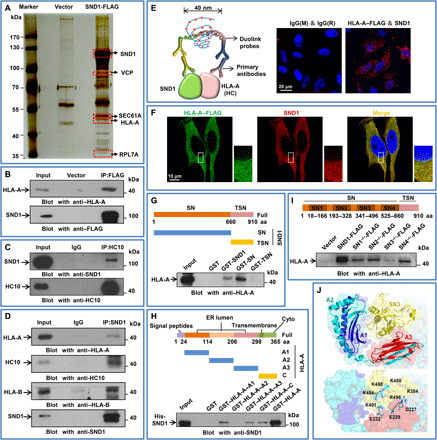
(A) Immunopurification and mass spectrometry of SND1-containing protein complexes. Cellular extracts from HeLa cells stably expressing SND1-FLAG were immunopurified with anti-FLAG affinity beads and eluted with FLAG peptide. The elutes were resolved on SDS-PAGE and silver-stained. The protein bands on the gel were recovered by trypsinization and analyzed by mass spectrometry. (B) Co-IP analysis of the association between SND1 and HLA-A. Whole-cell extracts from HeLa cells with SND1-FLAG expression were immunoprecipitated with anti-FLAG beads, followed by Western blot with antibodies against the HLA-A. (C) Co-IP analysis of the association between SND1 and HC10. Whole-cell extracts from HeLa cells were immunoprecipitated with anti-HC10, followed by immunoblot (IB) with antibodies against the SND1. (D) Cellular extracts from HeLa cells were immunoprecipitated with anti-SND1 antibody, followed by Western blot with antibodies against the indicated proteins. (E) Duolink in situ PLA was adopted for detecting the association between SND1 and HLA-A. Two PLA probes were designed to respectively recognize either mouse or rabbit antibody against SND1 or HLA-A. Immunoglobulin G (IgG) was used as staining control. Scale bar, 20 μm. (F) Immunostaining and confocal microscopic analysis of subcellular colocalization of SND1 and HLA-A–FLAG (C terminus) in HeLa cells. HeLa cells were fixed and immunostained with antibodies against the indicated proteins. Scale bar, 10 μm. (G) GST pull-down analysis of the bacterially produced GST-fusion protein containing full-length SND1 (GST-SND1), SN domain (GST-SN), and TSN domain (GST-TSN) involved in the interaction with in vitro–translated HLA-A from rabbit reticulocytes. Coomassie blue staining for GST-fusion proteins refers to fig. S1A. aa, amino acid. (H) GST pull-down analysis of the different domains of HLA-A involved in the interaction with SND1. The His-SND1 and sample of GST-tagged different domains of HLA-A were purified from E. coli bacteria cells. Coomassie blue staining for GST-fusion proteins refers to fig. S1B. (I) Immunoprecipitation analysis of the domains involved in the interaction between SND1 and HLA-A with FLAG-tagged deletion mutants of SND1 purified from HeLa SND1-KO cells. The immunoprecipitation of FLAG refers to fig. S1C. (J) The spatial conformation of SND1-HLA-A complex predicted by the database of ZDOCK (http://zdock.umassmed.edu/) was further analyzed using the Gromacs package. The structural stability and binding energy refer to fig. S1 (E and F).
SND1 is a novel ER-associated protein interacting with SEC61A on ER membrane
Since the nascent HC is synthesized on the ER membrane and “matured” in the ER lumen (9), it raises the question of where the interaction of SND1 and HC occurs. By analyzing and comparing our previous mass spectrometry data from Jurkat cells with the present data from HeLa cells, we found 221 proteins (fig. S2A) in the overlapped set of SND1-associated proteins, including ribosomal (RPLs or RPSs) or ER-associated proteins, such as HLA-A, SEC61A, ribosome binding protein 1 (RRBP1), VCP, signal recognition particle 72 (SRP72), etc. We then performed immunofluorescence to investigate the cellular colocalization of SND1 and ER-associated proteins. As shown in Fig. 2A, SND1 colocalized with RRBP1 (a ribosome receptor on ER; top) and SEC61A (a core component of ER translocation channel; middle). The colocalization of HLA-A and RRBP1 (bottom) was used as positive control. It indicated that SND1 was a potential ER-associated protein.
Fig. 2. SND1 is a novel ER-associated protein interacting with SEC61A on ER membrane via N-terminal peptide.
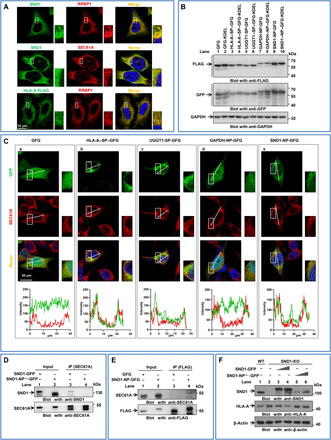
(A) Immunostaining for cellular colocalizations, followed by confocal microscopic analysis by using antibody against SND1, RRBP1, SEC61A, and HLA-A–FLAG. Scale bar, 10 μm. (B) HeLa cells were transfected with the ER reporter plasmids, GFG, HLA-SP-GFG, UGGT1-SP-GFG, GAPDH-NP-GFG, and SND1-NP-GFG, respectively. Western blot for molecular weight of these GFG-tagged fusion proteins expressed in HeLa cells. (C) Colocalizations of these GFG-tagged fusion proteins with SEC61A were detected by confocal microscopy. UGGT1 was used as a positive control for ER-associating protein, while GAPDH was used as a negative control. Scale bar, 20 μm. Fluorescence intensity profiles of regions indicated by short lines are shown in the bottom. (D) Co-IP by antibody against SEC61A for interaction with SND1-GFP or SND1-NP−/−-GFP in HeLa cells transfected with either SND1-GFP (lane 3) or SND1-NP−/−-GFP vector (lane 4). (E) Co-IP by antibody against FLAG for interaction with SND1-NP in HeLa cells transfected with either GFG (lane 3) or SND1-NP-GFG vector (lane 4). (F) Ectopically increased expression of either SND1-GFP or SND1-NP−/−-GFP in SND1-KO HeLa cells followed by Western blot for SND1 and HLA-A expression. WT, wild type.
ER lumen or secretory proteins containing an N-terminal signaling peptide (SP) composed of hydrophobic amino, which is recognized by SRP and subsequently cleaved by signal peptidase in the ER lumen (24), such as HLA-A and UGGT1 (a glucosyltransferase in ER lumen) (as illustrated in fig. S2B). We noticed a hydrophobic amino acid sequence at N terminus (NP) of SND1 protein. To investigate whether it is an SP, we developed an ER reporter assay by constructing a pair of two ER luminal reporter vectors. One is named GFG containing green fluorescent protein (GFP)–FLAG–GFP sequences after the multiple cloning sites that could insert the sequence of designated peptide. The other one is GFG-KDEL with an additional sequence of peptide KDEL following GFP-FLAG-GFP sequences. KDEL is a specific peptide sequence at the C terminus of ER lumen proteins that keeps the protein in the ER lumen (25). Using these vectors, we constructed a series of plasmids containing the SP of HLA-A (HLA-A–SP–GFG and HLA-A–SP–GFG-KDEL) or UGGT1 (UGGT1–SP–GFG and UGGT1–SP–GFG-KDEL), respectively, as positive control of ER-associated proteins. The N-terminal sequence of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (GAPDH-NP-GFG and GAPDH–NP–GFG-KDEL) was used as negative control of ER-associated proteins. The N-terminal sequence of SND1 was inserted to construct SND1-NP-GFG and SND1–NP–GFG-KDEL (fig. S2B). The plasmids were transfected into HeLa cells, respectively, and Western blot was performed to observe whether the S(N)P could be cleaved from the S(N)P-GFG fusion protein by signal peptidase (Fig. 2B). The molecular mass of SND1-NP-GFG(-KDEL) (lanes 7 and 8) was heavier than that of the GFG(-KDEL) control (lanes 1 and 2) (molecular mass is about 55 kDa), HLA-A–SP–GFG(-KDEL) (lanes 3 and 4), or UGGT1-SP-GFG(-KDEL) (lanes 5 and 6) but at the same level as negative control GAPDH-NP-GFG(-KDEL) (lanes 9 and10). It demonstrated that the SP of HLA-A–SP–GFG(-KDEL) and UGGT1-SP-GFG(-KDEL) was successfully cleaved from the fusion protein by signal peptidase, but not the NP of SND1-NP-GFG(-KDEL) or GAPDH-NP-GFG(-KDEL). All these data indicate that SND1 is an ER-associated protein but not an ER lumen protein since the NP of SND1 is not an SP. Therefore, it is likely that SND1 associates with HLA-A on ER membrane.
To further clarify the localization of SND1, the above X-S(N)P-GFG plasmids were transfected into HeLa cells, respectively; then, the colocalization of the X-S(N)P-GFG with endogenous SEC61A and RRBP1 was detected by immunofluorescence assay. Fluorescence intensity profiles were used to quantify the degree of localization. As shown in Fig. 2C and fig. S2C, the GFG control protein did not colocalize with SEC61A (Fig. 2C, a) or RRBP1 (fig. S2C, a). HLA-A–SP–GFG or UGGT1-SP-GFG was well colocalized with SEC61A (Fig. 2C, b and c) and RRBP1 (fig. S2C, b and c). There was no obvious colocalization of GAPDH-NP-GFG with SEC61A (Fig. 2C, d) or RRBP1 (fig. S2C, d). SND1-NP-GFG was colocalized with SEC61A (Fig. 2C, e) or RRBP1 (fig. S2C, e) respectively, which is in accordance with the results of S(N)P–GFG-KDEL (fig. S2D). It is understandable that the colocalization of HLA-A–SP–GFG or UGGT1-SP-GFG with SEC61A and RRBP1 is due to the distribution of the GFG in the ER lumen cleaved by SP. How to explain the colocalization of SND1-NP-GFG with SEC61A and RRBP1?
In eukaryotes, ER membrane–located SEC61 translocation complex is the core component of the translocon that transports proteins to the ER (26). We thus performed Co-IP assay to explore the potential interaction of SND1 with SEC61A. HeLa cells were transfected with the expression plasmid containing full-length SND1 tagged with GFP (SND1-GFP) or N-terminal peptide–deficient SND1 tagged with GFP (SND1-NP−/−-GFP), respectively. As shown in Fig. 2D, SEC61A coprecipitated with ectopically overexpressed SND1-GFP (lane 3) but not SND1-NP−/−-GFP (lane 4). In addition, the endogenous SEC61A was coprecipitated with anti-FLAG for SND1-NP-GFG (Fig. 2E, lane 4) but not the GFG alone (lane 3). All these data suggest that the NP of SND1 is required for the efficient interaction of SND1 and SEC61A. Consistently, fig. S2E showed that the full-length SND1-GFP, but not SND1-NP−/−-GFP, was well colocalized with endogenous SEC61A. In HeLa cells with deletion of endogenous SND1, the protein level of HLA-A was reduced with ectopically overexpressed SND1-GFP in a dose-dependent manner (Fig. 2F, lanes 3 and 4), while there was no obvious alteration with overexpressed SND1-NP−/−-GFP (lanes 5 and 6). Together, it is likely that SND1 is an ER-associated protein anchored on ER membrane by binding SEC61A upon where nascent MHC-I HC is caught.
The highly expressed SND1 promotes the degradation of MHC-I HC in tumor cells
To determine the relevance between SND1/HC association and the presentation of MHC-I molecules, we carried out flow cytometry to detect the protein level of HLA-A/B/C on the surface of tumor cells with deletion of SND1 (SND1-KO) by CRISPR-Cas9 system. As shown in Fig. 3A, the protein level of HLA-A/B/C molecules was increased in two different SND1-KO HeLa cell clones. Likewise, the similar changes were observed in two SKOV3 ovarian cancer cell clones with deletion of SND1 (shSND1#1 and shSND1#2) (fig. S3B). The Western blot further confirmed that the protein level of HLA-A was enhanced in the cells with deletion of SND1 (Fig. 3B, left), while it is reduced in the cells with ectopic overexpression of SND1 (Fig. 3B, right). However, there was no obvious alteration at the mRNA level (fig. S3A). The same results were observed in SKOV3 cells (fig. S3, B to F). We then detected the half-life of endogenous HLA-A in HeLa cells with treatment of cycloheximide (CHX). As shown in Fig. 3C, HLA-A was gradually degraded in parental HeLa cells [wild type (WT)]; however, the degradation of HLA-A was noticeably retarded in SND1-KO cells. In addition, the protein level of HLA-A was significantly increased in the cells pretreated with proteasome inhibitor MG132 (Fig. 3D, lane 2) but not in the cells pretreated with lysosomal inhibitor chloroquine (Fig. 3D, lane 3). Therefore, we examined the ubiquitylation of HLA-A to determine whether SND1-promoted HLA-A destabilization is via ubiquitin mediated–proteasome pathway. As shown in Fig. 3E, compared with the control cells (lanes 1 and 3), the ubiquitylation of ectopically overexpressed HLA-A was obviously decreased in SND1-KO cells (lane 4) but relatively increased in the cells with overexpression of SND1 (SND1-HA) (lane 2). On the basis of the aforementioned data, it is likely that SND1 leads to HLA-A degradation through the ubiquitin-proteasome pathway.
Fig. 3. SND1 promotes the degradation of MHC-I HC.
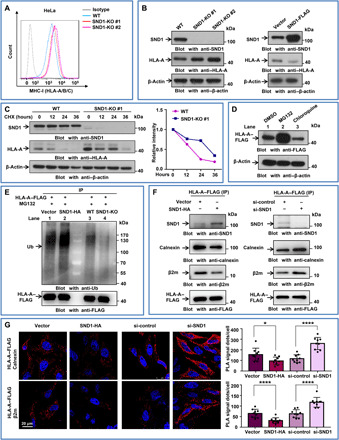
(A) Two clones of HeLa cells with stable depletion of SND1 by CRISPR-Cas9 system were analyzed by flow cytometry for human MHC-I using antibody simultaneously against HLA-A/B/C. (B) Two clones of HeLa cells with stable depletion of SND1 by CRISPR-Cas9 system and HeLa cells stably expressing SND1-FLAG were collected, followed by Western blot using antibodies against HLA-A. (C) The effect of KO SND1 on the half-life of HLA-A was evaluated in HeLa cells treated with CHX (50 μg/ml) and harvested at the indicated time point, followed by Western blot. The protein half-life curves were obtained by quantifying relative intensities. (D) HeLa cells with ectopic HLA-A expression were pretreated with proteasome inhibitor MG132 (10 mM) or lysosomal inhibitor chloroquine (100 mM) for 8 hours and subjected to Western blot with anti–HLA-A antibody. DMSO, dimethyl sulfoxide. (E) WT and SND1-KO HeLa cells were transfected with HLA-A–FLAG and treated with MG132 (10 mM) for 8 hours. Cellular extracts were immunoprecipitated with anti-FLAG, followed by Western blot with anti-ubiquitin (Ub) antibody. (F) HeLa cells were cotransfected with control vector or SND1-HA and HLA-A–FLAG or with control small interfering RNA (siRNA) or SND1 siRNA and HLA-A–FLAG, and whole-cell lysates were collected and immunoprecipitated with anti-FLAG, followed by Western blot with indicating antibodies. (G) The Duolink in situ PLA was adopted for detecting the direct association between HLA-A and calnexin or β2-microglobulin (β2m) in the presence of SND1-HA or in the absence of SND1. Scale bar, 20 μm. The signal dots were calculated and plotted. *P < 0.05 and ****P < 0.0001, by unpaired t test.
In ER lumen, the nascent unfolded HLA-A can be initially retained by a key chaperone, calnexin, to ensure proper folding and quality control before ultimate assembly with β2-microglobulin (β2m) to form mature MHC-I (27). The catching of nascent HLA-A by SND1 may have an impact on the association of calnexin and HLA-A and lead to a misfolding process of HLA-A. Immunoprecipitation assay revealed that the binding efficiency of HLA-A to calnexin, and HLA-A to β2m, was remarkably reduced in the presence of ectopically overexpressed SND1 (Fig. 3F, left) but was obviously increased in the absence of SND1 (right). According to the red dots of PLA probe signal from confocal images, Duolink assay (Fig. 3G) further validated that the binding efficiency of calnexin and β2m to HLA-A was decreased by overexpression of SND1 (SND1-HA), while it was significantly increased by deletion of SND1 (si-SND1). Moreover, glycosylation site–mutated HLA-A (N110Q) is not able to associate with both calnexin and β2m but sufficiently interacts with SND1 (fig. S3H). These observations suggest that SND1 hindering the normal assembly process of MHC-I in the ER lumen, consequently guiding the nascent HLA-A for degradation.
SND1 “hijacks” the nascent MHC-I HC to ERAD process in tumor cells
Misfolded or nascent HC, which fails to achieve the native conformation in complex with β2m, is dislocated from ER to cytosol and ubiquitinated for ERAD process (28). To investigate the underlying mechanisms of SND1-mediated HC degradation, we used affinity purification and mass spectrometry to identify HLA-A–associated proteins in HeLa cells with stable expression of HLA-A–FLAG. There were 278 affinity-purified proteins (fig. S4A) overlapping in both HLA-A–associated and SND1-associated proteins. These overlapped proteins were further filtered by Kyoto Encyclopedia of Genes and Genomes analysis (fig. S4B). The top-ranking proteins include SND1, HLA-A, VCP, calnexin (Fig. 4A), SEC61A, etc. (fig. S4C).
Fig. 4. SND1 hijacks the nascent MHC-I HC to ERAD process in tumor cells.
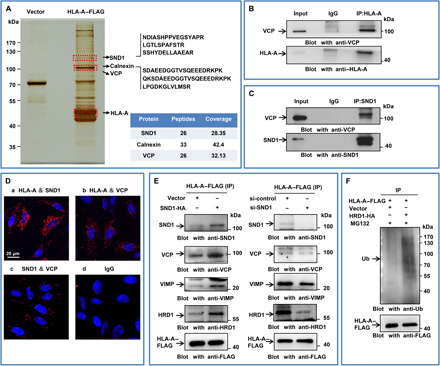
(A) Immunopurification and mass spectrometry of HLA-A–containing protein complexes. Cellular extracts from HeLa cells stably expressing HLA-A–FLAG were immunopurified with anti-FLAG affinity beads and eluted with FLAG peptide. The elutes were resolved on SDS–polyacrylamide gel electrophoresis (SDS-PAGE) and silver-stained. The protein bands on the gel were recovered by trypsinization and analyzed by mass spectrometry. HLA-A–interacted proteins were highlighted. (B) HeLa cells were coimmunoprecipitated by HLA-A antibody and subjected to Western blot by antibody against VCP. (C) HeLa cells were coimmunoprecipitated by SND1 antibody and subjected to Western blot by antibody against VCP. (D) Duolink assay followed by confocal microscopic analysis for direct molecular interactions among SND1, VCP, and HLA-A. IgG was used as a negative control. Scale bar, 20 μm. (E) HeLa cells were cotransfected with control vector or SND1-HA and HLA-A–FLAG or cotransfected with control siRNA or SND1 siRNA and HLA-A–FLAG, and whole-cell lysates were collected and immunoprecipitated with anti-FLAG, followed by Western blot with anti-SND1, anti-VCP, anti-VIMP, and anti-HRD1 antibodies. Results of input were shown in fig. S4D. (F) HeLa cells were cotransfected with vector or HRD1-HA and HLA-A–FLAG with the treatment of MG132. Cellular extracts were immunoprecipitated with anti-FLAG, followed by Western blot with anti-ubiquitin antibody.
As VCP plays essential roles in ERAD, we then validated the association of VCP, SND1, and HLA-A. Co-IP experiments revealed that VCP was able to physically interact with both HLA-A (Fig. 4B) and SND1 (Fig. 4C) in vivo. Moreover, Duolink assay (Fig. 4D) further demonstrated the association of these three proteins. Comparatively, the binding ability of SND1 and HLA-A (a) or HLA-A and VCP (b) was stronger than SND1 and VCP (c). It was reported that VIMP (the cofactor of VCP) and HRD1 (the E3 ligase) are the key participants in the degradation of HLA-A (29); meanwhile, the interaction of HRD1 with SND1 was found in our present study by Co-IP (fig. S4E). We therefore investigated the correlation of the SND1 expression and the association of VCP/VIMP/HRD1 with HLA-A. As shown in Fig. 4E, the interaction of VCP, VIMP, and HRD1 to HLA-A was remarkably increased in the presence of ectopically overexpressed SND1 (left) but was largely decreased in the absence of SND1 (right). Furthermore, compared with the control cells, the ubiquitylation of ectopically overexpressed HLA-A was obviously increased in the cells with overexpression of HRD1 (HRD1-HA) (Fig. 4F). These observations suggest that SND1 sequestrates the nascent HC of MHC-I and redirects it to the ERAD pathway for proteasomal degradation.
High expression of SND1 in tumor cells impairs CD8+ T cell response in mice
To explore the consequence of SND1-mediated HC degradation in vivo, we used murine syngeneic tumor models on the C57BL/6 background by using two murine cancer cell lines, B16F10 melanoma cells and MC38 colon adenocarcinoma cells. B16F10–SND1-KO and MC38–SND1-KO cell clones with deletion of SND1 were obtained by using CRISPR-Cas9 system. Consistent with previous results, both Western blot detection (Fig. 5A) and flow cytometry analysis (Fig. 5B) demonstrated that the protein level of H2Kb (HC of mouse MHC-I) was increased in different B16F10–SND1-KO cells. The same results were observed in MC38–SND1-KO cells (fig. S5, A and B). We then subcutaneously inoculated 5 × 105 parental or B16F10–SND1-KO cells into the flank of C57BL/6 mice, and the tumor growth was monitored in the following days. Compared with the parental B16F10 tumor, the growth of B16F10–SND1-KO cells with SND1 deficiency was markedly slow in terms of the developmental kinetics (Fig. 5C and fig. S5C). Consistently, the tumor size (Fig. 5D for B16F10 and fig. S5D for MC38) and weight (Fig. 5E for B16F10 and fig. S5E for MC38) were also smaller in SND1-KO cells with SND1 deletion than those in the control parental cells. As MHC-I molecules play essential roles in tumor antigen presentation for CD8+ T cell–mediated immune response, we thus investigated the infiltration of CD8+ T cells in tumor tissues by immunofluorescence and flow cytometry. Compared with the parental tumor tissue, there were more CD8+ T cells (red) infiltrated in B16F10–SND1-KO (Fig. 5F) and MC38–SND1-KO tumor tissues (fig. S5F). The flow cytometry analysis further revealed that the infiltration of CD45.2+ leukocytes (Fig. 5, G and H) and CD8+ T cells (Fig. 5, G and I) was significantly increased in the B16F10–SND1-KO tumor tissue and the MC38–SND1-KO tumor tissue (fig. S5, G to I) compared with the parental tumor tissue. Specifically, the proportion of CD8+ T cells among CD45.2+ leukocytes was significantly increased in the B16F10–SND1-KO tumors (Fig. 5J) and MC38–SND1-KO tumors (fig. S5J). Besides, we also detected the potential exhaustion of CD8+ T cells in tumor tissues. Compared with the control parental, there was no apparent discrepancy in the percentage of PD-1+ CD8+ T cells with deletion of SND1 (Fig. 5K). Furthermore, we clarified that SND1 deficiency in mice melanoma (fig. S6, A to D) and colon carcinoma (fig. S6, E to H) resulted in no significant changes in regulating cell proliferation, apoptosis, or cell cycle in vitro. We also performed the experiments by using RAG-1 (recombination activating gene 1) KO mice (Rag-1−/− mice) that lack mature T and B cells to investigate whether the absence of SND1 would affect tumor growth in vivo. We initially inoculated 5 × 105 WT or SND1-KO B16F10 cells subcutaneously into the flank of C57BL/6 WT mice and Rag-1−/− mice. By comparison between WT cells and SND1-KO B16F10 cells inoculated in either C57BL/6 WT mice or Rag-1−/− mice (fig. S6, I to K), we found that the size and weight of SND1-KO tumors were significantly decreased in the group of C57BL/6 WT mice, whereas in the group of Rag-1−/− mice, the tumors were comparable between WT and SND1-KO B16F10 cells in terms of the tumor size and weight. Together, these observations imply that deletion of SND1 in tumor cells is likely to promote CD8+ T cell–mediated cellular immune responses in the tumor microenvironment.
Fig. 5. Loss of SND1 in murine tumors limits tumor size with more CD8+ T cell infiltration in vivo.

(A) Three clones of B16F10 cells with stable depletion of SND1 by CRISPR-Cas9 system were collected, followed by IB for murine MHC-I (H2Kb). (B) Two clones of B16F10 cells with stable depletion of SND1 by CRISPR-Cas9 system were analyzed by flow cytometry for murine MHC-I (H2Kb/H2Db) using antibody simultaneously against H2Kb/H2Db. (C to E) 5 × 105 of either WT or SND1-KO B16F10 cells were subcutaneously transplanted into C57BL/6 mice. The tumor growth was monitored at the indicated times. C57BL/6 mice were sacrificed at day 11. Tumors were removed and photographed. The tumor tissues were weighed and plotted. Data are presented as means ± SD; n = 5 tumors for each group. *P < 0.05, two-tailed t test. (F) Immunofluorescence images of CD4+ T and CD8+ T cells in B16F10 tumor sections (scale bar, 20 μm). (G) C57BL/6 mice injected with equal numbers of WT or SND1-KO B16F10 cells were sacrificed at day 11. The digested tumor suspensions stained with antibodies against CD8 and CD45.2 (pan-leukocyte marker) were subjected to flow cytometry. (H to J) Percentages of infiltrating CD45.2+ cells and CD8+ T cells among total tumor tissue–derived cells and the percentage of infiltrating CD8+ T cells among total CD45+ leucocytes. n = 5 tumors for each group. *P < 0.05 and **P < 0.01, by unpaired t test. The experiments were performed and repeated at least three times, independently. (K) The percentage of infiltrating PD-1+ CD8+ T cells among total CD8+ T cells. n = 5 tumors for each group. n.s., not significant.
Moreover, in light of our observation that SN domain of SND1 is responsible for the association of SND1 with MHC-I HC, it is tempting to speculate the crucial function of SN domain in vivo. Our supplementary data support that the rescue of SN domain of SND1 significantly increased the tumor growth through mobilizing less CD8+ T cells infiltrating in tumors [fig. S7, A to H (B16F10) and I to P (MC38)].
To further clarify the influence of high expression of SND1 on CD8+ T cell–mediated cellular immune responses in tumor, we used transgenic OT-I mice. OT-I mice are ovalbumin (OVA)–specific T cell receptor transgenic (OT-I) mice whose CD8+ T cells could recognize the specific peptides (257 to 264 SIINFEKL) of chicken OVA, a surrogate tumor antigen that can be conveniently used to investigate CD8+ T cell–mediated immune responses directed against the OVA antigen (30). Meanwhile, B16F10-OVA cells and MC38-OVA cells were constructed by stably expressing the OVA antigen, which is able to be presented by MHC-I complex and specifically recognized by CD8+ T cells derived from OT-I mice. The identical expression of OVA was observed in both B16F10-OVA WT cells and B16F10-OVA SND1-KO cells with SND1 deletion (Fig. 6A), as well as in MC38-OVA WT and MC38-OVA SND1-KO cells (fig. S8A). The flow cytometry analysis demonstrated that higher level of MHC-I was detected in B16F10-OVA SND1-KO (Fig. 6B) or MC38-OVA SND1-KO cells (fig. S8B), which suggested that more OVA peptides might be presented in tumor cells in the absence of SND1. To interrogate the in vivo effect of antigen presentation, we subcutaneously inoculated equal numbers of B16F10-OVA WT or B16F10-OVA SND1-KO cells into OT-I mice. The tumor volume was monitored accordingly. The growth curve illustrated that SND1 deficiency in B16F10-OVA cells markedly inhibited the tumor growth in vivo (Fig. 6C). At day 19, the tumors were resected and the size and weight were measured. With deletion of SND1 (B16F10-OVA SND1-KO), the tumor size (Fig. 6D) and weight (Fig. 6E) were remarkably smaller than those of the control tumor (B16F10-OVA). The flow cytometry assay revealed that more CD45.2+ cells (Fig. 6F and G) and CD8+ T cells (Fig. 6F and H) were infiltrated in the tumor tissue of B16F10-OVA with SND1 deficiency (B16F10-OVA SND1-KO), and the ratio of CD8+ T cells to CD45.2+ leukocytes was significantly increased (Fig. 6I). These results further verified the impact of SND1 on MHC-I/antigenic peptide presenting in tumor cells and consequently affecting the infiltration of cytotoxic CD8+ T cells in tumor tissue.
Fig. 6. SND1 deletion promotes specific antigen presentation and enhances antitumor immunity.
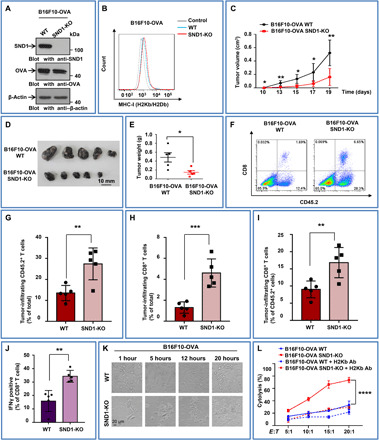
(A) B16F10 cells with stable depletion of SND1 by CRISPR-Cas9 system were stably transfected with OVA vector, followed by IB. (B) B16F10-OVA with SND1 deficiency was analyzed by flow cytometry for murine MHC-I (H2Kb/H2Db). (C to E) OT-I mice were injected with equal numbers of WT or SND1-KO B16F10-OVA cells, and tumor growth was observed over time. Then tumors were removed, photographed, and weighted. *P < 0.05 and **P < 0.01. (F) Flow cytometry was used for the analysis of CD45.2+ leucocyte and CD8+ T cell infiltration in tumor tissues. (G to I) Percentages of infiltrating CD45.2+ leucocytes and CD8+ T cells among total tumor tissue–derived cells and the percentage of infiltrating CD8+ T cells among total CD45.2+ leucocytes. n = 5 tumors for each group. **P < 0.01 and ***P < 0.001, by unpaired t test. (J) CD8+ T cells were purified from spleens of tumor-bearing OT-I mice and stimulated with 257 to 264 (SIINFEKL) peptide of OVA for 24 hours. Percentages of IFNγ+CD8+ T cells among total CD8+ T cells in the culture system were measured by flow cytometry. (n = 5, **P < 0.01). The experiments were repeated two times independently. (K) CD8+ T cells recognizing specific peptide of OVA (SIINFEKL) were purified from spleens of OT-I and then cocultured with WT or SND1-KO B16F10 cells stably expressing OVA (CD8+ T:B16F10-OVA, 10:1). Representative images were taken under a bright field at different time points. Scale bar, 20 μm. (L) In vitro comparison of cytolysis rates against CD8+ T cells purified from spleens of OT-I mice between WT and SND1-KO B16F10-OVA cells at different cell rates of CD8+ T (effector cells) to B16F10 (target cells) with/without anti–MHC class I antibodies (Ab) (E:T, 5:1, 10:1, 15:1, or 20:1). A lactate dehydrogenase–releasing cytotoxicity assay was performed to measure the cytolysis efficiency of CD8+ T cells on tumor cells. Each bar represents mean ± SD for biological triplicate experiments. ****P < 0.0001, two-way analysis of variance (ANOVA).
To further examine the contribution of SND1 to cytotoxic CD8+ T cell–mediated immune response in tumor, we isolated CD8+ T cells from spleen of OT-I mice bearing B16F10-OVA-WT or B16F10–OVA–SND1-KO tumor, respectively, and tested the cytotoxic CD8+ T cell population. As shown in Fig. 6J, the amount of interferon-γ (IFNγ)–producing CD8+ splenic T cells was significantly increased in SND1-KO group compared to WT control, suggesting that SND1 deficiency in tumor cells promoted and activated the OVA-specific CD8+ T cell response in the peripheral immune organ. To examine whether the enhanced antigen presentation in the SND1-KO cells results in robust recruitment and activation of CD8+ T cell–mediated cytotoxicity in the tumor environment, CD8+ T cells were isolated from OT-I mice and cocultured with tumor cells expressing OVA in the presence or absence of SND1. Representative time-lapse images showed that B16F10-OVA cells with SND1 deficiency were recognized and killed by more cytotoxic CD8+ T cells after 12 hours, while the parental B16F10-OVA cells survived after 12 hours with less aggregated CD8+ T cells (Fig. 6K). Furthermore, the amount of lactate dehydrogenase released from lysed target cells was used as indicator for cytolysis. Accordingly, the cytolysis value was higher in SND1-KO group in an E/T cell ratio–dependent manner and significantly abolished with the treatment of MHC-blocking antibodies (AF6-88.5) (Fig. 6L, red), which indicated an efficient cytotoxic effect mediated by CD8+ T cells. Consistently, the same results were also observed in MC38-OVA cells (fig. S8, C and D). Collectively, these findings illustrated that SND1 impaired tumor antigen presentation to cytotoxic CD8+ T cells and sabotaged the CD8+ T cell–mediated cellular immune response, supporting our previous data that deletion of SND1 in tumor cells promoted CD8+ T cell–mediated cellular immune response, and, as a result, inhibited the tumor growth in vivo.
To illustrate the significant relevance of high SND1 expression with tumor immune response in human, we screened the Tumor Immune Estimation Resource (TIMER) database at http://cistrome.org/TIMER/, a comprehensive resource for systematic analysis of immune infiltrates across diverse cancer types from The Cancer Genome Atlas (31). We analyzed the correlation of SND1 expression and the infiltration of CD8+ T cells in melanoma and colon adenocarcinoma (fig. S9, A and C). The results showed that SND1 expression was moderately negatively correlated with infiltration level of CD8+ T cells in melanoma (r = −0.247, P = 1.68 × 10−7) and colon adenocarcinoma (r = −0.393, P = 2.10 × 10−16).
In addition, we analyzed the correlation of SND1 with cancer patients’ prognosis by using PrognoScan database at http://dna00.bio.kyutech.ac.jp/PrognoScan/index.html (32). Notably, SND1 expression significantly affects the prognosis in melanoma. The cohort of melanoma (GSE19234) included 38 samples at different stages of melanoma and showed that high SND1 expression was significantly associated with poorer prognosis [Overall survival (OS) hazard ratio (HR) = 7.39, 95% confidence interval (CI) = 1.51 to 36.30, Cox P = 0.000912] (fig. S9B). Consistently, similar trend between SND1 expression with prognosis was observed in colorectal cancer (GSE17536; OS HR = 1.49, 95% CI = 0.69 to 3.22, Cox P = 0.314186) (fig. S9D). Since SND1 was negatively associated with the infiltration of CD8+ T cells and the survival of patients, we speculated that targeting SND1 might be a potentially therapeutic approach to enhance immune response and suppress tumor growth.
In conclusion, as shown in the working model (fig. S9E), SND1, localized on the membrane of ER in tumor cells, is able to hijack MHC-I HC from normally assembling and physically associating with its chaperone calnexin in the ER lumen at an early stage of ER processing, thereby leading MHC-I HC to the proteasomal pathway of ERAD promoted by VCP, cofactor VIMP, and E3 ligase HRD1 and sensitizing tumor to the diminished immune surveillance with decreased cytotoxic CD8+ T cells. Thus, SND1 profoundly facilitates immune evasion from tumor immune microenvironment through inhibition of antigen presentation and that this effect is mediated by down-regulation of MHC-I HC molecule (left). On the other hand, the absence of SND1 in either melanoma or colon carcinoma from subcutaneously tumor-bearing WT mouse results in growth inhibition and promotes tumor inflammation with more cytotoxic CD8+ T cells (right). Genetic ablation of SND1 in OVA-expressed tumors on OT-I mice induces sufficient antigen presentation to cytotoxic CD8+ T cells and enhances antitumor immunity. Therefore, SND1 determines the fate of MHC-I HC maturation and orchestrates a cancer-favored immune microenvironment. This model proposes the blockade of SND1–MHC-I HC axis in tumors as a viable option for immune system against cancer.
DISCUSSION
In the present study, we demonstrate that SND1 promotes immune escape of tumor cells through inhibition of MHC-I antigen presentation pathway, leading to impaired antitumor CD8+ T cell response in tumor microenvironment. Physiologically, the nascent unfolded HC of MHC-I would be stabilized by the chaperone calnexin before association with the β2m in ER. Here, we revealed that SND1 physically interacted with the nascent HC of MHC-I molecule in tumor cells. Instead of promoting the assembly of MHC-I molecule, SND1 recruits the nascent HC to VIMP/VCP complex for ERAD pathway. As a result, the MHC-I expression on tumor cell membrane is reduced, leading to impaired CD8+ T cell activation in tumor microenvironment.
SND1 is highly expressed in various cancers and is newly identified as a novel oncoprotein. We have previously reported that SND1 plays important physiological roles in a variety of cellular processes (17–19, 33). By using various methodologies, we identified cytoplasmic SND1 as an ER-associated protein and physically interacting with the nascent HC on the ER membrane in tumor cells. The biogenesis of transmembrane proteins requires the activity of the SEC61 complex, in which the subunit SEC61A has been proposed to act as a gate for the membrane insertion of nascent polypeptides (34). The MHC-I HC is synthesized on membrane-associated ribosomes and inserted cotranslationally into the ER through the translocon composed of SEC61 complex. In ER lumen, the nascent HC associates with its chaperones and glycosylation-related enzymes to generate a properly folded glycoprotein, and the formation of HC-β2m dimers indicates the maturity of MHC-I molecules (35). We have previously reported that SND1 protein is composed of SN and TSN domain (35). Here, we illustrated that SND1 protein is an ER-associated protein containing a functional N-terminal sequence (NP) that could associate with SEC61A on ER membrane. The predicted spatial conformation three-dimensional model of SND1-HLA-A complex is consistent with the mapping data, corroborating that SN domain of SND1 could physically interact with the A1 and A3 domain of HC. It indicates a fundamental and preceding role for SND1 at the early phase of HC assembly in the ER machinery. We further reveal that SND1 efficiently facilitates HC disassociation with calnexin and β2m, which interrupt the regular maturation and assembly of MHC-I molecule.
Previous studies have demonstrated that unfolded or misfolded HC that fails to form a proper structure can be recognized, dislocated, and degraded by the ERAD machinery (28). Accordingly, we, by far, are able to investigate how SND1 manages to hijack the nascent HLA-A for further degradation process on ER membrane. Although MHC-I HC is highly polymorphic and its potential ubiquitylation sites are variable (3), it is worthwhile to study the molecular mechanisms of how SND1 determines the fate of MHC-I HC precisely via the ubiquitin proteasome system. As a central element of the ubiquitin-proteasome system, VCP plays a key role in ERAD (36). During dislocation from the ER, misfolded or misassembled MHC-I HC as an ERAD substrate is ubiquitylated on the cytosolic side of the ER membrane and is degraded by the cytosolic proteasome (36). In the present study, we identified VCP, cofactor VIMP, and E3 ligase HRD1 of ERAD components as potential interactors of SND1, as revealed by mass spectrometric and coimmunoprecipitated analysis. Moreover, the results revealed that in the presence of SND1, more MHC-I HC was directed to the ERAD pathway for degradation.
We used mice model bearing tumors, especially transgenic OT-I mice, to illustrate the in vivo consequences of the tumor growth and immune response. With the absence of SND1 in either melanoma or colon carcinoma, the inoculated subcutaneous tumor growth was markedly inhibited but the amount of infiltrated CD8+ T cells in the tumor tissue was greatly increased. In accordance with the evidence from human database, we deduce that the highly expressed SND1 sabotages tumor antigen presentation to cytotoxic CD8+ T cell, thereby creating an immune niche with impaired surveillance that favors tumor growth.
Underlying the physiological relevance of our findings, the protein level of SND1 and MHC-I HC was negatively correlated with each other in human cervical and ovarian cancer cells, as well as in murine melanoma and colon cancerous cells. It is known that SND1 was robustly overexpressed in a variety of tumorigenic tissues and relatively highly expressed in normal tissues, and it was suggested that SND1 was an attractive target for anticancer therapy and a potent tumor marker (16). For more than a decade, it has been recognized that intact antigen presentation machinery, including MHC expression, in malignant cells is critical for T cell–dependent antitumor immunity because HLA-I antigen expression in tumors directly correlates with the degree of tumor T cell infiltration inside the tumor nests (3, 37). More recently, this knowledge has been underscored by findings showing that MHC class I molecule can be used as an independent prognostic factor for colorectal cancer and for predicting the efficacy of immunotherapy in bladder cancer and chemotherapy in ovarian cancer (1, 5, 38, 39). However, there is a long way to go before the different molecular mechanisms responsible for MHC-I alterations are precisely defined in different tumor types. For example, the mechanisms responsible for total MHC-I loss in about 60% of patients with breast cancer, in 50% of patients with prostate cancer, in 15% of patients with laryngeal cancer, or in 40% of patients with gastric cancer are yet to be identified (2, 6, 39, 40). However, it was proposed that during tumor development, tumors are heterogeneous with both HLA-positive and HLA-negative cells at early stages and are infiltrated by lymphocytes and M1 macrophages as a part of an active antitumor T helper 1 response (40). Thus, it is necessary to analyze tumor HLA expression and monitor HLA changes taking place during immunotherapy to understand how, when, and why the MHC/HLA alterations occur. In a way, our current study provides direct evidence for the idea of how the expression of MHC-I HC was regulated by an endogenously expressed protein SND1, the mechanism of which could be extendedly applied in the nonmalignant cells that may explain when and how MHC-I HC was altered during tumor initiation. Thus, SND1 could be a potential therapeutic target, at least for the treatment of malignancies with MHC-I defects in which the MHC-I is not genetically compromised.
To sum up, SND1 profoundly facilitates immune evasion from tumor immune microenvironment, and this effect is mediated by reducing the expression of MHC-I HC molecule. As a newly identified ER-associated protein, SND1 is able to hijack nascent MHC-I HC that is guided to ERAD-related proteasomal pathway, thereby impairing the proper assembling of HC with β2m in the ER lumen and sensitizing tumor cells to a diminished immune surveillance with abolished antigen presentation to cytotoxic CD8+ T cells. This novel finding may shed light on orchestrating the cancer-favored immune microenvironment via blockade of SND1–MHC-I HC axis in tumors as a viable option for immune system against cancer.
MATERIALS AND METHODS
Cell lines
HeLa, B16F10, and MC38 cells were obtained from the American Type Culture Collection (ATCC) and cultured using the standard conditions according to the ATCC instructions. B16F10 and HeLa cells were cultured in Dulbecco’s modified Eagle’s medium [Biological Industries (BI)] supplemented with 10% fetal bovine serum (FBS; BI), and MC38 cells were cultured with RPMI 1640 (BI) supplemented with 10% FBS. The human SKOV3 cell line was purchased from China Infrastructure of Cell Line Resources (Beijing, China), and SKOV3 cells were cultured with McCoy’s 5A Medium (Sigma-Aldrich) supplemented with 10% FBS. All cell lines were cultured under an atmosphere of 5% CO2 at 37°C. All of the cells were authenticated by examination of morphology and were confirmed to be mycoplasma-free.
Cells were transiently transfected with a Cas9 and single-guide RNA expression plasmid encoding puromycin resistance. The CRISPR-transfected cells will thus acquire transient resistance to puromycin, and the guide sequences were described as using the optimized CRISPR design at http://crispr.mit.edu. We confirmed that SND1-KO cells were not sensitive to puromycin after expansion, indicating a transient expression of CRISPR-Cas9 system in those cells.
Mice
Six- to 8-week-old male C57BL/6 mice were originally purchased from the Academy of Military Medical Sciences. OT-I transgenic mice, whose T cell receptor was designed to recognize OVA residues 257 to 264, were provided by Z. Dong from Tsinghua University. RAG-1 KO mice (Rag-1−/− mice) that lack mature T and B cells were purchased from Nanjing Biomedical Research Institute of Nanjing University. The gene phenotype was routinely confirmed by polymerase chain reaction (PCR) using specific primers. All animal procedures were approved by the Committee on the Use and Care of Animals of Tianjin Medical University.
For xenograft experiments, B16F10 cells or MC38 cells (5 × 105) (WT and SND1-KO) were subcutaneously transplanted into the flank of C57BL/6 mice or Rag-1−/− mice. Tumor height and width were measured with a caliper every 2 to 3 days to calculate tumor volume (= width2 × height × 0.523). Mice were sacrificed when tumors reached maximum allowed size (15 mm in diameter) or when signs of ulceration were evident. Likewise, 5 × 105 of B16-OVA or MC38-OVA cells were subcutaneously transplanted into the flank of OT-I mice. In all experiments, the initial implantation was conducted to animals at the age of 6 to 8 weeks.
Vector construction
Several plasmids were obtained from corporations including pCIpA102-G-HLA-A2-GFP (Addgene plasmid, no. 85162) and pcDNA3.1+ (Invitrogen, V79020), and several plasmids were gifts, including the following: pLV-IRES-Puro, pET-28a-c (+) vector from L. Shi (Tianjin Medical University), and CRISPR-Cas9 constructs px462 from X. Wu (Tianjin Medical University). Lentivirus plasmids expressing SND1 short hairpin RNA (shRNA) and the vector plasmid pLKO were purchased from MilliporeSigma (SHCLNG-NM_014390). The target sequences of shSND1 are shown as previously described (17). The FLAG-tagged or HA-tagged SND1 carried by pLV-IRES-Puro vector or pcDNA3.1+ were amplified from cDNA of HeLa cells or B16F10 cells with specific PCR primers. For exogenous HLA-A expression, the full length of HLA-A cloned from pCIpA102-G-HLA-A2-GFP with a FLAG tag at the C terminus was inserted into the lentiviral vector pLV-IRES-Puro. The HLA-A(N110Q)-FLAG mutant plasmid carried by pLV-IRES-Puro vector was constructed by the GENEWIZ Inc. The SN domain (SN-HA, 1 to 660 amino acids) or TSN domain (TSN-HA, 661 to 910 amino acids) of SND1 (mouse) protein with an HA tag at the C terminus was inserted into the lentiviral vector pLV-IRES-Puro. The FLAG-tagged full-length and truncation mutants of SND1 (human) were carried by pCMV-Blank vector for transient transfection. The pGEXT-4T-1 plasmids containing full length of SND1, SN domain, or TSN domain were generated as previously described (35). The pGEXT-4T-1 plasmids were inserted with full length of HLA-A(GST-HLA-A, 1 to 365 amino acids) and fragments (GST–HLA-A–A1, 25 to 114 amino acids; GST–HLA-A–A2, 115 to 206 amino acids; GST–HLA-A–A3, 207 to 298 amino acids; and GST–HLA-A–C, 299 to 365 amino acids) for GST pull-down assay.
An ER reporter plasmid was constructed with fusion protein (GFP-FLAG-GFP) and Eco RI/Xho I sites at the N terminus, which could be inserted with different signal peptides, such as HLA-A, UGGT, GAPDH, and SND1. KDEL, a target peptide sequence located on the C terminus, which prevents protein from secreting from the ER, was added at the N terminus of the fusion protein. Full length and deletion of N-terminal peptides (1 to 35 amino acids) of SND1 were inserted into the vector pcDNA3.1+ with a GFP tag at the C terminus.
Flow cytometry
Tumors were removed from sacrificed mice and digested by collagenase I (1.5 mg/ml) and deoxyribonuclease I (100 μg/ml; Solarbio) in RPMI 1640 for 1 hour at 37°C. The cell suspensions were passed through 70-μm filters (Falcon) to remove undigested tumor tissues, and then, the erythrocytes were removed by ACK lysis buffer. Cell suspensions were incubated in mouse Fc block (anti-CD16/CD32, BioLegend) before staining. Fluorochrome-conjugated anti-mouse CD45.2 (clone 104, eBioscience), CD8a (clone 53-6.7, BD Biosciences), IFNγ (clone XMG1.2, BioLegend), and PD-1 (clone RMP1-30, BioLegend) antibodies were used following the manufacturer’s protocol. Flow cytometry results were analyzed using FlowJo software.
Western blotting
Cells were collected and lysed with radioimmunoprecipitation assay buffer on ice. Protein concentration was determined by the bicinchoninic acid assay. Equal amounts of protein from each cell lysate were subjected to 10% SDS–polyacrylamide gel electrophoresis (SDS-PAGE). The resolved proteins were transferred to polyvinylidene difluoride membranes and blotted with indicated antibodies. β-Actin or GAPDH was used as an internal control.
Immunopurification and silver staining
Lysates from HeLa cells with or without SND1-FLAG expression were immunoprecipitated using Anti-FLAG M2 affinity beads (Sigma-Aldrich, A2220) for 12 hours at 4°C with constant rotation. After extensive washing with phosphate-buffered saline (PBS) plus 0.1% Tween, the bound proteins were eluted with excess FLAG peptides for 12 hours at 4°C with constant rotation, concentrated with Amicons (Ultra-0.5, 3 kDa, MilliporeSigma), and then visualized by silver staining on 8% SDS-PAGE. The distinct protein bands on the gel were recovered by trypsinization and analyzed by mass spectrometry.
Coimmunoprecipitation
Cellular lysates were prepared by incubating the cells in cell lysis buffer [50 mM tris-HCl (pH 8.0), 150 mM NaCl, 0.2% Nonidet P-40, and 2 mM EDTA] in the presence of protease inhibitor cocktail for 20 min at 4°C with vortex every 5 min, followed by centrifugation at 13,000 rpm for 10 min at 4°C. For immunoprecipitation, about 1 mg of protein was incubated with control [immunoglobulin G (IgG)] or specific antibodies (1 to 2 μg) for 12 hours at 4°C with constant rotation. A total of 50 μl of 50% protein A/G agarose beads was then added, and the incubation was continued for an additional 6 hours. Beads were then washed five times using the PBS with 0.1% Tween. Between washes, the beads were collected by centrifugation at 2000 rpm for 5 min at 4°C. The precipitated proteins were eluted from the beads by resuspending the beads in 2.5× SDS-PAGE loading buffer and boiling for 10 min. The boiled immune complexes were subjected to SDS-PAGE, followed by immunoblotting with appropriate antibodies.
Immunofluorescence and Duolink assay
Cells were fixed for 10 min at room temperature with 4% paraformaldehyde in PBS and permeabilized with 0.1% Triton X-100 in PBS for 10 min at room temperature. Samples were then blocked in 3% bovine serum albumin and incubated consecutively with primary antibodies to SND1/calnexin (ProteinTech, 60265-1-Ig/10427-2-AP) and the appropriate secondary antibodies coupled to Alexa Fluor 488 or 594 (Invitrogen). Cells were covered with a drop of 4′,6-diamidino-2-phenylindole (DAPI) for 5 min. After washing with PBS, slides were mounted for observation. Confocal images were captured on Leica DMi8 using a ×63 oil objective. The fluorescence intensity profiles of the targeted regions were obtained with a Leica DMi8 microscope and LAS X version 3.5.5.
Duolink assay was performed using the Duolink In Situ Red Starter Kit Mouse/Rabbit following the manufacturer’s instructions (Sigma-Aldrich, DUO92004), and its basic protocols of fixation, retrieval, and permeabilization are the same as immunofluorescence. Samples were incubated with blocking solution for 1 hour at 37°C in a humidity chamber and then overnight at 4°C with anti-SND1 mouse antibody and anti-calnexin rabbit antibody. Slides were then incubated for 1 hour at 37°C with a mix of the MINUS (anti-mouse) and PLUS (anti-rabbit) PLA probes. Hybridized probes were ligated using the ligation-ligase solution for 30 min at 37°C and then amplified using the amplification-polymerase solution for 100 min at 37°C. Slides were covered with a drop of DAPI (Invitrogen) for 5 min and mounted for observation with a Leica DMi8 confocal microscope.
Recombinant protein purification
GST-fusion proteins that contained full length or truncations of SND1 and HLA-A were produced in BL21 E. coli and purified using glutathione agarose beads. Recombinant proteins (HLA-A) were expressed with rabbit reticulocyte lysate (TNT systems, Promega) according to the manufacturer’s recommendation. His-SND1 was purified in BL21(DE3) E. coli and purified using HIS-Select Nickel Affinity Gel (Sigma-Aldrich) according to the standard procedures.
In GST pull-down assays, the bead-bound GST-fusion proteins were incubated with in vitro transcribed/translated products at 4°C for 10 hours. The beads were then washed five times with binding buffer containing 75 mM NaCl and detected by Western blotting.
Host immune response
OT-I mice were injected with WT or SND1-KO B16F10-OVA cells, and CD8+ T cells were collected by microbeads (no. 130-116-478, Miltenyi Biotec), restimulated ex vivo with SIINFEKL peptide (OVA-derived peptide being presented through the MHC class I molecules; no. S7951, Sigma-Aldrich) for 20 hours, and lastly cultured with brefeldin A (no. 420601, BioLegend) for 4 hours. Then, the cells were subjected to flow cytometric analysis, and the percentages of IFNγ+CD8+ T cells among total CD8+ T cells was measured by flow cytometry.
Cytolysis assay
A cytotoxicity detection kit (R&D Systems) was used to measure the cytolysis rate elicited by CD8+ T cells against different tumor cells. B16F10-OVA or MC38-OVA (WT or SND1-KO) cells (2 × 104) were cocultured with CD8+ T cells in an E/T cell ratio–dependent manner (1 × 105/2 × 105/3 × 105/4 × 105) isolated from OT-I mice with or without the treatment of MHC-blocking antibodies (AF6-88.5; 0.25 μg for 1 × 105 CD8+ T cells) in sterile 96-well tissue culture plates for 12 hours. The microplate was centrifuged at 250g for 10 min, and supernatant was removed to incubate with reaction mixture. The absorbance of the samples was measured at 490 or 492 nm with an enzyme-linked immunosorbent assay reader.
The amount of lactate dehydrogenase released from lysed target cells was used as an indicator for cytolysis. Cytolysis rate (percentage) was calculated on the basis of the following equation: cytotoxicity (%) = (effector/target cell mix − effector cell control − low control)/(high control − low control) × 100. Under the same conditions, B16F10-OVA or MC38-OVA (WT or SND1-KO) cells (1 × 105) were cocultured with CD8+ T cells (1 × 106) in sterile 24-well culture plates, and images were taken under a bright field using a Leica microscope at different time points.
Prediction of the spatial conformation of SND1-HLA-A complex
The three-dimensional structure of SND1 and HLA-A were generated using I-TASSER. Then, the ZDOCK website was used to predict the possible binding conformation between SND1 and HLA-A. The structure with the lowest binding free energy was chosen as the initial binding conformation, which was further optimized using molecular dynamics (MD) simulation. MD simulation was carried out with Gromacs package (version 5.0.1) and GROMOS96 54A7 force field. The binding complex of SND1 and HLA-A was first placed in a cube box with a minimum distance of 1.0 nm from the edge of box. Then, the complex was solvated with TIP3P water molecules and neutralized with the addition of Na+ and Cl− ions. A total of 1000 steps steepest descent energy minimization was conducted to remove local contacts, and the equilibration was performed including 1-ns NVT (Constant volume) and 5-ns NPT (Constant pressure) relaxations. Last, 50-ns MD simulations were performed at 300 K and 1.0-atm pressure under periodic boundary conditions. In all simulation steps, the SHAKE algorithm was applied to constrain all bonds involving hydrogen atoms, Particle mesh Ewald method was used to treat long-range electrostatics, and a cutoff distance of 1.0 nm was used for short-range electrostatic and van der Waals. On the basis of the resulting MD trajectory, we can analyze the structural stability of the complex, calculate the binding free energy using Molecular Mechanics Poisson Boltzmann Surface Area (MM-PBSA) method and identify key residues to the association process.
Database analysis
The correlation between SND1 and cancer immune infiltrates was investigated via TIMER (https://cistrome.shinyapps.io/timer/). The strength of correlations was evaluated using the Spearman correlation test; the Spearman coefficient was considered to indicate poor correlation if <0.2, moderate if <0.4, relatively strong if <0.6, strong if <0.8, and very strong if >0.8. P values <0.05 were considered statistically significant. The correlation between SND1 expression and survival in different cancers was analyzed by the PrognoScan database (http://dna00.bio.kyutech.ac.jp/PrognoScan/index.html). The threshold was adjusted to a Cox P < 0.05.
Statistics
Data from biological triplicate experiments are presented with error bars as means ± SD unless otherwise noted. Two-tailed unpaired t test was used to compare two groups of data, and analysis of variance (ANOVA) with Bonferroni’s correction was used to compare multiple groups of data used for statistical analysis. All of the statistical testing results were determined using GraphPad Prism 7.0 software.
Supplementary Material
Acknowledgments
We thank Z. Dong from Tsinghua University for sharing the OT-I mouse model. Funding: This work was supported by grant 31125012 from the National Science Foundation for Distinguished Young Scholars of China (to J.Y.); grant IRT13085 from the Innovation Team Development Plan of the Ministry of Education (to J.Y.); grant 31300709 (to X.W.), grants 31870747 and 31370749 (to J.Y.), and grant 81572882 (to Z.Y.) from the National Natural Science Foundation of China; and the High-Level Innovation and Entrepreneurship Team of Tianjin Talent Development Special Support Plan (to J.Y.). This work also received support from grant YJSCX201814 (to Y.W.) from the Postgraduate Innovation Fund of “13th Five-Year Comprehensive Investment,” Tianjin Medical University. Author contributions: Y.W., X.W., and J.Y. generated the initial idea and conducted key experiments. Y.W., X.C., H.L., C.H., and L.X. performed the research. Y.W., X.W., X.C., and J.Y. analyzed the data and wrote the manuscript. Y.Z., T.Z., and K.Z. helped with the bioinformatics analysis. J.Y., X.Y., and Z.Y. critically revised the manuscript for important intellectual content. L.G., X.L., J.H., Y.R., W.Z., and X.S. provided administrative, technical, or material support. J.Y. supervised the study. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/22/eaba5412/DC1
REFERENCES AND NOTES
- 1.Garrido F., Ruiz-Cabello F., Aptsiauri N., Rejection versus escape: The tumor MHC dilemma. Cancer Immunol. Immunother. 66, 259–271 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Morrison B. J., Steel J. C., Morris J. C., Reduction of MHC-I expression limits T-lymphocyte-mediated killing of Cancer-initiating cells. BMC Cancer 18, 469 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Garrido F., Aptsiauri N., Doorduijn E. M., Garcia Lora A. M., van Hall T., The urgent need to recover MHC class I in cancers for effective immunotherapy. Curr. Opin. Immunol. 39, 44–51 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Seliger B., Cabrera T., Garrido F., Ferrone S., HLA class I antigen abnormalities and immune escape by malignant cells. Semin. Cancer Biol. 12, 3–13 (2002). [DOI] [PubMed] [Google Scholar]
- 5.Cabrera T., López-Nevot M. A., Gaforio J. J., Ruiz-Cabello F., Garrido F., Analysis of HLA expression in human tumour tissues. Cancer Immunol. Immunother. 52, 1–9 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Garrido F., Romero I., Aptsiauri N., Garcia-Lora A. M., Generation of MHC class I diversity in primary tumours and selection of the malignant phenotype. Int. J. Cancer 138, 271–280 (2016). [DOI] [PubMed] [Google Scholar]
- 7.Kukita K., Tamura Y., Tanaka T., Kajiwara T., Kutomi G., Saito K., Okuya K., Takaya A., Kanaseki T., Tsukahara T., Hirohashi Y., Torigoe T., Furuhata T., Hirata K., Sato N., Cancer-associated oxidase ERO1-α regulates the expression of MHC class I molecule via oxidative folding. J. Immunol. 194, 4988–4996 (2015). [DOI] [PubMed] [Google Scholar]
- 8.Sadasivan B. K., Cariappa A., Waneck G. L., Cresswell P., Assembly, peptide loading, and transport of MHC class I molecules in a calnexin-negative cell line. Cold Spring Harb. Symp. Quant. Biol. 60, 267–275 (1995). [DOI] [PubMed] [Google Scholar]
- 9.Burr M. L., van den Boomen D. J. H., Bye H., Antrobus R., Wiertz E. J., Lehner P. J., MHC class I molecules are preferentially ubiquitinated on endoplasmic reticulum luminal residues during HRD1 ubiquitin E3 ligase-mediated dislocation. Proc. Natl. Acad. Sci. U.S.A. 110, 14290–14295 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.van den Boomen D. J., Timms R. T., Grice G. L., Stagg H. R., Skødt K., Dougan G., Nathan J. A., Lehner P. J., TMEM129 is a Derlin-1 associated ERAD E3 ligase essential for virus-induced degradation of MHC-I. Proc. Natl. Acad. Sci. U.S.A. 111, 11425–11430 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wiertz E. J., Tortorella D., Bogyo M., Yu J., Mothes W., Jones T. R., Rapoport T. A., Ploegh H. L., Sec61-mediated transfer of a membrane protein from the endoplasmic reticulum to the proteasome for destruction. Nature 384, 432–438 (1996). [DOI] [PubMed] [Google Scholar]
- 12.Ochoa B., Chico Y., Martínez M. J., Insights Into SND1 oncogene promoter regulation. Front. Oncol. 8, 606 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jariwala N., Rajasekaran D., Mendoza R. G., Shen X. N., Siddiq A., Akiel M. A., Robertson C. L., Subler M. A., Windle J. J., Fisher P. B., Sanyal A. J., Sarkar D., Oncogenic role of SND1 in development and progression of hepatocellular carcinoma. Cancer Res. 77, 3306–3316 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jariwala N., Rajasekaran D., Srivastava J., Gredler R., Akiel M. A., Robertson C. L., Emdad L., Fisher P. B., Sarkar D., Role of the staphylococcal nuclease and tudor domain containing 1 in oncogenesis (review). Int. J. Oncol. 46, 465–473 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sundström J. F., Vaculova A., Smertenko A. P., Savenkov E. I., Golovko A., Minina E., Tiwari B. S., Rodriguez-Nieto S., Zamyatnin A. A. Jr., Välineva T., Saarikettu J., Frilander M. J., Suarez M. F., Zavialov A., Ståhl U., Hussey P. J., Silvennoinen O., Sundberg E., Zhivotovsky B., Bozhkov P. V., Tudor staphylococcal nuclease is an evolutionarily conserved component of the programmed cell death degradome. Nat. Cell Biol. 11, 1347–1354 (2009). [DOI] [PubMed] [Google Scholar]
- 16.Gutierrez-Beltran E., Denisenko T. V., Zhivotovsky B., Bozhkov P. V., Tudor staphylococcal nuclease: Biochemistry and functions. Cell Death Differ. 23, 1739–1748 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xin L., Zhao R., Lei J., Song J., Yu L., Gao R., Ha C., Ren Y., Liu X., Liu Y., Yao Z., Yang J., SND1 acts upstream of SLUG to regulate the epithelial-mesenchymal transition (EMT) in SKOV3 cells. FASEB J. 33, 3795–3806 (2018). [DOI] [PubMed] [Google Scholar]
- 18.Su C., Zhang C., Tecle A., Fu X., He J., Song J., Zhang W., Sun X., Ren Y., Silvennoinen O., Yao Z., Yang X., Wei M., Yang J., Tudor staphylococcal nuclease (Tudor-SN), a novel regulator facilitating G1/S phase transition, acting as a co-activator of E2F-1 in cell cycle regulation. J. Biol. Chem. 290, 7208–7220 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cui X., Zhao C., Yao X., Qian B., Su C., Ren Y., Yao Z., Gao X., Yang J., SND1 acts as an anti-apoptotic factor via regulating the expression of lncRNA UCA1 in hepatocellular carcinoma. RNA Biol. 15, 1364–1375 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yu L., Liu X., Cui K., di Y., Xin L., Sun X., Zhang W., Yang X., Wei M., Yao Z., Yang J., SND1 acts downstream of TGFβ1 and upstream of Smurf1 to promote breast cancer metastasis. Cancer Res. 75, 1275–1286 (2015). [DOI] [PubMed] [Google Scholar]
- 21.Roy A., Kucukural A., Zhang Y., I-TASSER: A unified platform for automated protein structure and function prediction. Nat. Protoc. 5, 725–738 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pierce B. G., Wiehe K., Hwang H., Kim B. H., Vreven T., Weng Z., ZDOCK server: Interactive docking prediction of protein-protein complexes and symmetric multimers. Bioinformatics 30, 1771–1773 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang C., Nguyen P. H., Pham K., Huynh D., Le T. B. N., Wang H., Ren P., Luo R., Calculating protein-ligand binding affinities with MMPBSA: Method and error analysis. J. Comput. Chem. 37, 2436–2446 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kobayashi K., Jomaa A., Lee J. H., Chandrasekar S., Boehringer D., Shan S. O., Ban N., Structure of a prehandover mammalian ribosomal SRP.SRP receptor targeting complex. Science 360, 323–327 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zehner M., Marschall A. L., Bos E., Schloetel J.-G., Kreer C., Fehrenschild D., Limmer A., Ossendorp F., Lang T., Koster A. J., Dübel S., Burgdorf S., The translocon protein Sec61 mediates antigen transport from endosomes in the cytosol for cross-presentation to CD8+ T cells. Immunity 42, 850–863 (2015). [DOI] [PubMed] [Google Scholar]
- 26.Römisch K., A case for Sec61 channel involvement in ERAD. Trends Biochem. Sci. 42, 171–179 (2017). [DOI] [PubMed] [Google Scholar]
- 27.Meyer H., Bug M., Bremer S., Emerging functions of the VCP/p97 AAA-ATPase in the ubiquitin system. Nat. Cell Biol. 14, 117–123 (2012). [DOI] [PubMed] [Google Scholar]
- 28.Morito D., Nagata K., Pathogenic hijacking of ER-associated degradation: Is ERAD flexible? Mol. Cell 59, 335–344 (2015). [DOI] [PubMed] [Google Scholar]
- 29.van den Boomen D. J., Lehner P. J., Identifying the ERAD ubiquitin E3 ligases for viral and cellular targeting of MHC class I. Mol. Immunol. 68, 106–111 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Moroishi T., Hayashi T., Pan W.-W., Fujita Y., Holt M. V., Qin J., Carson D. A., Guan K.-L., The Hippo pathway kinases LATS1/2 suppress cancer immunity. Cell 167, 1525–1539.e17 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li B., Severson E., Pignon J.-C., Zhao H., Li T., Novak J., Jiang P., Shen H., Aster J. C., Rodig S., Signoretti S., Liu J. S., Liu X. S., Comprehensive analyses of tumor immunity: Implications for cancer immunotherapy. Genome Biol. 17, 174 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mizuno H., Kitada K., Nakai K., Sarai A., PrognoScan: A new database for meta-analysis of the prognostic value of genes. BMC Med. Genomics 2, 18 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yin J., Ding J., Huang L., Tian X., Shi X., Zhi L., Song J., Zhang Y., Gao X., Yao Z., Jing X., Yang J., SND1 affects proliferation of hepatocellular carcinoma cell line SMMC-7721 by regulating IGFBP3 expression. Anat. Rec. 296, 1568–1575 (2013). [DOI] [PubMed] [Google Scholar]
- 34.Meunier L., Usherwood Y. K., Chung K. T., Hendershot L. M., A subset of chaperones and folding enzymes form multiprotein complexes in endoplasmic reticulum to bind nascent proteins. Mol. Biol. Cell 13, 4456–4469 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Paulsson K., Wang P., Chaperones and folding of MHC class I molecules in the endoplasmic reticulum. Biochim. Biophys. Acta 1641, 1–12 (2003). [DOI] [PubMed] [Google Scholar]
- 36.Yang J., Aittomäki S., Pesu M., Carter K., Saarinen J., Kalkkinen N., Kieff E., Silvennoinen O., Identification of p100 as a coactivator for STAT6 that bridges STAT6 with RNA polymerase II. EMBO J. 21, 4950–4958 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Beatty G. L., Gladney W. L., Immune escape mechanisms as a guide for cancer immunotherapy. Clin. Cancer Res. 21, 687–692 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Garrido F., Algarra I., MHC antigens and tumour escape from immune surveillance. Adv. Cancer Res. 83, 117–158 (2001). [DOI] [PubMed] [Google Scholar]
- 39.Perea F., Bernal M., Sánchez-Palencia A., Carretero J., Torres C., Bayarri C., Gómez-Morales M., Garrido F., Ruiz-Cabello F., The absence of HLA class I expression in non-small cell lung cancer correlates with the tumor tissue structure and the pattern of T cell infiltration. Int. J. Cancer 140, 888–899 (2017). [DOI] [PubMed] [Google Scholar]
- 40.Aptsiauri N., Ruiz-Cabello F., Garrido F., The transition from HLA-I positive to HLA-I negative primary tumours: The road to escape from T-cell responses. Curr. Opin. Immunol. 51, 123–132 (2018). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/22/eaba5412/DC1