

Abstract
Immune cells use a variety of membrane-disrupting proteins [complement, perforin, perforin-2, granulysin, gasdermins, mixed lineage kinase domain-like pseudokinase (MLKL)] to induce different kinds of death of microbes and host cells, some of which cause inflammation. After activation by proteolytic cleavage or phosphorylation, these proteins oligomerize, bind to membrane lipids, and disrupt membrane integrity. These membrane disruptors play a critical role in both innate and adaptive immunity. Here we review our current knowledge of the functions, specificity, activation, and regulation of membrane-disrupting immune proteins and what is known about the mechanisms behind membrane damage, the structure of the pores they form, how the cells expressing these lethal proteins are protected, and how cells targeted for destruction can sometimes escape death by repairing membrane damage.
Keywords: complement, perforin, gasdermin, MLKL, granulysin, cytotoxic lymphocyte, necrosis, apoptosis, pyroptosis, necroptosis, microptosis
1. INTRODUCTION AND OVERVIEW
The immune system kills both microbial and host cells to control infection and tumors and sound alarms to recruit immune cells and activate a protective response to pathogens and other signs of danger. Until recently, cell death was divided into necrosis in which the cell membrane is rapidly ruptured as the primary event, leading to inflammatory release of intracellular contents, and apoptosis, a noninflammatory programmed cell death in which the dying cell actively participates. Programmed cell death has characteristic morphological changes (cell membrane blebbing, cell shrinkage, nuclear fragmentation, chromatin condensation) and is often mediated by disruption of the mitochondrial outer membrane and/or effector caspase activation, but it can be activated by other proteases, such as killer cell granzymes, independently of caspases. Cells undergoing these cell death programs are recognized and removed by phagocytic cells long before their cell membranes become permeabilized. Consequently, apoptosis and killer lymphocyte-mediated cell death have been thought of as noninflammatory, since secondary necrosis, if it occurs, happens inside phagosomes, and cellular contents are therefore not released into the extracellular milieu. The mechanistic basis for the eventual membrane disruption in secondary necrosis is not understood. In recent decades new forms of regulated cell death have emerged that include programmed inflammatory necrosis, necroptosis, and pyroptosis, as well as other forms of caspase-independent programmed cell death.
The execution of these diverse cell death pathways relies on a handful of molecules that permeabilize membranes of cells targeted for immune elimination—complement, perforin, perforin-2, granulysin, gasdermins, and mixed lineage kinase domain-like pseudokinase (MLKL) (1–6) (Figure 1; Table 1). Each of these proteins primarily activates a distinct type of cell death: Complement punches holes in microbial or mammalian membranes to cause necrosis; perforin delivers granzymes to mammalian cells to induce caspase-independent and -dependent programmed cell death that resembles classical apoptosis; granulysin triggers both necrosis and programmed cell death in microbes (microptosis), gasdermins trigger pyroptosis induced after sensing invasive infection or cytosolic danger signals that activate inflammatory caspases, and MLKL mediates necroptosis induced by death receptor signaling when caspase-8 is inhibited (Figure 2; Table 2). Some of these proteins multimerize and insert into the target membrane to form large pores in the shape of β barrels (complement, perforin-1 and −2, gasdermins); others are smaller α-helical proteins (granulysin, MLKL) that oligomerize and damage cell membranes through poorly understood mechanisms that may or may not involve transmembrane pores (7–14). Some of these proteins only efficiently disrupt microbial membranes (complement, granulysin), some only work on mammalian membranes (perforin), and some work on both (gasdermins) (2, 15–18). Some work primarily only by disrupting the targeted membrane (complement, gasdermins, MLKL), while others (perforin, granulysin) use their membrane-disrupting ability to deliver death-inducing enzymes that activate programmed cell death into target cells (2, 19). The strict separation of the death pathways in which these membrane-damaging molecules participate into inflammatory or noninflammatory cell death is no longer valid. For example, during classical apoptosis, caspase-3 activates gasdermin E (GSDME). GSDME processing converts relatively slow noninflammatory apoptosis to a more rapid inflammatory pyroptotic death in which the cell swells and forms bubbles, releases cytosolic molecules, and eventually bursts (20, 21). Another example of crossover between inflammatory and noninflammatory death pathways is caspase-8 activation by the inflammasome when caspase-1 is inhibited (22, 23). Cell membrane damage of any sort will activate secondary pyroptosis in any cell that expresses the NLRP3 NOD-like receptor, which senses ionic changes in membrane-damaged cells (24–26).
Figure 1.
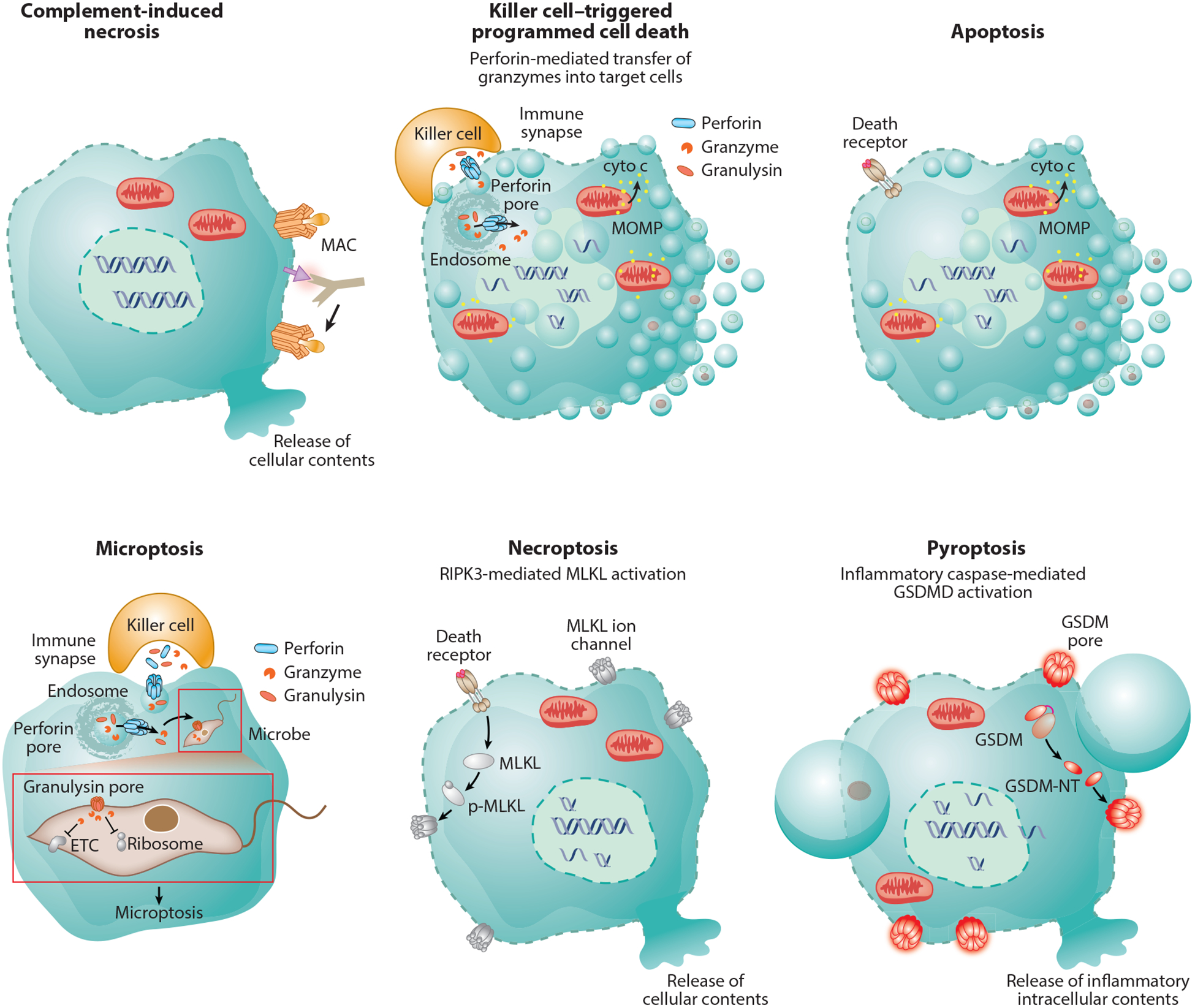
Immune membrane damage-mediated cell death. Complement-mediated lysis is a classic example of necrotic cell death characterized by cell membrane rupture. Perforin delivers granzymes to induce programmed target-cell death that resembles apoptosis morphologically with cell shrinkage, membrane blebbing, chromatin condensation, and nuclear fragmentation. Granzyme B also causes apoptosis—it activates caspase-3 and also directly cleaves many caspase-3 substrates to cause mitochondrial outer membrane permeabilization and oligonucleosomal double-stranded DNA breaks, visualized on gels as DNA ladders. Perforin also delivers granulysin into the target cell. Granulysin permeabilizes the cell membrane of intracellular microbes to cause microbial necrosis and allows the granzymes to enter the microbe to proteolytically cause microptosis, programmed cell death of bacteria, fungi, and parasites. Death receptor ligation causes classic apoptosis if caspase-8 or −10 is activated or necroptosis if these caspases are inhibited. In necroptosis, MLKL is phosphorylated, binds to membranes, and perforates them probably by forming ion channels that cause swelling and hypotonic lysis. During pyroptosis, activated in response to invasive pathogens and sterile danger signals, gasdermins are cleaved and the N-terminal fragment forms large plasma membrane pores. Pyroptotic membranes form large ballooning structures, while apoptotic membranes cause small blebs. Because proteins are released through gasdermin pores, the pyroptotic cell likely maintains osmolarity and does not burst. Abbreviations: cyto c, cytochrome C; ETC, electron transport chain; GSDM, gasdermin; GSDM-NT, GSDM N-terminal fragment; GSDMD, gasdermin D; MAC, membrane attack complex; MLKL, mixed lineage kinase domain-like pseudokinase; MOMP, mitochondrial outer membrane permeabilization; p-MLKL, phospho-MLKL.
Table 1.
Immune membrane-disrupting proteins
| Name | Type of membrane damage | Activation pathway | Inflammatory? | Type of cell death | Biological function | References |
|---|---|---|---|---|---|---|
| Complement | β-Barrel pore | Proteolytic complement cascade | Yes | Necrosis | Lyse bacteria and opsonized cells | 17, 32, 37, 45, 172, 173 |
| Perform | β-Barrel pore | Killer cell attack and degranulation Ca2 +-dependent |
No | Killer cell programmed cell death | Eliminate infected and cancerous cells | 13, 47, 59, 64, 69, 70 |
| Perforin-2 | Presumed β-barrel pore | Not known | Not known | Not known | Enhance clearance of invading bacteria | 31, 75, 77, 78, 79 |
| Gasdermins | β-Barrel pore | Inflammasome-inflammatory caspase-GSDM cleavage Apoptosis-capspase-3-GSDME |
Yes | Pyroptosis | Recruit immune cells and activate response to infection and sterile signs of danger | 7, 14, 18, 19, 23, 24 |
| MLKL | Ion channel? | TNFRSF-RIPK1/R1PK3 DAI-RIPK3 TLR-TRIF/RIPK3 |
Yes | Necroptosis | Cell death in setting of caspase inactivation Involved in infection, inflammation, sepsis, ischemia injury, neurodegeneration |
10, 16, 112, 113, 117, 118 |
| Granulysin | Not known | Killer cell degranulation | Yes | Microptosis | Kill intracellular and possibly cell-free bacteria, fungi, and parasites | 9, 11, 137, 150, 151 |
Abbreviations: GSDM, gasdermin; GSDME, gasdermin E; MLKL, mixed lineage kinase domain-like pseudokinase; TLR, Toll-like receptor; TNFRSF, tumor necrosis factor receptor superfamily.
Figure 2.
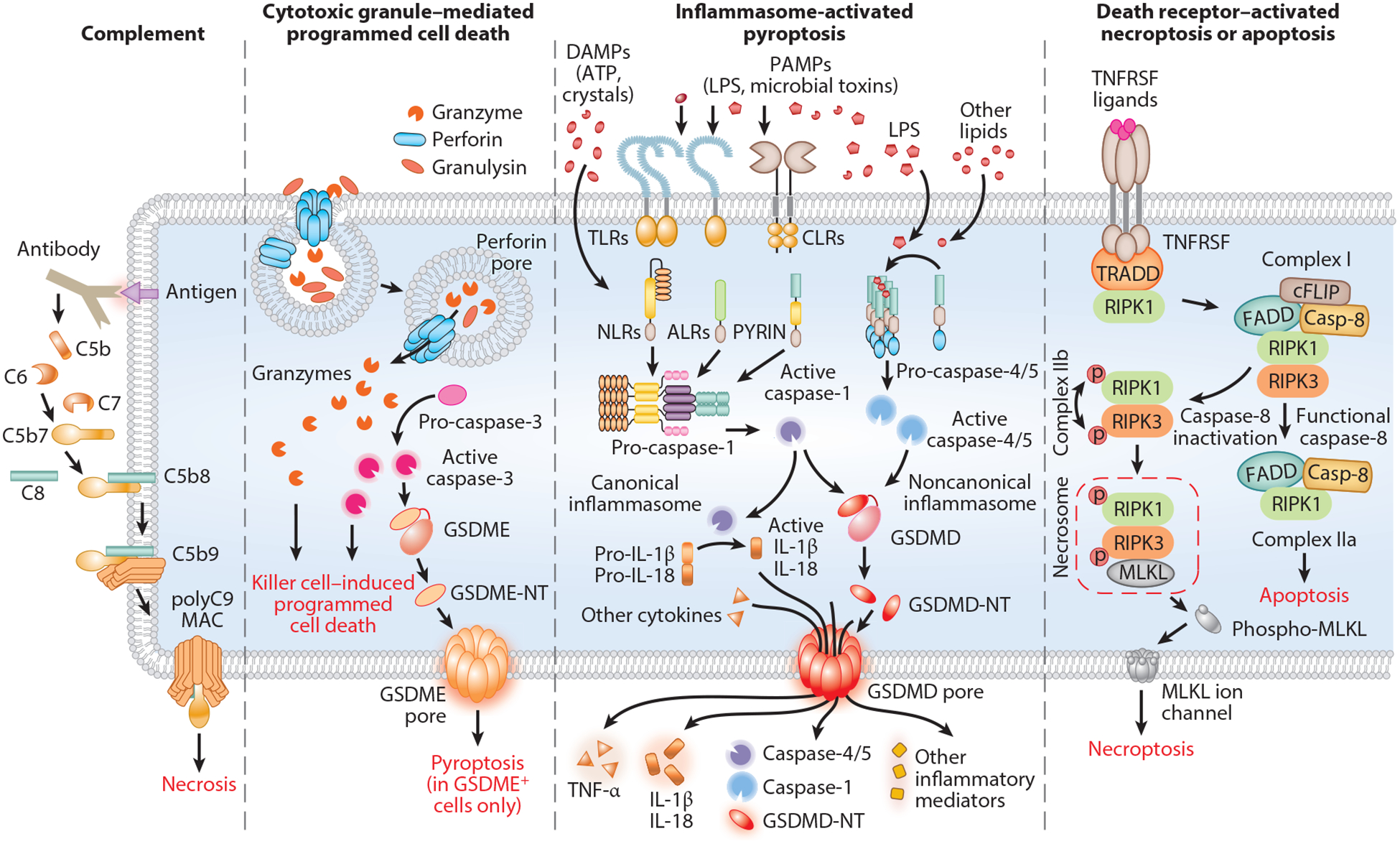
Molecular mechanisms that activate membrane damage. The complement system is activated by the classic, lectin, or alternative pathway, all of which share a common terminal pathway in which C5 is cleaved into the chemoattractant C5a and the pore-initiating protein C5b. C5b then sequentially binds C6, C7, and C8, which assemble on the target membrane and recruit C9, which oligomerizes to form the MAC pore. Killer cells release cytotoxic granules’ contents (perforin, granzymes, granulysin) into the immune synapse formed with the target cell. Perforin oligomerizes to form short-lived pores in the target cell membrane that trigger membrane repair, which activates endocytosis of the damaged membrane with bound perforin, granzymes, and granulysin. Perforin then forms pores in the endosome, releasing granzymes and granulysin into the target cell cytosol. Cytosolic granzymes induce both caspase-independent and -dependent programmed cell death by cleaving and activating downstream effector proteins. In target cells expressing GSDME, granzyme B-activated caspase-3 can cleave and activate GSDME to convert noninflammatory cell death to inflammatory pyroptosis. In response to cytosolic sensing of microbial infection or danger signals, canonical or noncanonical inflammasomes assemble to activate inflammatory caspases (caspase-1 and caspases-4/−5/−11, respectively). Activated inflammatory caspases cleave GSDMD, releasing its N-terminal fragment, GSDMD-NT, which forms pores in the plasma membrane to cause pyroptosis. Ligand-bound death receptors direct the recruitment and formation of distinct complexes that activate necroptosis or apoptosis. Complex I contains TRADD, RIPK1 and E3 ubiquitin ligases, which inhibit apoptosis by poly-ubiquitinating RIPK1. If RIPK1 is not ubiquitinated, complex IIa activates caspase-8 and initiates apoptosis, whereas when caspase-8 activation is blocked, complex IIb assembles when RIPK1 recruits and phosphorylates RIPK3, which initiates necroptosis by phosphorylating MLKL. Abbreviations: ALR, AIM2-like receptor; CLR, C-type lectin receptor; DAMP, danger-associated molecular pattern; GSDMD, gasdermin D; GSDMD-NT, GSDMD N-terminal fragment; GSDME, gasdermin E; GSDME-NT, GSDME N-terminal fragment; LPS, lipopolysaccharide; MAC, membrane attack complex; MLKL, mixed lineage kinase domain-like pseudokinase; NLR, nucleotide-binding oligomerization domain-like receptor; PAMP, pathogen-associated molecular pattern; TLR, Toll-like receptor; TNFRSF, tumor necrosis factor receptor superfamily; TRADD, TNF-receptor-associated death domain.
Table 2.
Types of cell death
| Type | Immune effectors | Morphological features | Molecular features | Characteristics |
|---|---|---|---|---|
| Complement-induced necrosis | Complement | Cell swelling, plasma membrane rupture | Cell membrane pores | Inflammatory |
| Killer cell-induced programmed cell death | Perforin/granzymes | Cell shrinkage, membrane blebbing, chromatin condensation, nuclear fragmentation | Caspase activation (granzyme B), mitochondrial ROS, loss of mi to Δψ, proteolytic cascade, global mRNA decay, MOMP, cytochrome C release, genomic DNA fragmentation | Noninflammatory |
| Apoptosis | Death receptors/effector caspases, granzyme B | Cell shrinkage, membrane blebbing, chromatin condensation, nuclear fragmentation | Caspase activation, mitochondrial ROS, loss of mito Δψ, proteolytic cascade, global mRNA decay, MOMP, cytochrome C release, genomic DNA fragmentation | Noninflammatory |
| Microptosis | Perforin/granulysin/granzymes | Parasites undergo cell death that morphologically resembles mammalian cell apoptosis | Generation of superoxide anion, disruption of antioxidant defenses, disruption of protein synthesis and central metabolism | Inflammatory |
| Pyroptosis | Inflammasome/inflammatory caspases/gasdermins | Membrane bubbles, cell flattening | Inflammatory caspase activation, gasdermin cleavage, cell membrane pore, release of inflammatory intracellular contents | Inflammatory |
| Necroptosis | Death receptors/MLKL | Cell swelling, cell membrane rupture | RIPK1/3, MLKL phosphorylation, membrane ion channel, release of cellular contents | Inflammatory |
Abbreviations: MLKL, mixed lineage kinase domain-like pseudokinase; MOMP, mitochondrial outer membrane permeabilization; ROS, reactive oxygen species.
Most of these membrane-disrupting proteins first arose when vertebrates appeared, about the same time as the antigen receptors that are the basis of adaptive immunity. However, the strategies used to disrupt membranes are similar to mechanisms used throughout evolution even in prokaryotes. None of these membrane-permeabilizing proteins, except some complement components, binds to a specific receptor. This means that microbes and cancer cells cannot readily evade immune elimination by mutating or downregulating a receptor, but it also means that systems need to be in place to avoid membrane damage to the cell that expresses these dangerous molecules and to enable selective targeting without damaging bystander cells. Each cell death-inducing protein needs a strategy to optimize disruption of membranes of cells to be killed and avoid harming membranes of cells that need to be left unharmed.
This review describes the membrane-disrupting proteins used in immune responses to infection, cancer, and danger, focusing on the newly identified members of this group (gasdermins, MLKL, perforin-2) and recent research that identifies unappreciated biological functions of granulysin. A homolog of perforin, perforin-2 (PFN-2 or MPEG1), constitutively expressed in myeloid cells and that damages the cell membranes of intracellular or phagocytosed bacteria, has recently been described (27, 28). Here we do not review the small antimicrobial peptides, like the defensins, produced by mucosal epithelia to control infection. We describe recent advances in understanding how these proteins damage membranes, made possible by new structural biology methods to study large membrane-associated complexes. Throughout we highlight the important unsolved questions in the field. We begin with an introductory description of each of the immune membrane-disrupting proteins and then describe how each of them addresses the biological challenges of selective disruption of target membranes and the target cell response to membrane damage.
2. THE PLAYERS
2.1. The Complement System
Complement was the first described immune pore-forming system, first identified by Paul Ehrlich in 1899 as a hemolytic activity in blood that complemented the activity of antibodies (29). It is a collection of mostly circulating precursor proteins that work together in a proteolytic cascade to form large membrane pores. Its main function is to destroy enveloped viruses and microbes, especially when they are opsonized by antibodies, although complement also removes damaged mammalian cells and immune complexes. The complement system is composed of more than 30 soluble and membrane-bound proteins that are activated by binding to microbial carbohydrates and immunoglobulins and by multiple proteolytic cascades that converge on cleaving soluble C5 complement into C5a, a powerful chemoattractant, and C5b, which triggers pore formation by nucleating a pore, the membrane attack complex (MAC) (30, 31) (Figures 3a,b and 4a). C5b, C6, and C7 first form a lipophilic complex that binds to and inserts in membranes when it recruits C8 (Figure 2). This complex recruits C9, which multimerizes with the membrane-inserted complex to form a 110-Å-diameter membrane pore, composed of C5b, C6, C7, heterotrimeric C8 (α, β, γ), and ~18 copies of C9. C6, C7, C8α, C8β, and C9 are members of the MAC/perforin (MACPF)/cholesterol-dependent cytolysin (CDC) protein superfamily, which includes complement, perforin, and many gram-positive bacterial toxins (1, 32, 33). More than 1,000 proteins from all kingdoms of life contain a MACPF domain that might form pores (34). The MAC is the only heteromeric immune pore-forming protein. MAC pores cause necrosis. Assembly of the complement MAC on bacteria is triggered when mannose-binding lectin (MBL) binds to bacterial carbohydrates or when antibody-opsonized bacteria are recognized by C1q in the C1 complex. The MAC kills bacteria by perforating bacterial cell membranes (35). Patients with complement deficiency are vulnerable to encapsulated bacteria, such as Neisseria meningitis, Streptococcus pneumoniae, Haemophilus influenzae, and Salmonella enterica serovar Typhimurium. Some organisms hijack the complement system to increase their virulence, for example, by using complement receptors to enter cells (36), while some viruses and intracellular bacteria bind complement regulatory proteins and receptors to escape complement-mediated death (37).
Figure 3.
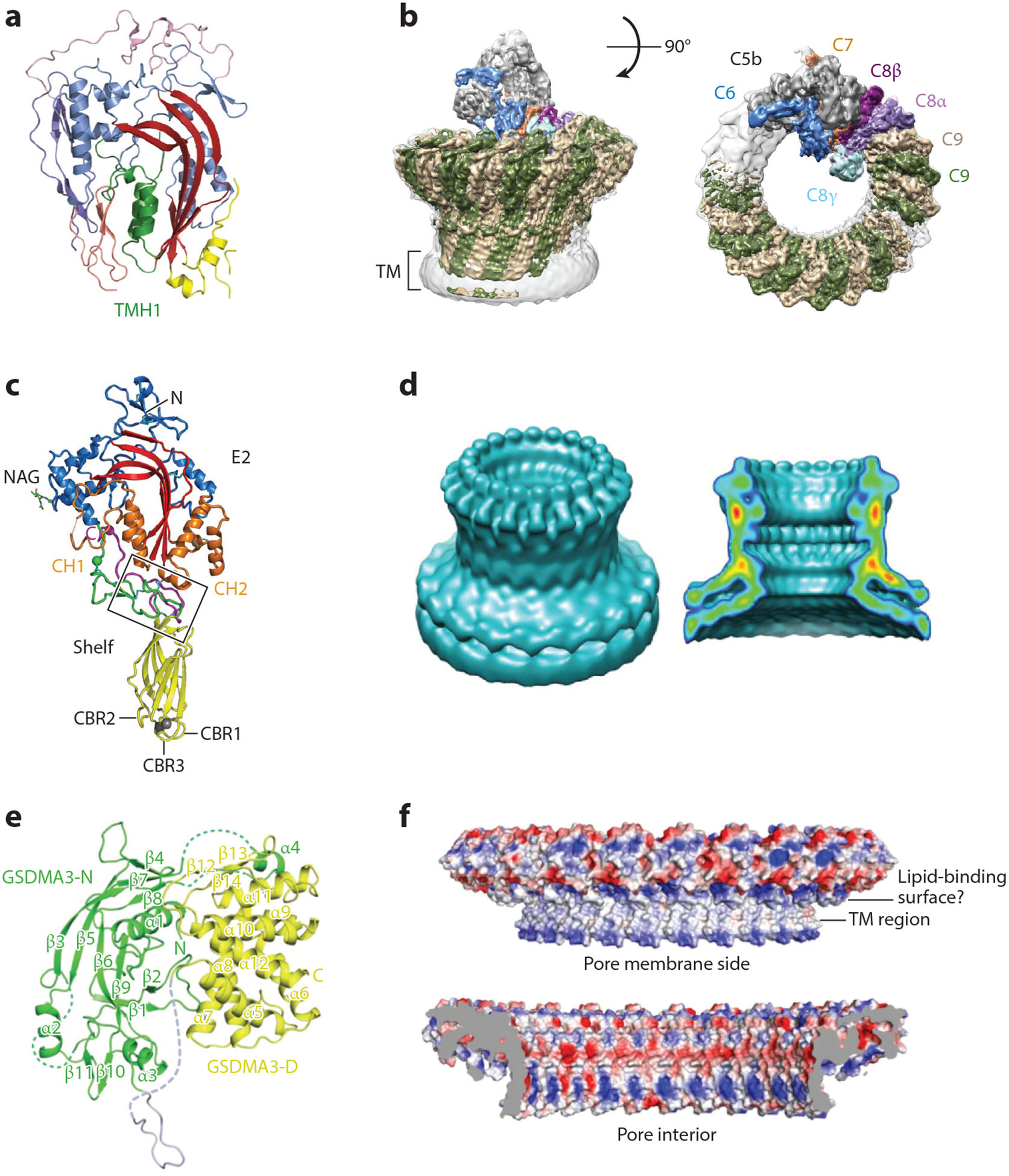
Structures of immune pore-forming proteins. (a,c,e) Crystal structures of the monomers of (a) complement C9, (c) perforin, and (e) GSDMA3. (b,d,f) Cryo-electron microscopy reconstruction of the membrane pores formed by (b) the complement membrane attack complex, (d) perforin, and (f) GSDMA3. Abbreviations: GSDMA3, gasdermin A3; TM, transmembrane. Panels a, b, e, and f adapted from References 10, 13, 15, and 163, respectively; panels c and d adapted from Reference 9.
Figure 4.
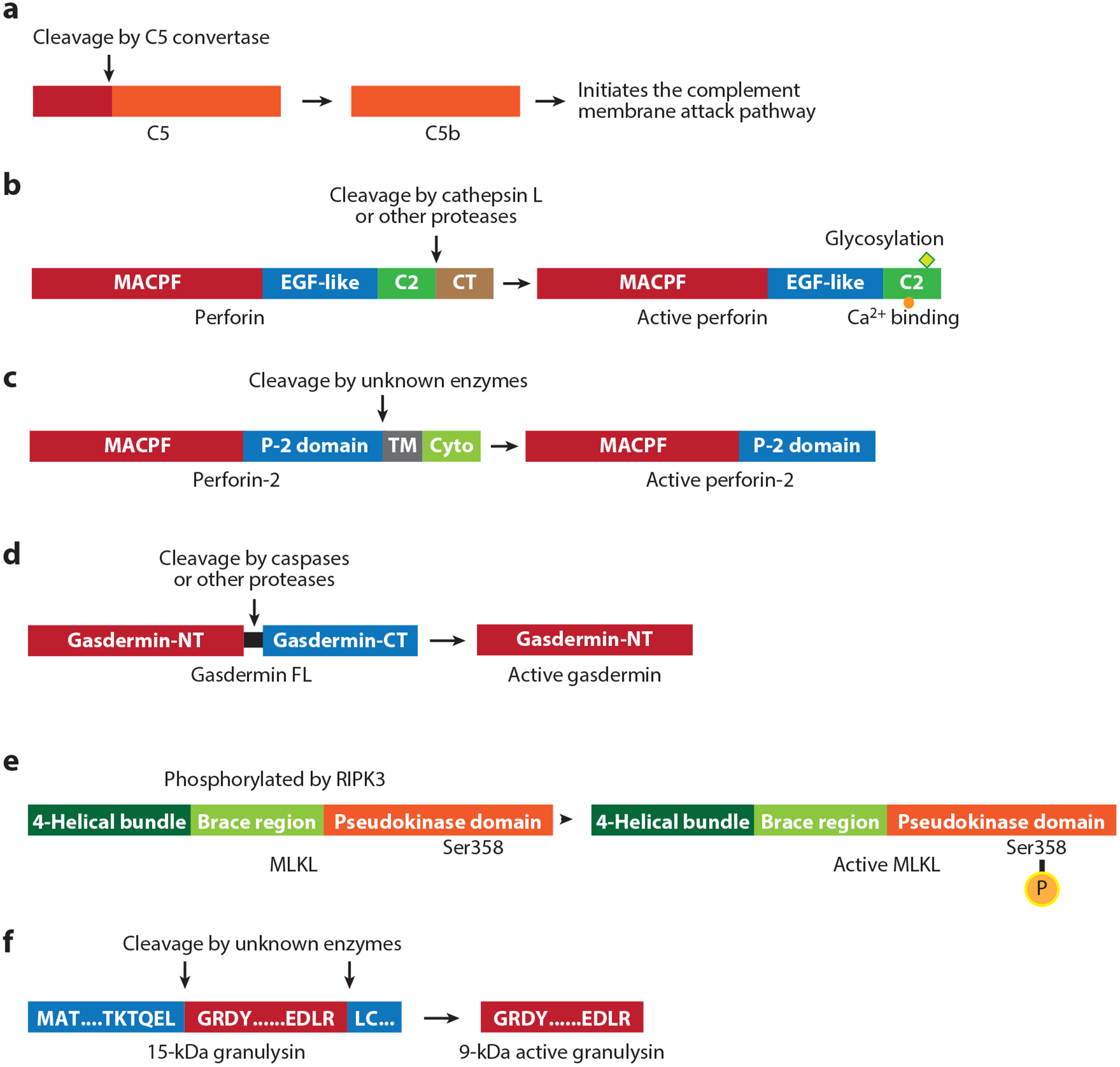
Activation of immune membrane-disrupting proteins. (a) The complement cascade is initiated when C5 is processed to generate C5b. (b) Perforin consists of a MACPF domain, an EGF-like domain, and a calcium-binding C2 domain followed by a C-terminal peptide. It is synthesized as an inactive precursor that is glycosylated in its C2 domain and processed in cytotoxic granules by cathepsin L or other unknown proteases. (c) Perforin-2, a membrane-bound protein, consists of a MACPF domain, conserved perforin-2 domain, transmembrane domain and cytoplasmic domain. Its pore-forming activity may be enhanced by proteolytic activation by unknown enzymes to remove the transmembrane domain and release it from vesicular membranes. (d) Gasdermins have N-terminal and C-terminal domains separated by a linker; the C-terminal domain folds back on the N-terminal domain to autoinhibit membrane pore formation. Cleavage in the linker generates the N-terminal pore-forming domain. (e) MLKL, composed of an N-terminal four-helical bundle domain followed by the brace region and a C-terminal pseudokinase inhibitory domain, is activated by phosphorylation of Ser358 by RIPK3. (f) The inactive 15-kDa granulysin propeptide is cleaved after Leu62 and Arg136 by unknown enzymes to produce active granulysin (9 kDa). Abbreviations: CT, C-terminal; Cyto, cytoplasmic; EGF, epidermal growth factor; FL, full length; MACPF, membrane attack complex/perforin; NT, N-terminal; MLKL, mixed lineage kinase domain-like pseudokinase; P-2, perforin-2; TM, transmembrane.
2.2. Perforin
Perforin, a pore-forming protein encoded by the PRF1 gene, has an N-terminal MACPF domain (5, 38, 39) that is similar to the pore-forming domains of the C6–C9 components of the complement MAC (especially C9) and bacterial CDC (40) (Figure 3c,d). Unlike the soluble complement components, which are expressed mostly by hepatocytes and secreted into the blood, perforin is expressed only in killer lymphocytes, which store it in cytotoxic granules, specialized secretory lysosomes (41). When a killer cell recognizes a target cell, its cytotoxic granules migrate along microtubules to the immune synapse, where they dock and fuse with the killer cell plasma membrane, releasing perforin and other cytotoxic effector proteins (granzymes and granulysin) into the immune synapse (42). Perforin then forms pores in the target cell membrane, which lead to cytosolic delivery of the other effector proteins. However, delivery does not occur directly through plasma membrane pores (43–45). Although like complement, perforin pokes holes in target cell membranes that would ordinarily cause necrosis, the membrane damage by killer cells is rapidly repaired by the ubiquitous cell membrane repair pathway, perhaps because damage is localized to the immune synapse. Membrane repair triggers endocytosis of perforin together with the death-inducing granzymes, which bind to the target cell membrane by charge interactions, which allows them to be coendocytosed with perforin (46, 47). Perforin then forms pores in the endosomes of target cells, which deliver the granzymes into the target cell cytosol, where they cause programmed cell death. Although most of the granzymes do not activate the caspases, granzyme B cleaves and activates caspase-3, which amplifies killer cell-mediated death (48). The perforin MACPF domain is followed by an EGF domain that contributes to the pore structure and a Ca2+-binding C2 domain, responsible for perforin’s Ca2+-dependent binding to target cell membranes (9, 49) (Figure 4b). Nineteen to twenty-four perforin monomers assemble (at least in lipid monolayers) into a pore with a lumen diameter of ~160 Å, large enough to deliver the granzymes (9). Perforin pore formation depends on membrane cholesterol; hence, perforin does not damage microbial membranes that lack cholesterol (2, 50). Why perforin forms pores only in cholesterol-containing membranes is not understood. At the immune synapse, perforin binding to the killer cell membrane does not harm the killer cell, for reasons that are not entirely clear. Following cytotoxic granule fusion with the killer cell plasma membrane, cytotoxic granule cathepsin B is exposed on the killer cell membrane at the synapse and proteolytically inactivates any perforin that binds to the killer cell (51). However, cathepsin B genetic deficiency does not lead to killer cell death during target cell attack, suggesting other uncharacterized protective mechanisms (52).
Prf1−/− mice are profoundly immunodeficient and are unable to protect themselves from viruses and prone to develop lymphoma (53, 54). Humans bearing hypomorphic PRF1 are impaired in handling intracellular infection and can develop an often-fatal inflammatory syndrome, familial hemophagocytic lymphohistiocytosis, due to unresolved infection, high levels of IFN-γ, and macrophage activation that can be treated by bone marrow transplantation or the recently approved anti-IFN-γ antibody emapalumab (55, 56). Individuals bearing less severe PRF1 mutations can be asymptomatic until adulthood and may develop lymphoma.
2.3. Perforin-2
Recently a weakly paralogous protein PFN-2 that contains a MACPF domain and is expressed from the MPEG1 gene mainly in macrophages and other myeloid cells has also been identified and is hypothesized to also form membrane pores (27, 28, 57) (Figure 4c). MPEG1 was the first MACPF domain-containing gene to appear in eukaryotes during evolution (in sponges, where it functions in antibacterial defense), and PRF1 may have arisen as a gene duplication of MPEG1. MPEG1 is constitutively expressed in professional phagocytic cells (macrophages, dendritic cells, neutrophils, microglia), in some innate or innate-like lymphocytes [γδ T cells, natural killer (NK) cells], and in some epithelia, including keratinocytes. It is broadly induced by interferons in epithelia, endothelia, fibroblasts, and lymphocytes, suggesting it may function in many cell types (58). Unlike other mammalian MACPF proteins, PFN-2 contains a transmembrane domain and may be bound to endosome and phagosome membranes oriented with its MACPF domain in the vesicular lumen (59). It has been proposed to disrupt membranes of phagocytosed bacteria to control bacterial infection (60), and MPEG1 knockdown reduces in vitro macrophage clearance of diverse bacterial species (61). A recent study linked heterozygotic mutations of MPEG1 to persistent atypical mycobacterial infection, providing support for an immune protective role in humans, especially for bacteria that infect macrophages (62). The MACPF domain of the protein might be proteolytically released into phagosomes to access bacterial cell membranes. Indeed, a cleaved PFN-2 was visualized by immunoblot after bacterial infection, although the responsible protease has not been identified (63). Overexpression of PFN-2 in HEK-293T cells or interferon-induced expression in bacteria-infected mouse embryo fibroblasts leads to pores in both bacterial and host cell membranes, visible by transmission electron microscopy (EM) (63). Pores are only formed if the membrane preparations are trypsinized, consistent with the hypothesis that an active soluble pore-forming protein is liberated by proteolytic digestion of the membrane-bound protein. The inner diameters of PRF-2 pores are ~100 Å, similar to the complement and PFN-1 MAC pores. A recent cryo-EM study of soluble PFN-2 lacking the C-terminal transmembrane and cytosolic domains visualized large oligomeric membrane-bound prepores (hypothesized to take shape before assembly into a membrane-spanning pore) formed by 16 monomers that associated with lipids by their juxtamembrane P-2 domain (14). Soluble PFN-2 permeabilized red blood cells, but only in acidic media, consistent with its need to function in acidic phagolysosomes. In this study rare images (not enough to solve the structure of the pore) captured a pore in one liposome connected to another liposome, suggesting that PFN-2 may not need to be cleaved from the membrane to form pores in phagocytosed bacteria. In one study MPEG1−/− mice were more susceptible than littermate control mice to bacterial challenges with Staphylococcus aureus and S. Typhimurium (63). These studies need to be considered as provisional, since the knockout mice were not backcrossed to the parental strain, but they suggest that PFN-2 is important in eliminating phagocytosed bacteria.
However, many questions remain. Does PFN-2 pore-forming activity depend on proteolytic activation, and what is the protease? On what membranes does PFN-2 localize, and what bacterial and eukaryotic membranes does the activated protein permeabilize? Does it permeabilize (phagolysosomal) membranes to release lysosomal proteases to kill host cells? If not, how is bacterial specificity regulated? Is PFN-2 active against other microbes, such as fungi and protozoan parasites, or enveloped viruses? Do any antimicrobial or cytolytic effector proteins enter through PFN-2 pores to contribute to killing? How important is PFN-2 in overall microbial defense in vivo?
2.4. Gasdermins
The gasdermins (GSDMs) are a family of homologous pore-forming proteins that cause pyroptosis, an inflammatory death associated with loss of cell membrane integrity and release of inflammatory mediators. In humans, this gene family consists of 6 genes, GSDMA-E and DFNB59. GSDME, also known as DFNA5, and DFNB59, also known as pejvakin, have both been linked by genetic mutation to hearing loss. Mice have three GSDMA counterparts (Gsdma1–3), four GSDMC counterparts (Gsdmc1–4), and no Gsdmb (64). As the name implies, the family members are constitutively expressed in the skin and mucosal epithelial cells. Although their expression overlaps to varying degrees, the GSDMs are differentially abundant in different sites: GSDMA in the skin, GSDMB in the lung, GSDMC in the gut. GSDMD is also highly expressed in immune antigen-presenting cells, macrophages, and dendritic cells. GSDMs can also be induced in many cell types, but how the expression of these genes is regulated is not known.
The GSDMs share highly conserved N- and C-terminal domains, connected by highly variable flexible linker regions (15) (Figures 3e and 4d). They are mostly cytosolic and are proteolytically activated to bind to membranes and form pores when the linker region is cut to separate the N-terminal pore-forming domain from the autoinhibitory C-terminal domain (15). The exceptions are GSDMB, which appears to be mostly nuclear, and DFNB59, which has a truncated C terminus. GSDMD membrane damage is triggered by cytosolic sensing of invasive infection or signs of danger that cause the formation of large amyloid-like complexes called inflammasomes, which activates inflammatory caspases (caspase-1 in mouse and human, −11 in mouse, −4 and −5 in humans) to cleave GSDMD and activate it to form pores. The inflammatory caspases cut GSDMD in the linker. Some myeloid cell proteases (neutrophil elastase and cathepsin G) also cleave GSDMD directly (65, 66). Two independent groups reported that TAK1 inhibition by the Yersinia effector protein YopJ triggers caspase-8-dependent GSDMD cleavage and activation, leading to pyroptosis (67, 68). However, it is unclear whether caspase-8 or another protease directly cleaves GSDMD.
GSDME is cut by caspase-3 after Asp270 and activated during apoptosis, which switches noninflammatory apoptotic death to pyroptosis in GSDME-expressing cells (20, 21). Although GSDME is widely expressed in epithelial cells, many cancer cell lines from the same tissues suppress its expression by promoter methylation, perhaps because pyroptosis of the cancer cell would recruit and activate antitumor immune cells to the tumor. How the other GSDMs are activated is not known. The GSDMs can permeabilize both bacterial and mammalian cell membranes (15, 16). A high-resolution structure of the pore formed by N-terminal GSDMA3 has been solved by cryo-EM (10) (Figure 3f; see below). Although the amino acid sequences of the GSDMs and the MACPF domains of complement and PFN are not at all homologous, the GSDMA3 pore assembles from approximately 27 monomers into a 180-Å pore in the form of a β barrel that closely resembles the structure of the complement and PFN pores. GSDM pores are examples of convergent evolution to a 3-D structure that resembles MACPF/CDC pores.
Up to now, little has been known about the physiological functions of gasdermins except for GSDMD. Polymorphisms or mutations in GSDM genes are associated with skin defects, asthma, and hearing loss (69–73). Gasdermin a3 (Gsdma3) was identified as a gene whose mutation causes developmental defects in hair follicle and sebaceous gland development. Postnatal activation of Gsdma3 causes alopecia. Three spontaneous (Dfl, Re-den, Rim3) and six ENU mutagenesis-induced Gsdma3 (Fgn, Bsk, Rco2, Ae, Gsdma3M1Btlr, Gsdma3I359N) mutant mouse lines are associated with skin abnormalities. These mice either bear mutations or loss of the C-terminal domain of Gsdma3 (71, 74–78). However, no skin defects are observed in Gsdma3−/− mice. Both the Gsdma3 mutant (Ae) and the N terminus of Gsdma3 similarly induce cell death. These findings suggest these Gsdma3 mutations are gain-of-function mutations in which the mutant C-terminal domain no longer fully inhibits the N-terminal domain (79). This idea is supported by the Gsdma3 structure, which showed that these mutations abolish autoinhibitory interactions between the domains to activate Gsdma3 constitutively (15).
GSDMB is highly expressed in bronchial epithelia, and its polymorphisms are associated with asthma susceptibility (80). However, the pathogenesis may not be related to inflammation. In one study, which needs to be confirmed, GSDMB was nuclear (unlike the other GSDMs) and GSDMB induced airway hyperresponsiveness and airway remodeling by activating the expression of TGF-β1 and 5-lipoxygenase (81).
DFNA5 and DFNB59 mutations are responsible for autosomal dominant and recessive nonsyndromic hearing impairment, respectively (72, 82). Unlike the other GSDM genes expressed primarily in immune and epithelial cells, DFNA5/GSDME is mainly expressed in heart, brain, and kidney, while the DFNB59 transcript is detected in brain, eye, inner ear, heart, lung, kidney, liver, intestine, and testis (72, 82). DFNA5-related hearing loss is attributed to gain-of-function mutation via aberrant splicing of the transcript. DFNB59 was found to modulate peroxisome function to defend against noise-induced oxidative stress in cochlear hair cells to maintain hair cell and auditory neuron homeostasis (83).
2.5. MLKL
Death receptor signaling, mediated by TNF-α, Fas ligand, or TRAIL binding, can trigger diverse outcomes: NF-κB activation of inflammation and cell proliferation, apoptosis, or programmed necrosis (necroptosis), depending on the posttranslational modifications (phosphorylation, ubiquitylation) and availability of receptor-associated proteins and formation of distinct receptor-associated complexes (4, 6). The death receptor-associated kinase RIPK1 integrates these signals. When caspase-8 activation is inhibited, RIPK1 is ubiquitylated and binds to RIPK3 via a homotypic interaction of their RIM homotypic interaction motif (RHIM) domains, which leads to the formation of the necrosome, the execution complex of necroptosis. Necroptosis is a form of programmed necrotic cell death, morphologically characterized by organelle and cell swelling and plasma membrane rupture (84). The necrosome, which appears to assemble into a large amyloid-like supramolecular organizing center (SMOC), phosphorylates and activates MLKL to bind to phosphoinositides on the inner leaflet of the cell membrane and permeabilize it (85–90). MLKL is composed of an N-terminal four α-helical bundle domain and a C-terminal pseudokinase inhibitory domain (Figure 4e). When RIPK3 phosphorylates the pseudokinase domain, the helical domain is exposed, binds to the cell membrane, and assembles into disulfide-bonded tetramers and probably higher-order oligomers that permeabilize it. How MLKL disrupts the cell membrane is unclear. There are many α-helical bundle small peptides expressed in prokaryotes and eukaryotes that oligomerize and disrupt microbial or cell membranes, including the antimicrobial cytotoxic granule peptide granulysin and the mitochondrial outer membrane-disrupting bax, but understanding how they cause membrane permeabilization will probably need to await structural information about the oligomers they form and how they assemble in membranes. RIPK3 can also phosphorylate and activate MLKL and necroptosis independently of death receptor signaling and RIPK1, such as by binding to TRIF or other mediators of interferon signaling or by binding to the intracellular DNA sensor ZBP1 (also known as DAI) (91, 92).
The biological roles of necroptosis are still not completely clear. Ripk3 and Mlkl are found in some vertebrates and mammals, but marsupials lack both genes and carnivores lack Mlkl, suggesting that necroptosis is not needed for normal development or effective immunity. In fact, mice deficient in Mlkl, the final executioner of necroptosis, develop normally and have no known phenotype or immunological deficit under homeostatic conditions (85, 93). Moreover, Mlkl deficiency has more modest effects than Ripk1 or Ripk3 deficiency in some of the challenge models that were used to show the importance of necroptosis, suggesting that some of the biological functions attributed to necroptosis are in fact not caused by necroptosis. Necroptosis has been implicated in a broad range of pathological conditions including infection, inflammation, sepsis, ischemia injury, and neurodegeneration (6, 94–98). The in vitro and in vivo models to probe the function of necroptosis generally are somewhat artificial, since they involve genetic deficiencies in key death receptor and apoptosis pathway molecules or other methods to inhibit caspase activation. Many of the studies assessing the importance of necroptosis have relied on inhibition of RIPK1, using inhibitors like necrostatin (Nec)-1 or the more specific inhibitor Nec-1s, or more recently developed inhibitors of RIPK3 (99–101). However, RIPK1 can trigger both apoptosis and necroptosis, and RIPK3 likely also has inflammatory functions that are independent of necroptosis (102–106). The MLKL inhibitor necrosulfonamide, which inhibits MLKL disulfide bonding required for pore formation, is a Cys-reactive drug, a class of drugs that are not specific because of their ability to modify cysteine residues on many proteins (107). Perhaps the most convincing (if indirect) evidence for the importance of necroptosis in disease pathogenesis comes from models of infection. Many microbes and cancer cells have elaborated mechanisms to block classical apoptosis, which could consequently favor necroptosis. The identification of viral and bacterial virulence factors that suppress necroptosis and in vivo data showing that Ripk3 deficiency alters infection in some models suggest that necroptosis contributes to host immunity, although this needs to be explored in Mlkl−/− mice to determine whether protective effects are in fact mediated by necroptosis or other downstream effects of the RIP kinases (108–111).
2.6. Granulysin
Granulysin (GNLY) is a saposin-like, five α-helical domain lipid-binding protein in killer lymphocyte cytotoxic granules that at high concentrations acts as a human antimicrobial peptide with broad activity against bacteria, fungi, and parasites (2, 112, 113) (Figure 4f). Granulysin is expressed in many mammalian species, but not in rodents, which indicates it is not essential for immune defense. Saposins assemble into α-helical proteins with a common fold that is held together by disulfide bonds. Granulysin is expressed exclusively in some innate (NK), innate-like (γδ T, NKT), or adaptive (CD4 and CD8 T) lymphocytes when they become cytotoxic. However, in healthy human donors, granulysin is only expressed in a minority of the cytotoxic lymphocytes that express granzymes or perforin. Granulysin is highly expressed in killer cells in the skin and in decidual NK cells in the placenta during the first trimester of pregnancy, again suggesting a role in protecting the barrier epithelia from infection (114, 115). Exposure of T cells to killed bacteria and IL-2 family cytokines that use the IL-2 receptor common γ chain (CD132) upregulates granulysin expression in vitro. A small N-terminal lipopeptide from a Mycobacterium leprae lipoprotein stimulated dendritic cells to secrete IL-12 and induce granulysin expression in cocultured T cells (116), suggesting that recognition of microbial pathogen-associated molecular patterns (PAMPs) may trigger GNLY expression. However, the microbial molecules (PAMPs), signaling pathways, and transcription factors that upregulate gene expression, other than Stat5, are not known.
Granulysin is processed from a 15-kDa inactive precursor molecule by cleavage at both ends to a 9-kDa active membrane-disrupting protein, probably by cathepsins in cytotoxic granules, where it is stored with perforin and granzymes (112, 113). The mechanism for membrane permeabilization is not known. The homologous, saposin-like amoebapore A protein from Entamoeba histolytica, which kills phagocytosed bacteria, dimerizes to expose a His residue near its C terminus that is believed to be important for membrane intercalation (117, 118). Gel filtration experiments suggest that higher-order oligomers (hexamers?) might oligomerize in the membrane. Granulysin has a very positively charged surface and binds to acidic phospholipids and cardiolipin, like gasdermins and MLKL. A patch of four basic Arg residues between helix 2 and helix 3 is important for lipid binding (119). Granulysin is about a thousand times more active at disrupting microbial membranes of bacteria, fungi, and parasites than mammalian membranes and is inhibited by cholesterol in mammalian membranes by an unknown mechanism (2, 119).
At micromolar concentrations granulysin permeabilizes microbial membranes. Such a high granulysin concentration is probably never reached in bodily fluids, except in the skin blisters of patients undergoing severe, often fatal, drug reactions (Stevens Johnson syndrome or toxic epidermal necrolysis) where disease pathology is linked to granulysin-dependent killing of keratinocytes (114, 120). Lower nanomolar concentrations of granulysin permeabilize bacterial membranes to deliver granzymes intracellularly and enable them to kill cell-free bacteria. However, it is unclear whether secreted granulysin, with or without secreted granzymes, efficiently kills cell-free bacteria in vivo. Yet, when killer lymphocytes recognize and kill cells harboring intracellular bacteria and parasites, they not only destroy the host cell but also kill the microbes, either in phagosomes or the cytosol, in a granulysin, perforin, and granzyme-dependent manner (121, 122). Granulysin binds to microbial cell membranes and delivers the death-inducing granzymes into the microbes, where they proteolytically disrupt essential pathways needed for viability (electron transport, antioxidant defenses, protein synthesis, and central metabolism) to cause microbial programmed cell death (microptosis) (123). Because of their complementary specificities for microbial and host cell membranes, granulysin and perforin work together to deliver the death-inducing granzymes into both the host cell and microbial cytosol. Some T and NK cells also bind directly to cell-free fungi, form an immune synapse with them, release their cytotoxic granules, and kill fungi in a granulysin-dependent manner (124, 125).
The importance of granulysin in immune defense has been examined by comparing wild-type mice that lack granulysin with GNLY-transgenic mice that express GNLY only in killer cells at levels similar to those in humans (121, 122, 126). In the three reported intracellular infection models (Listeria monocytogenes, Toxoplasma gondii, and Trypanosoma cruzi), GNLY-transgenic mice handled these infectious challenges much better than wild-type mice. In a nonfatal L. monocytogenes infection model, bacterial loads were about 2 logs lower in the livers and spleens of transgenic mice. In lethal challenges with the protozoan parasites, most GNLY-transgenic mice survived and cleared the infection. In two tumor models, GNLY protection from lymphoma was modest—in one lymphoma the GNLY transgene did not significantly change survival, while in another lymphoma, 20% of transgenic mice survived a tumor xenograft challenge that was lethal to all wild-type mice. The modest protection of GNLY in tumor models is consistent with its low activity in disrupting mammalian cell membranes.
3. ASSAYS THAT DISTINGUISH MEMBRANE-DISRUPTING CELL DEATH PATHWAYS
Studying membrane-disrupting pathways depends on having reliable ways to monitor and distinguish whether they are activated and cause cell death (Table 3). Overall cell death of any type can be quantified by counting the reduction in viable surviving colonies or by measuring the amount of intracellular ATP, which is proportional to the number of viable cells. Death is inflammatory or noninflammatory depending on how rapidly cell membrane integrity is disrupted compared to when the dying cell is recognized in vivo and phagocytosed and on whether membrane damage leads to protein release.
Table 3.
Assays that distinguish membrane-disrupting cell death pathways
| Assay | Apoptosis | Pyroptosis | Necroptosis |
|---|---|---|---|
| Count viable surviving colonies | ↓ | ↓ | ↓ |
| Measure intracellular ATP | ↓ (late) | ↓ (early) | ↓ (early) |
| Observe cell morphology | Cell shrinkage, membrane blebbing | Membrane bubbles, cell flattening | Cell swelling, cell membrane rupture |
| Detect the activation of mediators | Effector caspase cleavage | Inflammatory caspase/GSDM cleavage | MLKL phosphorylation |
| Uptake of cell-impermeable small molecule dyes (SYTOX Green or propidium iodide) | Late | Early | Early |
| LDH release | Late | Yes | Yes |
| HMGB1 release | No | Yes | Yes |
| Annexin V staining | Positive | Positive | Positive |
| DNA ladder assay, TUNEL assay | ++ | +/− | +/− |
Abbreviations: GSDM, gasdermin; MLKL, mixed lineage kinase domain-like pseudokinase; TUNEL, terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling.
Necroptosis, pyroptosis, and apoptosis can be distinguished morphologically by phase contrast light microscopy with time-lapse imaging or using fixed cells (127, 128) (Figure 5). Both apoptotic and pyroptotic adherent cells detach rapidly with similar kinetics as soon as the caspases or gasdermins are activated. In necroptosis, activated MLKL forms ion channels that cause osmotic swelling. The cell body swells, and the cell membrane suddenly bursts (129). In contrast during pyroptosis, large gasdermin cell membrane pores allow the cell to maintain its osmolarity (127). The damaged membrane forms large ballooning bubbles, which are much larger than apoptotic blebs and easy to distinguish, but the dying cell does not rupture and appears to flatten as its cytoplasmic contents are released and the cytoskeleton and intermediate filaments are proteolytically disrupted (127, 130). Small membrane blebs, cell shrinkage, nuclear contraction, and chromatin condensation are characteristics of apoptosis and killer cell-mediated cell death (5). The induction of each cell death program can be timed by measuring the specific activation of its mediators (cleavage of effector or inflammatory caspases and gasdermins, MLKL phosphorylation) by immunoblot or fluorescent inhibitor of caspase-3 or caspase-1 (FLICA) flow cytometry assays. Necroptosis or pyroptosis can also be confirmed or timed by assessing the translocation of GSDMD or MLKL, respectively, to the plasma membrane using fluorescence microscopy or cell fractionation (15, 16). In addition, downstream processing of important selective caspase-3, granzyme, or inflammatory caspase substrates, such as bid, PARP1, or pro-IL-1β, indicates which cell death pathways have been activated. The specific forms of cell death can also be verified by showing the effect of specific inhibitors, such as DEVD-fmk for classical apoptosis, EGTA for perforin-mediated killer cell death, Ac-YVAD-fmk for caspase-1, disulfiram for GSDMD pore formation, or necrosulfonamide or necrostatin-1 for necroptosis (107, 131, 132).
Figure 5.
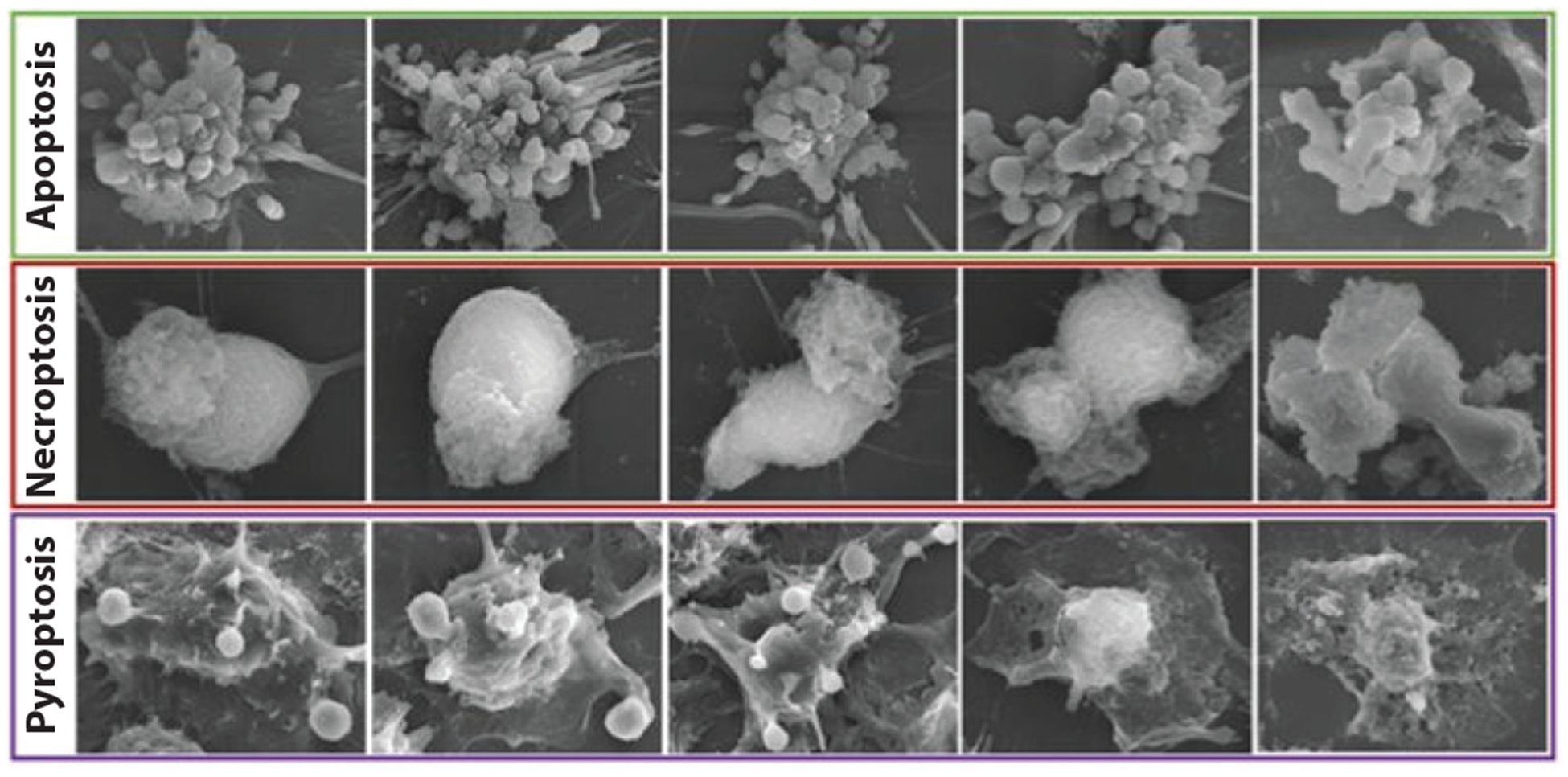
Morphological features of different types of cell death. Adapted from Reference 128.
DNA fragmentation is a hallmark of apoptosis that distinguishes apoptotic from necrotic death. DNA fragmentation is induced by the caspase-activated DNase (CAD) in classical apoptosis (133), which is also activated by granzyme B (134), and by NM23-H1 and Trex1, which are activated by granzyme A (135, 136). CAD activation can be detected by visualizing a DNA ladder assay on agarose gels. CAD- and granzyme-mediated DNA damage can also be visualized by terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) assay. An alternative method for examining DNA fragmentation is to measure DNA content by propidium iodide (PI) staining and flow cytometry. Apoptotic cells have sub-G0/G1 DNA content because DNA fragments are released. Although there are no known programs of DNA damage in necroptosis or pyroptosis, DNA breaks are sometimes observed, so detecting DNA damage does not necessarily indicate noninflammatory death.
Loss of plasma membrane integrity is easily assessed by detecting uptake of cell-impermeable small-molecule dyes, such as SYTOX Green or PI, by flow cytometry, plate-reader or fluorescence microscopy. Because these dyes bind nucleic acids, once taken up into cells they bind DNA and stably stain the cells. The release of LDH, a large ~140-kDa tetrameric enzyme that is too large to pass through smaller pores, detected by a colorimetric plate reader assay that measures enzymatic activity, may occur after small-dye uptake, for example, when a necrotic cell bursts. Inflammatory disruption of cell membrane integrity can also be assessed by measuring HMGB1 in culture supernatants by immunoblot or ELISA. Release of processed inflammatory cytokines from immune cells is an indicator of pyroptosis.
Phagocytic cells recognize early signs of membrane disruption by detecting externalized phosphatidylserine (PS) on the dying cell, which is experimentally assessed by annexin V binding. During apoptosis a scramblase transfers PS from the inner to outer leaflet of the plasma membrane; during other forms of cell death, PS binding may become detectable because the ~38-kDa annexin V protein can enter the cell when the cell membrane is no longer intact. Annexin V staining occurs early in programmed cell death at about the same time the effector caspases or MLKL is activated. Phagocytosis of apoptotic cells or of cells targeted by killer lymphocytes is a key mechanism for avoiding inflammation since cell lysis (i.e., secondary necrosis) of these dying cells is a late event that mostly occurs within phagosomes of phagocytic cells in vivo (137–140). By contrast, proteins that cause inflammatory death (complement, MLKL, and gasdermins) cause cytolysis more rapidly and presumably before or coincident with phagocytosis, which is inflammatory because endogenous danger signals or damage-associated molecular patterns (DAMPs) are released extracellularly instead of being contained within phagosomes (141). In the absence of phagocytic cells in in vitro cultures, all dying cells eventually lyse, so distinguishing the different forms of death requires careful attention to the timing of membrane disruption relative to caspase, gasdermin, or MLKL activation.
4. MOLECULAR MECHANISMS FOR REGULATING MEMBRANE DISRUPTION
Each immune pore-forming protein needs to control damage to the cells that express it. Three principal mechanisms are used: (a) expression of a propore protein that needs to be proteolytically cleaved before it can oligomerize (complement, perforin, perforin-2, granulysin, gasdermins); (b) storage in membrane-delimited compartments under conditions at which it is inactive (perforin, perforin-2, granulysin); and (c) requirement of posttranslational modifications for oligomerization [MLKL (phosphorylation) and possibly perforin (glycosylation)] (Figure 4).
Complement proteins are only activated outside the cell, so the cells that express them are not vulnerable to membrane damage. Extracellular complement activation occurs in three phases—recognition, activation of convertase enzymes that cleave the key complement components C3 and/or C5, and assembly of the MAC leading to cytolysis (142). A large variety of microorganisms, including bacteria, viruses, fungi, and protozoa, become complement targets when the mannan-binding lectin (MBL) recognizes surface glycans (143).
Perforin and granulysin are synthesized as proproteins that need to be cleaved to become active (112, 144). Both are only expressed once a lymphocyte gains cytotoxic capability. They are likely only processed into their active form by cathepsins within killer lymphocyte cytotoxic granules, where they are stored. Cytotoxic granules are very acidic (pH ~ 5); at this pH, perforin is inactive. Moreover, perforin insertion in membranes requires that the C2 domain binds to free Ca2+, which is reduced in cytotoxic granules, because they contain calreticulin, which chelates free calcium (144, 145), helping to safeguard the killer cell. Under some circumstances, such as in activated regulatory T cells (Tregs), which express granzymes and perforin, granzymes leak out of cytotoxic granules and kill the activated T cell (146). Self-killing reduces the half-life of activated Tregs, which may explain the transience of adoptively transferred Treg cell lines being developed as a therapy to block transplant rejection. Granzyme leakage might be caused by perforin pore formation in cytotoxic granules in the activated effector cell, but this has not been shown. GNLY is also stored in cytotoxic granules. However, because granulysin does not damage mammalian membranes except at extremely high and probably supraphysiological concentrations, granulysin leakage may not damage the killer cell. Secreted granulysin on its own (at high concentrations) or with granzymes (at lower concentrations) kills cell-free bacteria in vitro, but it is inactive against intracellular bacteria because it does not efficiently get into mammalian cells without perforin (2).
Gasdermins and MLKL are both cytosolic with ready access to cell and organelle membranes, but membrane damage is regulated because their membrane-damaging activity needs to be activated. The gasdermins are activated by proteolytic cleavage by caspases. Cleavage in the linker region triggers a radical conformational change of the N-terminal fragment that exposes membrane binding residues and induces oligomerization (15, 16, 127, 147, 148). It is not clear which happens first—membrane binding of a monomer followed by insertion and oligomerization in the membrane or oligomerization to form a prepore that then inserts in the membrane. The cryo-EM structure of GSDMA3 visualized both a membrane-penetrating pore and a 27-mer prepore structure in which the N-terminal domains had not yet much changed conformation (10). Similar prepores have been seen for the bacterial CDC, pneumolysin (149, 150), and perforin-2 (14). In prepore models, a prepore ring assembles at the membrane before membrane insertion. MLKL is activated by phosphorylation at T357 and S358 (90, 107) by activated RIPK3, which causes a conformational change that triggers formation of MLKL tetramers and possibly higher-order oligomers that organize into amyloid-like fibers and bind to cell membranes (12, 86–90, 151). For both gasdermins and MLKL, activation relocalizes these cytosolic proteins to the cell membrane. Recent evidence suggests that activated GSDMs and MLKL also disrupt mitochondria and other organelle membranes, including possibly the nuclear envelope (152–154). Disruption of these intracellular organelles, especially mitochondria, may promote cell death (153).
Membrane permeabilization by immune membrane-disrupting proteins other than MLKL is not known to be regulated by posttranslational modifications. However, a recent study indicated that GSDME Thr6, which is located in a region important for membrane binding, may be phosphorylated, which inhibits oligomerization, membrane binding, and pyroptosis (153). This Thr is conserved in GSDME, and phosphomimetic mutation of an analogous Thr (Thr8) in GSDMA also inhibits pore formation, suggesting that phosphorylation in this region may inhibit pyroptosis by multiple gasdermins. The kinase(s) that phosphorylate GSDME or pathways that trigger its phosphorylation are not yet known. Other posttranslational modifications of gasdermins, such as ubiquitylation, sumoylation or cysteine oxidation, which regulate other innate immune processes (155, 156), might also be important. GSDMD pore formation depends on a redox-sensitive Cys (Cys191 in humans, Cys192 in mice), whose oxidation will likely regulate pore formation (16, 132, 157).
5. LIPID BINDING AND MEMBRANE RECOGNITION
The binding of membrane-disrupting proteins to lipids and cellular proteins influences which membranes they permeabilize. Complement is the only system whose activity is regulated by binding to cell surface proteins, which helps provide target cell specificity. Since complement pore-forming constituents are soluble blood proteins that have access to many cells and tissues, a way to identify microbes or antibody-coated cell targets is needed. C3b complexes serve as both the assembly platform and protease to initiate pore formation (36). In the classical pathway, C3 is activated and targeted to target membranes by antibody opsonization; in the alternative pathway, other features of microbial membranes serve as recognition sites. The complement cascade is initiated when C5 binds to a cell surface enzyme complex that contains activated C3 (C3b), which cleaves C5 to generate a labile fragment C5b. C5b is stabilized by binding to C6 and then binds C7. The C5bC6C7 complex is responsible for irreversible binding to the membrane and then initiates MAC pore formation. The C5bC6C7 complex binds to lipid vesicles composed of any of the abundant phospholipids—phosphatidylcholine (PC), phosphatidylinositol, phosphatidic acid (PA), PS, and phosphatidylglycerol—found in all membranes of prokaryotes and eukaryotes. Therefore, complement can form pores in any membrane, including mammalian cell membranes. Specificity for bacterial versus mammalian membranes is achieved by the presence on mammalian membranes of CD55 (also called decay-accelerating factor) or CD46 (also called membrane cofactor protein), which degrade the C3 convertase or the activated subunit of C3, which initiates the complement cascade on cells; and CD59, which inhibits binding of C8 and C9 to the growing pore (158, 159). CD55 and CD59 are both GPI-anchored proteins. Individuals with genetic deficiency of PIGA, an X-linked gene whose product is needed to synthesize GPI anchors, or CD59 develop a hemolytic disease (paroxysmal nocturnal hemolytic anemia) caused by MAC pores in red blood cell membranes (160).
Perforin does not use accessory proteins for its Ca2+-dependent membrane binding. Perforin binds to PC in a Ca2+-dependent manner and also binds to immobilized phosphatidylethanolamine (PE) and the phosphatidylinositol phosphates (PIPs), but not phosphatidylserine (PS) (16). In principle, perforin could permeabilize mammalian cell membranes from either the inside (if it leaks from cytotoxic granules in the killer cell) or the outside (when it is released from cytotoxic granules). Although perforin can bind to microbial membranes, it does not form pores in them since pore formation by perforin, like the bacterial CDCs, depends on cholesterol, which is missing from microbial membranes. The other cytotoxic granule membrane-disrupting protein, granulysin, binds to the same phospholipids as perforin, but it also binds to cardiolipin, present on mitochondrial and bacterial inner membranes, and could be especially important for damaging microbial membranes (16). Although cardiolipin is initially transported to inner membranes, it also translocates to mitochondrial and gram-negative bacterial outer membranes, making them vulnerable to granulysin. In contrast to perforin, GNLY membrane damage is inhibited by cholesterol, making it 1,000-fold more active in microbial than mammalian membranes (119). It is not known how cholesterol inhibits granulysin membrane damage. Since granulysin binds to cardiolipin, it might also bind to and damage target cell mitochondrial membranes, as mitochondrial membranes are cholesterol poor and rich in cardiolipin. The distinct lipid preferences of perforin and granulysin mean that cytotoxic granule components could permeabilize and deliver granzymes into any cellular compartment to kill host cells and intracellular microbes, whether they are replicating in membrane-delimited phagosomes or the cytoplasm (2). Whether perforin or granulysin damage mitochondrial, nuclear, or organellar membranes during killer cell granule-mediated death has not been studied. However, the granzymes concentrate in target cell nuclei and mitochondria through not well appreciated mechanisms that do not require perforin (161, 162).
Full-length gasdermins and their C-terminal fragments do not bind to membranes, but their N-terminal fragments bind to cardiolipin and other acidic phospholipids (PIPs, PA, PS) found exclusively on the inner leaflet of the plasma membrane of live cells; however, they do not bind to outer leaflet phospholipids (PE, PC) (15, 16). Thus, N-terminal gasdermins rupture the cell membrane of the inflammatory cell only from the inside and do not damage membranes of bystander cells when they are released during pyroptosis. They may also bind to and damage some organelle membranes, including those of mitochondria (153). The affinity for cardiolipin is higher than for other phospholipids (15, 16). As a consequence, the GSDMD N-terminal domain kills both intracellular and extracellular bacteria, potentially helping control the infections that trigger pyroptosis.
MLKL preferentially binds to PIPs (restricted to the inner leaflet of the cell membrane) via a positively charged patch in the four-helix bundle domain of MLKL that becomes accessible after RIPK3 phosphorylation and oligomerization (88).
6. STRUCTURAL BASIS OF PORE FORMATION
The structure of pore-forming proteins in both monomer and pore forms illuminates how these pore-forming proteins function, including what conformational changes are required for membrane insertion, what residues mediate oligomerization and lipid binding to form the pore, and how these interactions may be targeted to disrupt pore formation by genetic mutations, posttranslational modifications, or inhibitors (Figure 3). Although solving the structures of monomeric proteins or soluble oligomers is usually not too difficult, solving the structures of large complexes, especially when they are membrane bound, is challenging. Indeed, structures of most of the immune membrane-disrupting monomers have been solved or can be modeled based on homologous proteins (7, 9, 14, 85, 163). However, solving the structure of the perforin monomer required mutating an amino acid required for multimerization to maintain the protein as monomers at high concentration (9). Until recently the membrane-bound oligomeric structures remained unknown. No structural information is available for oligomeric, membrane-bound granulysin or MLKL. The monomeric structure of both these molecules, like many antimicrobial peptides, consists of a handful of α helices, and membrane binding is localized to a small patch of positively charged amino acids. It is still not completely clear how many molecules assemble to disrupt the membrane or whether these assemble to span the membrane as a pore. A few biochemical studies suggest that phosphorylated MLKL assembles into octamers that span the membrane to create a small ion channel, but a structural model of membrane-associated oligomerized MLKL would provide a clearer picture (164, 165). Moreover, we need to understand whether formation of an MLKL ion channel causes osmotic swelling and cell membrane rupture.
The development and improvement of cryo-EM methods to tackle membrane-bound complexes has enabled high-resolution structures of three large (>100Å inner diameter) immune pores: complement, perforin, and gasdermin (8–11) (Figure 3). These monomers all undergo radical conformational changes to form similar large β-barrel structures that are hydrophobic on the outside and hydrophilic on the inside and that insert into and span the membrane to puncture it. Each monomer contributes two β hairpins (four β strands) to form the β barrel. These large channels are not ion selective and allow soluble mediators and small- and medium-sized proteins to exit the cell (without any known charge selectivity). Which molecules are released through these pores and which are retained in the dying cell is not known. However, these large pores can release proteins lacking secretion signals, including the processed proinflammatory cytokines IL-1β and IL-18, and other inflammatory mediators, such as HMGB1, and presumably many nonprotein components. Consequently, death by these large pores is highly inflammatory. The exception is killer cell death that generates perforin pores at immune synapses. In this setting, the target cell avoids necrosis by using its membrane repair capability (see below) to rapidly remove disrupted membrane and restore membrane integrity. Because all ions and many proteins can diffuse through these pores, the dying cell maintains its osmolarity and cells dying with these pores are unlikely to burst by hypotonic lysis, as was proposed for pyroptosis before the mechanism of cell lysis was identified.
The recent cryo-EM structure of the MAC (8, 13), solved to 4.9Å near-atomic resolution, consists of three parts: an asymmetric region (C5b, C6, C7, and C8), a hinge (C7, C8, and two C9 molecules), and a C9 oligomer (Figure 3b). The C5bC6C7 complex anchors to the membrane outer leaflet and lowers the activation energy for bending the membrane. C8 binding bends the membrane and C8 traverses the bilayer and recruits C9, which inserts into the membrane to oligomerize to complete the MAC pore, which contains about 18 C9 monomers and has an ~110-Å inner diameter. In the C9 transition from the soluble monomer to the pore form, two helical regions, TMH1 (transmembrane hairpin 1) and TMH2, unravel and insert into the membrane as amphipathic β hairpins (166). There is no evidence that complement MAC forms a prepore intermediate before membrane insertion.
Perforin multimerizes to form a homomeric cylindrical pore, whose structure was solved by cryo-EM at ~28Å resolution a decade ago (9) (Figure 3d). There is as yet no atomic resolution structure of the pore form of perforin. Most perforin pores in the early landmark study were composed of 19–24 subunits and had an inner diameter of 130–200 Å. The perforin MACPF domain at the N terminus of the molecule is followed by an epidermal growth factor (EGF) domain and a type II C2 Ca2+-binding domain that mediates membrane binding (9). The MACPF domain of perforin is formed from a bent and twisted four-stranded β sheet flanked by two clusters of amphipathic α helices named CH1 and CH2, which extend into amphipathic β hairpins that traverse the membrane to oligomerize and form pores.
Structures of monomeric full-length GSDMA3 and murine and human GSDMD revealed common features (15, 167) (Figure 3e). The globular pore-forming N-terminal domains and globular inhibitory C-terminal domains are connected by a disordered linker, which is the site of proteolytic cleavage, which frees the functional N-terminal gasdermin to form pores. Mutations that disrupt the interface between the two regions cause inflammatory syndromes. It is unclear whether because of weaker binding of the two halves of the protein, the full-length mutated protein assembles into pores or becomes more susceptible to cleavage to the active N-terminal fragment. The high-resolution (~3.8 Å) cryo-EM structure of the GSDMA3 pore in a cardiolipin-containing bilayer is the only resolved gasdermin pore structure (10). Most GSDMA3 pores showed 27-fold symmetry to form a large β barrel composed of 108 β strands with an ~180-Å inner and 280-Å outer diameter cylindrical structure. As with other pore-forming proteins, the monomer undergoes a radical conformational change from the globular N-terminal domain to a hand-shaped structure whose fingers are the β strands that penetrate the membrane in the pore form. Mutational studies indicated that a basic patch of three Arg residues in the N-terminal α1 helix binds to membrane lipids. Although the pores formed by GSDMA3 and MACPF domain proteins assume a similar final β-barrel structure, a comparison of the order of α helices and β sheets and the mechanism of membrane insertion of GSDMA3 and pneumolysin [a CDC whose pore structure has been solved (150)] suggests that GSDMs probably evolved independently of MACPF proteins to produce a protein with a similar final pore structure.
7. MEMBRANE REPAIR RESPONSES TO MEMBRANE DAMAGE
Not all cells in which pore-forming proteins are activated die. Eukaryotic cells respond to plasma membrane damage with a stereotypic membrane repair response, sometimes called cellular wound-healing, that can rapidly restore membrane integrity (168, 169). Many of the molecules that regulate vesicular trafficking in cells have Ca2+-sensing domains that react rapidly to the sudden rise in intracellular Ca2+ that occurs when membrane integrity is breached. If the membrane’s damage is not severe, its integrity can be restored in less than a minute by a combination of three processes: (a) patching of damaged membrane using membranes donated by intracellular organelles, especially lysosomes, that move to and fuse with the damaged membrane; (b) endocytosis of damaged membrane mediated by flotillin (caveolae)- and/or clathrin-dependent pathways; and (c) exocytosis of damaged membrane in a process that depends on the endosomal sorting complexes required for transport (ESCRT). Ca2+-dependent membrane repair is known to be mobilized in response to pores formed by bacterial CDCs, complement, perforin, gasdermins, and MLKL (43, 170–174). Without membrane repair, membrane permeabilization, especially by large pores like those formed by complement, perforin, and gasdermins, leads to necrosis and inflammation, because of the release of cytokines, chemokines, and other danger signals. However, in some circumstances, cell death can be averted or converted to noninflammatory apoptosis and the dying cell can continue to live and perform its functions or even become activated. After some inflammasome-activating stimuli in macrophages and dendritic cells, GSDMD is cleaved to form pores through which IL-1β is secreted, but the cells survive because of ESCRT-dependent membrane repair and become activated for phagocytosis and antigen presentation (172, 175–178). Because of their preserved functions and secretion of inflammatory cytokines and chemokines, these surviving immune sentinel cells have been termed hyperactivated. Similarly, cells triggered to undergo necroptosis that survive because of ESCRT-dependent membrane repair release more cytokines and chemokines than cells that die (173). As a consequence, adaptive immune responses are enhanced compared to those generated when membrane repair is inhibited. Complement and perforin-mediated necrotic death are very concentration dependent (43). At lower concentrations, damaged cells survive because of membrane repair. In the case of perforin-dependent killer lymphocyte-mediated death, the membrane repair process is required for delivering granzymes into the target cell—perforin pores in the plasma membrane lead to accelerated clathrin-dependent coendocytosis of granzymes and perforin from the damaged target cell membrane at the immune synapse (43–45). However, the target cell saved from necrosis still dies. Although the cell membrane barrier is restored, endocytosed perforin forms pores in endosomes, releasing granzymes into the target cell cytosol, where they activate programmed cell death that morphologically resembles apoptosis.
What determines whether the cell is resuscitated and avoids necrotic death is not understood. These outcomes presumably depend on the extent of membrane damage, perhaps on whether it is localized and the effectiveness of membrane repair. With perforin, plasma membrane repair seems to succeed in most target cells under physiological conditions and is critical for granzyme-mediated programmed cell death. In contrast, the outcome in cells attacked by complement seems to be mostly cell lysis without much of a role of plasma membrane repair. The outcome might be different because soluble complement components typically form pores scattered throughout the membrane, while perforin causes damage localized to a small area within the immune synapse. Localized damage may be easier to repair. Cell resuscitation by membrane repair presumably also depends on how robust membrane repair pathways are. However, whether these repair pathways are regulated has not been studied.
8. CONCLUDING REMARKS AND FUTURE PERSPECTIVES
Membrane-disrupting proteins play crucial roles in immune responses to infection, cancer, and signs of cellular damage. Recent studies identified novel membrane-disrupting proteins that act, either alone or in conjunction with other immune effector enzymes, especially granzymes, to cause a multiplicity of different forms of cell death. Some of these proteins (complement, perforin, perforin-2, and gasdermins) form large β-barrel pores, whose structures have just begun to be elucidated by cryo-EM. These pores release all sorts of cytosolic (and possibly noncytosolic) molecules that can cause inflammation and recruit other immune cells to sites of danger. The membrane-associated structure of others (MLKL, granulysin) is unknown; they may form smaller ion channels or perturb the membrane in other ways. Small channels can cause osmotic lysis, which can be highly inflammatory as cells burst, releasing their contents. In all cases of membrane damage to host cells, the targeted cell has the chance to survive if the membrane damage is repaired by the ubiquitous membrane repair response. As far as is known, bacterial cells have no comparable membrane defense. Some membrane-disrupting proteins are specific for bacterial versus host cell membranes, and some damage proteins from the outside and others from the inside. Cell target specificity is mostly (except for complement) not mediated by recognition of protein receptors, which means that target cells cannot escape by downmodulating expression of a receptor but have specificity determined by binding to specific membrane lipids or by the cholesterol content of the membrane, which may alter membrane stiffness.
There is still so much we do not know about these powerful proteins. Structural studies are needed to really understand how granulysin and MLKL damage membranes. Very little is known about how the expression of these membrane-damaging proteins is regulated and what cells express them. Additional mechanisms for protecting the expressing cell and regulating membrane damage likely exist and need to be investigated. Little is known about what happens to intracellular membranes of nuclei, mitochondria, endosomes, phagosomes, lysosomes, and other organelles when these molecules are activated. Are these membranes also damaged, causing organelle dysfunction, and does their damage contribute to cell death or inflammation? Which molecules are released from cells attacked by these membrane-disrupting proteins; do they differ according to the mediator of membrane damage, and which released molecules are most inflammatory? Importantly, little is known about how much the newly described membrane disruptors contribute to immune defense and pathology in vivo. For example, because in vivo models of necroptosis rely heavily on gene knockouts, the normal physiological role of necroptosis in normal immune defense and pathology is not well understood. Almost nothing is known about the functions of perforin-2. As we understand better the mechanisms behind membrane damage and the roles of these membrane-perturbing proteins in disease, can we develop small-molecule inhibitors or antibodies or harness their clever strategies for killing cells or delivering other molecules inside cells for therapeutic purposes? The next few years should see a lot of research to answer these questions.
ACKNOWLEDGMENTS
We thank past and current lab members and collaborators for useful discussions. This work was supported by NIH grants AI116577, AI123265, AI139914, AI131632, and AI145862 (J.L.), China National Youth Thousand Talents Program, National Natural Science Foundation of China (31972897), Key Research Program of the Chinese Academy of Sciences (ZDBS-LY-SM008), Strategic Priority Research Program of Chinese Academy of Sciences (XDB29030300), Rising-Star Program of Shanghai Science and Technology Committee (19QA1409800), and Shanghai Municipal Natural Science Foundation (18ZR1443900) (X.L.).
Footnotes
DISCLOSURE STATEMENT
J.L. is a cofounder of Ventus Therapeutics.
LITERATURE CITED
- 1.Rosado CJ, Kondos S, Bull TE, Kuiper MJ, Law RH, et al. 2008. The MACPF/CDC family of pore-forming toxins. Cell Microbiol 10:1765–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dotiwala F, Lieberman J. 2019. Granulysin: killer lymphocyte safeguard against microbes. Curr. Opin. Immunol 60:19–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liu X, Lieberman J. 2017. A mechanistic understanding of pyroptosis: the fiery death triggered by invasive infection. Adv. Immunol 135:81–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shan B, Pan H, Najafov A, Yuan J. 2018. Necroptosis in development and diseases. Genes Dev 32:327–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thiery J, Lieberman J. 2014. Perforin: a key pore-forming protein for immune control of viruses and cancer. Subcell. Biochem 80:197–220 [DOI] [PubMed] [Google Scholar]
- 6.Weinlich R, Oberst A, Beere HM, Green DR. 2017. Necroptosis in development, inflammation and disease. Nat. Rev. Mol. Cell Biol 18:127–36 [DOI] [PubMed] [Google Scholar]
- 7.Anderson DH, Sawaya MR, Cascio D, Ernst W, Modlin R, et al. 2003. Granulysin crystal structure and a structure-derived lytic mechanism. J. Mol. Biol 325:355–65 [DOI] [PubMed] [Google Scholar]
- 8.Dudkina NV, Spicer BA, Reboul CF, Conroy PJ, Lukoyanova N, et al. 2016. Structure of the poly-C9 component of the complement membrane attack complex. Nat. Commun 7:10588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Law RH, Lukoyanova N, Voskoboinik I, Caradoc-Davies TT, Baran K, et al. 2010. The structural basis for membrane binding and pore formation by lymphocyte perforin. Nature 468:447–51 [DOI] [PubMed] [Google Scholar]
- 10.Ruan J, Xia S, Liu X, Lieberman J, Wu H. 2018. Cryo-EM structure of the gasdermin A3 membrane pore. Nature 557:62–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Serna M, Giles JL, Morgan BP, Bubeck D. 2016. Structural basis of complement membrane attack complex formation. Nat. Commun 7:10587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Su L, Quade B, Wang H, Sun L, Wang X, Rizo J. 2014. A plug release mechanism for membrane permeation by MLKL. Structure 22:1489–500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Menny A, Serna M, Boyd CM, Gardner S, Joseph AP, et al. 2018. CryoEM reveals how the complement membrane attack complex ruptures lipid bilayers. Nat. Commun 9:5316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pang SS, Bayly-Jones C, Radjainia M, Spicer BA, Law RHP, et al. 2019. The cryo-EM structure of the acid activatable pore-forming immune effector Macrophage-expressed gene 1. Nat. Commun 10:4288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ding J, Wang K, Liu W, She Y, Sun Q, et al. 2016. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature 535:111–16 [DOI] [PubMed] [Google Scholar]
- 16.Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, et al. 2016. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 535:153–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pipkin ME, Lieberman J. 2007. Delivering the kiss of death: progress on understanding how perforin works. Curr. Opin. Immunol 19:301–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang J, Deobald K, Re F. 2019. Gasdermin D protects from melioidosis through pyroptosis and direct killing of bacteria. J. Immunol 202:3468–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chowdhury D, Lieberman J. 2008. Death by a thousand cuts: granzyme pathways of programmed cell death. Annu. Rev. Immunol 26:389–420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rogers C, Fernandes-Alnemri T, Mayes L, Alnemri D, Cingolani G, Alnemri ES. 2017. Cleavage of DFNA5 by caspase-3 during apoptosis mediates progression to secondary necrotic/pyroptotic cell death. Nat. Commun 8:14128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang Y, Gao W, Shi X, Ding J, Liu W, et al. 2017. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature 547:99–103 [DOI] [PubMed] [Google Scholar]
- 22.Antonopoulos C, Russo HM, El Sanadi C, Martin BN, Li X, et al. 2015. Caspase-8 as an effector and regulator of NLRP3 inflammasome signaling. J. Biol. Chem 290:20167–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mascarenhas DPA, Cerqueira DM, Pereira MSF, Castanheira FVS, Fernandes TD, et al. 2017. Inhibition of caspase-1 or gasdermin-D enable caspase-8 activation in the Naip5/NLRC4/ASC inflammasome. PLOS Pathog 13:e1006502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Laudisi F, Spreafico R, Evrard M, Hughes TR, Mandriani B, et al. 2013. Cutting edge: the NLRP3 inflammasome links complement-mediated inflammation and IL-1β release. J. Immunol 191:1006–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yao Y, Chen S, Cao M, Fan X, Yang T, et al. 2017. Antigen-specific CD8+ T cell feedback activates NLRP3 inflammasome in antigen-presenting cells through perforin. Nat. Commun 8:15402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Conos SA, Chen KW, De Nardo D, Hara H, Whitehead L, et al. 2017. Active MLKL triggers the NLRP3 inflammasome in a cell-intrinsic manner. PNAS 114:E961–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McCormack R, Bahnan W, Shrestha N, Boucher J, Barreto M, et al. 2016. Perforin-2 protects host cells and mice by restricting the vacuole to cytosol transitioning of a bacterial pathogen. Infect. Immun 84:1083–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Podack ER, Munson GP. 2016. Killing of microbes and cancer by the immune system with three mammalian pore-forming killer proteins. Front. Immunol 7:464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zipfel PF, Skerka C. 2009. Complement regulators and inhibitory proteins. Nat. Rev. Immunol 9:729–40 [DOI] [PubMed] [Google Scholar]
- 30.Guo RF, Ward PA. 2005. Role of C5a in inflammatory responses. Annu. Rev. Immunol 23:821–52 [DOI] [PubMed] [Google Scholar]
- 31.Ricklin D, Hajishengallis G, Yang K, Lambris JD. 2010. Complement: a key system for immune surveillance and homeostasis. Nat. Immunol 11:785–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Reboul CF, Whisstock JC, Dunstone MA. 2016. Giant MACPF/CDC pore forming toxins: a class of their own. Biochim. Biophys. Acta Biomembr 1858:475–86 [DOI] [PubMed] [Google Scholar]
- 33.Tschopp J, Muller-Eberhard HJ, Podack ER. 1982. Formation of transmembrane tubules by spontaneous polymerization of the hydrophilic complement protein C9. Nature 298:534–38 [DOI] [PubMed] [Google Scholar]
- 34.Anderluh G, Kisovec M, Krasevec N, Gilbert RJ. 2014. Distribution of MACPF/CDC proteins. Subcell. Biochem 80:7–30 [DOI] [PubMed] [Google Scholar]
- 35.Botto M, Kirschfink M, Macor P, Pickering MC, Wurzner R, Tedesco F. 2009. Complement in human diseases: lessons from complement deficiencies. Mol. Immunol 46:2774–83 [DOI] [PubMed] [Google Scholar]
- 36.Walport MJ. 2001. Complement: first of two parts. N. Engl. J. Med 344:1058–66 [DOI] [PubMed] [Google Scholar]
- 37.Lindahl G, Sjobring U, Johnsson E. 2000. Human complement regulators: a major target for pathogenic microorganisms. Curr. Opin. Immunol 12:44–51 [DOI] [PubMed] [Google Scholar]
- 38.Pipkin ME, Ljutic B, Cruz-Guilloty F, Nouzova M, Rao A, et al. 2007. Chromosome transfer activates and delineates a locus control region for perforin. Immunity 26:29–41 [DOI] [PubMed] [Google Scholar]
- 39.Voskoboinik I, Whisstock JC, Trapani JA. 2015. Perforin and granzymes: function, dysfunction and human pathology. Nat. Rev. Immunol 15:388–400 [DOI] [PubMed] [Google Scholar]
- 40.Shepard LA, Heuck AP, Hamman BD, Rossjohn J, Parker MW, et al. 1998. Identification of a membrane-spanning domain of the thiol-activated pore-forming toxin Clostridium perfringens perfringolysin O: an α-helical to β-sheet transition identified by fluorescence spectroscopy. Biochemistry 37:14563–74 [DOI] [PubMed] [Google Scholar]
- 41.Peters PJ, Borst J, Oorschot V, Fukuda M, Krahenbuhl O, et al. 1991. Cytotoxic T lymphocyte granules are secretory lysosomes, containing both perforin and granzymes. J. Exp. Med 173:1099–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stinchcombe JC, Bossi G, Booth S, Griffiths GM. 2001. The immunological synapse of CTL contains a secretory domain and membrane bridges. Immunity 15:751–61 [DOI] [PubMed] [Google Scholar]
- 43.Keefe D, Shi L, Feske S, Massol R, Navarro F, et al. 2005. Perforin triggers a plasma membrane-repair response that facilitates CTL induction of apoptosis. Immunity 23:249–62 [DOI] [PubMed] [Google Scholar]
- 44.Thiery J, Keefe D, Boulant S, Boucrot E, Walch M, et al. 2011. Perforin pores in the endosomal membrane trigger the release of endocytosed granzyme B into the cytosol of target cells. Nat. Immunol 12:770–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Thiery J, Keefe D, Saffarian S, Martinvalet D, Walch M, et al. 2010. Perforin activates clathrin- and dynamin-dependent endocytosis, which is required for plasma membrane repair and delivery of granzyme B for granzyme-mediated apoptosis. Blood 115:1582–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bird CH, Sun J, Ung K, Karambalis D, Whisstock JC, et al. 2005. Cationic sites on granzyme B contribute to cytotoxicity by promoting its uptake into target cells. Mol. Cell. Biol 25:7854–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shi L, Keefe D, Durand E, Feng H, Zhang D, Lieberman J. 2005. Granzyme B binds to target cells mostly by charge and must be added at the same time as perforin to trigger apoptosis. J. Immunol 174:5456–61 [DOI] [PubMed] [Google Scholar]
- 48.Darmon AJ, Nicholson DW, Bleackley RC. 1995. Activation of the apoptotic protease CPP32 by cytotoxic T-cell-derived granzyme B. Nature 377:446–48 [DOI] [PubMed] [Google Scholar]
- 49.Yagi H, Conroy PJ, Leung EW, Law RH, Trapani JA, et al. 2015. Structural Basis for Ca2+-mediated interaction of the perforin C2 domain with lipid membranes. J. Biol. Chem 290:25213–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gilbert RJ. 2010. Cholesterol-Dependent Cytolysins New York: Springer; [DOI] [PubMed] [Google Scholar]
- 51.Balaji KN, Schaschke N, Machleidt W, Catalfamo M, Henkart PA. 2002. Surface cathepsin B protects cytotoxic lymphocytes from self-destruction after degranulation. J. Exp. Med 196:493–503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Baran K, Ciccone A, Peters C, Yagita H, Bird PI, et al. 2006. Cytotoxic T lymphocytes from cathepsin B-deficient mice survive normally in vitro and in vivo after encountering and killing target cells. J. Biol. Chem 281:30485–91 [DOI] [PubMed] [Google Scholar]
- 53.Kagi D, Ledermann B, Burki K, Seiler P, Odermatt B, et al. 1994. Cytotoxicity mediated by T cells and natural killer cells is greatly impaired in perforin-deficient mice. Nature 369:31–37 [DOI] [PubMed] [Google Scholar]
- 54.van den Broek ME, Kagi D, Ossendorp F, Toes R, Vamvakas S, et al. 1996. Decreased tumor surveillance in perforin-deficient mice. J. Exp. Med 184:1781–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang K, Jordan MB, Marsh RA, Johnson JA, Kissell D, et al. 2011. Hypomorphic mutations in PRF1, MUNC13–4, and STXBP2 are associated with adult-onset familial HLH. Blood 118:5794–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jenkins MR, Rudd-Schmidt JA, Lopez JA, Ramsbottom KM, Mannering SI, et al. 2015. Failed CTL/NK cell killing and cytokine hypersecretion are directly linked through prolonged synapse time. J. Exp. Med 212:307–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Xiong P, Shiratsuchi M, Matsushima T, Liao J, Tanaka E, et al. 2017. Regulation of expression and trafficking of perforin-2 by LPS and TNF-α. Cell Immunol 320:1–10 [DOI] [PubMed] [Google Scholar]
- 58.McCormack R, de Armas LR, Shiratsuchi M, Ramos JE, Podack ER. 2013. Inhibition of intracellular bacterial replication in fibroblasts is dependent on the perforin-like protein (perforin-2) encoded by macrophage-expressed gene 1. J. Innate Immun 5:185–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.McCormack R, Podack ER. 2015. Perforin-2/Mpeg1 and other pore-forming proteins throughout evolution. J. Leukoc. Biol 98:761–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bai F, McCormack RM, Hower S, Plano GV, Lichtenheld MG, Munson GP. 2018. Perforin-2 breaches the envelope of phagocytosed bacteria allowing antimicrobial effectors access to intracellular targets. J. Immunol 201:2710–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fields KA, McCormack R, de Armas LR, Podack ER. 2013. Perforin-2 restricts growth of Chlamydia trachomatis in macrophages. Infect. Immun 81:3045–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.McCormack RM, Szymanski EP, Hsu AP, Perez E, Olivier KN, et al. 2017. MPEG1/perforin-2 mutations in human pulmonary nontuberculous mycobacterial infections. JCI Insight 2:89635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.McCormack RM, de Armas LR, Shiratsuchi M, Fiorentino DG, Olsson ML, et al. 2015. Perforin-2 is essential for intracellular defense of parenchymal cells and phagocytes against pathogenic bacteria. eLife 4:e06508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tamura M, Tanaka S, Fujii T, Aoki A, Komiyama H, et al. 2007. Members of a novel gene family, Gsdm, are expressed exclusively in the epithelium of the skin and gastrointestinal tract in a highly tissue-specific manner. Genomics 89:618–29 [DOI] [PubMed] [Google Scholar]
- 65.Burgener SS, Leborgne NGF, Snipas SJ, Salvesen GS, Bird PI, Benarafa C. 2019. Cathepsin G inhibition by Serpinb1 and Serpinb6 prevents programmed necrosis in neutrophils and monocytes and reduces GSDMD-driven inflammation. Cell Rep 27:3646–56.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sollberger G, Choidas A, Burn GL, Habenberger P, Di Lucrezia R, et al. 2018. Gasdermin D plays a vital role in the generation of neutrophil extracellular traps. Sci. Immunol 3:eaar6689. [DOI] [PubMed] [Google Scholar]
- 67.Sarhan J, Liu BC, Muendlein HI, Li P, Nilson R, et al. 2018. Caspase-8 induces cleavage of gasdermin D to elicit pyroptosis during Yersinia infection. PNAS 115:E10888–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Orning P, Weng D, Starheim K, Ratner D, Best Z, et al. 2018. Pathogen blockade of TAK1 triggers caspase-8-dependent cleavage of gasdermin D and cell death. Science 362:1064–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chao KL, Kulakova L, Herzberg O. 2017. Gene polymorphism linked to increased asthma and IBD risk alters gasdermin-B structure, a sulfatide and phosphoinositide binding protein. PNAS 114:E1128–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Collin RW, Kalay E, Oostrik J, Caylan R, Wollnik B, et al. 2007. Involvement of DFNB59 mutations in autosomal recessive nonsyndromic hearing impairment. Hum. Mutat 28:718–23 [DOI] [PubMed] [Google Scholar]
- 71.Runkel F, Marquardt A, Stoeger C, Kochmann E, Simon D, et al. 2004. The dominant alopecia phenotypes Bareskin, Rex-denuded, and Reduced Coat 2 are caused by mutations in gasdermin 3. Genomics 84:824–35 [DOI] [PubMed] [Google Scholar]
- 72.Van Laer L, Huizing EH, Verstreken M, van Zuijlen D, Wauters JG, et al. 1998. Nonsyndromic hearing impairment is associated with a mutation in DFNA5. Nat. Genet 20:194–97 [DOI] [PubMed] [Google Scholar]
- 73.Wu H, Romieu I, Sienra-Monge JJ, Li H, del Rio-Navarro BE, London SJ. 2009. Genetic variation in ORM1-like 3 (ORMDL3) and gasdermin-like (GSDML) and childhood asthma. Allergy 64:629–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Li J, Zhou Y, Yang T, Wang N, Lian X, Yang L. 2010. Gsdma3 is required for hair follicle differentiation in mice. Biochem. Biophys. Res. Commun 403:18–23 [DOI] [PubMed] [Google Scholar]
- 75.Tanaka S, Tamura M, Aoki A, Fujii T, Komiyama H, et al. 2007. A new Gsdma3 mutation affecting anagen phase of first hair cycle. Biochem. Biophys. Res. Commun 359:902–7 [DOI] [PubMed] [Google Scholar]
- 76.Kumar S, Rathkolb B, Budde BS, Nurnberg P, de Angelis MH, et al. 2012. Gsdma3I359N is a novel ENU-induced mutant mouse line for studying the function of Gasdermin A3 in the hair follicle and epidermis. J. Dermatol. Sci 67:190–92 [DOI] [PubMed] [Google Scholar]
- 77.Lunny DP, Weed E, Nolan PM, Marquardt A, Augustin M, Porter RM. 2005. Mutations in gasdermin 3 cause aberrant differentiation of the hair follicle and sebaceous gland. J. Investig. Dermatol 124:615–21 [DOI] [PubMed] [Google Scholar]
- 78.Porter RM, Jahoda CA, Lunny DP, Henderson G, Ross J, et al. 2002. Defolliculated (Dfl): a dominant mouse mutation leading to poor sebaceous gland differentiation and total elimination of pelage follicles. J. Investig. Dermatol 119:32–37 [DOI] [PubMed] [Google Scholar]
- 79.Shi P, Tang A, Xian L, Hou S, Zou D, et al. 2015. Loss of conserved Gsdma3 self-regulation causes autophagy and cell death. Biochem. J 468:325–36 [DOI] [PubMed] [Google Scholar]
- 80.Kang MJ, Yu HS, Seo JH, Kim HY, Jung YH, et al. 2012. GSDMB/ORMDL3 variants contribute to asthma susceptibility and eosinophil-mediated bronchial hyperresponsiveness. Hum. Immunol 73:954–59 [DOI] [PubMed] [Google Scholar]
- 81.Das S, Miller M, Beppu AK, Mueller J, McGeough MD, et al. 2016. GSDMB induces an asthma phenotype characterized by increased airway responsiveness and remodeling without lung inflammation. PNAS 113:13132–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Delmaghani S, del Castillo FJ, Michel V, Leibovici M, Aghaie A, et al. 2006. Mutations in the gene encoding pejvakin, a newly identified protein of the afferent auditory pathway, cause DFNB59 auditory neuropathy. Nat. Genet 38:770–78 [DOI] [PubMed] [Google Scholar]
- 83.Delmaghani S, Defourny J, Aghaie A, Beurg M, Dulon D, et al. 2015. Hypervulnerability to sound exposure through impaired adaptive proliferation of peroxisomes. Cell 163:894–906 [DOI] [PubMed] [Google Scholar]
- 84.Christofferson DE, Yuan J. 2010. Necroptosis as an alternative form of programmed cell death. Curr. Opin. Cell Biol 22:263–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Murphy JM, Czabotar PE, Hildebrand JM, Lucet IS, Zhang JG, et al. 2013. The pseudokinase MLKL mediates necroptosis via a molecular switch mechanism. Immunity 39:443–53 [DOI] [PubMed] [Google Scholar]
- 86.Cai Z, Jitkaew S, Zhao J, Chiang HC, Choksi S, et al. 2014. Plasma membrane translocation of trimerized MLKL protein is required for TNF-induced necroptosis. Nat. Cell Biol 16:55–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Chen X, Li W, Ren J, Huang D, He WT, et al. 2014. Translocation of mixed lineage kinase domain-like protein to plasma membrane leads to necrotic cell death. Cell Res 24:105–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Dondelinger Y, Declercq W, Montessuit S, Roelandt R, Goncalves A, et al. 2014. MLKL compromises plasma membrane integrity by binding to phosphatidylinositol phosphates. Cell Rep 7:971–81 [DOI] [PubMed] [Google Scholar]
- 89.Hildebrand JM, Tanzer MC, Lucet IS, Young SN, Spall SK, et al. 2014. Activation of the pseudokinase MLKL unleashes the four-helix bundle domain to induce membrane localization and necroptotic cell death. PNAS 111:15072–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wang H, Sun L, Su L, Rizo J, Liu L, et al. 2014. Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol. Cell 54:133–46 [DOI] [PubMed] [Google Scholar]
- 91.Kaiser WJ, Sridharan H, Huang C, Mandal P, Upton JW, et al. 2013. Toll-like receptor 3-mediated necrosis via TRIF, RIP3, and MLKL. J. Biol. Chem 288:31268–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Upton JW, Kaiser WJ, Mocarski ES. 2012. DAI/ZBP1/DLM-1 complexes with RIP3 to mediate virus-induced programmed necrosis that is targeted by murine cytomegalovirus vIRA. Cell Host Microbe 11:290–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wu J, Huang Z, Ren J, Zhang Z, He P, et al. 2013. Mlkl knockout mice demonstrate the indispensable role of Mlkl in necroptosis. Cell Res 23:994–1006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Chan FK. 2012. Fueling the flames: Mammalian programmed necrosis in inflammatory diseases. Cold Spring Harb. Perspect. Biol 4:a008805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Linkermann A, Hackl MJ, Kunzendorf U, Walczak H, Krautwald S, Jevnikar AM. 2013. Necroptosis in immunity and ischemia-reperfusion injury. Am. J. Transplant 13:2797–804 [DOI] [PubMed] [Google Scholar]
- 96.Sun L, Wang X. 2014. A new kind of cell suicide: mechanisms and functions of programmed necrosis. Trends Biochem. Sci 39:587–93 [DOI] [PubMed] [Google Scholar]
- 97.Pasparakis M, Vandenabeele P. 2015. Necroptosis and its role in inflammation. Nature 517:311–20 [DOI] [PubMed] [Google Scholar]
- 98.Mocarski ES, Guo H, Kaiser WJ. 2015. Necroptosis: The Trojan horse in cell autonomous antiviral host defense. Virology 479–480:160–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Chen X, Zhuang C, Ren Y, Zhang H, Qin X, et al. 2019. Identification of the Raf kinase inhibitor TAK-632 and its analogues as potent inhibitors of necroptosis by targeting RIPK1 and RIPK3. Br. J. Pharmacol 176:2095–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Hou J, Ju J, Zhang Z, Zhao C, Li Z, et al. 2019. Discovery of potent necroptosis inhibitors targeting RIPK1 kinase activity for the treatment of inflammatory disorder and cancer metastasis. Cell Death Dis 10:493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Vandenabeele P, Grootjans S, Callewaert N, Takahashi N. 2013. Necrostatin-1 blocks both RIPK1 and IDO: consequences for the study of cell death in experimental disease models. Cell Death Differ 20:185–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Bertrand MJ, Milutinovic S, Dickson KM, Ho WC, Boudreault A, et al. 2008. cIAP1 and cIAP2 facilitate cancer cell survival by functioning as E3 ligases that promote RIP1 ubiquitination. Mol. Cell 30:689–700 [DOI] [PubMed] [Google Scholar]
- 103.Kang TB, Yang SH, Toth B, Kovalenko A, Wallach D. 2013. Caspase-8 blocks kinase RIPK3-mediated activation of the NLRP3 inflammasome. Immunity 38:27–40 [DOI] [PubMed] [Google Scholar]
- 104.Lawlor KE, Khan N, Mildenhall A, Gerlic M, Croker BA, et al. 2015. RIPK3 promotes cell death and NLRP3 inflammasome activation in the absence of MLKL. Nat. Commun 6:6282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Moriwaki K, Chan FK. 2014. Necrosis-dependent and independent signaling of the RIP kinases in inflammation. Cytokine Growth Factor Rev 25:167–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Varfolomeev E, Blankenship JW, Wayson SM, Fedorova AV, Kayagaki N, et al. 2007. IAP antagonists induce autoubiquitination of c-IAPs, NF-κB activation, and TNFα-dependent apoptosis. Cell 131:669–81 [DOI] [PubMed] [Google Scholar]
- 107.Sun L, Wang H, Wang Z, He S, Chen S, et al. 2012. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 148:213–27 [DOI] [PubMed] [Google Scholar]
- 108.Mocarski ES, Kaiser WJ, Livingston-Rosanoff D, Upton JW, Daley-Bauer LP. 2014. True grit: programmed necrosis in antiviral host defense, inflammation, and immunogenicity. J. Immunol 192:2019–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Nogusa S, Thapa RJ, Dillon CP, Liedmann S, Oguin TH 3rd, et al. 2016. RIPK3 activates parallel pathways of MLKL-driven necroptosis and FADD-mediated apoptosis to protect against influenza A virus. Cell Host Microbe 20:13–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Robinson N, McComb S, Mulligan R, Dudani R, Krishnan L, Sad S. 2012. Type I interferon induces necroptosis in macrophages during infection with Salmonella enterica serovar Typhimurium. Nat. Immunol 13:954–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Sai K, Parsons C, House JS, Kathariou S, Ninomiya-Tsuji J. 2019. Necroptosis mediators RIPK3 and MLKL suppress intracellular Listeria replication independently of host cell killing. J. Cell Biol 218:1994–2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Pena SV, Hanson DA, Carr BA, Goralski TJ, Krensky AM. 1997. Processing, subcellular localization, and function of 519 (granulysin), a human late T cell activation molecule with homology to small, lytic, granule proteins. J. Immunol 158:2680–88 [PubMed] [Google Scholar]
- 113.Stenger S, Hanson DA, Teitelbaum R, Dewan P, Niazi KR, et al. 1998. An antimicrobial activity of cytolytic T cells mediated by granulysin. Science 282:121–25 [DOI] [PubMed] [Google Scholar]
- 114.Chung WH, Hung SI, Yang JY, Su SC, Huang SP, et al. 2008. Granulysin is a key mediator for disseminated keratinocyte death in Stevens-Johnson syndrome and toxic epidermal necrolysis. Nat. Med 14:1343–50 [DOI] [PubMed] [Google Scholar]
- 115.Veljkovic Vujaklija D, Dominovic M, Gulic T, Mahmutefendic H, Haller H, et al. 2013. Granulysin expression and the interplay of granulysin and perforin at the maternal-fetal interface. J. Reprod. Immunol 97:186–96 [DOI] [PubMed] [Google Scholar]
- 116.Maeda Y, Tamura T, Fukutomi Y, Mukai T, Kai M, Makino M. 2011. A lipopeptide facilitate induction of Mycobacterium leprae killing in host cells. PLOS Negl. Trop. Dis 5:e1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Leippe M, Ebel S, Schoenberger OL, Horstmann RD, Muller-Eberhard HJ. 1991. Pore-forming peptide of pathogenic Entamoeba histolytica. PNAS 88:7659–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Hecht O, Van Nuland NA, Schleinkofer K, Dingley AJ, Bruhn H, et al. 2004. Solution structure of the pore-forming protein of Entamoeba histolytica. J. Biol. Chem 279:17834–41 [DOI] [PubMed] [Google Scholar]
- 119.Barman H, Walch M, Latinovic-Golic S, Dumrese C, Dolder M, et al. 2006. Cholesterol in negatively charged lipid bilayers modulates the effect of the antimicrobial protein granulysin. J. Membr. Biol 212:29–39 [DOI] [PubMed] [Google Scholar]
- 120.Abe R, Yoshioka N, Murata J, Fujita Y, Shimizu H. 2009. Granulysin as a marker for early diagnosis of the Stevens-Johnson syndrome. Ann. Intern. Med 151:514–15 [DOI] [PubMed] [Google Scholar]
- 121.Dotiwala F, Mulik S, Polidoro RB, Ansara JA, Burleigh BA, et al. 2016. Killer lymphocytes use granulysin, perforin and granzymes to kill intracellular parasites. Nat. Med 22:210–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Walch M, Dotiwala F, Mulik S, Thiery J, Kirchhausen T, et al. 2014. Cytotoxic cells kill intracellular bacteria through granulysin-mediated delivery of granzymes. Cell 157:1309–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Dotiwala F, Sen Santara S, Binker-Cosen AA, Li B, Chandrasekaran S, Lieberman J. 2017. Granzyme B disrupts central metabolism and protein synthesis in bacteria to promote an immune cell death program. Cell 171:1125–37.e11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Li SS, Kyei SK, Timm-McCann M, Ogbomo H, Jones GJ, et al. 2013. The NK receptor NKp30 mediates direct fungal recognition and killing and is diminished in NK cells from HIV-infected patients. Cell Host Microbe 14:387–97 [DOI] [PubMed] [Google Scholar]
- 125.Vitenshtein A, Charpak-Amikam Y, Yamin R, Bauman Y, Isaacson B, et al. 2016. NK cell recognition of Candida glabrata through binding of NKp46 and NCR1 to fungal ligands Epa1, Epa6, and Epa7. Cell Host Microbe 20:527–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Huang LP, Lyu SC, Clayberger C, Krensky AM. 2007. Granulysin-mediated tumor rejection in transgenic mice. J. Immunol 178:77–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Chen X, He WT, Hu L, Li J, Fang Y, et al. 2016. Pyroptosis is driven by non-selective gasdermin-D pore and its morphology is different from MLKL channel-mediated necroptosis. Cell Res 26:1007–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Zhang Y, Chen X, Gueydan C, Han J. 2018. Plasma membrane changes during programmed cell deaths. Cell Res 28:9–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Newton K, Manning G. 2016. Necroptosis and inflammation. Annu. Rev. Biochem 85:743–63 [DOI] [PubMed] [Google Scholar]
- 130.Davis MA, Fairgrieve MR, Den Hartigh A, Yakovenko O, Duvvuri B, et al. 2019. Calpain drives pyroptotic vimentin cleavage, intermediate filament loss, and cell rupture that mediates immunostimulation. PNAS 116:5061–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, et al. 2005. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat. Chem. Biol 1:112–19 [DOI] [PubMed] [Google Scholar]
- 132.Hu J, Liu X, Zhao J, Xia S, Ruan J, et al. 2018. Identification of pyroptosis inhibitors that target a reactive cysteine in gasdermin D bioRxiv 365908. 10.1101/365908 [DOI]
- 133.Enari M, Sakahira H, Yokoyama H, Okawa K, Iwamatsu A, Nagata S. 1998. A caspase-activated DNase that degrades DNA during apoptosis, and its inhibitor ICAD. Nature 391:43–50 [DOI] [PubMed] [Google Scholar]
- 134.Thomas DA, Du C, Xu M, Wang X, Ley TJ. 2000. DFF45/ICAD can be directly processed by granzyme B during the induction of apoptosis. Immunity 12:621–32 [DOI] [PubMed] [Google Scholar]
- 135.Fan Z, Beresford PJ, Oh DY, Zhang D, Lieberman J. 2003. Tumor suppressor NM23-H1 is a granzyme A-activated DNase during CTL-mediated apoptosis, and the nucleosome assembly protein SET is its inhibitor. Cell 112:659–72 [DOI] [PubMed] [Google Scholar]
- 136.Chowdhury D, Beresford PJ, Zhu P, Zhang D, Sung JS, et al. 2006. The exonuclease TREX1 is in the SET complex and acts in concert with NM23-H1 to degrade DNA during granzyme A-mediated cell death. Mol. Cell 23:133–42 [DOI] [PubMed] [Google Scholar]
- 137.Nagata S 2018. Apoptosis and clearance of apoptotic cells. Annu. Rev. Immunol 36:489–517 [DOI] [PubMed] [Google Scholar]
- 138.Werfel TA, Cook RS. 2018. Efferocytosis in the tumor microenvironment. Semin. Immunopathol 40:545–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Poon IK, Lucas CD, Rossi AG, Ravichandran KS. 2014. Apoptotic cell clearance: basic biology and therapeutic potential. Nat. Rev. Immunol 14:166–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Lieberman J 2013. Cell-mediated cytotoxicity In Fundamental Immunology, ed. Paul WE, pp. 891–909. Philadelphia: Wolters Kluwer/Lippincott Williams Wilkins [Google Scholar]
- 141.Kono H, Rock KL. 2008. How dying cells alert the immune system to danger. Nat. Rev. Immunol 8:279–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Rus H, Cudrici C, Niculescu F. 2005. The role of the complement system in innate immunity. Immunol. Res 33:103–12 [DOI] [PubMed] [Google Scholar]
- 143.Kjaer TR, Thiel S, Andersen GR. 2013. Toward a structure-based comprehension of the lectin pathway of complement. Mol. Immunol 56:413–22 [DOI] [PubMed] [Google Scholar]
- 144.Uellner R, Zvelebil MJ, Hopkins J, Jones J, MacDougall LK, et al. 1997. Perforin is activated by a proteolytic cleavage during biosynthesis which reveals a phospholipid-binding C2 domain. EMBO J 16:7287–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Dupuis M, Schaerer E, Krause KH, Tschopp J. 1993. The calcium-binding protein calreticulin is a major constituent of lytic granules in cytolytic T lymphocytes. J. Exp. Med 177:1–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Sula Karreci E, Eskandari SK, Dotiwala F, Routray SK, Kurdi AT, et al. 2017. Human regulatory T cells undergo self-inflicted damage via granzyme pathways upon activation. JCI Insight 2:91599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Aglietti RA, Estevez A, Gupta A, Ramirez MG, Liu PS, et al. 2016. GsdmD p30 elicited by caspase-11 during pyroptosis forms pores in membranes. PNAS 113:7858–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Sborgi L, Ruhl S, Mulvihill E, Pipercevic J, Heilig R, et al. 2016. GSDMD membrane pore formation constitutes the mechanism of pyroptotic cell death. EMBO J 35:1766–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.van Pee K, Mulvihill E, Muller DJ, Yildiz O. 2016. Unraveling the pore-forming steps of pneumolysin from Streptococcus pneumoniae. Nano Lett 16:7915–24 [DOI] [PubMed] [Google Scholar]
- 150.van Pee K, Neuhaus A, D’Imprima E, Mills DJ, Kuhlbrandt W, Yildiz O. 2017. CryoEM structures of membrane pore and prepore complex reveal cytolytic mechanism of Pneumolysin. eLife 6:e23644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Liu S, Liu H, Johnston A, Hanna-Addams S, Reynoso E, et al. 2017. MLKL forms disulfide bond-dependent amyloid-like polymers to induce necroptosis. PNAS 114:E7450–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Karch J, Kanisicak O, Brody MJ, Sargent MA, Michael DM, Molkentin JD. 2015. Necroptosis interfaces with MOMP and the MPTP in mediating cell death. PLOS ONE 10:e0130520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Rogers C, Erkes DA, Nardone A, Aplin AE, Fernandes-Alnemri T, Alnemri ES. 2019. Gasdermin pores permeabilize mitochondria to augment caspase-3 activation during apoptosis and inflammasome activation. Nat. Commun 10:1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.de Vasconcelos NM, Van Opdenbosch N, Van Gorp H, Parthoens E, Lamkanfi M. 2019. Single-cell analysis of pyroptosis dynamics reveals conserved GSDMD-mediated subcellular events that precede plasma membrane rupture. Cell Death Differ 26:146–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Liu J, Qian C, Cao X. 2016. Post-translational modification control of innate immunity. Immunity 45:15–30 [DOI] [PubMed] [Google Scholar]
- 156.Liu X, Wang Q, Chen W, Wang C. 2013. Dynamic regulation of innate immunity by ubiquitin and ubiquitin-like proteins. Cytokine Growth Factor Rev 24:559–70 [DOI] [PubMed] [Google Scholar]
- 157.Rathkey JK, Zhao J, Liu Z, Chen Y, Yang J, et al. 2018. Chemical disruption of the pyroptotic pore-forming protein gasdermin D inhibits inflammatory cell death and sepsis. Sci. Immunol 3:eaat2738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Kimberley FC, Sivasankar B, Paul Morgan B. 2007. Alternative roles for CD59. Mol. Immunol 44:73–81 [DOI] [PubMed] [Google Scholar]
- 159.Kim DD, Song WC. 2006. Membrane complement regulatory proteins. Clin. Immunol 118:127–36 [DOI] [PubMed] [Google Scholar]
- 160.Brodsky RA. 2015. Complement in hemolytic anemia. Blood 126:2459–65 [DOI] [PubMed] [Google Scholar]
- 161.Blink EJ, Trapani JA, Jans DA. 1999. Perforin-dependent nuclear targeting of granzymes: a central role in the nuclear events of granule-exocytosis-mediated apoptosis? Immunol. Cell Biol 77:206–15 [DOI] [PubMed] [Google Scholar]
- 162.Martinvalet D, Dykxhoorn DM, Ferrini R, Lieberman J. 2008. Granzyme A cleaves a mitochondrial complex I protein to initiate caspase-independent cell death. Cell 133:681–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Spicer BA, Law RHP, Caradoc-Davies TT, Ekkel SM, Bayly-Jones C, et al. 2018. The first transmembrane region of complement component-9 acts as a brake on its self-assembly. Nat. Commun 9:3266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Huang D, Zheng X, Wang ZA, Chen X, He WT, et al. 2017. The MLKL channel in necroptosis is an octamer formed by tetramers in a dyadic process. Mol. Cell. Biol 37:e00497–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Xia B, Fang S, Chen X, Hu H, Chen P, et al. 2016. MLKL forms cation channels. Cell Res 26:517–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Tschopp J, Engel A, Podack ER. 1984. Molecular weight of poly(C9): 12 to 18 C9 molecules form the transmembrane channel of complement. J. Biol. Chem 259:1922–28 [PubMed] [Google Scholar]
- 167.Liu Z, Wang C, Yang J, Zhou B, Yang R, et al. 2019. Crystal structures of the full-length murine and human gasdermin D reveal mechanisms of autoinhibition, lipid binding, and oligomerization. Immunity 51:43–49.e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Andrews NW, Corrotte M. 2018. Plasma membrane repair. Curr. Biol 28:R392–97 [DOI] [PubMed] [Google Scholar]
- 169.McNeil PL, Kirchhausen T. 2005. An emergency response team for membrane repair. Nat. Rev. Mol. Cell Biol 6:499–505 [DOI] [PubMed] [Google Scholar]
- 170.Dal Peraro M, van der Goot FG. 2016. Pore-forming toxins: ancient, but never really out of fashion. Nat. Rev. Microbiol 14:77–92 [DOI] [PubMed] [Google Scholar]
- 171.Tegla CA, Cudrici C, Patel S, Trippe R 3rd, Rus V, et al. 2011. Membrane attack by complement: the assembly and biology of terminal complement complexes. Immunol. Res 51:45–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Ruhl S, Shkarina K, Demarco B, Heilig R, Santos JC, Broz P. 2018. ESCRT-dependent membrane repair negatively regulates pyroptosis downstream of GSDMD activation. Science 362:956–60 [DOI] [PubMed] [Google Scholar]
- 173.Gong YN, Guy C, Olauson H, Becker JU, Yang M, et al. 2017. ESCRT-III acts downstream of MLKL to regulate necroptotic cell death and its consequences. Cell 169:286–300.e16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Lieberman J, Wu H, Kagan JC. 2019. Gasdermin D activity in inflammation and host defense. Sci. Immunol 4:eaav1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Chen KW, Gross CJ, Sotomayor FV, Stacey KJ, Tschopp J, et al. 2014. The neutrophil NLRC4 inflammasome selectively promotes IL-1β maturation without pyroptosis during acute Salmonella challenge. Cell Rep 8:570–82 [DOI] [PubMed] [Google Scholar]
- 176.Gaidt MM, Ebert TS, Chauhan D, Schmidt T, Schmid-Burgk JL, et al. 2016. Human monocytes engage an alternative inflammasome pathway. Immunity 44:833–46 [DOI] [PubMed] [Google Scholar]
- 177.Wolf AJ, Reyes CN, Liang W, Becker C, Shimada K, et al. 2016. Hexokinase is an innate immune receptor for the detection of bacterial peptidoglycan. Cell 166:624–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Zanoni I, Tan Y, Di Gioia M, Broggi A, Ruan J, et al. 2016. An endogenous caspase-11 ligand elicits interleukin-1 release from living dendritic cells. Science 352:1232–36 [DOI] [PMC free article] [PubMed] [Google Scholar]