

Abstract
Phosphoglycerate dehydrogenase (PHGDH), the first rate-limiting enzyme of serine synthesis, is frequently overexpressed in human cancer. PHGDH overexpression activates serine synthesis to promote cancer progression. Currently, PHGDH regulation in normal cells and cancer is not well understood. Parkin, an E3 ubiquitin ligase involved in Parkinson’s disease, is a tumor suppressor. Parkin expression is frequently downregulated in many types of cancer, and its tumor-suppressive mechanism is poorly defined. Here, we show that PHGDH is a substrate for Parkin-mediated ubiquitination and degradation. Parkin interacted with PHGDH and ubiquitinated PHGDH at lysine 330, leading to PHGDH degradation to suppress serine synthesis. Parkin deficiency in cancer cells stabilized PHGDH and activated serine synthesis to promote cell proliferation and tumorigenesis, which was largely abolished by targeting PHGDH with RNA interference, CRISPR/Cas9 KO, or small-molecule PHGDH inhibitors. Furthermore, Parkin expression was inversely correlated with PHGDH expression in human breast cancer and lung cancer. Our results revealed PHGDH ubiquitination by Parkin as a crucial mechanism for PHGDH regulation that contributes to the tumor-suppressive function of Parkin and identified Parkin downregulation as a critical mechanism underlying PHGDH overexpression in cancer.
Keywords: Metabolism, Oncology
Keywords: Tumor suppressors, Ubiquitin-proteosome system

Introduction
Cancer cells reprogram their metabolism to meet their bioenergetic and biosynthetic needs for rapid growth and proliferation (1–3). Increased serine biosynthesis is an important metabolic adaptation in many human cancers that provides precursors for amino acids, nucleotides, and lipids to support bioenergetic and biosynthetic needs of cancer cells (4–9). Serine can be imported from the extracellular environment via transporters or synthesized by the serine synthesis pathway (4–6). Phosphoglycerate dehydrogenase (PHGDH) is the first rate-limiting enzyme in the serine synthesis pathway that produces 3-phosphohydroxypyruvate (3-PPyr) by NAD+-coupled oxidation of 3-phosphoglycerate (3-PG) (9–11). Recent studies show that PHGDH is frequently overexpressed in some types of cancers, including approximately 40% of breast cancers and 50% to 80% of lung cancers, and PHGDH overexpression activates serine synthesis to promote cancer progression (9, 10, 12–14). Importantly, targeting PHGDH in cancer cells with PHGDH overexpression greatly compromises cancer progression, suggesting that PHGDH is a potential therapeutic target in cancers with PHGDH overexpression (9–12, 15–17). Currently, the regulation of PHGDH in cells and mechanism underlying PHGDH overexpression in cancer are poorly defined. Gene amplification has been identified as a mechanism contributing to PHGDH overexpression in breast cancer and melanoma (10, 13). However, PHGDH gene amplification is observed in only approximately 6% of breast cancers, whereas PHGDH overexpression is observed in approximately 40% of breast cancers (10), suggesting that there are mechanisms other than gene amplification that contribute to PHGDH overexpression in breast cancer. In addition to gene amplification, NRF2, a transcription factor frequently activated in lung cancer, was reported to upregulate PHGDH in non-small cell lung cancer (18).
Parkin, encoded by the PARK2 gene, is an E3 ubiquitin ligase. Mutations in PARK2 have been linked to autosomal recessive juvenile Parkinson’s disease, a common familial form of Parkinson’s disease (PD) (19, 20). Ubiquitination activity of Parkin has been reported as contributing greatly to the role of Parkin in preventing PD (21–25). Interestingly, ample studies have shown that Parkin is a bona fide tumor suppressor. PARK2 mutations have been reported in different types of human cancers, including breast and lung cancers, although the mutation frequency of PARK2 is relatively low (less than 5% in both breast and lung cancers) (26–29). Parkin expression is frequently downregulated in many cancers, including 40% to 70% of breast cancers and over 30% of lung cancers, and this downregulation can be caused by different mechanisms, including loss of heterozygosity, loss of copy number, and the promoter hypermethylation of PARK2 (26, 30–34). Parkin downregulation in different types of cancers is frequently associated with poor prognosis of cancer patients (26, 27, 35–37). Mice deficient for Parkin are more susceptible to developing tumors, including spontaneous hepatocellular carcinoma and γ-irradiation–induced lymphoma (38, 39). In addition, Parkin deficiency promotes colorectal tumorigenesis in ApcMin/+ mice (31). Currently, the mechanism of the tumor-suppressive function of Parkin is poorly defined. The ubiquitination activity of Parkin has been suggested as being crucial for the tumor-suppressive function of Parkin. For instance, Parkin was reported to ubiquitinate cyclin D/E, HIF-1α, RIPK3, mitotic regulators, and mitochondrial iron importers, contributing to its tumor-suppressive function (35, 36, 40–43).
In this study, we identified Parkin as a critical binding partner and a negative regulator of PHGDH. Using co-immunoprecipitation (co-IP) followed by liquid chromatography–tandem mass spectrometry (LC-MS/MS), we found that Parkin binds to PHGDH and degrades it through ubiquitination to inhibit serine synthesis, which contributes greatly to the tumor-suppressive function of Parkin. Decreased Parkin expression in cancer cells leads to stabilization and accumulation of PHGDH to promote serine synthesis and cancer progression, which can be largely abolished by targeting PHGDH using RNAi, CRISPR/Cas9 KO, and small-molecule PHGDH inhibitors. These results reveal an important mechanism underlying the regulation of PHGDH and tumor-suppressive function of Parkin in cancers.
Results
Parkin interacts with PHGDH.
PHGDH is frequently overexpressed in human breast cancer and lung cancer. To reveal the mechanism of PHGDH regulation in cells and PHGDH overexpression in cancer, we screened for potential proteins interacting with PHGDH using co-IP followed by LC-MS/MS assays in normal human breast MCF10A cells transduced with or without a retroviral vector to express PHGDH-Flag. LC-MS/MS analysis identified Parkin as a potential binding protein for PHGDH-Flag (Supplemental Table 1; supplemental material available online with this article; https://doi.org/10.1172/JCI132876DS1). The interaction between exogenous PHGDH-Flag and Myc-Parkin was verified by co-IP, followed by Western blot assays in MCF10A cells with ectopic expression of PHGDH-Flag and Myc-Parkin (Figure 1A). The interaction between endogenous PHGDH and Parkin was observed in human Hs578T breast cancer (Figure 1B) and H1299 lung cancer cells (Figure 1C) that express high levels of PHGDH (Supplemental Figure 1) by co-IP and Western blot analysis. As a negative control, endogenous PHGDH was knocked down using 2 different shRNA vectors (Figure 1, B and C). To further support our observations, Hs578T cells with PHGDH KO using CRISPR/Cas9 were employed for co-IP assays. The interaction between endogenous PHGDH and Parkin was observed in control PHGDH WT Hs578T cells, but not in 2 different PHGDH KO clonal cell lines (Hs578T-PHGDH-KO) (Figure 1D).
Figure 1. PHGDH interacts with Parkin.

(A) PHGDH-Flag interacted with Myc-Parkin in MCF10A cells. Cells with ectopic expression of PHGDH-Flag and Myc-Parkin were employed for co-IP assays using the anti-Flag (left) and anti-Myc antibodies (right), respectively. (B and C) Endogenous PHGDH interacted with endogenous Parkin in Hs578T (B) and H1299 cells (C), as detected by co-IP assays. PHGDH was knocked down by shRNAs in cells as negative controls. (D) Co-IP analysis of interaction of endogenous PHGDH and Parkin in WT Hs578T cells and Hs578T cells with PHGDH KO by CRISPR/Cas9. (E) Parkin bound to PHGDH at its SBD2 domain. Left: schematic representation of vectors expressing WT or serial deletion mutants of PHGDH-Flag. (F) PHGDH bound to Parkin at its IBR domain. Left: schematic representation of vectors expressing WT or serial deletion mutants of Myc-Parkin. (G) Direct interaction of recombinant GST-Parkin and His-Trx-PHGDH proteins analyzed by in vitro GST pull-down assays.
Next, we sought to determine the domain of PHGDH required for its interaction with Parkin by expressing different deletion mutants of PHGDH-Flag and WT Myc-Parkin in Hs578T cells for co-IP assays (Figure 1E). The substrate binding domain 2 (SBD2) of PHGDH was found to be required and sufficient for PHGDH to bind to Parkin by co-IP assays (Figure 1E). By expressing different deletion mutants of Myc-Parkin and WT PHGDH-Flag in Hs578T cells for co-IP assays, we found that the in-between RING (IBR) domain of Parkin was required and sufficient for Parkin to interact with PHGDH (Figure 1F). To address the question of whether PHGDH directly interacts with Parkin, in vitro glutathione S-transferase (GST) pull-down assays were conducted using recombinant GST-Parkin and His-PHGDH proteins purified from bacteria. A direct interaction between GST-Parkin and His-PHGDH was detected in vitro (Figure 1G); this was disrupted by deletion of the IBR domain of Parkin (ΔIBR) (Figure 1G). These results collectively demonstrate that PHGDH interacts directly with Parkin in cells.
Parkin downregulates PHGDH protein levels in cells and mouse tissues.
As an E3 ubiquitin ligase, Parkin promotes ubiquitination and degradation of different protein substrates (23, 27, 29, 35). Given that Parkin interacts with PHGDH, we determined whether Parkin regulates PHGDH protein levels in cells. We first examined the effect of Parkin on PHGDH protein levels in cells by coexpressing Myc-Parkin and PHGDH-Flag in Hs578T and H1299 cells. Myc-Parkin expression downregulated PHGDH-Flag protein levels in a dose-dependent manner in both Hs578T and H1299 cells (Figure 2A). We then determined whether Myc-Parkin expression downregulates endogenous PHGDH protein levels by expressing Myc-Parkin in different human breast cancer cell lines (including Hs578T, HCC70, MDA-MB468, and T47D) and lung cancer cell lines (including H1299 and A549), which were reported to express higher levels of PHGDH protein compared with normal tissues (Supplemental Figure 1 and refs. 10, 12, 15, 18). Expression of Myc-Parkin greatly downregulated the endogenous PHGDH protein levels in all of these cell lines (Figure 2B). Further, knockdown of endogenous Parkin with shRNA vectors increased PHGDH protein levels in all of these cancer cell lines and normal breast MCF10A cells (Figure 2C). The downregulation of endogenous PHGDH by Parkin was confirmed in Hs578T and H1299 cells with Parkin KO by CRISPR/Cas9; Parkin-KO cells displayed much higher PHGDH protein levels compared with their control WT cells (Figure 2D). Further, mouse embryonicfibroblasts (MEFs) from Parkin–/– mice displayed higher PHGDH protein levels compared with Parkin+/+ MEFs (Figure 2E). Importantly, breast and lung tissues from Parkin–/– mice displayed higher PHGDH protein levels compared with these tissues from Parkin+/+ mice (Figure 2F). Results from quantitative real-time PCR assays showed that Parkin did not affect PHGDH mRNA levels in cells or mouse tissues (Supplemental Figure 2, A–E). Taken together, these results demonstrate that Parkin downregulates PHGDH protein levels in cells and mouse tissues.
Figure 2. Parkin negatively regulates PHGDH protein levels in cells and mice.

(A) Myc-Parkin expression reduced levels of PHGDH-Flag protein in Hs578T and H1299 cells. Cells were transfected with the PHGDH-Flag vector together with varying amounts of Myc-Parkin or control (Con) vectors. (B) Ectopic Myc-Parkin expression reduced levels of endogenous PHGDH protein in different human breast and lung cancer cells. (C) Knockdown of endogenous Parkin by 2 different shRNA vectors increased levels of endogenous PHGDH protein in different human breast and lung cells. (D) KO of Parkin by CRISPR/Cas9 increased levels of PHGDH protein in Hs578T and H1299 cells. (E) Higher PHGDH protein levels in Parkin−/− MEFs compared with Parkin+/+ MEFs. (F) Higher PHGDH protein levels in the breast and lung tissues of Parkin–/– mice compared with Parkin+/+ mice. n = 5 mice/group.
Parkin ubiquitinates and degrades PHGDH.
Given our finding that Parkin binds to and downregulates PHGDH, we speculated that Parkin downregulates PHGDH through ubiquitin-proteasome degradation. To test this hypothesis, Hs578T cells transduced with control or WT Myc-Parkin vectors were treated with the proteasome inhibitor MG132 and PHGDH levels were analyzed by Western blot assays. MG132 treatment increased PHGDH protein levels in cells and, importantly, abolished the inhibitory effect of WT Myc-Parkin on PHGDH levels (Figure 3A). Similar results were observed in WT and Parkin-KO Hs578T cells treated with MG132; MG132 treatment increased PHGDH protein levels and abolished the inhibitory effect of endogenous Parkin on PHGDH levels in cells (Figure 3A). Furthermore, expression of C431A Myc-Parkin, a mutant Parkin with impaired E3 ubiquitin ligase activity (44, 45), did not clearly affect PHGDH levels in cells (Figure 3A), suggesting that E3 ubiquitin ligase activity is required for Parkin to downregulate PHGDH levels in cells. Parkin is known to be involved in inducing mitophagy, the selective degradation of mitochondria by autophagy (21, 46, 47). To investigate whether autophagy and mitophagy also regulate PHGDH protein levels and contribute to PHGDH downregulation by Parkin in cells, WT and Parkin-KO Hs578T cells were treated with chloroquine and bafilomycin A1, 2 widely used autophagy inhibitors that inhibit autolysosome degradation (48), respectively. In contrast to the effect of MG132, chloroquine or bafilomycin A1 did not clearly affect the PHGDH protein levels or the effect of Parkin on PHGDH protein levels in cells (Supplemental Figure 3A). We further treated WT and Parkin-KO Hs578T and H1299 cells with carbonyl cyanide 3-chlorophenylhydrazone (CCCP), which induces mitophagy (48), and found that CCCP treatment also did not clearly affect PHGDH levels or the effect of Parkin on PHGDH levels in cells (Supplemental Figure 3B). Taken together, these results suggest that Parkin downregulates PHGDH mainly through ubiquitin-proteasomal degradation in cells.
Figure 3. Parkin promotes PHGDH protein degradation through ubiquitination.

(A) Treatment of proteasome inhibitor MG132 increased PHGDH protein levels and abolished inhibitory effect of Parkin on PHGDH levels in Hs578T cells. Hs578T cells with ectopic expression of WT or C431A Myc-Parkin (upper) and WT or Parkin-KO Hs578T cells (lower) were treated with MG132 (5 μM) or DMSO for 12 hours before Western blot assays. (B) Myc-Parkin expression decreased PHGDH protein half-life (left), whereas knockdown of endogenous Parkin increased PHGDH protein half-life (right) in Hs578T cells. Cells were treated with CHX or DMSO for different hours before Western blot assays. (C) KO of Parkin by CRISPR/Cas9 increased PHGDH protein half-life in Hs578T cells. In B and C, data are presented as mean ± SD. n = 3. (D) Effects of WT and mutant Myc-Parkin on ubiquitination of PHGDH-Flag in Hs578T cells analyzed by in vivo ubiquitination (Ub) assays. (E) GST-Parkin ubiquitinates His-Trx-PHGDH in vitro analyzed by in vitro ubiquitination assays using recombinant proteins. (F) Parkin knockdown by shRNA (left) or Parkin KO by CRISPR/Cas9 (middle and right) reduced ubiquitination of PHGDH-Flag in Hs578T and H1299 cells analyzed by in vivo ubiquitination assays. (G) Parkin knockdown by shRNA reduced ubiquitination of endogenous PHGDH in Hs578T cells analyzed by in vivo ubiquitination assays. (H) T240M and P294S mutations impaired Parkin’s ubiquitination activity toward PHGDH in Hs578T cells analyzed by in vivo ubiquitination assays. (I) T240M and P294S mutations impaired the ability of Myc-Parkin to downregulate PHGDH protein levels in Hs578T and H1299 cells.
Next, we determined whether Parkin affects protein stability of PHGDH by measuring the protein half-life of PHGDH in cells. Hs578T cells with or without Myc-Parkin expression were treated with protein synthesis inhibitor cyclohexamide (CHX) for different time periods before Western blot assays. Compared with control cells, Myc-Parkin expression clearly reduced the half-life of PHGDH protein (Figure 3B). In contrast, knockdown of Parkin by shRNAs increased the half-life of PHGDH protein in cells (Figure 3B). This result was verified in Hs578T-Parkin-KO cells; compared with what occurred in control WT cells, Parkin KO increased the half-life of PHGDH protein (Figure 3C).
To directly address the question of whether Parkin regulates PHGDH ubiquitination, in vivo ubiquitination assays were performed in Hs578T cells with ectopic expression of PHGDH-Flag, HA-ubiquitin, and WT, C431A, or ΔIBR Myc-Parkin. Expression of WT but not C431A Myc-Parkin markedly increased ubiquitination of PHGDH-Flag in cells (Figure 3D). The ΔIBR Myc-Parkin, which cannot bind to PHGDH, failed to promote PHGDH-Flag ubiquitination (Figure 3D). The effect of Parkin on PHGDH ubiquitination was confirmed by in vitro ubiquitination assays using recombinant GST-Parkin and His-PHGDH proteins; the WT GST-Parkin protein markedly induced His-PHGDH ubiquitination, whereas C431A and ΔIBR GST-Parkin proteins failed to induce His-PHGDH ubiquitination in vitro (Figure 3E). To further examine whether endogenous Parkin ubiquitinates PHGDH, Hs578T cells with Parkin knockdown or KO and H1299 cells with Parkin KO were transfected with PHGDH-Flag for in vivo ubiquitination assays. As shown in Figure 3F, Parkin knockdown or KO dramatically decreased PHGDH-Flag ubiquitination in these cells. Similarly, Parkin knockdown clearly reduced the ubiquitination of endogenous PHGDH in Hs578T cells (Figure 3G).
PARK2 mutations have been observed in breast cancer and lung cancer (26, 27, 29). T240M and P294S are 2 cancer-associated mutations in PARK2; these mutations impair the ubiquitin ligase activity and tumor-suppressive function of Parkin (49). We determined whether T240M and P294S mutations impair the function of Parkin in ubiquitination and degradation of PHGDH in cells. Compared with WT Myc-Parkin, these 2 mutations markedly reduced the ability of Parkin to ubiquitinate and degrade PHGDH as shown by in vivo ubiquitination (Figure 3H) and Western blot assays (Figure 3I), respectively. Collectively, these results indicate that Parkin downregulates PHGDH through ubiquitination and proteasome degradation.
Parkin ubiquitinates PHGDH at lysine 330.
To identify the target residue or residues in PHGDH modified by Parkin-dependent ubiquitination, in vivo ubiquitination assays were performed in Hs578T cells and PHGDH-Flag ubiquitinated by Myc-Parkin was immunoprecipitated for LC-MS/MS analysis. The lysines at 330, 310, and 364 of PHGDH (K330, K310, and K364) were identified as the top 3 putative ubiquitination sites for Parkin (Table 1 and Figure 4A). The lysines at these 3 sites in PHGDH-Flag were mutated to arginine (K330R, K310R, or K364R), respectively, and in vivo ubiquitination assays were performed to determine whether these mutations diminish PHGDH ubiquitination. Compared with WT PHGDH-Flag, the K330R mutation dramatically reduced the ability of Myc-Parkin to ubiquitinate PHGDH-Flag, whereas the K310R or K364R mutation did not clearly reduce the ability of Myc-Parkin to ubiquitinate PHGDH-Flag in Hs578T cells (Figure 4B). Compared with the K330R mutation, triple K to R mutations at these 3 sites (K310R/K330R/K364R) did not further reduce the ubiquitination of PHGDH by Parkin (Figure 4B). Results from co-IP assays showed that K330R mutation did not affect the Parkin-PHGDH interaction (Supplemental Figure 4). Notably, K330R mutation markedly reduced the ability of Parkin to downregulate PHGDH protein levels in cells (Figure 4C). Compared with WT PHGDH, K330R PHGDH displayed a longer protein half-life in cells (Figure 4D). Further, expression of Myc-Parkin greatly reduced the half-life of WT PHGDH, but not K330R PHGDH, protein in cells (Figure 4D). These results together indicate that K330 of PHGDH is a major ubiquitination site for Parkin.
Table 1. Sequences and counts of peptides containing potential ubiquitination sites for Parkin in PHGDH analyzed by LC-MS/MS.
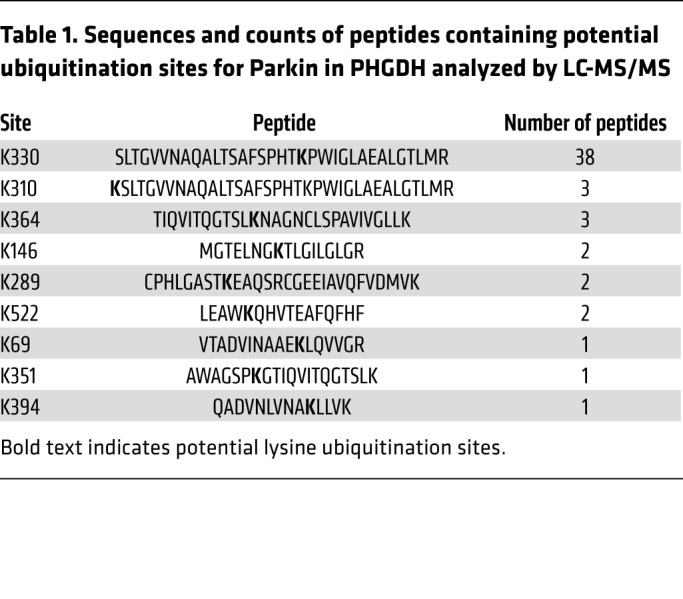
Figure 4. Ubiquitination of PHGDH at lysine 330 (K330) by Parkin.

(A) Top 3 potential lysine ubiquitination sites in PHGDH identified by LC-MS/MS analysis, including K310, K330, and K364. (B) K330 mutation (K330R) largely abolished ubiquitination of PHGDH by Parkin. Hs578T cells with expression of WT or indicated mutant PHGDH-Flag were used for in vivo ubiquitination assays. (C) K330R mutation largely abolished the negative regulation of PHGDH protein levels by Myc-Parkin in Hs578T and H1299 cells. Hs578T and H1299 cells were transduced with WT or K330R PHGDH-Flag vectors together with Myc-Parkin vectors for Western blot assays. (D) Myc-Parkin expression did not clearly affect the K330R PHGDH-Flag protein half-life in Hs578T cells. Data are presented as mean ± SD. n = 3.
Parkin inhibits serine synthesis through ubiquitination of PHGDH.
PHGDH is the first rate-limiting enzyme in serine synthesis that converts 3-PG to 3-PPyr. 3-PPyr is then converted to serine, and serine can be further converted to glycine to support the synthesis of amino acids, nucleotides, and lipids (Figure 5A and refs. 4, 5, 50). Our finding that Parkin ubiquitinates and degrades PHGDH suggests that Parkin may suppress serine synthesis. To investigate the effect of Parkin on serine synthesis, Hs578T and H1299 cells were cultured in the presence of uniformly carbon-13–labeled glucose (U-13C6-glucose) to trace its contribution to newly synthesized serine (with a mass shift of 3 [M + 3]) due to the incorporation of glucose-derived 13C via 3-PG and glycine (with a mass-shift of 2 [M + 2]) by LC-MS–based metabolomics analysis (Figure 5A). Expression of Myc-Parkin significantly decreased the incorporation of 13C into serine and glycine and levels of total serine and glycine in both Hs578T and H1299 cells (Figure 5B). In contrast, Parkin knockdown (Figure 5C) or KO (Figure 5D) significantly increased the incorporation of 13C into serine and glycine and levels of total serine and glycine in cells. Notably, PHGDH knockdown not only greatly decreased the incorporation of 13C into serine and glycine and levels of total serine and glycine, but also largely abolished the inhibitory effect of Myc-Parkin (Figure 5B) and the promoting effect of Parkin knockdown (Figure 5C) on the incorporation of 13C into serine and glycine and levels of total serine and glycine in cells. Further, PHGDH KO in Hs578T cells not only completely blocked the incorporation of 13C into serine and glycine, but also abolished the promoting effect of Parkin KO on levels of total serine and glycine (Figure 5D). These results together indicate that Parkin downregulates PHGDH to suppress serine synthesis and reduce levels of total serine and glycine in cells.
Figure 5. Parkin inhibits serine synthesis in cells through negative regulation of PHGDH.

(A) Schematic of U-13C-glucose incorporation into serine and glycine in cells. (B) Myc-Parkin expression reduced the incorporation of 13C into serine and glycine and the levels of total intracellular serine and glycine in Hs578T and H1299 cells, which was largely abolished by PHGDH knockdown. (C) Parkin knockdown increased the incorporation of 13C into serine and glycine and the levels of total serine and glycine in cells, which was largely abolished by PHGDH knockdown. (D) Parkin KO increased the incorporation of 13C into serine and glycine and the levels of total serine and glycine in cells, which was abolished by PHGDH KO. (E) Effects of Parkin mutants (C431A, T240M, and P294S) on the incorporation of 13C into serine and glycine and the levels of total serine and glycine in Hs578T cells. (F) PHGDH K330R mutation largely abolished the effect of Myc-Parkin on the incorporation of 13C into serine and glycine and the levels of total serine and glycine in PHGDH-KO Hs578T cells. (G) Parkin–/– MEFs displayed higher levels of 13C incorporation into serine and glycine as well as higher levels of total serine and glycine compared with Parkin+/+ MEFs. Three different MEF lines were used for assays. For B–G, cells were incubated with U-13C-glucose for 25 minutes for LC-MS analysis. (H) Parkin–/– mice displayed higher levels of serine and glycine in breast and lung tissues as well as serum compared with Parkin+/+ mice. Data are presented as mean ± SD. n = 3 for B, E–G; n = 5 for C, D; and n = 5 mice/group for H. #P < 0.05; ##P < 0.01; *P < 0.001; **P < 0.0001, heterogeneous variance model with Dunnett’s adjustment for multiple testing.
We further determined whether Parkin inhibits serine synthesis through ubiquitination of PHGDH. Compared with WT Parkin, C431A and cancer-associated T240M and P294S mutant Parkin displayed significantly decreased activities in suppressing the incorporation of 13C into serine and glycine and reducing levels of total serine and glycine in Hs578T cells (Figure 5E). To further support our observations, the WT or K330R PHGDH-Flag vector was expressed in Hs578T-PHGDH-KO cells (Figure 5F). Myc-Parkin expression significantly suppressed serine synthesis in cells expressing WT PHGDH-Flag, but not in cells expressing K330R PHGDH-Flag, which cannot be efficiently ubiquitinated and degraded by Parkin (Figure 5F).
LC-MS–based metabolomics analysis was further employed to analyze metabolites in MEFs as well as tissues from Parkin+/+ and Parkin–/– mice. Compared with Parkin+/+ MEFs, Parkin–/– MEFs displayed significantly higher levels of 13C incorporation into serine and glycine as well as higher levels of total serine and glycine (Figure 5G). Similarly, Parkin–/– mice displayed significantly higher levels of serine and glycine in breast and lung tissues as well as serum compared with Parkin+/+ mice (Figure 5H). Collectively, these results indicate that Parkin suppresses serine synthesis through its ubiquitination and downregulation of PHGDH in cells and mouse tissues.
Parkin inhibits proliferation of cancer cells through PHGDH downregulation.
PHGDH overexpression plays a critical role in promoting proliferation of cancer cells, including breast and lung cancer cells (4, 10, 18). Targeting PHGDH by RNAi or specific small-molecule PHGDH inhibitors (e.g., NCT-503 and CBR-5884) was reported to inhibit proliferation of cancer cells with PHGDH overexpression both in vitro and in vivo (10, 15, 16, 18). Here, we investigated whether Parkin suppresses proliferation of cancer cells through negative regulation of PHGDH in aforementioned different cancer cells, including Hs578T, H1299 (Figure 6), HCC70, MDA-MB468, T47D, and A549 cells (Supplemental Figure 5). Ectopic expression of Myc-Parkin significantly inhibited cell proliferation (Figure 6A and Supplemental Figure 5A), whereas knockdown of Parkin significantly promoted cell proliferation in these cell lines (Figure 6B and Supplemental Figure 5B). Notably, PHGDH knockdown greatly abolished the inhibitory effect of Myc-Parkin expression or the promoting effect of Parkin knockdown on cell proliferation (Figure 6, A and B, Supplemental Figure 5, A and B). Similarly, Parkin KO significantly promoted proliferation of both Hs578T and H1299 cells, which was greatly abolished by KO of PHGDH in these cells (Figure 6C). To further support our observations, we expressed the WT or K330R PHGDH-Flag together with Myc-Parkin in Hs578T and H1299 cells with PHGDH KO. Myc-Parkin expression significantly inhibited proliferation of PHGDH-KO cells expressing WT PHGDH-Flag, but displayed a much less pronounced inhibitory effect in PHGDH-KO cells expressing K330R PHGDH-Flag (Figure 6D). These results suggest that the ubiquitination and downregulation of PHGDH contributes greatly to Parkin’s inhibitory effect on cell proliferation.
Figure 6. Parkin inhibits proliferation of human breast and lung cancer cells through negative regulation of PHGDH.

(A) Myc-Parkin expression inhibited the proliferation of Hs578T and H1299 cells, which was greatly abolished by PHGDH knockdown. (B) Parkin knockdown promoted the proliferation of Hs578T and H1299 cells, which was largely abolished by PHGDH knockdown. (C) Parkin KO by CRISPR/Cas9 promoted the proliferation of Hs578T and H1299 cells, which was largely abolished by PHGDH KO. (D) Myc-Parkin expression greatly decreased proliferation of cells expressing WT but not K330R PHGDH-Flag in PHGDH-KO Hs578T and H1299 cells. (E and F) Effects of PHGDH inhibitors NCT-503 (E) and CBR-5884 (F) on proliferation of Hs578T and H1299 cells with Parkin knockdown and their control cells. (G and H) The effects of NCT-503 (G) and CBR-5884 (H) on proliferation of Parkin-KO Hs578T cells and control WT cells. For A–D, the same amounts of cells were plated, and cells were cultured for 4 days. For E–H, at 12 hours after cell seeding, cells were treated with NCT-503, CBR-5884, or DMSO (for 0 μM groups) for 4 days. For A–H, cell numbers were measured daily for 4 days by MTT assays. Relative cell numbers normalized to relevant controls at day 1 are presented. Data are presented as mean ± SD. n = 6. *P < 0.001; **P < 0.0001, mixed model analysis with Bonferroni’s adjusted P values.
Given that Parkin deficiency leads to the stabilization and accumulation of PHGDH in cancer cells, we tested to determine whether cancer cells with Parkin deficiency are sensitive to PHGDH inhibitors, including NCT-503 and CBR-5884. Consistent with previous reports (15, 16), NCT-503 and CBR-5884 significantly inhibited proliferation of the above-mentioned cancer cell lines, which express high levels of PHGDH (Figure 6, E and F, and Supplemental Figure 5, C and D). Further, the effects of NCT-503 and CBR-5884 on cell proliferation were largely PHGDH dependent, as shown in H1299 cells with PHGDH KO (Supplemental Figure 5, E and F). Importantly, our results show that NCT-503 and CBR-5884 significantly inhibited the proliferation of cancer cells with Parkin knockdown or Parkin KO and suggest that NCT-503 and CBR-5884 may display better inhibitory effects on proliferation in Parkin-deficient cells than in control cells (Figure 6, E–H, and Supplemental Figure 5, C–F).
It has been well known that Hs578T, HCC70, MDA-MB468, T47D, H1299, and A549 cells depend on serine synthesis, but not exogenous serine/glycine, in culture media for their needs on serine/glycine and are sensitive to PHGDH inhibition (15, 16). Interestingly, some cancer cells, including human breast cancer MCF7, ZR-75-1, and MDA-MB-231 cells, were reported to be dependent on exogenous serine/glycine for their needs on serine/glycine (15, 16). These cell lines are insensitive to PHGDH inhibition under the serine/glycine-replete condition, but become sensitive to PHGDH inhibition under the serine/glycine-depleted condition, although its underlying mechanism is not well understood (15, 16). Consistent with previous reports (15, 16), these 3 cell lines were dependent on exogenous serine/glycine for proliferation; depletion of serine/glycine in culture media dramatically inhibited their proliferation (Supplemental Figure 6A). Further, they were insensitive to PHGDH inhibition under the serine/glycine-replete condition; knockdown of PHGDH in these cell lines did not clearly affect cell proliferation under the serine/glycine-replete condition, but significantly inhibited cell proliferation under the serine/glycine-depleted condition (Supplemental Figure 6A). Here, we examined the effect of Parkin on PHGDH and cell proliferation in these 3 cell lines. We found that Parkin knockdown increased PHGDH protein levels in MCF7, ZR-75-1, and MDA-MB-231 cells (Supplemental Figure 6B). Although Parkin knockdown slightly promoted proliferation of these cells under the serine/glycine-replete condition, Parkin knockdown appeared to display a much more pronounced promoting effect on cell proliferation under the serine/glycine-depleted condition, which was largely abolished by PHGDH knockdown (Supplemental Figure 6C). Consistent with previous reports (15, 16), MCF7, ZR-75-1, and MDA-MB-231 cells were insensitive to NCT-503 and CBR-5884 under the serine/glycine-replete condition, and depletion of serine/glycine greatly increased the sensitivity of these cell lines to NCT-503 and CBR-5884 (Supplemental Figure 6, D and E). Parkin knockdown did not clearly affect the inhibitory effects of NCT-503 or CBR-5884 on proliferation of these cell lines under the serine/glycine-replete condition (Supplemental Figure 6, D and E). Furthermore, our results suggest that NCT-503 and CBR-5884 display better proliferation inhibitory effects on cells with Parkin knockdown than on control cells under the serine/glycine-depleted condition (Supplemental Figure 6, D and E). Currently, the mechanism of why these cell lines are dependent on exogenous serine/glycine and insensitive to PHGDH inhibition under the serine/glycine-replete condition is not well understood, and this requires further studies.
Parkin inhibits tumorigenesis through ubiquitination of PHGDH.
We further investigated whether negative regulation of PHGDH contributes to the tumor-suppressive function of Parkin in vivo using orthotopic breast tumorigenesis and s.c. xenograft tumorigenesis mouse models. Compared with control cells, Parkin KO in Hs578T cells significantly promoted the growth of orthotopic breast tumors formed by fat pat injection of cells (Figure 7A). Notably, the promoting effect of Parkin KO on tumor growth was greatly abolished by further KO of PHGDH in Hs578T cells (Figure 7A). Immunohistochemistry (IHC) staining of orthotopic breast tumor tissues showed that Parkin KO increased PHGDH levels and staining of Ki-67, a cell-proliferation marker (Figure 7B). PHGDH KO in cells significantly reduced Ki-67 staining in tumors and, importantly, largely abolished the promoting effect of Parkin KO on Ki-67 staining in tumors (Figure 7B). Furthermore, total serine and glycine levels were significantly higher in Hs578T-Parkin-KO breast tumors than in control WT tumors (Figure 7C). Importantly, the promoting effect of Parkin KO on serine and glycine levels in tumors was largely abolished by further KO of PHGDH (Figure 7C). Similar results were observed in s.c. xenograft tumors formed by H1299 cells; Parkin KO significantly enhanced tumor growth (Figure 7D) and levels of PHGDH and Ki-67 in tumors (Supplemental Figure 7), which were largely abolished by PHGDH KO in cells (Figure 7D and Supplemental Figure 7). In contrast, expression of Myc-Parkin in Hs578T cells significantly inhibited the growth of orthotopic breast tumors, which was greatly abolished by PHGDH KO in cells (Figure 7E). These results suggest that downregulation of PHGDH by Parkin contributes greatly to the tumor-suppressive function of Parkin.
Figure 7. Parkin inhibits tumorigenesis through negative regulation of PHGDH.

(A) Parkin KO promoted the growth of orthotopic breast tumors formed by Hs578T cells, which was greatly abolished by PHGDH KO. Left and middle: representative images of mice bearing tumors (left) and the collected tumors (middle) at day 34 after cell inoculation. Right: growth curves of tumors. Scale bars: 10 mm. n = 8. (B) IHC staining of Parkin, PHGDH, and Ki-67 in Hs578T orthotopic breast tumors described in A. Left panels: representative images. Scale bar: 20 μm. Right panel: percentage of Ki-67–positive cells in tumors. n = 6. (C) Levels of total serine and glycine in Hs578T orthotopic breast tumors described in A analyzed by LC-MS–based metabolomics analysis. n = 8. (D) Parkin KO promoted the growth of s.c. xenograft tumors formed by H1299 cells, which was greatly abolished by PHGDH KO in cells. n = 8. (E) Myc-Parkin expression in Hs578T cells inhibited the growth of orthotopic breast tumors, which was greatly abolished by PHGDH KO. n = 8. (F) Ectopic expression of K330R mutant PHGDH greatly abolished the inhibitory effect of Myc-Parkin on the growth of orthotopic breast tumors formed by PHGDH-KO Hs578T cells. n = 8. (G) Orthotopic breast tumors formed by Hs578T-Parkin-KO cells and s.c. tumors formed by H1299-Parkin-KO cells were more sensitive to NCT-503 treatment (i.p., 15 mg/kg; once daily for 4 weeks) compared with tumors formed by their control WT cells. n = 10. (H) Representative images of IHC staining of Parkin and PHGDH in human breast cancer specimens and lung cancer specimens. Scale bars: 20 μm. *P < 0.001; **P < 0.0001, mixed model analysis with Bonferroni’s adjusted P values for A and D–G and heterogeneous variance model with Dunnett’s adjusted P values for B and C.
To further support our observations, Hs578T-PHGDH-KO cells were cotransduced with the WT or K330R PHGDH-Flag vectors and Myc-Parkin vectors, and the cells were used for orthotopic breast tumorigenesis assays. Myc-Parkin expression significantly inhibited the growth of orthotopic tumors formed by cells expressing WT PHGDH-Flag, but displayed a much less pronounced inhibitory effect on the growth of tumors formed by cells expressing K330R PHGDH-Flag (Figure 7F), which suggests that ubiquitination of PHGDH contributes greatly to the tumor-suppressive function of Parkin.
It has been reported that PHGDH inhibitor NCT-503 can greatly suppress the growth of tumors with PHGDH overexpression in mouse models, whereas CBR-5884 is unstable in mouse plasma and unable to be used for in vivo studies (15, 16). Therefore, we used NCT-503 to investigate whether pharmacological inhibition of PHGDH can preferentially inhibit growth of tumors with Parkin deficiency. When the volume of Hs578T orthotopic breast tumors and H1299 s.c. xenograft tumors reached approximately 100 mm3, mice were treated with NCT-503 (i.p., 15 mg/kg) or vehicle once daily for 4 weeks. NCT-503 greatly inhibited the growth of Hs578T orthotopic breast tumors and H1299 s.c. xenograft tumors (Figure 7G). Importantly, orthotopic Hs578T-Parkin-KO tumors and s.c. H1299-Parkin-KO tumors were more sensitive to NCT-503 treatment compared with Hs578T-Parkin-WT and H1299-Parkin-WT tumors, respectively (Figure 7G).
Parkin expression is inversely associated with PHGDH expression in human cancers.
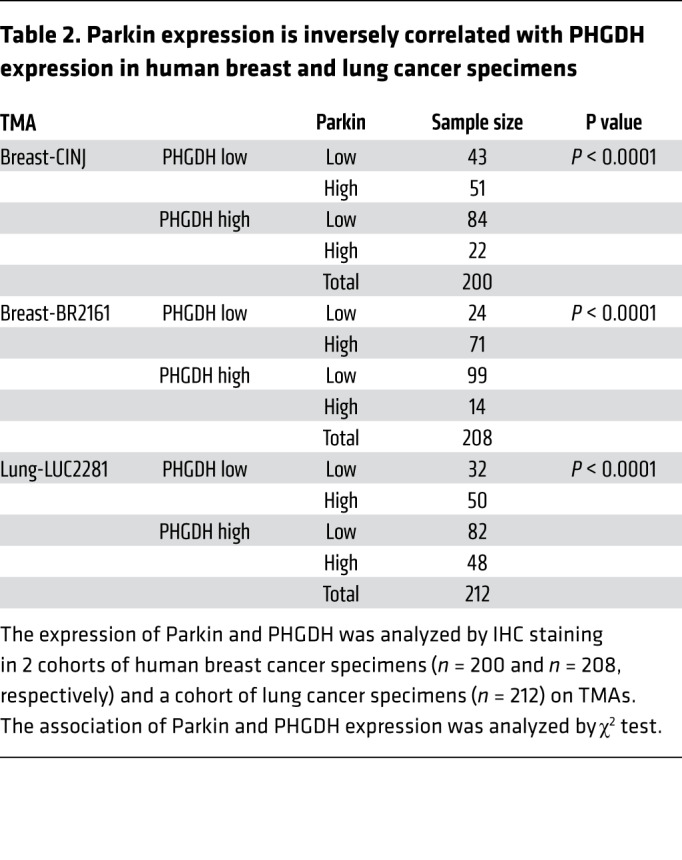
Parkin is frequently downregulated in human cancers, including 40% to 70% of breast cancers and over 30% of lung cancers (26, 30, 32, 33). Given our finding that Parkin is an important negative regulator of PHGDH protein levels, as shown in human cancer cells and mouse cells and tissues (Figure 2), we investigated the potential correlation between the expression of Parkin and PHGDH in human cancer specimens. The protein levels of Parkin and PHGDH in human breast cancer and lung cancer specimens on 3 tissue microarrays (TMAs), including 2 breast cancer TMAs (n = 200 and 208, respectively) and 1 lung cancer TMA (n = 212), were determined by IHC staining assays. A very significant reverse correlation between expression of Parkin and PHGDH was observed in both breast and lung cancer specimens in all of these 3 TMAs (Table 2 and Figure 7H). These results together with our results in cancer cells and mice (Figure 2) suggest that the frequent Parkin downregulation in human breast and lung cancers is an important mechanism contributing to the frequently observed PHGDH overexpression in human breast and lung cancers.
Table 2. Parkin expression is inversely correlated with PHGDH expression in human breast and lung cancer specimens.
In summary, our results strongly suggest that the ubiquitination and degradation of PHGDH by Parkin is a crucial mechanism for PHGDH regulation, which constitutes an important mechanism underlying the tumor-suppressive function of Parkin, and Parkin deficiency leads to PHGDH stabilization and accumulation in cancer cells to activate serine synthesis and promote tumorigenesis (Figure 8).
Figure 8. Schematic model showing that Parkin promotes PHGDH ubiquitination and degradation to inhibit serine synthesis and tumor progression.

Parkin binds to PHGDH and ubiquitinates it at lysine 330, leading to PHGDH degradation to suppress serine synthesis and tumorigenesis. Parkin deficiency results in PHGDH stabilization and accumulation in cancer cells, which in turn activates serine synthesis and promotes tumorigenesis. Targeting PHGDH with small-molecule inhibitors may benefit therapy of some cancers with Parkin deficiency.
Discussion
PHGDH is frequently overexpressed in human cancer, including breast and lung cancers. This overexpression activates serine synthesis to promote cancer progression and is associated with poor prognosis in cancer patients (10–16). However, the mechanism of PHGDH regulation and its overexpression in cancer are not well understood. Parkin is a bona fide tumor suppressor; however, its underlying mechanism is poorly understood. In this study, we found that Parkin is an E3 ubiquitin ligase for PHGDH; Parkin ubiquitinates PHGDH at lysine 330, leading to PHGDH degradation to suppress serine synthesis and tumorigenesis. Parkin deficiency promotes PHGDH protein stabilization and accumulation in cancer cells to activate serine synthesis and promote tumorigenesis, which can be greatly abolished by knockdown or KO of PHGDH and PHGDH inhibitors. These results strongly suggest that the negative regulation of PHGDH and serine synthesis by Parkin is an important mechanism contributing to the tumor-suppressive function of Parkin. Parkin expression is frequently downregulated in many types of cancers, including breast and lung cancers. Our results show that low Parkin expression is associated with high PHGDH expression in both breast and lung cancers, which suggests that the downregulation of Parkin expression is an important mechanism contributing to PHGDH overexpression in cancer. These results revealed an important mechanism of PHGDH regulation in cells and PHGDH overexpression in cancer (Figure 8).
The results in this study also suggest that enhanced serine synthesis resulting from PHGDH overexpression could be a metabolic vulnerability that occurs upon Parkin deficiency in cancer, and PHGDH inhibition may benefit therapy of some cancers with Parkin deficiency. As shown in Figure 7G, the PHGDH inhibitor NCT-503 displayed a better growth inhibitory effect on Hs578T and H1299 Parkin-KO tumors compared with control WT tumors. Considering that serine is an important metabolite and serine synthesis is a critical pathway for tumor cell growth and proliferation, it is possible that tumor cells gain resistance to PHGDH inhibitors during treatments through different mechanisms to provide serine and glycine for their growth and proliferation. These potential mechanisms may include the induction of PHGDH expression and increased serine uptake and from the extracellular environment via transporters, etc. Currently, we have no data for long-term tumor growth or recurrence after the treatment. It will be of interest to study the long-term effects of PHGDH inhibitors on Parkin-deficient cancer and the effects of PHGDH inhibitors on different types of cancers in addition to breast and lung cancers in future studies. Further understanding the specificity and effect of PHGDH-targeted therapy will be important for potential application of targeting PHGDH in cancer therapy in future. These future studies will help us understand whether and how PHGDH inhibition benefits therapy of some cancers with Parkin deficiency.
Our results also show that Parkin displayed some inhibitory effect, although limited, on proliferation of cancer cells with PHGDH knockdown both in vitro and in vivo (Figures 6 and 7), suggesting that the regulation of other protein substrates (such as cyclin D/E, mitotic regulators, HIF-1α, etc.) and pathways by Parkin in addition to PHGDH and the serine synthesis pathway may also contribute to the tumor-suppressive function of Parkin. Interestingly, in addition to our finding that Parkin suppresses serine synthesis, recent studies also show that Parkin suppresses glycolysis (35, 39, 42). Enhanced glycolysis (also known as the Warburg effect) is the most well-characterized metabolic change in cancer, which is crucial for cancer progression (1, 2). Currently, the mechanism underlying Parkin’s function in suppression of glycolysis is not well understood, which could be attributed to Parkin’s activity in ubiquitination of HIF-1α and inhibition of PI3K/AKT signaling (26, 35), since activation of HIF-1α and PI3K/AKT signaling activate glycolysis in cancer (1, 2). Recently, Parkin has also been reported to mediate degradation of mitochondrial iron importers to increase mitochondrial iron accumulation, leading to glycolysis (42). These findings together suggest that the role of Parkin in regulation of cellular metabolism, including serine and glucose metabolism, contributes to the tumor-suppressive function of Parkin.
Mutations of PARK2 are linked to autosomal recessive juvenile PD; approximately 50% of the individuals with autosomal recessive juvenile PD carry PARK2 mutations (19, 24). Parkin dysfunction has also been implicated in the more common sporadic form of PD (19, 51). Interestingly, many epidemiological studies have reported that PD is associated with reduced risks of different types of cancers, including lung, prostate, kidney, stomach, and colorectal cancers (52–56). In contrast, PD was also reported to be associated with increased risks of some types of cancers, including brain cancer and skin cancer (54, 56–58). The mechanism underlying the association of PD with the risk of cancers and the reason PD increases the risk of some types of cancers and reduces the risk of some other types of cancers remain unclear. Currently, it is also unclear whether the role of Parkin in regulation of metabolism, such as serine and glucose metabolism, is involved in its function in prevention of PD. Further studies are warranted to address these questions.
In summary, in this study, we identified PHGDH as a substrate for the E3 ubiquitin ligase Parkin, which provides an important mechanism of PHGDH regulation in cells. Through ubiquitination and degradation of PHGDH, Parkin suppresses serine synthesis, which contributes greatly to the tumor-suppressive function of Parkin. Parkin deficiency leads to PHGDH stabilization and accumulation in cancer cells to activate serine synthesis and promote tumorigenesis. These results also reveal that the frequent downregulation of Parkin expression in cancer is an important mechanism of PHGDH overexpression in cancer. Future studies will shed further light on whether and how PHGDH inhibition can benefit therapy of cancers with Parkin deficiency.
Methods
Cell lines, vectors, and reagents.
Hs578T, MDA-MB468, T47D, HCC70, MCF7, ZR-75-1, MDA-MB-231, MCF10A, H1299, and A549 cells were obtained from ATCC. MEFs were isolated from 13.5-day embryos according to the standard procedures (39). Cells were authenticated by short tandem repeat profiling. All cells used tested negative for Mycoplasma. The vectors expressing Myc-tagged WT Parkin (pLPCX-Myc-Parkin), deletion mutants, and point mutants, including C431A, T240M,and P294S, were constructed as described previously (35). Flag-tagged PHGDH (pLPCX-PHGDH-Flag) and their deletion mutants were constructed by PCR amplification. pLPCX-PHGDH-Flag vectors with different lysine residue mutations were constructed by site-directed mutagenesis using the QuikChange II XL Site-Directed Mutagenesis Kit (Agilent Technologies). Detailed information on the primer sequences for site-directed mutagenesis is provided in Supplemental Table 2. Two lentiviral shRNA vectors against Parkin (no. 1, V3LHS_327550, and no. 2, V3LHS_327555) and control shRNA vectors were obtained from Open Biosystems. Two shRNA vectors against PHGDH were constructed by inserting the following sequences for human PHGDH siRNA into the PLKO.1 hygro lentiviral shRNA vector (Addgene 24150). For PHGDH no. 1, sequence was 5′-CTTAGCAAAGAGGAGCTGATA-3′; for PHGDH no. 2, sequence was 5′-CAGACTTCACTGGTGTCAGAT-3′. To express and purify GST-Parkin and His-Trx-PHGDH proteins, human Parkin cDNA was cloned into the GST-fusion vector pGEX-4T-1 and human PHGDH cDNA was cloned into the His-trx-fusion vector pET-32a, respectively. NCT-503, CBR-5884, CCCP, chloroquine, and Bafilomycin A1 were purchased from MedChemExpress. CHX was purchased from Sigma-Aldrich. U-13C-glucose was purchased from Cambridge Isotope Laboratories.
Generation of Parkin and/or PHGDH KO cell lines using CRISPR/Cas9.
The sgRNAs were designed by the CRISPR sgRNA design web tool, as described previously (59). The sgRNA sequences were as follows: for Parkin, sgRNA-a: 5′-GTGTCAGAATCGACCTCCAC-3′ and sgRNA-b: 5′-TGCTAAGCGACAGGGGGTTC-3′; for PHGDH: sgRNA-a: 5′-GCTCTGAGCCTCCTTGGTGC-3′ and sgRNA-b: 5′-TGCTGTTCAGTTCGTGGACA-3′. The annealed oligonucleotides were ligated into the pSpCas9n(BB)-2A-GFP plasmid (Addgene 48140). Cells transfected with 2 sgRNAs were sorted by flow cytometry, and the GFP-positive single cells were seeded. Single-cell colonies were selected by sequencing PCR products of the edited regions. The deletion of Parkin and/or PHGDH was validated by Western blot assays.
Co-IP assays and LC-MS/MS proteomic assays.
The potential PHGDH-binding proteins in cells were identified by co-IP followed by LC-MS/MS assays. Assays were performed as described previously (35, 60). In brief, MCF10A cells were infected with the pLPCX-PHGDH-Flag vector, and control MCF10A cells were infected with the empty vector for co-IP assays. PHGDH-Flag was pulled down by co-IP using an anti-Flag antibody and eluted with the Flag peptide. Elutions were then used for a second round of co-IP using an anti-PHGDH antibody. LC-MS/MS analysis was performed at the Biological Mass Spectrometry facility of Rutgers University.
To identify the potential ubiquitination site or sites of PHGDH for Parkin, Hs578T cells were cotransfected with vectors expressing Myc-Parkin, PHGDH-Flag, and HA-Ub. Cells were lysed, and PHGDH-Flag was pulled down by co-IP using anti-Flag beads and subjected to LC-MS/MS analysis.
For co-IP of Myc-Parkin and PHGDH-Flag proteins in cells with ectopic expression of Myc-Parkin and PHGDH-Flag, anti-Myc (catalog sc-40, Santa Cruz Biotechnology Inc.) and anti-Flag (catalog A2220, Sigma-Aldrich) agarose beads were used to pull down Myc-Parkin and PHGDH-Flag in cell lysates, respectively. For co-IP of endogenous Parkin and PHGDH, the anti-Parkin antibody (catalog 4211, Cell Signaling Technology) and anti-PHGDH antibody (catalog 66350, Cell Signaling Technology) were used, respectively. The mouse or rabbit purified IgG was used as a negative control for co-IP assays.
Western blot assays.
Standard Western blot assays were used to analyze protein expression in cells. Cellular and mouse tissue proteins were extracted using RIPA lysis buffer (Thermo Fisher Scientific) containing protease inhibitor cocktail. Protein concentration in extracts was measured using Bradford reagent. Protein samples were resolved by SDS-PAGE, transferred onto PVDF membranes, and blocked with TBST containing 5% nonfat dry milk. The following antibodies were used: anti–Flag-M2 (catalog F1804, Sigma-Aldrich, 1:10000), anti-GAPDH (catalog sc-47724, Santa Cruz Biotechnology Inc., 1:10000), anti-Myc (catalog sc-40, Santa Cruz Biotechnology Inc., 1:1000), anti-Parkin (catalog 4211, Cell Signaling Technology, 1:500), anti-PHGDH (catalog HPA021241, Sigma-Aldrich, 1:5000), anti-HA (catalog 11867423001, Sigma-Aldrich, 1:1000), anti-Ub (catalog sc-8017, Santa Cruz Biotechnology Inc., 1:2000), anti-GST antibody (catalog sc-138, Santa Cruz Biotechnology Inc., 1:5000), anti-His (catalog sc-803, Santa Cruz Biotechnology Inc., 1:1000), and anti-LC3B (catalog 2775, Cell Signaling Technology, 1:2000). The band intensity of Western blot assays was quantified using ImageJ software (NIH).
Analysis of PHGDH protein half-life.
To determine the protein half-life of PHGDH in cells, cells were treated with CHX (50 μg/mL) or DMSO for indicated time periods (3–12 hours) before being collected for Western blot analysis of PHGDH protein levels using the anti-PHGDH antibody (catalog HPA021241, Sigma-Aldrich, 1:5000).
Quantitative real-time PCR assays.
Total RNA was prepared with the RNeasy Kit (QIAGEN). cDNA was prepared using a TaqMan Reverse Transcription Kit, and real-time PCR was performed with the TaqMan PCR Mixture (Applied Biosystems), as previously described (35). The expression of genes in cells was normalized to the expression of the GAPDH gene.
In vitro GST pull-down assays.
In vitro GST pull-down assays were performed as we previously described (35). In brief, E. coli (BL21 DE3 strain) transformed with GST-Parkin or His-trx-PHGDH vectors was induced with 0.4 mM IPTG for 16 hours at 16°C to express GST-Parkin or His-trx-PHGDH proteins. The purified GST-Parkin protein (200 ng) was immobilized on Glutathione-Sepharose beads, which were then incubated with purified His-Trx-PHGDH protein (200 ng). GST protein alone was used as a negative control. After washing, proteins bound to the beads were analyzed by Western blot assays using an anti-His (catalog sc-803, Santa Cruz Biotechnology Inc., 1:1000) or anti-GST antibody (catalog sc-138, Santa Cruz Biotechnology Inc., 1:5000).
In vivo and in vitro ubiquitination assays.
In vivo ubiquitination assays were performed as we previously described (35, 60). In brief, cells were transfected with vectors, including vectors expressing Myc-Parkin, PHGDH-Flag, and HA-Ub, respectively, for 24 hours. Cells were then treated with MG132 (5 μM) for 12 hours before being collected for assays. The levels of PHGDH-Flag ubiquitination were determined by IP of PHGDH-Flag using an anti-Flag antibody (catalog A2220, Sigma-Aldrich), followed by Western blot assays using an anti-HA antibody (catalog 11867423001, Sigma-Aldrich, 1:1000). To detect the ubiquitination of endogenous PHGDH, cells with or without Parkin knockdown by shRNAs were transfected with the HA-Ub expression vector. The levels of ubiquitination of endogenous PHGDH were determined by IP of PHGDH using an anti-PHGDH antibody (catalog 66350, Cell Signaling Technology) followed by Western blot assays using an anti-HA antibody (catalog 11867423001, Sigma-Aldrich, 1:1000).
In vitro ubiquitination assays were performed as previously described (61). The reaction mixtures (50 μL) contained 5 mM MgCl2, 50 mM Tris (pH 7.4), 1 mM DTT, 2 mM ATP, E1 (0.5 μg, Boston Biochem), E2 (0.5 μg; Boston Biochem), Ub (5 μg, Boston Biochem), PINK1 (0.5 μg, Boston Biochem), purified His-trx-PHGDH protein (0.5 μg), and purified GST-Parkin (0.5 μg). After incubation for 3 hours at 37°C, the mixtures were used for Western blot assays using an anti-Ub antibody (catalog sc-8017, Santa Cruz Biotechnology Inc., 1:2000).
LC-MS measurement of total and 13C-labeled metabolites.
For analysis of metabolites of mouse tissues, different tissues were snap-frozen in liquid nitrogen, then ground in liquid nitrogen using a mortar and pestle to obtain a homogenous powder. Approximately 20 mg of tissue powder was weighed and used for measurement. For analysis of metabolites in cells, cells (1 × 106 cells) were plated the day before labeling, and the medium was changed to glucose-free DMEM containing 10% dialyzed serum and 25 mM U-13C glucose for the indicated time points (15). Metabolites were extracted in ice-cold methanol:acetonitrile:water (40:40:20) with 0.5% formic acid and neutralized with 15% NH4HCO3. The cellular extracts were analyzed on the Q Exactive PLUS Hybrid Quadrupole-Orbitrap Mass Spectrometer (Thermo Fisher Scientific) coupled to hydrophilic interaction chromatography (HILIC). The LC separation was performed on an UltiMate 3000 UHPLC System with an XBridge BEH Amide Column (150 mm × 2.1 mm, 2.5 μM particle size, Waters) with the corresponding XP VanGuard Cartridge. The LC used a gradient of solvent A (95%:5% H2O:acetonitrile with 20 mM ammonium acetate, 20 mM ammonium hydroxide, pH 9.4), and solvent B (20%:80% H2O:acetonitrile with 20 mM ammonium acetate, 20 mM ammonium hydroxide, pH 9.4). The gradient was as follows: 0 minutes, 100% B; 3 minutes, 100% B; 3.2 minutes, 90% B; 6.2 minutes, 90% B; 6.5 minutes, 80% B; 10.5 minutes, 80% B; 10.7 minutes, 70% B; 13.5 minutes, 70% B; 13.7 minutes, 45% B; 16 minutes, 45% B; 16.5 minutes, 100% B. The flow rate was 300 μL/min. Injection volume was 5 μL, and column temperature was 25°C. The mass spectrometer was under negative ionization mode. Metabolite features including serine m/z 104.035 (M + 0) and 107.048 (M + 3) and glycine m/z 74.025 (M + 0) and 76.031 (M + 2) were extracted in MAVEN with a mass accuracy window of 5 ppm (62). The 13C isotope natural abundance and impurity of labeled substrate were corrected using AccuCor written in R as previously described (63).
Cell proliferation assays.
Cell proliferation was determined by MTT assays as previously described (60). In brief, the same amounts of cells were plated at a low density in 24-well plates (day 0) and cells were cultured for 4 days. For the treatment of PHGDH inhibitors, culture medium was aspirated at 12 hours after cell seeding, cells were washed with PBS, and fresh serine-replete or serine-depleted medium containing different doses of PHGDH inhibitors (NCT-503 and CBR-5884) or vehicle (DMSO) was added (day 0). Cells were cultured for 4 days, and fresh medium containing PHGDH inhibitors was added every day. Cell numbers were measured daily for 4 days by MTT assays.
IHC assays.
The breast cancer TMA composed of 200 primary breast tumors (TMA-Breast-CINJ) was obtained from Rutgers Cancer Institute of New Jersey. The breast cancer TMA composed of 208 breast tumor tissues (TMA-Breast-BR2161) and the lung cancer TMA composed of 212 lung tumor tissues (TMA-Lung-LUC2281) were obtained from US Biomax. All cancer specimens were deidentified samples. IHC staining was performed as described previously (35, 60). The anti-Parkin (catalog NB100-91921, Novus), anti-PHGDH (catalog HPA021241, MilliporeSigma), and anti–Ki-67 (catalog 556003, BD Biosciences) antibodies were used to detect the levels of Parkin, PHGDH, and Ki-67 in TMAs and tumor sections, respectively. The results were scored by using a scoring system from 0 to 9 as described (35, 60). The scores were obtained by multiplying the intensity of signals with the percentages of positive cells (signal: 0, no signal; 1, weak signal; 2, intermediate signal; 3, strong signal; percentages: 0, 0%; 1, ≤ 25%; 2, 25%–50%; 3, ≥ 50%). Low and high expression were defined as scores of less than 6 and 6 or more, respectively. For Ki-67 staining, sections of 6 different tumors were counted for each group. For each section, 5 fields of view at ×40 original magnification were randomly selected and counted. The number of Ki-67–positive cells was divided by the total cell number, and the percentages for each group were averaged.
Mice and tumorigenesis assays.
Eight-week-old female Parkin–/– mice (The Jackson Laboratory; stock number 006582) were used for analysis of levels of protein and metabolites. For orthotropic breast tumor models, Hs578T cells (5 × 106 in a 50:50 mix of DMEM: Matrigel) were injected into the mammary fat pads of 8-week-old female BALB/c athymic nude mice, as described previously (Taconic; n = 8 or 10 mice per group) (35). For s.c. xenograft tumor models, 5 × 106 H1299 cells in 0.2 mL of PBS were s.c. injected into flanks of 8-week-old BALB/c male athymic nude mice (64). NCT-503 was prepared in a vehicle of 5% ethanol, 35% PEG 300 (Sigma-Aldrich), and 60% of an aqueous 30% hydroxypropyl-β-cyclodextrin (Sigma-Aldrich) solution. When the tumor volume reached approximately 100 mm3, mice were injected with NCT-503 (i.p., 15 mg/kg) or vehicle once daily for 4 weeks. Mice were sacrificed at 2 days after last injection of NCT-503. Tumor volume was equal to ½ (length × width2).
Statistics.
Data are shown as mean ± SD. Sample sizes (n) indicate biological replicates within the same experiment. Differences in cell proliferation and tumor growth among groups were analyzed using mixed model analysis that adjusted for intracell or intramouse correlation between longitudinal measurements. Linear contrasts were made to compare growth curves using F tests, and P values were adjusted using Bonferroni’s correction. The association of Parkin and PHGDH expression in TMAs was analyzed by χ2 test. For other experiments, statistical significance was determined using heterogeneous variance model with Dunnett’s method for multiple testing. All statistical analyses were performed using SAS, version 9.4.
Study approval.
Animal maintenance and all experiments were performed in accordance with IACUC guidelines and with approval of the IACUC at Rutgers University. Cancer sample analysis was performed in accordance with the protocol approved by the Rutgers IRB. The breast cancer TMA was obtained from Rutgers Cancer Institute of New Jersey with IRB approval.
Author contributions
JL, CZ, HW, XXS, YL, SH, XY, and XS performed the experiments and analyzed data. SEL, BGH, EW, and ZS analyzed data and contributed important reagents. WH and ZF conceived and supervised the study. JL, ZF, and WH wrote the manuscript. JL and CZ contributed equally to this work. JL initiated this study, and therefore, JL is listed before CZ.
Supplementary Material
Acknowledgments
This work was supported in part by grants from the NIH (R01CA227912 and R01CA214746 to ZF, R01CA203965 to WH, and R01CA163591 to EW), from Congressionally Directed Medical Research Programs (W81XWH-16-1-0358 to WH), and from the Breast Cancer Research Foundation (to BGH). The Metabolomics Shared Resource of Rutgers Cancer Institute of New Jersey is supported in part with funding from NCI-CCSG P30CA072720-5923. LC-MS/MS proteomic analysis was performed at the Biological Mass Spectrometry facility of Rutgers University.
Version 1. 05/18/2020
Electronic publication
Version 2. 06/01/2020
Print issue publication
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Copyright: © 2020, American Society for Clinical Investigation.
Reference information: J Clin Invest. 2020;130(6):3253–3269.https://doi.org/10.1172/JCI132876.
See the related Commentary at Parkin on serine: a Parkinson disease gene suppresses serine synthesis in cancer.
Contributor Information
Juan Liu, Email: jl1530@cinj.rutgers.edu.
Cen Zhang, Email: cenzh1978@gmail.com.
Hao Wu, Email: hw288@cinj.rutgers.edu.
Xiao-Xin Sun, Email: sunx@ohsu.edu.
Yanchen Li, Email: sueb4603@gmail.com.
Shan Huang, Email: zhcen7917@gmail.com.
Xuetian Yue, Email: mazecheng650@gmail.com.
Shou-En Lu, Email: sl1020@sph.rutgers.edu.
Zhiyuan Shen, Email: shenzh@cinj.rutgers.edu.
Xiaoyang Su, Email: xs137@rwjms.rutgers.edu.
Eileen White, Email: epwhite@cinj.rutgers.edu.
Wenwei Hu, Email: wh221@cinj.rutgers.edu.
Zhaohui Feng, Email: zhfeng02@gmail.com.
References
- 1.Pavlova NN, Thompson CB. The emerging hallmarks of cancer metabolism. Cell Metab. 2016;23(1):27–47. doi: 10.1016/j.cmet.2015.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.DeNicola GM, Cantley LC. Cancer’s fuel choice: new flavors for a picky eater. Mol Cell. 2015;60(4):514–523. doi: 10.1016/j.molcel.2015.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wolpaw AJ, Dang CV. Exploiting metabolic vulnerabilities of cancer with precision and accuracy. Trends Cell Biol. 2018;28(3):201–212. doi: 10.1016/j.tcb.2017.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mattaini KR, Sullivan MR, Vander Heiden MG. The importance of serine metabolism in cancer. J Cell Biol. 2016;214(3):249–257. doi: 10.1083/jcb.201604085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yang M, Vousden KH. Serine and one-carbon metabolism in cancer. Nat Rev Cancer. 2016;16(10):650–662. doi: 10.1038/nrc.2016.81. [DOI] [PubMed] [Google Scholar]
- 6.Amelio I, Cutruzzolá F, Antonov A, Agostini M, Melino G. Serine and glycine metabolism in cancer. Trends Biochem Sci. 2014;39(4):191–198. doi: 10.1016/j.tibs.2014.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Reina-Campos M, et al. Increased serine and one-carbon pathway metabolism by PKCλ/ι deficiency promotes neuroendocrine prostate cancer. Cancer Cell. 2019;35(3):385–400.e9. doi: 10.1016/j.ccell.2019.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sullivan MR, et al. Increased serine synthesis provides an advantage for tumors arising in tissues where serine levels are limiting. Cell Metab. 2019;29(6):1410–1421.e4. doi: 10.1016/j.cmet.2019.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Reid MA, et al. Serine synthesis through PHGDH coordinates nucleotide levels by maintaining central carbon metabolism. Nat Commun. 2018;9(1):5442. doi: 10.1038/s41467-018-07868-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Possemato R, et al. Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature. 2011;476(7360):346–350. doi: 10.1038/nature10350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Locasale JW, et al. Phosphoglycerate dehydrogenase diverts glycolytic flux and contributes to oncogenesis. Nat Genet. 2011;43(9):869–874. doi: 10.1038/ng.890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang B, et al. PHGDH defines a metabolic subtype in lung adenocarcinomas with poor prognosis. Cell Rep. 2017;19(11):2289–2303. doi: 10.1016/j.celrep.2017.05.067. [DOI] [PubMed] [Google Scholar]
- 13.Mullarky E, Mattaini KR, Vander Heiden MG, Cantley LC, Locasale JW. PHGDH amplification and altered glucose metabolism in human melanoma. Pigment Cell Melanoma Res. 2011;24(6):1112–1115. doi: 10.1111/j.1755-148X.2011.00919.x. [DOI] [PubMed] [Google Scholar]
- 14.Zhu J, et al. High expression of PHGDH predicts poor prognosis in non-small cell lung cancer. Transl Oncol. 2016;9(6):592–599. doi: 10.1016/j.tranon.2016.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mullarky E, et al. Identification of a small molecule inhibitor of 3-phosphoglycerate dehydrogenase to target serine biosynthesis in cancers. Proc Natl Acad Sci U S A. 2016;113(7):1778–1783. doi: 10.1073/pnas.1521548113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pacold ME, et al. A PHGDH inhibitor reveals coordination of serine synthesis and one-carbon unit fate. Nat Chem Biol. 2016;12(6):452–458. doi: 10.1038/nchembio.2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang Q, et al. Rational design of selective allosteric inhibitors of PHGDH and serine synthesis with anti-tumor activity. Cell Chem Biol. 2017;24(1):55–65. doi: 10.1016/j.chembiol.2016.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.DeNicola GM, et al. NRF2 regulates serine biosynthesis in non-small cell lung cancer. Nat Genet. 2015;47(12):1475–1481. doi: 10.1038/ng.3421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Klein C, Westenberger A. Genetics of Parkinson’s disease. Cold Spring Harb Perspect Med. 2012;2(1):a008888. doi: 10.1101/cshperspect.a008888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Matsumine H, et al. Localization of a gene for an autosomal recessive form of juvenile Parkinsonism to chromosome 6q25.2-27. Am J Hum Genet. 1997;60(3):588–596. [PMC free article] [PubMed] [Google Scholar]
- 21.Pickrell AM, Youle RJ. The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron. 2015;85(2):257–273. doi: 10.1016/j.neuron.2014.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Winklhofer KF. Parkin and mitochondrial quality control: toward assembling the puzzle. Trends Cell Biol. 2014;24(6):332–341. doi: 10.1016/j.tcb.2014.01.001. [DOI] [PubMed] [Google Scholar]
- 23.Trempe JF, et al. Structure of parkin reveals mechanisms for ubiquitin ligase activation. Science. 2013;340(6139):1451–1455. doi: 10.1126/science.1237908. [DOI] [PubMed] [Google Scholar]
- 24.Kitada T, et al. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature. 1998;392(6676):605–608. doi: 10.1038/33416. [DOI] [PubMed] [Google Scholar]
- 25.Villa E, Marchetti S, Ricci JE. No Parkin zone: mitophagy without Parkin. Trends Cell Biol. 2018;28(11):882–895. doi: 10.1016/j.tcb.2018.07.004. [DOI] [PubMed] [Google Scholar]
- 26.Gupta A, et al. PARK2 depletion connects energy and oxidative stress to PI3K/Akt activation via PTEN S-nitrosylation. Mol Cell. 2017;65(6):999–1013.e7. doi: 10.1016/j.molcel.2017.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xu L, Lin DC, Yin D, Koeffler HP. An emerging role of PARK2 in cancer. J Mol Med. 2014;92(1):31–42. doi: 10.1007/s00109-013-1107-0. [DOI] [PubMed] [Google Scholar]
- 28.Veeriah S, et al. Somatic mutations of the Parkinson’s disease-associated gene PARK2 in glioblastoma and other human malignancies. Nat Genet. 2010;42(1):77–82. doi: 10.1038/ng.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu J, Zhang C, Hu W, Feng Z. Parkinson’s disease-associated protein Parkin: an unusual player in cancer. Cancer Commun (Lond) 2018;38(1):40. doi: 10.1186/s40880-018-0314-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cesari R, et al. Parkin, a gene implicated in autosomal recessive juvenile parkinsonism, is a candidate tumor suppressor gene on chromosome 6q25-q27. Proc Natl Acad Sci U S A. 2003;100(10):5956–5961. doi: 10.1073/pnas.0931262100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Poulogiannis G, et al. PARK2 deletions occur frequently in sporadic colorectal cancer and accelerate adenoma development in Apc mutant mice. Proc Natl Acad Sci U S A. 2010;107(34):15145–15150. doi: 10.1073/pnas.1009941107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tay SP, et al. Parkin enhances the expression of cyclin-dependent kinase 6 and negatively regulates the proliferation of breast cancer cells. J Biol Chem. 2010;285(38):29231–29238. doi: 10.1074/jbc.M110.108241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Picchio MC, et al. Alterations of the tumor suppressor gene Parkin in non-small cell lung cancer. Clin Cancer Res. 2004;10(8):2720–2724. doi: 10.1158/1078-0432.CCR-03-0086. [DOI] [PubMed] [Google Scholar]
- 34.Denison SR, et al. Alterations in the common fragile site gene Parkin in ovarian and other cancers. Oncogene. 2003;22(51):8370–8378. doi: 10.1038/sj.onc.1207072. [DOI] [PubMed] [Google Scholar]
- 35.Liu J, et al. Parkin targets HIF-1α for ubiquitination and degradation to inhibit breast tumor progression. Nat Commun. 2017;8(1):1823. doi: 10.1038/s41467-017-01947-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yeo CW, et al. Parkin pathway activation mitigates glioma cell proliferation and predicts patient survival. Cancer Res. 2012;72(10):2543–2553. doi: 10.1158/0008-5472.CAN-11-3060. [DOI] [PubMed] [Google Scholar]
- 37.Toma MI, et al. PARK2 and PACRG are commonly downregulated in clear-cell renal cell carcinoma and are associated with aggressive disease and poor clinical outcome. Genes Chromosomes Cancer. 2013;52(3):265–273. doi: 10.1002/gcc.22026. [DOI] [PubMed] [Google Scholar]
- 38.Fujiwara M, et al. Parkin as a tumor suppressor gene for hepatocellular carcinoma. Oncogene. 2008;27(46):6002–6011. doi: 10.1038/onc.2008.199. [DOI] [PubMed] [Google Scholar]
- 39.Zhang C, et al. Parkin, a p53 target gene, mediates the role of p53 in glucose metabolism and the Warburg effect. Proc Natl Acad Sci U S A. 2011;108(39):16259–16264. doi: 10.1073/pnas.1113884108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gong Y, et al. Pan-cancer genetic analysis identifies PARK2 as a master regulator of G1/S cyclins. Nat Genet. 2014;46(6):588–594. doi: 10.1038/ng.2981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lee SB, et al. Parkin regulates mitosis and genomic stability through Cdc20/Cdh1. Mol Cell. 2015;60(1):21–34. doi: 10.1016/j.molcel.2015.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li C, et al. PINK1 and PARK2 suppress pancreatic tumorigenesis through control of mitochondrial iron-mediated immunometabolism. Dev Cell. 2018;46(4):441–455.e8. doi: 10.1016/j.devcel.2018.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lee SB, et al. The AMPK-Parkin axis negatively regulates necroptosis and tumorigenesis by inhibiting the necrosome. Nat Cell Biol. 2019;21(8):940–951. doi: 10.1038/s41556-019-0356-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Riley BE, et al. Structure and function of Parkin E3 ubiquitin ligase reveals aspects of RING and HECT ligases. Nat Commun. 2013;4:1982. doi: 10.1038/ncomms2982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Spratt DE, et al. A molecular explanation for the recessive nature of parkin-linked Parkinson’s disease. Nat Commun. 2013;4:1983. doi: 10.1038/ncomms2983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol. 2008;183(5):795–803. doi: 10.1083/jcb.200809125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.McWilliams TG, Muqit MM. PINK1 and Parkin: emerging themes in mitochondrial homeostasis. Curr Opin Cell Biol. 2017;45:83–91. doi: 10.1016/j.ceb.2017.03.013. [DOI] [PubMed] [Google Scholar]
- 48.Klionsky DJ, et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition) Autophagy. 2016;12(1):1–222. doi: 10.1080/15548627.2015.1100356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lin DC, et al. Genomic and functional analysis of the E3 Ligase PARK2 in glioma. Cancer Res. 2015;75(9):1815–1827. doi: 10.1158/0008-5472.CAN-14-1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Achouri Y, Rider MH, Schaftingen EV, Robbi M. Cloning, sequencing and expression of rat liver 3-phosphoglycerate dehydrogenase. Biochem J. 1997;323(pt 2):365–370. doi: 10.1042/bj3230365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pilcher H. Parkin implicated in sporadic Parkinson’s disease. Lancet Neurol. 2005;4(12):798. doi: 10.1016/s1474-4422(05)70237-4. [DOI] [PubMed] [Google Scholar]
- 52.Wirdefeldt K, Adami HO, Cole P, Trichopoulos D, Mandel J. Epidemiology and etiology of Parkinson’s disease: a review of the evidence. Eur J Epidemiol. 2011;26(suppl 1):S1–58. doi: 10.1007/s10654-011-9581-6. [DOI] [PubMed] [Google Scholar]
- 53.Bajaj A, Driver JA, Schernhammer ES. Parkinson’s disease and cancer risk: a systematic review and meta-analysis. Cancer Causes Control. 2010;21(5):697–707. doi: 10.1007/s10552-009-9497-6. [DOI] [PubMed] [Google Scholar]
- 54.Catalá-López F, et al. Inverse and direct cancer comorbidity in people with central nervous system disorders: a meta-analysis of cancer incidence in 577,013 participants of 50 observational studies. Psychother Psychosom. 2014;83(2):89–105. doi: 10.1159/000356498. [DOI] [PubMed] [Google Scholar]
- 55.Tabarés-Seisdedos R, Rubenstein JL. Inverse cancer comorbidity: a serendipitous opportunity to gain insight into CNS disorders. Nat Rev Neurosci. 2013;14(4):293–304. doi: 10.1038/nrn3464. [DOI] [PubMed] [Google Scholar]
- 56.Zhang P, Liu B. Association between Parkinson’s disease and risk of cancer: a PRISMA-compliant meta-analysis. ACS Chem Neurosci. 2019;10(10):4430–4439. doi: 10.1021/acschemneuro.9b00498. [DOI] [PubMed] [Google Scholar]
- 57.Ye R, Shen T, Jiang Y, Xu L, Si X, Zhang B. The relationship between Parkinson disease and brain tumor: a meta-analysis. PLoS One. 2016;11(10):e0164388. doi: 10.1371/journal.pone.0164388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Feng DD, Cai W, Chen X. The associations between Parkinson’s disease and cancer: the plot thickens. Transl Neurodegener. 2015;4:20. doi: 10.1186/s40035-015-0043-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat Protoc. 2013;8(11):2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhang C, et al. Cullin3-KLHL25 ubiquitin ligase targets ACLY for degradation to inhibit lipid synthesis and tumor progression. Genes Dev. 2016;30(17):1956–1970. doi: 10.1101/gad.283283.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Xiong H, et al. Parkin, PINK1, and DJ-1 form a ubiquitin E3 ligase complex promoting unfolded protein degradation. J Clin Invest. 2009;119(3):650–660. doi: 10.1172/JCI37617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Melamud E, Vastag L, Rabinowitz JD. Metabolomic analysis and visualization engine for LC-MS data. Anal Chem. 2010;82(23):9818–9826. doi: 10.1021/ac1021166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Su X, Lu W, Rabinowitz JD. Metabolite spectral accuracy on orbitraps. Anal Chem. 2017;89(11):5940–5948. doi: 10.1021/acs.analchem.7b00396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhang C, et al. Tumour-associated mutant p53 drives the Warburg effect. Nat Commun. 2013;4:2935. doi: 10.1038/ncomms3935. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.