

Abstract
Although autophagy is generally protective, uncontrolled or excessive activation of autophagy can be detrimental. However, it is often difficult to distinguish death by autophagy from death with autophagy, and whether autophagy contributes to death in cardiomyocytes (CMs) is still controversial. Excessive activation of autophagy induces a morphologically and biochemically defined form of cell death termed autosis. Whether autosis is involved in tissue injury induced under pathologically relevant conditions is poorly understood. In the present study, myocardial ischemia/reperfusion (I/R) induced autosis in CMs, as evidenced by cell death with numerous vacuoles and perinuclear spaces, and depleted intracellular membranes. Autosis was observed frequently after 6 hours of reperfusion, accompanied by upregulation of Rubicon, attenuation of autophagic flux, and marked accumulation of autophagosomes. Genetic downregulation of Rubicon inhibited autosis and reduced I/R injury, whereas stimulation of autosis during the late phase of I/R with Tat–Beclin 1 exacerbated injury. Suppression of autosis by ouabain, a cardiac glycoside, in humanized Na+,K+-ATPase–knockin mice reduced I/R injury. Taken together, these results demonstrate that autosis is significantly involved in I/R injury in the heart and triggered by dysregulated accumulation of autophagosomes due to upregulation of Rubicon.
Keywords: Cardiology, Cell Biology
Keywords: Autophagy, Signal transduction

Introduction
Autophagy is an evolutionarily conserved catabolic mechanism for intracellular components, from cytosolic proteins to organelles, which are engulfed into double-membrane vesicles called autophagosomes and delivered to lysosomes for degradation (1). Autophagy eliminates misfolded proteins and dysfunctional organelles under both basal and stress conditions, including oxidative, ER, metabolic, and genotoxic stress (2–4). In addition, autophagy supplies sources of energy, including amino acids and fatty acids, under nutrient- and energy-depleted conditions.
Beclin 1, the ortholog of yeast Atg6, is one of the key regulators of the autophagy pathway. Beclin 1 associates with class III PI3K (PI3KC3) and vacuolar protein sorting 15 (VPS15) to form the PI3KC3 complex, which mediates vesicle trafficking and autophagy. In addition, several other components, including Atg14L, the UV radiation resistance–associated gene (UVRAG), and Rubicon, associate with the PI3KC3 complex in a context-dependent manner. Rubicon suppresses autophagy by interacting with Beclin1 and UVRAG during multiple steps of the autophagic process, including the autophagosome-lysosome fusion step (5).
Like many biological processes, such as proteasomal degradation and the ER stress response, autophagy is generally beneficial when its activity is maintained at appropriate levels. However, increasing lines of evidence suggest that dysregulated autophagy can be detrimental (6). Morphological studies using electron microscopy (EM) have suggested that in many contexts, cell death takes place when autophagy is observed (7). However, since autophagy is often activated by stress as an adaptive mechanism and may continue until cells die, it is challenging to determine whether cell death is triggered by autophagy or occurs concurrently with autophagy. In order to prove that cell death is caused by autophagy, it is necessary to establish a direct link between autophagy and cell death. Previous studies have suggested that some molecular mechanisms activating autophagy can directly stimulate cell death (8–10). However, the currently available evidence is somewhat inconclusive, in that many interventions to inhibit autophagy can directly affect mechanisms shared by other types of cell death. Thus, whether autophagy can directly kill cells under pathophysiologically relevant conditions has been a subject of debate in the field.
A study by Levine and colleagues described a novel form of cell death caused by autophagy, termed autosis (11). Cell death by autosis is induced by strong activation of autophagy by either Tat–Beclin 1 or starvation in HeLa cells and has unique morphological and biochemical features that distinguish it from other forms of cell death. Morphologically, autosis is characterized by accumulation of vacuoles, including autophagosomes, autolysosomes, and empty vacuoles, and by separation of the inner and outer nuclear membranes and swelling of the perinuclear space (PNS). In later stages of autosis, disappearance of intracellular organelles is observed (12). Autosis is inhibited by suppression of autophagy but not by suppression of apoptosis or necrosis. Importantly, unbiased compound library screenings showed that pharmacological inhibition or genetic inactivation of Na+,K+-ATPase inhibits autosis (11). Levine and colleagues showed that autosis is induced by hypoxia-ischemia in the neonatal mouse brain in vivo (11); however, it remains unclear whether autosis is observed under other pathophysiological conditions in vivo.
Autophagy generally plays an important role in maintaining cardiac function in the adult heart under baseline conditions, in response to fasting or ischemia, and during heart failure (13, 14). However, activation of autophagy in the heart can be detrimental under some conditions. For example, autophagy is strongly activated during ischemia/reperfusion (I/R), and suppression of autophagy due to haploinsufficiency of Beclin 1 reduces I/R injury (13, 15). Similarly, autophagy is activated during the acute phase of pressure overload, and its suppression improves cardiac function. The chemotherapy agent doxorubicin (DOX) also activates excessive autophagy in conjunction with cardiac injury, and inhibiting DOX-induced autophagy improves cardiac function (16, 17). However, whether autophagy induces detrimental effects in the heart, including induction of cardiomyocyte (CM) death, is controversial, since, as in other cell types, it has been difficult to establish a direct link between autophagy and cell death mechanisms in CMs.
Thus, the goals of this study were to investigate (i) whether autosis can be induced by activation of autophagy in CMs, (ii) whether autosis is induced by I/R in the heart in vivo, (iii) the molecular mechanism by which autosis is induced during I/R, and (iv) whether suppression of autosis protects the heart against I/R injury.
Results
High doses of Tat–Beclin 1 induce autosis in CMs.
Tat–Beclin 1 activates autophagy without cytotoxicity at low dosages, but it induces autosis at high dosages in HeLa cells (11). To evaluate the effect of Tat–Beclin 1 on autophagic flux in CMs, we employed an autophagy probe, GFP-LC3-RFP (18). GFP-LC3-RFP is cleaved into GFP-LC3 and RFP fragments in cells. Although the level of GFP-LC3 is decreased by autophagic degradation, the level of RFP is maintained even in the presence of autophagic flux. Thus, the GFP/RFP ratio is decreased when autophagic flux is increased (Figure 1A). Treatment of neonatal rat CMs (NRCMs) with Tat–Beclin 1 induced a significant decrease in the GFP/RFP ratio (yellow in the ratiometric image), indicating that Tat–Beclin 1 increased autophagic flux. Autophagic flux was not increased after treatment with a control peptide, Scrambled (Figure 1B and Supplemental Figure 1A; supplemental material available online with this article; https://doi.org/10.1172/JCI132366DS1). Treatment with Tat–Beclin 1 significantly increased the LC3-II/GAPDH ratio and decreased the level of p62 (Figure 1, C and D). Similar results were observed with adult mouse CMs (AMCMs) and human induced pluripotent stem cell–derived (iPSC-derived) CMs (Supplemental Figure 1, B and E). These results suggest that Tat–Beclin 1 increases autophagic flux in NRCMs, AMCMs, and human iPSC-derived CMs.
Figure 1. Tat–Beclin 1 induces autosis in CMs.

(A) The autophagic flux probe GFP-LC3-RFP. (B) NRCMs were transduced with Ad-GFP-LC3-RFP and treated with Tat–Beclin 1 (TB) for 3 hours. GFP/RFP ratio is shown. n = 5 each; scale bar: 20 μm. (C) NRCMs were treated with Tat–Beclin 1 for 2 hours and analyzed by Western blotting. See complete unedited blots in the supplemental material. (D) Ratios of LC3-II to GAPDH and p62 to GAPDH. Mean ± SD, n = 3; *P < 0.05, **P < 0.01, 1-way ANOVA with Tukey’s post hoc test. (E) NRCMs were treated with Tat–Beclin 1 for 3 hours and analyzed with CellTiter-Blue assays. Mean ± SD, n = 4 values were measured from more than 8 different wells per experiment; **P < 0.01, 1-way ANOVA with Dunnett’s post hoc test. (F) NRCMs were pretreated with Z-VAD, Nec1, or 3MA for 1 hour; treated with Tat–Beclin 1 for 3 hours; and analyzed with CellTiter-Blue assays. Mean ± SD, n = 11 (Scrambled, 5 μM Tat–Beclin 1), n = 8 (10 μM Tat-Beclin 1); *P < 0.05, **P < 0.01 vs. Scrambled, ##P < 0.01 vs. 10 μM Tat–Beclin 1 with vehicle (Veh.), 2-way ANOVA. (G–I) NRCMs were treated with Tat–Beclin 1 for 3 hours and subjected to EM analyses. (G) Nuclei (Nu), ballooning of the PNS, empty vacuoles (EVs), mitochondria (Mito), sarcomeres, and autophagic vacuoles (AVs) (scale bars: 2 μm). Number of AVs per cell (H) and percentage of cells with PNS (I) were evaluated from more than 10 different areas (H) and more than 20 CM nuclei (I) in each experiment. Mean ± SEM, n = 6 (Tat-Scrambled [TS], 5 μM Tat–Beclin 1), n = 3 (1 μM Tat–Beclin 1); **P < 0.01 vs. Tat-Scrambled; ##P < 0.01, 1-way ANOVA with Tukey’s post hoc test. (J) NRCMs were transduced with Ad–shNa+,K+-ATPase α1 and analyzed by Western blotting. (K) NRCMs were transduced with Ad-shNa+,K+-ATPase α1, then treated with Tat–Beclin 1 for 3 hours, and analyzed with CellTiter-Blue assays (mean ± SD, n = 4, values were measured from more than 16 wells per experiment; *P < 0.05, **P < 0.01 vs. Scrambled, ##P < 0.01, 2-way ANOVA). See also Supplemental Figure 1.
We next examined the cell death–promoting effect of Tat–Beclin 1 in CMs. Tat–Beclin 1 induced cell death in NRCMs at concentrations as low as 2.5 μM and increased cell death in a dose-dependent manner, as determined using CellTiter-Blue (Figure 1E), trypan blue exclusion (Supplemental Figure 1G), and SYTOX Green assays (Supplemental Figure 1H). Tat–Beclin 1 also induced cell death in AMCMs and human iPSC-derived CMs (Supplemental Figure 1, C, D, and F). Treatment with 3-methyladenine (3MA), an inhibitor of the PI3KC3 complex, inhibited Tat–Beclin 1–induced cell death; but neither Z-VAD, a pan-caspase inhibitor (19), nor necrostatin-1 (Nec1), an inhibitor of receptor-interacting serine/threonine-protein kinase 1–mediated (RIPK1-mediated) necrosis (20), attenuated Tat–Beclin 1–induced cell death, as indicated by CellTiter-Blue assays (Figure 1F). shRNA-mediated knockdown of regulators of autophagy including as ULK1, Beclin 1, and Atg7 significantly decreased Tat–Beclin 1–induced cell death in NRCMs (Supplemental Figure 1I). In addition, Tat–Beclin 1–induced cell death was significantly attenuated in AMCMs isolated from cardiac tissue-specific Atg7-knockout mice (21) compared with those from WT mice (Supplemental Figure 1, C and D). These results suggest that Tat–Beclin 1 triggers both autophagy and cell death in CMs, and that the cell death is autophagy dependent but necrosis and apoptosis independent.
We next examined whether Tat–Beclin 1–induced CM cell death is autosis (11). EM analyses showed that NRCMs treated with 5 μM Tat–Beclin 1 exhibited morphological features of autosis (12), including upregulation of autophagic vacuoles and empty vacuoles, and focal ballooning of the PNS, which were not observed in NRCMs treated with either a low dose of Tat–Beclin 1 or Scrambled (Figure 1, G–I). It has been shown that autosis is inhibited by cardiac glycosides, inhibitors of Na+,K+-ATPase α1 (11). Since Na+,K+-ATPase α1 in rodents is relatively insensitive to cardiac glycosides (22), we generated an adenovirus harboring shRNA against the α1 subunit of Na+,K+-ATPase (Figure 1J). Knockdown of the α1 subunit of Na+,K+-ATPase attenuated Tat–Beclin 1–induced cell death in NRCMs (Figure 1K). Together, these data show that Tat–Beclin 1–induced autophagic cell death in CMs exhibits both morphological and biochemical features of autosis.
In addition, we tested whether autosis can be induced in NRCMs by other forms of pathological stress. Hypoxia-induced cell injury can be aggravated by reoxygenation. Here, we tested whether autosis is involved in hypoxia/reoxygenation-induced cell death. Interestingly, knockdown of the α1 subunit of Na+,K+-ATPase significantly attenuated hypoxia/reoxygenation-induced cell death in NRCMs (Supplemental Figure 1J). Thus, autosis also partially contributes to hypoxia/reoxygenation-induced cell death.
Autosis in CMs is accompanied by increased consumption of intracellular membranes.
We next investigated additional features of death induced by Tat–Beclin 1 in CMs. Since Tat–Beclin 1 strongly activates autophagy, we hypothesized that CMs might degrade vital cellular components. To evaluate whether Tat–Beclin 1–induced cell death occurs through lysosomal degradation, we treated NRCMs with bafilomycin A1, a lysosomal inhibitor. Interestingly, suppression of lysosomal degradation did not rescue Tat–Beclin 1–induced cell death (Supplemental Figure 2A), suggesting that Tat–Beclin 1 does not induce cell death through lysosomal degradation.
Tat–Beclin 1–induced autosis is accompanied by accumulation of many autophagic vacuoles, including autophagosomes, autolysosomes, and empty vacuoles. In order to generate autophagic vacuoles, cells recruit membranes from intracellular organelles, including the ER, mitochondria, and plasma membrane (23, 24). Therefore, we speculated that autosis is triggered by excessive consumption of organelle membranes. We first performed subcellular fractionations to quantify the level of ER, mitochondria, and plasma membrane in NRCMs after treatment with Scrambled or Tat–Beclin 1. Compared with treatment with Scrambled, Tat–Beclin 1 treatment significantly and dose-dependently decreased the levels of calnexin, an ER marker protein; pyruvate dehydrogenase (PDH), a mitochondrial protein; and calcium pump of the plasma membrane (PMCA), a plasma membrane protein in the heavy membrane fraction that includes ER, mitochondria, and plasma membrane (Figure 2, A–D). In addition, although Scrambled-treated NRCMs showed normal ER and mitochondrion morphology, Tat–Beclin 1–treated NRCMs exhibited both abnormal ER patterns, namely shrinkage and fragmentation, and abnormal mitochondria patterns, including a dot scatter form (Supplemental Figure 2, B and C). To examine whether consumption of membrane affects membrane potential of mitochondria, we tested mitochondria membrane potential using JC1 dye in the presence of Tat–Beclin 1. Mitochondrial membrane potential was significantly decreased by Tat–Beclin 1 dose-dependently (Supplemental Figure 2, D and E). However, treatment with cyclosporin A, which blocks the mitochondrial permeability transition pore and prevents swelling of mitochondria, did not significantly affect Tat–Beclin 1–induced cell death in NRCMs (Supplemental Figure 2, F and G). These results suggest that strong activation of autophagy may facilitate alterations in organelle membranes, which may, in turn, lead to a shortage of essential membranes.
Figure 2. Tat–Beclin 1–treated CMs show decreased levels of cellular membranes.

(A–D) NRCMs were treated with Scrambled or Tat–Beclin 1 (5 and 10 μM) for 3 hours and subjected to membrane fractionation assays. The heavy membrane and cytosolic fractions were analyzed by Western blotting using anti-calnexin (ER), anti-PDH (mitochondria), anti-PMCA (plasma membrane), and anti–α-tubulin antibodies (A). Expression ratios of calnexin (B), PDH (C), and PMCA to α-tubulin (D) were quantified; mean ± SD, n = 5 (B), n = 4 (C and D); *P < 0.05, **P < 0.01, 1-way ANOVA with Dunnett’s post hoc test. (E and F) NRCMs were transfected with siControl (siCtrl) or siVAPA and siVAPB. After 60 hours, cells were treated with Scrambled or Tat–Beclin 1 at the indicated doses for 3 hours and subjected to membrane fractionation assays. (E) Whole cell lysates (WCL) and heavy membrane (mem.) fractions were used for immunoblot analyses with anti-calnexin, anti-VAPA, anti-LC3, and anti–α-tubulin antibodies. (F) Cell death induced by 10 μM Tat–Beclin 1 was quantified with CellTiter-Blue assays; mean ± SD, n = 4 values were measured from more than 4 different wells per experiment; **P < 0.01, 1-way ANOVA with Tukey’s post hoc test. See also Supplemental Figure 2.
The ER forms contacts with isolation membranes, precursors of autophagosomes, to facilitate their progression into autophagosomes. Vesicle-associated membrane protein–associated proteins (VAPs), integral ER proteins, participate in establishing ER contacts with isolation membranes, and depletion of VAPs impairs progression of isolation membranes into autophagosomes (25). Since VAPs directly control the interaction between the ER and isolation membranes, we reasoned that downregulation of VAPA and VAPB with siRNAs (Supplemental Figure 2H) might inhibit the consumption of ER membranes for autophagosome biogenesis. Thus, we investigated whether VAPA/B double knockdown prevents Tat–Beclin 1–induced decreases in the ER membrane. Although Tat–Beclin 1 substantially decreased the level of the ER marker protein calnexin, in the presence of siControl, VAPA/B double knockdown abolished Tat–Beclin 1–induced decreases in calnexin in CMs (Figure 2E). Furthermore, Tat–Beclin 1–induced cell death was significantly attenuated in the presence of VAPA/B double knockdown compared with siControl-treated CMs (Figure 2F). These results suggest that autosis is accompanied by increased consumption of ER membranes for autophagosome biogenesis in CMs.
I/R induces autosis of CMs in the mouse heart.
We have shown previously that autophagy is strongly activated during I/R and promotes myocardial injury (13). We next asked whether autosis occurs in the heart in vivo during I/R. To this end, the mouse heart was subjected to 30 minutes of ischemia, followed by reperfusion. First, we evaluated autophagic flux in the ischemic border region by Western blot analyses. The level of LC3-II was increased in a time-dependent manner during reperfusion (Figure 3, A and B). The level of p62 was decreased during the early phase (2–6 hours) of reperfusion but was increased 24 hours after reperfusion (Figure 3, A and C). To exclude the possibility that I/R affects the transcription of LC3 or p62, which could affect the protein level of LC3 or p62 independently of autophagy, we examined the mRNA levels of LC3 and p62 during I/R. LC3 mRNA levels were not altered during early reperfusion and were decreased 24 hours after reperfusion, while p62 mRNA levels were not altered during I/R (Supplemental Figure 3, A and B). These results suggest that autophagic flux is activated during the early phase (up to 6 hours) of I/R but is attenuated thereafter in the ischemic border region. To further evaluate how I/R affects autophagic flux, we used transgenic mice with CM-specific expression of GFP-LC3-RFP-LC3ΔG. GFP-LC3-RFP-LC3ΔG is cleaved by Atg4 to produce GFP-LC3 and RFP-LC3ΔG. Although GFP-LC3 binds to autophagosomes and is degraded by autophagy, RFP-LC3ΔG cannot bind to autophagosomes and remains undegraded in the cytosol, serving as an internal control. Thus, the ratio between GFP and RFP inversely correlates with autophagic flux. These mice are useful for evaluating the tissue distribution of autophagic flux via confocal microscopic analyses (18). In the ischemic border area, the GFP/RFP ratio was decreased 4 hours after reperfusion, whereas it was increased 24 hours after reperfusion, suggesting that autophagic flux was increased during the early phase but decreased thereafter (Figure 3, D–G). In the ischemic core area, autophagic flux was decreased compared with in the remote areas at both 4 and 24 hours after reperfusion. We also evaluated autophagic flux in the border region using transgenic mice with cardiac tissue–specific expression of Tg-tandem fluorescent-tagged LC3 (tf-LC3) (15). Both GFP/RFP double-positive (yellow) dots, indicating autophagosomes, and RFP-positive (red) dots, indicating autolysosomes, were significantly increased in number in hearts subjected to ischemia or 2 hours after I/R compared with sham-operated mouse hearts (Supplemental Figure 3, C and D). However, after 6 hours of reperfusion, there was more prominent accumulation of autophagosomes (Supplemental Figure 3, C and D), indicating that autophagic flux starts to be blunted 6 hours after reperfusion. EM analyses showed that the numbers of both autophagosomes and autolysosomes were increased after ischemia and reperfusion in a time-dependent manner (Figure 3, H and I). Interestingly, the number of autophagosomes containing nondegraded cellular material significantly increased starting 6 hours after reperfusion (Figure 3, J and K). Taken together, these results indicate that autophagic flux is increased by ischemia and during the early phase (up to 6 hours) of reperfusion. However, autophagic flux deteriorates during the late phase of reperfusion, which induces further accumulation of autophagic vacuoles in the border region of myocardial infarction (MI) (Supplemental Figure 3E).
Figure 3. Autophagic flux is increased during early reperfusion but is inhibited during late reperfusion in the mouse heart.

(A–C) Three-month-old C57BL/6J mice were subjected to 30 minutes of ischemia (Is.) followed by reperfusion for the indicated times, and the border region of infarction in the heart was analyzed by Western blotting using anti-p62, anti-LC3, and anti–α-tubulin antibodies (A). The ratios of LC3-II (B) and p62 to α-tubulin (C) were quantified (mean ± SEM, n = 6; *P < 0.05 vs. Sham, 1-way ANOVA with Dunnett’s post hoc test). (D–G) Three-month-old cardiac tissue–specific GFP-LC3-RFP-LC3ΔG transgenic mice were subjected to I/R for 4 and 24 hours. and hearts were analyzed by confocal microscopy to assess autophagic flux. Representative fluorescence ratio images of heart sections from cardiac tissue–specific GFP-LC3-RFP-LC3ΔG transgenic mice subjected to I/R for 4 (D) and 24 hours (E). Scale bars: 100 μm. The graph shows quantification of GFP/RFP fluorescence ratio intensities at 4 (F) and 24 hours (G) (mean ± SEM, n = 3; *P < 0.05, **P < 0.01 vs. remote region, 1-way ANOVA with Dunnett’s post hoc test). (H–K) Samples of hearts from mice subjected to I/R for various periods of time were subjected to EM analyses. Autolysosomes (ALs, arrow) and autophagosomes (APs, arrowhead) are indicated (H) and enlarged (J). Scale bars: 1 μm. Cytoplasmic ALs and APs (I) and enlarged APs with nondegraded materials (K) were counted. (I and K) Mean ± SEM, n = 7 for sham, n = 3 for ischemia, n = 5 for IR 2 hours, n = 4 for IR 6 hours, n = 7 for IR 24 hours; **P < 0.01 versus Sham, ##P < 0.01; values were measured from more than 10 different areas per mouse; 2-way ANOVA (I) and 1-way ANOVA with Dunnett’s post hoc test (K). See also Supplemental Figure 3.
Morphological features of autosis can be divided into 2 phases. In phase 1, the number of autophagic vacuoles and empty vacuoles is drastically increased; and fragmented ER, electron-dense mitochondria, and mild chromatin condensation are observed. In phase 2, ballooning of the PNS is detected (Supplemental Figure 4A and ref. 11). To evaluate whether autosis is activated in the heart during I/R, we conducted EM analyses. Typical signs of phase 1 autosis — namely autophagic vacuoles and empty vacuoles and electron-dense mitochondria that have undergone damage-induced calcium overload (26) — and mild chromatin condensation were observed in mouse hearts subjected to I/R but not sham operation (Figure 4, A–C). Ballooning of the PNS, indicating phase 2 autosis, was increased dramatically between 6 and 24 hours after reperfusion (Figure 4, D and E).
Figure 4. Autosis is induced in the heart during I/R.

(A–E) Mice were subjected to 30 minutes of ischemia with reperfusion for the indicated periods of time, and heart samples were subjected to EM analyses. (A) Autophagic vacuoles (AVs, arrowheads), empty vacuoles (EVs, arrow), and electron-dense mitochondria (asterisks) are indicated (scale bars: 2 μm). SR, sarcoplasmic reticulum. Cytoplasmic AVs and EVs (B) and electron-dense mitochondria (C) were counted. Representative images of CM nuclei showing ballooning of the PNS (scale bar: 2 μm) (D). (E) The percentage of cells with ballooning of the PNS was calculated. Mean ± SEM, n = 7 for sham and IR 24 hours; n = 3 for ischemia, IR 2 hours, and IR 6 hours. **P < 0.01 versus Sham; values were measured from more than 10 different areas (B and C) and more than 100 CM nuclei (E) per mouse; 2-way ANOVA (B), unpaired Student’s t test (C), and 1-way ANOVA with Dunnett’s post hoc test (E). See also Supplemental Figure 4.
It has been suggested that CM cell death is completed during the early phase of MI. To evaluate whether cardiac cell death is increased 6–24 hours after I/R, when the occurrence of autosis becomes more obvious, we conducted triphenyltetrazolium chloride (TTC) staining experiments at various time points after reperfusion. The MI area was significantly greater after 24 hours of reperfusion than after 6 hours of reperfusion (Supplemental Figure 4, B–D, and ref. 27). However, the number of TUNEL-positive CMs was significantly increased after 6 hours of reperfusion but was not further increased after 24 hours of reperfusion (Supplemental Figure 4, E and F). These results suggest that autosis may contribute to the progression of myocardial injury during the late phase of reperfusion.
Inhibition of autosis attenuates I/R injury in the heart.
If autosis contributes to death of CMs during I/R, selective suppression of autosis may reduce myocardial injury during I/R. Since one of the important characteristics of autosis is that it can be inhibited by cardiac glycosides, which inhibit Na+,K+-ATPase, we asked whether death of CMs during the late phase of I/R is sensitive to such intervention and, if so, whether it is possible to reduce infarct size through suppression of autosis. Since rodent Na+,K+-ATPase α1 is largely resistant to cardiac glycosides, we employed humanized Na+,K+-ATPase α1–knockin mice, which are sensitive to cardiac glycosides (Figure 5A and ref. 22). To inhibit Na+,K+-ATPase activity during late reperfusion, we injected ouabain, a cardiac glycoside, at 3 and 6 hours after reperfusion and then examined the MI (Figure 5B). The area at risk (AAR) was not significantly affected by ouabain treatment (Figure 5, C and E). However, injection with ouabain significantly reduced the size of MI/AAR after I/R compared with injection of vehicle in humanized Na+,K+-ATPase-knockin mice but not in WT mice (Figure 5, C and D). EM examination showed that levels of CMs with typical morphological features of autosis, including ballooning of the PNS and the presence of many vacuoles, were decreased in the heart 24 hours after I/R in Na+K+-ATPase–knockin mice treated with ouabain compared with hearts of ouabain-treated WT mice (Figure 5, F–J). These results suggest that inhibition of autosis by inhibiting Na+,K+-ATPase activity reduces I/R injury in vivo.
Figure 5. Inhibition of Na+,K+-ATPase α1 activity attenuates I/R injury in the mouse heart.

(A) Schematic diagram of the humanized sequence of Na+,K+-ATPase used to produce the Na+,K+-ATPase–knockin (NaK-KI) mice. (B) Schematic diagram of the experimental design for injection of ouabain during I/R. (C–E) Three-month-old WT and homozygous NaK-KI mice were subjected to 30 minutes of ischemia and 24 hours of reperfusion. They were injected with PBS or ouabain intraperitoneally as indicated in B. Hearts of PBS- or ouabain-injected WT and NaK-KI mice were subjected to TTC staining. (C) Representative images of LV myocardial sections after Alcian blue and TTC staining (scale bar: 1 mm). Ratios of AAR to total LV (E) and infarction area to AAR (D) were compared in WT and NaK-KI mice with PBS or ouabain injection (mean value ± SEM, n = 3 with PBS injected, n = 6 with ouabain injected; **P < 0.01, 2-way ANOVA). (F–J) Twenty-four hours after reperfusion, ouabain-injected WT and NaK-KI mice were subjected to EM analyses. (F) Representative images show AVs (arrowheads) and EVs (arrow) (scale bar: 2 μm). Cytoplasmic AVs and EVs (G) and electron-dense mitochondria (H) were counted. (I) Representative images of CM nuclei showing ballooning of the PNS (scale bar: 2 μm). (J) Percentage of cells with ballooning of the PNS was calculated. Mean ± SEM, n = 4; **P < 0.01 versus ouabain-injected WT; values were measured from more than 10 different areas (G and H) and more than 100 CM nuclei (J) per mouse; 2-way ANOVA (G) and unpaired Student’s t test (H and J). See also Supplemental Figure 5.
Na+,K+-ATPase contributes significantly to cardiac and vascular signaling and calcium homeostasis (28). Therefore, we examined whether inhibition of Na+,K+-ATPase affects other types of cell death besides autosis such as apoptosis or necrosis during the late phase of I/R. We evaluated apoptosis and necroptosis using humanized Na+,K+-ATPase–knockin mice treated with ouabain. Although ouabain-injected humanized Na+,K+-ATPase–knockin mice exhibited significantly smaller infarcts (Figure 5, C–E), the amount of apoptosis, evaluated by TUNEL assay, and necroptosis, evaluated via phosphorylation of Rip1, was not changed (Supplemental Figure 5, A–C). These results suggest that inhibition of Na+,K+-ATPase attenuates I/R injury through inhibition of autosis. Since inhibition of Na+,K+-ATPase can change the CM calcium environment, we next investigated whether calcium signaling is involved in the regulation of autosis. To this end, we pretreated NRCMs with Bapta-AM (29), a calcium chelator, in the presence or absence of Tat–Beclin 1. Pretreatment with Bapta-AM did not have any additional effect on Tat–Beclin 1–induced cell death in NRCMs (Supplemental Figure 5D), suggesting that the regulation of intracellular calcium level is not involved in autosis signaling.
Drastic accumulation of autophagosomes during the late phase of reperfusion is caused by upregulation of Rubicon.
We next focused on identifying the molecular mechanism by which cardiac autosis is triggered in response to I/R. In contrast to in Tat–Beclin 1–treated CMs in vitro, where autophagic flux was persistently increased (Supplemental Figure 6, A and B), autophagic flux was increased initially but then decreased during the late phase in response to I/R in the mouse heart in vivo. Interestingly, among several autophagy-related factors, the protein level of Rubicon was significantly increased 2 hours after reperfusion and was drastically increased starting 6 hours after reperfusion, a time course that coincided with that of autosis (Figure 6, A and B). Rubicon was initially identified as a component of the PI3KC3 complex and a negative regulator of autophagy and endosomal trafficking. Overexpression of Rubicon inhibits the autophagosome maturation step and induces accumulation of autophagosomes (5). To examine the effect of increasing Rubicon in CMs, we generated adenovirus harboring Rubicon and used it to overexpress Rubicon in NRCMs. Overexpression of Rubicon increased the level of LC3-II in both Tat-Scrambled- and Tat–Beclin 1–treated CMs. Overexpression of Rubicon induced further accumulation of autophagosomes, but not autolysosomes, in the presence of Tat–Beclin 1 in NRCMs (Supplemental Figure 6, C–F). Next, we investigated whether increasing the level of Rubicon triggers autosis in CMs. Overexpression of Rubicon alone was insufficient to induce autosis in NRCMs at baseline (Supplemental Figure 6G). However, Rubicon further enhanced cell death in the presence of hypoxia/reoxygenation (Supplemental Figure 6G). These results suggest that autophagic flux is inhibited by upregulation of Rubicon but that an additional stress triggering enhanced production of autophagosomes is needed for the induction of autosis.
Figure 6. Marked accumulation of autophagosomes during late reperfusion is accompanied by upregulation of Rubicon.

(A) Three-month-old mice were subjected to 30 minutes of ischemia with reperfusion for the times indicated, and heart lysates were analyzed by Western blot using anti-Rubicon, anti-Vps34, anti-Atg14L, and anti–α-tubulin antibodies. (B) The ratio of Rubicon to α-tubulin was quantified (mean ± SEM, n = 4; *P < 0.05, **P < 0.01 versus Sham, 1-way ANOVA with Dunnett’s post hoc test). (C–F) Three-month-old mice were subjected to 30 minutes of ischemia and 24 hours of reperfusion. The mice were injected with 5 mg/kg Scrambled or Tat–Beclin 1 in the jugular vein 3 hours before ischemia (Pre), at the time of reperfusion (At rep.), or 3 hours after reperfusion (Post). (C) Schematic diagram of Tat–Beclin 1 injection during I/R. (D) Representative images of LV myocardial sections after Alcian blue and TTC staining (scale bar: 1 mm). Ratios of AAR to total LV (F) and infarction area to AAR (E) were compared (mean ± SEM, indicated n; *P < 0.05, **P < 0.01 vs. Scrambled, 1-way ANOVA with Dunnett’s post hoc test [E] and Tukey’s post hoc test [F]). See also Supplemental Figure 6.
Stimulation of autophagosome formation when autophagic flux is impaired during the late stage of reperfusion may induce excessive accumulation of autophagosomes and thus become toxic. To test this hypothesis, we applied Tat–Beclin 1 to activate autophagy at different time points during I/R. Injection of a low dose of Tat–Beclin 1 into the jugular vein has been shown to increase autophagy without causing autosis, effectively activating cardiac autophagic flux without cardiac damage (30). To validate the effective duration for induction of autophagic flux by Tat–Beclin 1, we treated Tg-GFP-LC3 mice with Tat–Beclin 1. Autophagic flux induced by Tat–Beclin 1 was drastically activated at 3 hours and gradually declined time-dependently (Supplemental Figure 6, H and I). Therefore, we injected Tat–Beclin 1 three hours before surgery, right after reperfusion, or 3 hours after reperfusion (Figure 6C) to evaluate the effect of stimulation of autophagosome formation. Consistent with the aforementioned results, although application of Tat–Beclin 1 before I/R was protective, during the late phase of I/R it exacerbated myocardial injury (Figure 6, D–F). These results support our hypothesis that stimulation of autophagosome formation during the late phase of I/R, when autophagic flux is attenuated, is detrimental in the heart.
Cardiac tissue–specific knockout of Rubicon attenuates I/R injury and simultaneously inhibits autosis.
We next focused on Rubicon, which is drastically upregulated specifically during the late phase of I/R. To evaluate the role of endogenous Rubicon in the heart in mediating autosis during the late phase of I/R, we generated Rubicon floxed mice and crossed them with Myh6-Cre–transgenic mice to obtain cardiac tissue–specific Rubicon-KO (rubi-cKO) mice (Supplemental Figure 7A and ref. 31). rubi-cKO mice were born at the expected Mendelian ratios and showed normal cardiac function under basal conditions (Table 1). We confirmed heart-specific reduction of Rubicon in rubi-cKO mice (Supplemental Figure 7, B and C). Western blotting revealed that Rubicon protein is drastically increased by I/R in WT mice, whereas heterozygous rubi-cKO mice showed significantly attenuated upregulation during I/R (Supplemental Figure 7, E and F). Although the mRNA level of Rubicon was upregulated around 2-fold during I/R, the protein level of Rubicon was increased more than 5-fold (Supplemental Figure 7, D and F). This may suggest that the level of Rubicon was stabilized during the late phase of I/R. Moreover, autophagic flux was consistently attenuated 24 hours after reperfusion in WT mice. However, autophagic flux was normalized and increased at this time point in rubi-cKO mice (Supplemental Figure 7, G and H). EM analyses also revealed that the number of autolysosomes was significantly increased, indicating increased autophagic flux, even under basal conditions, in rubi-cKO mice (Supplemental Figure 7, I and J). Interestingly, the number of autophagosomes and enlarged autophagosomes containing nondegraded cellular material, which was increased in WT mice after 24 hours of I/R, was normalized in rubi-cKO mice (Supplemental Figure 7, I–K). Taken together, these results suggest that suppression of autophagic flux during the late phase of reperfusion and consequent accumulation of autophagosomes are normalized in rubi-cKO mice.
Table 1. Echocardiographic analyses of cardiac tissue–specific Rubicon-knockout mice at baseline.
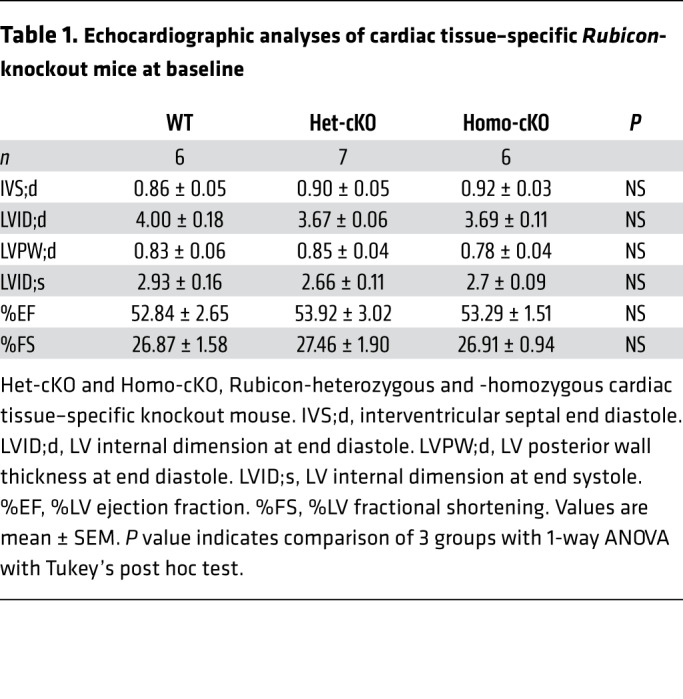
We next tested whether MI during I/R is reduced in rubi-cKO mice. Three-month-old WT and heterozygous rubi-cKO mice were subjected to I/R and then, after 24 hours, TTC staining was conducted to compare infarction volume. Although the AAR did not differ significantly between WT and rubi-cKO mice (Figure 7, A and C), rubi-cKO mice showed a drastically reduced infarction size/AAR ratio compared with WT mice (Figure 7, A and B). We examined whether the decreased infarction size in rubi-cKO mice was caused by inhibition of autosis. EM analyses showed that electron-dense mitochondria and ballooning of the PNS, typical morphological features of autosis, were almost completely abolished in the rubi-cKO mouse heart 24 hours after I/R (Figure 7, D–F). We also evaluated whether inhibition of Na+,K+-ATPase by ouabain in rubi-cKO mice would induce a further reduction in the size of MI. To this end, we crossed homozygous humanized Na+,K+-ATPase–knockin mice with heterozygous rubi-cKO mice, and applied I/R. AAR did not differ significantly between groups (Figure 7, G and I). Inhibition of Na+,K+-ATPase by ouabain injection into humanized Na+,K+-ATPase–knockin mice significantly reduced the size of MI compared with WT mice. However, injection of ouabain into humanized Na+,K+-ATPase–knockin mice/rubi-cKO failed to produce a further reduction in infarct size compared with rubi-cKO mice (Figure 7, G and H). Thus, inhibition of Na+,K+-ATPase and of Rubicon reduces the size of MI through a common mechanism, namely suppression of autosis. These results suggest that normalization of autophagic flux and consequent reduction of the number of autophagosomes through downregulation of Rubicon reduce I/R injury by inhibiting autosis in the mouse heart.
Figure 7. Cardiac tissue–specific knockout of Rubicon attenuated I/R injury and reduced autosis in the mouse heart.

Three-month-old WT (Rubiconfl/+ without αMHC-Cre) and cardiac tissue–specific Rubicon heterozygous knockout (rubi-cKO [hetero]) mice were subjected to 30 minutes of ischemia and 24 hours of reperfusion. Mice were subjected to TTC staining (A–C) or subjected to EM analyses (D–F). (A) Representative images of LV myocardial sections after Alcian blue and TTC staining (scale bar: 1 mm). Ratios of the infarction area to AAR (B) and AAR to total LV (C) were compared in WT and rubi-cKO mice (mean ± SEM, n = 3 WT and n = 5 rubi-cKO; **P < 0.01, unpaired Student’s t test). (D) Electron-dense mitochondria (arrows) and ballooning of the PNS are indicated (scale bars: 2 μm). Electron-dense mitochondria (E) and cells with PNS (F) were counted; mean ± SEM, n = 4; **P < 0.01 versus WT, unpaired Student’s t test; values were measured from more than 10 different areas (E) and more than 100 CM nuclei (F) per mouse. (G–I) Three-month-old WT (Rubiconfl/+ without αMHC-Cre), homozygous NaK-KI (humanized Na+,K+-ATPase–knockin with Rubiconfl/+ without αMHC-Cre), rubi-cKO (Rubiconfl/+ with αMHC-Cre), and homozygous NaK-KI/rubi-cKO (humanized Na+,K+-ATPase–knockin with Rubiconfl/+ with αMHC-Cre) mice were subjected to 30 minutes of ischemia and 24 hours of reperfusion. They received intraperitoneal injection of PBS or ouabain, as indicated in Figure 5B. (G) Hearts were subjected to TTC staining. Representative images of LV myocardial sections after Alcian blue and TTC staining (scale bar: 1 mm). Ratios of the AAR to total LV (I) and infarction area to AAR (H) were compared in WT and NaK-KI mice or rubi-cKO and NaK-KI/rubi-cKO mice with either PBS or ouabain injection (mean ± SEM; *P < 0.05, **P < 0.01, 2-way ANOVA). See also Supplemental Figure 7.
Discussion
Cardiomyocytes undergo autophagy-dependent cell death through autosis.
We show here that strong activation of autophagy induces cell death with both morphological and biochemical features consistent with autosis in NRCMs. Tat–Beclin 1–induced cell death was inhibited either by 3MA or downregulation of Atg7 but not by inhibitors of apoptosis or necrosis, suggesting that autophagy, but not apoptosis or necrosis, mediates Tat–Beclin 1–induced cell death in CMs. Tat–Beclin 1–induced death in CMs was also inhibited when Na+,K+-ATPase was downregulated. Thus, Tat–Beclin 1–induced cell death in CMs is accompanied by all the reported features of autosis. Since the features of Tat–Beclin 1–induced cell death were similar in AMCMs and human iPSC-derived CMs, it is likely that CMs can die by autosis.
How does autosis kill CMs?
Since autophagy degrades proteins and organelles at lysosomes, one might speculate that cell death by autophagy occurs through massive self-digestion in lysosomes. However, Tat–Beclin 1–induced cell death was not alleviated by bafilomycin A1, a lysosome inhibitor. How, then, does Tat–Beclin 1 induce cell death? One possibility is that excessive autophagosome formation causes cellular malfunction. Autosis is accompanied by a characteristic ballooning of the PNS, in which the structure of the nuclear envelope is disrupted, and disappearance of perinuclear organelles, including ER and mitochondria. We show that the levels of intracellular membrane proteins were decreased in the presence of Tat–Beclin 1 in a dose-dependent manner (Figure 2, A–D). Since the materials used to form the double-membrane structures of autophagosomes originate from intracellular membranes, including ER, mitochondria, and plasma membrane, we speculate that massive generation of autophagosomes depletes intracellular and plasma membranes, thereby leading to the degradation of intracellular organelles and consequently to organelle dysfunction.
Is autosis stimulated by I/R and involved in myocardial injury?
It has been shown previously that severe cerebral hypoxia-ischemia induces autosis in neonatal rat brain in vivo (11). Here we show that autosis of CMs is induced by I/R in the mouse heart in vivo. CM death induced by myocardial I/R exhibited all known morphological and biochemical features of autosis. Ballooning of the PNS, one of the most prominent morphological features of autosis, was observed as early as 2 hours after reperfusion but was further increased after 6 hours. Myocardial reperfusion after ischemia increased autophagic flux and lysosomal activity in the border zone within 2–4 hours, which increased the number of autophagosomes and autolysosomes in the myocardium. Although lysosomal activity returned to normal levels around 6 hours and the autophagic flux in the border zone decreased to below physiological levels thereafter, the number of autophagosomes continued increasing 24 hours after reperfusion. Interestingly, the individual vacuoles became larger during the late phase of reperfusion, and many of the vacuoles contained undigested cytosolic materials. Since the time at which the vacuoles increase in size correlates well with the appearance of ballooning of the PNS, it is likely that autosis occurs when CMs produce many autophagosomes and autolysosomes with attenuated recycling. The finding that endogenous Na+,K+-ATPase was upregulated 6–24 hours after reperfusion also supports activation of autosis during the late phase of reperfusion.
Furthermore, our data suggest that autosis contributes to myocardial injury after reperfusion. First, the extent of myocardial injury was significantly greater 24 than 6 hours after reperfusion. Thus, myocardial injury continues to expand between 6 and 24 hours. Since autosis becomes more prominent after 6 hours — in contrast to other forms of cell death, which develop within a short period of reperfusion — it may contribute to myocardial injury during the late phase of reperfusion. Second, treatment of humanized Na+,K+-ATPase–knockin mice with ouabain significantly inhibited both autosis and myocardial reperfusion injury. These results strongly support the notion that autosis contributes to myocardial injury during the late phase of reperfusion.
Upregulation of Rubicon during reperfusion stimulates autosis.
Autosis became more prominent during the late phase of reperfusion, when the number of autophagic vacuoles was dramatically increased but fusion of autophagosomes to lysosomes started to show attenuation. We found that the level of Rubicon, a negative regulator of autophagy, was significantly increased 6 hours after reperfusion when autophagic flux started to exhibit attenuation. Multiple lines of evidence suggest that marked accumulation of autophagosomes can be detrimental for the heart. Stimulation of autophagy with Tat–Beclin 1 three hours after reperfusion and thereafter exacerbated myocardial injury (Figure 6, C–F). Furthermore, downregulation of Rubicon improved autophagic flux during the late phase of I/R. This improvement was accompanied by alleviation of autophagosome accumulation, and reduced autosis and myocardial injury during the late phase of I/R. In addition, downregulation of Rubicon and inhibition of Na+,K+-ATPase failed to elicit additive effects in reducing MI size. Interestingly, infarct size in ouabain-treated humanized Na+,K+-ATPase–knockin/rubi-cKO mice was further reduced compared with that in ouabain-treated humanized Na+,K+-ATPase–knockin mice (Figure 7, G and H). This may be due to rescue of other types of cell death affected by knockdown of Rubicon (31). Although one might argue that CM death during the late phase of I/R may be caused by the decline in autophagic flux, injection of Tat-Beclin1, an intervention to stimulate autophagy, exacerbated CM death during the late phase of I/R, suggesting that excessive autophagosome formation, rather than impaired autophagic activity, is the cause of CM death in this condition. Thus, it would be important to eliminate the block caused by upregulation of Rubicon rather than enhance autophagosome formation in order to prevent autosis-mediated injury during the late phase of I/R injury. Alternatively, inhibiting excessive accumulation of autophagosomes by inhibiting autophagy might be equally effective in normalizing the flux to prevent autosis during the late phase of I/R (13).
Can autosis be inhibited by cardiac glycoside treatment?
Since autosis promotes myocardial injury during the late phase of reperfusion, effective suppression of autosis should reduce overall myocardial injury during reperfusion and act protectively to preserve cardiac function. Digitalis is known to be highly selective for the Na+,K+-ATPase α2/3 subunit rather than the α1 subunit, cause toxicity, and increase the risk of cardiac arrhythmia; thus, it may not be suitable for reducing autosis in patients with acute MI at risk of developing life-threatening ventricular arrhythmia. However, small molecules capable of targeting Na+,K+-ATPase without side effects could be effective. It is known that mammals endogenously produce digitalis-like substances (32). Interventions to induce production of these endogenous substances may help inhibit autosis during the late phase of reperfusion. Alternatively, since the endogenous Na+,K+-ATPase α1 subunit is upregulated during the late phase of reperfusion, interventions to inhibit its upregulation may also be effective.
Molecular mechanism of autosis.
The molecular mechanism through which Na+,K+-ATPase regulates autosis remains to be elucidated. Since Na+,K+-ATPase is an important regulator of calcium homeostasis in cardiomyocytes, one might speculate that autosis may be a calcium-sensitive event. However, a calcium chelator did not affect autosis in CMs (Supplemental Figure 5D). We have shown recently that Na+,K+-ATPase physically interacts with Beclin 1 to stimulate autosis. This interaction between Na+,K+-ATPase and Beclin 1 was observed in the mouse kidney during I/R. Moreover, enhancing this interaction with DigiFab, an antibody that effectively blocks endogenous digoxin-like molecules, aggravates autosis in the mouse heart during exercise (33). Since Rubicon also physically interacts with Beclin 1, it would be interesting to investigate how Rubicon and Na+,K+-ATPase affect the interactions between Beclin 1 and Na+,K+-ATPase and between Beclin 1 and Rubicon, respectively, and how they therefore regulate autosis.
It would appear that promotion of autophagy by Tat–Beclin 1 when autophagic flux is suppressed due to upregulation of Rubicon promotes autosis. However, the molecular mechanisms through which stimulation of autophagy and accumulation of autophagosomes induce autosis remain to be elucidated. In particular, it is necessary to analyze more precisely the mechanisms by which stimulation of autophagy leads to the disappearance of intracellular organelles and ballooning of the PNS. In addition, the causative involvement of organelle dysfunction in autotic cell death requires further clarification.
In summary, we show that CMs can die as a result of dysregulated autophagy in the presence of myocardial I/R in vivo, and that autosis contributes to myocardial injury during the late phase of reperfusion. Since the ultimate goal of interventions in patients with acute MI is to minimize the death of myocardial tissue, preventing CMs from dying by autosis is highly relevant. Thus, further study is warranted to investigate the underlying mechanism and identify effective interventions to selectively prevent autotic cell death of CMs during reperfusion.
Methods
Mouse models.
Humanized Na+,K+-ATPase α1–knockin mice were generated according to a previous report (22). Transgenic mice expressing mRFP-GFP-LC3 (tf-LC3) have been described (15). Cardiac tissue–specific Rubicon-KO mice were generated by crossing Rubiconfl/fl mice (31) with Myh6-Cre transgenic mice.
Generation of GFP-LC3-RFP-LC3ΔG mice.
A cDNA fragment was isolated from αMHC-pBluescriptII-GFP-LC3-RFP-LC3ΔG using NotI, purified using an endotoxin-free preparation kit from QIAGEN (catalog 12362), and microinjected into the pronucleus of 1-cell embryos from C57BL/6J mice. The transgenic founders were confirmed by genomic PCR using 5′-ATGACAGACAGATCCCTCCTATCTCC-3′ and 5′-GGGTAGCGGCTGAAGC-3′ as primers. Twenty-five mice were screened, of which 2 were positive for the transgene. The transgenic line showing the highest expression was used in this study.
CM cultures.
Primary cultures of CMs were prepared from 1-day-old Charles River Laboratory (Crl)/Wistar Institute (WI) BR-Wistar rats (Harlan Laboratories) as described previously (33). A CM-rich fraction was obtained by centrifugation through a discontinuous Percoll gradient. Human iPSCs were differentiated into beating human iPSC-CMs as described previously (34). Isolation and culture of AMCMs were conducted according to a protocol described previously (35). This method uses direct needle perfusion of the left ventricle (LV) ex vivo, which allows isolation, separation, and culture of AMCMs.
I/R in vivo.
C57BL/6J mice were obtained from the Jackson Laboratory. Mice were housed in a temperature-controlled environment with 12-hour light/12-hour dark cycles, where they received food and water ad libitum. Three-month-old C57BL/6J mice were anesthetized by intraperitoneal injection of pentobarbital sodium (60 mg/kg). A rodent ventilator (Minivent; Harvard Apparatus Inc.) was used with 65% oxygen during the surgical procedure. The animals were kept warm using heat lamps. Rectal temperature was monitored and maintained between 36.8°C and 37.2°C. The chest was opened by a horizontal incision at the fourth intercostal space. Ischemia was achieved by ligating the anterior descending branch of the left anterior descending artery (LAD) using an 8-0 Prolene suture, with silicon tubing (1 mm OD) placed on top of the LAD, 2 mm below the border between the left atrium and LV. Ischemia was confirmed by ECG change (ST elevation) and color change of myocardium. After occlusion for 30 minutes, the silicon tubing was removed to achieve reperfusion. When recovered from anesthesia, the mice were returned to their cages and housed in a climate-controlled environment.
Measurement of infarct size.
After I/R, the animals were anesthetized and intubated, and the chest was opened. To demarcate the ischemic AAR, Alcian blue dye (1%) was perfused into the aorta and coronary arteries. Hearts were excised, and LVs were sliced into 1-mm-thick cross sections. The heart sections were then incubated with a 1% TTC solution at 37°C for 15 minutes. The infarct area (white), AAR (not blue), and total LV area from both sides of each section were measured using ImageJ software (NIH), and the values obtained were averaged. The percentages of area of infarction and AAR of each section were multiplied by the weight of the section and then totaled from all sections. AAR/LV and infarct area/AAR were expressed as percentages (13).
Ouabain injection.
In order to maximize the possibility that ouabain would successfully inhibit Na+,K+-ATPase α1 activity, we injected ouabain twice, at 3 and 6 hours after reperfusion, when autosis is triggered. Digitalis is highly selective to the α2/3 compared with the α1 subunit. On the other hand, ouabain is moderately selective to the α1 subunit compared with the other subunits (36). In order to inhibit Na+,K+-ATPase in the humanized Na+,K+-ATPase α1 subunit–knockin mice, we applied ouabain instead of digitalis.
Viability of the cells.
Viability of the cells was measured by CellTiter-Blue assays (Promega). In brief, CMs were seeded onto 96-well dishes. The cells were preincubated with the indicated adenovirus for 48 hours. Viable cell numbers were measured after Scrambled or Tat–Beclin 1 treatment. The CellTiter-Blue assays were performed according to the supplier’s protocol. Staining of cultured CMs with SYTOX Green nucleic acid stain (Invitrogen, Thermo Fisher Scientific) to evaluate cell death was conducted according to the manufacturer’s instructions.
Tat–Beclin 1.
The Tat–Beclin 1 amino acid sequence was YGRKKRRQRRGGVWNATFHIWHD. The control peptide, Tat-Scrambled (TS), consisted of the Tat protein transduction domain, a GG linker, and a scrambled version of the 11 C-terminal amino acids of Tat–Beclin 1 (YGRKKRRQRRGGWNHADHTFVWI). For in vivo experiments, d-amino acid peptides were dissolved in H2O and stored at –80°C until use. For induction of autophagy in vitro, CMs were washed with PBS (without calcium or magnesium) and treated with peptides dissolved in OPTI-MEM acidified with (0.15% v/v) 6 N HCl (37). For in vivo induction, mice were injected with TS or Tat–Beclin 1 at 5 mg/kg through the jugular vein.
EM.
Conventional EM was performed as described previously (13). The average number of vacuoles or electron-dense mitochondria was calculated per 400-μm2 area.
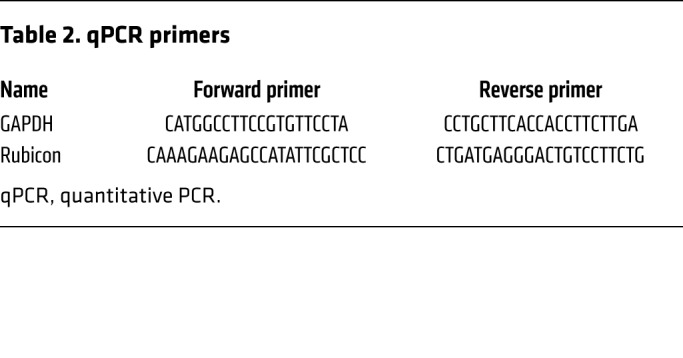
Real-time quantitative PCR.
Total RNA was extracted from mouse hearts using the RNeasy Plus Universal Kit (QIAGEN). Total RNA was converted to cDNA using PrimeScript RT Master Mix (Takara). Primer sequences are shown in Table 2.
Table 2. qPCR primers.
RNA interference.
For siRNA interference, cells were transfected using Lipofectamine RNAi MAX (Thermo Fisher Scientific) and cultured for 60 hours before analysis. siRNA oligonucleotides were purchased from Dharmacon (ON-TARGETplus SMARTpool).
AAV transduction.
Adeno-associated viruses (AAVs) were generated by Vector BioLabs. Doses of 1 × 1012 particles of each vector were administered intravenously through the jugular vein (30).
Evaluation of mitochondrial membrane potential and integrity.
In order to evaluate mitochondrial membrane potential and integrity, staining of cultured CMs with JC-1 (Molecular Probes) was conducted according to the manufacturer’s instructions.
Immunoblot analyses.
The methods used for preparation of cell lysates from in vitro and in vivo samples and for immunoblot analyses have been described previously.(14) The antibodies used include LC3 (Novus, NB100-2220), p62 (Abnova, H00008878-M01), GAPDH (Cell Signaling Technology, 2118S), Na+,K+-ATPase α1 (Santa Cruz Biotechnology, sc-21712), α-tubulin (Sigma-Aldrich, T6199), calnexin (Abcam, ab22595), Tom20 (Santa Cruz Biotechnology, sc-17764), Rubicon (MBL International, PD027), Vps34 (Cell Signaling Technology, 4263), Atg7 (Cell Signaling Technology, 8558), PDH (Cell Signaling Technology, 2784), VAPA (Proteintech, 15275-1-AP), PMCA (Abcam, ab2825), cleaved poly(ADP-ribose) polymerase (PARP) (Cell Signaling Technology, 5625), RIP1 (Cell Signaling Technology, 3493), phospho-RIP1 (Ser166) (Cell Signaling Technology, 65746), and Atg14 (Sigma-Aldrich, A6358).
Statistics.
Statistical analyses were conducted using GraphPad Prism 8.0 software. Data are presented as line graphs expressing mean ± SD of the indicated number of experiments or as dot plots expressing mean ± SEM for the indicated number of mice. The difference in means between 2 groups or multiple groups was evaluated using 2-tailed Student’s t test (paired or unpaired, as appropriate, for 2 groups); 1-way ANOVA with Tukey’s post hoc tests (for multiple comparisons); or 1-way ANOVA with Dunnett’s post hoc test (for multiple comparisons to control). A P value less than 0.05 was considered statistically significant. The reported significance values are 2-sided. Normal distribution of data was confirmed by the Shapiro-Wilk test. Non-normally distributed data were tested by the nonparametric Mann-Whitney U test after confirmation of equality of rank variances.
Study approval.
All experiments involving animals were approved by the Rutgers New Jersey Medical School’s Institutional Animal Care and Use Committee.
Author contributions
JN and JS conceptualized the study; developed methodology; and wrote the original draft. JN validated and engaged in formal analysis. JN, CYH, PZ, and SM performed investigations. AF, BL, and JS provided resources. JN, BL, and JS reviewed and edited the manuscript. JN was responsible for visualization. BL and JS supervised. JS administered the project and acquired funding.
Supplementary Material
Acknowledgments
The authors thank Masaaki Komatsu (Tokyo Metropolitan Institute of Medical Science) for Atg7fl/fl mice and Daniela Zablocki (Rutgers New Jersey Medical School) for critical reading of the manuscript. This work was supported in part by Postdoctoral Fellowship 18POST34050036 (to JN) and 2020 Merit Award 20 Merit35120374 (to JS) from the American Heart Association; US Public Health Service Grants HL67724, HL91469, HL112330, HL138720, and AG23039 (to JS); and Fondation Leducq Transatlantic Network of Excellence grant 15CBD04 (to JS and BL).
Version 1. 05/04/2020
Electronic publication
Version 2. 06/01/2020
Print issue publication
Footnotes
Conflict of interest: BL is a scientific cofounder of Casma Therapeutics Inc.
Copyright: © 2020, American Society for Clinical Investigation.
Reference information: J Clin Invest. 2020;130(6):2978–2991.https://doi.org/10.1172/JCI132366.
Contributor Information
Jihoon Nah, Email: gn111@njms.rutgers.edu.
Peiyong Zhai, Email: zhaipe@njms.rutgers.edu.
Chun-Yang Huang, Email: ch898@njms.rutgers.edu.
Álvaro F. Fernández, Email: ffernandezalvaro@gmail.com.
Satvik Mareedu, Email: sm1629@gsbs.rutgers.edu.
Beth Levine, Email: beth.levine@utsouthwestern.edu.
Junichi Sadoshima, Email: sadoshju@njms.rutgers.edu.
References
- 1.Yang Z, Klionsky DJ. Mammalian autophagy: core molecular machinery and signaling regulation. Curr Opin Cell Biol. 2010;22(2):124–131. doi: 10.1016/j.ceb.2009.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ogata M, et al. Autophagy is activated for cell survival after endoplasmic reticulum stress. Mol Cell Biol. 2006;26(24):9220–9231. doi: 10.1128/MCB.01453-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nah J, et al. Phosphorylated CAV1 activates autophagy through an interaction with BECN1 under oxidative stress. Cell Death Dis. 2017;8(5):e2822. doi: 10.1038/cddis.2017.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kroemer G, Mariño G, Levine B. Autophagy and the integrated stress response. Mol Cell. 2010;40(2):280–293. doi: 10.1016/j.molcel.2010.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhong Y, et al. Distinct regulation of autophagic activity by Atg14L and Rubicon associated with Beclin 1-phosphatidylinositol-3-kinase complex. Nat Cell Biol. 2009;11(4):468–476. doi: 10.1038/ncb1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Levine B, Yuan J. Autophagy in cell death: an innocent convict? J Clin Invest. 2005;115(10):2679–2688. doi: 10.1172/JCI26390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kroemer G, Levine B. Autophagic cell death: the story of a misnomer. Nat Rev Mol Cell Biol. 2008;9(12):1004–1010. doi: 10.1038/nrm2529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pyo JO, et al. Essential roles of Atg5 and FADD in autophagic cell death: dissection of autophagic cell death into vacuole formation and cell death. J Biol Chem. 2005;280(21):20722–20729. doi: 10.1074/jbc.M413934200. [DOI] [PubMed] [Google Scholar]
- 9.Shimizu S, et al. Role of Bcl-2 family proteins in a non-apoptotic programmed cell death dependent on autophagy genes. Nat Cell Biol. 2004;6(12):1221–1228. doi: 10.1038/ncb1192. [DOI] [PubMed] [Google Scholar]
- 10.Lamy L, et al. Control of autophagic cell death by caspase-10 in multiple myeloma. Cancer Cell. 2013;23(4):435–449. doi: 10.1016/j.ccr.2013.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu Y, et al. Autosis is a Na+/K+-ATPase-regulated form of cell death triggered by autophagy-inducing peptides, starvation, and hypoxia-ischemia. Proc Natl Acad Sci U S A. 2013;110(51):20364–20371. doi: 10.1073/pnas.1319661110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Galluzzi L, Bravo-San Pedro JM, Blomgren K, Kroemer G. Autophagy in acute brain injury. Nat Rev Neurosci. 2016;17(8):467–484. doi: 10.1038/nrn.2016.51. [DOI] [PubMed] [Google Scholar]
- 13.Matsui Y, et al. Distinct roles of autophagy in the heart during ischemia and reperfusion: roles of AMP-activated protein kinase and Beclin 1 in mediating autophagy. Circ Res. 2007;100(6):914–922. doi: 10.1161/01.RES.0000261924.76669.36. [DOI] [PubMed] [Google Scholar]
- 14.Hariharan N, Maejima Y, Nakae J, Paik J, Depinho RA, Sadoshima J. Deacetylation of FoxO by Sirt1 plays an essential role in mediating starvation-induced autophagy in cardiac myocytes. Circ Res. 2010;107(12):1470–1482. doi: 10.1161/CIRCRESAHA.110.227371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hariharan N, Zhai P, Sadoshima J. Oxidative stress stimulates autophagic flux during ischemia/reperfusion. Antioxid Redox Signal. 2011;14(11):2179–2190. doi: 10.1089/ars.2010.3488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhu H, et al. Cardiac autophagy is a maladaptive response to hemodynamic stress. J Clin Invest. 2007;117(7):1782–1793. doi: 10.1172/JCI27523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kobayashi S, Volden P, Timm D, Mao K, Xu X, Liang Q. Transcription factor GATA4 inhibits doxorubicin-induced autophagy and cardiomyocyte death. J Biol Chem. 2010;285(1):793–804. doi: 10.1074/jbc.M109.070037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kaizuka T, et al. An autophagic flux probe that releases an internal control. Mol Cell. 2016;64(4):835–849. doi: 10.1016/j.molcel.2016.09.037. [DOI] [PubMed] [Google Scholar]
- 19.Slee EA, Zhu H, Chow SC, MacFarlane M, Nicholson DW, Cohen GM. Benzyloxycarbonyl-Val-Ala-Asp (OMe) fluoromethylketone (Z-VAD.FMK) inhibits apoptosis by blocking the processing of CPP32. Biochem J. 1996;315(pt 1):21–24. doi: 10.1042/bj3150021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vandenabeele P, Grootjans S, Callewaert N, Takahashi N. Necrostatin-1 blocks both RIPK1 and IDO: consequences for the study of cell death in experimental disease models. Cell Death Differ. 2013;20(2):185–187. doi: 10.1038/cdd.2012.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nakai A, et al. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat Med. 2007;13(5):619–624. doi: 10.1038/nm1574. [DOI] [PubMed] [Google Scholar]
- 22.Dostanic I, Schultz Jel J, Lorenz JN, Lingrel JB. The alpha 1 isoform of Na,K-ATPase regulates cardiac contractility and functionally interacts and co-localizes with the Na/Ca exchanger in heart. J Biol Chem. 2004;279(52):54053–54061. doi: 10.1074/jbc.M410737200. [DOI] [PubMed] [Google Scholar]
- 23.Gatica D, Chiong M, Lavandero S, Klionsky DJ. Molecular mechanisms of autophagy in the cardiovascular system. Circ Res. 2015;116(3):456–467. doi: 10.1161/CIRCRESAHA.114.303788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tooze SA, Yoshimori T. The origin of the autophagosomal membrane. Nat Cell Biol. 2010;12(9):831–835. doi: 10.1038/ncb0910-831. [DOI] [PubMed] [Google Scholar]
- 25.Zhao YG, Liu N, Miao G, Chen Y, Zhao H, Zhang H. The ER Contact proteins VAPA/B interact with multiple autophagy proteins to modulate autophagosome biogenesis. Curr Biol. 2018;28(8):1234–1245.e4. doi: 10.1016/j.cub.2018.03.002. [DOI] [PubMed] [Google Scholar]
- 26.Solenski NJ, diPierro CG, Trimmer PA, Kwan AL, Helm GA, Helms GA. Ultrastructural changes of neuronal mitochondria after transient and permanent cerebral ischemia. Stroke. 2002;33(3):816–824. doi: 10.1161/hs0302.104541. [DOI] [PubMed] [Google Scholar]
- 27.Ojha N, et al. Characterization of the structural and functional changes in the myocardium following focal ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol. 2008;294(6):H2435–H2443. doi: 10.1152/ajpheart.01190.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tian J, Xie ZJ. The Na-K-ATPase and calcium-signaling microdomains. Physiology (Bethesda) 2008;23:205–211. doi: 10.1152/physiol.00008.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sadoshima J, Izumo S. Signal transduction pathways of angiotensin II--induced c-fos gene expression in cardiac myocytes in vitro. Roles of phospholipid-derived second messengers. Circ Res. 1993;73(3):424–438. doi: 10.1161/01.RES.73.3.424. [DOI] [PubMed] [Google Scholar]
- 30.Shirakabe A, et al. Drp1-dependent mitochondrial autophagy plays a protective role against pressure overload-induced mitochondrial dysfunction and heart failure. Circulation. 2016;133(13):1249–1263. doi: 10.1161/CIRCULATIONAHA.115.020502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tanaka S, et al. Rubicon inhibits autophagy and accelerates hepatocyte apoptosis and lipid accumulation in nonalcoholic fatty liver disease in mice. Hepatology. 2016;64(6):1994–2014. doi: 10.1002/hep.28820. [DOI] [PubMed] [Google Scholar]
- 32.Gruber KA, Whitaker JM, Buckalew VM. Endogenous digitalis-like substance in plasma of volume-expanded dogs. Nature. 1980;287(5784):743–745. doi: 10.1038/287743a0. [DOI] [PubMed] [Google Scholar]
- 33.Maejima Y, et al. Mst1 inhibits autophagy by promoting the interaction between Beclin1 and Bcl-2. Nat Med. 2013;19(11):1478–1488. doi: 10.1038/nm.3322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Baljinnyam E, et al. Effect of densely ionizing radiation on cardiomyocyte differentiation from human-induced pluripotent stem cells. Physiol Rep. 2017;5(15):e13308. doi: 10.14814/phy2.13308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ackers-Johnson M, Li PY, Holmes AP, O’Brien SM, Pavlovic D, Foo RS. A simplified, Langendorff-free method for concomitant isolation of viable cardiac myocytes and nonmyocytes from the adult mouse heart. Circ Res. 2016;119(8):909–920. doi: 10.1161/CIRCRESAHA.116.309202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Katz A, et al. Selectivity of digitalis glycosides for isoforms of human Na,K-ATPase. J Biol Chem. 2010;285(25):19582–19592. doi: 10.1074/jbc.M110.119248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shoji-Kawata S, et al. Identification of a candidate therapeutic autophagy-inducing peptide. Nature. 2013;494(7436):201–206. doi: 10.1038/nature11866. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.