

Abstract
Plasmacytoid dendritic cells (pDCs) are robust producers of IFNα and one of the first immune cells to respond to SIV infection. To elucidate responses to early HIV-1 replication, we studied blood pDCs in 29 HIV-infected participants who initiated antiretroviral therapy during acute infection and underwent analytic treatment interruption (ATI). We observed an increased frequency of partially activated pDCs in the blood before detection of HIV RNA. Concurrent with peak pDC frequency, we detected a transient decline in the ability of pDCs to produce IFNα in vitro, which correlated with decreased phosphorylation of IFN regulatory factory 7 (IRF7) and NF-κB. The levels of phosphorylated IRF7 and NF-κB inversely correlated with plasma IFNα2 levels, implying that pDCs were refractory to in vitro stimulation after IFNα production in vivo. After ATI, decreased expression of IFN genes in pDCs inversely correlated with the time to viral detection, suggesting that pDC IFN loss is part of an effective early immune response. These data from a limited cohort provide a critical first step in understanding the earliest immune response to HIV-1 and suggest that changes in blood pDC frequency and function can be used as an indicator of viral replication before detectable plasma viremia.
Keywords: AIDS/HIV, Immunology
Keywords: Dendritic cells, Innate immunity
Introduction
Plasmacytoid dendritic cells (pDCs) are important mediators of the early innate immune response to viral infection through robust production of type I IFNs, particularly IFNα (1, 2). pDCs recognize single-stranded RNA and unmethylated CpG-rich DNA through activation of TLR7 or TLR9 (3–5), respectively, which results in activation of NF-κB and IFN regulatory factory 7 (IRF7) to induce cytokine production. The constitutive expression of high levels of IRF7 by pDCs allows for rapid production of type I IFNs (6). Type I IFNs contribute to the immune response through inhibition of viral replication (7), as well as by mediating activation and survival of myeloid DCs (mDCs), macrophages, NK cells, and T cells (8). Loss of pDC function early after infection could thus have critical effects on the propagation of the immune response. To date, little is known about the pDC response during the earliest stages of HIV-1 infection or before detection of viral RNA, or how this response contributes to subsequent immune control or disease progression.
HIV-1 infection induces a marked alteration of pDC frequency and function after peak viremia that correlates with disease progression. Frequencies of pDCs are lower in the blood of individuals living with HIV-1 than in uninfected individuals and are only partially restored after antiretroviral therapy (ART) (9–14). In viremic individuals, pDC frequency positively correlates with CD4 count and inversely correlates with plasma viral load (VL) (15). pDCs from individuals with HIV-1 have a reduced capacity to produce IFNα following in vitro stimulation with TLR7 or TLR9 ligands, HIV-1, and other viruses (14, 16–18). HIV-1 infection affects IFNα production by altering both the intracellular signaling capability of pDCs and the phenotypic make-up of the pDC population in the blood. Exposure of pDCs from healthy donors to HIV-1 in vitro results in a delay and reduction of IFNα production compared with TLR7 agonists and other viruses (19, 20). This delay has been associated with increased phosphorylation of spleen-associated tyrosine kinase (SYK) due to engagement of gp-120 with the pDC marker CD303 (19) and decreased nuclear translocation of IRF7 as a result of decreased phosphorylation of Akt (21). Furthermore, decreased production of IFNα has been associated with an increase in T cell immunoglobulin and mucin domain–containing 3–positive (Tim-3+) exhausted pDCs present in the blood during HIV-1 infection (22), increased CD40 expression on blood pDCs during HIV-1 infection (23), and mobilization of immature pDCs from the bone marrow after SIV infection (24–26). These studies were mostly performed with pDCs from healthy donors exposed to HIV-1 in vitro or with pDCs obtained from individuals during the chronic stage of HIV-1 infection.
Limited knowledge about the function of pDCs during the earliest stages of infection, before detection of plasma viral RNA, has been derived from nonhuman primate models. pDCs are the first major cell population recruited to the vaginal mucosa after SIV infection in rhesus macaques, where they accumulate beneath the endocervical epithelium and produce IFNα within 1 day of infection (27). A transient increase in the frequency of pDCs in the blood has also been reported to occur before peak viremia after intravenous SIV infection (25, 26, 28). Frequent sampling of plasma after intravenous SIV inoculation of African green monkeys (AGMs) revealed a transient increase in the low plasma IFNα levels 2 days after infection, before the subsequent robust increase in plasma IFNα levels that accompanies viremia (29). It is logistically difficult to determine whether a similar increase in plasma IFNα levels occurs within days of HIV-1 infection, however, plasma IFNα levels are one of the first to increase during the initial cytokine storm that occurs after HIV-1 infection, similar to what has been described in SIV infection (28, 30). The importance of the early IFNα response has been shown by decreased SIV acquisition when rhesus macaques were treated with type I IFNs (31, 32) and more rapid disease progression when treated with an IFN-I receptor antagonist (31). However, the uncontrolled type I IFN response after acute HIV and pathogenic SIV infection is associated with chronic immune activation and disease progression even in treated individuals (29, 33–36).
Although it is challenging to measure the early pDC response during the eclipse phase of acute HIV-1 infection (AHI), when virus is not yet detectable in the blood, it is possible to monitor the immune response to the initial low-level viral replication in the tissues after the interruption of ART in aviremic individuals. The identification of post-treatment controllers, who maintain undetectable VLs for years after treatment cessation (37), has led to an increased interest in identifying therapeutic interventions to induce control after analytic treatment interruption (ATI). ATI also allows measurement of the early innate immune response to HIV replication. Three ATI studies have been completed with participants in the RV254 cohort, a unique group of individuals living with HIV-1 in Thailand who initiated ART during AHI (Fiebig stages I–V) (38, 39): (a) the single-arm RV411 study of ATI after very early ART initiation during Fiebig I (40); (b) the RV409 study of vorinostat, hydroxychloroquine, and maraviroc (VHM) with ART versus ART alone (control arm) followed by ATI (our unpublished observations); and (c) the RV397 study of the VRC01 broadly neutralizing Ab versus placebo administration during ATI (41). Here, we analyzed the mobilization, activation, and function of pDCs in the blood of participants in these 3 ATI studies to identify markers of viral replication before detection of viremia using standard commercial assays (≥20 copies/mL). Although these data are from 3 different clinical trials with limited numbers of participants, these participants are homogenous with regard to age, virus clade, and time of ART initiation.
Results
pDCs are mobilized to the blood before detection of viremia after ATI.
To study the innate response before detectable viremia, we analyzed longitudinal samples from participants enrolled in 3 separate ATI studies: RV411, RV409, and RV397 (Table 1 and Supplemental Figure 1; supplemental material available online with this article; https://doi.org/10.1172/JCI130597DS1). We measured the activation of innate cells, including pDCs, before ATI in the participants of the RV409 study who received VHM, so only participants in the control arm of RV409, who received no therapeutic intervention, were included in the current analysis. All participants in all 3 trials experienced viral rebound during ATI.
Table 1. Characteristics of the study participants.
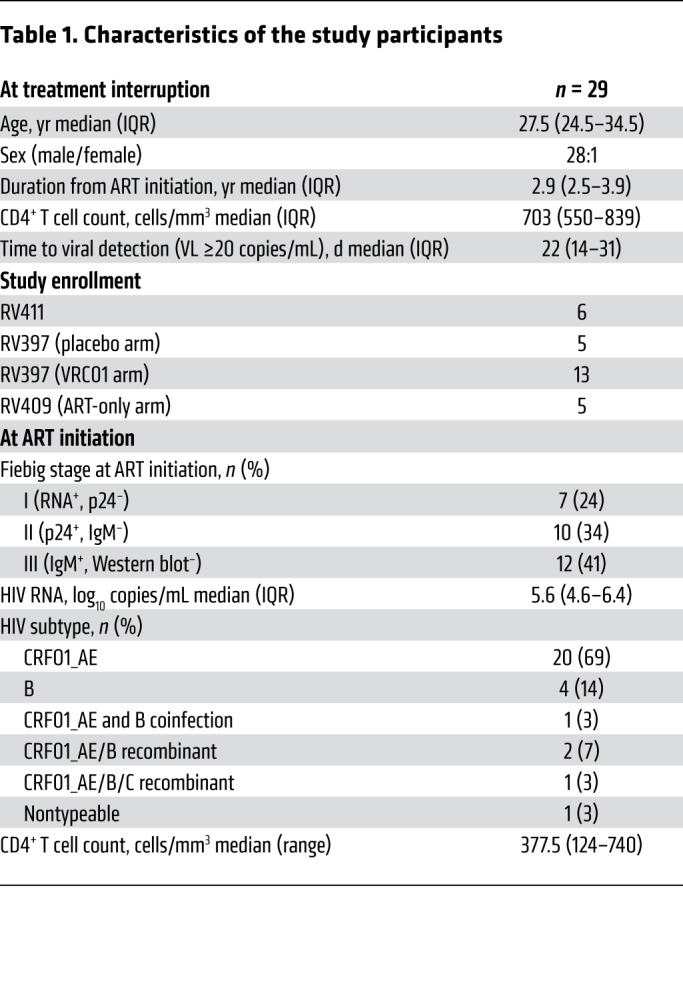
We examined the presence of pDCs in the blood after ATI to look for indications of pDC mobilization before detection of viremia. We observed a transient increase in the frequency of pDCs (Lin–HLA-DR+CD11c–CD303+) (Figure 1A) in the blood before the detection of plasma viral RNA (Figure 1B). In participants for whom the absolute pDC count could be calculated, we found that the transient increase in pDC frequency approximated a similar increase in the absolute pDC count (Supplemental Figure 2A). As pre-rebound samples from RV411 participants were limited to the baseline visit, when treatment was interrupted, and during the last visit at which the participant was aviremic (VL <20 copies/mL), we used these time points to quantify changes in pDC frequency before detection of viremia in the 3 ATI cohorts. We found that a significant increase in pDC frequency occurred between the baseline ATI visit and the last aviremic visit (median of 7.6% vs. median of 10.8%, P < 0.001) (Figure 1C). As administration of VRC01 Ab to participants in RV397 could have altered their pDC response, we confirmed that there was a significant increase in pDC frequencies in the blood of the participants who underwent ATI in the absence of therapeutic treatment (P < 0.001) (Supplemental Figure 2B) and that there was no significant difference in the fold change of pDC frequencies between the placebo and treatment arms of the RV397 study (Supplemental Figure 2C). In contrast, participants in RV409 treated with ART alone showed no increase in pDC frequencies during the 10 weeks before ATI (Figure 1D), suggesting that the transient increase seen after ATI was an early response to viral replication that followed ART interruption. Importantly, this increase was specific to pDCs, as a similar increase was not found in the frequencies of subsets of CD1c+ mDCs, CD141+ mDCs, CD14+CD16– classical monocytes, CD14+CD16+ intermediate monocytes, or CD14loCD16+ nonclassical monocytes before detection of viremia (Supplemental Figure 3).
Figure 1. Frequency of pDCs in the blood increases before detectable viremia.

(A) pDCs were identified as CD45+HLA-DR+CD303+ cells that were negative for CD14, CD11c, and CD1c and the lineage markers CD3, CD14, CD19, and CD56 (Lin–). (B) The frequency of pDCs in the blood was measured by flow cytometry as a percentage of lineage-negative cells. Representative graphs show the pDC frequency (blue) relative to the VL after ATI (black). (C) The pDC frequency at the time of ATI was compared with the frequency at the last aviremic time point to show increased pDC frequency before viral RNA was detected in the blood. ***P < 0.001, by Wilcoxon test. n = 25 (10 of whom received VRC01). (D) The frequency of pDCs in the blood before ATI is shown for the 5 participants in the placebo arm of RV409. Changes in pDC frequency were compared when measurements were taken at a 2- or 4-week time interval.
pDCs are partially activated before detection of viremia after ATI.
To ascertain the activation state of pDCs entering the blood before detectable viremia, we analyzed the expression of multiple markers by flow cytometry. Circulating pDCs showed evidence of activation, as measured by increased expression of the markers CD69, PD-L1, and CD40 (Figure 2A). Between the baseline time point of ATI and the last aviremic time point during ATI, we observed significant increases in the expression of CD69 (MFI of 256 vs. 299, P < 0.01), PD-L1 (MFI of 113 vs. 287, P < 0.01), and CD40 (190 vs. 239, P < 0.01) (Figure 2B).
Figure 2. Expression of activation markers increases on pDCs before detectable viremia.

(A) The MFI of CD69, PD-L1, and CD40 on pDCs was measured by flow cytometry. Representative graphs show CD69 (green), PD-L1 (orange), and CD40 (purple) MFI relative to the changes in pDC frequency (blue) and VL (black). (B) The MFI of CD69, PD-L1, and CD40 at ATI was compared with the MFI at the last time point at which viral RNA was undetectable in the plasma. **P < 0.01, by Wilcoxon test. (C) Changes in CD86 and CD83 MFI between baseline ATI and the last aviremic time point. n = 25 (10 of whom received VRC01).
Upon activation, pDCs can differentiate into more conventional DCs, losing the capacity to produce large amounts of IFNα but gaining the capacity to stimulate T cells (42–44). This maturation is characterized by increased expression of the costimulatory marker CD86 and the maturation marker CD83, among others (42, 43, 45). We found no difference in the expression levels of CD86 or CD83 on pDCs before detectable viremia after ATI (Figure 2C). There were also no significant differences in the expression of activation markers between study arms in RV397 (Supplemental Figure 4). These data indicate that after ATI, pDCs entering the blood before detection of viremia are activated but not differentiated.
pDCs are primed to traffic to sites of inflammation before detection of viremia.
To assess the destination of the pDCs entering the blood, we analyzed the expression of pDC surface proteins involved in cell trafficking. Because of sample availability, all remaining experiments were performed on samples from the RV397 study only. pDCs showed no changes in expression of the chemokine receptors CCR7 or CCR9 before detectable viremia but did have decreased expression of integrin β7, which mediates homing to mucosal sites (46), at the last aviremic time point compared with baseline ATI (392 vs. 299, P < 0.05) (Figure 3A). pDCs had increased expression of CD29 (integrin β1) (1905 vs. 2535, P < 0.05), which is used for firm adhesion before extravasation from the blood vessel (47), and CXCR4 (1010 vs. 1516, P < 0.01), which mediates chemotaxis to lymph nodes via high endothelial venules, particularly during inflammation (Figure 3A). We observed no differences between the placebo and treatment arms for any of the measured markers. Further, plasma levels of stromal cell–derived factor 1α (SDF-1α), the primary ligand for CXCR4, were also elevated relative to levels at baseline ATI before detectable viremia (126 vs. 255, P < 0.01) (Figure 3B). These data suggest that the pDCs were primed for recruitment to secondary lymph organs and extravasation at sites of inflammation.
Figure 3. Expression of migration markers increases on pDCs before detectable viremia.

(A) Changes in CCR7, CCR9, integrin β7, CXCR4, and CD29 MFI between baseline ATI and the last aviremic time point are shown. (B) SDF-1α levels were measured in the plasma of RV397 participants, and changes between baseline ATI and the last aviremic time point are shown. *P < 0.05 and **P < 0.01, by Wilcoxon test. n = 15 participants from RV397 (10 of whom received VRC01).
Transient loss of IFN production occurs in pDCs before detection of viremia.
As these changes to pDCs occurred before detection of viral RNA in the blood, we assessed their functional capacity to respond to virus by measuring IFNα and TNF-α production after stimulation with the TLR7 agonist imiquimod (Figure 4A). Before detectable viremia, we noted a transient decline in the percentage of pDCs that produced IFNα upon in vitro stimulation (Figure 4B). As this drop in IFNα production tended to correspond with peak pDC frequency, we compared the frequency of cells producing IFNα at the time of peak pDC frequency with that of the week before peak pDC frequency. We found a significant decline in the frequency of IFNα+ pDCs (19.6% vs. 14.1%, P < 0.05) (Figure 4C) and a trend toward a lower frequency of TNF-α+ pDCs (54.2% vs. 45.3%, P = 0.08) (Figure 4D and Supplemental Figure 5A) at the time of peak pDC frequency. It is important to note that the 4 participants with increased frequencies of IFNα+ pDCs at the time of peak pDC frequency had the lowest frequencies of IFNα+ pDCs at the prior visit. For 2 of these participants, the decreased capacity of pDCs to produce IFNα after stimulation occurred the week before peak pDC frequency, when the frequencies of pDCs in the blood had increased but not yet reached maximum levels. For the other 2 participants, rebound occurred very early, within just 2 weeks of treatment interruption, and the limited sampling before rebound may not have been sufficient to capture the transient decline. Moreover, we noted a trend toward decreased IFNα MFI at the time of the peak pDC frequency among all participants (7144 vs. 6442, P = 0.15) (Supplemental Figure 5B), suggesting that not only were fewer cells able to produce IFNα but also that less IFNα could be produced on a per-cell basis after in vitro stimulation.
Figure 4. pDCs have a transient decrease in the capacity to produce IFNα in vitro.

(A) Total PBMCs were stimulated with imiquimod for 6 hours, and IFNα production by pDCs was measured by flow cytometry. (B) The percentage of pDCs that produced IFNα in response to imiquimod (IQ) stimulation is shown (red). pDC frequency (blue) and HIV-1 VL (black) are included for reference. The percentage of pDCs producing IFNα (C) or TNF-α (D) in response to imiquimod were compared between the time point before viremia at which the highest pDC frequency occurred and the time point immediately prior. *P < 0.05, by Wilcoxon test. n = 14 participants from RV397 (11 of whom received VRC01).
pDC activation in participants with extended aviremia.
The 2 participants in RV397 with the longest period off ART before detection of viremia (≥20 copies/mL) had an increased frequency of pDCs in the blood after ATI, with transient increases in expression of activation markers (Figure 5A). Participant 7242 had a delayed rebound of 65 days but showed an increased frequency of pDCs in the blood with a decreased capacity to produce IFNα 1 week after ATI. pDC function in participant 7242 recovered 3 weeks after ATI but was lost again before detection of viremia. Although participant 3499 experienced the longest time to rebound (42 weeks), there was a viral blip identified by a high-sensitivity single-copy assay, and an increased frequency of pDCs in the blood was observed immediately before this viral blip. We also observed a transient impaired capacity to produce IFNα after in vitro stimulation in these 2 participants (Figure 5B). Despite a viral blip of fewer than 2 copies/mL 6 weeks after ATI, pDCs from participant 3499 still showed a decreased capacity to produce IFNα before the blip. Participant 7242 did not have a detectable viral blip by single-copy assay, but 6 of the other participants in RV397 had measurable virus of 2.1 copies/mL or fewer at the last time point at which the participant was considered aviremic (VL <20 copies/mL). One of these participants had a VL of just 21 copies/mL at the first visit that a VL was detectable by commercial assay but already had a VL of 1.5 copies/mL 1 week prior, as measured by the single-copy assay. These data indicate that viral replication may begin more than 1 week before it is detectable by commercial assays and suggest that the increased frequency and decreased function of pDCs after ATI may be a marker for low-level viral replication, even when detectable viremia does not occur until weeks or months later.
Figure 5. Increased frequency and activation of pDCs in participants with the longest period of aviremia.

(A) The frequency of pDCs as a percentage of lineage-negative cells is shown in blue for 2 participants in RV397 who had the longest period of aviremia (VL <20 copies/mL). The MFI of CD69 (green), PD-L1 (orange), and CD40 (purple) on pDCs is shown. The single-copy VL (black) is shown for participant 3499. (B) The frequency of pDCs that produced IFNα after in vitro imiquimod stimulation is shown (red), with pDC frequency and VL indicated for reference. Both participants received VRC01.
pDCs undergo a refractory state before detection of viremia.
Previous studies reported a decline in IFNα production by pDCs after peak viremia during acute SIV and HIV-1 infections or after detectable viremia during treatment interruption (14, 16–18, 22, 23, 48). In SIV infection, this decline was attributed to an egress of immature pDCs from the bone marrow into the blood after peak viremia (25). To determine whether the increased frequency of pDCs in the blood before detection of viremia was due to bone marrow egress, we measured the frequency of cells expressing Ki-67, a marker of immature pDCs (26). We detected no difference in the frequency of Ki-67+ pDCs between the week of peak pDC frequency and the week prior (Supplemental Figure 6A), suggesting that the decreased frequency of IFNα+ pDCs after in vitro stimulation was not due to an influx of immature cells into the blood. An inability to produce IFNα has also been attributed to exhaustion of pDCs over time during HIV-1 infection (22). To determine whether the decreased ability to produce IFNα at the time of peak pDC frequency was due to an influx of exhausted cells, characterized by Tim-3 expression, we measured the frequency of Tim-3+ pDCs. At the time of peak pDC frequency, we noted a decline in the frequency of pDCs expressing Tim-3 compared with frequencies the week prior (7.3% vs. 5.7%, P < 0.01) (Supplemental Figure 6B). Thus, the decrease in IFNα production we observed before detectable viremia was not a result of an influx of exhausted pDCs into the blood.
During in vitro activation of pDCs with HIV-1, there is a delayed production of IFNα that has been associated with increased phosphorylation of the signaling molecule SYK as a result of interactions between gp-120 and CD303 (19). To assess whether the decrease in IFNα production by pDCs was due to exposure to gp-120 in vivo, we measured the basal percentage of phosphorylated SYK (p-SYK) in 8 RV397 participants at a visit before (pre-ATI visit) and at a visit at the nadir of the IFNα response or at the only available visit remaining after ATI (ATI visit). There was a significant increase in the percentage of p-SYK at the ATI visit compared with the percentage at the pre-ATI visit (median 68.1% vs. 58.7%, P < 0.01) (Supplemental Figure 7A). However, we found no correlation between the basal levels of p-SYK and the capacity to produce IFNα. Thus, although these data suggest that a portion of the pDCs may have come in contact with HIV-1 in vivo, resulting in higher SYK phosphorylation, this phosphorylation did not appear to be the cause of the decreased IFNα production in vitro.
The decreased IFNα production in vitro could also be a result of the refractory state of the pDCs after in vivo activation by HIV-1 (18, 49). We measured the capability of pDCs from 8 participants in RV397 to phosphorylate IRF7 (p-IRF7) and NF-κB (p–NF-κB), key transcription factors in the production of IFNα and TNF-α, respectively. We assessed whether the capacity to activate IRF7 or NF-κB correlated with IFNα production and detected negative correlations between the levels of p-IRF7 or p–NF-κB after stimulation and loss of IFNα capacity, as measured by the fold decrease in IFNα+ pDCs after in vitro stimulation at the ATI visit compared with the week prior (r = –0.76, P < 0.05, r = 0.71, P = 0.06, respectively) (Figure 6A and Supplemental Figure 7, B and C). These data indicate that pDCs at the ATI visit were capable of producing IFNα when the upstream transcription factors were activated but lost IFNα capacity when signaling was hampered. We further hypothesized that if the pDCs were in a refractory state due to activation by HIV-1, then earlier production of IFNα in vivo would be indicated by higher plasma IFNα2 levels. Along these lines, we found that there was a negative correlation between plasma IFNα2 levels at the ATI visit and the levels of p-IRF7 (r = –0.78, P < 0.05) and p–NF-κB (r = –0.64, P = 0.10) in pDCs after imiquimod stimulation (Figure 6B and Supplemental Figure 7D). In contrast, the levels of p-IRF7 and p–NF-κB after imiquimod stimulation of a sample obtained before ATI correlated with the plasma IFNα2 levels measured at the ATI visit (r = 0.79, P < 0.05, r = 0.92, P < 0.01, respectively) (Figure 6C and Supplemental Figure 7E), indicating that the pDCs had the capacity to phosphorylate IRF7 before ATI. Cumulatively, these data suggest that prior to detectable viremia following ATI, pDCs are activated and produce IFNα before entering into a refractory state.
Figure 6. pDC signaling capacity negatively correlates with plasma IFNα2 levels.

(A) p-IRF7 levels were measured in pDCs by flow cytometry after in vitro imiquimod stimulation of PBMCs obtained at the visit with the lowest IFNα response or from the only remaining sample available after ATI (ATI visit). Spearman’s correlations were determined between the MFI of p-IRF7 and the loss of IFNα-producing capacity, as measured by the fold decrease in pDCs producing IFNα in vitro at the ATI visit compared with levels detected at the preceding visit. (B and C) Plasma IFNα2 levels in RV397 participants were measured by SIMOA assay at the ATI visit. Spearman’s correlations were performed between the plasma IFNα2 levels at the ATI visit and the MFI of p-IRF7 in pDCs after in vitro imiquimod stimulation of PBMCs obtained at the ATI visit (B) or a pre-ATI visit (C). n = 8 participants from RV397 (all of whom received VRC01).
It has been reported that IRF7 mRNA levels are lower in pDCs from individuals living with HIV-1 during both chronic, untreated infection and after ART, resulting in lower IFNα production (50, 51). To determine whether the decreased IFNα production measured after ATI was related to decreased expression of key signaling molecules, we measured the mRNA levels of IFNs, multiple cytokines and chemokines, surface receptors, and signaling genes in sorted pDCs from pre-ATI and ATI samples from 8 different RV397 participants (Figure 7A). Several genes showed lower expression in pDCs from the ATI visit compared with expression levels at the pre-ATI visit. In particular, at the ATI visit, we detected lower expression levels of genes related to type I IFN signaling, including IRF7 (median 1.44-fold, P < 0.05); TLR7 (median of 1.29-fold, P < 0.01); STAT1 (median of 1.57-fold, P < 0.01); and 2 IFN-stimulated genes (MX1, median of 1.77-fold, P < 0.01; MX2, median of 1.41-fold, P < 0.01), with the latter 4 genes maintaining significance after correction for multiple comparisons (Figure 7A). To determine whether the changes in IFN signaling genes were intrinsic to pDCs or were common to other innate cells, we also measured gene expression in sorted CD1c+ mDCs (Supplemental Figure 7F). We found that mDCs had very little change in gene expression between the pre-ATI and ATI time points, only showing a decrease in MX2 expression (1.34-fold, P < 0.05) and an increase in IRF1 expression (1.40-fold, P < 0.01), neither of which remained significant when corrected for multiple comparisons. These data suggest that there is a pDC-specific loss in expression of genes related to type I IFN signaling that may contribute to the loss of in vitro IFNα production.
Figure 7. Type I IFN gene expression negatively correlates with time to rebound.

(A) mRNA levels of cytokines, chemokines, surface receptors, and IFN signaling molecules were measured by BioMark in sorted pDCs obtained from the pre-ATI and ATI visits. Shown is the difference in CT values between the ATI and pre-ATI time points after normalization to GAPDH. Data indicate the mean ± SEM. *P < 0.05 and **P < 0.01, by 1-sample Wilcoxon test. Blue single asterisks indicate values that were no longer significant when corrected for a FDR of 10% by the Benjamini-Hochberg procedure. (B) Spearman’s correlation between plasma IFNα2 levels after viral rebound and the change in IFNA2 levels at the ATI time point. (C) Spearman’s correlation between the change in expression of IFN genes at the ATI time point and the days from the ATI time point to viral detection. n = 8 participants from RV397 (4 of whom received VRC01).
Consistent with the decreased in vitro IFNα production, we observed that pDCs had lower expression levels of type I IFN genes. IFNA5 and IFNB1 transcript levels were 2.14- and 1.87-fold lower in the pDCs obtained at the ATI visit, respectively (P < 0.05) (Figure 7A). Though the 2.53-fold decrease in IFNA2 expression did not remain significant after correcting for multiple comparisons, we noted a significant positive correlation between the expression levels of IFNA2 at the ATI visit and the subsequent plasma IFNα2 levels measured after rebound (r = 0.9048, P < 0.01) (Figure 7B). We did not observe a similar correlation between IFNA2 expression and rebound plasma IFNα2 levels in the mDCs (Supplemental Figure 7G), suggesting that pDC function after ATI drives early IFNα production after rebound. Interestingly, there were negative correlations between the expression levels of type I IFN genes and the time from the ATI visit to viral detection (VL ≥20 copies/mL) (Figure 7C). IFNA1, IFNA2, IFNA7, and IFNA8 levels were all significantly negatively correlated with the time to viral detection after correction for multiple comparisons (r = –0.9132, P < 0.01 for all), and IFNA21/1 and IFNB1 levels only reached significance without correction (r = –0.8498 and –0.7864, respectively, P < 0.05). These data suggest that the loss of type I IFN gene expression by pDCs after ATI may be indicative of a more effective early innate immune response, correlating with a longer time to viral detection.
Discussion
Using longitudinal samples from participants who interrupted ART as part of the ATI studies, we show that there was an increased frequency of activated pDCs in the blood before detection of plasma viremia. These data from clinical trials are in agreement with prior observations of increased frequencies of pDCs preceding peak plasma viremia during acute SIV infection in macaques (26, 28). As pDCs are one of the first innate cells recruited to the site of infection (27), we posit that the increased frequency and decreased function of pDCs measured here was an indicator of low-level viral replication in the tissues (Figure 8). After ATI, viral replication in CD4+ T cells occurs in a mono- or oligoclonal manner (step 1) (52). The location of these initial reactivation events is yet to be determined, but they probably occur in lymphoid tissues, where CD4+ T cells harboring HIV-1 DNA reside (53–55). Successful viral replication initiates a secondary immune response, presumably starting with the recruitment and activation of pDCs at the site of replication, similar to that seen during the primary infection (step 2) (27). Production of CCL4 and other cytokines by pDCs at the site of replication can then recruit other immune cells, including CD4+ T cells that can propagate virus through new infection or by cellular activation and recrudescence of quiescent virus (step 3). With increased viral replication and dissemination to distal sites, we hypothesized that mobilization of innate immune cells should be evident in the blood. Our data demonstrate the presence of an increased frequency of activated pDCs in the blood before detectable viremia after ATI (step 4). Peripheral blood pDCs were activated, as measured by expression of CD69, PD-L1, and CD40, but had not differentiated into antigen-presenting cells expressing CD83 and CD86. These cells also expressed increased levels of CXCR4 and CD29, suggesting that they are being trafficked to sites of inflammation (56, 57). In contrast, the lack of increase in CCR7 expression on pDCs prior to detectable viremia suggests that they would be responsive to the higher levels of SDF-1α in the plasma at this time, as demonstrated in an earlier reported study (58). A partially activated phenotype, similar to that seen here, has previously been associated with an ability to continuously produce IFNα by pDCs exposed to HIV-1 in vitro (44). However, pDCs in the blood of ATI participants showed a transient decline in the ability to produce IFNα after in vitro stimulation (step 5), similar to the refractory state that has been reported in pDCs obtained after peak viremia during AHI, after rebound in ATI, and in SIV-infected AGMs (14, 16–18, 59).
Figure 8. Schematic representation of pDC sensing of viral replication after ATI.

Step 1: After interruption of antiretroviral therapy, viral recrudescence from HIV-infected CD4+ T cells occurs in the tissues. Step 2: Successful viral replication leads to the recruitment of innate immune cells, including pDCs, which are one of the first to respond to initial HIV infection. pDC sensing of virus at the site of recrudescence results in their activation and rapid cytokine production. Step 3: Production of CCL4 by activated pDCs can recruit more CD4+ T cells to the site of viral replication, resulting in further viral propagation and, eventually, measurable virus in the blood. Step 4: Our data demonstrate an increased frequency of pDCs in the blood prior to detection of plasma viremia after ATI. These pDCs expressed the activation markers CD69, PD-L1, and CD40 as well as increased levels of CXCR4 and CD29, suggesting that they were trafficking to sites of inflammation. Step 5: pDCs in the blood showed a transient decline in the ability to produce IFNα after stimulation in vitro, indicating that they enter a state of refraction after in vivo activation.
Identification of a refractory state instead of a state of persistent IFNα production may depend on the timing of HIV-1 exposure and IFNα measurement. pDCs are refractory to IFNα production after multiple in vitro stimulations with influenza virus, R848, or CpGB, indicating a mechanism for controlling persistent activation and IFNα production (44, 60). However, IFNα production is delayed in pDCs stimulated with HIV-1 compared with these other ligands, with peak production not occurring until 24 to 48 hours after activation compared with just 4 hours with other viruses (19). Thus, induction of the refractory state after exposure to HIV-1 is also probably delayed until after peak IFNα production. The refractory state of pDCs in the blood before detectable viremia would then imply that these pDCs had already reached peak IFNα production in vivo. We found that there were increases in plasma IFNα2 levels in some participants at peak pDC frequency, an observation that may have been restricted by the limitations of weekly sampling to identify what would be expected to be a very low, transient increase in IFNα produced before viremia (29). However, the added observation that plasma IFNα2 levels negatively correlated with the ability of pDCs to phosphorylate IRF7 is consistent with a refractory state of the pDCs at this stage (step 5).
The observations made here were limited by the inclusion of participants enrolled in ATI studies performed in Thailand alone. Although it may be presumed that a similar increase in pDC frequency and decrease in function would be seen after ATI in other cohorts, further studies are needed to rule out any effects of genetic background or HIV-1 clade on these pDCs. We did include participants from RV397 who received VRC01 treatment during ATI in this study, after showing that there was no difference in changes in pDC frequency or phenotype compared with placebo-treated participants, but it is possible that VRC01 had an indirect effect on pDCs that we did not identify in our analysis. It has been shown that VRC01 did not alter IFNα production by pDCs exposed to HIV-1 in vitro, suggesting that VRC01 does not affect pDC sensing of HIV-1 (61, 62). However, pDCs can produce IFNα after sensing immune complexes (63), and although we did not see differences in pDC increases and activation between placebo- and VRCO1-treated participants, we cannot rule out the possibility that VRC01 formed immune complexes in vivo that could have enhanced viral sensing by pDCs in VRC01-treated participants. Though we combined participants from multiple ATI studies for this analysis, the number of participants who received no therapeutic intervention was limited, and analysis of pDC function and viral sensing in future ATI studies would further elucidate these findings. Sampling frequency was also a limitation of the ATI studies, particularly for the participants who had detectable VL within 2 weeks of ATI and thus only had one aviremic time point. Unfortunately, the current sampling regimen is already burdensome for participants, and nonhuman primate studies may be necessary for more frequent sampling to better characterize the innate response after ATI. The unavailability of tissue samples in this analysis prevented us from confirming that low-level viral replication in tissues was indeed the source of pDC activation. However, the identification of changes in blood pDCs as a potential indicator of HIV replication provides critical information to time the sampling in future studies in order to confirm when viral replication initiates in the tissue, investigate where early replication begins, and study the earliest immune responses to viral replication after ATI.
It has been estimated that viral replication is initiated once every 5 to 8 days after ATI, based on the time to detectable plasma viremia of more than 20 copies/mL (64). However, this calculation does not take into account the potential of local control of replicating virus by the immune system. One participant in RV397 did not have plasma VLs above 20 copies/mL until 42 weeks but showed an increased frequency of activated pDCs in the blood early after ATI, just before a low-level peripheral blood VL blip of 1.5 copies/mL. These data support our conjecture that pDC dynamics in the blood are an indicator of viral replication in the tissue and may be used to develop a more sensitive estimate of viral reactivation after ATI. Given the longer time intervals between sampling points during prolonged ATI, we were unable to determine whether multiple reactivation events occurred in the participants with the longest aviremia durations. It has previously been reported that the frequencies of CD30+ T cells increased months before viral detection in 2 participants who had an extended period of aviremia (65). Unfortunately, the levels of HLA-DR and CD38, which we used to measure T cell activation in the 3 ATI studies, did not change on the T cells before viral rebound, so we were unable to identify correlations between pDC activation and T cell activation in our analysis. Experiments in nonhuman primates may lead to a better understanding of the timing and mechanisms of these early innate immune responses to viral replication and to identifying aspects of the immune response that contribute to control of replication in individuals with extended aviremia.
Although our data suggest that pDCs are sensing low-level HIV-1 replication after ATI, other factors that could be causing pDC activation should also be considered. ART could have an off-target effect on pDCs, resulting in pDC activation after antiretroviral (ARV) clearance. However, given that pDC frequencies and function recover after ART initiation, this seems unlikely (13, 14). Further, participants who were on the same ART regimen at the time of ATI had varied times to pDC activation, from 1 to 3 weeks after ATI, suggesting that ARV clearance was not responsible for pDC activation. It is also conceivable that ARVs were having an indirect effect on the control of other viruses that were then reactivated after ARV clearance and caused pDC activation. However, a virome analysis to identify other viruses activated after ATI was outside the scope of this work. Finally, pDC activation could be a result of microbial translocation in the gut. As early as 2 weeks after SIV infection, damage to the intestinal epithelium results in microbial translocation that is associated with immune activation, including increased plasma IFNα levels and T cell activation (66, 67). Although low levels of IFNα were detected before viral detection in participants of the ATI cohorts, there was no evidence of T cell activation before viral detection (40). This does not exclude the potential for microbial translocation, but gut biopsies were not available before viral detection to investigate whether pDCs were being activated and mobilized as a result of microbial translocation.
Our data show that activated pDCs were mobilized to the blood before detectable viremia after ATI and that they entered a refractory state after in vivo production of IFNα. Whereas the early activation of pDCs in response to viral infections is well documented, less information is available on the mechanisms that blunt the pDC response to prevent excessive IFNα production. Decreased levels of pIRF7 after in vitro stimulation were associated with higher levels of plasma IFNα2, suggesting that the refractory state of the pDCs is a result of in vivo activation. Decreased expression of type I IFN genes before viral detection was associated with a longer time to reach a VL over 20 copies/mL, indicating that the refractory state of the pDCs may be indicative of a favorable immune response during ATI. Indeed, IRF7 levels decreased in pDCs over time after in vitro stimulation with the TLR7 agonist R848 (44). If the extent to which type I IFN gene expression is lost is a measure of the extent to which pDCs were activated in vivo, these data suggest that a robust and thorough activation of pDCs is associated with a longer time to detection of viremia after ATI.
Although the contribution of this pDC activation toward controlling viral replication is yet to be determined, it is known that the timing of IFNα production is important for viral clearance during acute viral infections. Early administration of IFNα protects against rectal SIV infection of rhesus macaques, but a delay in IFNα signaling results in more rapid disease progression (31). A delayed IFNα response to robust replication of LCMV CL-13 and SARS-CoV in mice is associated with a stronger cytokine storm and disease progression, but early introduction of IFNα is ameliorative (68, 69). Further, administration of pegylated IFNα just before and during ATI resulted in extended virus control and decreased integrated HIV DNA in a portion of recipients (70). Although many experiments suggest that pDCs are not the main producer of IFNα in later stages of acute and chronic HIV-1 and SIV infection (24, 59, 71, 72), an early role of pDC function before detection of viremia has not been extensively studied. Importantly, although pDCs are not the main producer of IFNα involved in the control of LCMV CL-13 viremia during the normal course of infection (68), knockout of the negative regulator OASL1 in LCMV CL-13–infected mice resulted in prolonged IFNα production by pDCs, which was associated with maintenance of CD8 T cell function and eventual LCMV CL-13 clearance (60). These studies show that early and sustained IFNα production is important for an effective immune response and viral clearance. Thus, although the early induction of IFNα production after ATI may have the potential to be beneficial, a blunting of the pDC response may hinder the mobilization of a successful immune response to clear the virus. Therefore, new strategies to boost and sustain the initial IFN-mediated response to replicating virus are needed to potentially sustain HIV-1 remission after treatment interruption.
Methods
ATI study design.
Twenty-nine individuals with HIV-1 who initiated ART during acute infection as part of the RV254 cohort in Thailand (ClinicalTrials.gov NCT00796146) and underwent ATI were included in this study (Table 1). Participants in the RV254 cohort initiated ART during the earliest stages of AHI, as staged according to a previously described sequential algorithm of pooled nucleic acid testing and immunoassays (73). Early initiation of treatment is associated with preserved immune function and decreased viral reservoir seeding, making individuals in the RV254 cohort ideal for enrollment in ATI studies to identify post-treatment controllers (74, 75). As such, participants included in this study were enrolled in 1 of 3 ATI studies: RV411, RV409, or RV397 (Supplemental Figure 1).
Eight participants who initiated ART during Fiebig stage I of AHI were enrolled in the RV411 study (ClinicalTrials.gov NCT02614950) (40). Participants in the RV411 study underwent ATI without additional interventions. Plasma VL was monitored weekly, except during weeks 2 to 6 after ATI when twice-weekly monitoring was performed. PBMC samples for RV411 participants were limited to the following time points: baseline, last aviremic point (VL <20 copies/mL), rebound, and 2 weeks after ART reinitiation. The RV409 study (ClinicalTrials.gov NCT02475915) included 15 participants who initiated ART during Fiebig stages III and IV of AHI, 14 of whom interrupted ART (our unpublished observations). During the 10 weeks before ATI, participants were treated with ART alone (n = 5) or with VHM to induce production of latent virus while maintaining ART (n = 10). Plasma VL was monitored weekly for the first 12 weeks after ATI and biweekly thereafter. PBMC samples were collected at baseline ATI, weekly through week 4, and biweekly thereafter. During the 10 weeks before ATI, PBMCs were collected biweekly. The RV397 study (ClinicalTrials.gov NCT02664415) included 18 participants who initiated ART during Fiebig stages I–III of AHI (41). Participants received intravenous injections of the broadly neutralizing Ab VRC01 (n = 13) or placebo (n = 5) every 3 weeks during ATI, starting on the day ART was ceased and continuing for 24 weeks or until ART resumption. Plasma VL was monitored weekly, with twice-weekly testing done during weeks 2 to 6 and 26 to 30. PBMC samples were collected from RV397 participants at baseline, weekly through week 3, and once every 3 weeks after that. All participants in these 3 studies experienced viral rebound, and ART was resumed after 2 consecutive VLs of greater than 1000 copies/mL. At the time of enrollment in the ATI studies, all participants had a CD4 count above 400 cells/μL and had maintained viral suppression (HIV-1 RNA <50 copies/mL) for at least 48 weeks.
RV409 participants receiving VHM and ART were not included in the current analysis because of detection of intervention-induced pDC activation before ATI. Furthermore, 2 participants in RV411 and 1 participant in the ART-only arm of RV409 were not included in any analysis because of the extended amount of time between the last aviremic sample collected and the detection of viral RNA in the blood (>2 weeks).
Measurement of HIV-1 RNA.
Plasma VL was measured quantitatively with the COBAS TaqMan HIV-1 Test (Roche Diagnostics) with a lower limit of detection of 20 copies/mL. Single-copy HIV-1 RNA levels were measured retrospectively in samples from RV397 using ultrasensitive hybrid real-time/digital PCR with a limit of detection of 0.45 copies/mL, as previously described (76, 77).
Abs and reagents for flow cytometry.
BV421-labeled anti-CCR7 (clone G043H7), phycoerythrin-Cy7–labeled (PE-Cy7–labeled) anti-CCR9 (clone L053E8), BV785-labeled anti-CD11c (clone 3.9), Pacific blue–labeled anti-CD11c (clone 3.9), BV510-labeled anti-CD14 (clone M5E2), BV785-labeled anti-CD14 (clone M5E2), BV510-labeled anti-CD19 (clone SJ25C1), PE/dazzle 594–labeled (PE/dazzle 594–labeled) anti-CD1c, allophycocyanin-Cy7–labeled (APC-Cy7–labeled) anti-CD29 (clone L161), BV510-labeled anti-CD3 (clone UCHT1), Alexa Fluor 700–labeled anti-CD303 (clone 201A), PE-labeled anti-CD303 (clone 201A), Alexa Fluor 700–labeled anti-CD45 (clone 2D1), BV510-labeled anti-CD56 (clone 5.1H11), BV605-labeled anti-CD86 (clone IT2.2), BV605-labeled anti-CXCR4 (clone 12G5), BV711-labeled anti–HLA-DR (clone L243), APC-labeled anti–integrin β7 (clone 29E.2A3), PE-Cy7–labeled anti-PDL1 (clone 29E.2A3), BV650-labeled anti–Tim-3 (clone F38-2E2), and PE-Cy7–labeled anti–TNF-α mAbs (clone MAb11) were obtained from BioLegend. PerCP-Cy5.5–labeled anti-CD123 (clone 7G3), APC-labeled anti-CD141 (clone 1A4), BUV496-labeled anti-CD16 (clone 3G8), BV650-labeled anti-CD3 (clone UCHT1), BUV395-labeled anti-CD40 (clone 5C3), BUV737-labeled anti-CD69 (clone FN50), BUV737-labeled anti-CD83 (clone HB15e), BUV395-labeled anti–Ki-67 (clone B56), AF488-labeled anti-IRF7 (pS477/pS479) (clone K47-671), GolgiPlug (catalog 555029), BD Perm/Wash Buffer (catalog 554723), and BD Phosflow Perm Buffer III (catalog 558050) were obtained from BD Biosciences. PE-labeled anti–p-NF-κB p65 (Ser536) (clone 93H1) and Alexa Fluor 647–labeled anti–p-SYK (Tyr352) (clone 65E4) Abs were obtained from Cell Signaling Technology. APC-labeled anti-IFNα Ab (clone LT27:295) was obtained from Miltenyi Biotec. The Foxp3/Transcription Factor Staining Buffer Set (catalog 50-112-8857) was purchased from Thermo Fisher Scientific (eBioscience).
Flow cytometric analysis.
Frozen PBMCs were thawed for phenotypic analysis by flow cytometry. Longitudinal samples from each participant were processed on the same day. Cells were stained with LIVE/DEAD (Thermo Fisher Scientific) for 10 minutes at room temperature before staining for surface markers at 4°C for 20 minutes. Cells were washed twice with PBS containing 2% FBS (wash buffer) before fixation with PBS containing 2% formaldehyde. For intracellular cytokine staining, cells were permeabilized in BD Perm/Wash Buffer at room temperature for 15 minutes after overnight fixation. The cells were stained with anti-IFNα and anti–TNF-α mAbs at room temperature for 30 minutes followed by 3 washes with Perm/Wash buffer. All cells were resuspended in PBS containing 2% formaldehyde before analysis on a LSR II (BD Biosciences). All flow cytometric data were analyzed with FlowJo software, version 10. For measurement of SYK phosphorylation, PBMCs were stained immediately after thawing. For measurement of p-IRF7 and p–NF-κB, PBMCs were stimulated with 2.5 μg/mL imiquimod as described below. Cells were stained with LIVE/DEAD and surface markers as described above. Cells were fixed in prewarmed PBS containing 2% formaldehyde and incubated for 10 minutes at 37°C. Cells were then permeabilized in BD Phosflow Perm Buffer III and incubated for 20 minutes on ice. Next, cells were washed 3 times in PBS containing 10% FBS, either immediately or after overnight incubation at –20°C. After incubation in wash buffer for 30 minutes on ice, cells were stained for 20 minutes at room temperature followed by 2 washes with wash buffer. For intracellular staining of Ki-67, cells were stained for surface markers as described above, followed by fixation and permeabilization with the Foxp3/Transcription Factor Staining Buffer Set. The cells were stained with anti–Ki-67 mAbs for 30 minutes at room temperature followed by washes with Foxp3 permeabilization buffer and wash buffer, respectively
Plasma SDF-1α levels.
Plasma SDF-1α levels were measured using Luminex technology with a ProcartaPlex Multiplex Immunoassay (MXH49YW, Life Technologies, Thermo Fisher Scientific). Samples were run according to the manufacturer’s instructions, and cytokine standards were provided by the manufacturer. A Bioplex-200 system was used to acquire samples, and the data were analyzed using BioPlex Manager Software (Bio-Rad Laboratories).
pDC stimulation.
Thawed PBMCs were reconstituted at 2 × 106 cells/mL in RPMI 1640 supplemented with 10% FBS, penicillin/streptomycin, and 20 ng/mL IL-3 (PeproTech) and then allowed to rest for 5 hours. Cells were stimulated with 2.5 μg/mL imiquimod (InvivoGen) or left in media alone for 6 hours, with GolgiPlug added for the last 4 hours of stimulation.
Plasma IFNα2 levels.
Plasma IFNα2 levels were measured using the SIMOA Human IFNα Kit (Quanterix) according to the manufacturer’s instructions. The lower limit of quantification of this assay is 4.7 fg/mL.
Quantitative reverse transcription PCR.
Thawed PBMCs were stained for flow cytometric sorting on a BD FACSAria (BD Biosciences). Replicates of 250 pDCs (Lin–HLA-DR+CD11c–CD123+CD303+) or 1000 mDCs (Lin–HLA-DR+CD11c +CD303–CD1c+) were sorted into 10 μL SuperScript III Platinum Taq One-Step qRT-PCR Master Mix (Invitrogen, Thermo Fisher Scientific) in 0.2-mL PCR tubes for immediate lysis. Direct reverse transcription (RT) and PCR preamplification with gene-specific primers was performed as previously described (78). Preamplification product was diluted 5-fold before use for multiplexed quantitative reverse RT-PCR (qRT-PCR) on a Fluidigm BioMark HD system (Fluidigm). Gene expression was measured with the 96.96 BioMark Dynamic Array Chip for Gene Expression (Fluidigm) using TaqMan assays (Life Technologies, Thermo Fisher Scientific) (Supplemental Table 1) according to the manufacturer’s instructions and as previously described (78). Data were analyzed with JMP Software (SAS Inc.).
Statistics.
All summary data are in the form of median values unless otherwise noted. Samples were excluded from analysis if taken more than 2 weeks before detection of viral RNA (last aviremic point) or the week of peak pDC frequency (week prior point). Statistical analyses were performed using the Wilcoxon matched-pairs, signed-rank test for comparisons between 2 visits, and the Mann-Whitney U test for treatment group comparisons. Gene expression data were analyzed with the 1-sample Wilcoxon test to identify changes in expression that were significantly different from 0. Correlations were performed with the nonparametric Spearman’s test. When multiple comparisons were made for gene expression or correlations, the Benjamini-Hochberg procedure was used to correct for a FDR of 10%. All statistical analyses were performed using GraphPad Prism (GraphPad Software), and significance was defined as a P value of less than 0.05 for 2-sided testing.
Study approval.
Informed consent was obtained from all participants before inclusion in the studies. All studies were approved by the IRBs of Chulalongkorn University and the Walter Reed Army Institute of Research. The investigators have adhered to the policies for protection of human subjects as prescribed in Army regulation 70-25 (AR-70-25).
Author contributions
JLM designed and performed the experiments, analyzed the data, and wrote the manuscript. HT helped design and perform the experiments and edited the manuscript. RM performed the Luminex assay. DJC, EK, TAC, CS, S. Puttamaswin, KB, and RT managed participant recruitment and follow-up in the studies. DJC, EK, and TAC edited the manuscript. S. Pinyakorn provided help with statistical analyses. RLT provided supervision. LF provided supervision and edited the manuscript. VRP and FM provided resources. DLB provided resources and edited the manuscript. EKH provided conceptual advice and edited the manuscript. SRL provided conceptual advice and edited the manuscript. PP, MLR, NLM, MDS, and NP provided support for the clinical studies. JA designed the clinical studies, provided conceptual advice, and edited the manuscript. LT designed the experiments, analyzed the data, and wrote the manuscript.
Supplementary Material
Acknowledgments
We thank our study participants and staff at the TRC-ARC, Chulalongkorn University, and the AFRIMS for their valuable contributions to this study. We wish to thank M. Creegan for sorting samples. We are grateful to the Thai Government Pharmaceutical Organization, Gilead, Merck and ViiV Healthcare for providing antiretroviral agents for these studies. This study was supported by the NIH (R01AI108433 and R01MH106466) and a cooperative agreement (W81XWH-07-2-0067 and W81XWH-11-2-0174) between the HJF and the US Department of Defense. The views expressed are those of the authors and should not be construed to represent the positions of the US Army, the Department of Defense, the NIH, the Department of Health and Human Services, or HJF. See Supplemental Acknowledgments for details on the RV397, RV411, RV409, and RV254 study groups.
Version 1. 02/04/2020
In-Press Preview
Version 2. 04/27/2020
Electronic publication
Version 3. 06/01/2020
Print issue publication
Footnotes
JLM, HT, and LT’s present address is: Vaccine and Gene Therapy Institute, Oregon Health and Science University, Hillsboro, Oregon, USA.
JA’s present address is: Bill and Melinda Gates Medical Research Institute, Cambridge, Massachusetts, USA.
Conflict of interest: JA has received honoraria from Merck, ViiV Healthcare, Roche, AbbVie, and Gilead for her participation in advisory meetings.
Copyright: © 2020, American Society for Clinical Investigation.
Reference information: J Clin Invest. 2020;130(6):2845–2858.https://doi.org/10.1172/JCI130597.
See the related Commentary at The immune response fails to control HIV early in initial virus spread.
Contributor Information
Julie L. Mitchell, Email: mijulie@ohsu.edu.
Hiroshi Takata, Email: takatah@ohsu.edu.
Roshell Muir, Email: Roshell.Muir@DrexelMed.edu.
Donn J. Colby, Email: donn.c@searchthailand.org.
Trevor A. Crowell, Email: tcrowell@hivresearch.org.
Carlo Sacdalan, Email: carlo.s@searchthailand.org.
Suteeraporn Pinyakorn, Email: spinyakorn@hivresearch.org.
Khunthalee Benjapornpong, Email: Khunthalee.b@searchthailand.org.
Rapee Trichavaroj, Email: rapeet@hiv-th.org.
Randall L. Tressler, Email: randall.tressler@nih.gov.
Lawrence Fox, Email: lfox@niaid.nih.gov.
Victoria R. Polonis, Email: vpolonis@hivresearch.org.
Diane L. Bolton, Email: dbolton@hivresearch.org.
Frank Maldarelli, Email: fmalli@mail.nih.gov.
Sharon R. Lewin, Email: sharon.lewin@unimelb.edu.au.
Elias K. Haddad, Email: ee336@drexel.edu.
Praphan Phanuphak, Email: Praphan.P@chula.ac.th.
Merlin L. Robb, Email: mrobb@hivresearch.org.
Nelson L. Michael, Email: nmichael@hivresearch.org.
Mark de Souza, Email: markdes@searchthailand.org.
Nittaya Phanuphak, Email: nittaya.p@prevention-trcarc.org.
Jintanat Ananworanich, Email: jananworanich@hivresearch.org.
Lydie Trautmann, Email: ltrautmann@hivresearch.org.
References
- 1.Siegal FP, et al. The nature of the principal type 1 interferon-producing cells in human blood. Science. 1999;284(5421):1835–1837. doi: 10.1126/science.284.5421.1835. [DOI] [PubMed] [Google Scholar]
- 2.Fitzgerald-Bocarsly P. Human natural interferon-alpha producing cells. Pharmacol Ther. 1993;60(1):39–62. doi: 10.1016/0163-7258(93)90021-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rasmussen SB, et al. Type I interferon production during herpes simplex virus infection is controlled by cell-type-specific viral recognition through toll-like receptor 9, the mitochondrial antiviral signaling protein pathway, and novel recognition systems. J Virol. 2007;81(24):13315–13324. doi: 10.1128/JVI.01167-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tabeta K, et al. Toll-like receptors 9 and 3 as essential components of innate immune defense against mouse cytomegalovirus infection. Proc Natl Acad Sci USA. 2004;101(10):3516–3521. doi: 10.1073/pnas.0400525101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lund J, Sato A, Akira S, Medzhitov R, Iwasaki A. Toll-like receptor 9-mediated recognition of Herpes simplex virus-2 by plasmacytoid dendritic cells. J Exp Med. 2003;198(3):513–520. doi: 10.1084/jem.20030162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kerkmann M, et al. Activation with CpG-A and CpG-B oligonucleotides reveals two distinct regulatory pathways of type I IFN synthesis in human plasmacytoid dendritic cells. J Immunol. 2003;170(9):4465–4474. doi: 10.4049/jimmunol.170.9.4465. [DOI] [PubMed] [Google Scholar]
- 7.Isaacs A, Lindenmann J. Virus interference. I. The interferon. Proc R Soc Lond, B, Biol Sci. 1957;147(927):258–267. doi: 10.1098/rspb.1957.0048. [DOI] [PubMed] [Google Scholar]
- 8.Reizis B. Plasmacytoid dendritic cells: development, regulation, and runction. Immunity. 2019;50(1):37–50. doi: 10.1016/j.immuni.2018.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Soumelis V, et al. Depletion of circulating natural type 1 interferon-producing cells in HIV-infected AIDS patients. Blood. 2001;98(4):906–912. doi: 10.1182/blood.V98.4.906. [DOI] [PubMed] [Google Scholar]
- 10.Feldman S, et al. Decreased interferon-alpha production in HIV-infected patients correlates with numerical and functional deficiencies in circulating type 2 dendritic cell precursors. Clin Immunol. 2001;101(2):201–210. doi: 10.1006/clim.2001.5111. [DOI] [PubMed] [Google Scholar]
- 11.Pacanowski J, et al. Reduced blood CD123+ (lymphoid) and CD11c+ (myeloid) dendritic cell numbers in primary HIV-1 infection. Blood. 2001;98(10):3016–3021. doi: 10.1182/blood.V98.10.3016. [DOI] [PubMed] [Google Scholar]
- 12.Donaghy H, et al. Loss of blood CD11c(+) myeloid and CD11c(-) plasmacytoid dendritic cells in patients with HIV-1 infection correlates with HIV-1 RNA virus load. Blood. 2001;98(8):2574–2576. doi: 10.1182/blood.V98.8.2574. [DOI] [PubMed] [Google Scholar]
- 13.Killian MS, Fujimura SH, Hecht FM, Levy JA. Similar changes in plasmacytoid dendritic cell and CD4 T-cell counts during primary HIV-1 infection and treatment. AIDS. 2006;20(9):1247–1252. doi: 10.1097/01.aids.0000232231.34253.bd. [DOI] [PubMed] [Google Scholar]
- 14.Kamga I, et al. Type I interferon production is profoundly and transiently impaired in primary HIV-1 infection. J Infect Dis. 2005;192(2):303–310. doi: 10.1086/430931. [DOI] [PubMed] [Google Scholar]
- 15.Lehmann C, et al. Increased interferon alpha expression in circulating plasmacytoid dendritic cells of HIV-1-infected patients. J Acquir Immune Defic Syndr. 2008;48(5):522–530. doi: 10.1097/QAI.0b013e31817f97cf. [DOI] [PubMed] [Google Scholar]
- 16.Martinson JA, et al. Dendritic cells from HIV-1 infected individuals are less responsive to toll-like receptor (TLR) ligands. Cell Immunol. 2007;250(1-2):75–84. doi: 10.1016/j.cellimm.2008.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang J, et al. Dendritic cell dysfunction during primary HIV-1 infection. J Infect Dis. 2011;204(10):1557–1562. doi: 10.1093/infdis/jir616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tilton JC, et al. Human immunodeficiency virus viremia induces plasmacytoid dendritic cell activation in vivo and diminished alpha interferon production in vitro. J Virol. 2008;82(8):3997–4006. doi: 10.1128/JVI.01545-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lo CC, et al. HIV delays IFN-α production from human plasmacytoid dendritic cells and is associated with SYK phosphorylation. PLoS ONE. 2012;7(5):e37052. doi: 10.1371/journal.pone.0037052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dhamanage A, Thakar M, Paranjape R. Human immunodeficiency virus-1 impairs IFN-alpha production induced by TLR-7 agonist in plasmacytoid dendritic cells. Viral Immunol. 2017;30(1):28–34. doi: 10.1089/vim.2016.0084. [DOI] [PubMed] [Google Scholar]
- 21.Dhamanage AS, Thakar MR, Paranjape RS. HIV-1-mediated suppression of IFN-α production is associated with Inhibition of IRF-7 translocation and PI3K/akt pathway in plasmacytoid dendritic cells. AIDS Res Hum Retroviruses. 2019;35(1):40–48. doi: 10.1089/aid.2018.0136. [DOI] [PubMed] [Google Scholar]
- 22.Schwartz JA, et al. Tim-3 is a marker of plasmacytoid dendritic cell dysfunction during HIV infection and is sssociated with the recruitment of IRF7 and p85 into lysosomes and with the Submembrane Displacement of TLR9. J Immunol. 2017;198(8):3181–3194. doi: 10.4049/jimmunol.1601298. [DOI] [PubMed] [Google Scholar]
- 23.Donhauser N, et al. Chronic immune activation in HIV-1 infection contributes to reduced interferon alpha production via enhanced CD40:CD40 ligand interaction. PLoS ONE. 2012;7(3):e33925. doi: 10.1371/journal.pone.0033925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kader M, et al. Blocking TLR7- and TLR9-mediated IFN-α production by plasmacytoid dendritic cells does not diminish immune activation in early SIV infection. PLoS Pathog. 2013;9(7):e1003530. doi: 10.1371/journal.ppat.1003530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bruel T, et al. Plasmacytoid dendritic cell dynamics tune interferon-alfa production in SIV-infected cynomolgus macaques. PLoS Pathog. 2014;10(1):e1003915. doi: 10.1371/journal.ppat.1003915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brown KN, Wijewardana V, Liu X, Barratt-Boyes SM. Rapid influx and death of plasmacytoid dendritic cells in lymph nodes mediate depletion in acute simian immunodeficiency virus infection. PLoS Pathog. 2009;5(5):e1000413. doi: 10.1371/journal.ppat.1000413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li Q, et al. Glycerol monolaurate prevents mucosal SIV transmission. Nature. 2009;458(7241):1034–1038. doi: 10.1038/nature07831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Malleret B, et al. Primary infection with simian immunodeficiency virus: plasmacytoid dendritic cell homing to lymph nodes, type I interferon, and immune suppression. Blood. 2008;112(12):4598–4608. doi: 10.1182/blood-2008-06-162651. [DOI] [PubMed] [Google Scholar]
- 29.Jacquelin B, et al. Nonpathogenic SIV infection of African green monkeys induces a strong but rapidly controlled type I IFN response. J Clin Invest. 2009;119(12):3544–3555. doi: 10.1172/JCI40093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stacey AR, et al. Induction of a striking systemic cytokine cascade prior to peak viremia in acute human immunodeficiency virus type 1 infection, in contrast to more modest and delayed responses in acute hepatitis B and C virus infections. J Virol. 2009;83(8):3719–3733. doi: 10.1128/JVI.01844-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sandler NG, et al. Type I interferon responses in rhesus macaques prevent SIV infection and slow disease progression. Nature. 2014;511(7511):601–605. doi: 10.1038/nature13554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Veazey RS, et al. Prevention of SHIV transmission by topical IFN-β treatment. Mucosal Immunol. 2016;9(6):1528–1536. doi: 10.1038/mi.2015.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sedaghat AR, et al. Chronic CD4+ T-cell activation and depletion in human immunodeficiency virus type 1 infection: type I interferon-mediated disruption of T-cell dynamics. J Virol. 2008;82(4):1870–1883. doi: 10.1128/JVI.02228-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rempel H, Sun B, Calosing C, Pillai SK, Pulliam L. Interferon-alpha drives monocyte gene expression in chronic unsuppressed HIV-1 infection. AIDS. 2010;24(10):1415–1423. doi: 10.1097/QAD.0b013e32833ac623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fernandez S, et al. CD4+ T-cell deficiency in HIV patients responding to antiretroviral therapy is associated with increased expression of interferon-stimulated genes in CD4+ T cells. J Infect Dis. 2011;204(12):1927–1935. doi: 10.1093/infdis/jir659. [DOI] [PubMed] [Google Scholar]
- 36.Hardy GA, et al. Interferon-α is the primary plasma type-I IFN in HIV-1 infection and correlates with immune activation and disease markers. PLoS ONE. 2013;8(2):e56527. doi: 10.1371/journal.pone.0056527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sáez-Cirión A, et al. Post-treatment HIV-1 controllers with a long-term virological remission after the interruption of early initiated antiretroviral therapy ANRS VISCONTI Study. PLoS Pathog. 2013;9(3):e1003211. doi: 10.1371/journal.ppat.1003211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ananworanich J, et al. Virological and immunological characteristics of HIV-infected individuals at the earliest stage of infection. J Virus Erad. 2016;2:43–48. doi: 10.1016/S2055-6640(20)30688-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fiebig EW, et al. Dynamics of HIV viremia and antibody seroconversion in plasma donors: implications for diagnosis and staging of primary HIV infection. AIDS. 2003;17(13):1871–1879. doi: 10.1097/00002030-200309050-00005. [DOI] [PubMed] [Google Scholar]
- 40.Colby DJ, et al. Rapid HIV RNA rebound after antiretroviral treatment interruption in persons durably suppressed in Fiebig I acute HIV infection. Nat Med. 2018;24(7):923–926. doi: 10.1038/s41591-018-0026-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Crowell TA, et al. Safety and efficacy of VRC01 broadly neutralising antibodies in adults with acutely treated HIV (RV397): a phase 2, randomised, double-blind, placebo-controlled trial. Lancet HIV. 2019;6(5):e297–e306. doi: 10.1016/S2352-3018(19)30053-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ahonen CL, et al. Dendritic cell maturation and subsequent enhanced T-cell stimulation induced with the novel synthetic immune response modifier R-848. Cell Immunol. 1999;197(1):62–72. doi: 10.1006/cimm.1999.1555. [DOI] [PubMed] [Google Scholar]
- 43.Loré K, et al. Toll-like receptor ligands modulate dendritic cells to augment cytomegalovirus- and HIV-1-specific T cell responses. J Immunol. 2003;171(8):4320–4328. doi: 10.4049/jimmunol.171.8.4320. [DOI] [PubMed] [Google Scholar]
- 44.O’Brien M, et al. Spatiotemporal trafficking of HIV in human plasmacytoid dendritic cells defines a persistently IFN-α-producing and partially matured phenotype. J Clin Invest. 2011;121(3):1088–1101. doi: 10.1172/JCI44960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hemmi H, et al. Small anti-viral compounds activate immune cells via the TLR7 MyD88-dependent signaling pathway. Nat Immunol. 2002;3(2):196–200. doi: 10.1038/ni758. [DOI] [PubMed] [Google Scholar]
- 46.Berlin C, et al. Alpha 4 beta 7 integrin mediates lymphocyte binding to the mucosal vascular addressin MAdCAM-1. Cell. 1993;74(1):185–195. doi: 10.1016/0092-8674(93)90305-A. [DOI] [PubMed] [Google Scholar]
- 47.Diacovo TG, Blasius AL, Mak TW, Cella M, Colonna M. Adhesive mechanisms governing interferon-producing cell recruitment into lymph nodes. J Exp Med. 2005;202(5):687–696. doi: 10.1084/jem.20051035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Barblu L, et al. Plasmacytoid dendritic cells (pDCs) from HIV controllers produce interferon-α and differentiate into functional killer pDCs under HIV activation. J Infect Dis. 2012;206(5):790–801. doi: 10.1093/infdis/jis384. [DOI] [PubMed] [Google Scholar]
- 49.Ito T, Kanzler H, Duramad O, Cao W, Liu YJ. Specialization, kinetics, and repertoire of type 1 interferon responses by human plasmacytoid predendritic cells. Blood. 2006;107(6):2423–2431. doi: 10.1182/blood-2005-07-2709. [DOI] [PubMed] [Google Scholar]
- 50.Sachdeva N, Asthana V, Brewer TH, Garcia D, Asthana D. Impaired restoration of plasmacytoid dendritic cells in HIV-1-infected patients with poor CD4 T cell reconstitution is associated with decrease in capacity to produce IFN-alpha but not proinflammatory cytokines. J Immunol. 2008;181(4):2887–2897. doi: 10.4049/jimmunol.181.4.2887. [DOI] [PubMed] [Google Scholar]
- 51.Chehimi J, et al. Inability of plasmacytoid dendritic cells to directly lyse HIV-infected autologous CD4+ T cells despite induction of tumor necrosis factor-related apoptosis-inducing ligand. J Virol. 2010;84(6):2762–2773. doi: 10.1128/JVI.01350-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Joos B, et al. HIV rebounds from latently infected cells, rather than from continuing low-level replication. Proc Natl Acad Sci USA. 2008;105(43):16725–16730. doi: 10.1073/pnas.0804192105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Horiike M, et al. Lymph nodes harbor viral reservoirs that cause rebound of plasma viremia in SIV-infected macaques upon cessation of combined antiretroviral therapy. Virology. 2012;423(2):107–118. doi: 10.1016/j.virol.2011.11.024. [DOI] [PubMed] [Google Scholar]
- 54.Perreau M, et al. Follicular helper T cells serve as the major CD4 T cell compartment for HIV-1 infection, replication, and production. J Exp Med. 2013;210(1):143–156. doi: 10.1084/jem.20121932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Estes JD, et al. Defining total-body AIDS-virus burden with implications for curative strategies. Nat Med. 2017;23(11):1271–1276. doi: 10.1038/nm.4411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Vanbervliet B, et al. The inducible CXCR3 ligands control plasmacytoid dendritic cell responsiveness to the constitutive chemokine stromal cell-derived factor 1 (SDF-1)/CXCL12. J Exp Med. 2003;198(5):823–830. doi: 10.1084/jem.20020437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Krug A, et al. IFN-producing cells respond to CXCR3 ligands in the presence of CXCL12 and secrete inflammatory chemokines upon activation. J Immunol. 2002;169(11):6079–6083. doi: 10.4049/jimmunol.169.11.6079. [DOI] [PubMed] [Google Scholar]
- 58.Penna G, Sozzani S, Adorini L. Cutting edge: selective usage of chemokine receptors by plasmacytoid dendritic cells. J Immunol. 2001;167(4):1862–1866. doi: 10.4049/jimmunol.167.4.1862. [DOI] [PubMed] [Google Scholar]
- 59.Jochems SP, et al. Modulation of type I interferon-associated viral sensing during acute simian immunodeficiency virus infection in African green monkeys. J Virol. 2015;89(1):751–762. doi: 10.1128/JVI.02430-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lee MS, Park CH, Jeong YH, Kim YJ, Ha SJ. Negative regulation of type I IFN expression by OASL1 permits chronic viral infection and CD8+ T-cell exhaustion. PLoS Pathog. 2013;9(7):e1003478. doi: 10.1371/journal.ppat.1003478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lederle A, et al. Neutralizing antibodies inhibit HIV-1 infection of plasmacytoid dendritic cells by an FcγRIIa independent mechanism and do not diminish cytokines production. Sci Rep. 2014;4:5845. doi: 10.1038/srep05845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Su B, et al. Broadly neutralizing antibody VRC01 prevents HIV-1 transmission from plasmacytoid dendritic cells to CD4 T lymphocytes. J Virol. 2014;88(18):10975–10981. doi: 10.1128/JVI.01748-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lövgren T, Eloranta ML, Båve U, Alm GV, Rönnblom L. Induction of interferon-alpha production in plasmacytoid dendritic cells by immune complexes containing nucleic acid released by necrotic or late apoptotic cells and lupus IgG. Arthritis Rheum. 2004;50(6):1861–1872. doi: 10.1002/art.20254. [DOI] [PubMed] [Google Scholar]
- 64.Pinkevych M, et al. Correction: HIV reactivation from latency after treatment interruption occurs on average every 5–8 days — implications for HIV remission. PLoS Pathog. 2016;12(8):e1005745. doi: 10.1371/journal.ppat.1005745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Henrich TJ, et al. HIV-1 persistence following extremely early initiation of antiretroviral therapy (ART) during acute HIV-1 infection: an observational study. PLoS Med. 2017;14(11):e1002417. doi: 10.1371/journal.pmed.1002417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Brenchley JM, et al. Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nat Med. 2006;12(12):1365–1371. doi: 10.1038/nm1511. [DOI] [PubMed] [Google Scholar]
- 67.Estes JD, et al. Damaged intestinal epithelial integrity linked to microbial translocation in pathogenic simian immunodeficiency virus infections. PLoS Pathog. 2010;6(8):e1001052. doi: 10.1371/journal.ppat.1001052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sullivan BM, Teijaro JR, de la Torre JC, Oldstone MB. Early virus-host interactions dictate the course of a persistent infection. PLoS Pathog. 2015;11(1):e1004588. doi: 10.1371/journal.ppat.1004588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Channappanavar R, et al. Dysregulated type I interferon and inflammatory monocyte-macrophage responses cause lethal pneumonia in SARS-CoV-infected mice. Cell Host Microbe. 2016;19(2):181–193. doi: 10.1016/j.chom.2016.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Azzoni L, et al. Pegylated Interferon alfa-2a monotherapy results in suppression of HIV type 1 replication and decreased cell-associated HIV DNA integration. J Infect Dis. 2013;207(2):213–222. doi: 10.1093/infdis/jis663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wonderlich ER, Wijewardana V, Liu X, Barratt-Boyes SM. Virus-encoded TLR ligands reveal divergent functional responses of mononuclear phagocytes in pathogenic simian immunodeficiency virus infection. J Immunol. 2013;190(5):2188–2198. doi: 10.4049/jimmunol.1201645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Nascimbeni M, et al. Plasmacytoid dendritic cells accumulate in spleens from chronically HIV-infected patients but barely participate in interferon-alpha expression. Blood. 2009;113(24):6112–6119. doi: 10.1182/blood-2008-07-170803. [DOI] [PubMed] [Google Scholar]
- 73.Ananworanich J, et al. A novel acute HIV infection staging system based on 4th generation immunoassay. Retrovirology. 2013;10:56. doi: 10.1186/1742-4690-10-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Takata MA, Soll SJ, Emery A, Blanco-Melo D, Swanstrom R, Bieniasz PD. Global synonymous mutagenesis identifies cis-acting RNA elements that regulate HIV-1 splicing and replication. PLoS Pathog. 2018;14(1):e1006824. doi: 10.1371/journal.ppat.1006824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Schuetz A, et al. Initiation of ART during early acute HIV infection preserves mucosal Th17 function and reverses HIV-related immune activation. PLoS Pathog. 2014;10(12):e1004543. doi: 10.1371/journal.ppat.1004543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Palmer S, et al. New real-time reverse transcriptase-initiated PCR assay with single-copy sensitivity for human immunodeficiency virus type 1 RNA in plasma. J Clin Microbiol. 2003;41(10):4531–4536. doi: 10.1128/JCM.41.10.4531-4536.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Somsouk M, et al. The immunologic effects of mesalamine in treated HIV-infected individuals with incomplete CD4+ T cell recovery: a randomized crossover trial. PLoS ONE. 2014;9(12):e116306. doi: 10.1371/journal.pone.0116306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bolton DL, McGinnis K, Finak G, Chattopadhyay P, Gottardo R, Roederer M. Combined single-cell quantitation of host and SIV genes and proteins ex vivo reveals host-pathogen interactions in individual cells. PLoS Pathog. 2017;13(6):e1006445. doi: 10.1371/journal.ppat.1006445. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.