

Abstract
Neutrophil accumulation is associated with lung pathology during active tuberculosis (ATB). However, the molecular mechanism or mechanisms by which neutrophils accumulate in the lung and contribute to TB immunopathology are not fully delineated. Using the well-established mouse model of TB, our new data provide evidence that the alarmin S100A8/A9 mediates neutrophil accumulation during progression to chronic TB. Depletion of neutrophils or S100A8/A9 deficiency resulted in improved Mycobacterium tuberculosis (Mtb) control during chronic but not acute TB. Mechanistically, we demonstrate that, following Mtb infection, S100A8/A9 expression is required for upregulation of the integrin molecule CD11b specifically on neutrophils, mediating their accumulation during chronic TB disease. These findings are further substantiated by increased expression of S100A8 and S100A9 mRNA in whole blood in human TB progressors when compared with nonprogressors and rapidly decreased S100A8/A9 protein levels in the serum upon TB treatment. Furthermore, we demonstrate that S100A8/A9 serum levels along with chemokines are useful in distinguishing between ATB and asymptomatic Mtb-infected latent individuals. Thus, our results support targeting S100A8/A9 pathways as host-directed therapy for TB.
Keywords: Immunology, Infectious disease
Keywords: Innate immunity, Neutrophils, Tuberculosis

Introduction
Mycobacterium tuberculosis (Mtb), the causative agent of the disease tuberculosis (TB), is estimated to infect one-fourth of the world’s population and results in approximately 1.6 million deaths each year (1). In addition, the emergence of multidrug and extensively drug-resistant Mtb strains and variable efficacy of the currently used vaccine, Mycobacterium bovis BCG, are barriers to global control of TB. Thus, more research is needed to fully understand the mechanisms of TB immunopathogenesis that can be targeted to improve host control of Mtb infection.
Neutrophil accumulation has been associated with TB disease in humans and mouse models of TB (2, 3). Neutrophils are the primary Mtb-infected cell population in the sputum, bronchoalveolar lavage (BAL) fluid, and cavities of Mtb-infected patients (4). In mouse models, neutrophils are among the first cells infected with Mtb and are capable of recognizing and responding to Mtb infection and displaying effector functions (5, 6). Calprotectin, also known as S100A8/A9, is mainly expressed by myeloid cells, specifically neutrophils and monocytes, and is a feature of chronic inflammatory diseases, such as autoimmune diseases and TB (7–9). S100A8 and S100A9 covalently form the heterocomplex protein S100A8/A9 and bind to receptors such as TLR4 and the receptor for advanced glycation end products (RAGE), which mediates a proinflammatory response through NF-κB and nuclear factor erythroid-derived 2-like 2 (Nrf2) signaling (10, 11). We recently demonstrated that S100A8/A9-expressing neutrophils accumulated within TB granulomas in human and animal models of TB and that S100A8/A9 serum levels coincided with increased neutrophil numbers and enhanced TB disease in active TB (ATB) patients (7). However, we did not fully understand the mechanism or mechanisms by which S100A8/A9 mediates neutrophil accumulation that may promote TB disease severity.
In the current study, we demonstrate that S100A8/A9 expression mediated accumulation of lung neutrophils in chronically Mtb-infected mice. Additionally, the observed accumulation was mirrored by increased S100A8 and S100A9 mRNA in whole blood transcriptional profiles of latently infected individuals (LTBI) who progressed to ATB disease (progressors) when compared with individuals who did not progress to TB disease (nonprogressors). Importantly, serum levels of S100A8/A9 protein decreased rapidly upon successful TB treatment. These studies suggest that, in animal models and human progressors, S100A8/A9 expression levels increase during TB progression or chronic TB disease. Consistent with a detrimental role for S100A8/A9, mice depleted of neutrophils or deficient in S100A9 exhibited decreased Mtb burden in the lungs during chronic infection and TB reactivation. The improved Mtb control in S100A9KO mice coincided with reduced lung neutrophil accumulation within TB granulomas and decreased expression of the integrin CD11b on neutrophils. Our mechanistic in vitro studies show that S100A9KO neutrophils have defective Mtb uptake as well as reduced ability to upregulate CD11b integrin expression. Importantly, we demonstrate that S100A8/A9 serum levels along with chemokines are useful for distinguishing between ATB patients and LTBI. Our results suggest that targeting S100A8/A9 as host-directed therapy in TB would provide pathways that can be used as adjunct therapy to improve Mtb control.
Results
Depletion of neutrophils during chronic but not acute TB improves Mtb control.
A neutrophil-associated transcriptional signature was reported in ATB patients, and neutrophil accumulation was associated with TB disease in mouse models (2, 3, 7). Consistent with this, neutrophils accumulated in the blood during ATB, but not in asymptomatic latency in macaques infected with Mtb (Figure 1A). However, it is unclear whether neutrophils play distinct roles during the acute and chronic stages of Mtb infection. Thus, we studied the kinetics of neutrophil accumulation in the well-established mouse model of low-dose aerosol Mtb infection in C57BL/6J (B6) mice. Neutrophils accumulated in the lung following infection with Mtb HN878 early, starting at 30 days post infection (dpi). Accumulation further increased during chronic stages of infection, starting around 60 dpi and continuing until 300 dpi (Figure 1B). As we observed increased neutrophil accumulation during both acute and chronic stages, we next addressed the functional role for neutrophils at the 2 distinct phases of Mtb infection. We found that treatment with 1A8 antibody, which specifically depletes neutrophils (12), during acute Mtb infection did not affect overall lung and spleen Mtb burden (Figure 1C). Treatment with 1A8 depleted lung neutrophils significantly, but also coincided with increased monocyte and macrophage populations and increased production of the chemokine G-CSF (Figure 1D and Supplemental Figure 1A; supplemental material available online with this article; https://doi.org/10.1172/JCI130546DS1). However, despite changes in myeloid cell accumulation, the total inflammatory area measured in the lung was not significantly altered (Figure 1E). In contrast, 1A8 treatment during chronic Mtb infection led to decreased Mtb burden in both the lung and spleen (Figure 1F). Neutrophil depletion during chronic TB did not affect other myeloid populations (Supplemental Figure 1B), but there was decreased production of proinflammatory cytokines, such as IL-6 and TNF-α (Figure 1G). The area of lung cellular inflammation was still not affected in mice when neutrophils were depleted during chronic infection (Figure 1H). These data together suggest that, despite early accumulation of neutrophils, neutrophils do not significantly affect Mtb control during acute stages of infection. In contrast, neutrophil accumulation during chronic Mtb infection may have a functional role in promoting higher Mtb burden and contribute to increased susceptibility.
Figure 1. Neutrophil depletion during chronic TB improves Mtb control.

(A) The numbers of blood neutrophils from Rhesus macaques with ATB (n = 8) or LTBI (n = 10) were measured at dpi following Mtb infection or at necropsy (N). B6 mice (n = 5–13) were aerosol infected with approximately 100 CFU Mtb HN878. (B) Neutrophil accumulation was determined by flow cytometry at dpi. B6 mice were infected with Mtb HN878 and administered IgG (n = 5–10) or 1A8 (n = 5–10, 300-μg/dose) i.p. every other day. (C) Bacterial burden in the lung and spleen was determined by plating on 21 dpi. (D) Lung homogenates were analyzed for G-CSF protein expression by ELISA on 21 dpi. (E) Pulmonary inflammation was quantified on formalin-fixed, paraffin-embedded (FFPE) lung sections from 21 dpi samples stained with H&E. B6 mice were infected with HN878 and administered IgG (n = 4–5) or 1A8 (n = 4–5, 300-μg/dose) i.p. every other day. (F) Bacterial burden in the lung and spleen was determined by plating on 106 dpi. (G) Lung homogenates were analyzed to measure levels of IL-6 and TNF-α proteins at 106 dpi using Luminex assays. (H) Pulmonary inflammation was quantified on FFPE lung sections from 106 dpi tissues stained with H&E. Figures depict 1 experiment representative of 2 or combined data from multiple experiments. Data points represent the mean ± SEM of values. (A) 2-way ANOVA; (B) Student’s t test between 0 and indicated dpi; (C–G) Student’s t test between isotype- and 1A8-treated mice. *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001; ****P ≤ 0.0001.
S100A8/A9 is induced during progression to TB disease in animal models and in humans.
S100A8/A9 makes up 40% of the cytoplasmic protein of a neutrophil (13). In addition, we have shown that increased serum S100A8/A9 levels correlate with increased neutrophils in peripheral blood of ATB patients (7). Accordingly, we found that accumulation of S100A8/A9 levels in Mtb-infected mouse lungs also closely followed the accumulation of neutrophil influx in the infected lung. Early increased production of S100A8/A9 was detected between 15 and 30 dpi, and sustained production during chronic infection was observed after 30 dpi (Figure 2A). As TB disease progressed, S100A8/A9 levels increased, coinciding with the increased accumulation of neutrophils observed in chronically Mtb-infected lungs (Figure 1B and Figure 2A), and the S100A8/A9 levels positively correlated with the number of lung neutrophils (Figure 2B). Thus, we next addressed whether S100A8/A9 expression also correlated with TB disease progression in humans with LTBI. Therefore, we analyzed the whole blood transcriptional mRNA levels of S100A8 and S100A9 in participants enrolled in the Adolescent Cohort Study (ACS) of TB progressors and matched Mtb-infected controls (14). Transcript levels of both S100A8 and S100A9 mRNA significantly increased in the TB progressors about 6 months before TB diagnosis compared with expression levels in matched healthy controls (HCs) (Figure 2C). Our results show that, in both mice and in humans, an increase in S100A8/A9 expression coincides with TB disease progression.
Figure 2. S100A8/A9 mRNA levels are indicators of TB disease progression in mice and humans with TB.

B6 mice (n = 3–5) were aerosol infected with approximately 100 CFU Mtb HN878. (A) Lung S100A8/A9 levels were determined by ELISA at dpi. (B) The number of neutrophils was correlated with the levels of S100A8/A9 using a linear regression of the means. (C) Kinetics of S100A8 and S100A9 mRNA expression over time, expressed as log2 fold change (FC) between bin-matched progressors (n = 44) and controls (n = 106) and modeled as nonlinear splines (dotted lines). Light green shading represents 99% CI and dark green shading 95% CI for the temporal trends, computed by performing 2000 spline fitting iterations after bootstrap resampling from the full data set. The deviation time (day), calculated as the time point at which the 99% CI deviates from a log2 fold change of 0, is indicated by the vertical red line. Pulmonary TB patients were enrolled and treated with standard first-line TB regimen. Serum samples were collected at time of diagnosis (0) and at weeks 2, 6, and 26 during treatment. S100A8/A9 levels were determined by ELISA in patients that were (D) successfully treated and cured (n = 34), (E) successfully treated but relapsed (n = 10), and (F) cured (n = 34) versus failed treatment (n = 10) at 2 weeks after treatment initiation. (A) Student’s t test between 0 and indicated dpi; (B) linear regression of the means; (D–F) mixed effects analysis of 1-way ANOVA with Dunnett’s posttest. Figures depict 1 experiment representative of 2 or combined data from multiple experiments. Data points represent the mean ± SEM of values. *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001; ****P ≤ 0.0001.
Thus, we next determined whether serum levels of S100A8/A9 protein declined upon successful treatment in ATB patients receiving standard first-line TB treatment. In ATB patients that were successfully treated and were either cured (n = 34; Figure 2D) or cured but then relapsed in the following 2 years after treatment (n = 10; Figure 2E), the S100A8/A9 levels decreased rapidly by 2 weeks after treatment and continued to decrease throughout treatment. However, in patients that failed TB treatment (n = 10; Figure 2F), the S100A8/A9 levels did not decrease significantly within the first 2 weeks after treatment. Together, our data show that S100A8/A9 increases during TB disease progression and that levels decline sharply upon successful TB treatment.
S100A9 deficiency improves Mtb control during chronic infection by limiting neutrophil accumulation.
Our new data here show that S100A8/A9 levels increase during chronic Mtb infection in mice and human TB progressors and that depletion of neutrophils during chronic infection in mice improves Mtb control (Figures 1 and 2). Therefore, we next assessed the long-term outcome of Mtb infection in B6 and S100A9KO mice (15) (which lack functional S100A8/A9) during chronic stages of infection with Mtb HN878. Although lung and spleen Mtb HN878 CFU was comparable at 50 dpi, we found that, during chronic infection from 100 dpi until 300 dpi, Mtb CFU in the lungs of S100A9KO mice were significantly lower when compared with those in B6 HN878-infected mice (Figure 3, A and B). This improved Mtb control was also observed during infection with another clinical strain, HN563, but not when the lab-adapted Mtb strain H37Rv was used (Supplemental Figure 2, A and B).
Figure 3. S100A8/A9 deficiency protects mice during chronic TB.

B6 (n = 5–10) and S100A9KO (n = 5–9) mice were aerosol infected with approximately 100 CFU Mtb HN878. (A and B) Lung and spleen bacterial burden were determined by plating at different dpi. Lung myeloid population cell counts were enumerated in B6 and S100A9KO HN878-infected mice using flow cytometry at 50, 100, 150, and 300 dpi. (C) Neutrophils and (D) other myeloid cells are shown. (E) FFPE lung sections were used to carry out immunofluorescence staining for Ly6G (green), MPO (red), and DAPI (blue) or B220 (green), CD3 (red), and DAPI (blue). (F) Total area occupied by B cell follicles was determined using the morphometric tool of the Zeiss Axioplan microscope. Original magnification, ×200. Figures depict 1 experiment representative of 2 or combined data from multiple experiments. Data points represent the mean ± SEM of values. (A–F) Student’s t test between B6 and S100A9KO at each indicated time point. *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001; ****P ≤ 0.0001.
S100A8/A9 plays a key role in neutrophil recruitment and accumulation (16–18), and thus the absence of S100A8/A9 may promote improved Mtb control by altering cellular accumulation to the lung and regulating inflammation. Coinciding with the timing of improved Mtb control in the lungs of S100A9KO mice at 100 dpi, we found that neutrophil accumulation was reduced, while other myeloid subsets, such as alveolar macrophages (AMs), recruited macrophages (RMs), monocytes, and DCs, were not significantly different in lungs of S100A9KO mice when compared with B6 infected mice (Figure 3, C and D). However, at the later stage of chronic disease, AMs, DCs, monocytes, and neutrophils were all reduced in S100A9KO mice when compared with B6 infected lungs (Figure 3, C and D). The localization of myeloperoxidase-expressing (MPO-expressing) neutrophils within TB granulomas during chronic infection was also decreased in S100A9KO mice, suggesting that S100A8/A9 expression promoted the accumulation of neutrophils in the lung, specifically localization within TB granulomas (Figure 3E). Presence and maintenance of B cell follicles within lung granulomas have been associated with effective Mtb control through optimal macrophage activation (19). Consistent with improved protection in the lungs of S100A9KO infected mice at chronic stages, lung B cell follicle area was significantly enhanced within TB granulomas (Figure 3F). However, despite altered accumulation of neutrophils within granulomas in S100A9KO mice, overall inflammatory area or the specific type of inflammation (myeloid or lymphocytic) in the lungs of S100A9KO Mtb-infected mice was not significantly different when compared with that in Mtb-infected B6 mice (Supplemental Figure 3, A and B).
S100A8/A9 binds to RAGE (20), and delivery of RAGE inhibitor (FPS-ZM1) to Mtb-infected mice during the chronic stages of infection significantly decreased Mtb burden so that it was similar to levels observed in S100A9KO mice (Figure 4A). Although inflammation was not significantly decreased in RAGE-treated mice, B cell follicle size was significantly increased within TB granulomas (Figure 4, B and C). Delivery of the RAGE inhibitor to Mtb-infected S100A9KO mice (4.89 ± 0.095 log10CFU) did not significantly decrease Mtb burden when compared with that in DMSO-treated S100A9KO Mtb-infected mice (5.28 ± 0.44 log10CFU), suggesting that the off-target effects of use of RAGE inhibitor are minimal (P = 0.1213). These results together suggest that absence of S100A8/A9 expression or inhibition of its receptor function both improve Mtb control during chronic TB. To further experimentally test the role of S100A8/A9 in TB reactivation and disease progression, we used the Cornell mouse model of TB reactivation in which mice were infected with Mtb and treated with antibiotics, following which reactivation of Mtb was assessed. Our data show that, while B6 mice that received antibiotic chemotherapy reactivated with significantly elevated Mtb CFU in the lung, S100A9KO mice exhibited significantly lower rates of reactivation and harbored overall lower lung Mtb CFU when compared with B6 mice, with 8 of 14 mice exhibiting no detectable lung Mtb CFU (Figure 4D). In conjunction with lower Mtb CFU, while inflammation was not altered, neutrophil accumulation was significantly decreased in lung granulomas of S100A9KO mice (Figure 4, E and F). These results suggest that, while S100A8/A9 expression coincides with neutrophil accumulation and improved Mtb control, S100A8/A9 expression did not directly regulate inflammation during TB.
Figure 4. Targeting the S100A8/A9 pathway limits susceptibility to chronic TB and TB reactivation.

Mtb-infected B6 and S100A9KO (n = 10) mice were treated with RAGE inhibitor (1 mg/kg RAGE-specific blocker FPS-ZM1 [n = 5] or DMSO in PBS [n = 5]) at 205 dpi for 15 days, and (A) lung bacterial burden at 220 dpi was determined by plating. (B) Pulmonary inflammation was quantified on FFPE lung sections stained with H&E, and (C) area occupied by B cell follicles was quantified histologically. B6 (n = 9) or S100A9KO (n = 13) infected mice were treated with rifabutin (100 mg/l) and INH (200 mg/l) for 6 weeks, and mouse organs were then harvested and homogenized at 140 dpi to determine reactivation of Mtb infection. (D) Lung bacterial burden was determined by plating, (E) pulmonary inflammation was quantified on FFPE lung sections stained with H&E, and (F) quantification of MPO+Ly6G+ neutrophils was carried out in FFPE lung sections. Figures depict 1 experiment representative of 2 or combined data from multiple experiments. Data points represent the mean ± SEM of values. (A) One-way ANOVA with Tukey’s posttest; (B–F) Student’s t test between groups. *P ≤ 0.05; **P ≤ 0.01; ****P ≤ 0.0001.
S100A8/A9 expression regulates CD11b expression on neutrophils during chronic TB.
As neutrophil accumulation was markedly decreased in S100A9KO Mtb-infected mice, we next determined whether the integrin CD11b, a key molecule in leukocyte adhesion and migration (21), was differentially expressed on the surface of myeloid cells in S100A9KO Mtb-infected mice when compared with B6 Mtb-infected mice. We observed that CD11b surface expression was significantly decreased within lung neutrophils in S100A9KO Mtb-infected mice during chronic stages of infection, whereas lung monocytes and RMs had no significant changes in CD11b expression (Figure 5A). Similar decreases in CD11b expression on lung neutrophils were observed when S100A9KO mice were infected with M. bovis BCG by the pulmonary route (Supplemental Figure 4). Gr-1 (Ly6G/Ly6C) surface expression was also assessed, but no significant changes were seen in the aforementioned other lung myeloid cell populations during both acute and chronic stages of infection in S100A9KO mice when compared with B6 Mtb-infected mice (data not shown). As myeloid cells, such as neutrophils and monocytes, are among the primary producers of S100A8/A9 (9, 22), we addressed whether adoptive transfer of CD11b+ cells producing S100A8/A9 could reverse the improved Mtb control seen in S100A9KO Mtb-infected mice. Accordingly, CD11b+ cells transferred into S100A9KO mice resulted in increased lung Mtb burden, suggesting that a CD11b myeloid population was contributing to the increased susceptibility during Mtb infection (Figure 5B). This was not because of transfer of Mtb within CD11b+ transferred cells, as colocalization of Mtb with S100A9 expression was minimal in transferred cells (Supplemental Figure 5). These data were corroborated by decreased lung protein levels of neutrophil-attracting chemokines CXCL-1 and CXCL-2 in S100A9KO mice during chronic infection (Figure 5C). Taken together, these data show that absence of S100A8/A9 during chronic Mtb infection reduced neutrophil localization and decreased expression of CD11b expression on neutrophils; adoptive transfer of CD11b cells reversed the protection observed in S100A9KO Mtb-infected lung.
Figure 5. CD11b+ cells mediate improved protection in S100A9KO Mtb-infected mice.

B6 (n = 5–24) and S100A9KO (n = 5–24) mice were aerosol infected with approximately 100 CFU Mtb HN878. (A) CD11b MFI on neutrophils, monocytes, and RMs was determined using flow cytometry at 50, 100, 150, and 300 dpi. S100A9KO (n = 6) infected mice received CD11b+ purified cells from B6 mice (50 μL containing 1 × 106 cells) intratracheally at 100 dpi. (B) Lung bacterial burden was determined by plating at 120 dpi. (C) Lung homogenates were analyzed by Luminex for CXCL-1 and CXCL-2 at 300 dpi. Figures depict 1 experiment representative of 2 or combined data from multiple experiments. Data points represent the mean ± SEM of values. (A and C) Student’s t test between B6 and S100A9KO per time point; (B) 1-way ANOVA with Tukey’s posttest. *P ≤ 0.05; ***P ≤ 0.001; ****P ≤ 0.0001.
S100A8/A9 regulates CD11b expression on neutrophils during Mtb infection.
Our data point to a critical role for S100A8/A9 in regulating CD11b expression specifically on neutrophils to modulate accumulation into the lung. To mechanistically address the role of S100A8/A9 in regulation of CD11b expression on neutrophils and Mtb control, bone marrow–derived neutrophils were infected with reporter-expressing Mtb and Mtb infection and neutrophil marker expression was assessed. Although B6 neutrophils were more readily infected with Mtb, S100A9KO neutrophils showed significantly decreased infected neutrophils (Figure 6A). Consistent with our data from lung neutrophils, CD11b expression was significantly upregulated on B6 Mtbhi neutrophils, but not as highly induced in S100A9KO Mtbhi neutrophils (Figure 6B). S100A9KO neutrophils expressed lower levels of CD11b even in uninfected controls, as previously shown (15), suggesting that while S100A8/A9 regulates the expression of CD11b at homeostatic levels, this effect is much more profound upon Mtb infection. However, in B6 neutrophils, the fold induction of CD11b upon Mtb infection was 1.96, compared with 2.42 in S100A9KO mice. Similar results were obtained when Gr-1 and CD18 levels were measured on S100A9KO Mtb-infected neutrophils when compared with B6 Mtb-infected neutrophils (Figure 6B). This coincided with increased induction of mRNA for signaling receptors, such as TLR2 and TLR4, and phagocytic receptors, such as C-type lectin receptor and macrophage inducible Ca2+-dependent lectin receptor (MINCLE), which play a role in phagocytosis of Mtb (23, 24), but not formyl peptide receptor 1 (FPR1) or DECTIN-1 (Figure 6C). This increased expression of phagocytic receptors on S100A9KO neutrophils also coincided with increased production of proinflammatory cytokines, such as IL-6 and TNF-α, in Mtb-infected neutrophil supernatants when compared with B6 Mtb-infected neutrophils (Figure 6D). We also assessed whether Mtb-infected S100A9KO neutrophils were deficient in chemotactic ability when compared with Mtb-infected B6 neutrophils and found that the ability of S100A9KO neutrophils to migrate toward supernatants from Mtb-infected epithelial cells was not impaired (Figure 6E). To address whether live Mtb infection was required for CD11b upregulation, we then tested to determine whether stimulation with heat-killed (HK) Mtb or components of Mtb would similarly regulate the S100A8/A9-dependent effects on CD11b regulation. We found that HK Mtb and stimulation with culture filtrate protein (CFP) and cell wall components (CW) of Mtb similarly induced an S100A9-dependent CD11b upregulation on neutrophils, suggesting that live Mtb infection was not necessary (Figure 6F). Since S100A8/A9 is known to induce NF-κB signaling, we also wanted to understand the role of NF-κB signaling in mediating CD11b upregulation. We isolated neutrophils from IKKfl/fl LysMCre mice, which lack canonical IKK and NF-κB signaling in LysM-expressing cells. We found that lack of NF-κB signaling reduced Mtb infection in neutrophils (Figure 6G) and that NF-κB signaling was required for upregulation of CD11b and Gr-1 on Mtb-infected neutrophils (Figure 6G). These results together demonstrate that absence of S100A8/A9 expression on neutrophils substantially affects Mtb uptake and CD11b induction following Mtb infection, which is in part dependent on NF-κB signaling.
Figure 6. S100A8/A9 regulates CD11b expression on neutrophils.

Bone marrow neutrophils were isolated and infected with mCherry-labeled HN878 (MOI of 1) for 3 hours. (A) Mtb uptake by neutrophils was determined in B6 (n = 5) and S100A9KO (n = 5) neutrophils using flow cytometry. (B) MFI of CD11b, Gr-1, and CD18 expression on uninfected (UI) and highly infected (Mtbhi) neutrophils was determined by flow cytometry. Bone marrow neutrophils were isolated and infected with HN878 (MOI of 1) for 3 hours. (C) Expression of phagocytic markers quantitated via RT-PCR and (D) TNF-α and IL-6 levels as quantitated by Luminex assays. Bone marrow neutrophils were isolated from B6 (n = 5) and S100A9KO (n = 5) mice and infected with HN878 (MOI of 1) for 1 hour. (E) Neutrophil chemotactic activity was assayed against gravity controls (Grav.) (circles) or supernatants from infected epithelial cells (Supe.) (squares). Neutrophils from B6 mice were left untreated or treated with HK Mtb, Mtb CFP, and Mtb CW. (F) CD11b and Gr-1 MFI were assessed using flow cytometry. Bone marrow neutrophils from IKKfl/fl LysMCre mice (n = 5) and IKKfl/fl (n = 5) mice were infected with Mtb (MOI 1). (G) Mtb uptake and MFI of CD11b and Gr-1 expression on uninfected or infected neutrophils was determined by flow cytometry. Figures depict 1 experiment representative of 2 or combined data from multiple experiments. Data points represent the mean ± SEM of values. (A–G) Student’s t test. *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001; ****P ≤ 0.0001.
S100A8/A9 along with chemokines can distinguish between ATB and LTBI.
Our data demonstrate that S100A8/A9 mRNA levels increased during progression to TB in humans and rapidly decreased upon successful TB treatment. Thus, we wanted to next address whether serum S100A8/A9 levels can be used to distinguish ATB patients from healthy LTBI and uninfected HCs. S100A8/A9 serum protein levels were significantly higher in ATB patients compared with HCs (Table 1 and Figure 7A). We also found increased S100A8/A9 serum levels in LTBI tuberculin skin test+ (TST+) and quantiferon+ (QFT+) household contacts of ATB patients and LTBI TST+QFT+ individuals with occupational exposure to TB when compared with uninfected HCs (Figure 7A). These results suggest that S100A8/A9 levels could serve as an easily measurable surrogate of TB disease progression in LTBI individuals. Additionally, we assessed whether increased S100A8/A9 serum protein was only increased during ATB or was also increased in other acute and chronic inflammatory pulmonary diseases. Although S100A8/A9 serum protein levels were not higher in patients with chronic obstructive pulmonary disease (COPD), S100A8/A9 serum levels were much higher in influenza-infected patients when compared with levels in ATB patients (Figure 7A and Table 1). These results suggest that S100A8/A9 serum measurements may distinguish between ATB and LTBI, while taking into consideration clinical symptoms that may confound the measurements.
Table 1. Descriptive statistics (median and IQR) of biomarkers in ATB, latent TB, HCs, and influenza-infected patients.

Figure 7. S100A8/A9 proteins can distinguish between ATB and HCs.

Healthy uninfected controls (TST–QFT–) (n = 26), LTBI household contacts (TST+QFT+LTBI) (n = 36), LTBI occupational-exposed individuals who are TST+QFT+ (n = 19), LTBI occupational-risk individuals who are TST+QFT– (n = 9), ATB (n = 52), patients with COPD (n = 16), and influenza (Flu) (n = 18) patient sera were collected and (A) S100A8/A9 levels were determined by ELISA. Groups were compared with 1-way ANOVA with Dunnett’s posttest. (B) ROC curves of single (top) and top 3 biomarker combinations (bottom) for ATB versus HC (left) and ATB versus LTBI (right) shown. *P ≤ 0.05; ****P ≤ 0.0001.
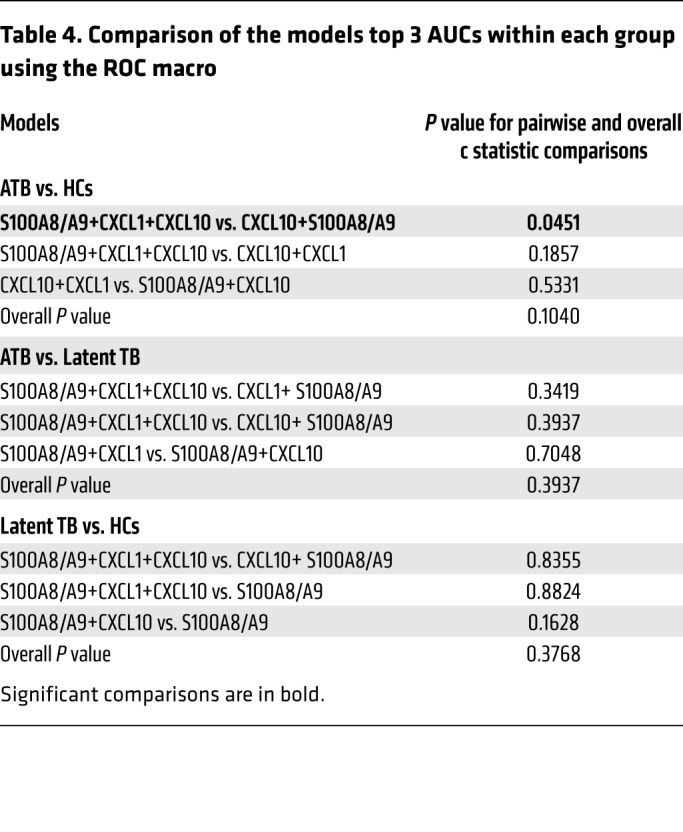
As CXCL-1 and CXCL-10 serum levels were additionally increased in ATB patients (Table 1), we next sought to understand whether combining S100A8/A9 with CXCL-1 and CXCL-10 serum protein levels will improve biomarker performance for distinguishing ATB patients from LTBI and HC than S100A8/A9 alone (7). Median serum biomarker concentrations, with IQRs, across the 3 groups of participants are included in Table 2. The medians were the lowest in the uninfected HCs for all biomarkers, followed by levels in LTBI, and the highest expression was noted in the ATB group. When influenza and ATB groups were compared, expression of both CXCL-1 and S100A8/A9 was higher in influenza-infected patients when compared with levels in ATB patients (Table 2). Overall and pairwise comparisons are listed in Table 3, where the differences in the distribution of the biomarkers across all levels were significant when comparisons were made between ATB and LTBI as well as ATB and HC. For a threshold of 114 pg/mL, CXCL-1 could differentiate between ATB and HC groups with a sensitivity of 88.4% (95% CI, 0.78–0.96) and a specificity of 61.5% (95% CI, 0.42–0.80), while for CXCL-10, a threshold of 302 pg/mL would differentiate between ATB and HC with a sensitivity of 86.5% (95% CI, 0.76–0.94) and a specificity of 57.7% (95% CI, 0.38–0.76). For S100A8/A9, a threshold of 1805 pg/mL would differentiate between ATB and HC with a sensitivity of 92.3% (95% CI, 0.84–0.98) and a specificity of 76.9% (95% CI, 0.57–0.92). Notably, S100A8/A9 levels were the only significant determination when LTBI and HCs were compared pairwise and not when CXCL-1 and CXCL-10 were determined for pairwise comparisons between the LTBI versus HC (Table 2). Furthermore, receiver operating characteristic (ROC) analysis was applied to appraise the diagnostic values of the 3 biomarkers individually and their combination (Tables 3 and 4). Combining S100A8/A9 along with CXCL-1 and CXCL-10 into a biomarker signature improved differentiation between ATB and HCs (0.9467, 95% CI, 0.88–1.0) when compared with using CXCL-10 and S100A8/A9 as combined biomarker signatures (0.9268, 95% CI, 0.85–0.99; Tables 3 and 4 and Figure 7B).
Table 2. Differences in the distribution across the 3 individual biomarkers.

Table 3. AUC of model biomarker combinations.
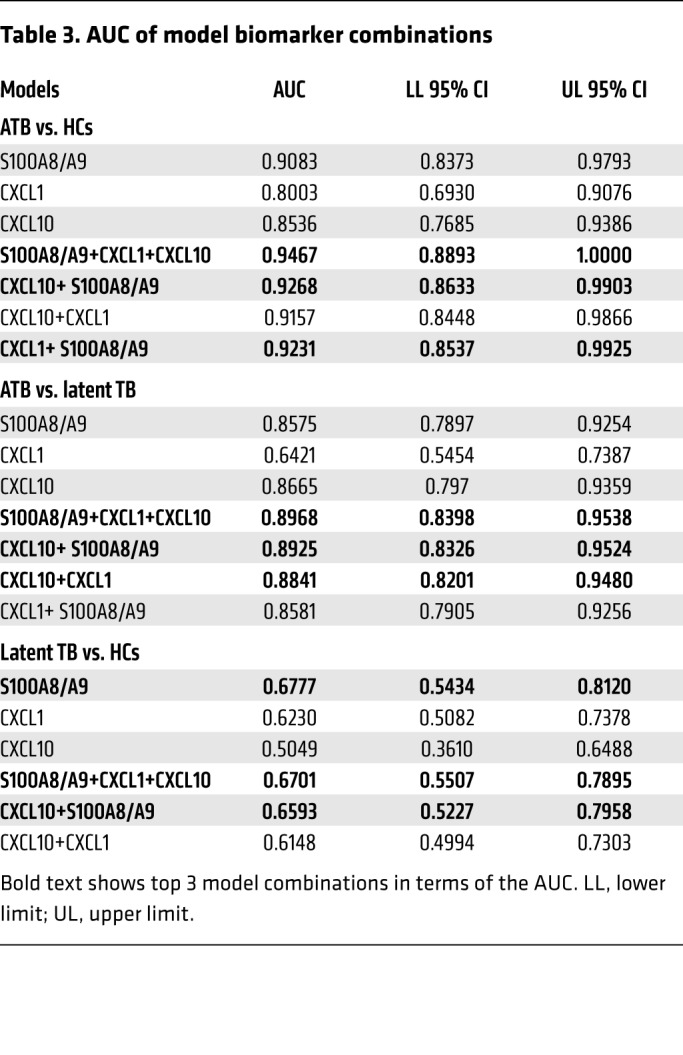
Table 4. Comparison of the models top 3 AUCs within each group using the ROC macro.
Discussion
Neutrophils have recently been implicated as drivers of immunopathogenesis of TB in human and animal models (2, 7, 25). However, the molecular mechanisms by which neutrophils regulate TB immunopathogenesis are not clearly understood. In this study, we demonstrate that neutrophils accumulate during progression to TB disease in Mtb-infected humans and mice and play a detrimental role during chronic TB disease in mice. Additionally, S100A8/A9 levels mirror neutrophil accumulation in the lungs of mice, and S100A9 deficiency in mice results in improved Mtb control during chronic disease. We show that the mechanism by which S100A8/A9 contributes to increased TB disease pathogenesis operated by regulating the expression of the integrin CD11b, which is required for neutrophil accumulation in the lung (15, 17). Furthermore, S100A9 deficiency also reduces TB reactivation in mice, and use of RAGE inhibition results in improved Mtb control. Finally, our human studies suggest that S100A8/A9 levels along with expression of chemokines such as CXCL-1 and CXCL-10 may serve to distinguish between LTBI and ATB from HCs. Together, our results demonstrate that S100A8/A9 play pivotal functions in regulating neutrophil accumulation during TB, primarily through their effects on CD11b expression, thus providing insights into the immunopathogenesis of TB.
Although neutrophil accumulation has been associated with increased disease in mouse and nonhuman primate (NHP) models, as well as human TB, it is still unclear whether neutrophils have protective or pathological functions in Mtb infection (2, 26–29). Despite neutrophils being one of the first cell types infected with Mtb (5), our results show that neutrophil depletion during acute Mtb infection in the B6 resistant strain has no significant impact on Mtb control or TB pathology. Indeed, in highly susceptible mouse strains, such as I/St inbred mice, neutrophil depletion during acute stages may similarly improve Mtb control and pathology (30). Our data show that depletion of neutrophils during chronic Mtb infection improves Mtb control and clearly establish a detrimental role for neutrophils during the chronic stages of Mtb infection. Additionally, that the levels of lung S100A8/A9 correlate with neutrophil accumulation during chronic TB in mice suggests a threshold of neutrophil accumulation may likely be required for the detrimental effects of neutrophils and S100A8/A9 proteins during TB. Additionally, during very late stages of infection, it is possible that the threshold is no longer needed, as other compensatory effects may mediate immunopathogenesis. During chronic inflammation, S100A8/A9 proteins are important in the generation and recruitment of myeloid-derived suppressor cells (MDSCs) (31). Accumulation of mycobacterial permissive and immunosuppressive polymorphonuclear and mononuclear MDSCs has also been reported in chronic Mtb infection in both mice and humans (32, 33). These immunoregulatory cells localize to the perinecrotic regions of chronic granulomas in the lungs of murine and NHP TB models, signifying host failure of Mtb replication control (33–35). Although the presence of CD11bloGr-1hiLy6GhiLy6Clo polymorphonuclear MDSCs (PMN-MDSCs) was not specifically investigated here, it is possible that the MDSCs are included in the CD11b+Gr-1+ gate used in the current study. Interestingly, while both neutrophil depletion and S100A8/A9 deficiency improved Mtb control, overall inflammatory lung disease was not affected. In the case of neutrophil deficiency, it is possible that transient depletion of neutrophils was not sufficient to affect overall inflammation and that prolonged neutrophil depletion may be required to affect lung pathology. During S100A8/A9 deficiency, while overall inflammatory disease was trending toward decreased inflammation, these data were not significant. However, the reduced accumulation of neutrophils into TB granulomas coincided with increased formation of B cell–containing granulomas, which our studies in mice and NHPs have previously shown to be associated with improved Mtb control through enhanced macrophage activation (19, 36, 37). Furthermore, increased expression of S100A8 and S100A9 mRNA in LTBI who progress to pulmonary TB is likely due to inflammatory events associated with progression of disease. These data along with the overall reduced TB reactivation seen in S100A9KO in the Cornell mouse model implicate S100A8/A9 expression in mediating TB disease progression. Together, our results demonstrate that neutrophils and S100A8/A9 may play key roles in modulating TB immunopathogenesis by driving neutrophil accumulation.
A mechanism by which S100A8/A9 regulates TB pathogenesis is likely direct regulation of CD11b expression on neutrophils. Although S100A9 deficiency did not affect CD11b expression on neutrophils during the early stages of Mtb infection, deficient mice expressed lower CD11b levels on neutrophils during chronic TB and BCG infection. In contrast, in S100A9KO bone marrow neutrophils, CD11b expression was markedly lower in uninfected controls, and upon Mtb infection, upregulated CD11b to a lesser extent than B6 bone marrow neutrophils. Considering the baseline differences in CD11b expression in bone marrow neutrophils, it is intriguing that differences in CD11b expression did not occur in S100A9KO mice until day 100 following Mtb infection. The nature of the microenvironment (e.g., presence of other proinflammatory chemokines, prolonged Mtb infection, and stimulation, etc.) may contribute to differences observed in in vitro studies and ex vivo isolated neutrophils. Additionally, it is of considerable interest that, while S100A9KO mice were protected from infection with a clinical Mtb isolate, HN878, S100A9KO mice did not show any protection when infected with a lab-adapted Mtb strain, H37Rv. Thus, our data suggest that S100A8/A9 may play homeostatic roles in regulating CD11b expression in bone marrow neutrophils, and following infection and likely resultant induction of inflammatory signals, these effects may be amplified. Our data showing that S100A9KO neutrophils upon Mtb infection induce higher levels of phagocytic receptor expression, including TLR2, TLR4, and MINCLE, and coincident increased expression of IL-6 and TNF-α suggest that one mechanism by which S100A9KO mice exhibit better Mtb control in vivo during chronic TB could be increased Mtb uptake and role for neutrophils in Mtb clearance. Neutrophil chemotaxis has been associated with cytoskeletal reorganization and actin polymerization and regulation. S100A9-deficient neutrophils had actin defects when responding to IL-8 stimulation, suggesting that S100A9 has a role in cytoskeletal dynamics and reorganization (15). Despite our studies showing that S100A9KO neutrophils do not exhibit any defects in chemotactic migration toward Mtb-infected epithelial cell supernatants, it is possible that functional actin coordinates cell surface integrin regulation (e.g., CD11b/CD18) and could affect neutrophil migration and adhesion in vivo; this will be tested in future studies. Together, these data suggest that S100A8/A9 expression in the lung may induce chemokines that mediate neutrophil accumulation, potentially directly acting as a chemoattractant for neutrophils (17), but also by upregulating the expression of CD11b to amplify neutrophil migration into the lung. In addition, upon recruitment to the lung, Mtb infection–induced S100A8/A9 proteins may regulate phagocytic receptor expression on neutrophils, enabling increased Mtb infection of neutrophils. Together, these S100A8/A9-dependent mechanisms increase Mtb susceptibility and likely mediate immunopathology.
S100A8/A9 is known to interact with TLR4 and RAGE, while CD11b expression can be driven by interactions with TLR4 on neutrophils (38, 39). Thus, S100A8/A9 may engage TLR4 and RAGE to upregulate CD11b expression on neutrophils. Adoptive transfer of CD11b+ cells increased susceptibility in S100A9KO mice, suggesting that CD11b+ immune populations, namely neutrophils or monocytes expressing S100A8/A9, are the likely cellular population mediating increased susceptibility during chronic TB. The utilization of RAGE inhibitor FPS-ZM1 is protective in animal models of emphysema and Alzheimer’s disease (11, 40). We show that, during chronic TB, transient use of RAGE inhibitors is protective and is sufficient to decrease lung Mtb burden, but not lung inflammation. Additionally, our data showing that RAGE inhibitors do not further improve Mtb control in S100A9KO chronically Mtb-infected mice suggest that the effect is S100A8/A9 dependent rather than through the interaction of RAGE, with its other ligands, such as advanced glycation end products (AGE) and high-mobility group protein (B) 1 (HMGB1). Whether use of RAGE inhibitor limits neutrophil accumulation and thus reduces the Mtb niche is not fully explored and should be the focus of future studies. It is exciting that our data showing that S100A8/A9 deficiency delays TB reactivation suggest that RAGE inhibitors may be potentially used as host-directed therapeutics in combination with current antibiotic regimens to improve Mtb control and should be further studied.
The rapid diagnostic test for detection of TB recommended by the WHO is an automated PCR assay that detects mycobacteria in sputum expectorated by patients. However, access remains restricted in low-resource settings. Sputum-based microscopy to identify Mtb still remains the most commonly used diagnostic for TB, but this method is primarily capable of identifying ATB patients. Therefore, new nonsputum-based screening tools for identifying individuals with ATB are required so that they can be prioritized for clinical investigation and treatment. Blood biomarkers that can differentiate progression of LTB to subclinical disease and ATB, failure of TB treatment, and TB relapse would provide additional tools to improve TB diagnosis and combat the global TB epidemic. Several studies have explored the use of host transcriptional biosignatures as diagnostic biomarkers for progression of TB and for predicting risk of human TB disease (2, 41). Although these studies propose the use of transcriptional gene signatures as screening or triage tests for TB, the costs and technology associated with reverse transcription PCR (RT-PCR) or RNA-Seq may still limit use of these biomarkers as diagnostic in a low-resource setting. Our published studies have shown that ATB patients in India, Mexico, and South Africa all reliably exhibit increased expression of serum S100A8/A9 proteins when compared with HC (7) and now with QFT+LTB individuals. Serum is more amenable to developing a simple screening test that may be easier to translate into a point-of-care format (e.g., the Quantum Blue assay, which works for serum and feces). Additionally, our new studies also show that serum protein levels of S100A8/A9 along with chemokines CXCL-1 and CXCL-10 are reliably able to differentiate between ATB and HC and, to a lesser extent, can still discriminate between ATB and QFT+ LTB individuals. Furthermore, that S100A8/A9 is not significantly increased during other chronic pulmonary diseases, such as COPD, suggests that S100A8/A9 may be useful as a biomarker for a triage test to rule out individuals who do not have TB so that biomarker-positive individuals then get followed up for further TB diagnosis. One limitation of the use of S100A8/A9 and chemokines is that acute respiratory infections such as influenza also result in high expression of S100A8/A9 and CXCL-1. Detailing the expression of these biomarkers in other acute bacterial infections will be useful criteria for future studies. Interestingly, S100A8/A9 could also show initial potential as an early indicator for successful TB treatment, as S100A8/A9 levels decreased significantly (P ≤ 0.01) 2 weeks after treatment initiation in cured TB patients and did not decrease in TB patients who failed treatment. Thus, further validation of the use of S100A8/A9, CXCL-1, and CXCL-10 in different geographical cohorts and in treatment failure and relapse studies will be useful and timely.
In summary, our studies have mechanistically described a pivotal role for S100A8/A9 proteins in mediating TB pathogenesis through regulation of CD11b and neutrophil recruitment. Additionally, our experimental studies targeting the S100A8/A9 signaling pathway project a pathway for host-directed therapeutics for TB. A more mechanistic understanding of the role of S100A8/A9 proteins further validates the development of S100A8/A9 and related biomarkers as diagnostics for TB.
Methods
Mice and NHP Mtb infection.
C57BL/6J (B6) mice were purchased from the Jackson Laboratory. S100A9KO was obtained in-house (15), and mice were bred at the Washington University in St. Louis animal facility. Both sexes were used, and mice were between the ages of 6 and 8 weeks. Mtb strains were cultured, and mice were aerosol infected with approximately 100 CFUs, as described previously (19). At specific time points after infection, lungs were harvested and homogenized, and serial dilutions of tissue homogenates were plated on 7H11 agar plates to determine Mtb CFU. For reactivation experiments, mice were treated with rifabutin (MilliporeSigma) (100 mg/L) and isoniazid (INH) (MilliporeSigma) (200 mg/L) for 6 weeks and mouse tissue was then harvested at 140 dpi. For rhesus macaques infected with Mtb, we used clinical data stored in the Animal Records System at the Tulane National Primate Research Center. Macaques were chosen if they were experimentally infected with Mtb (either CDC1551, Erdman, or H37Rv strains) during 2007 to 2015 and exhibited either ATB or were latently infected. After infection, data were obtained weekly from animals until euthanasia or necropsy. Neutrophil percentages in the blood were obtained from complete blood counts performed at the same time as serum chemistry.
Adoptive transfer of CD11b+ cells.
Lung cell suspensions were prepared from the lungs of B6 mice 100 dpi following HN878 infection. CD11b+ cells were enriched from the lung suspension using magnetic selection with CD11b microbeads (Miltenyi Biotec) per the manufacturer’s instructions, and 50 μL (1 × 106 cells) of this suspension (72.4% purity) was administered intratracheally to HN878-infected mice at 100 dpi. These mice were harvested on 120 dpi as indicated.
RAGE-inhibitor treatment.
RAGE signaling was inhibited starting 205 or 245 dpi as previously described (11) through daily i.p. injection of 1 mg/kg RAGE-specific blocker FPS-ZM1 (Tocris) or 0.1% (v/v) DMSO in PBS (control). Mice were euthanized 15 days after treatment.
Neutrophil depletion.
Neutrophils were depleted as described (42) using 300 μg anti-mouse Ly6G (Bio X Cell) or isotype IgG (MilliporeSigma). Briefly, in acute infections (less than 21 dpi), mice were given 300 μg of anti-mouse Ly6G i.p. every other day between 10 and 20 dpi. In chronic infections, mice were given 300 μg of anti-mouse Ly6G every other day between 95 to 105 dpi via i.p. injections.
Lung cell preparation and flow cytometry.
Lung cell suspensions were prepared, stained, collected, and analyzed for flow cytometry as described before (19). Fluorochrome-labeled antibodies specific for CD11b (M1/70, BD), CD11c (HL3, BD), and Gr-1 (RB6-8C5, eBioscience) were used in this study. Cells were collected using a FACSJazz with FACS Sortware software (BD). Cell populations were gated based on their forward by side scatter characteristics, and the frequency of specific cell types was analyzed using FlowJo, version 7.6.5 (Tree Star Inc.). Lung AMs were gated as CD11c+CD11b–, lung myeloid DCs were gated on CD11c+ CD11b+, neutrophils were gated as CD11b+ Gr-1hi, RMs were annotated as CD11b+ Gr-1lo, and monocytes were gated on CD11b+ Gr1int cells as in Dunlap et al. (43).
In vitro neutrophil infections.
Neutrophils were isolated from the bone marrow using the mouse neutrophil isolation kit (Miltenyi Biotec). Neutrophils were infected at an MOI of 1 for 3 hours. Neutrophil uptake was derived from neutrophil infection with Mtb HN878-mCherry. CD11b MFI was also assessed in neutrophils treated with HK Mtb HN878 (1 × 106 CFU) or Mtb HN878 CFP and CW (10 μg/mL). Fluorochrome-labeled antibodies specific for CD11b (M1/70, BD), CD18 (M18/2, BioLegend), and Gr-1 (RB6-8C5, eBioscience) were used in this study.
In vitro neutrophil chemotaxis assay.
Neutrophils were isolated from the bone marrow of B6 or S100A9KO using the Mouse Neutrophil Isolation Kit (Miltenyi Biotec). Neutrophils were infected at an MOI of 1 for 1 hour and then transferred to the upper compartments of a Transwell plate separated by a 5-μm polycarbonate membrane (Corning). Supernatants from Mtb-infected epithelial cells or HBSS supplemented with 1% FCS (gravity control) was added to the lower chambers and the cells incubated for 1 hour at 37°C. Chemotaxis was assessed by flow cytometry by determining the number of neutrophils found in the lower chamber after incubation. Fluorochrome-labeled antibodies specific for CD11b (M1/70, BD) and Gr-1 (RB6-8C5, eBioscience) were used in this study.
RNA extraction and quantitative RT-PCR.
Total RNA was isolated from neutrophils using an RNeasy Mini Kit (QIAGEN). cDNA was synthesized using ABI reverse transcription reagents (Thermo Fisher) on a Bio-Rad DNA Engine Thermal Cycler. Gene expression was assessed using primers from IDT and ABI and run on a Viia7 Real-Time PCR System (Life Technologies, Thermo Fisher). Expression of genes of interest (FPR1, TLR2, TLR4, DECTIN [Clec7a], and MINCLE [Clec4e]) was normalized to GAPDH expression, and log10 fold induction over the control group was assessed using the ΔΔCt calculation.
Lung histology.
Lungs from Mtb-infected mice were perfused with 10% neutral buffered formalin and were paraffin embedded. Lung sections were stained with H&E and processed for light microscopy. Images were obtained using a Zeiss Axioplan 2 microscope and were recorded with a Zeiss AxioCam digital camera. Sections were probed with rabbit anti-MPO (PB9057, BosterBio; dilution 1:100) and biotinylated rat anti-mouse Ly6G (clone IA8, BioLegend; dilution 1:100) to detect neutrophils or with anti-B220 (clone RA3-6B2, BD Biosciences — Pharmingen; dilution: 1/100) to detect B cells. Primary antibodies were detected with Cy3 donkey anti-rabbit IgG (711-166-152, Jackson ImmunoResearch Laboratories; dilution 1:200) and Alexa Fluor 488 streptavidin (A21208, Thermo Fisher Scientific; dilution 1:200). B cell follicles were assessed through the automated tool of the Zeiss Axioplan 2 microscope, and total area and average size were calculated in squared microns. Myeloid and lymphocyte areas were acquired with stained slide images using a Hamamatsu Nanozoomer 2.0 HT system with NDP scan image acquisition software and were quantified using Visiomorph image-processing software (Visiopharm).
In order to calculate the percentage of inflammation per lobe, all clusters of inflammatory cells in an individual lobe were systematically outlined with the automated tool of the Zeiss Axioplan microscope. The total area covered by inflammation was calculated by adding all the individual areas occupied by clusters of inflammatory cells located in the interstitial, peribronchial, and perivascular regions. Next, a picture of the whole lobe was obtained with the stich tool of the Zeiss Axioplan microscope. Lung parenchyma, excluding empty spaces (alveoli, bronchi, and blood vessel lumens), was outlined with the automated tool of the Zeiss Axioplan microscope to determine the area of the lobe. Finally, the percentage of the area covered by inflammation per lobe was calculated by dividing the total area covered by inflammatory cells by the total area occupied by lung tissue, multiplied by 100.
RNA-Seq data from ACS progressors and controls.
We compared gene-level mRNA expression levels of S100A8 and S100A9 in Mtb-infected adolescents enrolled into the ACS who remained healthy during 2 years of study follow-up (controls) (n = 106) or who progressed to microbiologically confirmed ATB (progressors) (n = 44), as described (14, 41). Adolescents who were Mtb+ at enrollment or developed ATB disease more than 6 months after Mtb infection was first detected were included in our analyses. Each progressor had 2 healthy matched controls based on age, sex, ethnicity, school of attendance, and presence or absence of prior episodes of TB disease. Participants were excluded if they were ATB+ within 6 months of enrollment or QFT and/or TST conversion or if they were HIV infected. Participants with diagnosed or suspected TB disease were referred to a study-independent public health physician for treatment according to national TB control programs of the country involved. RNA was extracted from PAX gene tubes and RNA-Seq performed as described (14, 41). Prospective RNA-Seq data of progressors were realigned to the time point at which ATB was diagnosed. Differences in gene-level mRNA expression between each progressor sample and the average of demographically matched control samples were computed using the published ACS metadata (14, 41). Time-to-diagnosis values were assigned to each sample according to the original definitions. The log2 fold-change values between progressor and control biomarkers were modeled as a nonlinear function of time to diagnosis for the entire population using the smooth-spline function in R with 3 degrees of freedom. Ninety-nine percent CIs for the temporal trends were computed by performing 2000 iterations of spline fitting after bootstrap resampling from the full data set.
ATB cohort.
We recruited patients (n = 52) with confirmed ATB. The diagnosis of ATB was made by the conventional microscopic detection of acid-alcohol–resistant bacteria as well as culture of Mtb in Lowenstein-Jensen medium in serial, nonconcentrated sputum samples as well as radiological studies, physical examination, and clinical history. All patients with ATB were residents of the urban and metropolitan areas of Mexico City, and some patients were residents from different states of the south and central area of Mexico, including Veracruz, Oaxaca, Puebla, and Chiapas. Patients with ATB, excluding patients with HIV and cancer, were recruited at the Instituto Nacional de Enfermedades Respiratorias Ismael Cosio Villegas, Tuberculosis clinic in Mexico City, Mexico, in a period between 2015 and 2017.
At the same time, household contacts of ATB patients who are TST+QFT+ LTBI (n = 36) were recruited in the first 2 months of diagnosis. The criteria for inclusion of contacts of ATB patients were as follows: (a) subjects of legal age (18 to 54) willing to sign the letter of consent, (b) subjects in close contact with the index case of ATB, and (c) subjects willing to provide the blood samples needed to participate in the study. In addition, a group of individuals (n = 28) who were health personnel (resident physicians of the pulmonology specialty) and administrative medical staff of the INER, considered an occupational risk population, were included; these encompassed both TST+QFT+ (n = 19) and TST+QFT– (n = 9) individuals. Also, a group of uninfected healthy individuals (TST–QFT–) were also recruited (n = 26). After the consent signature, 20 mL of blood anticoagulated with EDTA and 10 mL of blood without anticoagulant was used to isolate serum for the purpose of performing the experimental analysis.
TB treatment cohort.
Study participants, 34 cured and 10 relapse TB patients and 10 patients who failed TB treatment, were enrolled and treated from TB clinics surrounding Tygerberg Hospital in Cape Town, South Africa, as a substudy to the Pulmonary TB Cohort Study (April 2010 and April 2013) and the Action TB Study (May 1999 and July 2002) (44, 45). Serum samples were used for the quantification of circulating S100A8/A9 concentrations. Pulmonary TB patients were all untreated at the time of enrollment, with a first episode of TB. All TB patients received directly observed treatment, which consisted of an intensive phase (2 months) of rifampicin (RIF), INH, pyrazinamide, and ethambutol, followed by a continuation phase (4 months) of RIF and INH. Patients were between 20 and 65 years of age, and clinical information on age, sex, weight, and height (BMI) was recorded. Mycobacterial culture was performed using the automated BACTEC 12B liquid radiometric method. Individuals who had 2 consecutive negative culture results at the end of treatment were regarded as successfully cured. Bacteriological relapse following anti-TB treatment was determined within 24 months after treatment completion. Standardized restriction fragment length polymorphism (RFLP) banding patterns generated by Southern hybridization with the insertion sequence (IS) 6110 probe17 were determined in patients with recurrent TB. Episodes were classified as relapse when the strain pattern was the same. TB treatment failure was defined by a positive sputum culture at month 6 after initiation of standard treatment of drug-sensitive TB. All individuals adhered to treatment (>80% of drugs taken) and were infected with drug-sensitive Mtb strains. No distinct differences in strain types were observed between failed and cured patients. Patients were excluded if they previously had TB; had multidrug-resistant TB; were HIV positive; presented with diabetes, malignancy, lung cancer, chronic bronchitis, or sarcoidosis; were on steroid treatment; or were pregnant. Chest x-rays (CXRs) were only obtained at baseline and were read independently in a blinded fashion by a pulmonologist or clinician.
G-CSF, IL-6, TNF-α, CXCL-2, S100A8/A9, CXCL-1, and CXCL-10 protein quantification.
Levels of G-CSF, IL-6, and TNF-α, CXCL-1, and CXCL-2 in mouse lung homogenates were measured using a mouse Luminex assay (Linco/Millipore). Circulating levels of S100A8/A9 were measured in serum samples by ELISA using the DuoSet ELISA Development Kit for human S100A8/S100A9 heterodimer (catalog DY8226, R&D Systems Inc.), according to the manufacturer’s instructions. Serum samples were diluted 1:4000 in reagent diluent (1% BSA in PBS). Absorbance was read at 450 nm and 540 nm (for wavelength correction) using the Synergy HT Microplate ELISA reader (BioTek Inc.). Serum levels of CXCL-1 were measured by the CXCL-1 Quantikine ELISA Kit (catalog DGR00B, R&D Systems Inc.) according to the manufacturer’s instructions. For this assay, samples were diluted 1:2. Absorbance was read at 450 nm and 540 nm using the Synergy HT microplate ELISA reader (BioTek Inc.). Serum levels of CXCL-10 were assessed by Luminex using the Bio-Plex Pro CXCL10 Set (Bio-Rad) according to the manufacturer’s instructions. The samples were read in a Bio-Plex 200 instrument. The results were analyzed using Bio-Plex software, version 4.1 (Bio-Rad).
Statistics.
Differences between the means of 2 groups were analyzed using the 2-tailed Student’s t test in GraphPad Prism 5 or GraphPad Prism 8. Multiple groups were analyzed using 1-way ANOVA with Tukey’s or Dunnett’s posttest analysis or mixed effects analysis of 1-way ANOVA with Dunnett’s posttest, as indicated. P ≤ 0.05 was considered significant.
The distribution of the biomarkers in human samples was examined and expressed as a median and IQR. The differences in the distributions across the groups were evaluated using the Kruskal-Wallis nonparametric test where the Dwass, Steel, Critchlow-Fligner method was used to check for pairwise 2-sided multiple comparison analysis (46–48). The diagnostic efficiency of the individual biomarkers and their combinations was assessed by ROC analysis where the AUC and associated 95% CIs were determined. A nonparametric approach was used to compare the correlated ROC curves without adjustments for pairwise comparisons (49). Logistic regression analysis was performed to determine the predictive probability of these biomarkers on each of the outcome combinations. The level of significance was set to 0.05, and all analyses were 2 sided. Statistical analyses and figures were created with SAS 9.4 (SAS Institute Inc.).
Study approval.
For human studies, written informed consent from participants and parents or legal guardians where applicable was received under protocols approved by the Ethics Committee of the Instituto Nacional de Enfermedades Respiratorias Ismael Cosio Villegas, the Human Research Ethics Committee of the Faculty of Health Sciences, University of Cape Town, and the City of Cape Town City Health. The study was reviewed and approved by the Institutional Research Committee (protocol number B04-15) of Instituto Nacional de Enfermedades Respiratorias Ismael Cosio Villegas. Protocols involving the use of animals were approved by the IACUC at Washington University in St. Louis and Tulane National Primate Research Center. All the experiments were performed in accordance with the protocols.
Author contributions
SAK conceptualized the study. NRS, RVS, RDG, JRM, SD, MA, and PT performed mouse experiments. ANB and SM performed NHP experiments. NRS, NA, BAK, ACL, LJA, MMT, KBL, GW, and NDP were responsible for human analysis. NRS, NA, RDG, and SAK wrote the original draft of the manuscript. NRS, RVS, NA, JRM, SD, TV, NDP, DK, TJS, JZ, and SAK reviewed and edited the manuscript. DK, TJS, JZ, and SAK acquired funding. SAK supervised the study.
Supplementary Material
Acknowledgments
This work was supported by Washington University in St. Louis and NIH grants HL105427 (to SAK), AI123780 (to SAK, TJS, and DK), AI111914 (to SAK and DK), and NIH/NHLBI T32 HL007317-40 (to NRS) as well as NIH Shared Instrumentation Grant S10 RR0227552. This work was supported by grants from the Interdisciplinary Center of Clinical Research of the University of Münster (Vo2/014/09) and the German Research Foundation (DFG) CRC 1009 B8 (to TV). This work was supported by the National Council of Science and Technology of Mexico (Consejo Nacional de Tecnología de México, CONACYT, Grant FONSEC SSA/IMSS/ISSSTE/S0008-2017-1/290512). We thank Shyamala Thirunavukkarasu and Nicole Howard, and Lan Lu and Misty Veschak (Washington University in St. Louis) for technical support.
Version 1. 03/05/2020
In-Press Preview
Version 2. 05/11/2020
Electronic publication
Version 3. 06/01/2020
Print issue publication
Funding Statement
This work was supported by Washington University in St. Louis, NIH grant HL105427 to S.A.K., AI123780 to S.A.K., T.S., and D.K., AI111914 to S.A.K. and D.K., NIH/NHLBI T32 HL007317-40 to NS, and NIH Shared Instrumentation Grant S10 RR0227552. This work was supported by Grants from the Interdisciplinary Center of Clinical Research of the University of Muenster (Vo2/014/09) and the German Research Foundation (DFG) CRC 1009 B8 to T.V. We thank Lan Lu, Shyamala Thirunavukkarasu, Misty Veschak (Washington University in St. Louis) for technical support.
Footnotes
Conflict of interest: TJS is coinventor of a patent of the 16-gene ACS signature (patent reference number 15/775323).
Copyright: © 2020, American Society for Clinical Investigation.
Reference information: J Clin Invest. 2020;130(6):3098–3112.https://doi.org/10.1172/JCI130546.
Contributor Information
Ninecia R. Scott, Email: ninecia.scott@wustl.edu.
Rosemary V. Swanson, Email: rswanson@wustl.edu.
Noor Al-Hammadi, Email: noor@wubios.wustl.edu.
Racquel Domingo-Gonzalez, Email: racqueld@stanford.edu.
Javier Rangel-Moreno, Email: Javier_Rangel-Moreno@URMC.Rochester.edu.
Belinda A. Kriel, Email: belindak@sun.ac.za.
Allison N. Bucsan, Email: abucsan@tulane.edu.
Shibali Das, Email: shibalidas@wustl.edu.
Mushtaq Ahmed, Email: mushtaqahmed@wustl.edu.
Smriti Mehra, Email: smehra@tulane.edu.
Puthayalai Treerat, Email: junetreerat@gmail.com.
Alfredo Cruz-Lagunas, Email: alfredocl@gmail.com.
Luis Jimenez-Alvarez, Email: jimenez.alvarez.l@gmail.com.
Marcela Muñoz-Torrico, Email: dra_munoz@hotmail.com.
Karen Bobadilla-Lozoya, Email: karenbolo@hushmail.com.
Thomas Vogl, Email: vogl@uni-muenster.de.
Gerhard Walzl, Email: gwalzl@sun.ac.za.
Nelita du Plessis, Email: nelita@sun.ac.za.
Deepak Kaushal, Email: dkaushal@txbiomed.org.
References
- 1. WHO. Global Tuberculosis Report. Geneva, Switzerland: WHO; 2018. [Google Scholar]
- 2.Berry MP, et al. An interferon-inducible neutrophil-driven blood transcriptional signature in human tuberculosis. Nature. 2010;466(7309):973–977. doi: 10.1038/nature09247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dorhoi A, et al. The adaptor molecule CARD9 is essential for tuberculosis control. J Exp Med. 2010;207(4):777–792. doi: 10.1084/jem.20090067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Eum SY, et al. Neutrophils are the predominant infected phagocytic cells in the airways of patients with active pulmonary TB. Chest. 2010;137(1):122–128. doi: 10.1378/chest.09-0903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Blomgran R, Ernst JD. Lung neutrophils facilitate activation of naive antigen-specific CD4+ T cells during Mycobacterium tuberculosis infection. J Immunol. 2011;186(12):7110–7119. doi: 10.4049/jimmunol.1100001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ernst JD. The immunological life cycle of tuberculosis. Nat Rev Immunol. 2012;12(8):581–591. doi: 10.1038/nri3259. [DOI] [PubMed] [Google Scholar]
- 7.Gopal R, et al. S100A8/A9 proteins mediate neutrophilic inflammation and lung pathology during tuberculosis. Am J Respir Crit Care Med. 2013;188(9):1137–1146. doi: 10.1164/rccm.201304-0803OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rammes A, Roth J, Goebeler M, Klempt M, Hartmann M, Sorg C. Myeloid-related protein (MRP) 8 and MRP14, calcium-binding proteins of the S100 family, are secreted by activated monocytes via a novel, tubulin-dependent pathway. J Biol Chem. 1997;272(14):9496–9502. doi: 10.1074/jbc.272.14.9496. [DOI] [PubMed] [Google Scholar]
- 9.Voganatsi A, Panyutich A, Miyasaki KT, Murthy RK. Mechanism of extracellular release of human neutrophil calprotectin complex. J Leukoc Biol. 2001;70(1):130–134. [PubMed] [Google Scholar]
- 10.Goyette J, Geczy CL. Inflammation-associated S100 proteins: new mechanisms that regulate function. Amino Acids. 2011;41(4):821–842. doi: 10.1007/s00726-010-0528-0. [DOI] [PubMed] [Google Scholar]
- 11.Lee H, et al. Blockade of RAGE ameliorates elastase-induced emphysema development and progression via RAGE-DAMP signaling. FASEB J. 2017;31(5):2076–2089. doi: 10.1096/fj.201601155R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Daley JM, Thomay AA, Connolly MD, Reichner JS, Albina JE. Use of Ly6G-specific monoclonal antibody to deplete neutrophils in mice. J Leukoc Biol. 2008;83(1):64–70. doi: 10.1189/jlb.0407247. [DOI] [PubMed] [Google Scholar]
- 13.Edgeworth J, Gorman M, Bennett R, Freemont P, Hogg N. Identification of p8,14 as a highly abundant heterodimeric calcium binding protein complex of myeloid cells. J Biol Chem. 1991;266(12):7706–7713. [PubMed] [Google Scholar]
- 14.Scriba TJ, et al. Sequential inflammatory processes define human progression from M. tuberculosis infection to tuberculosis disease. PLoS Pathog. 2017;13(11):e1006687. doi: 10.1371/journal.ppat.1006687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Manitz MP, et al. Loss of S100A9 (MRP14) results in reduced interleukin-8-induced CD11b surface expression, a polarized microfilament system, and diminished responsiveness to chemoattractants in vitro. Mol Cell Biol. 2003;23(3):1034–1043. doi: 10.1128/MCB.23.3.1034-1043.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Raquil MA, Anceriz N, Rouleau P, Tessier PA. Blockade of antimicrobial proteins S100A8 and S100A9 inhibits phagocyte migration to the alveoli in streptococcal pneumonia. J Immunol. 2008;180(5):3366–3374. doi: 10.4049/jimmunol.180.5.3366. [DOI] [PubMed] [Google Scholar]
- 17.Ryckman C, Vandal K, Rouleau P, Talbot M, Tessier PA. Proinflammatory activities of S100: proteins S100A8, S100A9, and S100A8/A9 induce neutrophil chemotaxis and adhesion. J Immunol. 2003;170(6):3233–3242. doi: 10.4049/jimmunol.170.6.3233. [DOI] [PubMed] [Google Scholar]
- 18.Vandal K, Rouleau P, Boivin A, Ryckman C, Talbot M, Tessier PA. Blockade of S100A8 and S100A9 suppresses neutrophil migration in response to lipopolysaccharide. J Immunol. 2003;171(5):2602–2609. doi: 10.4049/jimmunol.171.5.2602. [DOI] [PubMed] [Google Scholar]
- 19.Slight SR, et al. CXCR5+ T helper cells mediate protective immunity against tuberculosis. J Clin Invest. 2013;123(2):712–726. doi: 10.1172/JCI65728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ehlermann P, et al. Increased proinflammatory endothelial response to S100A8/A9 after preactivation through advanced glycation end products. Cardiovasc Diabetol. 2006;5:6. doi: 10.1186/1475-2840-5-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ding ZM, et al. Relative contribution of LFA-1 and Mac-1 to neutrophil adhesion and migration. J Immunol. 1999;163(9):5029–5038. [PubMed] [Google Scholar]
- 22.Pechkovsky DV, Zalutskaya OM, Ivanov GI, Misuno NI. Calprotectin (MRP8/14 protein complex) release during mycobacterial infection in vitro and in vivo. FEMS Immunol Med Microbiol. 2000;29(1):27–33. doi: 10.1111/j.1574-695X.2000.tb01501.x. [DOI] [PubMed] [Google Scholar]
- 23.Lee WB, et al. Neutrophils promote mycobacterial trehalose dimycolate-induced lung inflammation via the mincle pathway. PLoS Pathog. 2012;8(4):e1002614. doi: 10.1371/journal.ppat.1002614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gopalakrishnan A, Dietzold J, Verma S, Bhagavathula M, Salgame P. Toll-like receptor 2 prevents neutrophil-driven immunopathology during infection with Mycobacterium tuberculosis by curtailing CXCL5 production. Infect Immun. 2019;87(3):e00760-18. doi: 10.1128/IAI.00760-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Keller C, Hoffmann R, Lang R, Brandau S, Hermann C, Ehlers S. Genetically determined susceptibility to tuberculosis in mice causally involves accelerated and enhanced recruitment of granulocytes. Infect Immun. 2006;74(7):4295–4309. doi: 10.1128/IAI.00057-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Eruslanov EB, et al. Neutrophil responses to Mycobacterium tuberculosis infection in genetically susceptible and resistant mice. Infect Immun. 2005;73(3):1744–1753. doi: 10.1128/IAI.73.3.1744-1753.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kisich KO, Higgins M, Diamond G, Heifets L. Tumor necrosis factor alpha stimulates killing of Mycobacterium tuberculosis by human neutrophils. Infect Immun. 2002;70(8):4591–4599. doi: 10.1128/IAI.70.8.4591-4599.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lombard R, et al. IL-17RA in non-hematopoietic cells controls CXCL-1 and 5 critical to recruit neutrophils to the lung of mycobacteria-infected mice during the adaptive immune response. PLoS One. 2016;11(2):e0149455. doi: 10.1371/journal.pone.0149455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tan BH, et al. Macrophages acquire neutrophil granules for antimicrobial activity against intracellular pathogens. J Immunol. 2006;177(3):1864–1871. doi: 10.4049/jimmunol.177.3.1864. [DOI] [PubMed] [Google Scholar]
- 30.Yeremeev V, Linge I, Kondratieva T, Apt A. Neutrophils exacerbate tuberculosis infection in genetically susceptible mice. Tuberculosis (Edinb) 2015;95(4):447–451. doi: 10.1016/j.tube.2015.03.007. [DOI] [PubMed] [Google Scholar]
- 31.Sinha P, Okoro C, Foell D, Freeze HH, Ostrand-Rosenberg S, Srikrishna G. Proinflammatory S100 proteins regulate the accumulation of myeloid-derived suppressor cells. J Immunol. 2008;181(7):4666–4675. doi: 10.4049/jimmunol.181.7.4666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.du Plessis N, et al. Increased frequency of myeloid-derived suppressor cells during active tuberculosis and after recent mycobacterium tuberculosis infection suppresses T-cell function. Am J Respir Crit Care Med. 2013;188(6):724–732. doi: 10.1164/rccm.201302-0249OC. [DOI] [PubMed] [Google Scholar]
- 33.Knaul JK, et al. Lung-residing myeloid-derived suppressors display dual functionality in murine pulmonary tuberculosis. Am J Respir Crit Care Med. 2014;190(9):1053–1066. doi: 10.1164/rccm.201405-0828OC. [DOI] [PubMed] [Google Scholar]
- 34.Obregón-Henao A, Henao-Tamayo M, Orme IM, Ordway DJ. Gr1(int)CD11b+ myeloid-derived suppressor cells in Mycobacterium tuberculosis infection. PLoS One. 2013;8(11):e80669. doi: 10.1371/journal.pone.0080669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Domingo-Gonzalez R, et al. Interleukin-17 limits hypoxia-inducible factor 1α and development of hypoxic granulomas during tuberculosis. JCI Insight. 2017;2(19):92973. doi: 10.1172/jci.insight.92973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kaushal D, et al. Mucosal vaccination with attenuated Mycobacterium tuberculosis induces strong central memory responses and protects against tuberculosis. Nat Commun. 2015;6:8533. doi: 10.1038/ncomms9533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Foreman TW, et al. CD4+ T-cell-independent mechanisms suppress reactivation of latent tuberculosis in a macaque model of HIV coinfection. Proc Natl Acad Sci U S A. 2016;113(38):E5636–E5644. doi: 10.1073/pnas.1611987113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pruenster M, et al. Extracellular MRP8/14 is a regulator of β2 integrin-dependent neutrophil slow rolling and adhesion. Nat Commun. 2015;6:6915. doi: 10.1038/ncomms7915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vogl T, et al. Mrp8 and Mrp14 are endogenous activators of Toll-like receptor 4, promoting lethal, endotoxin-induced shock. Nat Med. 2007;13(9):1042–1049. doi: 10.1038/nm1638. [DOI] [PubMed] [Google Scholar]
- 40.Hong Y, Shen C, Yin Q, Sun M, Ma Y, Liu X. Effects of RAGE-specific inhibitor FPS-ZM1 on amyloid-β metabolism and AGEs-induced inflammation and oxidative stress in rat hippocampus. Neurochem Res. 2016;41(5):1192–1199. doi: 10.1007/s11064-015-1814-8. [DOI] [PubMed] [Google Scholar]
- 41.Zak DE, et al. A blood RNA signature for tuberculosis disease risk: a prospective cohort study. Lancet. 2016;387(10035):2312–2322. doi: 10.1016/S0140-6736(15)01316-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kang DD, Lin Y, Moreno JR, Randall TD, Khader SA. Profiling early lung immune responses in the mouse model of tuberculosis. PLoS One. 2011;6(1):e16161. doi: 10.1371/journal.pone.0016161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dunlap MD, et al. A novel role for C-C motif chemokine receptor 2 during infection with hypervirulent Mycobacterium tuberculosis. Mucosal Immunol. 2018;11(6):1727–1742. doi: 10.1038/s41385-018-0071-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hesseling AC, et al. Mycobacterial genotype is associated with disease phenotype in children. Int J Tuberc Lung Dis. 2010;14(10):1252–1258. [PubMed] [Google Scholar]
- 45.Ronacher K, et al. Distinct serum biosignatures are associated with different tuberculosis treatment outcomes. Tuberculosis (Edinb) 2019;118:101859. doi: 10.1016/j.tube.2019.101859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Douglas CE, Michael FA. On distribution-free multiple comparisons in the one-way analysis of variance. Commun Stat Theory Methods. 1991;20(1):127–139. doi: 10.1080/03610929108830487. [DOI] [Google Scholar]
- 47. Olkin I. Contributions to Probability and Statistics; Essays in Honor of Harold Hotelling. Stanford, California, USA: Stanford University Press; 1960. [Google Scholar]
- 48.Steel RGD. A rank sum test for comparing all pairs of treatments. Technometrics. 1960;2(2):197–207. doi: 10.1080/00401706.1960.10489894. [DOI] [Google Scholar]
- 49. Zhou X-H, Obuchowski NA, McClish DK. Statistical Methods in Diagnostic Medicine, 2nd ed. Hoboken, New Jersey, USA: John Wiley & Sons; 2011. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.