

Abstract
Mycobacterium tuberculosis (M. tuberculosis) has coevolved with humans for millennia and developed multiple mechanisms to evade host immunity. Restoring host immunity in order to improve outcomes and potentially shorten existing therapy will require identification of the full complement by which host immunity is inhibited. Perturbation of host DNA methylation is a mechanism induced by chronic infections such as HIV, HPV, lymphocytic choriomeningitis virus (LCMV), and schistosomiasis to evade host immunity. Here, we evaluated the DNA methylation status of patients with tuberculosis (TB) and their asymptomatic household contacts and found that the patients with TB have DNA hypermethylation of the IL-2/STAT5, TNF/NF-κB, and IFN-γ signaling pathways. We performed methylation-sensitive restriction enzyme–quantitative PCR (MSRE-qPCR) and observed that multiple genes of the IL-12/IFN-γ signaling pathway (IL12B, IL12RB2, TYK2, IFNGR1, JAK1, and JAK2) were hypermethylated in patients with TB. The DNA hypermethylation of these pathways was associated with decreased immune responsiveness with decreased mitogen-induced upregulation of IFN-γ, TNF, IL-6, CXCL9, CXCL10, and IL-1β production. The DNA hypermethylation of the IL-12/IFN-γ pathway was associated with decreased IFN-γ–induced gene expression and decreased IL-12–inducible upregulation of IFN-γ. This study demonstrates that immune cells from patients with TB are characterized by DNA hypermethylation of genes critical to mycobacterial immunity resulting in decreased mycobacteria-specific and nonspecific immune responsiveness.
Keywords: Immunology, Infectious disease
Keywords: Cytokines, Epigenetics, Tuberculosis

Introduction
Tuberculosis (TB) continues to be a global problem, with 10 million cases and over 1 million deaths each year (1). Anti-TB therapy (ATT) requires 6 months of antibiotics, despite the fact that the majority of individuals become culture negative within the first 2 months of therapy. Although multiple mechanisms of TB-induced immunosuppression have been identified, it is unknown why ATT must be continued for months after an individual becomes culture negative. Further, after successful TB therapy, patients have a 13-fold increased risk of disease recurrence compared with the general population (2). Along with strain virulence and environmental risk factors, persistent perturbation of host immunity is one of the likely causes of recurrent TB disease. It is therefore critical to identify mechanisms of host immunosuppression that persist despite successful TB therapy.
TB has been recognized as an archetypical chronic infection since at least 1882. Since 1984, the clone 13 strain of murine lymphocytic choriomeningitis virus (LCMV) has emerged as the prototypical animal model of chronic infection (3). Chronic viral infections with viruses such as HIV, hepatitis B and C, and partially with EBV and CMV, and epitomized by clone 13 LCMV, induce immune exhaustion, which is defined by decreased antigen-induced immune cell proliferation, decreased immune effector function, and increased expression of immune checkpoint inhibitors (4–7). Immune exhaustion is mediated by epigenetic alterations, conformational changes in immune cell DNA methylation, and a chromatin architecture that inhibits robust cell-mediated immunity (8). These epigenetic changes are mediated by DNA methyltransferase (DNMT) and the enhancer of zeste 2 polycomb-repressive (EZH2) complex that imbues a repressive chromatin state, resulting in an immune-nonresponsive state (9–12). DNA methylation is a common and stable epigenetic alteration that blocks transcription factor binding and inhibits gene expression and mediates immune exhaustion (10). Once exhausted, the epigenetic perturbations persist, and the immune phenotype remains subdued even if cells are rested for 3 weeks, or if they are transferred into healthy, noninfected mice (11, 13). Similarly, patients with chronic HIV infection or schistosomiasis have DNA methylation and immune exhaustion phenotypes that persist for months to years after successful therapy (9, 14, 15).
Given that chronic antigen stimulation from HIV, schistosomiasis, or LCMV induces long-lasting epigenetic-mediated immune exhaustion (9, 11, 14–16), we were concerned that TB disease may also result in persistent, detrimental epigenetic changes. Since both TB and LCMV are chronic infections characterized by decreased immune function, we hypothesized that patients with TB would have a DNA methylation landscape similar to that described in chronic LCMV and after schistosomiasis. Mouse models of chronic LCMV have shown persistent epigenetic perturbations, with hypomethylating agents capable of restoring host immunity (10, 11, 13). We thought it pertinent to investigate the longitudinal DNA methylation of host immune cells before and after successful ATT. In a cohort of adults with microbiologically confirmed pulmonary TB from Eswatini, patients with TB exhibited a hyporesponsive immune phenotype with decreased immune proliferation and decreased mycobacterium-induced and mitogen-induced cytokine production in association with DNA hypermethylation of multiple critical immune genes and pathways. These DNA methylation perturbations persisted 6 months after successful ATT and identify a plausible mechanisms of suppressed host immunity.
Results
We previously described a cohort of asymptomatic, TB-exposed children from Eswatini (formerly known as Swaziland) with schistosomiasis-induced DNA hypermethylation and associated inhibition of bacillus Calmette–Guérin (BCG) and Mycobacterium tuberculosis (M. tuberculosis) immunity (15). Here, to evaluate whether TB induces similar DNA methylation perturbations that correspond with immune exhaustion, we assessed adults with TB symptoms and microbiologically confirmed pulmonary TB (by culture and/or GeneXpert). We compared their immune phenotype and DNA methylation status with their asymptomatic, healthy household contacts who remained asymptomatic for 12 months after initial exposure (Figure 1 and Supplemental Table 1; supplemental material available online with this article; https://doi.org/10.1172/JCI134622DS1). All controls were negative for ascaris and schistosomiasis by urine and stool microscopy and quantitative PCR (qPCR) as previously described (15). All study participants were BCG vaccinated, as determined via either vaccine records and/or a BCG scar. An HIV screen was performed at the time of the study’s initiation and then annually thereafter. All included participants had TB treatment success as defined by both WHO criteria and a simplified clinically relevant definition (17).
Figure 1. Patients with TB exhibit an exhausted immune phenotype.

PBMCs from individuals with TB (n = 29) and their asymptomatic household contacts (HC) (n = 49) were subjected to antigenic stimulation with BCG sonicate followed by flow cytometry–based multidimensional immune profiling. CITRUS clustering was implemented on CD3, CD4, CD8, and CD56 surface markers with a minimum cluster size of 10% per node. Lines indicate the median and interquartile range, and P values were determined by nonparametric Mann-Whitney U test. (A) Citrus clustering identified Th (CD3+CD4+), CTL (CD3+CD8+), and NK (CD3–CD56+) cell populations, with CD3 signal depicted by a heatmap. Gray-shaded subclusters have an FDR of less than 1% and depict decreased IFN-γ and Ki-67 expression among Th cells (CD3+CD4+), CTLs (CD3+CD8+), and NK cells (CD3–CD56+). Representative dot plots of the MFI clusters are depicted from the yellow highlighted clusters. (B) Reanalysis of CITRUS, with exclusion of HIV-coinfected individuals, for patients with TB (n = 20) and controls (n = 40) shows CD4+, CD8+, and CD56+ lymphocyte subsets with decreased Ki-67 and IFN-γ (gray-shaded clusters have decreased IFN-γ and Ki-67, with an FDR <1%; representative dot plots of MFI clusters are depicted). Mann-Whitney U test P values describe MFI differences between controls and patients with TB.
Patients with TB demonstrate immune exhaustion phenotypes.
To evaluate the cell-specific and mycobacterium-specific immunity of patients with TB, PBMCs were stimulated with ESAT-6 and CFP-10 (M. tuberculosis–specific antigens) or BCG sonicate followed by flow cytometry–based multidimensional immune profiling (MDIP). Cells were stained subsequently for viability and cell surface and intracellular markers. Multidimensional immune changes were evaluated using CITRUS (clustering identification, characterization, and regression) (18). Clustering was implemented via CITRUS using the surface markers CD3, CD4, CD8, and CD56 to identify subsets of Th cells (CD3+CD4+), cytotoxic T lymphocytes (CTLs) (CD3+CD8+), and NK cells (CD3–CD56+) (Figure 1). The median fluorescence intensity (MFI) of each node was characterized between healthy controls (HCs) and individuals with TB. CITRUS revealed that, compared with controls, participants with TB had Th cells, CTLs, and NK cells with decreased IFN-γ and proliferative capacity in response to both M. tuberculosis–specific antigens (ESAT-6 and CFP-10) and BCG (Figure 1A, Supplemental Figure 1A, and Supplemental Table 1). Compared with controls, individuals with TB were more likely to be coinfected with HIV, however, after excluding people living with HIV (PLWH), participants with TB still had lymphocytes (CD4+, CD8+, and CD3–CD56+) with decreased IFN-γ and proliferative capacity (Figure 1B and Supplemental Figure 1). Study participants with TB who were not HIV coinfected had a 21% and 25% decrease in BCG-induced IFN-γ production compared with HIV-uninfected controls, respectively, for CD4+ and CD8+ T cell populations (Figure 1B). Similarly, participants with TB who were not infected with HIV had a 16% and 14% decrease in BCG-induced Ki-67 expression, respectively, for CD4+ and CD8+ T cell populations (Figure 1B). In response to the M. tuberculosis–specific peptides ESAT-6 and CFP-10, patients with TB had CD4+ and CD8+ T cell populations with 11% and 45% decreased IFN-γ production and 12% and 19% decreased Ki-67 expression, respectively (Supplemental Figure 1A).
The decrease in proliferative capacity was not mycobacterium-specific, as the superantigen staphylococcal enterotoxin B (SEB) also induced decreased Ki-67 upregulation (SEB; Supplemental Figure 1B). The decrease in SEB-induced IFN-γ was not statistically significant. Increased expression of immune checkpoint blockade, as measured by programmed cell death 1 (PD-1), cytotoxic T lymphocyte–associated protein 4 (CTLA4), and/or T cell immunoglobulin mucin-3 (TIM3), is another characteristic of immune exhaustion and similar to what has been described in previous reports (19, 20). Further, compared with asymptomatic HCs, we found that patients with TB had an increased abundance of PD-1–expressing NK cells (Supplemental Figure 1C).
The TB DNA methylome resembles the immune exhaustion epigenetic landscape.
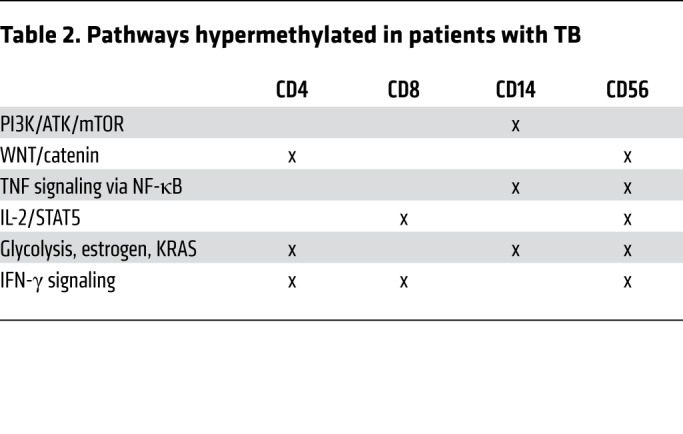
Epigenetic mechanisms mediate immune exhaustion (4, 5, 10, 21), and we therefore evaluated the DNA methylation status of participants with TB using the EPIC array. From bulk PBMCs, we determined cell-specific DNA methylation using epigenetic deconvolution (EDEC) (Figure 2A) (22). EDEC identified global differential methylation changes in Th cells, CTLs, NK cells, and monocytes both at baseline and 6 months after completion of successful ATT, 12 months after study enrollment (Figure 2D). Pathway enrichment (Figure 2B) indicated that the IL-2/STAT5 pathway, a critical component of cell proliferation, was differentially methylated at baseline and 6 months after successful therapy in all lymphocyte subsets from patients with TB compared with controls. The PI3K/AKT pathway, which modulates intracellular signaling of the immunometabolic pathway in both adaptive and innate cells, was differentially methylated in all cell types except CD4+ T cells. Similarly, the TNF/NF-κB signaling pathway was differentially methylated in all evaluated cell types except Th cells. IFN-γ signaling, critical to antimycobacterial immunity, was differentially methylated in all cell types at baseline and 6 months after the completion of ATT (Figure 2B). Specifically, compared with HCs, patients with TB had DNA hypermethylation of the IFN-γ signaling pathway for all lymphocyte subsets (CD4+ Th cells, CD3+CD8+ CTLs, and CD3–CD56+ NK cells), whereas this pathway was hypomethylated in monocytes (Figure 3, Tables 1 and 2, and Supplemental Table 2).
Figure 2. Global DNA methylation perturbations persist 6 months after successful ATT.

The Illumina DNA Methylation EPIC array was performed on bulk PBMCs from asymptomatic household contacts (n = 10) and individuals with TB (n = 15; TB/HIV– = 7; TB/HIV+ = 8) at baseline. All individuals with TB had treatment success. For individuals without HIV coinfection, DNA methylation status was evaluated at baseline and 12 months later, 6 months after completion of successful ATT. (A) To ascertain the immune cell–specific DNA methylation changes, cell-specific DNA methylation reference profiles were downloaded from public archives, informative loci were identified, and cell type–specific methylation profiles were identified. (B) Cell-specific DNA methylation differences are shown for TB patients (base) and TB/HIV (HIV) patients at baseline and for TB patients 12 months after baseline (12 mo), 6 months after completion of successful ATT. All results were compared with HC data. GO pathway analysis for monocytes (CD14+), Th cells (CD3–CD4+), NK cells (CD3–CD56+), and CTLs (CD3+CD8+). (C and D) Purified Th cells (CD3+CD4+) (n = 2) and EDEC-identified Th cells (CD3+CD4+) (n = 8 TB patients; n = 10 controls) were compared with cells from controls, and common hypermethylated pathways and genes were identified.
Figure 3. The TB epigenetic landscape is similar to LCMV-induced immune exhaustion.

(A) DNA hypermethylation of genes in CD3+CD8+ CTLs in TB patients (green), and that reported by Ghoneim et al. (10) (red) and Sen et al. (4) (blue). (B) Genes and GO pathways of CD8+ CTLs with DNA hypermethylation for patients with TB from this cohort were compared with genes with DNA hypermethylation, reported by Ghoneim et al. (10), and ChARs induced by chronic LCMV–induced immune exhaustion, reported by Sen et al. (4).
Table 1. Genes hypermethylated in patients with TB.
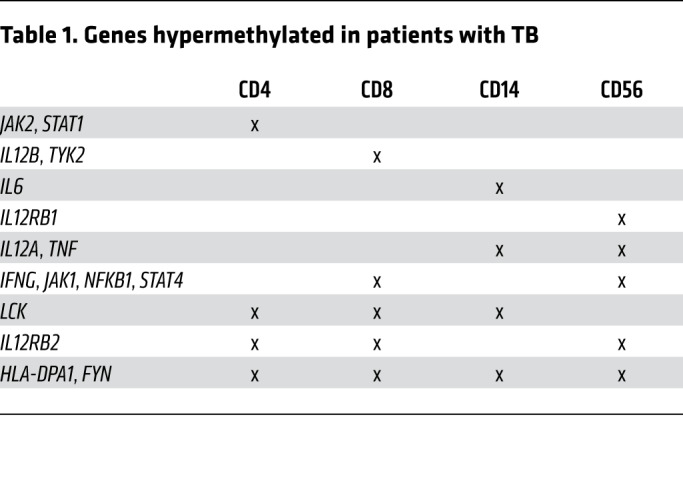
Table 2. Pathways hypermethylated in patients with TB.
To validate the EDEC results, we isolated CD4+ T cells and evaluated DNA methylation at baseline and 6 months after successful ATT. Of note, cell proliferation, intracellular calcium signaling, and IL-2/STAT5 and IFN-γ pathways were hypermethylated in both isolated CD4+ T cells and the deconvoluted CD4+ T cells (Figure 2C). Analysis of isolated CD4+ T cells confirmed the results from EDEC for 82% of hypermethylated genes (probability of overlap = 9.3 × 10–5). Baseline DNA hypermethylation, as identified by EDEC and in isolated CD4+ T cells, was observed in JAK1, IL12RB2, STAT1, FYN, GATA2, IKZF2, and TOX (Figure 2D).
Previous studies of chronic LCMV, cancer, HIV, and schistosomiasis identified persistent epigenetic-mediated immune exhaustion long after removal of the chronic antigen stimulation (9, 11, 14, 15). DNA hypermethylation in patients with TB, both at baseline and 6 months after successful ATT, resembled the closed chromatin confirmation induced by chronic LCMV infection models. We evaluated the similarities between the epigenetic landscape of murine LCMV and that of patients with TB by comparing the closed chromatin–accessible regions (ChARs) previously published by Sen et al. (4) and the DNA hypermethylated regions previously published by Ghoneim et al. (10) from chronic LCMV models with the CD8 DNA hypermethylated regions of TB patients from this study. In particular, we observed DNA hypermethylation and closed chromatin conformation of the IFN-γ pathway (IL12RB2, JAK1, STAT4) in both TB and chronic LCMV (Figure 3, A and B). We performed immunologic gene set enrichment analysis and found that CD8+ T cells from patients with TB were enriched (P = 4.41 × 10–11) to a degree similar to that of genes downregulated in LCMV-induced immune exhaustion (23). In addition to DNA methylation changes being induced by chronic antigenic exposure, M. tuberculosis itself releases epigenetic-modifying enzymes that target host chromatin and DNA methylation. For example, Rv2966c is a DNMT that is secreted by M. tuberculosis and chaperoned into host immune cell nuclei (24). In in vitro models, Rv2966c induces DNA hypermethylation of host immune cells (25) in a manner similar to that seen in this cohort of TB patients, including hypermethylation of IL12RB2, STAT4, IFNG, IRF1, and JAK1 (Supplemental Figure 2).
Patients with TB exhibit DNA hypermethylation of the IL-12/IFN-γ signaling pathway.
To validate the DNA methylome results, we implemented targeted methylation-specific restriction endonuclease qPCR (MSRE-qPCR) on genes previously relevant to mycobacterial biology (26). Although the EPIC array evaluates DNA methylation via bisulfite conversion, MSRE quantifies the methylation status using endonuclease isochizomers that degrade DNA on the basis of methylation status. MSRE-qPCR of isolated CD4+ T cells confirmed DNA hypermethylation of the IL-12/IFN-γ pathway (Figure 4). Specifically, IL12B demonstrated 6.1-fold increased methylation (P = 0.007); IL12RB2 was hypermethylated 35.2-fold (P < 0.03); TYK2 demonstrated 17.7-fold increased methylation (P = 0.008); IFNGR1 was hypermethylated 24.0-fold (P = 0.007); and JAK1 and JAK2 demonstrated 33.3- and 11.7-fold increased DNA methylation (P = 0.004 and 0.03), respectively (Figure 4). STAT1 and IRF1 were not statistically hypermethylated (P = 0.21 and 0.15, respectively; Figure 4B). Regulation of immunity is epigenetically controlled at specific pathways, but also by a network of transcription factors (27). MSRE-qPCR revealed that CD4+ T cells from patients with TB had DNA hypermethylation of the transcription factors TOX, NFATC1, BATF3, ID2, PPARG, RUNX2, IRF5, and IKZF1 (P < 0.001; Figure 4B).
Figure 4. Targeted DNA methylation confirms DNA hypermethylation of the IL-12/IFN-γ pathway.

(A) Graphic representation of the IL-12/IFN-γ pathway. (B) DNA methylation was evaluated using MSRE-qPCR in non–HIV-coinfected TB patients (n = 6) and their asymptomatic healthy household contacts (n = 5). *P < 0.01, by Mann-Whitney U test.
Patients with TB have decreased mitogen responsiveness.
Patients with TB had DNA hypermethylation of IL-2, IL-6, TNF, and IFN-γ signaling pathways (Figures 2–4). Notably, the TNF/NF-κB signaling pathway and the TNF gene were hypermethylated in CD14+, CD8+, and CD56+ cells from patients with TB (Figure 3). Given that these pathways result in activation of MAPK and are also downstream of mitogen activation, we hypothesized that patients with TB would have decreased mitogen responsiveness. Freshly collected whole blood was stimulated overnight with mitogen (phytohemagglutinin), and a multiplexed ELISA was performed on the supernatant. Although the baseline production of TNF was similar (3.7 vs 1.4 pg/mL; P = 0.2), after mitogen stimulation, the patients with TB exhibited 72% decreased upregulation of TNF by ELISA (80.5 vs. 293 pg/mL; P = 0.01; Figure 5). The IFN-γ signaling pathway was hypermethylated in all lymphocytes, and the IFNG gene was hypermethylated in CD8 and CD56 cells from patients with TB, and although patients with TB had increased IFN-γ at baseline by ELISA (1.31 vs. 0.58 pg/mL, P = 0.007), they had a 35.4% decreased capacity to upregulate IFN-γ upon mitogen activation (increase of 246.9 vs. 382.4 pg/mL; P = 0.03). The chemokines CXCL9 and CXCL10 are downstream of IFN-γ, and like IFN-γ, they were increased at baseline (42.5 vs. 10.6 and 238.3 vs. 117.4 pg/mL; P < 0.01) but had 30.7% and 30.1% decreased upregulation after mitogen-induced activation (P = 0.01 and 0.002, respectively). IL-6 and IL-1β levels were comparable at baseline, but patients with TB had 40.5% and 60.7% decreased mitogen-induced upregulation of IL-6 (8870 vs. 14,917 pg/mL; P < 0.001) and IL-1β (1968 vs. 5974 pg/mL, P = 0.0005), respectively.
Figure 5. Decreased mitogen-induced responsiveness.

Fresh whole blood (0.8–1.2 mL) from patients with TB (n = 40) and asymptomatic healthy household contacts (n = 39) was stimulated overnight with and without mitogen, and the resultant plasma was evaluated for cytokine and chemokine upregulation using a custom-designed, bead-based multiplex ELISA. P values were determined by nonparametric Mann-Whitney U test. FC, fold change.
Patients with TB have decreased IFN-γ–induced gene upregulation.
An intact IL-12/IFN-γ pathway is necessary, but not sufficient, for control of mycobacterial infections (26, 28). Since patients with TB exhibited DNA hypermethylation of the IL-12/IFN-γ signaling pathway in CD4+ Th cells, NK cells, and CTLs, we postulated that they would also exhibit decreased IFN-γ–inducible gene upregulation using a previously described 10-gene IFN-γ–inducible score (15, 29, 30). In healthy immune cells, IFN-γ stimulation induces gene expression of CITTA, GBP1, STAT1, CXCL9, CXCL10, CXCL11, IL15, SERPING1, IDO1, and FCGR1A/B (29); however, previous studies have demonstrated decreased IFN-γ–inducible gene expression in cancer (30) and chronic schistosomiasis infection (15). In this cohort, compared with controls, the PBMCs from patients with TB had 69.7% decreased IFN-γ–inducible gene upregulation (17.9- vs. 59.2-fold increase; P = 0.02; Figure 6).
Figure 6. Decreased IFN-γ–inducible gene expression.

PBMCs (1 × 106) from patients with TB (n = 10) and asymptomatic healthy household contacts (n = 10) were cultured overnight with and without 50 ng IFN-γ. Then, RNA was isolated, and IFN-γ–inducible gene expression was evaluated by microarray. P = 0.02, by Mann-Whitney U test.
Patients with TB have decreased IL-12–inducible IFN-γ production.
Lymphocytes are primed with IL-12 for robust IFN-γ production (31). Since patients with TB showed DNA hypermethylation of the IL-12 pathway (IL12A, IL12B, IL12RB1, and IL12RB2), we evaluated the functionality of the IL-12/IFN-γ axis by stimulating PBMCs overnight with BCG, with and without IL-12. Although Th cell (CD3+CD4+) IFN-γ production was similar, patients with TB had a muted IL-12–induced production of IFN-γ in CTLs and NK cells. Specifically, NK cells had 62.5% decreased IL-12–inducible upregulation of IFN-γ (P = 0.008), whereas CTLs had a 43.4% decrease in IL-12–induced IFN-γ upregulation (P = 0.03; Figure 7 and Supplemental Figure 3).
Figure 7. Decreased IL-12–inducible IFN-γ production.

PBMCs from patients with TB (n = 15) and asymptomatic healthy household contacts (n = 10) were treated overnight with PBS media control (Nil stim), or were stimulated with BCG sonicate (5 μg) or IL-12 (20 ng/mL) and BCG (minimum of 0.5 × 106 PBMCs per condition) followed by multidimensional flow cytometry. See Supplemental Figure 3 for the gating strategy and representative dot plots of CD3+CD4+ and CD3+CD8+ T cells. (A) Representative dot plots of NK cell (CD3–CD56+) production of IFN-γ and TNF-α after no stimulation or stimulation with BCG or with IL-12 plus BCG. (B) t-distributed stochastic neighbor embedding (tSNE) dimension reduction clustering of PBMCs from a control and from patients with TB demonstrated the cell-specific (CD3, CD4, CD8, and CD56) contribution of IFN-γ production after combined IL-12 and BCG stimulation, with blue indicating no protein expression, and green, yellow, and red indicating increasing protein expression. (C and D) Dot plots demonstrate the percentage of CTLs (CD3+CD8+) and NK cells (CD3–CD56+) that produced IFN-γ after stimulation with IL-12 and BCG. P = 0.0308 and 0.008, by Mann-Whitney U test, for CTLs and NK cells, respectively.
Discussion
Despite becoming culture negative after 2 months of antituberculosis therapy, patients with TB require 6 months of antibiotics (32). However, even after these 6 months of antibiotics, patients with successful ATT have an increased risk of recurrent TB compared with the general population (2). Epigenetic-mediated immune exhaustion has been demonstrated to impede immunotherapy in murine LCMV experimental models (10), but it has not previously been evaluated as a potential etiology of TB-induced immune exhaustion. Here we demonstrate that patients with TB have DNA hypermethylation in critical immune pathways including the IL-12/IFN-γ pathway and that this is associated with decreased mitogen, mycobacterial, IL-12, and IFN-γ immune responsiveness.
Depending on the definition applied for anergy, approximately 5%–25% of patients with TB fail to mount a positive skin reaction to tuberculin, or fail to release IFN-γ after exposure to mycobacterial antigens (33–39). Patients with TB who fail to produce M. tuberculosis–specific IFN-γ have increased mortality (40), potentially indicating more advanced disease. The TB-induced anergy is not specific to mycobacterial antigens, as patients with TB also exhibit decreased responsiveness to Candida and histoplasmin antigens (41). HIV increases the likelihood of anergy (36), but in some cohorts, up to 46% of HIV– TB patients are anergic (42). Modern immune analysis has characterized TB-induced immune exhaustion demonstrating that patients with TB have decreased IFN-γ and IL-2 expression and proliferative capacity and increased expression of immune checkpoint inhibitors such as PD-1, TIM3, and CTLA4 (19, 20, 43, 44). In addition to limiting critical cytokines, previous studies have demonstrated that patients with TB have dysfunctional intracellular signaling downstream of IFN-γ with decreased upregulation of IFN-γ–inducible gene expression (45, 46). DNA hypermethylation has been shown to inhibit robust immunity in LCMV models, and here we demonstrate that DNA hypermethylation was associated with decreased immune responsiveness during TB.
Immune exhaustion after chronic antigen stimulation is not limited to TB and has been observed in many instances of persistent inflammation due to chronic infection, sepsis, or cancer (6, 47–49). Continuously expressed tumor antigens induce immune exhaustion via upregulation of DNMTs and the transcriptionally repressive EZH2 complex, both known moderators of epigenetic-mediated immune exhaustion (13, 21). Epigenetic-mediated immune exhaustion persists even if cells are rested for 3 weeks, or if they are transferred into healthy, noninfected mice (11, 13). In clinical cohorts, even 2 years after successful aviremia, patients infected with HIV retain detrimental epigenetic marks (9, 14). Similarly, 6 months after successful deworming, children previously infected with schistosomiasis retained detrimental DNA methylation marks (15). In this report, 6 months after completing successful ATT, the immune system showed persistent DNA hypermethylation perturbations. Patients in this cohort were only followed 6 months after completion of ATT, and longitudinal studies will need to assess whether persistent detrimental DNA methylation marks correlate with recurrent TB disease.
Chronic LCMV models have demonstrated that immune exhaustion is moderated by broad remodeling of the epigenetic landscape and that these epigenetic perturbations encumber immune checkpoint blockade–based immunotherapy (4, 5). Ghoneim et al. demonstrated that immune exhaustion is moderated by DNA methylation with hypomethylating agents dosed before immune checkpoint inhibitor therapy capable of reversing epigenetic-mediated immune exhaustion (10). These data demonstrate that the DNA hypermethylation landscape of patients with TB before and after successful ATT resembles murine LCMV-induced immune exhaustion regions of closed chromatin by chronic LCMV (45) and DNA hypermethylation (10). In addition to chronic antigen-induced epigenetic-mediated immune exhaustion, an alternative and nonmutually exclusive mechanism of host epigenetic perturbations is pathogen production of host epigenetic–modifying enzymes. Leishmania, HIV, HPV, and M. tuberculosis all produce epigenetic-modifying enzymes that target host epigenetic status (24, 25, 46, 50–54). M. tuberculosis produces a functional DNMT that is secreted and chaperoned into the nucleus, inducing host DNA methylation changes in the IL-12/IFN-γ signaling pathway (24, 25) similar to the DNA methylation changes observed in this cohort (Supplemental Figure 2). The current study did not evaluate the M. tuberculosis strain for DNMT production, but future studies should evaluate the effect of strain on host DNA methylation in clinical cohorts.
DNA hypermethylation of the IL-12/IFN-γ signaling pathway, especially among CD8+ T cell and NK cell populations, may partially explain the complexity of identifying IFN-γ–mediated immune correlates of protection. The Mendelian susceptibility to mycobacterial diseases (MSMDs) are a rare collection of gene mutations that increase an individual’s risk for TB and other intracellular infections. The MSMDs highlight the critical nature and complexity of this pathway. Upstream mutations of IL12B, IL12RB, or NEMO result in decreased IFN-γ, decreased IFN-γ–inducible gene expression, and decreased mycobacterial killing capacity. In contrast, mutations of IFNGR1, IFNGR2, or STAT1 downstream of IFN-γ result in increased IFN-γ but decreased IFN-γ–inducible gene expression and decreased mycobacterial killing capacity. Here, we demonstrate that patients with TB have increased DNA methylation in the canonical Il-12/IFN-γ pathway (Figure 4A) both upstream (in IL12, IL12RB2, and STAT4) and downstream (in IFNG, JAK1, and STAT1) of IFN-γ (Figures 3 and 4). However, the IFNG gene expression response is dependent on both canonical and noncanonical signaling. Noncanonical modulators of IFN-γ–inducible gene expression include FYN, MAL, and the mTOR pathway, which were also hypermethylated in patients with TB. DNA hypermethylation of the canonical and noncanonical IL-12/IFN-γ pathways was associated with reduced IFN-γ–inducible gene expression (Figure 6), reduced mitogen-induced cytokine production (Figure 5), and reduced IL-12–inducible IFN-γ production (Figure 7).
Host-directed immunotherapy has been identified as a priority research area to improve TB treatment outcomes for the 10 million people afflicted each year. TB is an archetypical chronic infection known to inhibit host immunity, and here we demonstrate that decreased immune responsiveness to IL-12, IFN-γ, mitogen, and mycobacterial antigens is associated with DNA hypermethylation of critical immune transcription factors and the canonical and noncanonical IL-12/IFN-γ signaling pathways. The persistent DNA methylation perturbations we have identified are a plausible explanation for why successful TB therapy must be continued for months after M. tuberculosis culture conversion. These data suggest that it is critical to evaluate whether modulating DNA methylation could effectively augment host antimycobacterial immunity.
Methods
Multidimensional immune profiling.
PBMCs were isolated using Ficoll separation and cryopreserved in liquid nitrogen. Upon thawing, greater than 70% viability was determined by flow cytometry using trypan blue staining and lymphocyte amine reactive dye. PBMCs were stimulated under the following conditions: (a) DMSO vehicular control, (b) 2.5 μg each of ESAT-6 and CFP-10 overlapping peptide pools, (c) 5 μg BCG sonicate, and (d) 0.2 μL staphylococcal enterotoxin B. Stimulations took place in the presence of costimulation (CD28 and CD49) for 18 hours with monensin and brefeldin A for the last 12 hours. Cells were stained with an amine reactive dye (Ghost Dye), surface antibodies (CD4, CD8, CD56, and PD-1), and intracellular antibodies (CD3, Ki-67, IFN-γ, TNF, IL-4, IL-10, IL-13, perforin, T-bet, and GATA-3) and acquired on a BD LSR II Fortessa. Complete antibody information can be found in Supplemental Table 3. The following reagents were obtained through BEI Resources, NIAID, NIH: peptide array, M. tuberculosis ESAT-6 protein, NR-50711; and peptide array, M. tuberculosis CFP-10 protein, NR-50712.
DNA methylation.
Genomic DNA (gDNA) was isolated from 1 × 106 PBMCs or magnetic-bead isolated CD4+ T cells with a minimum of 80% viability using the DNeasy kit (QIAGEN). Nucleic acid quantity and quality were examined using a Qubit 3.0 fluorometer and an Agilent 4200 TapeStation System, and 500 ng gDNA was treated with bisulfite before running the Illumina Infinium MethylEPIC array. Methylation IDAT files were preprocessed and normalized using Bioconductor’s R statistical minfi package. Probes with greater than 2-fold or less than 0.5-fold differential methylation with P values of less than 0.05 were considered significant. Gene ontology (GO) and Gene Set Enrichment Analysis were implemented using the Molecular Signature Database (MSigDB) with hypergeometric distribution that accounted for multiple comparisons (55). Epigenetic deconvolution was performed as previously described (56). The EpiTect II DNA Methylation Enzyme kit (QIAGEN) was used to validate methylome results as previously described (15). In brief, gDNA underwent methylation-specific enzymatic digestion, and qPCR was performed to determine the percentage of methylated and unmethylated DNA relative to mock and double digestion (15, 57). The DNA MethylEPIC data were deposited in the NCBI’s Gene Expression Omnibus (GEO) database (GEO GSE145714).
ELISA.
Fresh whole blood (0.4 mL per condition) from patients with TB and their asymptomatic, healthy household contacts was stimulated overnight with and without mitogen (phytohemagglutinin), and the resultant supernatant was evaluated for IFN-γ, TNF, IL-6, CXCL9, CXCL10, and IL-1β using a customized bead-based multiplex ELISA (LEGENDplex kits, BioLegend).
Gene expression.
PBMCs were stimulated overnight with IFN-γ (50 ng/mL) or media control (PBS) for 16 hours followed by RNA isolation, and gene expression was evaluated using the NanoString nCounter Human Immunology 2 probe set. Data were normalized using 8 negative controls and 10 housekeeping genes.
Statistics.
Flow cytometric data were processed using FlowJo (Tree Star) and GraphPad Prism 6.0 (GraphPad Software). CITRUS was implemented using CytoBank (http://www.cytobank.org) using nearest shrunken centroid classification (PAMR) with a minimum cluster size of 10%, a cross validation fold of 10, and a FDR of 1%. DNA methylEPIC probes were considered significant if they had greater than 2- or less than 0.5-fold differences compared with controls, with a P value of less than 0.05 considered significant. Gene expression, flow cytometry, ELISA, and targeted MSRE DNA methylation data were evaluated using a nonparametric Mann-Whitney U test, with a P value of less than 0.5 considered significant and violin plots showing the data distribution with the median and quartiles delineated.
Study approval.
The study protocol was approved by the Baylor College of Medicine Children’s Foundation-Swaziland and the Eswatini National Health Research and Review Board. All participation was voluntary and implemented in accordance with institutional and Declaration of Helsinki guidelines for the protection of human subjects, and written informed consent was obtained from all participants or their guardians. In this analysis, immune cells from patients with TB were analyzed if they had pulmonary disease as defined by symptoms of TB (cough, fevers, weight loss, etc.) and microbiologically confirmed disease by sputum Gene Xpert test and/or culture. All participants were screened for HIV. Individuals with helminth coinfection (by urine or stool microscopy and urine and stool qPCR) were excluded from this analysis. HCs were household contacts of the TB patients with a TB contact score (58) of 5 or greater and excluded if they progressed to active TB and did not remain asymptomatic for 12 months. All individuals were BCG vaccinated as confirmed by BCG scar and/or vaccine card.
Author contributions
ARD and AMM designed the study. G Mtetwa, G Maphalala, AK, ARD, QD, and JK implemented cohort enrollment and designed the clinical study. TN, ARD, QD, and JK performed the immunology and DNA methylation studies. SM, SLG, and ARD performed CITRUS analysis. KR, SLG, and CC conducted the bioinformatic and statistical analyses. ARD, AMM, G Makedonas, EMM, JDC, CC, MGN, and RVC assisted with data interpretation and designing of the biologic validation studies.
Supplementary Material
Acknowledgments
This work was supported by Texas Children’s Hospital, the Burroughs Wellcome Fund/American Society of Tropical Medicine and Hygiene Postdoctoral Fellowship in Tropical Infectious Diseases, the Thrasher Research Fund, and the NIH (K23AI141681A). This project was supported by the Genomic and RNA Profiling Core at Baylor College of Medicine and the Cancer Prevention Institute of Texas (CPRIT) (grant RP170005), the NIH (P30 shared resource grant CA125123), and the National Institute of Environmental Health Sciences (NIEHS), NIH (P30 Center Core grant 1P30ES030285).
Version 1. 03/03/2020
In-Press Preview
Version 2. 05/11/2020
Electronic publication
Version 3. 06/01/2020
Print issue publication
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Copyright: © 2020, American Society for Clinical Investigation.
Reference information: J Clin Invest. 2020;130(6):3113–3123.https://doi.org/10.1172/JCI134622.
Contributor Information
Kimal Rajapakshe, Email: Kimal.Rajapakshe@bcm.edu.
Tomoki Nishiguchi, Email: nishiguc@bcm.edu.
Sandra L. Grimm, Email: sgrimm@bcm.edu.
Godwin Mtetwa, Email: godwinmtetwa@yahoo.com.
Qiniso Dlamini, Email: qinisod@baylorswaziland.org.sz.
Sanjana Mahapatra, Email: sanjanam@buffalo.edu.
Gugu Maphalala, Email: gpmaph@hotmail.com.
Emily M. Mace, Email: em3375@cumc.columbia.edu.
George Makedonas, Email: George.Makedonas@bcm.edu.
Jeffrey D. Cirillo, Email: jdcirillo9@gmail.com.
Reinout van Crevel, Email: reinout.vancrevel@radboudumc.nl.
Cristian Coarfa, Email: coarfa@bcm.edu.
Anna M. Mandalakas, Email: anna.mandalakas@bcm.edu.
References
- 1. WHO. Global Tuberculosis Report 2016. Geneva, Switzerland: World Health Organization; 2016. [Google Scholar]
- 2.Millet JP, et al. Tuberculosis recurrence after completion treatment in a European city: reinfection or relapse? PLoS One. 2013;8(6):e64898. doi: 10.1371/journal.pone.0064898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ahmed R, Salmi A, Butler LD, Chiller JM, Oldstone MB. Selection of genetic variants of lymphocytic choriomeningitis virus in spleens of persistently infected mice. Role in suppression of cytotoxic T lymphocyte response and viral persistence. J Exp Med. 1984;160(2):521–540. doi: 10.1084/jem.160.2.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sen DR, et al. The epigenetic landscape of T cell exhaustion. Science. 2016;354(6316):1165–1169. doi: 10.1126/science.aae0491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pauken KE, et al. Epigenetic stability of exhausted T cells limits durability of reinvigoration by PD-1 blockade. Science. 2016;354(6316):1160–1165. doi: 10.1126/science.aaf2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Martinez GJ, et al. The transcription factor NFAT promotes exhaustion of activated CD8+ T cells. Immunity. 2015;42(2):265–278. doi: 10.1016/j.immuni.2015.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kahan SM, Wherry EJ, Zajac AJ. T cell exhaustion during persistent viral infections. Virology. 2015;479–480:180–193. doi: 10.1016/j.virol.2014.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pauken KE, Wherry EJ. SnapShot: T cell exhaustion. Cell. 2015;163(4):1038–1038.e1. doi: 10.1016/j.cell.2015.10.054. [DOI] [PubMed] [Google Scholar]
- 9.Nakayama-Hosoya K, et al. Epigenetic repression of interleukin 2 expression in senescent CD4+ T cells during chronic HIV type 1 infection. J Infect Dis. 2015;211(1):28–39. doi: 10.1093/infdis/jiu376. [DOI] [PubMed] [Google Scholar]
- 10.Ghoneim HE, et al. De novo epigenetic programs inhibit PD-1 blockade-mediated T cell rejuvenation. Cell. 2017;170(1):142–157.e19. doi: 10.1016/j.cell.2017.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ahn E, Youngblood B, Lee J, Lee J, Sarkar S, Ahmed R. Demethylation of the PD-1 promoter is imprinted during the effector phase of CD8 T cell exhaustion. J Virol. 2016;90(19):8934–8946. doi: 10.1128/JVI.00798-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Miller BC, et al. Subsets of exhausted CD8+ T cells differentially mediate tumor control and respond to checkpoint blockade. Nat Immunol. 2019;20(3):326–336. doi: 10.1038/s41590-019-0312-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schietinger A, et al. Tumor-specific T cell dysfunction Is a dynamic antigen-driven differentiation program Initiated early during tumorigenesis. Immunity. 2016;45(2):389–401. doi: 10.1016/j.immuni.2016.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Youngblood B, et al. Cutting edge: prolonged exposure to HIV reinforces a poised epigenetic program for PD-1 expression in virus-specific CD8 T cells. J Immunol. 2013;191(2):540–544. doi: 10.4049/jimmunol.1203161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.DiNardo AR, et al. Schistosomiasis induces persistent DNA methylation and tuberculosis-specific immune changes. J Immunol. 2018;201(1):124–133. doi: 10.4049/jimmunol.1800101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ringeard M, Marchand V, Decroly E, Motorin Y, Bennasser Y. FTSJ3 is an RNA 2’-O-methyltransferase recruited by HIV to avoid innate immune sensing. Nature. 2019;565(7740):500–504. doi: 10.1038/s41586-018-0841-4. [DOI] [PubMed] [Google Scholar]
- 17.Günther G, et al. Treatment outcomes in multidrug-resistant tuberculosis. N Engl J Med. 2016;375(11):1103–1105. doi: 10.1056/NEJMc1603274. [DOI] [PubMed] [Google Scholar]
- 18.Bruggner RV, Bodenmiller B, Dill DL, Tibshirani RJ, Nolan GP. Automated identification of stratifying signatures in cellular subpopulations. Proc Natl Acad Sci USA. 2014;111(26):E2770–E2777. doi: 10.1073/pnas.1408792111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Singh A, Mohan A, Dey AB, Mitra DK. Inhibiting the programmed death 1 pathway rescues Mycobacterium tuberculosis-specific interferon γ-producing T cells from apoptosis in patients with pulmonary tuberculosis. J Infect Dis. 2013;208(4):603–615. doi: 10.1093/infdis/jit206. [DOI] [PubMed] [Google Scholar]
- 20.Day CL, et al. PD-1 expression on Mycobacterium tuberculosis-specific CD4 T cells is associated with bacterial load in human tuberculosis. Front Immunol. 2018;9:1995. doi: 10.3389/fimmu.2018.01995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Viré E, et al. The Polycomb group protein EZH2 directly controls DNA methylation. Nature. 2006;439(7078):871–874. doi: 10.1038/nature04431. [DOI] [PubMed] [Google Scholar]
- 22.Onuchic V, et al. Epigenomic deconvolution of breast tumors reveals metabolic coupling between constituent cell types. Cell Rep. 2016;17(8):2075–2086. doi: 10.1016/j.celrep.2016.10.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wherry EJ, et al. Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity. 2007;27(4):670–684. doi: 10.1016/j.immuni.2007.09.006. [DOI] [PubMed] [Google Scholar]
- 24.Sharma G, Upadhyay S, Srilalitha M, Nandicoori VK, Khosla S. The interaction of mycobacterial protein Rv2966c with host chromatin is mediated through non-CpG methylation and histone H3/H4 binding. Nucleic Acids Res. 2015;43(8):3922–3937. doi: 10.1093/nar/gkv261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sharma G, et al. Genome-wide non-CpG methylation of the host genome during M. tuberculosis infection. Sci Rep. 2016;6:25006. doi: 10.1038/srep25006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bustamante J, Boisson-Dupuis S, Abel L, Casanova JL. Mendelian susceptibility to mycobacterial disease: genetic, immunological, and clinical features of inborn errors of IFN-γ immunity. Semin Immunol. 2014;26(6):454–470. doi: 10.1016/j.smim.2014.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Evans CM, Jenner RG. Transcription factor interplay in T helper cell differentiation. Brief Funct Genomics. 2013;12(6):499–511. doi: 10.1093/bfgp/elt025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Orange JS, et al. Human nuclear factor kappa B essential modulator mutation can result in immunodeficiency without ectodermal dysplasia. J Allergy Clin Immunol. 2004;114(3):650–656. doi: 10.1016/j.jaci.2004.06.052. [DOI] [PubMed] [Google Scholar]
- 29.Waddell SJ, et al. Dissecting interferon-induced transcriptional programs in human peripheral blood cells. PLoS One. 2010;5(3):e9753. doi: 10.1371/journal.pone.0009753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ayers M, et al. IFN-γ-related mRNA profile predicts clinical response to PD-1 blockade. J Clin Invest. 2017;127(8):2930–2940. doi: 10.1172/JCI91190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wysocka M, et al. Interleukin-12 is required for interferon-gamma production and lethality in lipopolysaccharide-induced shock in mice. Eur J Immunol. 1995;25(3):672–676. doi: 10.1002/eji.1830250307. [DOI] [PubMed] [Google Scholar]
- 32.Gillespie SH, et al. Four-month moxifloxacin-based regimens for drug-sensitive tuberculosis. N Engl J Med. 2014;371(17):1577–1587. doi: 10.1056/NEJMoa1407426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zeitz SJ, Ostrow JH, Van Arsdel PP. Humoral and cellular immunity in the anergic tuberculosis patient. A prospective study. J Allergy Clin Immunol. 1974;53(1):20–26. doi: 10.1016/0091-6749(74)90095-5. [DOI] [PubMed] [Google Scholar]
- 34.Holden M, Dubin MR, Diamond PH. Frequency of negative intermediate-strength tuberculin sensitivity in patients with active tuberculosis. N Engl J Med. 1971;285(27):1506–1509. doi: 10.1056/NEJM197112302852704. [DOI] [PubMed] [Google Scholar]
- 35.Furcolow ML, Hewell B, Nelson WE, Palmer CE. Quantitative studies of the tuberculin reaction: I. Titration of tuberculin sensitivity and its relation to tuberculous infection. Public Health Reports. 1941;56(21):1082–1100. doi: 10.2307/4583745. [DOI] [Google Scholar]
- 36.Cobelens FG, Egwaga SM, van Ginkel T, Muwinge H, Matee MI, Borgdorff MW. Tuberculin skin testing in patients with HIV infection: limited benefit of reduced cutoff values. Clin Infect Dis. 2006;43(5):634–639. doi: 10.1086/506432. [DOI] [PubMed] [Google Scholar]
- 37.Nash DR, Douglass JE. Anergy in active pulmonary tuberculosis. A comparison between positive and negative reactors and an evaluation of 5 TU and 250 TU skin test doses. Chest. 1980;77(1):32–37. doi: 10.1378/chest.77.1.32. [DOI] [PubMed] [Google Scholar]
- 38.Sollai S, Galli L, de Martino M, Chiappini E. Systematic review and meta-analysis on the utility of Interferon-gamma release assays for the diagnosis of Mycobacterium tuberculosis infection in children: a 2013 update. BMC Infect Dis. 2014;14(Suppl 1):S6. doi: 10.1186/1471-2334-14-S1-S6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Metcalfe JZ, et al. Interferon-γ release assays for active pulmonary tuberculosis diagnosis in adults in low- and middle-income countries: systematic review and meta-analysis. J Infect Dis. 2011;204(Suppl 4):S1120–S1129. doi: 10.1093/infdis/jir410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nguyen DT, Teeter LD, Graves J, Graviss EA. Characteristics associated with negative interferon-γ release assay results in culture-confirmed tuberculosis patients, Texas, USA, 2013-2015. Emerging Infect Dis. 2018;24(3):534–540. doi: 10.3201/eid2403.171633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McMurray DN, Echeverri A. Cell-mediated immunity in anergic patients with pulmonary tuberculosis. Am Rev Respir Dis. 1978;118(5):827–834. doi: 10.1164/arrd.1978.118.5.827. [DOI] [PubMed] [Google Scholar]
- 42.Sousa AO, et al. An epidemic of tuberculosis with a high rate of tuberculin anergy among a population previously unexposed to tuberculosis, the Yanomami Indians of the Brazilian Amazon. Proc Natl Acad Sci USA. 1997;94(24):13227–13232. doi: 10.1073/pnas.94.24.13227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sahiratmadja E, et al. Dynamic changes in pro- and anti-inflammatory cytokine profiles and gamma interferon receptor signaling integrity correlate with tuberculosis disease activity and response to curative treatment. Infect Immun. 2007;75(2):820–829. doi: 10.1128/IAI.00602-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Harari A, et al. Dominant TNF-α+ Mycobacterium tuberculosis-specific CD4+ T cell responses discriminate between latent infection and active disease. Nat Med. 2011;17(3):372–376. doi: 10.1038/nm.2299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kincaid EZ, Ernst JD. Mycobacterium tuberculosis exerts gene-selective inhibition of transcriptional responses to IFN-gamma without inhibiting STAT1 function. J Immunol. 2003;171(4):2042–2049. doi: 10.4049/jimmunol.171.4.2042. [DOI] [PubMed] [Google Scholar]
- 46.Pennini ME, Liu Y, Yang J, Croniger CM, Boom WH, Harding CV. CCAAT/enhancer-binding protein beta and delta binding to CIITA promoters is associated with the inhibition of CIITA expression in response to Mycobacterium tuberculosis 19-kDa lipoprotein. J Immunol. 2007;179(10):6910–6918. doi: 10.4049/jimmunol.179.10.6910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Doering TA, Crawford A, Angelosanto JM, Paley MA, Ziegler CG, Wherry EJ. Network analysis reveals centrally connected genes and pathways involved in CD8+ T cell exhaustion versus memory. Immunity. 2012;37(6):1130–1144. doi: 10.1016/j.immuni.2012.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mognol GP, et al. Exhaustion-associated regulatory regions in CD8+ tumor-infiltrating T cells. Proc Natl Acad Sci USA. 2017;114(13):E2776–E2785. doi: 10.1073/pnas.1620498114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Foster SL, Hargreaves DC, Medzhitov R. Gene-specific control of inflammation by TLR-induced chromatin modifications. Nature. 2007;447(7147):972–978. doi: 10.1038/nature05836. [DOI] [PubMed] [Google Scholar]
- 50.Yaseen I, Kaur P, Nandicoori VK, Khosla S. Mycobacteria modulate host epigenetic machinery by Rv1988 methylation of a non-tail arginine of histone H3. Nat Commun. 2015;6:8922. doi: 10.1038/ncomms9922. [DOI] [PubMed] [Google Scholar]
- 51.Pennini ME, Pai RK, Schultz DC, Boom WH, Harding CV. Mycobacterium tuberculosis 19-kDa lipoprotein inhibits IFN-gamma-induced chromatin remodeling of MHC2TA by TLR2 and MAPK signaling. J Immunol. 2006;176(7):4323–4330. doi: 10.4049/jimmunol.176.7.4323. [DOI] [PubMed] [Google Scholar]
- 52.Marr AK, MacIsaac JL, Jiang R, Airo AM, Kobor MS, McMaster WR. Leishmania donovani infection causes distinct epigenetic DNA methylation changes in host macrophages. PLoS Pathog. 2014;10(10):e1004419. doi: 10.1371/journal.ppat.1004419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Durzynska J, Lesniewicz K, Poreba E. Human papillomaviruses in epigenetic regulations. Mutat Res Rev Mutat Res. 2017;772:36–50. doi: 10.1016/j.mrrev.2016.09.006. [DOI] [PubMed] [Google Scholar]
- 54.Chandran A, Antony C, Jose L, Mundayoor S, Natarajan K, Kumar RA. Mycobacterium tuberculosis infection induces HDAC1-mediated suppression of IL-12B gene expression in macrophages. Front Cell Infect Microbiol. 2015;5:90. doi: 10.3389/fcimb.2015.00090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liberzon A, Birger C, Thorvaldsdóttir H, Ghandi M, Mesirov JP, Tamayo P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015;1(6):417–425. doi: 10.1016/j.cels.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Reinius LE, et al. Differential DNA methylation in purified human blood cells: implications for cell lineage and studies on disease susceptibility. PLoS One. 2012;7(7):e41361. doi: 10.1371/journal.pone.0041361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lindqvist BM, Wingren S, Motlagh PB, Nilsson TK. Whole genome DNA methylation signature of HER2-positive breast cancer. Epigenetics. 2014;9(8):1149–1162. doi: 10.4161/epi.29632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mandalakas AM, et al. Well-quantified tuberculosis exposure is a reliable surrogate measure of tuberculosis infection. Int J Tuberc Lung Dis. 2012;16(8):1033–1039. doi: 10.5588/ijtld.12.0027. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.