

Abstract
Chiral sugar derivatives are potential cyclitol surrogates of the Ca2+-mobilizing intracellular messenger d-myo-inositol 1,4,5-trisphosphate [Ins(1,4,5)P3]. Six novel polyphosphorylated analogues derived from both d- and l-glucose were synthesized. Binding to Ins(1,4,5)P3 receptors [Ins(1,4,5)P3R] and the ability to release Ca2+ from intracellular stores via type 1 Ins(1,4,5)P3Rs were investigated. β-d-Glucopyranosyl 1,3,4-tris-phosphate, with similar phosphate regiochemistry and stereochemistry to Ins(1,4,5)P3, and α-d-glucopyranosyl 1,3,4-tris-phosphate are full agonists, being equipotent and 23-fold less potent than Ins(1,4,5)P3, respectively, in Ca2+-release assays and similar to Ins(1,4,5)P3 and 15-fold weaker in binding assays. They can be viewed as truncated analogues of adenophostin A and refine understanding of structure-activity relationships for this Ins(1,4,5)P3R agonist. l-Glucose-derived ligands, methyl α-l-glucopyranoside 2,3,6-trisphosphate and methyl α-l-glucopyranoside 2,4,6-trisphosphate, are also active, while their corresponding d-enantiomers, methyl α-d-glucopyranoside 2,3,6-trisphosphate and methyl α-d-glucopyranoside 2,4,6-trisphosphate, are inactive. Interestingly, both l-glucose-derived ligands are partial agonists: they are among the least efficacious agonists of Ins(1,4,5)P3R yet identified, providing new leads for antagonist development.
Introduction
d-myo-Inositol 1,4,5-trisphosphate [Ins(1,4,5)P3, 1] is a second messenger that binds to tetrameric d-myo-inositol 1,4,5-trisphosphate receptors [Ins(1,4,5)P3Rs] on the endoplasmic reticulum. Ins(1,4,5)P3Rs are Ca2+ channels that open to release Ca2+ to the cytosol.1,2 The resulting local or global increases in cytosolic Ca2+ concentration regulate diverse cellular processes, including mitochondrial metabolism, cell proliferation, differentiation, smooth muscle contraction, secretion, exocytosis, and ion channel opening.3
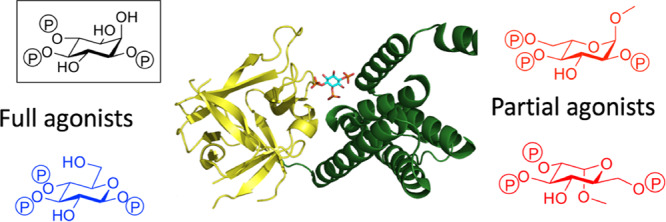
Ins(1,4,5)P3 (Figure 1a) binds to the Ins(1,4,5)P3-binding core (IBC; residues 224–604) close to the N terminus of each of the four Ins(1,4,5)P3R subunits (Figure 1b,c). The IBC consists of an α-helical domain and a β-trefoil domain between which there is a cleft rich in basic amino acid residues.4 Ins(1,4,5)P3 binding within the cleft allows phosphates at positions 1 and 5 to interact with the α-domain, while the 4-phosphate interacts with the β-domain (Figure 1c).5 As Ins(1,4,5)P3 interacts with both domains, it pulls the two sides of the clam-like IBC together.6,7 The clam closure leads to channel opening, possibly by rearranging Ca2+-binding sites such that Ca2+ can bind to the Ins(1,4,5)P3R and trigger conformational changes that lead to opening of the Ca2+-permeable pore.1,6,7
Figure 1.

Structures of the Ins(1,4,5)3R IBC and its agonists. (a) Structure of Ins(1,4,5)P3 (1). (b) Binding mode of 1 to the IBC of Ins(1,4,5)P3R with key amino acid residues involved in binding labeled. (c) Crystal structure of the IBC of type 1 InsP3R with Ins(1,4,5)P3 (1) bound (PDB: 1N4K, file available in the Associated Content)4. The α-domain is shown in green and the β-domain in yellow. (d) Structure of the Ins(1,4,5)P3R agonist adenophostin A (AdA).
Modulators of Ins(1,4,5)P3R activity are highly sought after, and many studies have examined structure–activity relationships (SARs) of ligands binding to Ins(1,4,5)P3R, attempting to identify partial agonists or antagonists.8−10 Synthetic analogues have played a key role in this process, including those of the potent glyconucleotides, the adenophostins5,11 (Figure 1d), with a recent synthetic study reporting the effects of replacing the glucose moiety of adenophostin A (AdA) with a chiro-inositol core.12 This also highlights developing interest in examining less explored isomers of inositol than the myo-form.13
The phosphates attached to the 4- and 5-positions of Ins(1,4,5)P3 (Figure 1a) are thought to be essential to agonist activity as each interacts with a different domain of the IBC.4,14 The 1-phosphate increases affinity, but it is not essential for receptor activation.5 The hydroxyl group attached to the 6-position of Ins(1,4,5)P3 appears to be important for optimal activity but it is not essential,15 while hydroxyl groups attached to the 2- and 3-positions are less involved in ligand binding.16
A few Ins(1,4,5)P3R antagonists have been identified, but these suffer major drawbacks including poor target selectivity, cell impermeability (heparin),17 inconsistent effectiveness in assays (2-aminoethoxydiphenylborane, 2-APB),17 low potency (caffeine),17,18 and disputed activity (xestospongins).17 For decades, analogues of Ins(1,4,5)P3 have been synthesized and investigated in attempts to discover a selective antagonist for Ins(1,4,5)P3R that could be made, at least temporarily, cell permeable with enzyme-cleavable or photolabile protecting groups.19,20 Very recently, there has also been work carried out by Li et al. that describes the delivery of inositol phosphates into cells via lysosomes, rendering phosphate protecting groups unnecessary.21 To date, however, only a small number of analogues of Ins(1,4,5)P3 with minor structural modifications have been identified as partial agonists or antagonists at Ins(1,4,5)P3R. Most of these compounds demonstrate that conservative modifications to the phosphates attached to positions 4 and 5 and the hydroxyl group attached to position 3 can lead to degrees of antagonist activity.22−25 However, many analogues of Ins(1,4,5)P3 with modifications to the same regions are inactive.26 Attaching a bulky substituent to the axial 2-position hydroxyl can also lead to partial agonist activity,27 and interestingly, even using a simple benzene ring as a surrogate for inositol in a benzene polyphosphate approach, as a dimer or biphenyl, can provide low-affinity antagonists.28,29
Stimulated in part by the discovery of the adenophostins,5,10,30 there have been a number of studies to investigate polyphosphates of d-glucose and of other sugars30−35 as inositol phosphate analogues. By using such carbohydrates, the need for optical resolution of protected cyclitol precursors or resulting phosphate regioisomers is bypassed as chiral starting materials are readily available. Also, the structural features of carbohydrates offer additional opportunities for synthetic versatility. In this study, we return to and expand upon the use of d-glucose to try to identify novel Ins(1,4,5)P3R ligands. We also investigate, perhaps counterintuitively, the use of l-glucose as a starting material.
Although several studies have generated d-glucose-based ligands of Ins(1,4,5)P3R,30,31,34 no ligands based on the l-glucose enantiomer have been synthesized and nor is it known whether ligands with this scaffold would bind to Ins(1,4,5)P3R. In a previous study,5 Ins(4,5)P2 [albeit a low-affinity Ins(1,4,5)P3R ligand] was effectively mimicked by a d-Gluc(3,4)P2 surrogate. We noted that both d- and l-glucose offer three hydroxyl groups of the requisite relative configuration that could, in principle, be used to mimic the 4,5,6-hydroxyl groups in myo-inositol. Thus, we anticipated that we could use this similarity (Figure 2, highlighted in red), alongside intrinsic structural differences of l- and d-glucose, to prepare diverse chiral ligands with appropriately located phosphates. These would present different structural motifs to the Ins(1,4,5)P3R and allow further investigation of the binding site and perhaps identify novel activity. With this in mind, we designed and synthesized six novel ligands based on l-glucose and d-glucose (Figure 3) and evaluated their activity at Ins(1,4,5)P3R.
Figure 2.
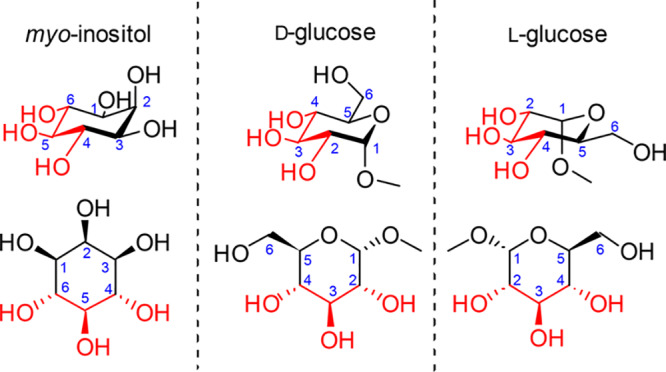
Structures of methyl α-d-glucopyranoside and methyl α-l-glucopyranoside relative to myo-inositol with the shared stereochemistry of the hydroxyl groups highlighted in red.
Figure 3.

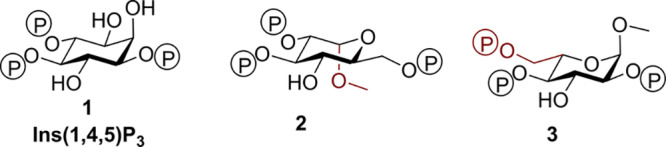
Structures of Ins(1,4,5)P3 (1) analogues: methyl α-l-glucopyranoside 2,3,6-trisphosphate (2) and methyl α-l-glucopyranoside 2,4,6-trisphosphate (3), methyl α-d-glucopyranoside 2,3,6-trisphosphate (4) and methyl α-d-glucopyranoside 2,4,6-trisphosphate (5), α-d-glucopyranosyl 1,3,4-trisphosphate (6), and β-d-glucopyranosyl 1,3,4-trisphosphate (7).
We ensured that the designed ligands retained structures equivalent to the critical 4,5-bisphosphate motif of Ins(1,4,5)P3 and had no major structural modifications in regions believed to be necessary for ligand binding; modifications in regions equivalent to the 2-O-position were permitted as these were expected to remain outside the binding pocket. We hypothesized that the l-glucose-based ligands [methyl α-l-glucopyranoside 2,3,6-trisphosphate (2) and methyl α-l-glucopyranoside 2,4,6-trisphosphate (3)] might have sufficiently conservative structural changes relative to Ins(1,4,5)P3 such that they could still bind to Ins(1,4,5)P3R, possibly with novel activity. The d-glucose-based ligands [methyl α-d-glucopyranoside 2,3,6-trisphosphate (4) and methyl α-d-glucopyranoside 2,4,6-trisphosphate (5)] were not expected to adopt orientations that position the phosphate groups in appropriate regions of the IBC (see the Supporting Information SI-1, S1 for details of all possible predicted binding modes). Their bioassay was designed to enable confirmation of receptor enantioselectivity. From a practical standpoint, the synthesis of ligands 4 and 5 was optimized first with d-glucose before the commercially available, but considerably more costly, l-glucose was used to make the respective l-enantiomers, 2 and 3. We also anticipated that the two ligands with phosphates at the anomeric carbon, α-d-glucopyranosyl 1,3,4-trisphosphate (6) and β-d-glucopyranosyl 1,3,4-trisphosphate (7), would help to elucidate the basis for the high affinity of AdA.5,12,36 We expected both novel truncated analogues to be agonists due to their structural similarity to Ins(1,4,5)P3 but were interested to compare their activities with AdA and the previously analyzed truncated analogue Gluc(3,4)P2.5
Results
Chemistry
Methyl α-l-glucopyranoside 2,3,6-trisphosphate (2) was synthesized in a five-step route from readily available l-glucose (Scheme 1). Refluxing l-glucose in an acidic methanol solution resulted in the protection of the anomeric hydroxyl with a methyl group. Multiple recrystallizations from ethanol afforded methyl α-l-glucopyranoside 8 in 45% yield. The 4- and 6-position hydroxyls were protected with a benzylidene group to form 9 in 91% yield. This benzylidene group was reduced regioselectively, opening to form methyl 4-O-benzyl-α-d-glucopyranoside (10). The benzylidene reduction was first attempted with borane/THF and AlCl3, but this was found to be insufficiently regioselective and produced inseparable regioisomers. The reaction was successfully carried out with borane/THF and La(Tf)3 following the method of Shie et al.37 to yield 10 in 31% yield following purification. The hydroxyl groups in triol 10 were then phosphitylated with dibenzyl diisopropylphosphoramidite, and subsequent oxidation with mCPBA formed 11 in 86% yield. The benzyl protecting groups on phosphates and O-4 were then removed by stirring a solution of 11 with Pearlman’s catalyst under hydrogen overnight. After filtration to remove the catalyst and evaporation of the solvent, 2 was collected as the triethylammonium salt in 53% yield.
Scheme 1. Synthesis of l-Glucose-Derived Ligands: Methyl α-l-Glucopyranoside 2,3,6-Trisphosphate (2) and Methyl α-l-Glucopyranoside 2,4,6-Trisphosphate (3).

Reagents and conditions: (a) MeOH, AcCl, reflux, 5 days; (b) (1) TMSCl, py, 22 h and (2) DCM, benzaldehyde, FeCl2·6H2O, MeCN, triethylsilane, 0 °C to room temperature, 1.5 h; (c) MeOH, H2O, 1 M HCl(aq), reflux, 3 h; (d) (1) DCM, 5-phenyl-1H-tetrazole, (BnO)2PN(iPr)2, 20 h and (2) mCPBA, −78 °C to room temperature; (e) MeOH/H2O (10:1 v/v), cat. Pd(OH)2/C, H2, 24 h; (f) MeCN, benzaldehyde dimethyl acetal, cat. CSA, 24 h; (g) BH3/THF, La(Tf)3, 7 days.
Methyl α-l-glucopyranoside 2,4,6-trisphosphate (3) was also synthesized in five steps from l-glucose, diverging from the synthesis of 2 after the initial methylation step. Methyl α-l-glucopyranoside (8) was protected in a regioselective one-pot reaction that involved persilylation followed by FeCl2-catalyzed benzylidene protection as described by Bourdreux et al.38 to yield 12 in 79% yield. The acid-labile benzylidene group was removed through reflux with HCl(aq) to form 13 in 93% yield. Phosphorylation using the standard phosphoramidite methodology39 was then employed to give 14 in 40% yield. After debenzylation with hydrogen and Pearlman’s catalyst and filtration and evaporation of the solvent, the final product, 3, was collected as its triethylammonium salt in 90% yield.
Both α-d-glucopyranosyl 1,3,4-trisphosphate (6) and β-d-glucopyranosyl 1,3,4-trisphosphate (7) were synthesized via a divergent route, starting with allyl 2,6-di-O-Bn-α-d-glucopyranoside (15) (Scheme 2).34 Palladium chloride-catalyzed deallylation yielded 16, although purification of this compound was found to be very difficult at this step and purification after phosphorylation proved to be much more effective. Thus, slightly impure 16 was phosphorylated to yield a pure, partially separable mixture of the epimeric phosphates 17 and 18 (56% yield total: 14% 17, 19% 18, and 23% mixed epimers). It was unclear how stable 17 and especially the phosphorylated β-epimer 18 would be as there are reports of compounds with phosphate groups at the anomeric carbon atom being unable to survive purification by silica column chromatography in some cases and not in others.40−42
Scheme 2. Synthesis of Epimeric Ligands with a Phosphate Group at the Anomeric Carbon: α-d-Glucopyranosyl 1,3,4-Trisphosphate (6) and β-d-Glucopyranosyl 1,3,4-Trisphosphate (7).

Reagents and conditions: (a) MeOH, cat. PdCl2, 6 h; (b) (1) DCM, 5-phenyl-1H-tetrazole, (BnO)2PN(iPr)2, 20 h and (2) mCPBA, −78 °C to room temperature; (c) MeOH/H2O (10:1 v/v), cat. Pd(OH)2/C, H2, NaHCO3, 24 h.
We found in this case that both compounds survived silica gel chromatography, although 18 showed slight degradation by 31P NMR over time (approx. 30% after 3 weeks at 4 °C). Catalytic hydrogenolysis of both compounds 18 and 19 was carried out in the presence of sodium bicarbonate to prevent acidic hydrolysis of the potentially labile C-1 phosphates. The final products 6 and 7 were purified by reversed-phase ion-pair chromatography and collected as their triethylammonium salts.
Methyl α-d-glucopyranoside 2,3,6-trisphosphate (4) and methyl α-d-glucopyranoside 2,4,6-trisphosphate (5) were synthesized using the same methods as described for their enantiomers (2 and 3), starting the route with d-glucose (Scheme 3).
Scheme 3. Synthesis of d-Glucose-Derived Ligands: Methyl α-d-Glucopyranoside 2,3,6-Trisphosphate (4) and Methyl α-d-Glucopyranoside 2,4,6-Trisphosphate (5).

Reagents and conditions: (a) MeOH, reflux, 5 days; (b) (1) TMSCl, py, 22 h and (2) DCM, benzaldehyde, Fe(II)Cl2·6H2O, MeCN, triethylsilane, 0 °C to room temperature, 1.5 h; (c) MeOH, H2O, 1 M HCl(aq), reflux, 3 h; (d) (1) DCM, 5-phenyl-1H-tetrazole, (BnO)2PN(iPr)2, 20 h and (2) mCPBA, −78 °C to room temperature; (e) MeOH/H2O (10:1 v/v), cat. Pd(OH)2/C, H2, 24 h; (f) MeCN, benzaldehyde dimethyl acetal, cat. CSA, 24 h; (g) BH3/THF, La(Tf)3, 7 days.
The relative stabilities of α-d-glucopyranosyl 1,3,4-tris-phosphate (6) and β-d-glucopyranosyl 1,3,4-trisphosphate (7) were first investigated by allowing each compound (as triethylammonium salts) to remain in a solution of D2O at room temperature at pH 7. Over the course of 2 months, neither isomer showed any sign of degradation. Following this, a mixture of the compounds in a known starting ratio was exposed to increasingly harsh conditions, and relative isomer degradation was monitored through 1H NMR spectroscopy (see the Supporting Information, SI-1 Figure S5). From the results of the hydrolysis study, we determined that the β-epimer (7) degraded more readily than the corresponding α-epimer (6). Both compounds were, however, surprisingly durable and required strongly acidic conditions to be fully hydrolyzed (i.e., at pH 1 for 1 day), while strongly basic conditions (i.e., at pH 14 for 1 day) only produced limited degradation (and at pH 10 for a week, there was no change). We are, therefore, confident that both compounds remained intact during the near-neutral conditions of the biological assays. All final compounds were assessed by HPLC for purity (SI-1 Figure S6), and products from acid hydrolysis were examined by HPLC using a synthetic standard of d-glucose 3,4-bisphosphate5 to confirm the expected hydrolysis product of both compounds (see the Supporting Information, SI-1 Figure S7).
Biology
Permeabilized HEK-Ins(1,4,5)P3R1 cells were used to determine the ability of compounds 2–7 (with Ins(1,4,5)P3 and AdA as controls) to evoke Ca2+ release from intracellular stores (Figure 4 and Table 1). Maximally effective concentrations of Ins(1,4,5)P3, α-d-glucopyranosyl 1,3,4-tris-phosphate (6), or β-d-glucopyranosyl 1,3,4-trisphosphate (7) released the same fraction (ca. 80%) of the intracellular Ca2+ stores, suggesting that these two epimeric compounds are both full agonists (Figure 4). Compound 7 was equipotent with Ins(1,4,5)P3, and 6 was ca. 20 times less potent than Ins(1,4,5)P3.
Figure 4.
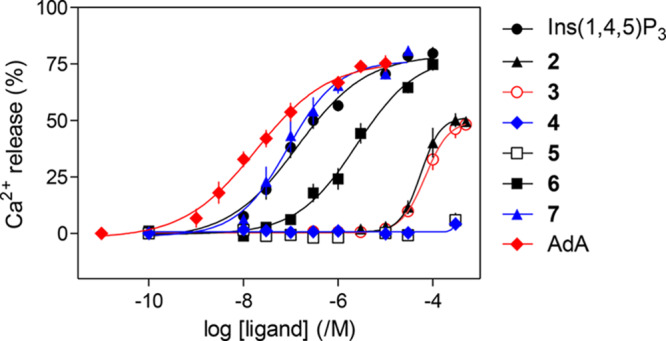
Concentration-dependent effects of Ins(1,4,5)P3 and related ligands on Ca2+ release from intracellular stores of permeabilized HEK-Ins(1,4,5)P3R1 cells. Results are means ± SEM from 5 to 11 independent experiments, each with duplicate determinations.
Table 1. Ins(1,4,5)P3R Binding and Ca2+ Release Mediated by Ins(1,4,5)P3, AdA, and Compounds 2–7a.
| Ca2+ release |
binding |
||||||
|---|---|---|---|---|---|---|---|
| ligand | pEC50; EC50 | release (%) | h | pKd; Kd (nM) | h | EC50/Kd | EC39/Kdb |
| Ins(1,4,5)P3 | 6.90 ± 0.12; 126 nM | 78.8 ± 1.3 | 0.7 ± 0.1 | 8.06 ± 0.03; 8.7 | 1.1 ± 0.2 | 14 (5–46) | 17 (5–63) |
| 2 | 4.06 ± 0.09c; 87.7 μM | 56.2 ± 2.6c | 1.4 ± 0.2 | 5.91 ± 0.03c; 1230 | 0.9 ± 0.1 | 71 (34–148) | 132 (49–355) |
| 3 | 3.98 ± 0.04c; 104 μM | 53.1 ± 5.0c | 1.3 ± 0.2 | 6.26 ± 0.07c; 549 | 0.9 ± 0.1 | 191c (123–295) | 462c (128–1667) |
| 4 | ND | 4.3 ± 2.1d | ND | 48 ± 12e | ND | ND | |
| 5 | ND | 5.9 ± 2.1d | ND | 53 ± 3e | ND | ND | |
| 6 | 5.53 ± 0.20c; 2.96 μM | 78.8 ± 3.0 | 0.9 ± 0.2 | 6.89 ± 0.09c; 129 | 0.7 ± 0.1 | 23 (5–105) | |
| 7 | 7.09 ± 0.18; 80 nM | 75.2 ± 1.4 | 1.0 ± 0.1 | 7.95 ± 0.05; 11.2 | 1.1 ± 0.2 | 7 (2–29) | |
| AdA | 7.62 ± 0.12c; 24 nM | 77.8 ± 4.5 | 0.8 ± 0.1 | 8.86 ± 0.14c; 1.4 | 1.2 ± 0.2 | 17 (5–51) | |
Effects of ligands on Ca2+ release from the intracellular stores of permeabilized HEK-Ins(1,4,5)P3R1 cells and on [3H]-Ins(1,4,5)P3 binding to cerebellar membranes are summarized. Results from functional assays are means ± SEM (pEC50 (−log of the half-maximally effective concentration), Ca2+ release (%), and Hill coefficient (h)) and means (EC50) from 5 to 11 independent experiments, each performed in duplicate. Results from binding experiments are means ± SEM (pKd (−log of the equilibrium dissociation constant) and h) and means (Kd) from three independent experiments. The pKd values for Ins(1,4,5)P3 and AdA have been published (Mills et al.)43 and are reproduced with permission. Final columns show EC50/Kd or (for partial agonists and Ins(1,4,5)P3) EC39/Kd (mean and 95% CI). ND, not determined.
EC39 reports the concentration of ligand required to evoke the same Ca2+ release (39% of the intracellular stores) as evoked by a half-maximally effective concentration of Ins(1,4,5)P3.
P < 0.05 relative to Ins(1,4,5)P3.
Ca2+ release evoked by 300 μM ligand.
Specific binding of [3H]-Ins(1,4,5)P3 in the presence of 30 μM competing ligand.
The l-glucose-based ligands methyl α-l-glucopyranoside 2,3,6-trisphosphate (2) and methyl α-l-glucopyranoside 2,4,6-trisphosphate (3) were much less potent than Ins(1,4,5)P3 (Figure 4), while their enantiomers methyl α-d-glucopyranoside 2,3,6-trisphosphate (4) and methyl α-d-glucopyranoside 2,4,6-trisphosphate (5) were, as predicted, inactive. The Ca2+ release evoked by maximally effective concentrations of 2 or 3 was only ca. 70% of that evoked by Ins(1,4,5)P3, suggesting that 2 and 3 are partial agonists. Since partial agonists bind to Ins(1,4,5)P3Rs but activate them less effectively than full agonists, a partial agonist must bind to more Ins(1,4,5)P3Rs than a full agonist to evoke comparable Ca2+ release. We performed equilibrium competition binding assays using [3H]-Ins(1,4,5)P3 and the active ligands to examine relationships between ligand binding and functional responses. The affinities of 6 and 7 for Ins(1,4,5)P3R aligned with their potencies in functional assays, with 7 having an affinity indistinguishable from that of Ins(1,4,5)P3, while 6 had ca. 15-fold lower affinity (Figure 5 and Table 1). The EC50/Kd values for Ins(1,4,5)P3, AdA, 6, and 7 were similar, consistent with each being a full agonist (Table 1). Comparison of the concentrations of 2 and 3 required to occupy 50% of binding sites (Kd) and to evoke release of 39% of the Ca2+ stores (EC39, i.e., the Ca2+ release evoked by a half-maximally effective Ins(1,4,5)P3 concentration) confirmed that 2 and 3 are weak partial agonists: their EC39/Kd values (132 and 462, respectively) were much greater than that of Ins(1,4,5)P3 (17). HPLC was used to confirm the purity of the compounds used in the biological assays (details in the Supporting Information SI-1, S6).
Figure 5.
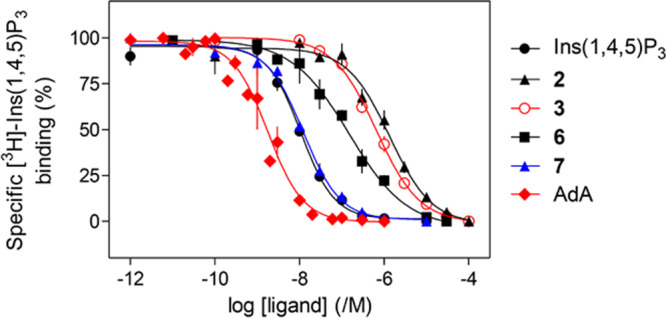
Equilibrium competition binding to cerebellar membranes using [3H]-Ins(1,4,5)P3 (1.5 nM) and the indicated concentrations of competing ligands. Results are means ± SEM from three independent experiments. The results for Ins(1,4,5)P3 and AdA have been published (Mills et al.).43
Discussion
Of the glucose polyphosphates considered in this study, the two that bound to Ins(1,4,5)P3R with the highest affinity were α-d-glucopyranosyl 1,3,4-trisphosphate (6) and β-d-glucopyranosyl 1,3,4-trisphosphate (7). Both compounds, which can be considered as truncated analogues of adenophostin A (AdA, Figure 6),44 were found to be full agonists of Ins(1,4,5)P3R, and the β-epimer (7) was equipotent with Ins(1,4,5)P3.
Figure 6.

Structural comparison of Ins(1,4,5)P3 (1), AdA, and some of its truncated analogues including α-d-glucopyranosyl 1,3,4-trisphosphate (6) and β-d-glucopyranosyl 1,3,4-trisphosphate (7). The conserved regions of the structures involved in binding are drawn in blue, while the differing auxiliary phosphate is shown in red.
Compounds containing a phosphate group attached to the anomeric carbon atom, as featured in 6 and 7, have not been previously investigated as Ins(1,4,5)P3R ligands, presumably due to concerns over their stability, at least in the case of the β-epimer.31 Nevertheless, we found that both α-d-glucopyranosyl 1,3,4-trisphosphate (6) and β-d-glucopyranosyl 1,3,4-trisphosphate (7) were surprisingly durable as their triethylammonium salts and neither compound showed signs of degradation after 2 months in neutral aqueous solution at room temperature. Both 6 and 7 were eventually degraded under strongly acidic conditions, and HPLC traces were taken to confirm their hydrolysis to glucose 3,4-bisphosphate (details in the Supporting Information SI-1, S7).
Previous studies using synthetic analogues of AdA30−34,45 demonstrated that the adenine moiety significantly increases potency of the agonist, the vicinal phosphates are crucial to activity, and minor adjustments to the placement of the auxiliary phosphate can be tolerated.5,34 The general consensus for AdA binding is that the ligand interacts with the binding site of Ins(1,4,5)P3R with the 3″-, 4″-, and 2′-phosphates mimicking the 5, 4, and 1 phosphates of Ins(1,4,5)P3, respectively.27,46 However, these previous studies employed analogues that differed from Ins(1,4,5)P3 in several ways, and it has, therefore, been difficult to isolate the specific impact of replacing the myo-inositol ring with d-glucopyranose.
This has implications for the possible mode of action of AdA and related compounds. Indeed, a cryo-EM study47 of tetrameric Ins(1,4,5)P3R1 has recently proposed that AdA interacts with the IBC in a completely different way from Ins(1,4,5)P3, with the two domains of the IBC being pulled together by the 3″- and 4″-phosphate groups of AdA interacting with one domain and the adenine moiety interacting with the other.47 In this model of AdA binding to Ins(1,4,5)P3R, the glucose bisphosphate structure of AdA only coincidentally resembles the myo-inositol 4,5-bisphosphate of Ins(1,4,5)P3, and there is no structural correspondence between the glucose ring of AdA and the inositol ring of Ins(1,4,5)P3. However, this conclusion does not support the observed activities of the compounds in this study and other AdA analogues; it should, therefore, be viewed with caution.12,47
In the present study, we found that the closest possible glucose-containing analogue of Ins(1,4,5)P3, namely, compound 7, is effectively indistinguishable from Ins(1,4,5)P3 in our assays of Ins(1,4,5)P3R binding and Ca2+ release. This is entirely consistent with the idea that, in the Ins(1,4,5)P3 binding site, the glucopyranoside ring of 7 is closely analogous to the myo-inositol ring of Ins(1,4,5)P3. In turn, this establishes that the flexible Ins(1,4,5)P3 binding site can accommodate the equatorial glucopyranoside hydroxymethyl (CH2OH) group and pyranoside ring oxygen in place of the myo-inositol 3-OH group and C-2, respectively, with no impact on activity. The α-epimer 6 is approximately 27-fold less potent than β-epimer 7 in Ca2+ release. This shows that the axial phosphate group in 6 can still contribute to binding [Gluc(3,4)P2 is much less potent] and is also consistent with an earlier report that d-chiro-Ins(1,3,4)P3, the C-1 epimer of Ins(1,4,5)P3 having an axial 1-phosphate group, had 25-fold lower potency than Ins(1,4,5)P3.48
Thus, the effects of trisphosphates 6 and 7 provide strong support for the argument that compounds containing the d-glucopyranosyl 3,4-bisphosphate structure mimic Ins(1,4,5)P3 due to the direct structural analogy between glucopyranosyl and myo-inositol rings depicted in Figures 2 and 6. In such compounds, it is highly likely that the glucose 3,4-bisphosphate structure simply pulls together the two domains of the IBC in the manner proposed for the inositol 4,5-bisphosphate motif of Ins(1,4,5)P3. Ligands 6 and 7 were docked into the IBC to explore potential binding site interactions (see Supporting Information SI-1 Figures S2 A and B, respectively, molecular docking files of 6 and 7 in 1N4K available in the Associated Content). We recently reported studies in which the glucose ring of AdA12 and ribophostin43 was replaced by d-chiro-inositol, leading to both modest and significant increases in biological activity. It therefore remains to be conclusively established whether the additional components present in the AdA molecule can, counterintuitively, induce a completely unrelated role for the glucose bisphosphate component of AdA itself, as suggested in the cryo-EM study.47
Previous studies of C-glycosidic truncated analogues of AdA with different chain lengths tethering the third, auxiliary phosphate group (Figure 6) have demonstrated that positioning of this phosphate has a significant effect on the affinity of the agonist.30−34 It has been observed that the Ca2+-releasing potency of these analogues at Ins(1,4,5)P3R decreases as the length and flexibility of the linkage to the auxiliary phosphate increases.34 The orientation of the linkage also plays a role, with axial linkages usually resulting in more potent compounds. However, even the most potent of these compounds (Figure 6, Terauchi et al.,31 axial linkage, n = 2) is still weaker than Ins(1,4,5)P3 and compound 7.
Methyl α-l-glucopyranoside 2,3,6-trisphosphate (2) was found to be a partial agonist of Ins(1,4,5)P3R1, while its d-glucose-based enantiomer, compound 4, was inactive. This supports the structural alignment of trisphosphate 2 with Ins(1,4,5)P3 shown in Figure 7 and in our molecular modeling in SI-1 Figure S3 (molecular docking file of 2 in 1N4K available in the Associated Content). No such alignment is possible for compound 4 because it does not possess a vicinal bisphosphate motif whose stereochemistry matches that of Ins(1,4,5)P3.
Figure 7.
Ins(1,4,5)P3 (1) and analogues methyl α-l-glucopyranoside 2,3,6-trisphosphate (2) and methyl α-l-glucopyranoside 2,4,6-trisphosphate (3) with their structural differences contribute to their Ins(1,4,5)P3R partial agonist activity in dark red.
In the predicted binding conformation of methyl α-l-glucopyranoside 2,3,6-trisphosphate (2) (SI-1 Figure S3 molecular docking file of 2 in 1N4K available in the Associated Content), the axial methyl group is positioned in a region of the binding site normally occupied by the 3-hydroxyl of Ins(1,4,5)P3. In the design of 2, we further anticipated that the phosphate group at C-6 of l-glucose would mimic the auxiliary 1-phosphate of Ins(1,4,5)P3 to some extent as there is evidence from previous studies showing that the Ins(1,4,5)P3R can accommodate more sterically demanding groups in this region of the binding site.5,49−51 A very recent example of this is that the replacement of the Ins(1,4,5)P3 1-phosphate by a pyrophosphate, which increases both charge and steric bulk, does not affect activity.49 In addition, trisphosphate 2 contains an hydroxyl group appropriately placed to mimic the important 6-OH group of Ins(1,4,5)P3.
In studies of Ins(1,4,5)P3 analogues as partial agonists, it has been shown that perturbations in the equivalent of the 3-hydroxyl of Ins(1,4,5)P3 can result in partial agonist activity,52 especially when this disruption (often by means of stereochemical inversion) occurs in conjunction with a modification to the vicinal phosphate pair or other region of the ligand.18,22,25,52 It has been observed in multiple studies that limited, equatorial extension of substituents from the 3-position equivalent can be tolerated,53−55 but larger groups hinder binding56,57 and inversion of the 3-hydroxyl to axial results in a slight decrease in ligand activity.22,58−60Figure 7 therefore suggests that the axial O-methyl group is the most likely component of 2 that causes it to display partial agonist activity, perhaps by interfering with the ligand binding to the β-domain of the IBC or by reducing the extent of domain closure.
Methyl α-l-glucopyranoside 2,4,6-trisphosphate (3) was designed to bind to the Ins(1,4,5)P3R in a manner that would potentially satisfy the essential binding requirements by positioning the pyranoside ring oxygen in place of the nonessential 3-OH group of Ins(1,4,5)P3, while the axial 1-methoxy group occupied the place of the unimportant 2-OH group of Ins(1,4,5)P3 (Figure 7 and SI-1 Figure S4, molecular docking file of 3 in 1N4K available in the Associated Content). In this docked binding mode, the l-glucose 6-phosphate group would enter the region of the binding site usually occupied by the 4-phosphate of Ins(1,4,5)P3.
In previous studies, it has been shown that conservative modifications to the phosphates attached to the 4 and 5 equivalent positions (and sometimes in conjunction with a modification to the 3-position equivalent) can produce partial agonists and even low-affinity antagonists.22−25,52,56,61 Bello et al.26 hypothesized that if a ligand could bind to only one side of the IBC (through disruption of the interactions of either the 4 or 5 equivalent phosphates), it would be unable to pull the clam-like structure of the binding site closed and would therefore be unable to activate the receptor. Thus, a suitable modification to the 4-phosphate of Ins(1,4,5)P3 might weaken the important interaction with the β-domain of the clam shell structure and induce suboptimal activation of Ins(1,4,5)P3R. However, previous studies attempting to generate partial agonists with modifications solely to the 4-phosphate have failed to identify any active Ins(1,4,5)P3 analogues.26
Pleasingly, our assays show that l-glucose trisphosphate 3 also behaves as a partial agonist of Ins(1,4,5)P3R1, with improved binding affinity and a higher EC50/Kd ratio than partial agonist 2. The fact that the d-enantiomer 5 is inactive supports the structural alignment of 3 with Ins(1,4,5)P3 depicted in Figure 7. The “extended” 4-phosphate group equivalent in 3 may thus disrupt the interaction of the ligand with the β-domain of the IBC as theorized.26 It is likely that the absence in 3 of an equivalent to the 3-OH group in Ins(1,4,5)P3 also contributes to a decreased interaction between the β-domain of the IBC and the ligand. Indeed, activity for 3-deoxy-Ins(1,4,5)P3 at Ins(1,4,5)P3R has been reported to drop up to 40-fold.10
The two partial agonists, α-l-glucopyranoside 2,3,6-trisphosphate (2) and α-l-glucopyranoside 2,4,6-trisphosphate (3), indicate that perturbations on the ring structure of the ligand are sufficient to induce partial agonism. Both ligands suffer from low affinity and, as a result, structurally related compounds are currently being developed that will incorporate similar structural differences to Ins(1,4,5)P3 and hopefully maintain the desired decreased efficacy while increasing affinity. The most promising avenue seems to be adapting the structure of 3 by generating other ligands with extended 4-position phosphate equivalents. This could hypothetically be continued with l-glucose, but inositol could also prove to be a useful starting material as ligands could be synthesized with a similar extension of the 4-position hydroxyl without the loss of an equivalent hydroxyl to position 3 in Ins(1,4,5)P3, perhaps thereby improving ligand affinity while retaining partial agonist activity. Such work is in progress.
Conclusions
We have synthesized four novel active ligands for the Ins(1,4,5)P3R based on both d-glucose and l-glucose templates as inositol surrogates. The two ligands based on l-glucose, namely, methyl α-l-glucopyranoside 2,3,6-trisphosphate (2) and methyl α-l-glucopyranoside 2,4,6-trisphosphate (3), are low-affinity, low-efficacy partial agonists of Ins(1,4,5)P3R, while their respective d-glucose-based enantiomers 4 and 5 are inactive. Two further synthetic d-glucose-based trisphosphates, α-d-glucopyranosyl 1,3,4-trisphosphate (6) and β-d-glucopyranosyl 1,3,4-trisphosphate (7), can be regarded as close analogues of Ins(1,4,5)P3, but they are also related structurally to the naturally occurring glyconucleotide Ins(1,4,5)P3R agonist adenophostin A (AdA). They can, therefore, further our understanding of how AdA binds to Ins(1,4,5)P3R. Both 6 and 7 were found to be full agonists of Ins(1,4,5)P3R, with the perhaps surprisingly stable β-epimer 7 being equipotent to Ins(1,4,5)P3 itself and potentially useful as a chemical biology tool under physiological conditions (with degradation induced only under extremes of pH). The potency of 7 demonstrates that the structural differences between myo-inositol and d-glucose need not result in any decrease in ligand activity. This is consistent with the d-glucopyranosyl 3,4-bisphosphate moiety of AdA directly mimicking the d-myo-inositol 4,5-bisphosphate structure of Ins(1,4,5)P3 at the binding site of Ins(1,4,5)P3Rs. All four active ligands 2, 3, 6, and 7 were docked into the IBC to explore potential binding site interactions.
Partial agonists 2 and 3 are the first l-glucose-derived ligands that have been synthesized for Ins(1,4,5)P3R. Both compounds provide evidence for the viability of generating partial agonists and potential antagonists of Ins(1,4,5)P3R by deliberately disrupting the crucial moieties involved in binding to the IBC clam shell and pulling the domains together upon ligand binding. We hypothesize that the axial O-methyl group of compound 2 and the extended phosphate in the equivalent of the 4-position phosphate in Ins(1,4,5)P3 of compound 3 cause the partial agonist activity of these compounds either by disrupting the interactions of the ligand with the β-domain of the IBC or by preventing complete closure of the IBC upon binding. These partial agonists could prove to be interesting starting points to generate structurally similar compounds with even lower efficacy and higher affinity that could result in the generation of improved partial agonists or antagonists.
Experimental Section
General Synthesis
Chemicals were purchased from Sigma-Aldrich, Acros, or Alfa Aesar. Anhydrous solvents were purchased from Sigma-Aldrich. TLC was performed on precoated plates (Merck aluminum sheets, silica 60 F254, art no. 5554). Chromatograms were visualized under UV light and by dipping plates into phosphomolybdic acid in EtOH followed by heating. Flash column chromatography was performed using RediSep Rf disposable flash columns on an ISCO CombiFlash Rf automated flash chromatography machine. Reversed-phase chromatography was performed on LiChroprep RP-18 (25–40 μm, Merck) using a BioLogic LP system (BioRad), eluting at 5 mL min–1 with a gradient of 0–10% MeCN in 0.05 M triethylammonium bicarbonate (TEAB) buffer, collecting 7 mL fractions. Fractions containing the target polyphosphate were identified using a modification of the Briggs phosphate assay.62 The purity of all of the final compounds used in biological assays was assessed by HPLC and found to be >95% pure (vide infra and HPLC data in the Supporting Information). Proton 1H NMR and COSY spectra were recorded on a Bruker Avance III (400 MHz) spectrometer. Proton chemical shifts are reported in ppm (δ) relative to internal tetramethylsilane (TMS, δ 0.0 ppm) or with the solvent reference relative to TMS employed as the internal standard (CDCl3, δ 7.26 ppm; CD3OD, δ 3.31 ppm). The following abbreviations are used to describe the multiplicity of the chemical shifts: br, broad; s, singlet; d, doublet; dd, double doublet; q, quartet; m, multiplet; and t, triplet. 13C and HSQC spectra were recorded on a Bruker Avance III (100 MHz) spectrometer with complete proton decoupling. Carbon chemical shifts are reported in ppm (δ) relative to TMS with the respective solvent resonance as the internal standard (CDCl3, δ 77.0 ppm, CD3OD, δ 49.0 ppm). 31P NMR spectra were recorded on a Bruker Avance III (162 MHz) spectrometer with complete proton decoupling. Phosphorus chemical shifts are reported in ppm (δ) relative to an 85% H3PO4 external standard (H3PO4, δ 0.0 ppm). All NMR data were collected at 25 °C. Optical rotations were measured at ambient temperature using an Optical Activity Ltd. AA-10 polarimeter in a cell volume of 5 cm3, and specific rotation is given in degmLg−1dm−1. Melting points were determined using a Stanford Research Systems Optimelt MPA100 automated melting point system and are uncorrected. Mass spectra were recorded on a Thermo Orbitrap Exactive mass spectrometer. All reactions were carried out under an argon atmosphere employing oven-dried glassware unless stated otherwise.
Methyl α-l-Glucopyranoside (8)
In a modified version of the Li et al.63 procedure, l-glucose (850 mg, 4.7 mmol) was dissolved in anhydrous MeOH (6.5 mL). A solution of hydrogen chloride was prepared by adding acetyl chloride (0.25 mL) to anhydrous MeOH (1.5 mL) at 0 °C, and this solution was added dropwise to the glucose reaction solution. The reaction was refluxed for 5 days while under nitrogen before the MeOH was evaporated to yield the crude product. The product was recrystallized from EtOH as a white crystalline solid, which contained approximately 5% of the β-anomer. The product was recrystallized from EtOH again to yield the pure α-anomer of the product as white crystals (414 mg, 2.13 mmol, 45% yield). mp (EtOH) 167.2–168.1 °C (lit.64 mp (EtOH) 161–163 °C). [α]D23 –168.4 (c = 1.00, MeOH) [lit.64 [α]D –161 (c = 1.0, MeOH)]. 1H NMR (CD3OD, 400 MHz): δ 4.67 (d, J = 3.8 Hz, 1H, H-1), 3.81 (dd, J = 11.8, 2.4 Hz, 1H, H-6), 3.67 (dd, J = 11.8, 5.8 Hz, 1H, H-6), 3.61 (t, J = 9.2 Hz, 1H, H-3), 3.55–3.50 (m, 1H, H-5), 3.41 (s, 3H, OMe), 3.38 (dd, J = 9.7, 3.8 Hz, 1H, H-2), 3.27 (dd, J = 10.0, 9.0 Hz, 1H, H-4). 13C NMR (CD3OD, 100 MHz): δ 101.2 (C-1), 75.1 (C-3), 73.54 (C-5), 73.53 (C-2), 71.8 (C-4), 62.7 (C-6), 55.5 (OMe).
Methyl 3-O-Benzyl-4,6-O-benzylidene-α-l-glucopyranoside (12)
Methyl α-l-glucopyranoside (8) (93 mg, 0.480 mmol) was dissolved in dry pyridine (1 mL) and put under argon. To this solution, trimethylsilyl chloride (0.30 mL, 2.39 mmol, 5 equiv) was added dropwise. The solution was allowed to stir for 22 h at room temperature. The reaction was then diluted with EtOAc and washed with water (2 × 30 mL). The organic phase was dried over MgSO4 and concentrated in vacuo to yield the persilylated glucopyranoside crude product. The persilylated product was not purified, but 1H NMR was used to confirm that the reaction had proceeded to completion. The persilylated product was dissolved and co-evaporated twice with toluene before being dissolved in dry DCM (0.7 mL) and put under argon. To this solution, benzaldehyde (0.15 mL, 1.42 mmol, 3 equiv) was added, and the reaction was cooled in an ice bath. To this chilled solution, iron(III) chloride hexahydrate (3.4 mg, 0.013 mg, 0.026 equiv) dissolved in MeCN (0.12 mL) was added dropwise. Triethylsilane (0.08 mL, 0.528 mmol, 1.1 equiv) was then added dropwise, and the reaction was allowed to warm to room temperature and stir for 1.5 h. After this time, the solution was diluted with EtOAc (50 mL), washed with sat. NaHCO3 solution (50 mL), and extracted from the aqueous phase twice more with EtOAc (2 × 30 mL). The combined organic phases were dried over MgSO4 and concentrated in vacuo to yield the crude product. The product was purified with flash chromatography (petroleum ether/EtOAc, 0–100%) to yield the pure product as a white solid (140.7 mg, 0.378 mmol, 79% yield). mp 184.0–185.3 °C. [α]D21 –87.5 (c = 1.00, CHCl3). 1H NMR (CDCl3, 400 MHz): δ 7.51–7.27 (m, 10H, Ar), 5.57 (s, 1H, H-7), 4.97 (d, J = 11.6 Hz, 1H, CH2Ph), 4.82 (d, J = 3.9 Hz, 1H, H-1), 4.79 (d, J = 11.6 Hz, 1H, CH2Ph), 4.30 (dd, J = 4.3, 9.8 Hz, 1H, H-6), 3.87–3.71 (m, 4H, H-2, H-3, H-5, H-6), 3.65 (t, J = 9.2 Hz, 1H, H-4), 3.45 (s, 3H, OMe), 2.29 (d, J = 7.2 Hz, 1H, OH). 13C NMR (CDCl3, 100 MHz): δ 138. 6 (Ar), 137.5 (Ar), 129.1 (Ar), 128.6 (Ar), 128.4 (Ar), 128.2 (Ar), 127.9 (Ar), 126.2 (Ar), 101.4 (C-7), 100.0 (C-1), 82.1 (C-4), 79.0 (C-2 or 3 or 5), 75.0 (CH2Ph), 72.6 (C-2 or 3 or 5), 69.2 (C-6), 62.7 (C-2 or 3 or 5), 55.6 (OMe). HRMS (ESI) m/z: [M + Na]+ calcd for C21H24O6, 395.14651; found, 395.14658.
Methyl 3-O-Benzyl-α-l-glucopyranoside (13)
Methyl 3-O-benzyl-4,6-O-benzylidene-α-l-glucopyranoside (12) (135.7 mg, 0.364 mmol) was dissolved in MeOH (3 mL). To this solution, water (0.15 mL) and 1 M HCl(aq) (0.3 mL) were added. The reaction was heated to reflux for 3 h. After this time, the reaction was quenched with the addition of NaHCO3(aq) (25.2 mg in 5 mL of water). The solution was concentrated in vacuo, and the residue was dissolved and co-evaporated with toluene twice to yield the crude product. The product was purified with flash chromatography (petroleum ether/EtOAc, 0–100%) to yield the pure product as a white solid (96.3 mg, 0.339 mmol, 93% yield). mp 91.2–93.8 °C. [α]D22 –89.5 (c = 0.93, CHCl3). 1H NMR (CDCl3, 400 MHz): δ 7.39–7.29 (m, 5H, Ar), 5.03 (d, J = 11.5 Hz, 1H, CH2Ph), 4.76 (d, J = 3.9 Hz, 1H, CH2Ph), 4.73 (d, J = 11.5 Hz, 1H, H-1), 3.88–3.75 (m, 2H, H-6 ×2), 3.70–3.52 (m, 4H, H-2, H-3, H-4, H-5), 3.44 (s, 3H, OMe), 2.30 (d, J = 2.4 Hz, 1H, OH), 2.14 (d, J = 9.2 Hz, 1H, OH), 1.93 (dd, J = 5.7, 7.2 Hz, 1H, OH). 13C NMR (CDCl3, 100 MHz): δ 138.6 (Ar), 128.8 (Ar), 128.1 (Ar), 99.7 (C-1), 82.8 (C-3), 75.1 (CH2Ph), 72.9 (C-2), 71.1 (C-5), 70.2 (C-4), 62.5 (C-6), 55.5 (OMe). HRMS (ESI) m/z: [M + Na]+ calcd for C14H20O6, 307.11521; found, 307.11515.
Methyl 3-O-Benzyl-α-l-glucopyranoside 2,4,6-Tris(dibenzyl phosphate) (14)
Methyl 3-O-benzyl-α-l-glucopyranoside (13) (96.3 mg, 0.339 mmol) was dissolved in dry DCM (4 mL), and the solution was put under argon. 5-Phenyl-1H-tetrazole (297 mg, 2.03 mmol, 6 equiv) was added to the solution followed by dibenzyl diisopropylphosphoramidite (0.55 mL, 1.52 mmol, 4.5 equiv). The reaction was allowed to stir at room temperature overnight. The next day, after the confirmation of successful phosphitylation with 31P NMR, the reaction flask was cooled to −78 °C, and mCPBA (502 mg, 70% purity, 2.03 mmol, 6 equiv) was added. The reaction was allowed to stir at room temperature for 10 min before the solution was diluted with EtOAc (50 mL), washed with 10% Na2SO3 solution (2 × 30 mL), dried over MgSO4, and concentrated to yield the crude product. The crude product was purified with flash chromatography (petroleum ether/EtOAc, 0–100%) to yield the pure product as a colorless oil (144.2 mg, 0.135 mmol, 40% yield). [α]D21 –38.8 (c = 1.01, CHCl3). 1H NMR (CDCl3, 400 MHz): δ 7.34–7.08 (m, 35H, Ar), 5.03 (s, 2H, CH2Ph ×2), 5.01 (s, 2H, CH2Ph ×2), 4.98 (d, J = 3.2 Hz, 1H, CH2Ph), 4.96 (d, J = 3.2 Hz, 1H, CH2Ph), 4.94–4.85 (m, 6H, H-1, CH2Ph ×5), 4.80–4.75 (m, 3H, CH2Ph ×3), 4.42–4.18 (m, 4H, H-2, H-3, H-6 ×2), 3.98 (t, J = 9.4 Hz, 1H, H-4), 3.86 (dd, J = 10.0, 5.2 Hz, 1H, H-5), 3.28 (s, 3H, OMe). 31P NMR (CDCl3, 162 MHz): δ −0.99, −1.74, −1.86. 13C NMR (CDCl3, 100 MHz): δ 138.1 (Ar), 136.0 (Ar), 135.9 (Ar), 135.8–135.6 (m, Ar), 128.7–128.5 (m, Ar), 128.3 (Ar), 128.0 (m, Ar), 127.9 (Ar), 127.7 (Ar), 127.5 (Ar), 97.6 (C-1), 78.4–78.3 (m, C-4), 76.8–76.7 (m, C-2 or 3), 75.1 (C-2 or 3, CH2Ph), 69.8–69.4 (m, CH2Ph), 69.0–68.9 (m, C-5), 66.1 (d, J = 5.1 Hz, C-6), 55.7 (OMe). HRMS (ESI) m/z: [M + H]+ calcd for C56H59O15P3, 1065.31396; found, 1065.31280.
Methyl α-l-Glucopyranoside 2,4,6-Trisphosphate Triethylammonium Salt (3)
Methyl 3-O-benzyl-α-l-glucopyranoside 2,4,6-tris(dibenzyl phosphate) (14) (141.9 mg, 0.133 mmol) was dissolved in MeOH (7.1 mL). Ultrapure water (0.71 mL) was added dropwise to the solution, ensuring that the precipitate formed upon addition was able to dissolve back into solution. Pd(OH)2/C (20%, ≥50% wet, 71.0 mg) was added to the solution, and the reaction flask was flushed with hydrogen. The reaction was allowed to stir at room temperature for 24 h after which the catalyst was filtered off and the collected filtrate was evaporated to yield the product as a free acid. No purification steps were deemed to be necessary, but triethylamine was added to sharpen the 31P NMR signals and to convert the product from the free acid into the triethylammonium salt. The product was concentrated in vacuo, lyophilized, and collected as a colorless glass (96.1 mg, 0.120 mmol, 90% yield). [α]D22 –46.5 (c = 0.88, MeOH). 1H NMR (CD3OD, 400 MHz): δ 4.90 (d, J = 3.0 Hz, 1H, H-1), 4.29–4.18 (m, 2H, H-3, H-6), 4.05–3.97 (m, 3H, H-2, H-4, H-6), 3.68 (d, J = 10.0 Hz, 1H, H-5), 3.38 (s, 3H, OMe), 3.14 (q, J = 7.3 Hz, approx. 18H, TEA CH2CH3), 1.30 (t, 7.3 Hz, approx. 27H, TEA CH2CH3). 31P NMR (CD3OD, 162 MHz): δ 1.90, 1.71, 1.28. 13C NMR (CD3OD, 100 MHz): δ 100.2 (d, J = 4.0 Hz, C-1), 76.3 (d, J = 5.1 Hz, C-2 or 4), 74.6 (d, J = 4.0 Hz, C-2 or 4), 73.5 (d, J = 4.9 Hz, C-3), 71.0–71.2 (m, C-5), 63.9 (d, J = 4.7 Hz, C-6), 55.6 (OMe), 46.9 (TEA CH2CH3), 9.4 (TEA CH2CH3). HRMS (ESI) m/z: [M – H]− for C7H17O15P3, 432.97075; found, 432.97065.
Methyl 4,6-O-Benzylidene-α-l-glucopyranoside (9)
In a version of the Tseberlidis et al.65 method, methyl α-l-glucopyranoside (8) (100 mg, 0.514 mmol) was suspended in dry MeCN (1.7 mL) and put under a nitrogen atmosphere. To this suspension, benzaldehyde dimethyl acetal (0.24 mL, 1.55 mmol, 3 equiv) and catalytic camphor-10-sulfonic acid (1.6 mg, 0.0068 mmol) were added, and the reaction was allowed to stir at room temperature overnight. After 24 h, the reaction was neutralized with a few drops of triethylamine and evaporated to yield the crude product as a white crystalline solid. The crude product was purified through flash chromatography (petroleum ether/EtOAc, 0–100%), and the pure product was collected as a white solid (132.6 mg, 0.470 mmol, 91% yield). mp 163.1–164.4 °C (lit.64 mp 161–162 °C). [α]D21 –110.5 (c = 0.69, CDCl3) [lit.64 [α]D –95 (c = 1.0, MeOH)]. 1H NMR (CDCl3, 400 MHz): δ 7.51–7.48 (m, 2H, Ar), 7.40–7.34 (m, 3H, Ar), 5.53 (s, 1H, H-7), 4.79 (d, J = 4.0 Hz, 1H, H-1), 4.29 (dd, J = 9.6, 4.3 Hz, 1H, H-6), 3.93 (td, J = 9.3, 2.2 Hz, 1H, H-3), 3.84–3.78 (m, 1H, H-5), 3.75 (q, J = 10.3 Hz, 1H, H-6), 3.63 (td, J = 9.3, 3.9 Hz, 1H, H-2), 3.49 (t, J = 9.3 Hz, 1H, H-4), 3.46 (s, 3H, OMe), 2.76 (d, J = 2.2 Hz, 1H, OH), 2.30 (d, J = 9.5 Hz, 1H, OH). 13C NMR (CDCl3, 100 MHz): δ 137.2 (Ar), 129.4 (Ar), 128.5 (Ar), 126.4 (Ar), 102.1 (C-7), 99.9 (C-1), 81.1 (C-4), 73.0 (C-2), 72.0 (C-3), 69.1 (C-6), 62.5 (C-5), 55.7 (OMe).
Methyl 4-O-Benzyl-α-l-glucopyranoside (10)
Methyl 4-O-benzyl-α-l-glucopyranoside was made as described for methyl 4-O-benzyl-α-d-glucopyranoside (21) using both method A to generate large amounts of impure product and method B to generate smaller amounts of pure product (26 mg, 37% yield). mp 128.5–132.0 (lit.66 mp 125–127 °C). [α]D25 –142.5 (c = 0.3, MeOH) [lit.66 [α]D –144.2 (c = 1.2, MeOH)]. 1H NMR (CDCl3, 400 MHz): δ 7.37–7.27 (m, 5H, Ar), 4.87 (d, J = 11.5 Hz, 1H, CH2Ph), 4.75 (d, J = 3.9 Hz, 1H, H-1), 4.72 (d, J = 11.4 Hz, 1H, CH2Ph), 3.86 (t, J = 9.2 Hz, 1H, H-3), 3.83 (dd, J = 11.8, 2.6 Hz, 1H, H-6), 3.75 (dd, J = 11.8, 3.8 Hz, H-6), 3.67–3.62 (m, 1H, H-5), 3.50 (dd, J = 3.9, 9.4 Hz, 1H, H-2), 3.45 (t, J = 9.4 Hz, 1H, H-4), 3.40 (s, 3H, OMe). 13C NMR (CDCl3, 100 MHz): δ 138.3 (Ar), 128.7 (Ar), 128.2 (Ar), 127.9 (Ar), 99.2 (C-1), 77.3 (C-4), 75.3 (C-3), 74.8 (CH2Ph), 72.9 (C-2), 70.9 (C-5), 62.1 (C-6), 55.5 (OMe).
Methyl 4-O-Benzyl-α-l-glucopyranoside 2,3,6-Tris(dibenzyl phosphate) (11)
Methyl 4-O-benzyl-α-l-glucopyranoside (10) (65.5 mg, 0.230 mmol) was dissolved in dry DCM (2 mL) and put under argon. 5-Phenyl-1H-tetrazole (202 mg, 1.38 mmol, 6 equiv) was added to the solution followed by dibenzyl diisopropylphosphoramidite (0.36 mL, 1.04 mmol, 4.5 equiv). The reaction was allowed to stir at room temperature overnight. The following day, the reaction was cooled to −78 °C, and mCPBA (70% pure, 342 mg, 1.38 mmol, 6 equiv) was added. The reaction was allowed to stir for 10 min at room temperature before it was diluted with EtOAc (50 mL) and washed twice with 10% Na2SO3 solution (2 × 30 mL). The organic layer was dried over MgSO4 and concentrated to yield the crude product. The residue was purified with flash chromatography (petroleum ether/EtOAc, 0–100%) to yield the pure product as a colorless oil (210.5 mg, 0.198 mmol, 86% yield). [α]D21 –42.3 (c = 1.06, CHCl3). 1H NMR (CDCl3, 400 MHz): δ 7.33–7.09 (m, 35H, Ar), 5.05–4.81 (m, 16H, H-1, H-3, H-6, CH2Ph ×13), 4.46 (d, J = 10.7 Hz, 1H, CH2Ph), 4.25 (ddd, J = 13.3, 6.2, 3.6 Hz, 1H, H-2), 4.19–4.16 (m, 1H, H-6), 3.75 (dq, J = 10.0, 2.3 Hz, 1H, H-5), 3.50 (t, J = 9.5 Hz, 1H, H-4), 3.24 (s, 3H, OMe). 31P NMR (CDCl3, 162 MHz): δ −0.76, −1.32, −2.01. 13C NMR (CDCl3, 100 MHz): δ 137.6 (Ar), 136.1–135.7 (m, Ar), 128.7–127.8 (m, Ar), 97.6 (C-1), 78.4 (dd, J = 8.7, 6.5 Hz, C-3), 76.2 (C-4), 75.3 (dd, J = 4.7, 3.2 Hz, C-2), 74.6 (CH2Ph), 69.8 (d, J = 5.6 Hz, CH2Ph), 69.6–69.4 (m, CH2Ph), 69.1 (d, J = 8.3 Hz, C-5), 65.8 (d, J = 5.6 Hz, C-6), 55.5 (OMe). HRMS (ESI) m/z: [M + H]+ calcd for C56H59O15P3, 1065.31396; found, 1065.31297.
Methyl α-l-Glucopyranoside 2,3,6-Trisphosphate Triethylammonium Salt (2)
Methyl 4-O-benzyl-α-l-glucopyranoside 2,3,6-tris(dibenzyl phosphate) (11) (60 mg, 0.056 mmol) was dissolved in MeOH (3 mL). To this solution, ultrapure water (0.3 mL) was added dropwise, ensuring that the white precipitate that formed returned to solution. Pd(OH)2/C (20%, ≥50% wet, 30 mg) was added to the solution, and the flask was flushed with hydrogen. The reaction was left to stir under hydrogen at room temperature overnight. The palladium catalyst was filtered off with a PTFE filter, and the solution was concentrated to yield the product as a free acid. No purification was deemed necessary. The free acid was converted to the triethylammonium salt through the addition of triethylamine to the free acid followed by concentration in vacuo. The product was lyophilized and collected as a colorless glass (44.5 mg, 0.056 mmol, 100% yield). [α]D21 –37.6 (c = 1.00, MeOH). 1H NMR (CD3OD, 400 MHz): δ 4.92 (d, J = 3.6 Hz, 1H, H-1), 4.35 (q, J = 8.7 Hz, 1H, H-3), 4.17 (ddd, J = 11.1, 5.2, 2.0 Hz, 1H, H-6), 4.07–3.98 (m, 2H, H-2, H-6), 3.73–3.68 (m, 1H, H-5), 3.55 (dd, J = 9.8, 8.7 Hz, 1H, H-4), 3.39 (s, 3H, OMe), 3.09 (q, J = 7.3 Hz, approx. 18H, TEA CH2CH3), 1.27 (t, J = 7.3 Hz, approx. 27H, TEA CH2CH3). 31P NMR (CD3OD, 162 MHz): δ 2.50, 1.53, 1.15. 13C NMR (CD3OD, 100 MHz): δ 100.5 (C-1), 78.5 (C-3), 75.5 (C-2), 72.4 (d, J = 8.7 Hz, C-5), 72.0 (C-4), 65.7 (d, J = 4.9 Hz, C-6), 55.4 (OMe), 47.0 (TEA CH2CH3), 9.4 (TEA CH2CH3). HRMS (ESI) m/z: [M – H]− calcd for C7H17O15P3, 432.97075; found, 432.97067.
2,6-Di-O-benzyl-d-glucopyranose (16)
Allyl 2,6-di-O-benzyl-d-glucopyranoside (15) (60 mg, 0.133 mmol) as synthesized in the method outlined by Jenkins and Potter30 was dissolved in dry MeOH (1.54 mL). To this solution, PdCl2 (6.2 mg, 0.03 mmol, 0.25 equiv) was added, and the reaction was allowed to stir at room temperature for 6 h with a drying tube affixed to the flask. After this time, the reaction was quenched with the addition of excess NaHCO3 and allowed to stir for 5 min before being filtered through Celite and concentrated to yield the crude product. The product of this reaction could not be successfully purified; although following phosphorylation (see below), the products could be successfully isolated.
2,6-Di-O-benzyl-α-d-glucopyranosyl 1,3,4-Tris(dibenzyl phosphate) (17) and 2,6-Di-O-benzyl-β-d-glucopyranosyl 1,3,4-Tris(dibenzyl phosphate) (18)
2,6-Di-O-benzyl-d-glucopyranose (16) (154.7 mg, 0.429 mmol) was added to dry DCM (4.5 mL). To this suspension, 5-phenyl-1H-tetrazole (376 mg, 2.58 mmol, 6 equiv) was added, followed by dibenzyl diisopropylphosphoramidite (0.67 mL, 1.93 mmol, 4.5 equiv). The reaction was allowed to stir under argon at room temperature overnight after which it was cooled to −78 °C and mCPBA (70% purity, 636 mg, 2.58 mmol, 6 equiv) was added. The reaction was then diluted with EtOAc (100 mL) and washed twice with 10% Na2SO3 solution (2 × 30 mL). The organic phase was dried over MgSO4 and concentrated to yield the crude product. The product was purified using flash chromatography (petroleum ether/EtOAc, 0–100%). The stereoisomers of the product were partially isolated and collected as colorless oils (total: 276.6 mg, 0.242 mmol, 56% yield; α-epimer: 70.4 mg, 0.062 mmol, 14% yield; β-epimer: 92.4 mg, 0.081 mmol, 19% yield; remaining unseparated mix of epimers: 113.8 mg, 0.100 mmol, 23% yield).
2,6-Di-O-benzyl-α-d-glucopyranosyl 1,3,4-Tris(dibenzyl phosphate) (17)
Rf (EtOAc), 0.63. [α]D20 +35.7 (c = 0.71, chloroform). 1H NMR (CDCl3, 400 MHz): δ 7.35–7.09 (m, 40H, Ar), 5.91 (dd, J = 3.3, 7.0 Hz, 1H, H-1), 5.07–4.81 (m, 13H, H-3, CH2Ph ×12), 4.72–4.61 (m, 3H, H-4, CH2Ph ×2), 4.44 (d, J = 12.0 Hz, 1H, CH2Ph), 4.31 (d, J = 12.0 Hz, 1H, CH2Ph), 3.96 (dq, J = 10.0, 1.6 Hz, 1H, H-5), 3.70 (dd, J = 11.2, 3.9 Hz, 1H, H-6), 3.65 (dt, J = 9.7, 3.1 Hz, 1H, H-2), 3.54 (dd, J = 11.2, 1.8 Hz, 1H, H-6). 31P NMR (CDCl3, 162 MHz): δ −1.66, −2.31, −2.50. 13C NMR (CDCl3, 100 MHz): δ 138.0 (Ar), 137.0 (Ar), 136.2–135.6 (m, Ar), 128.6–127.6 (m, Ar), 94.8 (d, J = 5.6 Hz, C-1), 77.9–77.8 (m, C-3), 77.0 (m, C-2), 73.7–73.6 (m, C-4), 73.3 (CH2Ph), 73.0 (CH2Ph), 71.6 (C-5), 70.0–69.9 (m, CH2Ph), 69.7–69.6 (m, CH2Ph), 69.5–69.4 (m, CH2Ph), 67.5 (C-6). HRMS (ESI) m/z: [M + Na]+ calcd for C62H63O15P3, 1163.32720; found, 1163.32495.
2,6-Di-O-benzyl-β-d-glucopyranosyl 1,3,4-Tris(dibenzyl phosphate) (18)
Rf (EtOAc), 0.57. [α]D21 +2.68 (c = 1.01, chloroform). 1H NMR (CDCl3, 400 MHz): δ 7.31–7.03 (m, 40H, Ar), 5.31 (t, J = 7.2 Hz, 1H, H-1), 5.04–4.87 (m, 11H, CH2Ph ×11), 4.81–4.60 (m, 5H, H-3, H-4, CH2Ph ×3), 4.42 (d, J = 12.0 Hz, 1H, CH2Ph), 4.31 (d, J = 12.0 Hz, 1H, CH2Ph), 3.77–3.55 (m , 4H, H-2, H-5, H-6 ×2). 31P NMR (CDCl3, 162 MHz): δ −1.90, −2.22, −2.59. 13C NMR (CDCl3, 100 MHz): δ 138.0 (Ar), 137.7 (Ar), 136.1–135.4 (m, Ar), 128.6–127.6 (m, Ar), 98.6 (C-1), 80.1 (m, C-3 or 4), 79.7–79.6 (m, C-2 or 5), 74.5 (d, J = 3.5 Hz, C-3 or 4), 74.1 (CH2Ph), 74.0–73.9 (m, C-2 or 5), 73.3 (CH2Ph), 70.0 (d, J = 5.8 Hz, CH2Ph), 69.8–69.7 (m, CH2Ph), 69.6–69.5 (m, CH2Ph), 68.1 (C-6). HRMS (ESI) m/z: [M + Na]+ calcd for C62H63O15P3, 1163.32720; found, 1163.32505.
α-d-Glucopyranosyl 1,3,4-Trisphosphate Triethylammonium Salt (6)
α-d-Glucopyranosyl 1,3,4-trisphosphate was prepared with 2,6-di-O-benzyl-α-d-glucopyranosyl 1,3,4-tris(dibenzyl phosphate) (17) (31.9 mg, 0.028 mmol) using the same hydrogenation method as described for the synthesis of β-d-glucopyranosyl 1,3,4-trisphosphate (7). The crude sodium salt of the product was purified by ion-pair column chromatography on RP-18 and lyophilized to yield the triethylamine salt of the product as a colorless glass (8.0 mg, 0.011 mmol, 39% yield). [α]D20 +33.9 (c = 0.97, methanol). 1H NMR (CD3OD, 400 MHz): δ 5.58 (dd, J = 7.0, 3.6 Hz, 1H, H-1), 4.45 (q, J = 9.0 Hz, 1H, H-3), 4.12 (q, J = 9.8 Hz, 1H, H-4), 3.98–3.91 (m, 2H, H-5, H-6), 3.76–3.72 (m, 1H, H-6), 3.61 (ddd, J = 9.5, 3.5, 2.4 Hz, 1H, H-2), 3.16 (q, J = 7.3 Hz, approx. 18H, TEA CH2CH3), 1.31 (t, J = 7.3 Hz, approx. 27H, TEA CH2CH3). 31P NMR (CD3OD, 162 MHz): δ 2.06, 2.06, −0.62. 13C NMR (CD3OD, 100 MHz): δ 96.3 (d, J = 5.4 Hz, C-1), 79.2–79.1 (m, C-3), 73.8–73.6 (m, C-2, C-4, C-5), 62.1 (C-6), 47.3 (TEA CH2CH3), 9.2 (TEA CH2CH3). HRMS (ESI) m/z: [M – H]− calcd for C6H15O15P3, 418.9551; found, 418.95422.
β-d-Glucopyranosyl 1,3,4-Trisphosphate Triethylammonium Salt (7)
2,6-Di-O-benzyl-β-d-glucopyranosyl 1,3,4-tris(dibenzyl phosphate) (18) (23.3 mg, 0.020 mmol) was dissolved in MeOH (1.5 mL). To this solution, ultrapure water (0.15 mL) was added dropwise, ensuring that the precipitate that formed upon addition returned to solution. NaHCO3 (5.15 mg, 0.061 mmol, 3 equiv) was then added, followed by Pd(OH)2/C (20%, ≥50% wet, 11.7 mg). The reaction flask was flushed with hydrogen and left to stir at room temperature for 24 h. The catalyst was then filtered off, and the collected filtrate was evaporated to yield the crude product as a sodium salt. The product was purified by ion-pair column chromatography on RP-18 and lyophilized to yield the triethylamine salt of the product as a colorless glass (6.7 mg, 0.008 mmol, 40% yield). [α]D21 +3.76 (c = 0.61, methanol). 1H NMR (CD3OD, 400 MHz): δ 4.98 (t, J = 7.6 Hz, 1H, H-1), 4.23 (q, J = 8.8 Hz, 1H, H-3), 4.08 (q, J = 9.8 Hz, 1H, H-4), 3.89 (dd, J = 12.7, 4.4 Hz, 1H, H-6), 3.84 (dd, J = 12.7, 2.1 Hz, 1H, H-6), 3.43–3.38 (m, 2H, H-2, H-5), 3.13 (q, J = 7.0 Hz, approx. 18H, TEA CH2CH3), 1.29 (t, J = 7.2 Hz, approx. 27H, TEA CH2CH3). 31P NMR (CD3OD, 162 MHz): δ 1.38, 1.06, −0.77. 13C NMR (CD3OD, 125 MHz): δ 99.3 (C-1), 81.4 (C-3), 77.9 (C-2 or 5), 76.2 (C-2 or 5), 73.8 (C-4), 62.4 (C-6), 47.2 (TEA CH2CH3), 9.3 (TEA CH2CH3). HRMS (ESI) m/z:[M – H]− calcd for C6H15O15P3, 418.9551; found, 418.95425.
Methyl 4-O-Benzyl-α-d-glucopyranoside (21)
Method A
In a version of the Daragics et al.67 method, methyl 4,6-O-benzylidene-α-d-glucopyranoside (100 mg, 0.354 mmol) was dissolved in dry DCM (5.3 mL) and put under argon. The solution was cooled in an ice bath, and borane/THF (1 M, 1.8 mL, 1.77 mmol, 5 equiv) was added, followed by a solution of AlCl3 (94.4 mg, 0.708 mmol, 2 equiv) in dry diethyl ether (0.9 mL). The solution was allowed to gradually warm to room temperature and then stir for 24 h. After this time, the reaction was quenched with the addition of triethylamine (0.2 mL) followed by MeOH (0.9 mL). The reaction solution was concentrated in vacuo to form a solid residue. This residue was dissolved in DCM (50 mL) and washed with 1 M HCl(aq), sat. NaHCO3(aq), and water. The combined aqueous washes were also extracted three times with EtOAc, and the organic layers were combined, dried over MgSO4, and concentrated to yield the crude product. The crude product was purified through silica column chromatography using petroleum ether and EtOAc. It should be noted that this product was not entirely pure as a very small amount of methyl 6-O-benzyl-α-d-glucopyranoside was generated as well. This regioisomer could not be separated from the desired product (although separation of the regioisomers post-phosphorylation was achievable).
Method B
Using a version of the Shie et al.37 procedure, methyl 4,6-O-benzylidene α-d-glucopyranoside (100 mg, 0.355 mmol) was added to borane/THF (1 M, 1.8 mL, 1.8 mmol, 5 equiv), and the reaction was put under argon. The solution was allowed to stir for 10 min before lanthanum triflate (31.2 mg, 0.053 mmol, 0.15 equiv) was added, and the reaction was allowed to stir at room temperature for a week. The reaction was then cooled to 0 °C, and the reaction was quenched with triethylamine (0.5 mL, 1 equiv) followed by MeOH (0.7 mL). The reaction was concentrated in vacuo and co-evaporated with MeOH twice before the crude product was isolated as a white solid. The product was purified with flash chromatography ((1) petroleum ether/EtOAc, 0–100% and (2) DCM/EtOAc, 0–100%). As impurities were still present, an aqueous workup was carried out. The product was dissolved in EtOAc and washed with 1 M HCl(aq), sat. NaHCO3, and water. The aqueous washes were extracted with EtOAc again, and the organic phases were combined, dried over MgSO4, and concentrated to yield the pure product as a white solid (30.8 mg, 0.108 mmol, 31% yield). mp 123.9–129.0 °C (lit.68 mp 126–127 °C). [α]D21 +116.2 (c = 1.47, CHCl3) [lit.68 [α]D +154.1 (c = 1, CHCl3)]. 1H NMR (CDCl3, 400 MHz): δ 7.36–7.28 (m, 5H, Ar), 4.86 (d, J = 11.4 Hz, 1H, CH2Ph), 4.75 (d, J = 3.9 Hz, 1H, H-1), 4.72 (d, J = 11.4 Hz, 1H, CH2Ph), 3.86 (t, J = 9.2 Hz, 1H, H-3), 3.83 (dd, J = 11.6, 2.6 Hz, 1H, H-6), 3.75 (dd, J = 11.9, 3.6 Hz, H-6), 3.63 (dt, J = 9.8, 3.3 Hz, 1H, H-5), 3.51 (brs, 1H, H-2), 3.45 (t, J = 9.4 Hz, 1H, H-4), 3.39 (s, 3H, OMe). 13C NMR (CDCl3, 100 MHz): δ 138.3 (Ar), 128.7 (Ar), 128.2 (Ar), 128.1 (Ar), 99.2 (C-1), 77.2 (C-4), 75.1 (C-3), 74.8 (CH2Ph), 72.8 (C-2), 70.9 (C-5), 62.0 (C-6), 55.5 (OMe).
Methyl 4-O-Benzyl-α-d-glucopyranoside 2,3,6-Tris(dibenzyl phosphate) (22)
Methyl 4-O-benzyl-α-d-glucopyranoside (21) (50 mg, 0.176 mmol) was dissolved in dry DCM (2 mL), and the solution was put under argon. 5-Phenyl-1H-tetrazole (154 mg, 1.06 mmol, 6 equiv) was added to the solution, followed by dibenzyl diisopropylphosphoramidite (0.27 mL, 0.792 mmol, 4.5 equiv). The reaction was allowed to stir at room temperature overnight. The following day, the reaction was cooled to −78 °C, and mCPBA (70% pure, 261 mg, 1.06 mmol, 6 equiv) was added. The reaction was allowed to stir for 10 min at room temperature before it was diluted with EtOAc (50 mL) and washed twice with 10% Na2SO3 solution (2 × 30 mL). The organic layer was dried over MgSO4 and concentrated to yield the crude product. The product was purified with flash chromatography ((1) petroleum ether/EtOAc, 0–100% and (2) DCM/EtOAc, 0–100%). The pure product was collected as a colorless oil (81.9 mg, 0.077 mmol, 44% yield). [α]D22 +35.7 (c = 1.00, CHCl3). 1H NMR (CDCl3, 400 MHz): δ 7.33–7.09 (m, 35H, Ar), 5.05–4.82 (m, 16H, H-1, H-3, H-6, CH2Ph ×13), 4.47 (d, J = 10.7 Hz, 1H, CH2Ph), 4.26 (ddd, J = 9.6, 6.2, 3.6 Hz, 1H, H-2), 4.19–4.16 (m, 1H, H-6), 3.76 (dq, J = 9.6, 2.7 Hz, 1H, H-5), 3.50 (t, J = 9.4 Hz, 1H, H-4), 3.25 (s, 3H, OMe). 31P NMR (CDCl3, 162 MHz): δ −0.76, −1.32, −2.01. 13C NMR (CDCl3, 100 MHz): δ 137.6 (Ar), 136.1–135.7 (m, Ar), 128.7–128.0 (m, Ar), 97.6 (C-1), 78.5–78.4 (m, C-3), 76.2 (C-4), 75.4–75.3 (m, C-5), 74.6 (CH2Ph), 69.8 (d, J = 5.6 Hz, CH2Ph), 69.6–69.4 (m, CH2Ph), 69.1 (d, J = 8.1 Hz, C-5), 65.8 (d, J = 5.5 Hz, C-6), 55.5 (OMe); HRMS (ESI) m/z: [M + H]+ calcd for C56H59O15P3, 1065.31396; found, 1065.31364.
Methyl α-d-Glucopyranoside 2,3,6-Trisphosphate Triethylammonium Salt (4)
Methyl 4-O-benzyl-α-d-glucopyranoside 2,3,6-tris(dibenzyl phosphate) (22) (55.5 mg, 0.052 mmol) was dissolved in MeOH (2.8 mL). To this solution, ultrapure water (0.28 mL) was added dropwise, ensuring that the white precipitate that formed returned to solution. Pd(OH)2/C (20%, ≥50% wet, 27.8 mg) was added to the solution, and the flask was flushed with hydrogen. The reaction was left to stir under hydrogen at room temperature overnight. The palladium catalyst was filtered off with a PTFE filter, and the solution was concentrated to yield the product as a free acid. No purification was deemed necessary. The free acid was converted to the triethylammonium salt through the addition of triethylamine to the free acid followed by concentration in vacuo. The product was lyophilized and collected as a colorless glass (24.8 mg, 0.034 mmol, 65% yield). [α]D22 +38.2 (c = 1.00, MeOH). 1H NMR (CD3OD, 400 MHz): δ 4.91 (d, J = 3.6 Hz, 1H, H-1), 4.35 (q, J = 8.7 Hz, 1H, H-3), 4.17 (ddd, J = 11.0, 5.2, 2.0 Hz, 1H, H-6), 4.07–3.98 (m, 2H, H-2, H-6), 3.72–3.68 (m, 1H, H-5), 3.54 (dd, J = 9.8, 8.8 Hz, 1H, H-4), 3.39 (s, 3H, OMe), 3.11 (q, J = 7.3 Hz, approx. 18H, TEA CH2CH3), 1.28 (t, J = 7.3 Hz, approx. 27H, TEA CH2CH3). 31P NMR (CD3OD, 162 MHz): δ 2.21, 1.25, 0.94. 13C NMR (CD3OD, 100 MHz): δ 100.2 (C-1), 78.5 (t, J = 5.7 Hz, C-3), 75.3 (t, J = 4.9 Hz, C-2), 72.2 (d, J = 8.3 Hz, C-5), 71.7 (C-4), 65.6 (d, J = 5.2 Hz, C-6), 55.5 (OMe), 47.2 (TEA, CH2CH3), 9.1 (TEA, CH2CH3). HRMS (ESI) m/z: [M – H]− calcd for C7H17O15P3, 432.97075; found, 432.97076.
Methyl 3-O-Benzyl-4,6-O-benzylidene-α-d-glucopyranoside (23)
As described by Bourdreux et al.,3823 was synthesized using methyl α-d-glucopyranoside (100 mg, 0.515 mmol). The crude product was purified with flash chromatography (petroleum ether/EtOAc, 0–100%), and the pure product was collected as a white solid (143 mg, 0.384 mmol, 74% yield). mp 178.4–181.0 °C (lit.69 mp 184 °C). [α]D22 +87.7 (c = 0.70, CHCl3) [lit.69 [α]D +77 (c = 0.91, CHCl3)]. 1H NMR (CDCl3, 400 MHz): δ 7.52–7.27 (m, 10H, Ar), 5.57 (s, 1H, H-7), 4.97 (d, J = 11.6 Hz, 1H, CH2Ph), 4.82 (d, J = 3.8 Hz, 1H, H-1), 4.79 (d, J = 11.6 Hz, 1H, CH2Ph), 4.30 (dd, J = 4.3, 9.8 Hz, 1H, H-6), 3.87–3.71 (m, 4H, H-2, H-3, H-5, H-6), 3.65 (t, 1H, J = 9.1 Hz, H-4), 3.45 (s, 3H, OMe), 2.29 (d, 1H, J = 7.4 Hz, OH). 13C NMR (CDCl3, 100 MHz): δ 138.6 (Ar), 137.4 (Ar), 129.1 (Ar), 128.5 (Ar), 128.4 (Ar), 128.1 (Ar), 127.9 (Ar), 126.1 (Ar), 101.4 (H-7), 100.0 (C-1), 82.1 (C-4), 79.0 (C-2 or 3 or 5), 74.9 (CH2Ph), 72.5 (C-2 or 3 or 5), 69.1 (C-6), 62.7 (C-2 or 3 or 5), 55.5 (OMe).
Methyl 3-O-Benzyl-α-d-glucopyranoside (24)
As described by Boettcher et al.,7024 was synthesized using methyl 4,6-O-benzylidene-3-O-benzyl-α-d-glucopyranoside (23) (120 mg, 0.322 mmol). The product was purified with flash chromatography (petroleum ether/EtOAc, 0–100%) to yield the pure product as a white solid (75.7 mg, 0.266 mmol, 83% yield). mp 92.1–93.9 °C (lit.71 mp 85–86 °C). [α]D23 +89.9 (c = 1.05, CHCl3) [lit.71 [α]D +95.1 (c = 1.16, CHCl3)]. 1H NMR (CDCl3, 400 MHz): δ 7.39–7.27 (m, 5H, Ar), 4.99 (d, J = 11.6, 1H, CH2Ph), 4.74 (d, J = 11.6 Hz, 1H, CH2Ph), 4.73 (d, J = 3.7 Hz, 1H, H-1), 3.82–3.74 (m, 2H, H-6 ×2), 3.67–3.52 (m, 4H, H-2, H-3, H-4, H-5), 3.42 (s, 3H, OMe), 2.74 (d, J = 2.5 Hz, 1H, OH), 2.32 (d, J = 8.7 Hz, 1H, OH), 2.27 (t, J = 6.4 Hz, 1H, OH). 13C NMR (CDCl3, 100 MHz): δ 138.6 (Ar), 128.7 (Ar), 128.10 (Ar), 128.07 (Ar), 99.7 (C-1), 82.8 (C-3), 75.1 (CH2Ph), 72.9 (C-2), 71.2 (C-5), 70.1 (C-4), 62.3 (C-6), 55.5 (OMe).
Methyl 3-O-Benzyl-α-d-glucopyranoside 2,4,6-Tris(dibenzyl phosphate) (25)
Methyl 3-O-benzyl-α-d-glucopyranoside (24) (51.9 mg, 0.183 mmol) was dissolved in dry DCM (2 mL), and the solution was put under argon. 5-Phenyl-1H-tetrazole (160 mg, 1.10 mmol, 6 equiv) was added to the solution, followed by dibenzyl diisopropylphosphoramidite (0.30 mL, 0.82 mmol, 4.5 equiv). The reaction was allowed to stir at room temperature overnight. The next day, after the confirmation of successful phosphitylation with 31P NMR, the reaction flask was cooled to −78 °C, and mCPBA (270.7 mg, 70% purity, 1.10 mmol, 6 equiv) was added. The reaction was allowed to stir at room temperature for 10 min before the solution was diluted with EtOAc (50 mL), washed with 10% Na2SO3 solution (2 × 30 mL), dried over MgSO4, and concentrated to yield the crude product. The product was purified with flash chromatography (petroleum ether/EtOAc, 0–100%) to yield the pure product as a colorless oil (138.1 mg, 0.130 mmol, 71% yield). [α]D21 +41.4 (c = 0.61, CHCl3). 1H NMR (CDCl3, 400 MHz): δ 7.35–7.08 (m, 35H, Ar), 5.03 (s, 2H, CH2Ph ×2), 5.01 (s, 2H, CH2Ph ×2), 4.98 (d, J = 3.2 Hz, 1H, CH2Ph), 4.97 (d, J = 3.2 Hz, 1H, CH2Ph), 4.94–4.85 (m, 6H, H-1, CH2Ph ×5), 4.81–4.75 (m, 3H, CH2Ph ×3), 4.43–4.18 (m, 4H, H-2, H-3, H-6 ×2), 3.98 (t, J = 9.3 Hz, 1H, H-4), 3.86 (dd, J = 10.0, 5.2 Hz, 1H, H-5), 3.28 (s, 3H, OMe). 31P NMR (CDCl3, 162 MHz): δ −0.98, −1.73, −1.85. 13C NMR (CDCl3, 100 MHz): δ 138.1 (Ar), 136.0 (Ar), 135.9 (Ar), 135.8–135.6 (m, Ar), 128.7–128.5 (m, Ar), 128.3 (Ar), 128.0 (m, Ar), 127.8 (Ar), 127.7 (Ar), 127.5 (Ar), 97.6 (C-1), 78.3 (m, C-4), 76.7 (m, C-2 or 3), 75.0 (C-2 or 3, CH2Ph), 69.6 (m, CH2Ph), 69.4 (m, CH2Ph), 68.9 (m, C-5), 66.1 (m, C-6), 55.7 (OMe). HRMS (ESI) m/z: [M + H]+ calcd for C56H59O15P3, 1065.31396; found, 1065.31336.
Methyl α-d-Glucopyranoside 2,4,6-Trisphosphate Triethylammonium Salt (5)
Methyl 3-O-benzyl-α-d-glucopyranoside 2,4,6-tris(dibenzyl phosphate) (25) (132.4 mg, 0.124 mmol) was dissolved in MeOH (6.6 mL). Ultrapure water (0.66 mL) was added dropwise to the solution, ensuring that the precipitate formed upon addition was able to dissolve back into solution. Pd(OH)2/C (20%, ≥50% wet, 66.2 mg) was added to the solution, and the reaction flask was flushed with hydrogen. The reaction was allowed to stir at room temperature for 24 h after which the catalyst was filtered off and the collected filtrate was evaporated to yield the product as a free acid. No purification steps were deemed to be necessary, but triethylamine was added to sharpen the 31P NMR signals and to convert the product from the free acid into the triethylammonium salt. The product was concentrated in vacuo before being lyophilized and collected as a colorless glass (86 mg, 0.10 mmol, 94% yield). [α]D22 +39.3 (c = 0.43, MeOH). 1H NMR (CD3OD, 400 MHz): δ 4.91 (d, J = 2.8 Hz, 1H, H-1), 4.26–4.16 (m, 2H, H-3, H-6), 4.05–3.98 (m, 3H, H-2, H-4, H-6), 3.70 (brd, J = 9.8 Hz, 1H, H-5), 3.39 (s, 3H, OMe), 3.12 (q, J = 7.5 Hz, approx. 18H, TEA CH2CH3), 1.29 (t, J = 7.5 Hz, approx. 27H, TEA CH2CH3). 31P NMR (CD3OD, 162 MHz): δ 1.99, 1.75, 1.32. 13C NMR (CD3OD, 100 MHz): δ 100.2 (C-1), 76.3 (d, 5.6 Hz, C-2 or 4), 74.6 (d, 3.4 Hz, C-2 or 4), 73.5 (d, 5.5 Hz, C-3), 71.1 (m, C-5), 63.9 (C-6), 55.6 (OMe), 47.0 (TEA CH2CH3), (TEA, CH2CH3) 9.3. HRMS (ESI) m/z: [M – H]− calcd for C7H17O15P3, 432.97075; found, 432.97063.
HPLC
For analysis and stability experiments, the sugar phosphates were resolved by anion-exchange HPLC on a 3 × 250 mm CarboPac PA200 column (Dionex) fitted with 3 × 50 mm guard cartridge of the same material. Compounds were eluted with a gradient derived from buffer reservoirs containing water (A) and 0.6 M methanesulfonic acid (B) delivered at a flow rate of 0.4 mL min–1 according to the following schedule: time (min), % B; 0, 0; 20, 80; 21, 0; and 31, 0. Compounds were detected with the phosphate detection reagent of Phillippy and Bland.72 For this purpose, the column eluate was mixed in a mixing Tee with a solution of 0.1% (w/v) ferric nitrate nonahydrate in 2% (w/v) perchloric acid delivered at a flow rate of 0.2 mL min–1 and passed through a 0.192 mL internal volume knitted reaction coil before being transferred to a UV detector set at 290 nm. Typically, samples of 40 μL of 500 μM solutions in water were injected. Data were exported from the ChromNav2 software as x,y data files and redrawn in GraFit.v7.73
Biology Methods
Materials
HEK-293 cells with all three Ins(1,4,5)P3R subtypes disrupted using CRISPR/Cas9 technology (HEK-3KO)74 were from Kerafast (Boston, USA). Dulbecco’s modified Eagle’s medium/nutrient mixture F-12 with GlutaMAX (DMEM/F-12 GlutaMAX) and Mag-Fluo-4 AM were from Thermo Fisher. TransIT-LT1 transfection reagent was from GENEFLOW (Elmhurst, Lichfield, UK). Most chemicals and fetal bovine serum (FBS) were from Sigma-Aldrich (Gillingham, UK). Cyclopiazonic acid (CPA) was from Tocris (Bristol, UK). G418 was from Formedium (Norfolk, UK). Protease inhibitor cocktail tablets were from Roche. Half-area 96-well black-walled plates were from Greiner. Ins(1,4,5)P3 was from Enzo (Exeter, UK). [3H]-Ins(1,4,5)P3 was from PerkinElmer.
Cell Culture and Transfection
HEK cells were cultured in DMEM/F-12 GlutaMAX medium supplemented with 10% FBS (37 °C in 95% air and 5% CO2). Cells were either passaged or used for experiments when they reached confluence. HEK cells expressing only Ins(1,4,5)P3R1 (HEK-Ins(1,4,5)P3R1) were generated by transfecting HEK-3KO cells with the gene encoding rat Ins(1,4,5)P3R1 (lacking the S1 splice site)27 cloned into pcDNA3.1(−)/Myc-His B plasmid75 using the TransIT-LT1 reagent following the manufacturer’s instructions. To generate stable cell lines, cells were passaged 48 h after transfection in medium with G418 (1 mg mL–1). Selection was maintained for 2 weeks, and the medium was changed every 3 days. Monoclonal cell lines were selected by plating cells (∼1 cell well–1) into 96-well plates in medium containing G418 (1 mg mL–1). After 4 days, wells with only one cell were identified, and the cells were allowed to reach confluence. These cell lines were then expanded, and their expression of Ins(1,4,5)P3R1 was confirmed by western blot using an anti-Ins(1,4,5)P3R1 antibody.27
Ca2+-Release Assays
The free [Ca2+] within the lumen of the endoplasmic reticulum (ER) was measured using the low-affinity Ca2+ indicator Mag-Fluo-4.76,77 The ER of HEK-Ins(1,4,5)P3R1 cells was loaded with the Ca2+ indicator by incubating cells (2.4 × 107 cells mL–1, 1 h, 22 °C) in HEPES-buffered saline (HBS; 135 mM NaCl, 5.9 mM KCl, 11.6 mM HEPES, 1.5 mM CaCl2, 11.5 mM glucose, 1.2 mM MgCl2, pH 7.3) supplemented with BSA (1 mg mL–1), Pluronic F127 (0.4 mg mL–1), and Mag-Fluo-4 AM (20 μM). Cells were then suspended in the Ca2+-free cytosol-like medium (CLM: 20 mM NaCl, 140 mM KCl, 1 mM EGTA, 20 mM Pipes, 2 mM MgCl2, pH 7.0) and permeabilized with saponin (10 μg mL–1, 3 min, 37 °C). Permeabilized cells were centrifuged (650 × g, 3 min) and incubated in CLM (7 min, 37 °C) to allow the Ca2+ stores to empty. Cells were then centrifuged (650 × g, 3 min) and resuspended in CLM without Mg2+ but supplemented with 375 μM CaCl2 to give a final free [Ca2+] of 220 nM after the addition of 1.5 mM MgATP. Cells (∼4 × 105 well–1) were added to poly-l-lysine-coated half-area 96-well black-walled plates. Fluorescence was recorded at 20 °C at intervals of 1.44 s using a FlexStation III plate reader (Molecular Devices, Sunnyvale, CA, USA) with excitation and emission wavelengths of 485 and 520 nm, respectively. MgATP (1.5 mM) was added to initiate Ca2+ uptake, and when the ER had loaded to the steady state (∼2.5 min), cyclopiazonic acid (CPA, 10 μM) was added to inhibit the ER Ca2+ pump. Ins(1,4,5)P3 or other ligands were added after a further 60 s. The amount of Ca2+ release was calculated as a percentage of the fluorescence signal from fully loaded stores (Ffull) minus the signal from nonloaded stores (Ffull – Fempty). Results are presented as means ± SEM from 5 to 11 independent experiments, each run in duplicate.
[3H]-Ins(1,4,5)P3 Binding to Cerebellar Membranes
Cerebellar membranes, which are rich in Ins(1,4,5)P3R1, were prepared from the cerebella of adult Wistar rats. Frozen cerebella were homogenized at 4 °C in the homogenization medium (HM: 1 mM EDTA, 50 mM Tris, protease inhibitors, pH 8.3) supplemented with 100 mM NaCl. After centrifugation (130,000 × g, 1 h, 4 °C), the membranes were resuspended in HM (∼6 mg protein mL–1) and stored at −80 °C. Equilibrium competition binding assays were performed at 4 °C in a final volume of 500 μL of Tris/EDTA medium (TEM: 50 mM Tris, 1 mM EDTA, pH 8.3) with [3H]-Ins(1,4,5)P3 (19.3 Ci mmol–1, 1.5 nM), competing ligands, and 25 μL of membranes.27 After 5 min, during which equilibrium was attained, bound and free ligands were separated by centrifugation (20,000 × g, 5 min, 4 °C), and the pellet was then rinsed and resuspended in TEM (200 μL) before liquid scintillation counting (1 mL, Ecoscint A). Nonspecific binding, determined by the addition of 10 μM Ins(1,4,5)P3, was always <10% of total binding, and <10% of the added [3H]-Ins(1,4,5)P3 was bound. Results are presented as means ± SEM from three independent experiments without replicates.
Data Analysis
Equilibrium binding results and concentration–effect relationships were fitted to Hill equations (GraphPad Prism, version 5) from which −log IC50 (pIC50) and −log EC50 (pEC50) values were obtained. For equilibrium competition binding assays pKd values were calculated using the Cheng and Prusoff equation.78 Because pEC50 and pKd values are normally distributed, these results are presented as means ± SEM from n independent experiments. For comparisons of the ratios between mean values (EC50/Kd), statistical analyses compared the differences between their log values (pEC50 and pKd),79 with the SEM calculated as follows, assuming that the population variances are the same (confirmed using an F test)80
where sp is the estimate of the population variance
where s1 and s2 are the sample standard deviations, and n1 and n2 are the sample sizes. Although all analyses were performed using log values, for greater clarity, we present ratios as the antilogs of the means and the 95% confidence interval.
Statistical analysis used ANOVA followed by Bonferroni’s multiple comparison test (GraphPad Prism, version 5). P < 0.05 was considered significant.
EC39.4% release/Kd was calculated because some ligands did not fully release the Ins(1,4,5)P3-sensitive stores. The ratio was calculated using the concentration of each ligand that caused a release of 39.4% of the total content of the stores (which is the % released by Ins(1,4,5)P3 at its EC50).
Molecular Docking
The X-ray crystal structure of the N-terminal IBC of the Type 1 Ins(1,4,5)P3R in complex with Ins(1,4,5)P3 (PDB: 1N4K)4 was used for the molecular docking experiments of ligands (1–7). Compounds 1–7 were built and minimized using Chem3D version 15.1 and Mercury version 3.10. Docking methods were optimized by docking Ins(1,4,5)P3 into the IBC structure with GOLD81 version 5.6.1 to reproduce the bound Ins(1,4,5)P3 conformation. The most successful docking runs were achieved with two water molecules in the binding site (1139 and 1198) being allowed to toggle and spin while the remaining water molecules were removed.82 The lysine residues in the binding site (K412, K508, and K569) were permitted constrained movement and internal H-bonds were allowed. Compounds 2–7 were docked 100 times and scored using the GoldScore scoring function. The highest scoring solutions for 2, 3, 6, and 7 were exported, and figures were prepared using PyMOL (DeLano Scientific LLC). More details are given in Supporting Information SI-1.
Acknowledgments
B.V.L.P. (grant 101010) and C.W.T. (grant 101844) are Wellcome Trust Senior Investigators.
Glossary
Abbreviations
- AdA
adenophostin A
- ATP
adenosine triphosphate
- COSY
correlation spectroscopy
- CPA
cyclopiazonic acid
- mCPBA
meta-chloroperoxybenzoic acid
- cryo-EM
cryogenic electron microscopy
- CSA
camphorsulfonic acid
- DCM
dichloromethane
- EDTA
ethylenediaminetetraacetic acid
- ER
endoplasmic reticulum
- FBS
fetal bovine serum
- Gluc(3,4)P2
d-glucose 3,4-bisphosphate
- HBS
HEPES-buffered saline
- HEK
human embryonic kidney
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- HPLC
high-performance liquid chromatography
- HRMS
high-resolution mass spectrometry
- HSQC
heteronuclear single quantum coherence
- IBC
Ins(1,4,5)P3-binding core
- Ins(1,4,5)P3
d-myo-inositol 1,4,5-trisphosphate
- Ins(1,4,5)P3R
d-myo-inositol 1,4,5-trisphosphate receptor
- NMR
nuclear magnetic resonance
- SAR
structure–activity relationship
- THF
tetrahydrofuran
- TMS
tetramethylsilane
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.0c00215.
Overlays, molecular docking, and ligand interaction diagrams for 2, 3, 6, and 7; HPLC data for compounds 2–7; stability studies of compounds 6 and 7 in SI-1 (PDF)
NMR spectral data for all compounds in SI-2 (PDF)
Molecular formula strings for all compounds (CSV)
Data for 1N4K (PDB)
Molecular docking file for methyl α-l-glucopyranoside 2,3,6-trisphosphate (2) (PDB)
Molecular docking file for methyl α-l-glucopyranoside 2,4,6-trisphosphate (3) (PDB)
Molecular docking file for α-d-glucopyranosyl 1,3,4-trisphosphate (6) (PDB)
Molecular docking file for β-d-glucopyranosyl 1,3,4-trisphosphate (7) (PDB)
The authors declare no competing financial interest.
Supplementary Material
References
- Rossi A. M.; Taylor C. W. IP3 Receptors – Lessons from Analyses Ex Cellula. J. Cell Sci. 2018, 132, jcs222463. 10.1242/jcs.222463. [DOI] [PubMed] [Google Scholar]
- Foskett J. K.; White C.; Cheung K.-H.; Mak D.-O. D. Inositol Trisphosphate Receptor Ca2+ Release Channels. Physiol. Rev. 2007, 87, 593–658. 10.1152/physrev.00035.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berridge M. J. Inositol Trisphosphate and Calcium Signalling Mechanisms. Biochim. Biophys. Acta 2009, 1793, 933–940. 10.1016/j.bbamcr.2008.10.005. [DOI] [PubMed] [Google Scholar]
- Bosanac I.; Alattia J.-R.; Mal T. K.; Chan J.; Talarico S.; Tong F. K.; Tong K. I.; Yoshikawa F.; Furuichi T.; Iwai M.; Michikawa T.; Mikoshiba K.; Ikura M. Structure of the Inositol 1, 4, 5-Trisphosphate Receptor Binding Core in Complex with Its Ligand. Nature 2002, 420, 696–700. 10.1038/nature01268. [DOI] [PubMed] [Google Scholar]
- Sureshan K. M.; Riley A. M.; Thomas M. P.; Tovey S. C.; Taylor C. W.; Potter B. V. L. Contribution of Phosphates and Adenine to the Potency of Adenophostins at the IP3 Receptor: Synthesis of All Possible Bisphosphates of Adenophostin A. J. Med. Chem. 2012, 55, 1706–1720. 10.1021/jm201571p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo M.-D.; Velamakanni S.; Ishiyama N.; Stathopulos P. B.; Rossi A. M.; Khan S. A.; Dale P.; Li C.; Ames J. B.; Ikura M.; Taylor C. W. Structural and Functional Conservation of Key Domains in InsP3 and Ryanodine Receptors. Nature 2012, 483, 108–112. 10.1038/nature10751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paknejad N.; Hite R. K. Structural Basis for the Regulation of Inositol Trisphosphate Receptors by Ca2+ and IP3. Nat. Struct. Mol. Biol. 2018, 25, 660–668. 10.1038/s41594-018-0089-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potter B. V. L.; Lampe D. Chemistry of Inositol Lipid Mediated Cellular Signaling. Angew. Chem., Int. Ed. Engl. 1995, 34, 1933–1972. 10.1002/anie.199519331. [DOI] [Google Scholar]
- Best M. D.; Zhang H.; Prestwich G. D. Inositol Polyphosphates, Diphosphoinositol Polyphosphates and Phosphatidylinositol Polyphosphate Lipids: Structure, Synthesis, and Development of Probes for Studying Biological Activity. Nat. Prod. Rep. 2010, 27, 1403–1430. 10.1039/b923844c. [DOI] [PubMed] [Google Scholar]
- Saleem H.; Tovey S. C.; Rahman T.; Riley A. M.; Potter B. V. L.; Taylor C. W. Stimulation of Inositol 1,4,5-Trisphosphate (IP3) Receptor Subtypes by Analogues of IP3. PLoS One 2013, 8, e54877 10.1371/journal.pone.0054877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saleem H.; Tovey S. C.; Riley A. M.; Potter B. V. L.; Taylor C. W. Stimulation of Inositol 1,4,5-Trisphosphate (IP3) Receptor Subtypes by Adenophostin A and Its Analogues. PLoS One 2013, 8, e58027 10.1371/journal.pone.0058027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dohle W.; Su X.; Mills S. J.; Rossi A. M.; Taylor C. W.; Potter B. V. L. A Synthetic Cyclitol-Nucleoside Conjugate Polyphosphate Is a Highly Potent Second Messenger Mimic. Chem. Sci. 2019, 10, 5382–5390. 10.1039/c9sc00445a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas M. P.; Mills S. J.; Potter B. V. L. The “Other” Inositols and Their Phosphates: Synthesis, Biology, and Medicine (with Recent Advances in Myo-Inositol Chemistry). Angew. Chem., Int. Ed. 2016, 55, 1614–1650. 10.1002/anie.201502227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor C. W.; Da Fonseca P. C. A.; Morris E. P. IP3 Receptors: The Search for Structure. Trends Biochem. Sci. 2004, 29, 210–219. 10.1016/j.tibs.2004.02.010. [DOI] [PubMed] [Google Scholar]
- Safrany S. T.; Wojcikiewicz R. J. H.; Strupish J.; Nahorski S. R.; Dubreuil D.; Cleophax J.; Gero S. D.; Potter B. V. L. Interaction of Synthetic d-6-Deoxy-Myo-Inositol 1,4,5-Trisphosphate with the Ca2+-Releasing d-Myo-Inositol 1,4,5-Trisphosphate Receptor, and the Metabolic Enzymes 5-Phosphatase and 3-Kinase. FEBS Lett. 1991, 278, 252–256. 10.1016/0014-5793(91)80128-P. [DOI] [PubMed] [Google Scholar]
- Wilcox R. A.; Primrose W. U.; Nahorski S. R.; Challiss R. A. J. New Developments in the Molecular Pharmacology of the Myo-Inositol 1,4,5-Trisphosphate Receptor. Trends Pharmacol. Sci. 1998, 19, 467–475. 10.1016/S0165-6147(98)01260-7. [DOI] [PubMed] [Google Scholar]
- Saleem H.; Tovey S. C.; Molinski T. F.; Taylor C. W. Interactions of Antagonists with Subtypes of Inositol 1,4,5-Trisphosphate (IP3) Receptor. Br. J. Pharmacol. 2014, 171, 3298–3312. 10.1111/bph.12685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konieczny V.; Stefanakis J. G.; Sitsanidis E. D.; Ioannidou N.-A. T.; Papadopoulos N. V.; Fylaktakidou K. C.; Taylor C. W.; Koumbis A. E. Synthesis of Inositol Phosphate-Based Competitive Antagonists of Inositol 1,4,5-Trisphosphate Receptors. Org. Biomol. Chem. 2016, 14, 2504–2514. 10.1039/C5OB02623G. [DOI] [PubMed] [Google Scholar]
- Li W.; Schultz C.; Llopis J.; Tsien R. Y. Membrane-Permeant Esters of Inositol Polyphosphates, Chemical Syntheses and Biological Applications. Tetrahedron 1997, 53, 12017–12040. 10.1016/S0040-4020(97)00714-X. [DOI] [Google Scholar]
- Conway S. J.; Miller G. J. Biology-Enabling Inositol Phosphates, Phosphatidylinositol Phosphates and Derivatives. Nat. Prod. Rep. 2007, 24, 687–707. 10.1039/b407701f. [DOI] [PubMed] [Google Scholar]
- Li X.; Gu C.; Hostachy S.; Sahu S.; Wittwer C.; Jessen H. J.; Fiedler D.; Wang H.; Shears S. B. Control of XPR1-Dependent Cellular Phosphate Efflux by InsP8 Is an Exemplar for Functionally-Exclusive Inositol Pyrophosphate Signaling. Proc. Natl. Acad. Sci. U. S. A. 2020, 117, 3568–3574. 10.1073/pnas.1908830117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcox R. A.; Fauq A.; Kozikowski A. P.; Nahorski S. R. Defining the Minimal Structural Requirements for Partial Agonism at the Type I Myo-Inositol 1,4,5-Trisphosphate Receptor. FEBS Lett. 1997, 402, 241–245. 10.1016/S0014-5793(96)01540-2. [DOI] [PubMed] [Google Scholar]
- Keddie N. S.; Ye Y.; Aslam T.; Luyten T.; Bello D.; Garnham C.; Bultynck G.; Galione A.; Conway S. J. Development of Inositol-Based Antagonists for the d-Myo-Inositol 1,4,5-Trisphosphate Receptor. Chem. Commun. 2011, 47, 242–244. 10.1039/C0CC03003A. [DOI] [PubMed] [Google Scholar]
- Mills S. J.; Riley A. M.; Murphy C. T.; Bullock A. J.; Westwick J.; Potter B. V. L. Myo-Inositol 1,4,6-Trisphosphorothioate and Myo-Inositol 1,3,4-Trisphosphorothioate: New Synthetic Ca2+-Mobilising Partial Agonists at the Inositol 1,4,5-Trisphosphate Receptor. Bioorg. Med. Chem. Lett. 1995, 5, 203–208. 10.1016/0960-894X(95)00012-I. [DOI] [Google Scholar]
- Murphy C. T.; Riley A. M.; Mills S. J.; Lindley C. J.; Potter B. V. L.; Westwick J. Myo-Inositol 1,4,6-Trisphosphorothioate and Myo-Inositol 1,3,6-Trisphosphorothioate: Partial Agonists with Very Low Intrinsic Activity at the Platelet Myo-Inositol 1,4,5-Trisphosphate Receptor. Mol. Pharmacol. 2000, 57, 595–601. 10.1124/mol.57.3.595. [DOI] [PubMed] [Google Scholar]
- Bello D.; Aslam T.; Bultynck G.; Slawin A. M. Z.; Roderick H. L.; Bootman M. D.; Conway S. J. Synthesis and Biological Action of Novel 4-Position-Modified Derivatives of d-Myo-Inositol 1,4,5-Trisphosphate. J. Org. Chem. 2007, 72, 5647–5659. 10.1021/jo070611a. [DOI] [PubMed] [Google Scholar]
- Rossi A. M.; Riley A. M.; Tovey S. C.; Rahman T.; Dellis O.; Taylor E. J. A.; Veresov V. G.; Potter B. V. L.; Taylor C. W. Synthetic Partial Agonists Reveal Key Steps in IP3 Receptor Activation. Nat. Chem. Biol. 2009, 5, 631–639. 10.1038/nchembio.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills S. J.; Luyten T.; Erneux C.; Parys J. B.; Potter B. V. L. Multivalent Benzene Polyphosphate Derivatives Are Non-Ca2+-Mobilizing Ins(1,4,5)P3 Receptor Antagonists. Messenger 2012, 1, 167–181. 10.1166/msr.2012.1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandeput F.; Combettes L.; Mills S. J.; Backers K.; Wohlkönig A.; Parys J. B.; De Smedt H.; Missiaen L.; Dupont G.; Potter B. V. L.; Erneux C. Biphenyl 2,3′,4,5′,6-Pentakisphosphate, a Novel Inositol Polyphosphate Surrogate, Modulates Ca2+ Responses in Rat Hepatocytes. FASEB J. 2007, 21, 1481–1491. 10.1096/fj.06-7691com. [DOI] [PubMed] [Google Scholar]
- Jenkins D. J.; Potter B. V. L. A Ca2+-Mobilising Carbohydrate-Based Polyphosphate: Synthesis of 2-Hydroxyethyl α-d-Glucopyranoside 2′,3,4-Trisphosphate. Carbohydr. Res. 1996, 287, 169–182. 10.1016/0008-6215(96)00078-X. [DOI] [PubMed] [Google Scholar]
- Terauchi M.; Abe H.; Tovey S. C.; Dedos S. G.; Taylor C. W.; Paul M.; Trusselle M.; Potter B. V. L.; Matsuda A.; Shuto S. A Systematic Study of C-Glucoside Trisphosphates as Myo-Inositol Trisphosphate Receptor Ligands. Synthesis of β-C-Glucoside Trisphosphates Based on the Conformational Restriction Strategy. J. Med. Chem. 2006, 49, 1900–1909. 10.1021/jm051039n. [DOI] [PubMed] [Google Scholar]
- Shuto S.; Tatani K.; Ueno Y.; Matsuda A. Synthesis of Adenophostin Analogues Lacking the Adenine Moiety as Novel Potent IP3 Receptor Ligands: Some Structural Requirements for the Significant Activity of Adenophostin A. J. Org. Chem. 1998, 63, 8815–8824. 10.1021/jo980925l. [DOI] [Google Scholar]
- Van Straten N. C. R.; van der Marel G. A.; Van Boom J. H. An Expeditious Route to the Synthesis of Adenophostin A. Tetrahedron 1997, 53, 6509–6522. 10.1016/S0040-4020(97)00307-4. [DOI] [Google Scholar]
- Rosenberg H. J.; Riley A. M.; Correa V.; Taylor C. W.; Potter B. V. L. C-Glycoside Based Mimics of d-Myo-Inositol 1,4,5-Trisphosphate. Carbohydr. Res. 2000, 329, 7–16. 10.1016/S0008-6215(00)00175-0. [DOI] [PubMed] [Google Scholar]
- Moitessier N.; Chrétien F.; Chapleur Y.; Humeau C. Synthesis and Biological Activities of Inositol 1,4,5-Trisphosphate Mimics Related to Xylopyranosides. Tetrahedron Lett. 1995, 36, 8023–8026. 10.1016/0040-4039(95)01677-A. [DOI] [Google Scholar]
- Rossi A. M.; Riley A. M.; Potter B. V. L.; Taylor C. W. Adenophostins: High-Affinity Agonists of IP3 Receptors. Curr. Top. Membr. 2010, 66, 209–233. 10.1016/s1063-5823(10)66010-3. [DOI] [PubMed] [Google Scholar]
- Shie C.-R.; Tzeng Z.-H.; Wang C.-C.; Hung S.-C. Metal Trifluoromethanesulfonate Catalyzed Regioselective Reductive Ring Opening of Benzylidene Acetals. J. Chin. Chem. Soc. 2009, 56, 510–523. 10.1002/jccs.200900076. [DOI] [Google Scholar]
- Bourdreux Y.; Lemétais A.; Urban D.; Beau J.-M. Iron(III) Chloride-Tandem Catalysis for a One-Pot Regioselective Protection of Glycopyranosides. Chem. Commun. 2011, 47, 2146–2148. 10.1039/c0cc04398b. [DOI] [PubMed] [Google Scholar]
- Nurminen E.; Lönnberg H. Mechanisms of the Substitution Reactions of Phosphoramidites and Their Congeners. J. Phys. Org. Chem. 2004, 17, 1–17. 10.1002/poc.681. [DOI] [Google Scholar]
- Fylaktakidou K. C.; Duarte C. D.; Koumbis A. E.; Nicolau C.; Lehn J. M. Polyphosphates and Pyrophosphates of Hexopyranoses as Allosteric Effectors of Human Hemoglobin: Synthesis, Molecular Recognition, and Effect on Oxygen Release. ChemMedChem 2011, 6, 153–168. 10.1002/cmdc.201000366. [DOI] [PubMed] [Google Scholar]
- Plante O. J.; Andrade R. B.; Seeberger P. H. Synthesis and Use of Glycosyl Phosphates as Glycosyl Donors. Org. Lett. 1999, 1, 211–214. 10.1021/ol9905452. [DOI] [PubMed] [Google Scholar]
- Carrel F. R.; Seeberger P. H. Protecting Group Manipulations on Glycosyl Phosphate Triesters. J. Carbohydr. Chem. 2007, 26, 125–139. 10.1080/07328300701298204. [DOI] [Google Scholar]
- Mills S. J.; Rossi A. M.; Konieczny V.; Bakowski D.; Taylor C. W.; Potter B. V. L. d-Chiro-Inositol Ribophostin: A Highly Potent Agonist at d-Myo-Inositol 1,4,5-Trisphosphate Receptors: Synthesis and Biological Activities. J. Med. Chem. 2020, 63, 3238–3251. 10.1021/acs.jmedchem.9b01986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi M.; Kagasaki T.; Hosoya T.; Takahashi S. Adenophostins A and B: Potent Agonists of Inositol-1,4,5-Trisphosphate Receptor Produced by Penicillium brevicompactum. J. Antibiot. (Tokyo). 1993, 46, 1643–1647. 10.7164/antibiotics.46.1643. [DOI] [PubMed] [Google Scholar]
- Rosenberg H. J.; Riley A. M.; Laude A. J.; Taylor C. W.; Potter B. V. L. Synthesis and Ca2+-Mobilizing Activity of Purine-Modified Mimics of Adenophostin A: A Model for the Adenophostin−Ins(1,4,5)P3 Receptor Interaction. J. Med. Chem. 2003, 46, 4860–4871. 10.1021/jm030883f. [DOI] [PubMed] [Google Scholar]
- Takahashi M.; Tanzawa K.; Takahashi S. Adenophostins, Newly Discovered Metabolites of Penicillium brevicompactum, Act as Potent Agonists of the Inositol 1,4,5-Trisphosphate Receptor. J. Biol. Chem. 1994, 269, 369–372. [PubMed] [Google Scholar]
- Fan G.; Baker M. R.; Wang Z.; Seryshev A. B.; Ludtke S. J.; Baker M. L.; Serysheva I. I. Cryo-EM Reveals Ligand Induced Allostery Underlying InsP3R Channel Gating. Cell Res. 2018, 28, 1158–1170. 10.1038/s41422-018-0108-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tegge W.; Denis G. V.; Ballou C. E. Synthesis and Ca2+-Release Activity of d- and l-Myo-Inositol 2,4,5-Trisphosphate and d- and l-Chiro-Inositol 1,3,4-Trisphosphate. Carbohydr. Res. 1991, 217, 107–116. 10.1016/0008-6215(91)84121-T. [DOI] [PubMed] [Google Scholar]
- Riley A. M.; Unterlass J. E.; Konieczny V.; Taylor C. W.; Helleday T.; Potter B. V. L. A Synthetic Diphosphoinositol Phosphate Analogue of Inositol Trisphosphate. MedChemComm 2018, 9, 1105–1113. 10.1039/c8md00149a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchant J. S.; Beecroft M. D.; Riley A. M.; Jenkins D. J.; Marwood R. D.; Taylor C. W.; Potter B. V. L. Disaccharide Polyphosphates Based upon Adenophostin A Activate Hepatic d-Myo-Inositol 1,4,5-Trisphosphate Receptors. Biochemistry 1997, 36, 12780–12790. 10.1021/bi971397v. [DOI] [PubMed] [Google Scholar]
- Beecroft M. D.; Marchant J. S.; Riley A. M.; Van Straten N. C. R.; Van Der Marel G. A.; Van Boom J. H.; Potter B. V. L.; Taylor C. W. Acyclophostin: A Ribose-Modified Analog of Adenophostin A with High Affinity for Inositol 1,4,5-Trisphosphate Receptors and pH-Dependent Efficacy. Mol. Pharmacol. 1999, 55, 109–117. 10.1124/mol.55.1.109. [DOI] [PubMed] [Google Scholar]
- Safrany S. T.; Wilcox R. A.; Liu C.; Dubreuil D.; Potter B. V.; Nahorski S. R. Identification of Partial Agonists with Low Intrinsic Activity at the Inositol-1,4,5-Trisphosphate Receptor. Mol. Pharmacol. 1993, 43, 499–503. [PubMed] [Google Scholar]
- Riley A. M.; Murphy C. T.; Lindley C. J.; Westwick J.; Potter B. V. L. 6-Deoxy-6-Hydroxymethyl Scyllo-Inositol 1,2,4-Trisphosphate: A Potent Agonist at the Inositol 1,4,5-Trisphosphate Receptor. Bioorg. Med. Chem. Lett. 1996, 6, 2197–2200. 10.1016/0960-894X(96)00399-X. [DOI] [Google Scholar]
- Wilcox R. A.; Challiss R. A.; Traynor J. R.; Fauq A. H.; Ognayanov V. I.; Kozikowski A. P.; Nahorski S. R. Molecular Recognition at the Myo-Inositol 1,4,5-Trisphosphate Receptor. 3-position substituted myo-inositol 1,4,5-trisphosphate analogues reveal the binding and Ca2+ release requirements for high affinity interaction with the myo-inositol 1,4,5-trisphosphate receptor. J. Biol. Chem. 1994, 269, 26815–26821. [PubMed] [Google Scholar]
- Fauq A. H.; Kozikowski A. P.; Ognyanov V. I.; Wilcox R. A.; Nahorski S. R. Probing the d-1,4,5-IP3/d-1,3,4,5-IP4 Functional Interface. Synthesis and Pharmacology of Novel d-3-Modified Myo-Inositol Trisphosphate Analogues. J. Chem. Soc., Chem. Commun. 1994, 1301–1302. 10.1039/c39940001301. [DOI] [Google Scholar]
- Burford N. T.; Nahorski S. R.; Chung S.-K.; Chang Y.-T.; Wilcox R. A. Binding and Activity of the Nine Possible Regioisomers of Myo-Inositol Tetrakisphosphate at the Inositol 1,4,5-Trisphosphate Receptor. Cell Calcium 1997, 21, 301–310. 10.1016/S0143-4160(97)90118-4. [DOI] [PubMed] [Google Scholar]
- Liu C.; Potter B. V. L. Synthesis of 3-Position-Modified Analogues of Myo-Inositol 1,4,5-Trisphosphate, Tools for Investigation of the Polyphosphoinositide Pathway of Cellular Signaling. J. Org. Chem. 1997, 62, 8335–8340. 10.1021/jo970926y. [DOI] [PubMed] [Google Scholar]
- Safrany S. T.; Wilcox R. A.; Liu C.; Potter B. V. L.; Nahorski S. R. 3-Position Modification of Myo-Inositol 1,4,5-Trisphosphate: Consequences for Intracellular Ca2+ Mobilisation and Enzyme Recognition. Eur. J. Pharmacol. 1992, 226, 265–272. 10.1016/0922-4106(92)90071-3. [DOI] [PubMed] [Google Scholar]
- Liu C.; Nahorski S. R.; Potter B. V. L. Synthesis from Quebrachitol of 1l-Chiro-Inositol 2,3,5-Trisphosphate, an Inhibitor of the Enzymes of 1d-Myo-Inositol 1,4,5-Trisphosphate Metabolism. Carbohydr. Res. 1992, 234, 107–115. 10.1016/0008-6215(92)85042-X. [DOI] [Google Scholar]
- Hirata M.; Watanabe Y.; Yoshida M.; Koga T.; Ozaki S. Roles for Hydroxyl Groups of d-Myo-Inositol 1,4,5-Trisphosphate in the Recognition by Its Receptor and Metabolic Enzymes. J. Biol. Chem. 1993, 268, 19260–19266. [PubMed] [Google Scholar]
- Dreef C. E.; Schiebler W.; van der Marel G. A.; van Boom J. H. Synthesis of 5-Phosphonate Analogues of Myo-Inositol 1,4,5-Trisphosphate: Possible Intracellular Calcium Antagonists. Tetrahedron Lett. 1991, 32, 6021–6024. 10.1016/S0040-4039(00)79454-8. [DOI] [Google Scholar]
- Lampe D.; Liu C.; Potter B. V. L. Synthesis of Selective Non-Ca2+-Mobilizing Inhibitors of d-Myo-Inositol 1,4,5-Trisphosphate 5-Phosphatase. J. Med. Chem. 1994, 37, 907–912. 10.1021/jm00033a007. [DOI] [PubMed] [Google Scholar]
- Li C.-W.; Dong H.-J.; Cui C.-B. The Synthesis and Antitumor Activity of Twelve Galloyl Glucosides. Molecules 2015, 20, 2034–2060. 10.3390/molecules20022034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medgyes A.; Farkas E.; Lipták A.; Pozsgay V. Synthesis of the Monosaccharide Units of the O-Specific Polysaccharide of Shigella Sonnei. Tetrahedron 1997, 53, 4159–4178. 10.1016/S0040-4020(97)00145-2. [DOI] [Google Scholar]
- Tseberlidis G.; Zardi P.; Caselli A.; Cancogni D.; Fusari M.; Lay L.; Gallo E. Glycoporphyrin Catalysts for Efficient C-H Bond Aminations by Organic Azides. Organometallics 2015, 34, 3774–3781. 10.1021/acs.organomet.5b00436. [DOI] [Google Scholar]
- D’Alonzo D.; Guaragna A.; Napolitano C.; Palumbo G. Rapid Access to 1,6-Anhydro-β-l-hexopyranose Derivatives via Domino Reaction: Synthesis of l-Allose and l-Glucose. J. Org. Chem 2008, 73, 5636–5639. 10.1021/jo800775v. [DOI] [PubMed] [Google Scholar]
- Daragics K.; Szabó P.; Fügedi P. Some Observations on the Reductive Ring Opening of 4,6-O-Benzylidene Acetals of Hexopyranosides with the Borane Trimethylamine-Aluminium Chloride Reagent. Carbohydr. Res. 2011, 346, 1633–1637. 10.1016/j.carres.2011.04.046. [DOI] [PubMed] [Google Scholar]
- Satomura S.; Iwata T.; Sakata Y.; Omichi K.; Ikenaka T. Synthesis of p-nitrophenyl 65-O-benzyl-α-maltopentaoside, a substrate for α-amylases. Carbohydr. Res. 1988, 176, 107–115. 10.1016/0008-6215(88)84062-X. [DOI] [PubMed] [Google Scholar]
- Bauder C. A Convenient Synthesis of Orthogonally Protected 2-Deoxystreptamine (2-DOS) as an Aminocyclitol Scaffold for the Development of Novel Aminoglycoside Antibiotic Derivatives against Bacterial Resistance. Org. Biomol. Chem. 2008, 6, 2952–2960. 10.1039/b804784g. [DOI] [PubMed] [Google Scholar]
- Boettcher S.; Matwiejuk M.; Thiem J. Acceptor-Influenced and Donor-Tuned Base-Promoted Glycosylation. Beilstein J. Org. Chem. 2012, 8, 413–420. 10.3762/bjoc.8.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liau B. B.; Milgram B. C.; Shair M. D. Total Syntheses of HMP-Y1, Hibarimicinone, and HMP-P1. J. Am. Chem. Soc. 2012, 134, 16765–16772. 10.1021/ja307207q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillippy B. Q.; Bland J. M. Gradient Ion Chromatography of Inositol Phosphates. Anal. Biochem. 1988, 175, 162–166. 10.1016/0003-2697(88)90374-0. [DOI] [PubMed] [Google Scholar]
- Leatherbarrow R. J.GraFit Version 7; Erithacus Software Ltd.: Horley, UK, 2009. [Google Scholar]
- Alzayady K. J.; Wang L.; Chandrasekhar R.; Wagner L. E. II; Van Petegem F.; Yule D. I. Defining the Stoichiometry of Inositol 1,4,5-Trisphosphate Binding Required to Initiate Ca2+ Release. Sci. Signal. 2016, 9, ra35. 10.1126/scisignal.aad6281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo F.; Chiang M. Y.; Wang Y.; Zhang Y. Z. An In Vitro Recombination Method to Convert Restriction- and Ligation-Independent Expression Vectors. Biotechnol. J. 2008, 3, 370–377. 10.1002/biot.200700170. [DOI] [PubMed] [Google Scholar]
- Laude A. J.; Tovey S. C.; Dedos S. G.; Potter B. V. L.; Lummis S. C. R.; Taylor C. W. Rapid Functional Assays of Recombinant IP3 Receptors. Cell Calcium 2005, 38, 45–51. 10.1016/j.ceca.2005.04.001. [DOI] [PubMed] [Google Scholar]
- Tovey S. C.; Sun Y.; Taylor C. W. Rapid Functional Assays of Intracellular Ca2+ Channels. Nat. Protoc. 2006, 1, 259–263. 10.1038/nprot.2006.40. [DOI] [PubMed] [Google Scholar]
- Cheng Y.-C.; Prusoff W. H. Relationship between the Inhibition Constant (KI) and the Concentration of Inhibitor Which Causes 50 Per Cent Inhibition (I50) of an Enzymatic Reaction. Biochem. Pharmacol. 1973, 22, 3099–3108. 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- Colquhoun D.Lectures on Biostatistics: An Introduction to Statistics with Applications in Biology and Medicine; Oxford University Press: London, UK, 1971. [Google Scholar]
- Ott R. L.; Longnecker M.. An Introduction to Statistical Methods and Data Analysis; Cengage Learning: Boston, USA, 2010. [Google Scholar]
- Jones G.; Willett P.; Glen R. C; Leach A. R; Taylor R. Development and Validation of a Genetic Algorithm for Flexible Docking. J. Mol. Biol. 1997, 267, 727–748. 10.1006/jmbi.1996.0897. [DOI] [PubMed] [Google Scholar]
- Verdonk M. L.; Chessari G.; Cole J. C.; Hartshorn M. J.; Murray C. W.; Nissink J. W. M.; Taylor R. D.; Taylor R. Modeling Water Molecules in Protein-Ligand Docking Using GOLD. J. Med. Chem. 2005, 48, 6504–6515. 10.1021/jm050543p. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.