

Abstract
Purpose:
Although Common Variable Immunodeficiency (CVID) is considered the most prevalent symptomatic primary antibody deficiency (PAD), there is a population with symptomatic PADs that do not meet criteria for CVID. We analyzed clinical and immunological profiles of patients with different PADs to better understand the differences and similarities between CVID and other PADs.
Methods:
We extracted clinical and laboratory data of patients with PADs from electronic medical records. Patients were categorized into CVID, IgG subclass 2 deficiency (IgG2D), IgG deficiency (IgGD) and selective antibody deficiency (sAbD) based on basal immunoglobulin levels and pneumococcal vaccine responses. We compared clinical and immunological characteristics in these groups.
Results:
All patients, regardless of PAD types, showed similar frequencies of infections, bronchiectasis, and interstitial lung disease (ILD). Hematopoietic malignancies were more frequently found in the CVID than in the IgG2D, IgGD and sAbD groups, while the latter groups trended towards an increased frequency of connective tissue diseases (CTD). Low counts of natural killer (NK) cells were associated with malignancy, autoimmunity, and ILD in CVID but not in other PAD groups.
Conclusions:
Higher frequency of hematopoietic malignancy in CVID than in the other PADs and association of lower NK cell counts with non-infectious complications in CVID suggest a relationship between immune alterations and the development of non-infectious manifestations in PADs.
Keywords: Primary antibody deficiency, Common Variable Immunodeficiency, IgG subclass 2 deficiency, Selective IgG subclass deficiency, IgG deficiency, Specific antibody deficiency
Introduction:
Primary antibody deficiencies (PADs) are a diverse group of disorders that account for 50–60% of all primary immunodeficiencies [1–3]. PADs include common variable immune deficiency (CVID), IgG subclass 2 deficiency (IgG2D), IgG deficiency (IgGD), and specific antibody deficiency (sAbD). Individuals with PADs are unable to produce an effective antibody response to pathogens [3, 4], which may arise from mutations in single genes critical for the proper development and function of B lymphocytes [1, 2].
Although the majority of single gene disorders manifest during childhood [2], some patients with PADs such as CVID may not develop symptoms until adulthood [3, 5]. With a prevalence of 1:25,000 in Caucasians, CVID is known as the most symptomatic PAD [1, 2, 6–8]. Patients with CVID can present with a broad range of clinical manifestations including sinopulmonary infections, autoimmune diseases, and malignancy [7–10]. A recent study reported less severe immune defects and a lower frequency of non-infectious manifestations in IgGD compared to CVID [11]. However, there is a scarcity of literature characterizing immunological and clinical characteristics of patients with less well characterized PADs including IgG2D and sAbD [5, 11].
Here, we determined the relationship between immunological characteristics and distinct clinical manifestations in patients with CVID, IgGD, IgG2D, and sAbD followed at a single tertiary medical center. The results of our study showed that patients with CVID presented with significantly higher frequency of hematopoietic malignancy than IgGD, IgG2D and sAbD. In addition, CVID patients with malignancy, autoimmunity and/or interstitial lung disease presented with lower NK cell counts. These results support the relationship between immune alterations and the development of non-infectious manifestations in patients with PADs.
Methods:
Subjects
Patients seen at the Yale Allergy & Immunology clinic between January 1, 2012 and December 31, 2018 and assigned ICD-9 codes (279.06, 279.03, 279.00) and ICD-10 codes for PADs (D83.9, D80.3, D80.6 and/or D80.1) were eligible for inclusion in the study. Medical records were examined using EPIC electronic medical record (Epic Systems Corporation Verona, Wisconsin USA). Protocols were reviewed and approved by the Institutional Review Board (IRB) of Yale University. Laboratory data were reviewed to assign subjects into different PAD subgroups; these included quantitative immunoglobulins and response to pneumococcal polysaccharide (PNA) vaccination. For PNA vaccine response, we used either 14 or 23 serotype panel that was available for both pre- and post- vaccination. Patients who had protective titers (≥ 1.3 microgram/mL) in less than 70% of serotypes were considered to have impaired PNA vaccine response [12].
Diagnoses were assigned using the criteria described in Table 1. CVID was defined with markedly decreased levels of IgG, IgA and/or IgM (2 standard deviations (SDs) below the mean for age) and low immune response to a PNA vaccine [7, 13, 14]. IgG deficiency was assigned if IgG levels were lower than 2 SD below the mean for age without meeting the criteria for CVID [5, 11, 15]. IgG2 deficiency was assigned if IgG subclass 2 levels were lower than 2 SD below the mean of age, with normal levels of total IgG and other IgG subclasses [16–18]. Specific antibody deficiency (sAbD) was assigned if a patient had a low immune response to a PNA vaccine but normal levels of IgG, IgA, and IgM [12]. Patients were excluded from analysis if chart review revealed an alternative cause for hypogammaglobulinemia such as intestinal loss, chronic corticosteroids use, pre-existing diagnoses of hematopoietic malignancies prior to the diagnosis of PADs or rituximab treatment. Patients without sufficient laboratory data (immunoglobulin levels and PNA vaccine response) were excluded.
Table 1.
Diagnoses criteria for each PAD.
| PAD | Diagnoses Criteria |
| CVID | IgG less than 2SD, IgA or IgM less than 2SD and <70% of Pneumovax serotype response |
| IgGD | IgG less than 2SD with IgA and IgM within normal limits |
| IgG2D | Total IgG within normal range and IgG subclass less than 2SD |
| SAbD | Total IgG, IgM and IgA within normal range and <70% of Pneumovax serotype response |
In our institution, two standard deviations (2SD) for IgG, IgA, IgM and IgG subclass 2 for age equal and greater than 18 years old were defined as 768–1623, 70–408, 40–263 and 241–700 mg/dL, respectively.
Laboratory data
Medical records were examined for levels of serum IgA, IgM, and IgG (including subclasses). Effective PNA vaccine response was defined as antibody titers to more than 70% of serotypes equal to or greater than 1.3 ug/mL [12]. The patients’ first results of flow cytometric analysis for absolute counts of T (CD3+, CD3+CD4+ and CD3+CD8+), B (CD19+ and CD19+CD27+IgD−IgM−), NKT (CD3+CD56+), and NK (CD3−CD56+) cells were recorded. These flow cytometric analyses were conducted prior to initiation of immunoglobulin therapy.
Clinical data
Clinical data were collected by reviewing problem list sections of medical records and clinic notes recorded by physicians in the specialties of primary care, allergy/immunology, oncology/hematology, rheumatology, and/or pulmonology (Figure 4). In addition to demographic information (age and gender), the collected clinical data included diagnoses of infections (recurrent sinopulmonary, latent viral or solid organ infection), interstitial lung disease (ILD), bronchiectasis, autoimmunity (sarcoidosis, splenomegaly/cytopenia, rheumatoid arthritis/atrophic arthritis, connective tissue diseases (systemic lupus erythematous, Sjögren’s disease, mixed connective tissue disorders and nonspecific connective tissues disorders), and malignancy (hematopoietic malignancy and solid organ malignancy).
Figure 4. Patients with co-morbid conditions have different immune parameters in CVID and IgG2D.
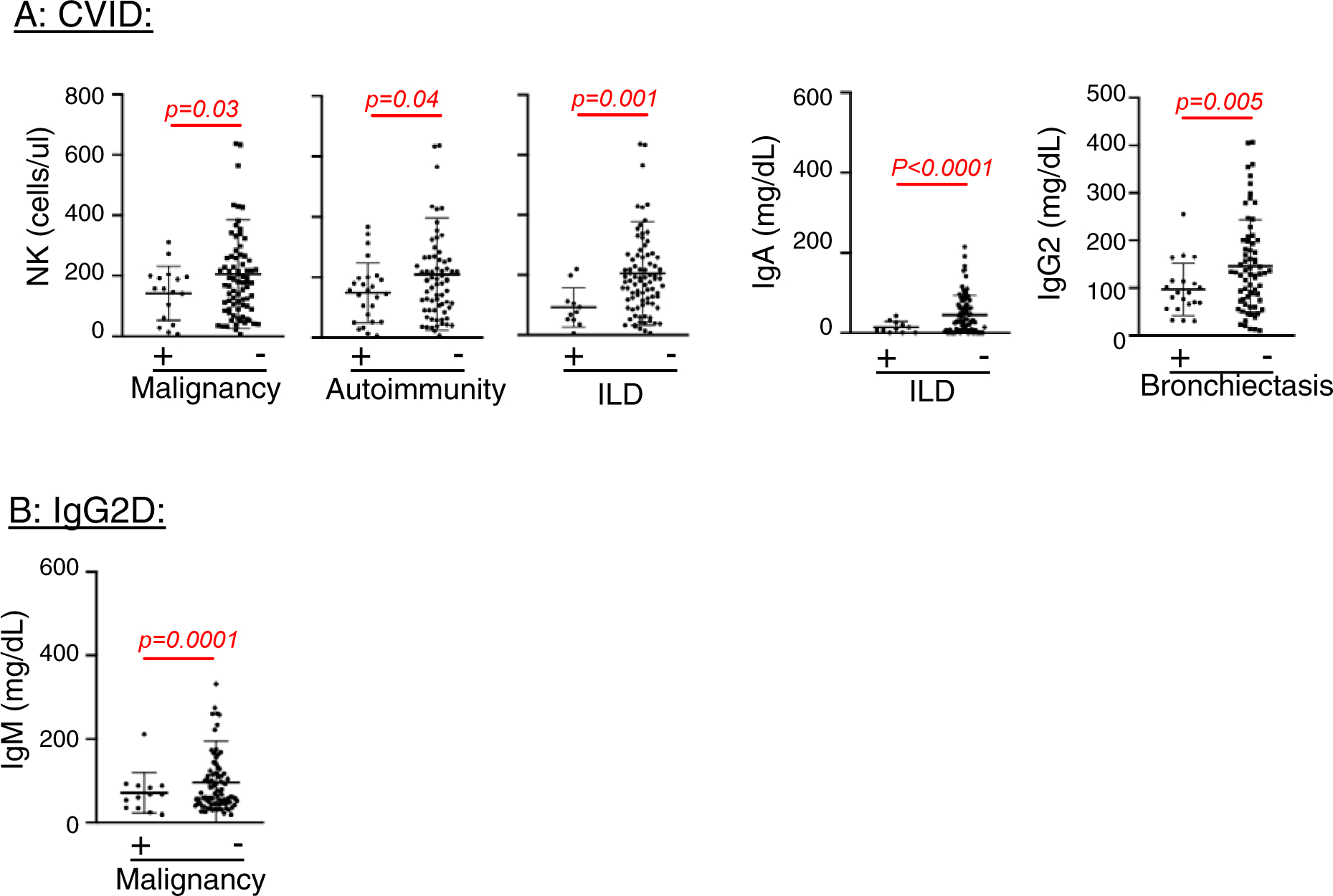
Immune parameters were compared between patients with or without indicated co-morbid conditions in CVID and IgG2D. Only the parameters which were different (P < 0.05) between the comparison groups are shown here.
Statistical analysis
Descriptive data were presented as means ± SDs. Continuous variables were analyzed using the Student t-test or one-way ANOVA with the Dunnett’s test for multiple comparisons, as appropriate. Categorical variables were analyzed using the Chi-square test. Data were analyzed with SPSS (IBM, Armonk, New York) and Prism 7 (GraphPad Software, Inc, San Diego, Calif). P values of 0.05 or less were considered statistically significant.
Results:
Characterization of patients in four groups of primary antibody deficiencies.
Six hundred and seventy-three subjects were identified with ICD-9 and ICD-10 codes relevant for PAD. The majority of them carried more than one diagnosis. They were then categorized into four groups as outlined in the methods section based on antibody levels and PNA vaccine response. After excluding patients with insufficient laboratory data and secondary causes of antibody deficiencies, the remaining 386 patients were categorized as follows: 105 CVID, 108 IgG2D, 129 IgGD, and 44 sAbD (Figure 1). The mean ages were similar in the five groups, (range of 19–93, mean of 56). Within each group, there was a female to male predominance with higher proportion of female gender (70–77%).
Figure 1. Extraction and grouping of patients by the diagnoses of primary antibody immunodeficiency.
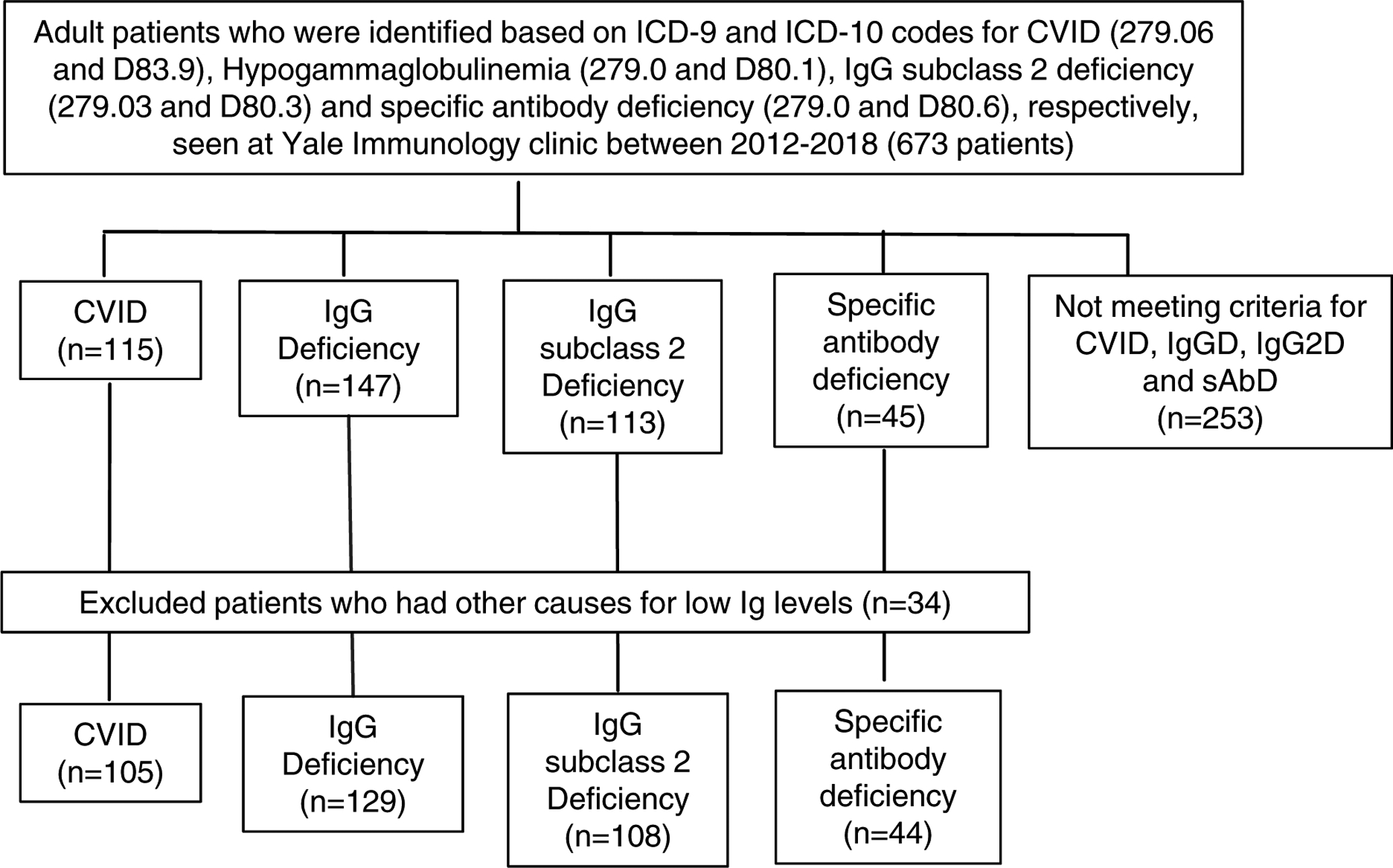
Patients were extracted from the electronic medical record based on PAD ICD-9 and ICD-10 codes. Subsequently, the patients with any secondary reasons for antibody deficiency were excluded. The remaining patients were classified into four different groups of PADs based on basal immunoglobulin levels and pneumococcal vaccine responses.
Humoral immune parameters were appropriately different in four PAD groups.
The immunoglobulin levels and PNA vaccine responses in the four groups were different as anticipated. IgG levels in the CVID and IgGD groups were significantly lower than that in the rest of the groups (Figure 2A). IgG levels of CVID were significantly lower than that in IgGD (Figure 2A, mean ± SD: 356.7 ± 143.3 vs 565.8 ± 123.6, p<0.0001). IgA and IgM levels in the CVID group were significantly lower than that in other groups (Figure 2A). The IgG subclass 1, 3, and 4 levels were significantly lower in the CVID and IgGD groups compared to those in the other groups (Figure 2A). IgG subclass 2 levels were similarly lower in CVID and IgG2D groups as compared to the other groups, respectively (Figure 2A). Of interest, IgG subclass 2 was the only IgG subclass whose levels were significantly lower in the CVID group than in the IgGD group (Figure 2A, 144.9 ± 106.0 vs 173.6 ± 70.5, p=0.02).
Figure 2. Analysis of serum immunoglobulin levels and peripheral immune cells subsets in patients with different PADs.
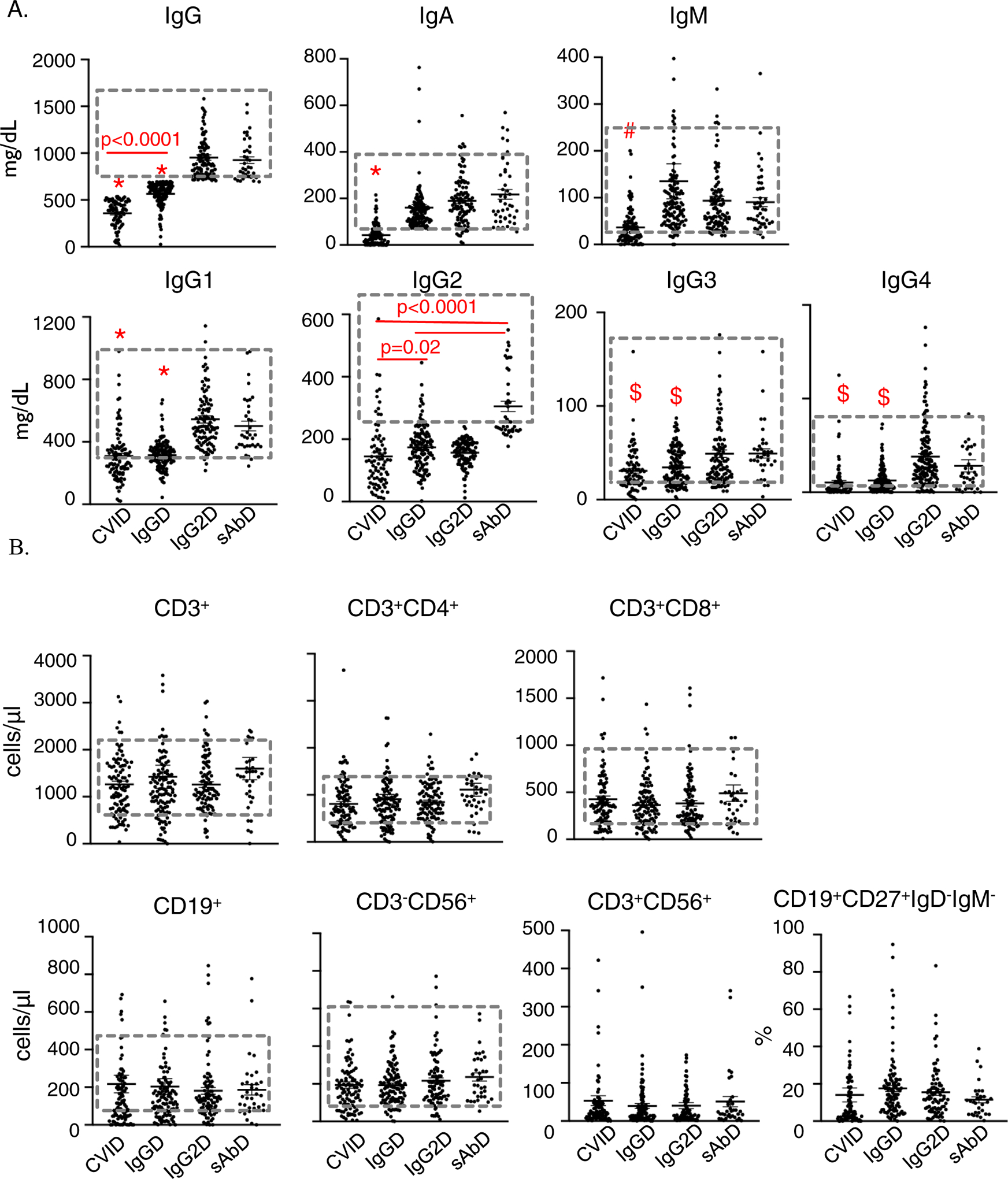
(A) *IgG, IgG1 and IgA levels of CVID are significantly lower than the levels of IgGD, IgG2D and sAbD (p<0.0001). *IgG and IgG1 level of IgGD is significantly lower than the level of IgG2D and sAbD (p<0.0001). #IgM level of CVID is significantly lower than the level of IgGD (p=0.035), *IgA level of CVID is significantly lower than IgA levels of IgGD (p<0.0001). $IgG3 and IgG4 levels of CVID and IgGD are significantly lower than the levels in IgG2D and sAbD (p<0.02). (B) Flow cytometric analysis of peripheral immune cell subsets in patients with different PADs were done. Absolute cell counts of peripheral immune cell subsets were determined by flow cytometry. Statistical analysis was done by the one-way ANOVA with the Dunnett’s test for multiple comparisons. Dotted boxes denote the normal range (2 standard deviations from the mean).
T-, B-, and NK- cell subsets in peripheral blood were similar among the four groups of PADs.
Of the 386 patients with available immunoglobulin levels and PNA vaccine response results, 325 patients had flow cytometric analysis on their peripheral blood (Figure 2B). The absolute cell counts for CD3+, CD3+CD4+, CD3+CD8+, CD19+, CD19+CD27+IgD−IgM−, CD3−CD56+ and CD3+CD56+ cells were compared among patients with CVID (n=92), IgGD (n=113), IgG2D (n=86) and sAbD (n=34) (Figure 2B). There was no difference in the absolute counts of all subsets of cells in the four groups of PADs (Figure 2B).
Comparison of clinical manifestations.
Clinical data on infections, autoimmunity, malignancy, ILD, and bronchiectasis were examined in all PAD groups (Figure 3). The frequencies of all infectious and non-infectious manifestations were similar in all groups of PADs except for hematopoietic malignancies. Reported malignancies in these groups include hematopoietic (leukemia and lymphoma), and solid organ tumors such as lung and gastrointestinal malignancies (Figure 3A). Our study showed a higher frequency of hematopoietic malignancies in the CVID group compared to other groups (IgGD, IgG2D, and sAbD) (Figure 3A, p=0.02). Most hematopoietic malignancies were lymphoma, with only 4 cases of leukemia. This finding of the increased frequency of hematopoietic malignancies in the CVID group is consistent with the results of published studies [11].
Figure 3. The proportions of co-morbid conditions are distinct in patients with different PADs.

The proportions of patients with malignancies (A), autoimmunity (B), infections (C), interstitial lung disease (ILD) (D) and bronchiectasis (E) were compared in individual groups of PADs. Statistical analysis was done by the Chi-square analysis.
Rates of autoimmune diseases were similar in all groups (Figure 3B), however there was a trend towards increased occurrences of CTDs in the IgGD, IgG2D, and sAbD groups as compared to the CVID group (Figure 3B). Infections including recurrent sinusitis, bronchitis, and pneumonia were found across all groups at a prevalence of 74–78% (Figure 3C). The frequencies of solid organ infections and viral infections with herpes simplex virus (HSV), Epstein-Barr virus (EBV), and cytomegalovirus (CMV) were low across all groups (Figure 3C). The frequencies of ILD and bronchiectasis were similar in the CVID, IgGD, and IgG2D groups, all seemingly higher than that in the sAbD group. However, these differences were not statistically significant. (Figure 3D–E). Overall, our findings support the consideration of careful clinical evaluations for non-infectious manifestations in both CVID and non-CVID PADs.
Patients with different clinical manifestations have distinct immune profiles.
Immunological profiles of patients in the CVID, IgGD, and IgG2D groups were each examined by clinical manifestations of malignancy, autoimmune disease, ILD, infections, and bronchiectasis. The sAbD group was excluded from this analysis due to small patient numbers (n=44). In the CVID group, NK cell counts were significantly lower in patients with malignancy (mean ± SD; 141 ± 87 vs. 205 ± 181 cells/μl, p=0.03), autoimmunity (148 ± 99 vs. 210 ± 186 cells/μl, p=0.04), and ILD (92 ± 69 vs. 205 ± 173 cells/μl, p=0.001) compared to those without these conditions (Figure 4A). In the same group, IgA levels were lower in patients with ILD than those without it (15 ± 14 vs. 46 ± 48 cells/μl, p<0.0001), while IgG2 levels were lower in patients with bronchiectasis than those without it (mean 97 ± 65 vs. 152 ± 110 mg/dL, p=0.005, Figure 4A). In the IgGD group, patients with infection had lower levels of IgG compared to those without infection (553.9 ± 131 vs. 615.5 ± 67.82mg/dL, p=0.024, Supplemental figure 1). In the IgG2D group, patients with malignancy had decreased IgM levels compared to those without malignancy (54 ± 19 vs 100 ± 125 mg/dL, p=0.0001, Figure 4B). These findings suggest a relationship between clinical manifestations of PAD and distinct immune alterations.
Individual patients with PAD may have multiple non-infectious manifestations. We thus explored whether certain non-infectious manifestations could occur together in CVID, IgGD and/or IgG2D. Compared to those without autoimmunity, CVID patients with autoimmunity were more likely to have malignancy (Figure 5A) although this relationship was not noticed in the IgGD and IgG2D. Regarding autoimmunity and ILD, patients with autoimmunity in the IgGD group were more likely to have ILD compared to those without autoimmunity (Figure 5B). There was no similar relationship noticed in the CVID and IgG2D groups. We also noticed the positive relationship between bronchiectasis and ILD in the IgGD and IgG2D groups (Figure 5C).
Figure 5. Autoimmunity, malignancy, interstitial lung disease and/or bronchiectasis occur together in patients with CVID, IgG2D and IgGD.
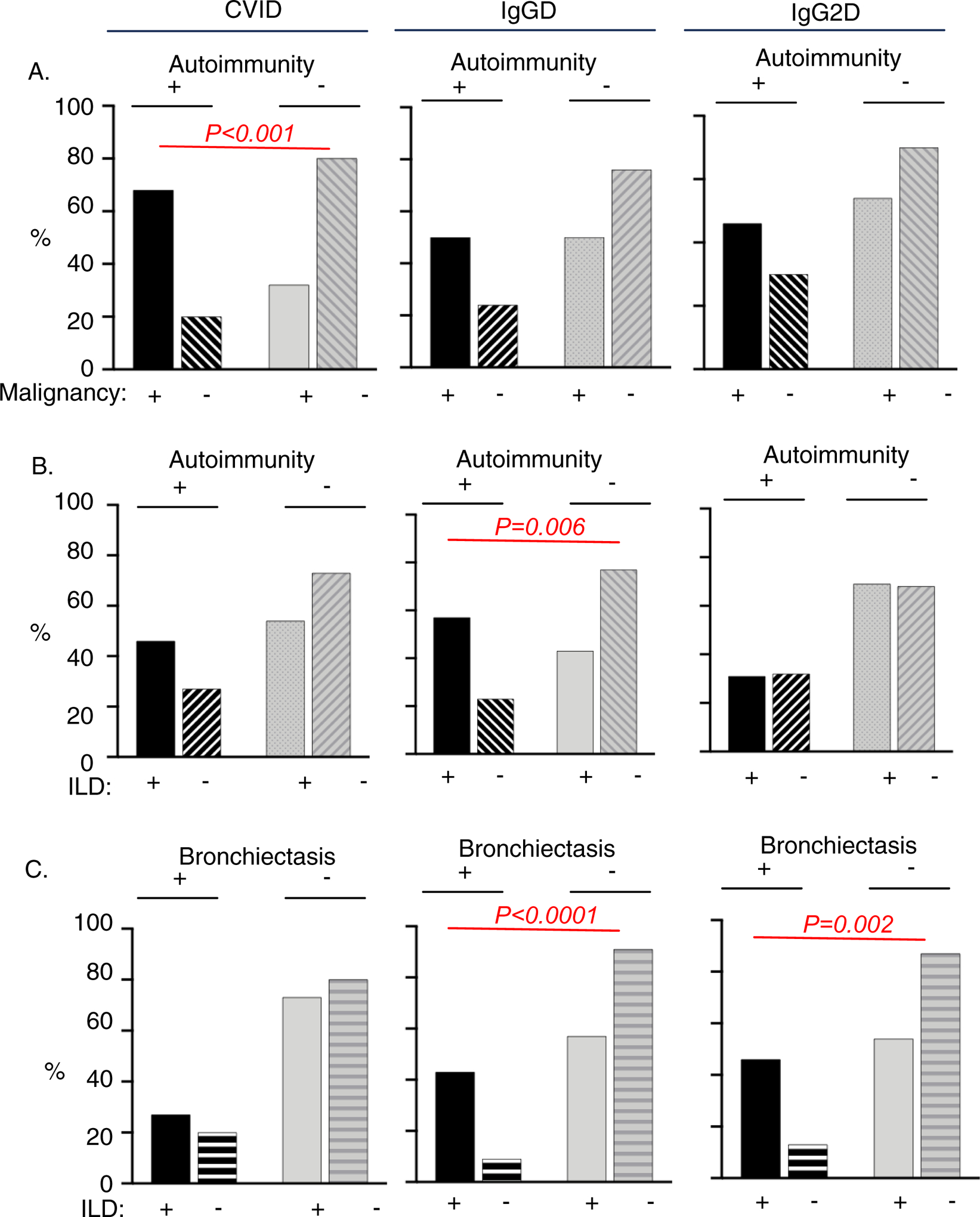
The proportions of patients with or without indicated co-morbid conditions were compared in CVID (A), IgG2D (B), and IgGD using the Chi-square analysis.
Discussion:
PADs represent a heterogeneous group of disorders including CVID, IgG subclass deficiency, IgGD, and sAbD. There is a paucity of literature exploring potential variation in the clinical and immunological characteristics in patients with different PADs. Here, we analyzed clinical and immunological data of a large cohort of patients with PADs followed at a tertiary medical center.
We found that the frequency of hematopoietic malignancies and CTDs, but not infectious diseases, differed by type of PAD. These results suggest that patients with IgGD, IgG2D and sAbD should be also routinely monitored and screened for possible non-infectious manifestations including autoimmunity, ILD and bronchiectasis as it is already emphasized in the patients with CVID. In a recent study, Filion et al. reported a higher frequency of non-infectious manifestations in patients with CVID than in patients with IgGD. Of note, the frequency of non-infectious manifestations was higher in this study than in ours (58.6% vs. 40%), which could account for our observation of similar frequencies of non-infectious manifestations in CVID and IgGD except hematologic malignancy. In patients with CVID, NK cell counts were lower in patients with malignancy, autoimmunity and/or ILD compared to those without these conditions. Also, in patients with CVID, IgGD, and IgG2D, immunoglobulin levels were associated with distinct clinical manifestations, including malignancy, ILD, infection, and bronchiectasis. These results support our hypothesis of an existing association between immune alterations and the development of infectious and non-infectious manifestations in patients with different types of PADs.
As expected, IgG levels were lower in the CVID and IgGD than the other groups in our study. However, IgG levels were significantly lower in CVID than IgGD, which are in line with the results of a recent study [11]. We also found lower IgG2 levels in the CVID group than in the IgGD group, although both groups had similar levels of other IgG subclasses (1, 3, and 4). This finding suggests the possible utility in measuring IgG subclasses for diagnosis and management of patients with PADs.
In the literature, IgG subclass 2 has been associated with specific antibody production to the polysaccharide pneumococcal vaccine [19], and patients with selective IgA deficiency in combination with IgG subclass 2 deficiency have been more symptomatic with recurrent sinopulmonary infections, autoimmunity, and malabsorption [20]. Our results suggest that patients with concurrent IgGD diagnosis and low levels of IgG subclass 2 and IgA, who do not meet the criteria for CVID diagnosis, should be monitored closely for progression to CVID and development of infectious and non-infectious complications.
In analyzing flow cytometry data, there was no difference in the counts of all the measured immune cell subsets in our four studied groups. These findings indicate the limited utility of routine flow cytometric analysis for CD4, CD8, B, NK, and NKT cells in differentiating different types of PADs; rather, in-depth flow cytometric analysis is needed to characterize immune cell subsets in PADs. Indeed, detailed immune phenotyping studies identified an increased frequency of CD45RO+CD27− memory CD4+ and CD8+ T cells in CVID [21] as well as an association of circulating follicular helper T cells with autoimmune cytopenia in CVID [22], as compared to other subgroups. Also, patients with various PADs had different numbers of memory B and plasma cells that expressed distinct immunoglobulin subclasses [23]. Two recent studies reported decreased isotype-switched memory B cells in CVID patients [11, 23]. A similar finding was not noticed in our study by comparing the CVID, IgGD and IgG2D groups (Fig 2B). However, in analyzing patients with infections, patients with CVID had lower levels of switched memory B cells compared to those with IgGD and IgG2D (mean (%) ± SD, 8 ± 9 vs. 16 ± 15 P = 0.01; 8 ± 9 vs.15 ± 15 P = 0.01) (Supplement Table 4).
Further examining the flow cytometry data, it is possible that increased frequency of hematopoietic malignancy in our patients with CVID could be related to alterations in immune cells, including T and NK cells. In fact, CVID patients with malignancy had a decreased frequency of NK cells which play a critical role in tumor immunity, compared to those without malignancy. Of interest, the frequency of NK cells was also lower in CVID patients with autoimmunity and ILD than in CVID patients without these conditions [24–26]. The decreased frequency of NK cells could be secondary to the possible co-existence of autoimmunity and malignancy in our CVID patients [24]. Alternatively, the altered NK cell counts could contribute to both autoimmunity and ILD in that NK cells can either promote or suppress autoimmunity and inflammation depending on the immune microenvironment, including the cytokine milieu [27].
CVID patients with ILD and bronchiectasis had decreased levels of IgA and IgG2, respectively. A previous study reported decreased IgA levels in patients with granulomatous lymphocytic interstitial lung disease (GLILD) in CVID [28]. GLILD, a noninfectious complication occurring in 10–30% of patients with CVID, is diagnosed with high-resolution computed tomography (CT) and/or lung biopsy [28, 29]. In most of our patients, detailed information on high resolution chest CT was unavailable since our electronic medical record system includes the results of imaging studies starting from 2012. Despite this limitation, our data still suggest that low IgA levels could be a predictor for ILD in patients with CVID. In addition, studying genetic mutations in these patients needs to be considered since both CVID and IgAD have been associated with genetic defects of transmembrane activator and CAML interaction (TACI), one of the mutations associated with ILD [30, 31].
Our finding of the association between low IgG2 levels and bronchiectasis in CVID patients is also supported by previous studies. In a previously published study of 65 patients with IgG2 deficiency, 19 (26%) presented with bronchiectasis [32]. Also, 90 percent of PI3Kδ mutation-associated CVID patients with low IgG2 levels had bronchiectasis [32, 33]. The association of immunoglobulin levels with ILD and bronchiectasis supports the current guideline of keeping a higher trough IgG level for CVID patients with pulmonary complications [34]. In IgGD, patients with a history of infections presented with significantly lower IgG levels compared to those without infections, suggesting the implication of IgG levels in developing infectious complications. In our study, patients with IgG2D who had malignancy also had decreased IgM levels. Both B cell counts and IgM secretion are known to decrease with aging; therefore, this finding could be related to the increased propensity to develop malignancy with aging [35]. Measuring IgM levels in IgG2D may be useful for monitoring non-infectious manifestations including malignancy.
We noticed a trend towards increased frequency of CTDs in the IgGD, IgG2D, and sAbD groups compared to the CVID group. It has been well-recognized that cytopenias, splenomegaly, and granulomatous disease are the most common autoimmune manifestations in CVID [36–38]. The United States Immunodeficiency Network (USIDNET) CVID cohort study found rheumatologic disorders in 5.8% of CVID patients [39]. Also, an Iranian PID registry study reported that 10.1% of CVID patients had a rheumatologic disease [40]. However, the prevalence of rheumatologic disorders in the non-CVID PADs is largely unknown. Autoimmunity was reported in 15% of patients with IgG deficiency [11]. Janssen et.al. reported an increased frequency of pain, fatigue, cognitive dysfunction, and vitality, which are commonly associated with rheumatic diseases in patients with unclassified PADs including deficiencies in IgG, selective IgM, IgA or IgG subclasses, and specific antibody deficiency [5].
Limitations of our study include its design as a retrospective, single center, cohort study. The immunology clinic is located at a multidisciplinary immune center, which receives referrals from rheumatology and hematology-oncology in addition to community physicians and other specialties. Thus, our patient population may represent a biased group with higher cases of non-infectious diseases including rheumatologic, hematologic and/or oncological conditions. Our cohort had a higher proportion of women compared to other studies reporting a similar proportion of men and women with PAD [3, 6]. However, female predominance (70.7% females) was also found in the Partner’s CVID cohort [41]. We also had the limitation of collecting medical records of these patients which were done prior to 2012.
To the best of our knowledge, this is the first study comparing multiple immunological parameters and clinical characteristics in the four groups of PADs including CVID, IgG2D, IgGD, and sAbD from a large cohort. Our findings suggest the association of immune alterations with critical non-infectious manifestations in less frequently studied PADs such as IgGD, IgG2D, and sAbD. In fact, both CVID and non-CVID PID groups had similar frequencies of non-infectious manifestations except for hematologic malignancies, supporting the importance of careful clinical evaluations for non-infectious diseases such as autoimmunity, malignancies, and lung diseases in patients with any type of PIDs. Our study also suggests that in-depth immune profiling may be necessary to identify immune-pathogenesis underlying unique clinical manifestations in patients with different PADs.
Supplementary Material
Acknowledgements:
This work was supported in part by grants from the National Institutes of Health (1R01AG056728, and R01AG055362 to IK).
We thank Caitlin Partridge in the Joint Data Analytics Team of the Yale Center for Clinical Investigation for her assistance in collecting data from electronic medical records. We also thank Dr. Veronika Shabanova for her advice on statistical analysis.
Footnotes
Disclosure of conflicts of interest:
The authors declare no competing financial interests.
References:
- 1.Picard C, Al-Herz W, Bousfiha A, Casanova JL, Chatila T, Conley ME, et al. Primary Immunodeficiency Diseases: an Update on the Classification from the International Union of Immunological Societies Expert Committee for Primary Immunodeficiency 2015. J Clin Immunol. 2015;35(8):696–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Picard C, Bobby Gaspar H, Al-Herz W, Bousfiha A, Casanova JL, Chatila T, et al. International Union of Immunological Societies: 2017 Primary Immunodeficiency Diseases Committee Report on Inborn Errors of Immunity. J Clin Immunol. 2018;38(1):96–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Durandy A, Kracker S, Fischer A. Primary antibody deficiencies. Nat Rev Immunol. 2013;13(7):519–33. [DOI] [PubMed] [Google Scholar]
- 4.Wood P, Stanworth S, Burton J, Jones A, Peckham DG, Green T, et al. Recognition, clinical diagnosis and management of patients with primary antibody deficiencies: a systematic review. Clin Exp Immunol. 2007;149(3):410–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Janssen LMA, Bassett P, Macken T, van Esch J, Pruijt H, Knoops A, et al. Mild Hypogammaglobulinemia Can Be a Serious Condition. Front Immunol. 2018;9:2384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cunningham-Rundles C Human B cell defects in perspective. Immunol Res. 2012;54(1–3):227–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bonilla FA, Barlan I, Chapel H, Costa-Carvalho BT, Cunningham-Rundles C, de la Morena MT, et al. International Consensus Document (ICON): Common Variable Immunodeficiency Disorders. J Allergy Clin Immunol Pract. 2016;4(1):38–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chapel H, Cunningham-Rundles C. Update in understanding common variable immunodeficiency disorders (CVIDs) and the management of patients with these conditions. Br J Haematol. 2009;145(6):709–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Singh K, Chang C, Gershwin ME. IgA deficiency and autoimmunity. Autoimmun Rev. 2014;13(2):163–77. [DOI] [PubMed] [Google Scholar]
- 10.Yel L Selective IgA deficiency. J Clin Immunol. 2010;30(1):10–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Filion CA, Taylor-Black S, Maglione PJ, Radigan L, Cunningham-Rundles C. Differentiation of Common Variable Immunodeficiency From IgG Deficiency. J Allergy Clin Immunol Pract. 2019;7(4):1277–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Orange JS, Ballow M, Stiehm ER, Ballas ZK, Chinen J, De La Morena M, et al. Use and interpretation of diagnostic vaccination in primary immunodeficiency: a working group report of the Basic and Clinical Immunology Interest Section of the American Academy of Allergy, Asthma & Immunology. J Allergy Clin Immunol. 2012;130(3 Suppl):S1–24. [DOI] [PubMed] [Google Scholar]
- 13.Ameratunga R, Brewerton M, Slade C, Jordan A, Gillis D, Steele R, et al. Comparison of diagnostic criteria for common variable immunodeficiency disorder. Front Immunol. 2014;5:415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ameratunga R, Gillis D, Steele R. Diagnostic criteria for common variable immunodeficiency disorders. J Allergy Clin Immunol Pract. 2016;4(5):1017–8. [DOI] [PubMed] [Google Scholar]
- 15.Gathmann B, Grimbacher B, Beaute J, Dudoit Y, Mahlaoui N, Fischer A, et al. The European internet-based patient and research database for primary immunodeficiencies: results 2006–2008. Clin Exp Immunol. 2009;157 Suppl 1:3–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Primary immunodeficiency diseases. Report of an IUIS Scientific Committee. International Union of Immunological Societies. Clin Exp Immunol. 1999;118 Suppl 1:1–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Olinder-Nielsen AM, Granert C, Forsberg P, Friman V, Vietorisz A, Bjorkander J. Immunoglobulin prophylaxis in 350 adults with IgG subclass deficiency and recurrent respiratory tract infections: a long-term follow-up. Scand J Infect Dis. 2007;39(1):44–50. [DOI] [PubMed] [Google Scholar]
- 18.Agarwal S, Cunningham-Rundles C. Assessment and clinical interpretation of reduced IgG values. Ann Allergy Asthma Immunol. 2007;99(3):281–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Edwards E, Razvi S, Cunningham-Rundles C. IgA deficiency: clinical correlates and responses to pneumococcal vaccine. Clin Immunol. 2004;111(1):93–7. [DOI] [PubMed] [Google Scholar]
- 20.Oxelius VA, Laurell AB, Lindquist B, Golebiowska H, Axelsson U, Bjorkander J, et al. IgG subclasses in selective IgA deficiency: importance of IgG2-IgA deficiency. N Engl J Med. 1981;304(24):1476–7. [DOI] [PubMed] [Google Scholar]
- 21.Nechvatalova J, Pavlik T, Litzman J, Vlkova M. Terminally differentiated memory T cells are increased in patients with common variable immunodeficiency and selective IgA deficiency. Cent Eur J Immunol. 2017;42(3):244–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Le Coz C, Bengsch B, Khanna C, Trofa M, Ohtani T, Nolan BE, et al. Common variable immunodeficiency-associated endotoxemia promotes early commitment to the T follicular lineage. J Allergy Clin Immunol. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Blanco E, Perez-Andres M, Arriba-Mendez S, Serrano C, Criado I, Del Pino-Molina L, et al. Defects in memory B-cell and plasma cell subsets expressing different immunoglobulin-subclasses in patients with CVID and immunoglobulin subclass deficiencies. J Allergy Clin Immunol. 2019;144(3):809–24. [DOI] [PubMed] [Google Scholar]
- 24.Ebbo M, Gerard L, Carpentier S, Vely F, Cypowyj S, Farnarier C, et al. Low Circulating Natural Killer Cell Counts are Associated With Severe Disease in Patients With Common Variable Immunodeficiency. EBioMedicine. 2016;6:222–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bateman EA, Ayers L, Sadler R, Lucas M, Roberts C, Woods A, et al. T cell phenotypes in patients with common variable immunodeficiency disorders: associations with clinical phenotypes in comparison with other groups with recurrent infections. Clin Exp Immunol. 2012;170(2):202–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kellner ES, Fuleihan R, Cunningham-Rundles C, Consortium U, Wechsler JB. Cellular Defects in CVID Patients with Chronic Lung Disease in the USIDNET Registry. J Clin Immunol. 2019;39(6):569–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gianchecchi E, Delfino DV, Fierabracci A. NK cells in autoimmune diseases: Linking innate and adaptive immune responses. Autoimmun Rev. 2018;17(2):142–54. [DOI] [PubMed] [Google Scholar]
- 28.Hartono S, Motosue MS, Khan S, Rodriguez V, Iyer VN, Divekar R, et al. Predictors of granulomatous lymphocytic interstitial lung disease in common variable immunodeficiency. Ann Allergy Asthma Immunol. 2017;118(5):614–20. [DOI] [PubMed] [Google Scholar]
- 29.Maglione PJ, Overbey JR, Cunningham-Rundles C. Progression of Common Variable Immunodeficiency Interstitial Lung Disease Accompanies Distinct Pulmonary and Laboratory Findings. J Allergy Clin Immunol Pract. 2015;3(6):941–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Castigli E, Wilson SA, Garibyan L, Rachid R, Bonilla F, Schneider L, et al. TACI is mutant in common variable immunodeficiency and IgA deficiency. Nat Genet. 2005;37(8):829–34. [DOI] [PubMed] [Google Scholar]
- 31.Pan-Hammarstrom Q, Salzer U, Du L, Bjorkander J, Cunningham-Rundles C, Nelson DL, et al. Reexamining the role of TACI coding variants in common variable immunodeficiency and selective IgA deficiency. Nat Genet. 2007;39(4):429–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.De Gracia J, Rodrigo MJ, Morell F, Vendrell M, Miravitlles M, Cruz MJ, et al. IgG subclass deficiencies associated with bronchiectasis. Am J Respir Crit Care Med. 1996;153(2):650–5. [DOI] [PubMed] [Google Scholar]
- 33.Angulo I, Vadas O, Garcon F, Banham-Hall E, Plagnol V, Leahy TR, et al. Phosphoinositide 3-kinase delta gene mutation predisposes to respiratory infection and airway damage. Science. 2013;342(6160):866–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Busse PJ, Farzan S, Cunningham-Rundles C. Pulmonary complications of common variable immunodeficiency. Ann Allergy Asthma Immunol. 2007;98(1):1–8; quiz −11, 43. [DOI] [PubMed] [Google Scholar]
- 35.Rodriguez-Zhurbenko N, Quach TD, Hopkins TJ, Rothstein TL, Hernandez AM. Human B-1 Cells and B-1 Cell Antibodies Change With Advancing Age. Front Immunol. 2019;10:483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Resnick ES, Moshier EL, Godbold JH, Cunningham-Rundles C. Morbidity and mortality in common variable immune deficiency over 4 decades. Blood. 2012;119(7):1650–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Feuille EJ, Anooshiravani N, Sullivan KE, Fuleihan RL, Cunningham-Rundles C. Autoimmune Cytopenias and Associated Conditions in CVID: a Report From the USIDNET Registry. J Clin Immunol. 2018;38(1):28–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wehr C, Kivioja T, Schmitt C, Ferry B, Witte T, Eren E, et al. The EUROclass trial: defining subgroups in common variable immunodeficiency. Blood. 2008;111(1):77–85. [DOI] [PubMed] [Google Scholar]
- 39.Gutierrez MJ, Sullivan KE, Fuleihan R, Consortium U, Bingham CO 3rd., Phenotypic characterization of patients with rheumatologic manifestations of common variable immunodeficiency. Semin Arthritis Rheum. 2018;48(2):318–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Azizi G, Kiaee F, Hedayat E, Yazdani R, Dolatshahi E, Alinia T, et al. Rheumatologic complications in a cohort of 227 patients with common variable immunodeficiency. Scand J Immunol. 2018;87(5):e12663. [DOI] [PubMed] [Google Scholar]
- 41.Farmer JR, Ong MS, Barmettler S, Yonker LM, Fuleihan R, Sullivan KE, et al. Common Variable Immunodeficiency Non-Infectious Disease Endotypes Redefined Using Unbiased Network Clustering in Large Electronic Datasets. Front Immunol. 2017;8:1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.