

Abstract
Objectives:
Non-immune hydrops fetalis (NIHF) accounts for 90% of hydrops fetalis cases. 15–29% of unexplained NIHF cases are caused by lysosomal storage diseases (LSD). We review the spectrum of LSD in NIHF in patients referred to our institution.
Methods:
We present a retrospective case-control study of NIHF cases referred for LSD biochemical testing at a single center. Cases with LSD were matched to controls with NIHF and negative LSD testing and analyzed according to the STROBE criteria
Results:
Between January 2006 and December 2018, 28 patients with NIHF were diagnosed with a LSD. Eight types of LSD were diagnosed: Galactosialidosis 8/28 (28.6%), Sialic Acid Storage Disease (SASD) 5/28 (17.9%), Mucopolysaccharidosis VII 5/28 (17.9%), Gaucher 4/28 (14.3%), Sialidosis 2/28 (7.1%), GM1 Gangliosidosis 2/28 (7.1%), Niemann-Pick disease type C 1/28 (3.6%), and Mucolipidosis II/III 1/28 (3.6%). Associated clinical features were hepatomegaly 16/21 (76.2%) vs 22/65 (33.8%), p < 0.05, splenomegaly 12/20 (60.0%) vs 14/58 (24.1%), p < 0.05 and hepatosplenomegaly 10/20 (50.0%) vs 13/58 (22.4%) p < 0.05.
Conclusion:
The most common LSD in NIHF were Galactosialidosis, SASD, Mucopolysaccharidosis VII, and Gaucher disease. LSD should be considered in unexplained NIHF cases, particularly if hepatomegaly, splenomegaly or hepatosplenomegaly is visualized on prenatal ultrasound.
Introduction
Hydrops Fetalis (HF) is a life-threatening fetal condition defined as an abnormal accumulation of fluid in two or more fetal compartments. This accumulation can be diagnosed by prenatal ultrasound and is characterized by the presence of ascites, pleural effusion, pericardial effusion, or generalized skin edema [1]. In addition, HF may be associated with polyhydramnios and placental edema [2].
The pathophysiology of HF can be classified according to immune and nonimmune etiology [3]. The prevalence of immune etiologies has dramatically decreased with the development of effective Rh(D) immunization in mothers at risk. Consequently, nonimmune hydrops fetalis (NIHF) now accounts for almost 90% of hydrops cases [1]. NIHF is most commonly associated with cardiovascular anomalies, chromosomal abnormalities, infectious diseases, hematological disorders and metabolic diseases. Cases without a clear etiology are considered unexplained, or idiopathic [4].
Inborn errors of metabolism (IEM) should be considered as a cause of NIHF once common etiologies of NIHF are ruled out by evaluation with ultrasound, laboratory testing, karyotype and microarray. Most of these IEM are classified as lysosomal storage diseases (LSD) accounting for approximately 1–15% of NIHF cases, and up to almost 30% of unexplained (idiopathic) NIHF cases [5–7].
We reviewed the spectrum of LSD diagnosed after the identification of NIHF in samples referred to a single center, the Lysosomal Diseases Testing Laboratory (LDTL) at Thomas Jefferson University. We also compared the presence of physician reported hepatomegaly, splenomegaly, and other clinical findings in NIHF with LSD positive cases to LSD negative controls.
Methods
This is a retrospective case-control study of all cases with NIHF referred for workup of LSD to the LDTL at Thomas Jefferson University in Philadelphia, PA. Included cases were from amniocentesis and neonatal blood specimens. Samples submitted for LSD testing between January 2006 and December 2018 were included if testing was positive. Comparison cases with NIHF and LSD negative testing were selected from January 2015 to December 2018 providing 3 controls for each LSD positive case. Clinical information for LSD negative NIHF patients tested before 2015 was no longer available. Clinical information abstracted on chart review of test requisitions included maternal demographics, prenatal ultrasound findings, maternal and fetal workup for NIHF, LSD testing, and fetal and neonatal outcomes. The clinical information provided to the testing lab varied greatly between samples and information not recorded in the records was excluded from analysis. Thomas Jefferson University Institutional Review Board approval #18D.651 was obtained. This study was performed according to the STROBE statement for observational studies to the extent the retrospective nature of this study allowed [8].
NIHF was defined, for the purpose of this study, by the presence of any of the following phrases in the patient chart: “hydrops”, “born with hydrops”, “fetal hydrops”, “hydropic newborn”, “hydrops in utero”, “hydrops detected prenatally”, “hydrops at birth”, “hydrops fetalis”, “non-immune hydrops”, and “non immune hydrops fetalis.” All submitted samples with a clinical diagnosis of NIHF undergo testing with a hydropic panel. Isolated leukocytes were evaluated for β-galactosidase for GM1 gangliosidosis and galactosialidosis, β-glucuronidase for mucoploysaccharidosis VII, β-glucocerebrosidase for Gaucher disease, acid sphingomyelinase for Niemann-Pick disease types A/B, α-iduronidase for Mucopolysaccharidosis I, and sialic acid content for galactosialidosis, sialidosis and sialic acid storage disease (SASD). Plasma was isolated from the original blood sample permitting the diagnosis of mucolipidosis II/III and was used to help differentiate GM1 gangliosidosis (low β-galactosidase in leukocytes and plasma) from galactosialidosis (low β-galactosidase in leukocytes and normal β-galactosidase in plasma). In some cases, cultured amniotic fluid cells (AFC) or cultured skin fibroblasts were received. Niemann-Pick type C was diagnosed by demonstrating an increase in free cholesterol within lysosomes as evidenced by strong filipin staining. All assays were performed following the methods described in Wenger and Williams [9].
The primary outcome of this study was the characterization of the spectrum of LSD diagnosed in NIHF. Secondary outcomes included hepatomegaly, splenomegaly and any additional clinical features on ultrasound or in the patient’s history that were associated with LSD diagnosis for patients with NIHF compared to LSD negative controls.
Statistical analysis was performed using IBM SPSS Statistics 24, Armonk, NY. Percentages and Chi-squared tests were performed for categorical values. Means (SD) and independent samples t-tests were performed for continuous variables. Fisher’s exact tests were performed when the expected values in any of the cells of a contingency table were below 5 for categorical variables. Moreover, Mann-Whitney U tests were performed as non-parametric alternative tests to the independent sample t-tests. We used a 2-sided level of statistical significance for alpha level of 0.05 for all statistical tests. Missing data was excluded from the analysis.
Results
Twenty-eight patients with NIHF were diagnosed with LSD by biochemical testing between January 2006 and December 2018 at our LDTL. Samples were a combination of neonatal blood (27) and amniotic fluid cells (1). From January 2015 to December 2018, 98 samples with NIHF were negative for LSD. Samples were a combination of neonatal blood (95), amniotic fluid cells (2), and cord blood (1). There was no statistically significant difference in the distribution of ethnicities, sex, parity, or gestational age at delivery between LSD positive NIHF and LSD negative NIHF (Table 1). Maternal age was younger in the LSD positive group 24.3 (±2.5) years vs 32.1 (±4.5) years, p < 0.05. Though gestational age was reported in a relatively small number of the cases, fetal hydrops was diagnosed at an earlier gestational age in the LSD positive group, 23.0 (±7.0) weeks vs 31.2 (±3.4) weeks, p = 0.049. Infants were older at the time of enzyme testing in the LSD positive group 24 [13.5, 100] days vs 16 [10, 35], p < 0.05.
Table 1.
Demographics
| Chracteristics | LSD Positive n (%) or mean (SD) | LSD Negative n (%) or mean (SD) | P value |
|---|---|---|---|
| Race/Ethnicitya | |||
| Caucasian | 6 (28.6%) | 30 (41.7%) | 0.28 |
| African Americanh | 2 (9.5%) | 11 (15.3%) | 0.73 |
| Asianh | 4 (l9.0%) | 6 (8.3%) | 0.23 |
| Hispanic/Latino | 6 (28.6%) | 16 (22.2%) | 0.55 |
| Other/Mixed Raceh | 3 (14.3%) | 9 (12.5%) | 1.00 |
| Sexb | 0.67 | ||
| Female | 14 (50.0%) | 44 (45.4%) | |
| Male | 14 (50.0%) | 53 (54.6%) | |
| MaLernal Age (years)c,i | 24.3 (2.5) | 32.1 (4.5) | 0.01 |
| Parityd | 1.5 (0.7) | 1.9 (1.6) | 1.00 |
| Gestational Age at NIHF diagnosis (weeks)e,i | 23.0 (7.0) | 31.2 (3.4) | 0.049 |
| Gestational Age at delivery (weeks)f | 34.5 (3.2) | 33.1 (5.1) | 0.28 |
| Age at time of enzyme testing| (days)g,i | 24 (13.5–100) | 16 (10–35) | 0.04 |
N=21,72
N=28,97
N=3,15
N=2, 14
N=3, 10
N=18, 58
Median and inter quartile ranges were calculated, N= 27, 94
Fisher’s Exact test used for expected cell counts <5
Mann- Whitney U test used for small sample
Limited data were available for physical exam or sonographic findings in each group. The referring providers were not always specific about the timing of these findings (prenatal or neonatal period) and did not consistently document the presence or absence of a finding. The following findings were reported. Hepatic enlargement was noted in 16/21 (76.2%) patients in the LSD positive group compared with 22/65 (33.8%) in the LSD negative group (p < 0.05). Similarly, splenic enlargement was noted in 12/20 (60.0%) patients in the LSD positive group compared with 14/58 (24.1%) in the LSD negative group (p < 0.05). Moreover, hepatosplenomegaly was noted in 10/20 (50.0%) patients in the LSD positive group compared with 13/58 (22.4%) in the LSD negative group (p < 0.05). In the LSD positive group, three families reported consanguinity and two families reported a history of prior hydrops, of which only one was in a consanguineous family. While no families in the LSD negative group reported consanguinity, four families reported a prior history of hydrops. Records for additional clinical features were available for some patients in the LSD positive group (n=19; 67%). In this group, 16 (84.2%) had ascites, 8 (42.1%) had skin edema, 3 (15.8%) had pleural effusion, and 1 (5.3%) had polyhydramnios. No cases of pericardial effusion were reported. In the LSD negative group, records were available for 59 samples (70%). In this group 37 (62.7%) had ascites, 28 (47.5%) had pleural effusion, 14 (23.7%) had skin edema, 9 (15.3%) had pericardial effusion, and 7 (11.9%) had polyhydramnios (Table 2). There was no statistically significant difference for any of these clinical findings other than pleural effusion, a finding that the LSD negative group was more likely to exhibit than the LSD positive group (p < 0.05). There was not a significant difference in record availability between the LSD positive group and the LSD negative group.
Table 2.
Clinical features of hydrops
| Chracteristics | LSD Positive n (%) | LSD Negative n (%) | P value |
|---|---|---|---|
| Hepatomegalya | 16 (76.2%) | 22 (33.8%) | < 0.01 |
| Splenomegalyb | 12 (60.0%) | 14 (24.1%) | < 0.01 |
| Hepatosplenomegalyb | 10 (50.0%) | 13 (22.4%) | 0.02 |
| N=19 | N=59 | ||
| Ascites | 16 (84.2%) | 37 (62.7%) | 0.08 |
| Skin Edema | 8 (42.1%) | 14 (23.7%) | 0.12 |
| Pleural Effusionc | 3 (15.8%) | 28 (47.5%) | 0.02 |
| Polyhydramniosc | 1 (5.3%) | 7 (11.9%) | 0.67 |
| Pericardial Effusion | 0 (0.0%) | 9 (15.3%) | 0.10 |
N= 21, 65
N=20, 58
Mann-Whitney U test used for small sample
The spectrum of LSD (Figure 1) diagnosed in NIHF at our LDTL was as follows: Galactosialidosis 8/28 (28.6%), SASD 5/28 (17.9%), Mucopolysaccharidosis VII 5/28 (17.9%), Gaucher 4/28 (14.3%), Sialidosis 2/28 (7.1%), GM1 Gangliosidosis 2/28 (7.1%), Niemann-Pick disease type C 1/28 (3.6%), and Mucolipidosis II/III 1/28 (3.6%). Enzyme levels relevant to each specific disorder can be found in Table 3.
Figure 1:
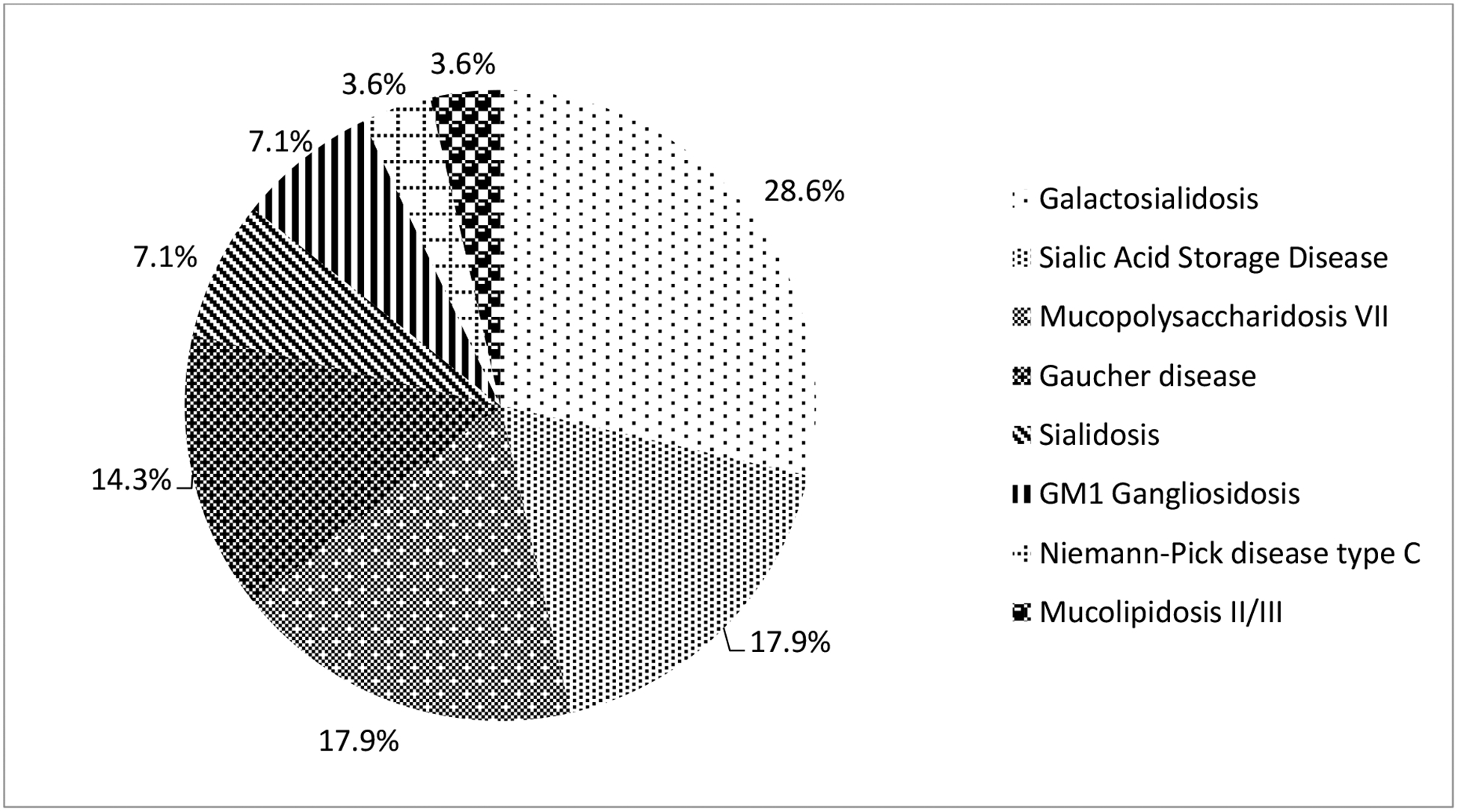
Spectrum of LSD in NIHF
Table 3.
Values tested in samples (in leukocytes unless otherwise specified)
| Patient | LSD | Test | Value Obtained* | Normal Range |
|---|---|---|---|---|
| LH3 | Galactosialidosis | Beta-galactosidase | 3.0 | 35–120 |
| Beta-galactosidase (plasma) | 13.1 | 10–40 | ||
| Total sialic acid content | 23.1 | 8–22 | ||
| LH8 | Galactosialidosis | Beta-galactosidase | 5.1 | 35–120 |
| Beta-galactosidase (plasma) | 17.6 | 10–40 | ||
| Total sialic acid content | 36.4 | 8–22 | ||
| LH12 | Galactosialidosis | Beta-galactosidasea | 8.5 | 35–120 |
| Beta-galactosidase (plasma)a | 14.2 | 10–40 | ||
| Total sialic acid contenta | 34.0 | 8–22 | ||
| LH16 | Galactosialidosis | Beta-galactosidase | 11.5 | 35–120 |
| Beta-galactosidase (plasma) | 8.4 | 10–40 | ||
| Total sialic acid content | 29.2 | 8–22 | ||
| LH17 | Galactosialidosis | Beta-galactosidase | 5.3 | 35–120 |
| Beta-galactosidase (plasma) | 10.1 | 10–40 | ||
| Total sialic acid content | 41.4 | 8–22 | ||
| LH18 | Galactosialidosis | Beta-galactosidase | 3.0 | 35–120 |
| Beta-galactosidase (plasma) | 4.0 | 10–40 | ||
| Total sialic acid content | 36.4 | 8–22 | ||
| LH22 | Galactosialidosis | Beta-galactosidase | 3.9 | 35–120 |
| Beta-galactosidase (plasma) | 22.0 | 10–40 | ||
| Total sialic acid content | 35.8 | 8–22 | ||
| LH30 | Galactosialidosis | Beta-galactosidase | 8.0 | 35–120 |
| Beta-galactosidase (plasma) | 33.0 | 10–40 | ||
| Total sialic acid content | 34.7 | 8–22 | ||
| LH1 | SASD | Total sialic acid contentb | 37.3 | 8–22 |
| Free sialic acid contentb | 12.3 | 10–20% of total | ||
| LH2 | SASD | Total sialic acid contentb | 32.4 | 8–22 |
| Free sialic acid contentb | 13.0 | 10–20% of total | ||
| LH11 | SASD | Total sialic acid contentc | 48.5 | 8–22 |
| Free sialic acid contentc | 38.4 | 10–20% of total | ||
| LH23 | SASD | Total sialic acid contentb | 26.5 | 8–22 |
| Free sialic acid contentb | 17.8 | 10–20% of total | ||
| LH28 | SASD | Total sialic acid content | 34.7 | 8–22 |
| Free sialic acid content | 20.8 | 10–20% of total | ||
| LH4 | Mucopolysaccharidosis VII | Beta-glucuronidase | 0.2 | 100–400 |
| LH19 | Mucopolysaccharidosis VII | Beta-glucuronidase | 0 | 100–400 |
| LH21 | Mucopolysaccharidosis VII | Beta-glucuronidase (AFC) | 0 | 100–400 |
| LH25 | Mucopolysaccharidosis VII | Beta-glucuronidase | 0.7 | 100–400 |
| LH26 | Mucopolysaccharidosis VII | Beta-glucuronidase | 0 | 100–400 |
| LH5 | Gaucher Disease | Glucocerebrosidase | 0.5 | 3.0–12.0 |
| LH6 | Gaucher Disease | Glucocerebrosidase | 0.7 | 3.0–12.0 |
| LH20 | Gaucher Disease | Glucocerebrosidase | 1.5 | 3.0–12.0 |
| LH24 | Gaucher Disease | Glucocerebrosidase | 1.0 | 3.0–12.0 |
| LH9 | Sialidosis | Total sialic acid content | 38.2 | 8–22 |
| Free sialic acid content | 3.9 | 10–20% of total | ||
| LH15 | Sialidosis | Total sialic acid contentc | 46.3 | 8–22 |
| Sialidasec | 1.2 | 50–120 | ||
| LH7 | GM1 Gangliosidosis | Beta-galactosidase | 2.8 | 35–120 |
| Beta-galactosidase (plasma) | 0.5 | 10–40 | ||
| LH14 | GM1 Gangliosidosis | Beta-galactosidase | 0.5 | 35–120 |
| Beta-galactosidase (plasma) | 0.1 | 10–40 | ||
| LH13 | Niemann-Pick Disease Type C | Excess free cholesterolc,d | ||
| LH29 | Mucolipidosis II/III | Beta-galactosidase (plasma) | 169.4 | 10–40 |
| Alpha-n-acglusaminidase (plasma) | 50.0 | 10–40 |
Enzyme values are expressed as nmol/hour/mg protein; sialic acid content is expressed as nmol/mg protein
Confirmed by measuring low sialidase activity in cultured fibroblasts
Confirmed with elevated total and free sialic acid in urine
Cultured fibroblasts
Filipin staining for free cholesterol was positive indicating a diagnosis of Niemann-Pick type C
AFC: amniotic fluid cells
Discussion
Principal Findings
From January 2006 to December 2018, twenty-eight fetuses and neonates with NIHF obtained a causative LSD diagnosis. Twenty-seven had one of eight disorders tested for in blood samples or cultured cells, and one had a diagnosis of Niemann-Pick type C which required cultured cells. The most frequently diagnosed LSD were galactosialidosis followed by SASD and mucopolysaccharidosis VII.
Results
Since 1973, we have made nearly 5,000 diagnoses from more than 60,000 samples, yielding a diagnostic rate of about 8%. The diagnoses made by our lab include cases referred for many indications, only a subset of which are NIHF. The LDTL at Thomas Jefferson University currently has the highest volume of clinical specimen submission for LSD evaluation both nationally and internationally. We receive samples from individuals with limited clinical information supplied by their physician. We included all the cases for which we had information that the indication for testing was hydrops. All patients presenting with NIHF are screened with the same group of tests, the hydropic panel, which includes all the LSD that have been associated with NIHF.
Our group has done a systematic literature review of LSD and NIHF starting January 2014 using the same methodology as reported by Gimovsky et al up to February 2019 [5]. Ultimately, 3 additional cohorts were identified. The largest had 108 fetuses with both NIHF and LSD [10], followed by a cohort of 7 fetuses and another cohort of 5 fetuses [11],[12].
The most common LSD diagnosed in our cohort are similar to the groups described by Vianey-Saban et al. [10] and Stone and Sidransky [7]. However, they differ from the prior meta-analysis by Gimovsky et al [5], which includes Mucopolysaccharidosis type VII among the most common LSD diagnosed in NIHF followed by Gaucher disease and GM1-gangliosidosis. It is interesting that the two most common disorders resulting in NIHF in our cohort are related to the metabolism of sialic acid. Sialic acid is a major component of glycoproteins and this could be related to a shared mechanism leading to NIFH in congenital disorders of glycosylation [13].
Clinical Implications
Lysosomes are membrane-enclosed cytoplasmic organelles that contain a variety of hydrolytic enzymes. LSD are a group of genetic conditions caused by defects in the hydrolysis of complex lipids or carbohydrates, the transport of metabolites from lysosomes, or the packaging of lysosomal enzymes via the mannose-6-phosphate pathway. This resulting dysfunction leads to an accumulation of upstream products that eventually overwhelms normal cellular function [14]. Diagnosis of LSD is based on enzymatic testing of chorionic villus or amniotic fluid samples in the prenatal period, or testing may be performed in the postnatal period using neonatal blood samples [1]. Currently, there are at least 10 different types of LSD associated with NIHF[5–7]. Fetal hydrops is a known manifestation of LSD. In a review by Sláma et al. they found 16 cases of fetal hydrops associated with 142 cases of galactosialidosis.[15] In a review by Zielonka et al. they found 40 cases of fetal hydrops associated with 88 cases of mucopolysaccharidosis type VII.[16] In another review by Zielonka et al. they found 24 cases of fetal hydrops associated with 116 cases of free sialic acid storage disease.[17]
The cause of excessive fluid accumulation within the peritoneal cavity in LSD seems to be multifactorial [18]. One mechanism involves obstruction of venous blood return, from visceromegaly secondary to accumulated storage materials [19]. In addition, excessive fluid collection may result from anemia caused by hypersplenism and the reduction of erythropoietic stem cells due to infiltrating storage cells. Other physiologies of ascites in LSD include congestive heart failure, hypoproteinemia, and liver dysfunction [18].
Our study highlights important prenatal markers that may raise suspicion for LSD in the setting of NIHF. Prenatal liver evaluation is currently not part of the routine workup for NIHF and this can be done easily [20]. The median age of enzyme testing for LSD positive NIHF cases was 24 days suggesting the hepatomegaly was likely to be preexisting, before birth. While our dataset is composed predominately of postnatal blood samples, prenatal detection of hepatomegaly should prompt a LSD workup.
All the LSD identified as causes of NIHF in our cohort are genetic disorders inherited in an autosomal recessive manner. Definitive identification of the metabolic defect facilitates genetic counseling on recurrence risks and the utility of performing further testing on family members. Early identification of LSD also allows earlier initiation of enzyme replacement therapies for disorders where this is available. While biochemical testing is accurate and provides a cause for the presence of NIHF, molecular analysis of the responsible gene would aid in prenatal testing of future pregnancies and carrier testing in family members. Diagnostic testing is also paramount to guiding providers in their discussions with families on the appropriateness of palliative versus therapeutic treatment in utero and after birth. Multiple in utero treatments are emerging for different genetic disorders. This includes an ongoing study of in utero stem cell transplant for fetuses affected with alpha thalassemia major (https://clinicaltrials.ucsf.edu/trial/NCT02986698) and intra-amniotic administration of recombinant ectodysplasin A protein to X-linked hypohidrotic ectodermal dysplasia (XLHED)-affected human fetuses.[21] A recent review by Almeida-Porada et al. showed that in utero transplantation has been performed for at least 14 different genetic disorders including hemoglobinopathies, chronic granulomatous disease, Chediak–Higashi syndrome, and inborn errors of metabolism.[22] None of the LSD listed in our study has an in-utero therapy in clinicaltrials.gov (accessed 1/27/2020). While in utero treatment such as in utero enzyme replacement therapy or stem cells remains theoretical at this time, our goal with this study is to increase awareness of these disorders among maternal fetal medicine providers. Our hope is that with increased diagnosis and earlier diagnosis, this will permit future studies of the utility of in utero treatments.
LSD diagnosis has implications for termination of pregnancy and preimplantation genetic testing for future pregnancies. It is interesting that the diagnosis of NIHF for the LSD positive group occurred at an earlier gestational age (23 weeks) compared to the LSD negative group (31 weeks). This earlier onset again highlights the possibility of early prenatal diagnosis in these cases. We can only hypothesize why maternal age and gestational age can be different in the LSD positive group. Certainly, the bias due to the retrospective nature of the study can be a factor. It is also possible that the LSD fetuses were more severely affected, and that is why they developed prenatal ultrasound findings of fetal hydrops at an earlier gestational age.
LSD have different phenotypic presentations based on their specific metabolic deficiency and the nature of the mutations present. Some of the clinical findings in the fetus may include hepatosplenomegaly and skeletal anomalies, while neonates may also show coarse facial features, spasticity, ataxia, seizures, developmental delay, ocular and facial abnormalities [14]. While most individuals with LSD are diagnosed after birth when clinical features become evident, others manifest in utero as NIHF. The complex and diverse metabolic pathways involved in NIHF pathology are often overlooked which might be the reason why the incidence of LSD is underestimated in comparison to the high incidence of idiopathic NIHF cases. The difficulty in establishing a diagnosis is further supported by the age of metabolic workup for our two groups.
Strengths and Limitations
To our knowledge this is the largest NIHF LSD positive cohort in the US and is the second largest series of LSD in NIHF [17]. It is the first case control study comparing NIHF LSD cases to NIHF negative LSD cases. Prenatal diagnosis will allow earlier initiation of therapy postnatally. Table 4 discusses every LSD in our study and the treatment options when available.
Table 4.
Therapies for types of LSD in our study
| Lysosomal Storage Disease | Approved therapies1 | Potential therapies in Clinical Trials* |
|---|---|---|
| Galactosialidosis | Symptomatic and supportive therapy | None |
| SASD | Symptomatic and supportive therapy | None |
| Mucopolysaccharidosis VII | ERT | ERT, Stem Cell Transplant in children and adults |
| Gaucher | ERT and SRT (types I, II and III), none (perinatal lethal form) | Stem Cell T ransplant in children and adults |
| Sialidosis | Symptomatic and supportive therapy | None |
| GM1 Gangliosidosis | Symptomatic and supportive therapy | Stem Cell Transplant in children and adults |
| Niemann-Pick disease type C | SRT | Stem Cell Transplant in children and adults |
| Mucolipidosis II/III | Symptomatic and supportive therapy | Stem Cell Transplant in children and adults |
ERT, enzyme replacement therapy; SRT, substrate reduction therapy.
Platt FM, d’Azzo A, Davidson BL, Neufeld EF, Tifft CJ. Lysosomal storage diseases. Nat Rev Dis Primers. 2018 Oct 1;4(1):27.
Clinicaltrials.gov accessed on 1/27/2020
Our study is limited by the clinical information available on each sample. Moreover, we did not have the actual ultrasound reports that made the diagnosis of NIHF. Additionally our controls do not span the same time period as the LSD cases. However, the testing methodology at our laboratory remained unchanged and the availability of clinical information was not significantly different between LSD cases and controls. Our study is limited as a retrospective study of cases where physicians suspected LSD. Further prospective prenatal ultrasound studies are needed to validate the finding of prenatal hepatomegaly in LSD as this was incompletely assessed in our study.
Conclusion
The most commonly associated LSD in NIHF in our study were Galactosialidosis, SASD, Mucopolysaccharidosis VII, and Gaucher disease. Three of the LSD (Galactosialidosis, Sialidosis, and SASD), accounting for 15/28 (54%) cases, share a common etiology of altered sialic acid metabolism. Diagnosis of LSD is vital for the management of NIHF as well as counseling parents on the risk of recurrence in subsequent pregnancies. The prenatal ultrasound finding of hepatomegaly, splenomegaly, or hepatosplenomegaly in NIHF should prompt a workup for LSD. Prenatal pleural effusion is more likely associated with other mechanisms of NIHF.
Contribution
What’s already known about this topic?
There are many causes of nonimmune hydrops fetalis including genetic, structural, and infectious causes.
Lysosomal storage diseases are likely an under diagnosed cause of nonimmune hydrops fetalis.
What does this study add?
The most common lysosomal storage diseases diagnosed in nonimmune hydrops fetalis include Galactosialidosis, Sialic Acid Storage Disease (SASD), Mucopolysaccharidosis VII, and Gaucher disease. This is the second largest cohort of patients in recent years describing the spectrum of lysosomal storage disease in nonimmune hydrops fetalis.
Hepatomegaly is a common finding on prenatal ultrasound in patients with hydrops fetalis and lysosomal storage disease.
Acknowledgments
Financial Support:
No financial support was received for this study; Results contributed by Neeta Vora funded by NICHD K23 HD088742 (PI: Vora)
Footnotes
Disclosure: The authors report no conflict of interest
Data availability statement: The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1.Norton ME, Chauhan SP, and Dashe JS, Society for maternal-fetal medicine (SMFM) clinical guideline #7: nonimmune hydrops fetalis. Am J Obstet Gynecol, 2015. 212(2): p. 127–39. [DOI] [PubMed] [Google Scholar]
- 2.Whybra C, et al. , Lysosomal storage disorder in non-immunological hydrops fetalis (NIHF): more common than assumed? Report of four cases with transient NIHF and a review of the literature. Orphanet J Rare Dis, 2012. 7: p. 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Santolaya J, et al. , Antenatal classification of hydrops fetalis. Obstet Gynecol, 1992. 79(2): p. 256–9. [PubMed] [Google Scholar]
- 4.Burin MG, et al. , Investigation of lysosomal storage diseases in nonimmune hydrops fetalis. Prenat Diagn, 2004. 24(8): p. 653–7. [DOI] [PubMed] [Google Scholar]
- 5.Gimovsky AC, Luzi P, and Berghella V, Lysosomal storage disease as an etiology of nonimmune hydrops. Am J Obstet Gynecol, 2015. 212(3): p. 281–90. [DOI] [PubMed] [Google Scholar]
- 6.Piraud M, et al. , Amniotic fluid for screening of lysosomal storage diseases presenting in utero (mainly as non-immune hydrops fetalis). Clinica Chimica Acta, 1996. 248(2): p. 143–155. [DOI] [PubMed] [Google Scholar]
- 7.Stone DL and Sidransky E, Hydrops fetalis: lysosomal storage disorders in extremis. Adv Pediatr, 1999. 46: p. 409–40. [PubMed] [Google Scholar]
- 8.Vandenbroucke JP, Von Elm E, Altman DG, Gøtzsche PC, Mulrow CD, Pocock SJ, Poole C, Schlesselman JJ, Egger M Strengthening the Reporting of Observational Studies in Epidemiology (STROBE): explanation and elaboration. Annals of internal medicine. 2007. 147(8):W–163. [DOI] [PubMed] [Google Scholar]
- 9.Wenger DA and WC, Screening for lysosomal disorders, in Techniques in Diagnostic Human Biochemical Genetics. A Laboratory Manual, Hommes FA, Editor. 1991, Wiley-Liss: New York: p. 587–617. [Google Scholar]
- 10.Vianey-Saban C, et al. , Antenatal manifestations of inborn errors of metabolism: biological diagnosis. J Inherit Metab Dis, 2016. 39(5): p. 611–624. [DOI] [PubMed] [Google Scholar]
- 11.Sheth J, et al. , Lysosomal Storage Disorders in Nonimmune Hydrops Fetalis (NIHF): An Indian Experience. JIMD Rep, 2017. 35: p. 47–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sudrie-Arnaud B, et al. , Metabolic causes of nonimmune hydrops fetalis: A next-generation sequencing panel as a first-line investigation. Clin Chim Acta, 2018. 481: p. 1–8. [DOI] [PubMed] [Google Scholar]
- 13.van de Kamp JM, et al. , Congenital disorder of glycosylation type Ia presenting with hydrops fetalis. J Med Genet, 2007. 44(4): p. 277–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ferreira CR and Gahl WA, Lysosomal storage diseases. Transl Sci Rare Dis, 2017. 2(1–2): p. 1–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sláma T, et al. , Quantitative natural history characterization in a cohort of 142 published cases of patients with galactosialidosis-A cross-sectional study. Journal of Inherited Metabolic Disease, 2019. 42(2): p. 295–302. [DOI] [PubMed] [Google Scholar]
- 16.Zielonka M, et al. , Quantitative clinical characteristics of 53 patients with MPS VII: a cross-sectional analysis. Genet Med, 2017. 19(9): p. 983–988. [DOI] [PubMed] [Google Scholar]
- 17.Zielonka M, et al. , A cross-sectional quantitative analysis of the natural history of free sialic acid storage disease-an ultra-orphan multisystemic lysosomal storage disorder. Genetics in Medicine, 2019. 21(2): p. 347–352 [DOI] [PubMed] [Google Scholar]
- 18.Staretz-Chacham O, et al. , Lysosomal storage disorders in the newborn. Pediatrics, 2009. 123(4): p. 1191–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hutchison AA, et al. , Nonimmunologic hydrops fetalis: a review of 61 cases. Obstet Gynecol, 1982. 59(3): p. 347–52. [PubMed] [Google Scholar]
- 20.Tongprasert F, et al. , Normal length of the fetal liver from 14 to 40 weeks of gestational age. J Clin Ultrasound, 2011. 39(2): p. 74–7. [DOI] [PubMed] [Google Scholar]
- 21.Schneider H, et al. , Prenatal Correction of X-Linked Hypohidrotic Ectodermal Dysplasia. New England Journal of Medicine, 2018. 378(17): p. 1604–1610. [DOI] [PubMed] [Google Scholar]
- 22.Almeida-Porada G, Atala A, and Porada CD, In utero stem cell transplantation and gene therapy: rationale, history, and recent advances toward clinical application. Molecular therapy. Methods & clinical development, 2016. 5: p. 16020–16020. [DOI] [PMC free article] [PubMed] [Google Scholar]