

Abstract
Class IIa histone deacetylases (HDACs) repress cardiomyocyte hypertrophy through association with the prohypertrophic transcription factor (TF) myocyte enhancer factor-2 (MEF2). The four class IIa HDACs — HDAC4, -5, -7, and -9 — are subject to signal-dependent phosphorylation by members of the Ca2+/calmodulin-dependent protein kinase (CaMK) group. In response to stress, HDAC4, HDAC5, and HDAC9 undergo phosphorylation-induced nuclear export in cardiomyocytes, freeing MEF2 to stimulate progrowth genes; it was generally assumed that HDAC7 is also antihypertrophic. However, in this issue of the JCI, Hsu and colleagues demonstrate that, in sharp contrast to the other class IIa HDACs, HDAC7 is constitutively localized to the cardiomyocyte cytoplasm, where it promotes cardiac hypertrophy. Phosphorylation of HDAC7 by the CaMK group member salt-inducible kinase 1 (SIK1) stabilized the deacetylase, leading to increased expression of c-Myc, which in turn stimulated a pathological gene program. These unexpected findings highlight the SIK1/HDAC7 signaling axis as a promising target for the treatment of cardiac hypertrophy and heart failure.
Cardiac hypertrophy and HF
Approximately six million Americans suffer from heart failure (HF), placing an economic liability on the United States that is projected to rise to approximately $70 billion annually by 2030 (1). HF is associated with poor quality of life and a five-year mortality rate nearing 50%, exceeding that of many malignancies (2). HF is a global problem that will likely become even more onerous given the ever-increasing prevalence of risk factors such as aging and metabolic disease.
One hallmark of HF is cardiomyocyte hypertrophy, a growth response driven by increased intracellular protein abundance in the absence of cell division. Cardiac hypertrophy has traditionally been viewed as a compensatory response that normalizes wall stress and maximizes ventricular performance in the face of a pathogenic insult. However, chronic hypertrophy is associated with poor outcomes in patients with cardiovascular disease (3–5), and in animal models of pressure overload, inhibition of hypertrophy is beneficial rather than detrimental (6, 7). Thus, there is a need to elucidate molecular mechanisms that control cardiomyocyte growth, with the goal of defining auspicious nodes for HF drug development (8).
Class IIa HDACs as repressors of cardiac hypertrophy
Cardiac hypertrophy is associated with a stereotypical pattern of aberrant myocyte gene expression (9, 10). A robust area of investigation over the last approximately 20 years has focused on how stress signals emanating from the cardiomyocyte cell surface transmit to the nucleus to trigger this common gene program. Insights into this process were uncovered by the discovery that a subset of class IIa histone deacetylases (HDACs), which are epigenetic regulators that typically repress gene expression, block cardiac hypertrophy by associating with the myocyte enhancer factor-2 (MEF2) transcription factor (TF) (11).
Although each of the four class IIa HDACs contains conserved MEF2 binding domains, initial work in cardiomyocytes focused on HDAC5 and HDAC9. A model emerged in which HDAC5/9 are normally bound to MEF2 in cardiomyocyte nuclei, squelching expression of MEF2 target genes that drive hypertrophy (11, 12). In response to stress signals, HDAC5/9 are phosphorylated on two serine residues, leading to association with the phospho-serine binding protein 14-3-3 and subsequent nuclear export, freeing MEF2 to stimulate prohypertrophic gene expression. HDAC4 also represses MEF2 and cardiac hypertrophy and undergoes phosphorylation-dependent nuclear export in cardiomyocytes (13). Kinases belonging to the calmodulin-dependent protein kinase (CaMK) group, including CaMKII, protein kinase D (PKD), microtubule-associated kinase (MARK), and SIK, can effectively phosphorylate the 14-3-3 target sites on class IIa HDACs (13–17). Remarkably, despite HDAC7 having a MEF2 binding domain and the ability to undergo phosphorylation-dependent nuclear export, a possible role for this class IIa HDAC in the control of cardiac hypertrophy remained unstudied during this 20-year span.
SIK1 promotes cardiac hypertrophy by stabilizing HDAC7
In this issue of the JCI, Hsu and colleagues (18) demonstrated that HDAC7 actually stimulates cardiac hypertrophy. They began by investigating a previously unexplored role for SIKs in the heart. Nonselective pharmacological inhibitors of SIK catalytic activity blocked agonist-induced hypertrophy and reexpression of the fetal gene program in cultured neonatal rat ventricular myocytes (NRVMs) and human cardiomyocytes derived from induced pluripotent stem cells. RNAi-mediated knockdown of the three SIK family members revealed that SIK1 is prohypertrophic. Consistent with this in vitro work, mice lacking SIK1 globally were protected against cardiac hypertrophy induced by left ventricular pressure overload and also had reduced cardiac fibrosis and improved systolic function compared with controls. Notably, SIK1-null animals appeared healthy under physiological conditions, with normal cardiac structure and function, suggesting the potential for developing SIK1-selective inhibitors that are well tolerated in humans.
SIK1 was originally shown to phosphorylate HDAC5 in skeletal muscle (14) and more recently was found to phosphorylate HDAC4 to trigger vascular calcification (19). Hsu et al. (18) addressed the possibility that SIK1 stimulates cardiac hypertrophy by targeting HDAC7. Surprisingly, HDAC7 abundance was substantially diminished when SIK1 was absent or pharmacologically inhibited. Conversely, HDAC7 mRNA levels were unaffected by SIK1 inhibition, suggesting a posttranscriptional mechanism of regulation. Indeed, SIK1 was subsequently shown to stabilize HDAC7 protein by phosphorylating the two 14-3-3 binding sites, thereby reducing ubiquitination and proteasome-dependent degradation of the deacetylase (Figure 1).
Figure 1. A model for regulation of cardiac hypertrophy by SIK1-dependent phosphorylation of HDAC7.
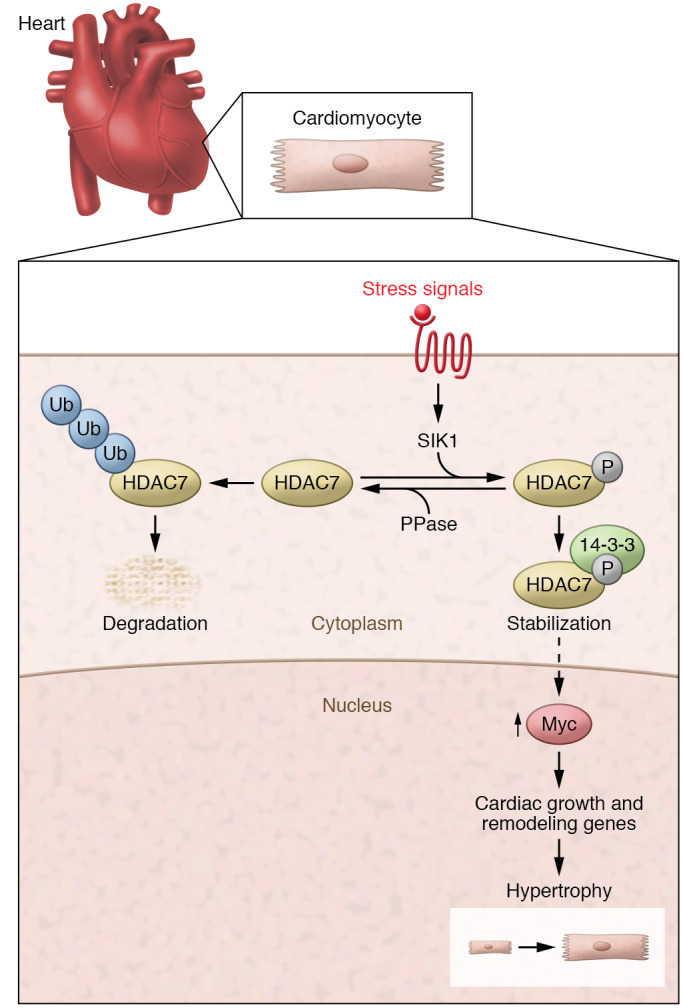
In response to stress signals, SIK1 phosphorylates HDAC7, leading to association with 14-3-3 protein and stabilization of the deacetylase. HDAC7 promotes c-Myc protein expression through an undefined mechanism. c-Myc stimulates genes that drive cardiomyocyte hypertrophy. Dephosphorylation of HDAC7 by a protein phosphatase (PPase) leads to ubiquitin-dependent (Ub-dependent) degradation of the deacetylase, thereby dampening this prohypertrophic signaling cascade.
The most unanticipated finding from the Hsu et al. (18) study is that HDAC7 stimulates, rather than inhibits, cardiomyocyte hypertrophy. Knockdown of HDAC7 in cultured NRVMs blocked agonist-mediated hypertrophy and fetal gene induction, while HDAC7 overexpression stimulated these processes. Mechanistically, as opposed to functioning in the nucleus to repress MEF2 transcriptional activity, HDAC7 is constitutively cytoplasmic in NRVMs and appears to promote hypertrophy by enhancing expression of the c-Myc TF. Indeed, ectopic expression of HDAC7 dramatically increased endogenous c-Myc protein expression and knockdown of c-Myc attenuated HDAC7-induced hypertrophy of NRVMs (18).
Future considerations and clinical implications
The most exciting discoveries in the lab are those that are unforeseen, and this one certainly fits the bill, opening up a plethora of interesting questions and translational angles. First, what is HDAC7 doing in the cytoplasm to promote c-Myc expression? HDAC7 overexpression in cardiomyocytes profoundly increases c-Myc protein abundance, but only modestly augments c-Myc mRNA expression, suggesting that the deacetylase is targeting the TF through a posttranscriptional mechanism. c-Myc is predominantly a nuclear protein, and thus it is likely that HDAC7 indirectly enhances its expression. Given that c-Myc stability is tightly controlled by the ubiquitin-proteasome system (UPS) (20), it is possible that HDAC7 regulates the localization or function of proteins that govern degradation of the TF.
Is HDAC7 catalytic activity required to increase c-Myc expression in cardiomyocytes? Despite having conserved catalytic domains that have high affinity for acetyl-lysine, class IIa HDACs are unable to deacetylate histones, leading some to speculate that their deacetylase domains simply serve as acetyl-lysine readers (21). However, class IIa HDAC catalytic activity can be monitored using an artificial substrate, which led to the discovery of highly selective small molecule inhibitors of this HDAC class (22). These compounds, as well as catalytically dead versions of HDAC7, should be used to determine the role of HDAC7 enzymatic activity in the control of c-Myc expression in cardiomyocytes. If HDAC7 catalytic activity is indeed involved in c-Myc induction, then defining HDAC7 substrates/binding partners should yield insights into pathogenic signaling in the heart. It is also essential that future studies employ cardiomyocyte-specific gain and loss of function to address whether HDAC7 stimulates cardiac hypertrophy in vivo.
Does SIK1 promote cardiac hypertrophy by phosphorylating substrates in addition to HDAC7? The demonstration that a SIK1 inhibitor blocks NRVM hypertrophy in response to HDAC7 overexpression suggests that the answer is yes, since SIK1 lies upstream of HDAC7. Studies to determine the impact that cardiomyocyte-specific SIK1 deletion has on cardiac and phospho-proteome remodeling are needed.
Do other kinases target HDAC7? Although SIK1 knockout reduces HDAC7 expression to undetectable amounts in unstressed hearts, total and phospho-HDAC7 are present, albeit at diminished levels, in knockouts subjected to pressure overload, suggesting that redundant kinases target HDAC7 in response to pathological stress. Thus, effective targeting of this pathway for therapeutic purposes could require combined inhibition of SIK1 and the redundant kinase(s), which is likely another member of the CaMK group.
Is SIK1/HDAC7 a druggable pathway for HF therapy? HDAC7 is essential for maintaining vascular integrity, and global deletion of HDAC7 in mice results in embryonic lethality (23). Furthermore, conditional deletion of HDAC7 in pro-B cells results in severe lymphopenia (24). While these data suggest that inhibiting HDAC7 would be toxic, it is important to keep in mind that knocking out a gene/protein is not the same as inhibiting it pharmacologically. If selective class IIa HDAC inhibitors reduce c-Myc expression in cardiomyocytes, they should be tested for efficacy in preclinical HF models. SIK1 has also been implicated in the regulation of diverse biological processes (25), so it is unknown whether study subjects would tolerate SIK1 inhibition. The fact that global deletion of SIK1 fails to elicit an overt phenotype at baseline is encouraging, but a pharmacological inhibitor of SIK1 needs to be tested. In the absence of a currently available SIK1-selective inhibitor, a tool compound pan-SIK inhibitor should be evaluated in HF models.
The elegant discovery of HDAC7 as the black sheep class IIa HDAC that promotes rather than inhibits cardiac hypertrophy (18) reinforces the notion that in science, as in life, one should never assume one knows the answer without having the data. As they say in Missouri, “Show me.” Even after 20 years of work, it is unquestionable that there are still many exciting discoveries to be made regarding epigenetic and nongenomic functions of HDACs in the heart as well as the therapeutic potential of targeting distinct HDAC isoforms and HDAC kinases in the context of HF.
Acknowledgments
TAM is supported by NIH grants HL116848, HL147558, DK119594, HL127240, and HL150225. JGT is supported by NIH grant HL147463. TAM is, and JGT was, supported by the American Heart Association (16SFRN31400013).
Version 1. 05/04/2020
Electronic publication
Version 2. 06/01/2020
Print issue publication
Footnotes
Conflict of interest: TAM received funding from Italfarmaco.
Copyright: © 2020, American Society for Clinical Investigation.
Reference information: J Clin Invest. 2020;130(6):2811–2813. https://doi.org/10.1172/JCI137074.
See the related article at Salt-inducible kinase 1 maintains HDAC7 stability to promote pathologic cardiac remodeling.
Contributor Information
Joshua G. Travers, Email: joshua.travers@cuanschutz.edu.
Tianjing Hu, Email: tianjing.hu@cuanschutz.edu.
References
- 1.Virani SS, et al. Heart disease and stroke statistics–2020 update: a report from the American Heart Association. Circulation. 2020;141(9):e139–e596. doi: 10.1161/CIR.0000000000000757. [DOI] [PubMed] [Google Scholar]
- 2.Mamas MA, et al. Do patients have worse outcomes in heart failure than in cancer? A primary care-based cohort study with 10-year follow-up in Scotland. Eur J Heart Fail. 2017;19(9):1095–1104. doi: 10.1002/ejhf.822. [DOI] [PubMed] [Google Scholar]
- 3.Devereux RB, et al. Prognostic significance of left ventricular mass change during treatment of hypertension. JAMA. 2004;292(19):2350–2356. doi: 10.1001/jama.292.19.2350. [DOI] [PubMed] [Google Scholar]
- 4.Gardin JM, Lauer MS. Left ventricular hypertrophy: the next treatable, silent killer? JAMA. 2004;292(19):2396–2398. doi: 10.1001/jama.292.19.2396. [DOI] [PubMed] [Google Scholar]
- 5.Levy D, Garrison RJ, Savage DD, Kannel WB, Castelli WP. Prognostic implications of echocardiographically determined left ventricular mass in the Framingham Heart Study. N Engl J Med. 1990;322(22):1561–1566. doi: 10.1056/NEJM199005313222203. [DOI] [PubMed] [Google Scholar]
- 6.Esposito G, et al. Genetic alterations that inhibit in vivo pressure-overload hypertrophy prevent cardiac dysfunction despite increased wall stress. Circulation. 2002;105(1):85–92. doi: 10.1161/hc0102.101365. [DOI] [PubMed] [Google Scholar]
- 7.Hill JA, et al. Targeted inhibition of calcineurin in pressure-overload cardiac hypertrophy. Preservation of systolic function. J Biol Chem. 2002;277(12):10251–10255. doi: 10.1074/jbc.M110722200. [DOI] [PubMed] [Google Scholar]
- 8.Schiattarella GG, Hill TM, Hill JA. Is load-induced ventricular hypertrophy ever compensatory? Circulation. 2017;136(14):1273–1275. doi: 10.1161/CIRCULATIONAHA.117.030730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lowes BD, et al. Myocardial gene expression in dilated cardiomyopathy treated with beta-blocking agents. N Engl J Med. 2002;346(18):1357–1365. doi: 10.1056/NEJMoa012630. [DOI] [PubMed] [Google Scholar]
- 10.Zhao M, Chow A, Powers J, Fajardo G, Bernstein D. Microarray analysis of gene expression after transverse aortic constriction in mice. Physiol Genomics. 2004;19(1):93–105. doi: 10.1152/physiolgenomics.00040.2004. [DOI] [PubMed] [Google Scholar]
- 11.Zhang CL, McKinsey TA, Chang S, Antos CL, Hill JA, Olson EN. Class II histone deacetylases act as signal-responsive repressors of cardiac hypertrophy. Cell. 2002;110(4):479–488. doi: 10.1016/S0092-8674(02)00861-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chang S, McKinsey TA, Zhang CL, Richardson JA, Hill JA, Olson EN. Histone deacetylases 5 and 9 govern responsiveness of the heart to a subset of stress signals and play redundant roles in heart development. Mol Cell Biol. 2004;24(19):8467–8476. doi: 10.1128/MCB.24.19.8467-8476.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Backs J, Song K, Bezprozvannaya S, Chang S, Olson EN. CaM kinase II selectively signals to histone deacetylase 4 during cardiomyocyte hypertrophy. J Clin Invest. 2006;116(7):1853–1864. doi: 10.1172/JCI27438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Berdeaux R, et al. SIK1 is a class II HDAC kinase that promotes survival of skeletal myocytes. Nat Med. 2007;13(5):597–603. doi: 10.1038/nm1573. [DOI] [PubMed] [Google Scholar]
- 15.McKinsey TA. Derepression of pathological cardiac genes by members of the CaM kinase superfamily. Cardiovasc Res. 2007;73(4):667–677. doi: 10.1016/j.cardiores.2006.11.036. [DOI] [PubMed] [Google Scholar]
- 16.Monovich L, et al. A novel kinase inhibitor establishes a predominant role for protein kinase D as a cardiac class IIa histone deacetylase kinase. FEBS Lett. 2010;584(3):631–637. doi: 10.1016/j.febslet.2009.12.014. [DOI] [PubMed] [Google Scholar]
- 17.Vega RB, et al. Protein kinases C and D mediate agonist-dependent cardiac hypertrophy through nuclear export of histone deacetylase 5. Mol Cell Biol. 2004;24(19):8374–8385. doi: 10.1128/MCB.24.19.8374-8385.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hsu A, et al. Salt-inducible kinase 1 maintains HDAC7 stability to promote pathologic cardiac remodeling. J Clin Invest. 2020;130(6):2966–2977. doi: 10.1172/JCI133753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Abend A, Shkedi O, Fertouk M, Caspi LH, Kehat I. Salt-inducible kinase induces cytoplasmic histone deacetylase 4 to promote vascular calcification. EMBO Rep. 2017;18(7):1166–1185. doi: 10.15252/embr.201643686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Farrell AS, Sears RC. MYC degradation. Cold Spring Harb Perspect Med. 2014;4(3):a014365. doi: 10.1101/cshperspect.a014365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bradner JE, et al. Chemical phylogenetics of histone deacetylases. Nat Chem Biol. 2010;6(3):238–243. doi: 10.1038/nchembio.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lobera M, et al. Selective class IIa histone deacetylase inhibition via a nonchelating zinc-binding group. Nat Chem Biol. 2013;9(5):319–325. doi: 10.1038/nchembio.1223. [DOI] [PubMed] [Google Scholar]
- 23.Chang S, Young BD, Li S, Qi X, Richardson JA, Olson EN. Histone deacetylase 7 maintains vascular integrity by repressing matrix metalloproteinase 10. Cell. 2006;126(2):321–334. doi: 10.1016/j.cell.2006.05.040. [DOI] [PubMed] [Google Scholar]
- 24.Azagra A, et al. In vivo conditional deletion of HDAC7 reveals its requirement to establish proper B lymphocyte identity and development. J Exp Med. 2016;213(12):2591–2601. doi: 10.1084/jem.20150821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wein MN, Foretz M, Fisher DE, Xavier RJ, Kronenberg HM. Salt-inducible kinases: physiology, regulation by cAMP, and therapeutic potential. Trends Endocrinol Metab. 2018;29(10):723–735. doi: 10.1016/j.tem.2018.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]