

Abstract
Mendelian susceptibility to mycobacterial disease (MSMD) is characterized by a selective predisposition to clinical disease caused by the Bacille Calmette-Guérin (BCG) vaccine and environmental mycobacteria. The known genetic etiologies of MSMD are inborn errors of IFN-γ immunity due to mutations of 15 genes controlling the production of or response to IFN-γ. Since the first MSMD-causing mutations were reported in 1996, biallelic mutations in the genes encoding IFN-γ receptor 1 (IFN-γR1) and IFN-γR2 have been reported in many patients of diverse ancestries. Surprisingly, mutations of the gene encoding the IFN-γ cytokine itself have not been reported, raising the remote possibility that there might be other agonists of the IFN-γ receptor. We describe 2 Lebanese cousins with MSMD, living in Kuwait, who are both homozygous for a small deletion within the IFNG gene (c.354_357del), causing a frameshift that generates a premature stop codon (p.T119Ifs4*). The mutant allele is loss of expression and loss of function. We also show that the patients’ herpesvirus Saimiri–immortalized T lymphocytes did not produce IFN-γ, a phenotype that can be rescued by retrotransduction with WT IFNG cDNA. The blood T and NK lymphocytes from these patients also failed to produce and secrete detectable amounts of IFN-γ. Finally, we show that human IFNG has evolved under stronger negative selection than IFNGR1 or IFNGR2, suggesting that it is less tolerant to heterozygous deleterious mutations than IFNGR1 or IFNGR2. This may account for the rarity of patients with autosomal-recessive, complete IFN-γ deficiency relative to patients with complete IFN-γR1 and IFN-γR2 deficiencies.
Keywords: Genetics, Immunology
Keywords: Genetic diseases
Introduction
Mendelian susceptibility to mycobacterial disease (MSMD) (OMIM #209950) is characterized by a predisposition to severe disease caused by weakly virulent mycobacteria, including Mycobacterium bovis Bacille Calmette-Guérin (BCG) vaccines and environmental mycobacteria (1, 2). Patients are typically otherwise healthy, normally resistant to most other microbes, and have no overt hematological or immunological abnormalities in routine tests. Nevertheless, some patients can suffer from infections due to more virulent mycobacteria, such as Mycobacterium tuberculosis (3). Moreover, extraintestinal, nontyphoidal salmonellosis has also been documented in approximately half of the patients (2, 4–6). Other infections caused by intramacrophagic bacteria, fungi, and parasites occur more rarely (2, 7). In particular, chronic mucocutaneous candidiasis can be seen in patients with specific genetic etiologies (8). MSMD was first described in the 1950s, with various clinical manifestations ranging from recurrent localized disease to progressive disseminated infections (9–11). The clinical outcome and response to treatment also vary considerably from patient to patient. The overall prevalence of MSMD has been estimated at approximately 1 of 50,000 births, and cases have been diagnosed or reported in almost all countries. The occurrence of familial cases segregating as Mendelian disorders led to this condition being named MSMD (12).
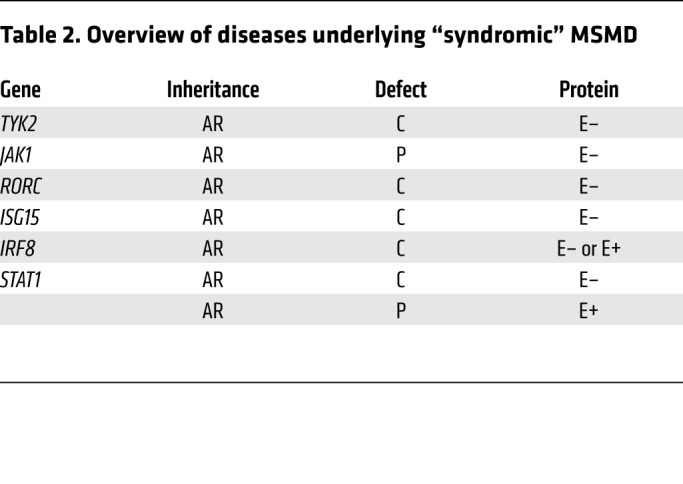
The first genetic etiology was discovered in 1996 in patients with IFN-γ receptor 1 (IFN-γR1) deficiency (13, 14). Causal mutations have been identified in 10 autosomal (IFNGR1, IFNGR2, STAT1, IRF8, IL12RB1, IL12RB2, IL23R, IL12B, SPPL2A, and TYK2) and 2 X-linked (XR) (NEMO and CYBB) genes (2, 7, 15–18). Allelic heterogeneity at these loci has led to the definition of 23 genetic etiologies (Table 1 and refs. 13–31). In addition, autosomal-recessive (AR) complete STAT1, IRF8, TYK2, ISG15, and RORγ/RORγT and AR partial JAK1 deficiencies almost invariably underlie mycobacterial diseases in the context of other clinical phenotypes; these deficiencies can be considered syndromic forms of MSMD (Table 2 and refs. 32–36). Collectively, there are, thus, 15 MSMD-causing genes and 30 genetic etiologies.
Table 1. Overview of diseases underlying “isolated” MSMD.
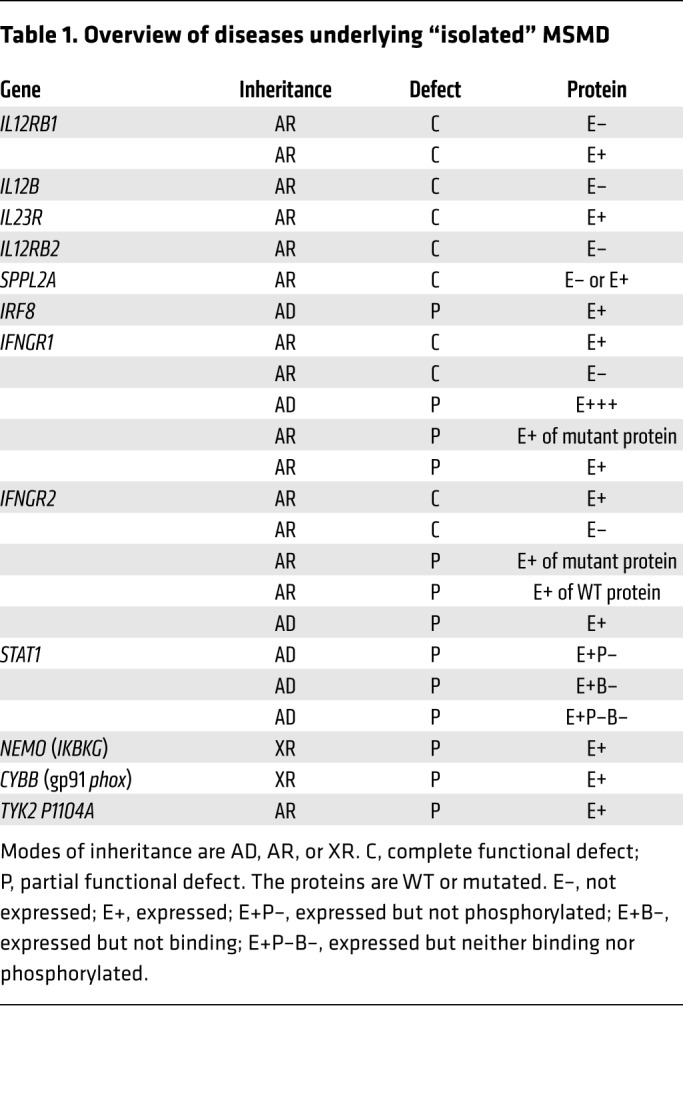
Table 2. Overview of diseases underlying “syndromic” MSMD.
Despite their high levels of locus and allelic heterogeneity, isolated and syndromic MSMDs display striking physiological homogeneity: all known genetic etiologies are inborn errors of IFN-γ immunity. Some genetic etiologies (ISG15, IL-12Rβ1, IL-12Rβ2, IL-23R, TYK2, RORγ/RORγT, and IL-12p40 deficiencies) impair the production of IFN-γ, whereas others (STAT1, IFN-γR1, IFN-γR2, JAK1, and gp91phox deficiencies) impair cellular responses to this cytokine. A few disrupt both the induction of IFN-γ and the response pathway for this cytokine (IRF8, SPPL2A, and NEMO deficiencies). IFN-γ is mostly secreted by NK cells and T lymphocytes (37). First identified in 1966 as a leukocyte antiviral IFN (38), IFN-γ was shown in 1983 to be the macrophage-activating factor (39). Mice lacking IFN-γ were shown to be prone to infection by both viruses and intramacrophagic pathogens (40–43). The study of MSMD has shown that human IFN-γ is much more a macrophage-activating factor than an antiviral IFN. The genetic etiology and immunological basis of MSMD remain unknown in about half of the patients identified to date.
Surprisingly, germline mutations of the IFN-γ cytokine itself have not been reported, despite the identification of hundreds of patients with mutations of the genes encoding the chains of its receptor (refs. 7, 13, 14, 16, 23–25, 31, 34, 44–56 and our unpublished data). There are 10 types of IFN-γR1 and IFN-γR2 deficiencies (Table 1), including recessive and dominant disorders, complete and partial functional defects, forms without surface receptors or with dysfunctional surface receptors, and different types of mechanisms by which mutations disrupt the function of surface receptors (7, 13, 14, 16, 23–25, 31, 34, 44–56). By contrast, in 25 years of study, to our knowledge, no patient has yet been reported to have any form of inherited IFN-γ deficiency. This surprising observation raised the remote but finite possibility that there might be other agonists of the human IFN-γ receptor. We set out to search for mutations of IFNG in patients with unexplained MSMD referred to our laboratory.
Results
Clinical description of 2 related patients with MSMD.
We studied 2 patients from 2 related consanguineous families from Lebanon living in Kuwait (Figure 1A). Patient 1 (P1, IV.3) was the third child born to consanguineous parents (second degree) of Arab origin. She was born at full term, with no perinatal complications. The child was vaccinated with BCG at the age of 3 months. Three weeks later, she developed a prolonged fever associated with a large left axillary mass (8 × 8 cm) that was erythematous and tender. She also had hepatosplenomegaly, maculopapular rashes, occasional diarrhea, and failure to thrive. The infant was treated with multiple courses of antibiotics, without improvement. Laboratory tests showed leukocytosis, anemia, thrombocytosis, and high levels of liver enzymes. Acid-fast bacilli (AFB) were detected on biopsy of an excised lymph node, confirming the diagnosis of disseminated mycobacterial disease (BCG-osis), which was initially treated with amikacin and levofloxacin. This treatment was later replaced by isoniazid, rifampin, and ethambutol after a return to normal liver enzyme levels. The immunological assays are shown in Supplemental Table 1 (Supplemental material available online with this article; https://doi.org/10.1172/JCI135460DS1). The patient had no other severe infections caused by other bacteria or viruses. Human recombinant IFN-γ (IMUKIN) treatment (50 μg/m2, 3 times per week) was started 18 months after the patient’s initial presentation. Fourteen months after the diagnosis of BCG-osis, the patient presented with prolonged, unremitting high fever, splenomegaly, pancytopenia, very high ferritin levels, and high fasting triglyceride levels. Hemophagocytic lymphohistiocytosis (HLH) was suspected, but no evidence of hemophagocytosis was found in the bone marrow. Extensive microbiological studies revealed no infectious cause for this episode. The patient was treated with dexamethasone and etoposide (VP-16), in accordance with the HLH-04 protocol, and experienced both clinical and biochemical remission. However, no mutations in known HLH genes were identified by whole-exome sequencing (WES). Two weeks after completing the 40-week HLH-04 treatment protocol, the patient presented with intermittent fever, severe irritability, headache, and possible convulsions. Cerebrospinal fluid studies were normal, and brain MRI showed a white-matter lesion with enhancing nodules and leptomeningeal enhancement. The patient’s electroencephalogram was normal. We found no signs of systemic HLH (cytopenia, hepatosplenomegaly, low fibrinogen or high ferritin levels). We initiated treatment with dexamethasone, intrathecal methotrexate, and prednisone in accordance with the HLH-04 protocol. The patient’s relapse of HLH led us to perform allogeneic hematopoietic stem cell transplantation (HSCT) with cells from a matched donor (her sister) at the age of 3 years. Nine days after HSCT, the patient experienced a prolonged convulsion and died.
Figure 1. IFN-γ deficiency.

(A) Pedigree of 2 related kindreds, showing familial segregation of the c.354_357del (p.T119Ifs*4) mutant (M) IFNG allele. The affected patients are represented by black circles, and an arrow indicates the proband. Each generation is designated by a roman numeral (I–IV). E?, individuals of unknown genotype. (B) Linkage analysis. (C) An analysis of WES data identified IFNG as a candidate MSMD gene carrying a rare homozygous mutation common to P1 and P2 within the linkage regions. SNVs, single nucleotide variants. (D) Alamut viewer presentation of the region of the genome containing the mutation, in the sense orientation, for P1 and P2. (E) CADD score and MAF of all homozygous coding variants previously reported in public databases (gnomAD v2.1 and TOPMED/BRAVO) (http://gnomad.broadinstitute.org; https://bravo.sph.umich.edu) and in our in-house (HGID) database. The dotted line corresponds to the MSC with its 95% CI. The c.354_357del mutation is shown as a red diamond. (F) Sanger sequencing of the region containing the IFNG mutation from a healthy control, patients, and the patients’ heterozygous relatives.
Patient 2 (P2, IV.5) was born to consanguineous parents at full term and with no perinatal complications. She is the cousin of P1. Six weeks after initial vaccination with BCG at the age of 3 months, she presented with fever associated with axillary swelling and an ulcerated lesion at the injection site. The swelling measured 4 × 5 cm and was erythematous but not tender. The patient also had hepatosplenomegaly, with normal growth parameters and no dysmorphic features. Laboratory tests at presentation showed leukocytosis, anemia, thrombocytosis, a low erythrocyte sedimentation rate, and high levels of liver enzymes. Serological tests showed the patient to be seropositive for respiratory syncytial virus (RSV) at the age of 5 months. A CT of the chest showed moderate infiltration in the left mid-lung field, with no pleural effusion or hilar or mediastinal masses. A biopsy of an excised lymph node confirmed the presence of AFB. Accordingly, the patient was diagnosed with BCG-osis. She was treated with isonizid, rifampin, and ethambutol, but her condition was complicated by conjugated hyperbilirubinemia and an increase in liver enzyme levels, with normalization after the cessation of all antimycobacterial drugs. The same drugs were then sequentially reintroduced, without adverse effects. Ethambutol was stopped after the completion of 2 months of treatment. The immunological test results obtained at the age of 5 months are shown in Supplemental Table 1.
Homozygosity for a frameshift in the IFNG gene.
We performed WES and genome-wide linkage analysis (GWL) for both families. Principal component analysis (PCA) of the WES data confirmed the Middle Eastern ancestries of the patients, and their parental consanguinity was confirmed by their homozygosity rates (6.8% for P1 and 4.8% for P2). No rare heterozygous or homozygous nonsynonymous variants were identified in any of the 15 known MSMD-causing genes (Tables 1 and 2). We performed multipoint linkage analysis with homozygosity mapping under an AR model with a very rare disease-causing allele (minor allele frequency [MAF] <10–3) and complete penetrance. We identified 6 chromosomal regions linked to MSMD on chromosomes 7, 8, 11, 12, 14, and 17, with a lod score (>2.65) close to the maximum expected value (2.959) (Figure 1B). These regions covered 12.16 Mb. We found only 2 homozygous coding variants that were nonsynonymous or affected an essential splice site and had a MAF higher than 0.001 in the linked chromosomal intervals (Figure 1C). The first variant, in the CD36 gene, was located on chromosome 7 and was probably irrelevant to MSMD (see Supplemental Data). The second variant was located on chromosome 12 and was a frameshift small deletion c.354_357del in exon 3 of IFNG, which encodes the IFN-γ protein (Figure 1D). The deletion of 4 nucleotides in this gene was predicted to generate a premature stop codon at position 119, located 48 amino acids upstream from the canonical stop codon and following a stretch of 4 novel amino acids. The combined annotation-dependent depletion (CADD) score of 25 obtained (c.354_357del; Figure 1E) was well above the IFNG mutation significance cutoff (MSC) of 20.877 (57–59). We confirmed the c.354_357del IFNG mutation by Sanger sequencing of genomic DNA from whole blood, peripheral blood mononuclear cells (PBMCs), EBV-transformed B cells (EBV-B cells), herpesvirus saimiri–transformed T cells (HVS-T cells), and fibroblasts from P1 and P2 (Figure 1F). The parents of both patients and the siblings of P1 were heterozygous for the same variant, consistent with an AR trait having complete penetrance in each family. This variant was not found in our WES database (>6000 patients with various infectious diseases) or in public databases (gnomAD, version 2.1 and TOPMed BRAVO). The MAF of the allele was therefore less than 3 × 10–6. Moreover, there were no homozygous variants of IFNG predicted to be loss of function (LOF) in the general population (Figure 1E). Only 2 missense variants (p.G54D, CADD 0.004 and p.R152Q, CADD 28.2) in the homozygous state are present in the gnomAD (version 2.1) and TOPMed BRAVO databases. Overall, both the familial segregation of the frameshift c.354_357del IFNG allele and the results of population genetic analyses strongly suggested that these 2 related MSMD patients from Lebanon had AR IFN-γ deficiency.
Strong purifying selection on IFNG.
We investigated the reasons for the identification of only 2 related MSMD families with putative AR IFN-γ deficiency, contrasting with the hundreds of kindreds diagnosed with various forms of IFN-γR1 or IFN-γR2 deficiency over the past 25 years, a problem that is perhaps as puzzling as the rarity of AR IL-12Rβ2 and IL-23R deficiencies relative to AR IL-12Rβ1 deficiency (17). We analyzed 2 selection parameters (see Methods) to estimate the negative evolutionary pressure on the IFNG, IFNGR1, and IFNGR2 loci. According to the f parameter from SnIPRE (selection inference using Poisson random effects), IFNG is at 0.318, ranking it in the top 8.9% of genes under the strongest evolutionary pressure (Figure 2A). By contrast, their f values placed IFNGR1 and IFNGR2 outside the top 28% and 65%, respectively, of genes under the strongest negative selection (Figure 2A). The gene damage index (GDI) value was also much lower for IFNG (ranked among the 1.3% of genes with the lowest GDI values) than for IFNGR1 and IFNGR2 (ranked in the top 62% and 59%, respectively, of genes with the lowest GDI values) (Figure 2B). Moreover, numerous missense and predicted LOF variants are present in the heterozygous state in IFNGR1 and IFNGR2, including 4 with a MAF above 0.01 (2 in each gene), whereas far fewer such variants are observed in IFNG, with a maximum individual frequency of 6 × 10–5 and a maximum cumulative frequency of 5.30 × 10–4. Finally, taking into account the size of the corresponding coding region, the ratio of nonsynonymous variants per bp for IFNG (0.09/bp) was approximately half that for IFNGR1 (0.20/bp) and IFNGR2 (0.17/bp) (Supplemental Table 2). When considering the noncoding adjacent regions covered by WES, this variants-per-bp ratio was higher for IFNG (0.12/bp) than for IFNGR1 (0.06/bp) and IFNGR2 (0.08/bp), indicating that IFNG does not lie in a region with a particularly low mutation rate (Supplemental Table 2). Finally, autosomal-dominant (AD) IFN-γR1 deficiency is caused exclusively by specific deletions that are dominant-negative through truncation of both the recycling and STAT1-binding sites in the intracellular tail, but not the transmembrane domain (2, 7, 23, 48), whereas AD IFN-γR2 deficiency acts by haploinsufficiency with very low penetrance for MSMD (25). Overall, these results indicate that IFNG has evolved under stronger negative selection than IFNGR1 and IFNGR2, implying that IFNG is less tolerant to heterozygous mutations than IFNGR1 and IFNGR2. These results are consistent with a previous study based on a sample of 186 individuals from sub-Saharan Africa, Europe, and East Asia that used, in particular, the classical McDonald-Kreitman Poisson random field test (60, 61). These findings probably account for the greater rarity of AR IFN-γ deficiency than of IFN-γR1 and IFN-γR2 deficiencies. They also suggest that patients heterozygous for deleterious IFNG mutations may display related life-threatening infectious phenotypes, such as tuberculosis (18).
Figure 2. List of variants and strength of the purifying selection acting on IFNG.

Genome-wide distribution of the strength of purifying selection acting on genes, estimated by (A) the f parameter, for which lower values in the interval [0,1] correspond to stronger purifying selection, and (B) GDI rank, for which higher values correspond to highly tolerant genes, on bar plots for the IFNG, IFNGR1, and IFNGR2 genes. The positions of IFNG, IFNGR1, and IFNGR2 are indicated by 1 red arrow and 2 orange vertical bars, respectively.
The mutant IFNG allele is LOF.
The IFNG gene encodes a glycoprotein of 20 to 25 kDa and 166 amino acids (62–65). The mutant protein in the patients’ cells is predicted to contain 123 amino acids, including 119 original and 4 novel amino acids (Figure 3A). We transiently transfected human embryonic kidney 293T (HEK293T) cells with plasmids encoding WT IFNG or mutant c.354_357del IFNG, with a C-terminal DDK tag. We also tested the 2 missense coding variants found in the homozygous state in public databases (p.G54D and p.R152Q IFNG). The IFNG mRNAs were produced in similar amounts (Figure 3B). Western blotting with a mouse mAb against amino acids 25–50 of the human IFN-γ protein detected a WT protein product with an apparent molecular weight (MW) between 20 and 30 kDa in the cell lysate and supernatant (Figure 3C). However, we detected no mutant IFN-γ protein (Figure 3C). Moreover, Western blotting with an antibody against the C-terminal DDK tag detected no proteins, ruling out a reinitiation of translation downstream from the premature stop codon. The proteins encoded by the 2 public missense variants had a normal MW. The active form of human IFN-γ is a homodimer (66, 67). We evaluated the function of the mutant proteins by assessing HLA-DR induction in SV-40 fibroblasts with supernatants from transfected HEK293T cells. Recombinant IFN-γ and supernatants from HEK293T cells transfected with the WT, p.G54D, or p.R152Q cDNA induced HLA-DR expression on the surface of SV-40 fibroblasts from a healthy control, but not on those from a patient with AR complete IFN-γR1 deficiency (Figure 3D). By contrast, supernatants from HEK293T cells transfected with the mutant c.354_357del or with a previously described LOF cDNA (p.R143*) (68) did not induce HLA-DR. These results suggest that the mutant allele found in the homozygous state in these 2 related patients with MSMD is LOF and that there are no homozygotes for LOF alleles in current public databases. These data further suggest that these 2 patients have MSMD due to AR complete IFN-γ deficiency.
Figure 3. Levels of RNA and protein produced from the IFNG allele in an overexpression system and in vitro characterization.

(A) Schematic representation of the WT protein and predicted proteins for P1 and P2. (B) qPCR on cDNA from HEK293T cells nontransfected (NT) or transfected with an empty plasmid (EV), WT-IFNG, or mutated IFNG. GUSB was used for normalization. The results shown are representative of 2 independent experiments. (C) Western blot analysis of IFN-γ in HEK293T cells left NT or that were transfected with an EV, WT-IFNG, or mutated IFNG, all inserted into p.CMV6 with a C-terminal DDK tag, with (left) or without (middle) brefeldin treatment and the addition of supernatants from HEK293T-transfected cells (right). The anti–IFN-γ antibodies used were a monoclonal mouse anti-IFN-γ antibody recognizing an N-terminal epitope between amino acids 20 and 50, and an antibody directed against the C-terminal DDK tag. An antibody against GAPDH (α-GAPDH) was used as a protein-loading control. The results shown are representative of 2 independent experiments. Different exposure times were used for each Western blot. (D) Induction of HLA-DR on SV-40 fibroblasts from a healthy control and from a patient with AR complete IFN-γR1 deficiency. Cells were activated with commercial IFN-γ or supernatants obtained from HEK293T cells transfected with different constructs. The results shown are representative of 2 independent experiments. (E) qPCR on cDNA from the HVS-T cells from healthy travel controls (C1 and C2), a heterozygous individual, and P1. GUSB was used for normalization. The results shown are representative of 2 independent experiments. (F) RT-PCR of exons 1–4 of the IFNG cDNA in PHA blasts from a healthy control (C+), 2 patients (P1 and P2), their relatives (Het1 and Het2), and a negative control (C–). The ACTB gene was used as a cDNA loading control.
Impaired IFNG mRNA expression in patients’ cells.
We measured IFNG transcript levels in phytohemagglutinin (PHA) blasts and HVS-T cells from P1 and healthy travel controls using quantitative real-time RT-PCR (qPCR) (Figure 3E and data not shown). Cells from the patient produced IFNG transcripts that were detectable by qPCR. The levels of IFNG mRNAs were diminished when compared with those in unrelated travel controls, suggesting that transcripts carrying the mutation are subject to some degree of mRNA decay. We assessed the consequences of the c.354_357del mutation of IFNG at the mRNA level by performing qPCR on PBMCs and HVS-T cells from patients and healthy controls (Figure 3E). We amplified a segment spanning exons 1–4 from healthy controls, P1, P2, and the patients’ parents. Cells from the patients yielded PCR products of MWs similar to those obtained in PCR products from healthy controls and heterozygous carriers (Figure 3F). TA cloning and Sanger sequencing of these PCR products confirmed that all molecular clones analyzed for each patient (P1 and P2) lacked the 4 nucleotides corresponding to c.354_357del, thereby causing a frameshift premature stop codon (p.T119Ifs4*) (Figure 3A). The C-terminal domain of human IFN-γ is important for its biological activity, including interaction with its specific receptor (68–71). The predicted truncated protein, if translated and expressed, would therefore probably not be functional (68). These findings indicate that the patients’ T cells expressed reduced amounts of the IFNG mRNAs encoded by the frameshift c.354_357del allele.
Lack of IFN-γ and secretion by the patients’ cells.
IFN-γ is produced predominantly by activated primary NK and T cells (37). It can also be produced by HVS-T cell lines (17, 34). HVS-T cells from 3 healthy controls secreted IFN-γ, whereas such cells from P1 did not (Figure 4A). Furthermore, the retroviral transduction of P1 cells with the WT IFNG cDNA rescued the cellular phenotype, whereas transduction with an empty plasmid or vector (EV) did not (Figure 4A). We studied the functional consequences of the homozygous IFNG mutation in primary lymphocytes by measuring cytokine secretion in whole-blood assays (72, 73). After stimulation with BCG plus IL-12, the whole blood of healthy controls secreted large amounts of IFN-γ. By contrast, the patients’ cells secreted no detectable IFN-γ under the same conditions as those for the local and travel controls. Cells from heterozygous relatives apparently produced smaller amounts of IFN-γ than did cells from the controls, although the difference was not statistically significant (Figure 4B). Similar results were also obtained for the stimulation of whole blood with lipopolysaccharide (LPS) (Supplemental Figure 1A). The amounts of IL-12p40 produced in assays on whole blood from patients were in the normal range after stimulation with BCG plus IFN-γ or LPS plus IFN-γ (Figure 4C and data not shown). The amounts of IL-12p70 produced by the patients’ PBMCs after stimulation with LPS plus IFN-γ were within the normal range (data not shown). We also used flow cytometry to assess the intracellular production of IFN-γ by PHA–IL-2–activated T cells. PHA-activated CD3+ T cells from healthy controls produced large amounts of IFN-γ after additional stimulation with PMA plus ionomycin. By contrast, cells from P1 and P2 produced no detectable IFN-γ in these conditions (Supplemental Figure 1B). We also analyzed the production of IFN-γ by 3 subsets of PHA-activated T cells (CD4+, CD8+, and δ T cells) (Figure 4D) after stimulation with IL-12, IL-23, or phorbol myristate acetate/ionomycin (PMA/ionomycin). Under these conditions, none of the subsets of PHA T cells from the patients produced any detectable IFN-γ. The amounts of TNF produced by PHA-activated CD4+, CD8+, and δ T cells following stimulation with IL-12, IL-23, or PMA/ionomycin were low but detectable in the patients’ cells (Figure 4D), as previously reported in patients with inherited AR complete IFN-γR1 deficiency (14, 74). Cells from heterozygous relatives produced normal amounts of IFN-γ and TNF. Collectively, these experiments indicate that P1 and P2 have AR complete IFN-γ deficiency.
Figure 4. Abolition of the production and secretion of IFN-γ by cells from the IFN-γ–deficient patients.

(A) IFN-γ secretion by controls (C1, C2, C3) and patient HVS-T cell lines untransduced or transduced with empty plasmid (pLZRS-IRES-ΔNGFR-EV) or WT-IFNG plasmid (pLZRS-IRES-ΔNGFR-WT-IFNG). The results shown are representative of 2 independent experiments. (B) Induction of IFN-γ secretion in a whole-blood assay for in-house (local) controls, travel controls, patients (P1 and P2), and heterozygous relatives (Het), following stimulation with live BCG alone or in combination with IL-12. (C) Induction of IL-12p40 secretion in a whole-blood assay for local controls, travel controls, patients (P1 and P2), and heterozygous relatives, following stimulation with live BCG alone or in combination with IFN-γ. NS, no stimulation. A nonparametric Wilcoxon test was performed for the data in B and C to calculate the P value between heterozygous relatives and controls (local or travel controls). (D) IFN-γ production by PHA-activated CD3+, CD4+, and CD8+ T cells from P1 and P2 and from a healthy donor in the absence of stimulation or following stimulation with PHA alone, PHA plus IL-12 or IL-23, or PMA-ionomycin (PMA/Iono) in the presence of brefeldin A.
Frequencies of leukocyte subsets in IFN-γ–deficient patients.
We analyzed the patients’ circulating leukocyte subsets by flow cytometry and found that they had normal counts of polymorphonuclear cells and monocytes (data not shown). The frequencies of total T, B, and NK cells were also within normal ranges. The proliferation of T cells in vitro in response to stimulation with PHA and tuberculin was normal (Supplemental Table 1). In addition, the patients had higher proportions of naive CD4+ and CD8+ T cells than did healthy controls, normal proportions of all subsets of CD4+ and CD8+ memory T cells, and normal proportions of mucosal-associated invariant T (MAIT) cells (Supplemental Figure 2, A and B). We also observed normal proportions of Tregs and γδ T cells in both P1 and P2 (Supplemental Figure 2B). The frequency of CD56bright NK cells was similar in these 2 patients compared with healthy controls, whereas the frequency of invariant NKT (iNKT) cells was lower (Supplemental Figure 2B). Total CD27+ memory B cell levels were low when compared with levels in non–age-matched controls (Supplemental Figure 2D), with low IgA+– and IgM+–specific, normal IgD+–specific, and high IgG+–specific memory B cell levels (Supplemental Figure 2E). Serum IgG, IgM, and IgA levels were also normal in both patients (Supplemental Table 1). These findings indicate that IFN-γ is not essential for the development of the known leukocyte subsets in humans, as previously documented in IFN-γR1– and IFN-γR2–deficient humans (2, 13, 14, 31, 48, 49). These findings also suggested that these patients had no other immunological deficits that could account for their mycobacterial disease.
Immunity to nonmycobacterial microorganisms.
Finally, we assessed the patients’ history of microbial exposure by measuring their repertoires of plasma antibodies specific for a wide array of nonmycobacterial species, using phage immunoprecipitation–sequencing (PhIP-Seq). The results obtained suggest that the patients (P1 and P2) had been exposed to various common respiratory and enteric pathogens by the age of 2 years but that this exposure did not result in significant disease. In particular, the patients had been exposed to human adenoviruses A and C, human RSV, human metapneumovirus, rhinoviruses A and B, human herpesviruses (HHVs) 6 and 7, and opportunistic bacterial pathogens, such as Streptococcus pneumoniae and Staphylococcus aureus (Figure 5A). We also performed a PCA of the peptide/antibody specificities significantly enriched in at least 1 of the plasma samples obtained from the patients at the age of 2 years. P1 and P2 differed in terms of their antimicrobial antibody repertoires and, presumably, exposure histories, and differences with respect to IgG-depleted sera and other unrelated control subjects were observed in both patients (Figure 5B), indicating that they were able to mount normal humoral responses. The difference in humoral immunity to common infections in these patients relative to the other subjects tested appeared to be driven principally by the presence of antibodies against human adenoviruses B and C, rhinoviruses A and B, and enteroviruses A and C (Figure 5C). These findings confirmed that protective immunity to a wide range of microorganisms, including common viruses, can be achieved in the absence of IFN-γ.
Figure 5. Microbial exposure in the IFN-γ–deficient patients.

Antibody profiles of patients (P1, P2) and unrelated control subjects (HC2 and HC3 are healthy adult controls, whereas HC1 is a healthy pediatric control). Pooled plasma used for intravenous immunoglobulin (IVIg) therapy and IgG-depleted serum served as additional controls and were used for comparison. (A) Heatmap plot depicting score values equivalent to the count of significantly enriched peptides for a given species sharing less than a continuous 7-residue subsequence, the estimated size of a linear epitope. The species shown are those for which at least 1 of the patients had a score above the threshold set for this analysis (≥3). The color indicates the score values, with scores above the threshold shown in darker shades of blue. Max, maximum. (B) Result from a PCA of peptide enrichment score values above the significance threshold for at least 1 of the patients. More than 70% of the variance was accounted for by the first 2 components and separated the various controls (mockIP, IVIg, IgG-depleted), healthy controls (HC1, HC2, and HC3), and patients (P1 and P2, shown in red). PC1, principal component 1; PC2, principal component 2. (C) Scatter plots showing the contribution of the enriched peptides to the principal components (PC1 and PC2). Peptide groups from microbial species containing at least 3 differentially enriched nonhomologous peptides are shown in color.
Discussion
We describe AR complete IFN-γ deficiency as a genetic etiology of MSMD in 2 patients from 2 related families. This is the first characterization to our knowledge of a genetic deficiency of human IFN-γ, a cytokine essential for protective immunity to weakly virulent mycobacteria. IFN-γ binds to IFN-γR1 and IFN-γR2. Over the past 25 years, 5 allelic forms of IFN-γR1 deficiency and 5 allelic forms of IFN-γR2 deficiency, 8 of which are recessive, have been characterized in studies of MSMD. Hundreds of patients with these deficiencies have been identified worldwide, with no particular ethnic preponderance, except for a higher frequency of recessive forms in areas with high rates of consanguineous unions (7, 13, 14, 16, 23, 25, 31, 44–53, 56, 75–79). The response to IFN-γ is impaired or abolished in affected patients. By contrast, we report here the first 2 patients, to our knowledge, with biallelic IFNG LOF mutations, suggesting that this deficiency is extremely rare.
One plausible explanation for this rarity is that the IFNG gene has evolved under stronger purifying selection, preventing the accumulation of deleterious monoallelic amino acid variants. There are fewer heterozygous missense variants of IFNG than of IFNGR1 and IFNGR2 in the general population. Moreover, most variants are predicted in silico to be neutral, and the only 2 missense variants common enough to be found in the homozygous state in the general population were shown to be neutral in vitro. The physiological reason for the different patterns of selection at the IFNG locus and at the IFNGR1 and IFNGR2 loci is probably related to IFN-γ immunity. The 2 chains of the receptor are ubiquitously expressed, as is IFN-γR1, and even IFN-γR2, the expression of which may be regulated in some cell types to enhance or attenuate the response to IFN-γ. By contrast, the production of IFN-γ is tightly regulated. Only a few cell types, all of the lymphoid lineage, produce this cytokine abundantly (NK cells, γδ T cells, CD8+ T cells, CD4+ T cells, innate lymphoid cells [ILCs], and MAIT cells). Moreover, IFN-γ is produced under stringent transcriptional and posttranscriptional control (17, 37, 62, 69). This strict regulation ensures effective immunity to infection, while preventing autoinflammation. Excessive IFN-γ production was recently proposed to result in fulminant viral hepatitis in a patient with AR IL-18BP deficiency (80). In other words, the interaction between this cytokine and its 2 receptor chains is modulated by regulation of the cytokine, which serves as the limiting factor.
In this context, heterozygous deleterious mutations at the IFNG locus are expected to be less tolerated than deleterious mutations at the IFNGR1 and IFNGR2 loci. We previously reported that haploinsufficiency for deleterious mutations at the IFNGR2 locus can underlie MSMD with very low penetrance, but that this is not the case for such mutations at the IFNGR1 locus (25). Our findings suggest that heterozygosity for IFNG mutations may underlie severe infectious phenotypes, including MSMD with low penetrance or tuberculosis with higher penetrance, as recently shown in humans homozygous for the P1104A variant of TYK2 (18, 81). It will be of interest to search for heterozygous IFNG variants in large cohorts of patients with tuberculosis and related infections. Our study confirms that MSMD is actually caused by inborn errors of IFN-γ. This experiment of nature rules out the remote but finite possibility that there are other agonists of the IFN-γ receptor, as documented for other receptors such as IL-4Rα, which can be stimulated by IL-4 and IL-13 (82–84), and IL-7Rα, which can be stimulated by IL-7 and thymic stromal lymphopoietin (TSLP) (85). Mutations of IFNG should thus be considered in patients with MSMD or related infectious diseases, including tuberculosis and other diseases caused by intramacrophagic microorganisms. Early diagnosis according to this approach may be advantageous, as patients would be predicted to benefit from recombinant IFN-γ treatment. Indeed, treatment with this cytokine has already been shown to be useful in patients with MSMD who have impaired IFN-γ production, such as those with IL-12Rβ1 deficiency (2, 86, 87).
Methods
Additional details are provided in the Supplemental Data.
Genomic DNA extraction, WES and genome-wide linkage analysis.
Genomic DNA was isolated from whole blood of patients, their relatives, and healthy donors, using iPrep instruments from Thermo Fisher Scientific. We used 3 μg DNA from P1 and P2 for WES. The Agilent 50-Mb SureSelect Exome Kit was used in accordance with the manufacturer’s instructions. Burrows-Wheeler Aligner (BWA) (http://bio-bwa.sourceforge.net/) was used to align the reads with the human reference genome hg19, before recalibration and annotation with the Genome Analysis Toolkit (GATK) (https://gatk.broadinstitute.org/hc/en-us), Picard (https://broadinstitute.github.io/picard/), and ANNOVAR (https://doc-openbio.readthedocs.io/projects/annovar/en/latest/). Our in-house software was used to filter the variants. For GWL analysis, the 2 patients were genotyped using the Affimetrix Genome-wide Mapping 6.0 chip, and the parents were genotyped using the Affymetrix Genome-wide Human Mapping 250 K chip with the Affymetrix Power Tools Software Package (http://www.affymetrix.com/estore/partners_programs/programs/developer/tools/powertools.aff) for genotype calling. SNPs presenting more than 1 Mendelian inconsistency were discarded. PLINK software (http://zzz.bwh.harvard.edu/plink/) was used to further filter SNPs with population-based filters according to the ethnicity of the kindred. A subset of 163,807 autosomal SNP markers common to the 2 genotyping arrays and optimized to decrease their linkage disequilibrium (LD) was used for linkage analysis, as previously described (88). Parametric multipoint linkage analyses were performed with MERLIN (http://csg.sph.umich.edu/abecasis/merlin/tour/input_files.html), assuming AR inheritance with complete penetrance and a deleterious allele frequency of 10–3 (89). The family founders and unrelated individuals from HapMap CEU (https://www.sanger.ac.uk/resources/downloads/human/hapmap3.html) were used to estimate allele frequencies for the 2 kindreds and to define linkage clusters, with an r2 threshold of 0.4. PCR products were analyzed by electrophoresis in 1% agarose gels, sequenced with the BigDye Terminator v3.1 Cycle Sequencing Kit (Thermo Fisher Scientific), and analyzed on an ABI Prism 3700 machine (Applied Biosystems).
Assessment of the selective pressures on the IFNG gene.
We used an approach based on 2 methods to estimate the strength of purifying selection acting on IFNG in humans. SnIPRE (selection inference using a Poisson random effects model) makes use of a generalized linear mixed model to model genome-wide variability for categories of mutations (90). It estimates 2 key population genetic parameters for each gene, one of which, f, quantifies the strength of purifying selection acting on human genes, with 0 corresponding to strong negative selection and 1 to neutral selection. We used the alignment of the hg19 human genome and the PanTro3 chimp genome from the UCSC Genome Browser (https://genome.ucsc.edu/). All differences between the 2 species were annotated functionally by SnpEff 50 (http://snpeff.sourceforge.net/SnpEff_manual.html), using the GRCh37.65 build (https://m.ensembl.org/info/website/tutorials/grch37.html). We retrieved all human coding sequences (CDSs) of more than 20 bp in length and considered the longest available transcript for each gene. We then retrieved all polymorphisms identified in the WES data for phase 1 of the 1000 Genomes Project. We analyzed SNPs annotated as nonsynonymous or synonymous, located outside of gaps in the human-chimp alignment and shown to be polymorphic in at least 1 human population. We also used the dbSNP build 136 chimp database (https://www.ncbi.nlm.nih.gov/projects/SNP/snp_summary.cgi?build_id=136) to remove positions that were polymorphic in humans or chimps from the list of positions divergent between humans and chimps. Finally, we determined the number of divergent and polymorphic synonymous and nonsynonymous mutations and the proportion of synonymous and nonsynonymous sites for a total of 18,969 genes. SnIPRE was then used to estimate the f parameter. The intolerance of each gene to damaging mutations was also assessed by the GDI (91). It uses all annotated alleles with a global MAF of less than 0.5 in the 1000 Genomes Project to estimate a gene damaging score. High GDI values are associated with highly tolerant genes. Genes are ranked according to their GDI value. We downloaded GDI values from The Rockefeller University’s site (http://lab.rockefeller.edu/casanova/GDI).
Plasmid cloning, mutagenesis, and Western blotting.
Empty plasmid (EV) and the WT-IFNG plasmid (RC209993) were purchased from OriGene. Mutated plasmids (p.G54D, p.R152Q, c.354_357del and p.R143*) were generated by site-directed mutagenesis using the QuikChange II XL Site-Directed Mutagenesis Kit (200521-5, Agilent Technologies) or by PCR followed by blunting (Quick Blunting Kit, New England BioLabs) and ligation (Quick Ligation, New England BioLabs). HEK293T cells were transfected by incubation for 48 hours in a 6-well plate with 2 mL DMEM supplemented with 10% FBS (Sigma-Aldrich) in the presence of X-tremeGENE-9 DNA Transfection Reagent (6365787001, Roche), with 1 μg plasmid containing EV, WT, or the mutant cDNA studied here. When indicated, cells were treated 6 hours before harvesting with brefeldin A diluted to 1:1000 (GolgiPlug, BD).
Proteins were extracted in whole-cell lysis buffer containing 20 mM Tris-HCl, pH 7.4, 140 mM NaCl, 2 mM EDTA, 50 mM NaF, and 1% NP40 supplemented with protease inhibitor cocktail (cOmplete Mini, Roche) and quantified with the Bradford assay. Lysate (50 μg protein) was mixed with Laemmli loading buffer and incubated at 95°C for 5 minutes before being subjected to electrophoresis in a precast 4%–12% acrylamide SDS-PAGE gel (Bio-Rad). The following antibodies were used for Western blotting: mouse monoclonal anti–human IFN-γ N-terminus (sc-373727, E-10, Santa Cruz Biotechnology), anti-GAPDH (2118, 14C10, Cell Signaling Technology), and HRP-conjugated anti-DDK (clone M2, Sigma-Aldrich). Uncoupled antibodies were detected by incubation with HRP-conjugated goat anti-mouse or antirabbit secondary antibodies (Bio-Rad).
Intracellular staining of cytokines.
Human PBMCs were isolated from heparinized whole blood by Ficoll-Hypaque density gradient centrifugation. IFN-γ production by PHA-IL-2–activated T cells was assessed by intracellular staining. Briefly, in 96-well plates containing RPMI medium supplemented with 10% FBS, PHA-IL-2–activated T cells were either left unstimulated or were stimulated for 6 hours in the presence of PMA (40 ng/mL) and ionomycin (10–5 M), or for 16 hours with PHA (1 μg/mL) and IL-12 (20 ng/mL) or IL-23 (100 ng/mL), as indicated. All incubations were performed in the presence of brefeldin (Golgi Plug, 1:1000, BD Biosciences). Cells were then harvested and stained for extracellular markers (CD3, CD8, CD4) and with a viability dye (LIVE/DEAD Aqua Cell Stain Kit, Thermo Fisher Scientific), before fixation and permeabilization with a fixation/permeabilization kit (eBioscience). Cells were then stained for intracellular IFN-γ (clone B27, BD Biosciences) and TNF (clone cA2, Miltenyi Biotec) and analyzed with a Gallios flow cytometer (Beckman Coulter) and FlowJo 10.1r5 software.
Retroviral transduction.
PBMCs from healthy controls and patients and their relatives were infected with Herpes saimiri for the generation of HVS-T cells, as previously described (34). HVS-T cells were cultured with a 1:1 mixture (by volume) of RPMI and Panserin 401 supplemented with 10% FBS, 350 μg/mL glutamine, 100 μg/mL gentamycin, and 10 IU/mL human recombinant IL-2 (hrIL-2) (Roche). HVS-T cells were transduced with retroviral particles, as previously described (34). Briefly, 6-well nontissue culture–treated plates were coated by incubation for 24 hours with RetroNectin (100 ng/mL). The next day, the wells were washed with PBS, and the viral supernatants were thawed and added to the empty wells. The volume in each well was made up to 2 mL with medium. Plates were centrifuged at 800 × g for 30 minutes, and 106 cells were added to each well. The plates were centrifuged for 5 minutes at 300 × g and incubated at 37°C for 5 days. Transduced cells were purified by magnetic cell separation (MACS) with a magnetic bead–conjugated anti-NGFR antibody (Miltenyi Biotec), in accordance with the manufacturer’s protocol. We used flow cytometry to check that the purity of the transduced cells exceeded 95%. ELISA was performed on the collected supernatants using the Human IFN-γ (Sanquin) and Deluxe Set Human IFN-γ (BioLegend) ELISA kits, according to the manufacturers’ instructions.
Statistics.
Statistical tests for Figure 4, B and C, and Supplemental Figure 1A were performed using a nonparametric Mann-Whitney Wilcoxon Test. Samples were compared pairwise to assess the null hypothesis that the levels of cytokine production were the same in the 2 samples tested. A P value below 0.05 was used to reject the null hypothesis.
Study approval.
The patients were referred to the Laboratory of Human Genetics of Infectious Diseases (HGID) for cellular and genetic diagnosis. Written informed consent forms were signed by the parents of the patients. This study was conducted in accordance with the Declaration of Helsinki, and approval was obtained from the French ethics committee Comité de Protection des Personnes (CPP), the French National Agency for Medicine and Health Product Safety (ANSM), the INSERM, and The Rockefeller University IRB.
Author contributions
JB and JLC designed experiments and supervised the study. AG, VJJ, JR, COQ, CD, DG, VB, TK, FAA, M. Rahman, M. Roynard, RMB, and MJM conducted the experiments. GK, YS, F. Rapaport, and LA performed statistical and bioinformatics analyses. SBD, AG, RY, VJJ, JR, COQ, CD, RSG, VB, TK, M. Rahman, FAA, NM, JB, and JLC analyzed and/or interpreted data. F. Rozenberg performed viral serological tests. BF generated the HVS-T cells. AAK and WAH provided samples and were responsible for the clinical diagnosis and follow-up of the patients. JYD, CYC, and CLK performed the tests for autoantibody detection. We included in the list of authors performing experiments JB and JLC wrote the manuscript and designed the figures, with contributions from all of the authors. All of the authors edited the manuscript and approved the final version for submission. The contributions of the co–first authors were as follows: GK analyzed WES, identified the mutation in patients, performed linkage and statistical analysis. AG performed Sanger sequencing verification, constructed the plasmid, generated the first overexpression results, and performed the flow cytometric analysis of PBMCs. JR verified the overexpression system and qPCR results, rescued and evaluated functional tests in the overexpression system, and prepared some of the figures. AAK was responsible for the clinical diagnosis and follow-up of the patients and provided the samples for the genetic diagnoses for both families. GK is the first author because he identified the mutation in this family and, using bioinformatic approaches, studied the cohort of patients with MSMD and conducted all the studies on this family.
Supplementary Material
Acknowledgments
We would like to thank the patients and their families, whose cooperation was essential for the collection of the data used in this study. We thank all members of the Laboratory of Human Genetics of Infectious Diseases for helpful discussions. We wish to thank Céline Desvallées, Tatiana Kochetkov, Dominick Papandrea, Cécile Patissier, Mark Woollett, and Yelena Nemirovskaya for their assistance. We also thank Steven Elledge for providing the VirScan phage library that was used in this study for antibody profiling. The Laboratory of Human Genetics of Infectious Diseases is supported in part by institutional grants from the INSERM, Paris Descartes University, the St. Giles Foundation, The Rockefeller University Center for Clinical and Translational Science (8UL1TR000043); the National Center for Research Resources and the National Center for Advancing Sciences (NCATS); the National Institute of Allergy and Infectious Diseases (NIAID), NIH (5R01AI089970-02 and 5R37AI095983); the French Foundation for Medical Research (FRM) (EQU201903007798); the SCOR Corporate Foundation for Science; and the French National Research Agency (ANR) under the “Investments for the Future” program (ANR-10-IAHU-01) and GENMSMD (human genetic dissection of Mendelian susceptibility to mycobacterial disease) (ANR-16-CE17-0005-01, to JB). NM, TK, MR, and FA were supported by Sidra Medicine and the Qatar National Research Fund (funding ID: PPM1-1220-150017). COQ was supported by ANR-HGDIFD (ANR-14-CE15-006-01). AG was supported by the ANR-IFNGPHOX (ANR13-ISV3-0001-01), GENMSMD (ANR-16-CE17-0005-01), and the Imagine Institute. VJJ was supported by the Integrative Biology of Emerging Infectious Diseases Laboratory of Excellence (ANR-10-LABX-62-IBEID). JR was supported by INSERM (“poste d’accueil”). GK was supported by the Imagine Institute.
Version 1. 03/12/2020
In-Press Preview
Version 2. 05/11/2020
Electronic publication
Version 3. 06/01/2020
Print issue publication
Funding Statement
ANR-IFNGPHOX
GENMSMD
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Copyright: © 2020, American Society for Clinical Investigation.
Reference information: J Clin Invest. 2020;130(6):3158–3171.https://doi.org/10.1172/JCI135460.
Contributor Information
Gaspard Kerner, Email: gaspard.kerner@inserm.fr.
Jérémie Rosain, Email: jeremie.rosain@gmail.com.
Antoine Guérin, Email: a.guerin@garvan.org.au.
Carmen Oleaga-Quintas, Email: coleaga@gmail.com.
Franck Rapaport, Email: frapaport@mail.rockefeller.edu.
Michel J. Massaad, Email: mm257@aub.edu.lb.
Jing-Ya Ding, Email: starview03@gmail.com.
Taushif Khan, Email: tkhan@sidra.org.
Fatima Al Ali, Email: falali@sidra.org.
Mahbuba Rahman, Email: mrahman1@sidra.org.
Caroline Deswarte, Email: caroline.deswarte@inserm.fr.
Raif S. Geha, Email: raif.geha@childrens.harvard.edu.
Valentine Jeanne-Julien, Email: valentine.jeannejulien@gmail.com.
Chih-Yu Chi, Email: cychyi@gmail.com.
Rui Yang, Email: ryang@mail.rockefeller.edu.
Manon Roynard, Email: manon.roynard@institutimagine.org.
Bernhard Fleckenstein, Email: bernhard.fleckenstein@fau.de.
Flore Rozenberg, Email: flore.rozenberg@aphp.fr.
Stéphanie Boisson-Dupuis, Email: stbo603@rockefeller.edu.
Cheng-Lung Ku, Email: clku@mail.cgu.edu.tw.
Yoann Seeleuthner, Email: yoann.seeleuthner@inserm.fr.
Nico Marr, Email: nmarr@sidra.org.
Laurent Abel, Email: laurent.abel@inserm.fr.
Waleed Al-Herz, Email: wemh@hotmail.com.
Jean-Laurent Casanova, Email: Jean-Laurent.Casanova@rockefeller.edu.
Jacinta Bustamante, Email: jacinta.bustamante@inserm.fr.
References
- 1.Casanova JL, Abel L. Genetic dissection of immunity to mycobacteria: the human model. Annu Rev Immunol. 2002;20:581–620. doi: 10.1146/annurev.immunol.20.081501.125851. [DOI] [PubMed] [Google Scholar]
- 2.Bustamante J, Boisson-Dupuis S, Abel L, Casanova JL. Mendelian susceptibility to mycobacterial disease: genetic, immunological, and clinical features of inborn errors of IFN-γ immunity. Semin Immunol. 2014;26(6):454–470. doi: 10.1016/j.smim.2014.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boisson-Dupuis S, et al. Inherited and acquired immunodeficiencies underlying tuberculosis in childhood. Immunol Rev. 2015;264(1):103–120. doi: 10.1111/imr.12272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zahid MF, et al. Recurrent salmonellosis in a child with complete IL-12Rβ1 deficiency. J immunodefic Disord. 2014;3:1000109. doi: 10.4172/2324-853X.1000109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.de Beaucoudrey L, et al. Revisiting human IL-12Rβ1 deficiency: a survey of 141 patients from 30 countries. Medicine (Baltimore) 2010;89(6):381–402. doi: 10.1097/MD.0b013e3181fdd832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Prando C, et al. Inherited IL-12p40 deficiency: genetic, immunologic, and clinical features of 49 patients from 30 kindreds. Medicine (Baltimore) 2013;92(2):109–122. doi: 10.1097/MD.0b013e31828a01f9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rosain J, et al. Mendelian susceptibility to mycobacterial disease: 2014-2018 update. Immunol Cell Biol. 2019;97(4):360–367. doi: 10.1111/imcb.12210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ouederni M, et al. Clinical features of candidiasis in patients with inherited interleukin 12 receptor β1 deficiency. Clin Infect Dis. 2014;58(2):204–213. doi: 10.1093/cid/cit722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mimouni J. [Our experiences in three years of BCG vaccination at the center of the O.P.H.S. at Constantine; study of observed cases (25 cases of complications from BCG vaccination)] Alger Medicale. 1951;55(8):1138–1147. [PubMed] [Google Scholar]
- 10.Casanova JL, et al. Idiopathic disseminated bacillus Calmette-Guérin infection: a French national retrospective study. Pediatrics. 1996;98(4 Pt 1):774–778. [PubMed] [Google Scholar]
- 11.Casanova JL, Jouanguy E, Lamhamedi S, Blanche S, Fischer A. Immunological conditions of children with BCG disseminated infection. Lancet. 1995;346(8974):581. doi: 10.1016/s0140-6736(95)91421-8. [DOI] [PubMed] [Google Scholar]
- 12.Reichenbach J, Rosenzweig S, Döffinger R, Dupuis S, Holland SM, Casanova JL. Mycobacterial diseases in primary immunodeficiencies. Curr Opin Allergy Clin Immunol. 2001;1(6):503–511. doi: 10.1097/00130832-200112000-00003. [DOI] [PubMed] [Google Scholar]
- 13.Jouanguy E, et al. Interferon-gamma-receptor deficiency in an infant with fatal bacille Calmette-Guérin infection. N Engl J Med. 1996;335(26):1956–1961. doi: 10.1056/NEJM199612263352604. [DOI] [PubMed] [Google Scholar]
- 14.Newport MJ, et al. A mutation in the interferon-gamma-receptor gene and susceptibility to mycobacterial infection. N Engl J Med. 1996;335(26):1941–1949. doi: 10.1056/NEJM199612263352602. [DOI] [PubMed] [Google Scholar]
- 15.Kong XF, et al. Disruption of an antimycobacterial circuit between dendritic and helper T cells in human SPPL2a deficiency. Nat Immunol. 2018;19(9):973–985. doi: 10.1038/s41590-018-0178-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Oleaga-Quintas C, et al. A purely quantitative form of partial recessive IFN-γR2 deficiency caused by mutations of the initiation or second codon. Hum Mol Genet. 2019;28(3):524. doi: 10.1093/hmg/ddy357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Martínez-Barricarte R, et al. Human IFN-γ immunity to mycobacteria is governed by both IL-12 and IL-23. Sci Immunol. 2018;3(30):eaau6759. doi: 10.1126/sciimmunol.aau6759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Boisson-Dupuis S, et al. Tuberculosis and impaired IL-23-dependent IFN-γ immunity in humans homozygous for a common TYK2 missense variant. Sci Immunol. 2018;3(30):eaau8714. doi: 10.1126/sciimmunol.aau8714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dorman SE, Holland SM. Mutation in the signal-transducing chain of the interferon-gamma receptor and susceptibility to mycobacterial infection. J Clin Invest. 1998;101(11):2364–2369. doi: 10.1172/JCI2901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.de Jong R, et al. Severe mycobacterial and Salmonella infections in interleukin-12 receptor-deficient patients. Science. 1998;280(5368):1435–1438. doi: 10.1126/science.280.5368.1435. [DOI] [PubMed] [Google Scholar]
- 21.Altare F, et al. Impairment of mycobacterial immunity in human interleukin-12 receptor deficiency. Science. 1998;280(5368):1432–1435. doi: 10.1126/science.280.5368.1432. [DOI] [PubMed] [Google Scholar]
- 22.Altare F, et al. Inherited interleukin 12 deficiency in a child with bacille Calmette-Guérin and Salmonella enteritidis disseminated infection. J Clin Invest. 1998;102(12):2035–2040. doi: 10.1172/JCI4950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jouanguy E, et al. A human IFNGR1 small deletion hotspot associated with dominant susceptibility to mycobacterial infection. Nat Genet. 1999;21(4):370–378. doi: 10.1038/7701. [DOI] [PubMed] [Google Scholar]
- 24.Döffinger R, et al. Partial interferon-gamma receptor signaling chain deficiency in a patient with bacille Calmette-Guérin and Mycobacterium abscessus infection. J Infect Dis. 2000;181(1):379–384. doi: 10.1086/315197. [DOI] [PubMed] [Google Scholar]
- 25.Kong XF, et al. Haploinsufficiency at the human IFNGR2 locus contributes to mycobacterial disease. Hum Mol Genet. 2013;22(4):769–781. doi: 10.1093/hmg/dds484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hambleton S, et al. IRF8 mutations and human dendritic-cell immunodeficiency. N Engl J Med. 2011;365(2):127–138. doi: 10.1056/NEJMoa1100066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fieschi C, et al. A novel form of complete IL-12/IL-23 receptor beta1 deficiency with cell surface-expressed nonfunctional receptors. Blood. 2004;104(7):2095–2101. doi: 10.1182/blood-2004-02-0584. [DOI] [PubMed] [Google Scholar]
- 28.Bustamante J, et al. Germline CYBB mutations that selectively affect macrophages in kindreds with X-linked predisposition to tuberculous mycobacterial disease. Nat Immunol. 2011;12(3):213–221. doi: 10.1038/ni.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dupuis S, et al. Impairment of mycobacterial but not viral immunity by a germline human STAT1 mutation. Science. 2001;293(5528):300–303. doi: 10.1126/science.1061154. [DOI] [PubMed] [Google Scholar]
- 30.Filipe-Santos O, et al. X-linked susceptibility to mycobacteria is caused by mutations in NEMO impairing CD40-dependent IL-12 production. J Exp Med. 2006;203(7):1745–1759. doi: 10.1084/jem.20060085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vogt G, et al. Gains of glycosylation comprise an unexpectedly large group of pathogenic mutations. Nat Genet. 2005;37(7):692–700. doi: 10.1038/ng1581. [DOI] [PubMed] [Google Scholar]
- 32.Bogunovic D, et al. Mycobacterial disease and impaired IFN-γ immunity in humans with inherited ISG15 deficiency. Science. 2012;337(6102):1684–1688. doi: 10.1126/science.1224026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang X, et al. Human intracellular ISG15 prevents interferon-α/β over-amplification and auto-inflammation. Nature. 2015;517(7532):89–93. doi: 10.1038/nature13801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Martinez-Barricarte R, et al. Transduction of herpesvirus saimiri-transformed T cells with exogenous genes of interest. Curr Protoc Immunol. 2016;115:7.21C.1–7.21C.12. doi: 10.1002/cpim.15. [DOI] [PubMed] [Google Scholar]
- 35.Kreins AY, et al. Human TYK2 deficiency: mycobacterial and viral infections without hyper-IgE syndrome. J Exp Med. 2015;212(10):1641–1662. doi: 10.1084/jem.20140280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Eletto D, et al. Biallelic JAK1 mutations in immunodeficient patient with mycobacterial infection. Nat Commun. 2016;7:13992. doi: 10.1038/ncomms13992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schoenborn JR, Wilson CB. Regulation of interferon-gamma during innate and adaptive immune responses. Adv Immunol. 2007;96:41–101. doi: 10.1016/S0065-2776(07)96002-2. [DOI] [PubMed] [Google Scholar]
- 38.Wheelock EF. Interferon-like virus-inhibitor induced in human leukocytes by phytohemagglutinin. Science. 1965;149(3681):310–311. doi: 10.1126/science.149.3681.310. [DOI] [PubMed] [Google Scholar]
- 39.Nathan CF, Murray HW, Wiebe ME, Rubin BY. Identification of interferon-gamma as the lymphokine that activates human macrophage oxidative metabolism and antimicrobial activity. J Exp Med. 1983;158(3):670–689. doi: 10.1084/jem.158.3.670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Harty JT, Bevan MJ. Specific immunity to Listeria monocytogenes in the absence of IFN gamma. Immunity. 1995;3(1):109–117. doi: 10.1016/1074-7613(95)90163-9. [DOI] [PubMed] [Google Scholar]
- 41.John B, Rajagopal D, Pashine A, Rath S, George A, Bal V. Role of IL-12-independent and IL-12-dependent pathways in regulating generation of the IFN-gamma component of T cell responses to Salmonella typhimurium. J Immunol. 2002;169(5):2545–2552. doi: 10.4049/jimmunol.169.5.2545. [DOI] [PubMed] [Google Scholar]
- 42.Huang S, et al. Immune response in mice that lack the interferon-gamma receptor. Science. 1993;259(5102):1742–1745. doi: 10.1126/science.8456301. [DOI] [PubMed] [Google Scholar]
- 43.Dalton DK, Pitts-Meek S, Keshav S, Figari IS, Bradley A, Stewart TA. Multiple defects of immune cell function in mice with disrupted interferon-gamma genes. Science. 1993;259(5102):1739–1742. doi: 10.1126/science.8456300. [DOI] [PubMed] [Google Scholar]
- 44.Jouanguy E, et al. In a novel form of IFN-gamma receptor 1 deficiency, cell surface receptors fail to bind IFN-gamma. J Clin Invest. 2000;105(10):1429–1436. doi: 10.1172/JCI9166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jouanguy E, et al. Partial interferon-gamma receptor 1 deficiency in a child with tuberculoid bacillus Calmette-Guérin infection and a sibling with clinical tuberculosis. J Clin Invest. 1997;100(11):2658–2664. doi: 10.1172/JCI119810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Martínez-Barricarte R, et al. Mycobacterium simiae infection in two unrelated patients with different forms of inherited IFN-γR2 deficiency. J Clin Immunol. 2014;34(8):904–909. doi: 10.1007/s10875-014-0085-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Moncada-Vélez M, et al. Partial IFN-γR2 deficiency is due to protein misfolding and can be rescued by inhibitors of glycosylation. Blood. 2013;122(14):2390–2401. doi: 10.1182/blood-2013-01-480814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dorman SE, et al. Clinical features of dominant and recessive interferon gamma receptor 1 deficiencies. Lancet. 2004;364(9451):2113–2121. doi: 10.1016/S0140-6736(04)17552-1. [DOI] [PubMed] [Google Scholar]
- 49.Vogt G, et al. Complementation of a pathogenic IFNGR2 misfolding mutation with modifiers of N-glycosylation. J Exp Med. 2008;205(8):1729–1737. doi: 10.1084/jem.20071987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rosenzweig SD, et al. A novel mutation in IFN-gamma receptor 2 with dominant negative activity: biological consequences of homozygous and heterozygous states. J Immunol. 2004;173(6):4000–4008. doi: 10.4049/jimmunol.173.6.4000. [DOI] [PubMed] [Google Scholar]
- 51.Rosenzweig S, Dorman SE, Roesler J, Palacios J, Zelazko M, Holland SM. 561del4 defines a novel small deletion hotspot in the interferon-gamma receptor 1 chain. Clin Immunol. 2002;102(1):25–27. doi: 10.1006/clim.2001.5135. [DOI] [PubMed] [Google Scholar]
- 52.Rose DM, Atkins J, Holland SM, Infante AJ. A novel mutation in IFN-γ receptor 1 presenting as multisystem Mycobacterium intracellulare infection. J Allergy Clin Immunol. 2014;133(2):591–592. doi: 10.1016/j.jaci.2013.07.054. [DOI] [PubMed] [Google Scholar]
- 53.van de Vosse E, van Dissel JT. IFN-γR1 defects: mutation update and description of the IFNGR1 variation database. Hum Mutat. 2017;38(10):1286–1296. doi: 10.1002/humu.23302. [DOI] [PubMed] [Google Scholar]
- 54.Humblet-Baron S, et al. IFN-γ and CD25 drive distinct pathologic features during hemophagocytic lymphohistiocytosis. J Allergy Clin Immunol. 2019;143(6):2215–2226.e7. doi: 10.1016/j.jaci.2018.10.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kong XF, et al. A novel form of cell type-specific partial IFN-gammaR1 deficiency caused by a germ line mutation of the IFNGR1 initiation codon. Hum Mol Genet. 2010;19(3):434–444. doi: 10.1093/hmg/ddp507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rosain J, et al. LINE-1-mediated AluYa5 insertion underlying complete autosomal recessive IFN-γR1 deficiency. J Clin Immunol. 2019;39(7):739–742. doi: 10.1007/s10875-019-00667-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Itan Y, et al. The mutation significance cutoff: gene-level thresholds for variant predictions. Nat Methods. 2016;13(2):109–110. doi: 10.1038/nmeth.3739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46(3):310–315. doi: 10.1038/ng.2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang P, et al. PopViz: a webserver for visualizing minor allele frequencies and damage prediction scores of human genetic variations. Bioinformatics. 2018;34(24):4307–4309. doi: 10.1093/bioinformatics/bty536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Manry J, et al. Evolutionary genetic dissection of human interferons. J Exp Med. 2011;208(13):2747–2759. doi: 10.1084/jem.20111680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Manry J, et al. Evolutionary genetics evidence of an essential, nonredundant role of the IFN-γ pathway in protective immunity. Hum Mutat. 2011;32(6):633–642. doi: 10.1002/humu.21484. [DOI] [PubMed] [Google Scholar]
- 62.Gray PW, Goeddel DV. Structure of the human immune interferon gene. Nature. 1982;298(5877):859–863. doi: 10.1038/298859a0. [DOI] [PubMed] [Google Scholar]
- 63.Devos R, Cheroutre H, Taya Y, Degrave W, Van Heuverswyn H, Fiers W. Molecular cloning of human immune interferon cDNA and its expression in eukaryotic cells. Nucleic Acids Res. 1982;10(8):2487–2501. doi: 10.1093/nar/10.8.2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Taya Y, Devos R, Tavernier J, Cheroutre H, Engler G, Fiers W. Cloning and structure of the human immune interferon-gamma chromosomal gene. EMBO J. 1982;1(8):953–958. doi: 10.1002/j.1460-2075.1982.tb01277.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mizrahi A, O’Malley JA, Carter WA, Takatsuki A, Tamura G, Sulkowski E. Glycosylation of interferons. Effects of tunicamycin on human immune interferon. J Biol Chem. 1978;253(21):7612–7615. [PubMed] [Google Scholar]
- 66.Ealick SE, et al. Three-dimensional structure of recombinant human interferon-gamma. Science. 1991;252(5006):698–702. doi: 10.1126/science.1902591. [DOI] [PubMed] [Google Scholar]
- 67.Bach EA, Aguet M, Schreiber RD. The IFN gamma receptor: a paradigm for cytokine receptor signaling. Annu Rev Immunol. 1997;15:563–591. doi: 10.1146/annurev.immunol.15.1.563. [DOI] [PubMed] [Google Scholar]
- 68.Nacheva G, Todorova K, Boyanova M, Berzal-Herranz A, Karshikoff A, Ivanov I. Human interferon gamma: significance of the C-terminal flexible domain for its biological activity. Arch Biochem Biophys. 2003;413(1):91–98. doi: 10.1016/S0003-9861(03)00113-9. [DOI] [PubMed] [Google Scholar]
- 69.Szente BE, Soos JM, Johnson HW. The C-terminus of IFN gamma is sufficient for intracellular function. Biochem Biophys Res Commun. 1994;203(3):1645–1654. doi: 10.1006/bbrc.1994.2375. [DOI] [PubMed] [Google Scholar]
- 70.Green MM, Larkin J, Subramaniam PS, Szente BE, Johnson HM. Human IFN gamma receptor cytoplasmic domain: expression and interaction with HuIFN gamma. Biochem Biophys Res Commun. 1998;243(1):170–176. doi: 10.1006/bbrc.1998.8077. [DOI] [PubMed] [Google Scholar]
- 71.Lin CH, et al. Identification of a major epitope by anti-interferon-γ autoantibodies in patients with mycobacterial disease. Nat Med. 2016;22(9):994–1001. doi: 10.1038/nm.4158. [DOI] [PubMed] [Google Scholar]
- 72.Feinberg J, et al. Bacillus Calmette Guerin triggers the IL-12/IFN-gamma axis by an IRAK-4- and NEMO-dependent, non-cognate interaction between monocytes, NK, and T lymphocytes. Eur J Immunol. 2004;34(11):3276–3284. doi: 10.1002/eji.200425221. [DOI] [PubMed] [Google Scholar]
- 73.Esteve-Solé A, et al. Laboratory evaluation of the IFN-γ circuit for the molecular diagnosis of Mendelian susceptibility to mycobacterial disease. Crit Rev Clin Lab Sci. 2018;55(3):184–204. doi: 10.1080/10408363.2018.1444580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Levin M, et al. Familial disseminated atypical mycobacterial infection in childhood: a human mycobacterial susceptibility gene? Lancet. 1995;345(8942):79–83. doi: 10.1016/S0140-6736(95)90059-4. [DOI] [PubMed] [Google Scholar]
- 75.Shabani M, et al. A novel recessive mutation of interferon-γ receptor 1 in a patient with Mycobacterium tuberculosis in bone marrow aspirate. J Clin Immunol. 2019;39(2):127–130. doi: 10.1007/s10875-019-00595-7. [DOI] [PubMed] [Google Scholar]
- 76.Prando C, et al. Paternal uniparental isodisomy of chromosome 6 causing a complex syndrome including complete IFN-gamma receptor 1 deficiency. Am J Med Genet A. 2010;152A(3):622–629. doi: 10.1002/ajmg.a.33291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sologuren I, et al. Partial recessive IFN-γR1 deficiency: genetic, immunological and clinical features of 14 patients from 11 kindreds. Hum Mol Genet. 2011;20(8):1509–1523. doi: 10.1093/hmg/ddr029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Taramasso L, et al. Pineal germinoma in a child with interferon-γ receptor 1 deficiency. case report and literature review. J Clin Immunol. 2014;34(8):922–927. doi: 10.1007/s10875-014-0098-0. [DOI] [PubMed] [Google Scholar]
- 79.Hoyos-Bachilogue R, et al. A digenic human immunodeficiency characterized by IFNAR1 and IFNGR2 mutations. J Clin Invest. 2017;127(12):4415–4420. doi: 10.1172/JCI93486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Belkaya S, et al. Inherited IL-18BP deficiency in human fulminant viral hepatitis. J Exp Med. 2019;216(8):1777–1790. doi: 10.1084/jem.20190669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kerner G, et al. Homozygosity for TYK2 P1104A underlies tuberculosis in about 1% of patients in a cohort of European ancestry. Proc Natl Acad Sci USA. 2019;116(21):10430–10434. doi: 10.1073/pnas.1903561116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hilton DJ, Zhang JG, Metcalf D, Alexander WS, Nicola NA, Willson TA. Cloning and characterization of a binding subunit of the interleukin 13 receptor that is also a component of the interleukin 4 receptor. Proc Natl Acad Sci USA. 1996;93(1):497–501. doi: 10.1073/pnas.93.1.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Obiri NI, Debinski W, Leonard WJ, Puri RK. Receptor for interleukin 13. Interaction with interleukin 4 by a mechanism that does not involve the common gamma chain shared by receptors for interleukins 2, 4, 7, 9, and 15. J Biol Chem. 1995;270(15):8797–8804. doi: 10.1074/jbc.270.15.8797. [DOI] [PubMed] [Google Scholar]
- 84.Mueller TD, Zhang JL, Sebald W, Duschl A. Structure, binding, and antagonists in the IL-4/IL-13 receptor system. Biochim Biophys Acta. 2002;1592(3):237–250. doi: 10.1016/s0167-4889(02)00318-x. [DOI] [PubMed] [Google Scholar]
- 85.Milford TA, et al. TSLP or IL-7 provide an IL-7Rα signal that is critical for human B lymphopoiesis. Eur J Immunol. 2016;46(9):2155–2161. doi: 10.1002/eji.201646307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Holland SM. Cytokine therapy of mycobacterial infections. Adv Intern Med. 2000;45:431–452. [PubMed] [Google Scholar]
- 87.Alangari AA, et al. Treatment of disseminated mycobacterial infection with high-dose IFN-γ in a patient with IL-12Rβ1 deficiency. Clin Dev Immunol. 2011;2011:691956. doi: 10.1155/2011/691956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.de Jong SJ, et al. The human CIB1-EVER1-EVER2 complex governs keratinocyte-intrinsic immunity to β-papillomaviruses. J Exp Med. 2018;215(9):2289–2310. doi: 10.1084/jem.20170308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Abecasis GR, Cherny SS, Cookson WO, Cardon LR. Merlin — rapid analysis of dense genetic maps using sparse gene flow trees. Nat Genet. 2002;30(1):97–101. doi: 10.1038/ng786. [DOI] [PubMed] [Google Scholar]
- 90.Eilertson KE, Booth JG, Bustamante CD. SnIPRE: selection inference using a Poisson random effects model. PLoS Comput Biol. 2012;8(12):e1002806. doi: 10.1371/journal.pcbi.1002806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Itan Y, et al. The human gene damage index as a gene-level approach to prioritizing exome variants. Proc Natl Acad Sci USA. 2015;112(44):13615–13620. doi: 10.1073/pnas.1518646112. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.