

Abstract
miR-155 is associated with the promotion of tumorigenesis. Herein, we indicate that abnormal miR-155 was negatively correlated with the expression of P21WAF1/Cip1. Our results suggest that miR-155 alters the transcriptome and inhibits the expression of H3F3A in liver cancer cells. Therefore, miR-155 inhibits the methylation modification of histone H3 on the 27th lysine. Notably, on the one hand, miR-155-dependent CTCF loops cause the CDK2 interacting with cyclin E in liver cancer cells; on the other hand, miR-155 promotes the phosphorylation modification of CDK2 by inhibiting H3F3A. Subsequently, miR-155 competitively blocks the binding of RNA polymerase II (RNA Pol II) to the P21WAF1/CIP1 promoter by increasing the phosphorylation of CDK2, inhibiting the transcription and translation of P21WAF1/CIP1. Strikingly, excessive P21WAF1/CIP1 abolishes the cancerous function of miR-155. In conclusion, miR-155 can play a positive role in the development of liver cancer and influence a series of gene expression through epigenetic regulation.
Keywords: liver cancer, miR-155, P21WAF1/Cip1, H3F3A, CDK2
Graphical Abstract
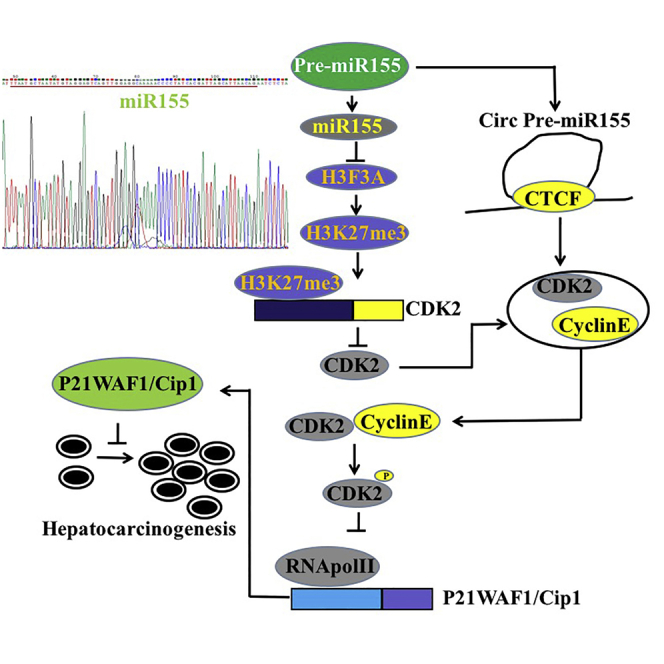
miR-155 promotes the phosphorylation modification of CDK2 and inhibits P21WAF1/CIP1 by inhibiting H3F3A. Therefore, miR-155 can play a positive role in the development of liver cancer and influence a series of gene expression through epigenetic regulation.
Introduction
MicroRNAs (miRNAs) are a class of small non-coding RNAs that are endogenous regulators of gene expression.1, 2, 3, 4, 5 At present, miRNAs have been shown to play an important role in many biological responses, and the dysfunction of miRNAs is associated with several human diseases, especially tumors.1,6
Several studies have reported that miR-155 is closely related to inflammation.7,8 Other studies have found that miR-155 can promote the progression of human leukemia and is one of the key factors for leukemia. Moreover, miR-155 is also closely related to tumors.9, 10, 11, 12, 13, 14, 15 Especially, it has been found that miR-155 is upregulated in the liver tissues of patients.16, 17, 18 So far, the role of miR-155 in the development of human liver cancer has not been clarified. Therefore, it is important to clarify the mechanism of miR-155 in the hepatocarcinogenesis.
Studies have shown that epigenetic regulation is closely related to tumorigenesis. Histone H3 modification is the most widely studied, and its biological significance of some modifications has been well characterized.19 For example, acetylation of lysine (H3K14) at position 14 of H3, methylation of lysine at position 4 (H3K4), and phosphorylation of serine at position 10 (H3S10) cause activation of gene transcription.20, 21, 22 In contrast, methylation of H3K9 and H3K27 is often associated with gene suppression.23, 24, 25, 26, 27, 28, 29, 30 Recently, it has been found that miR-155 can change the transcription initiation site of the serine/arginine-rich splicing factor 2 (SRSF2) promoter and regulate the protein kinase A (PKA) signaling pathway.31 These findings suggest that miR-155 is directly or indirectly involved in epigenetic regulation.
In this study, we found that miR-155 promotes the growth of human hepatoma cell Hep3B. miR-155 inhibits the expression of H3F3A and then reduces the modification level of H3K27me3. These findings provide a theoretical basis for the treatment and diagnosis of human liver cancer.
Results
miR-155 Promotes Progression of Human Liver Cancer Cells
To investigate the effect of miR-155 on hepatoma cells, the miR-155 (RefSeq: MI00000681) precursor sequence (CUGUUAAUGCUAAUCGUGAUAGGG GUUUUUGCCUCCAACUGACUCCUACAUAUUAGCAUUAACAG) was cloned into the lentiviral vector pLVX-ZsGreen-miRNA-Puro to construct a recombinant lentiviral framework plasmid pLVX-ZsGreen-miR-155-puro (pLVX-miR-155). The recombinant plasmid pLVX-miR-155 was then packaged into a lentivirus, resulting in a high-titer lentiviral rLV-ZsGreen-miR-155-Puro (rLV-miR-155) and rLV-miR-155. To investigate the effect of miR-155 on human hepatoma cells, rLV-miR-155 or rLV was infected into liver cancer cell line Hep3B. The two stable cell lines (rLV-Hep3B and rLV-miR-155-Hep3B) expressed green fluorescent protein (Figure 1A, Green). The miR-155 precursor and mature miR-155 were significantly increased in the rLV-miR-155 group compared with those in the rLV group (Figures 1B–1D). Next, the proliferation ability was significantly increased in the rLV-miR-155 group compared with that in the rLV group (24 h: p = 0.00033 < 0.01; 48 h: p = 0.000462 < 0.01; 72 h: p = 0.00323 < 0.01) (Figure 1E). The positive rate of bromodeoxyuridine (BrdU) (29.47% ± 3.59% versus 58.75% ± 6.59%, p = 0.00438 < 0.01) (Figure 1F), the number of plate colonies formed (21 ± 4.58 versus 840 ± 52.92, p = 0.000803 < 0.01) (Figure 1G), the sphere formation rate (3.04% ± 0.187% versus 8.03% ± 0.743%, p = 0.0031 < 0.01) (Figure 1H), and the rate of autophagy (6.34% ± 1.07% versus 22.49% ± 2.89%, p = 0.00955 < 0.01) (Figures 1Ia and 1Ib) were significantly increased in the rLV-miR-155 group compared with those in the rLV group. Furthermore, Luc activity, as recorded with the Autophagy LC3 HiBiT Reporter Assay, was significantly decreased in the rLV-miR-155 group compared with that in the rLV group (184.24 ± 34.05 versus 14.73 ± 2.24, p = 0.0069 < 0.01) (Figure 1Ic). Moreover, the senescence rate (5.07% ± 1.41% versus 17.15% ± 2.41%, p = 0.0092 < 0.01) (Figures 1Ja and 1Jb) and the senescence factor FBLN1 (1.5 ± 0.19 versus 3.44 ± 0.514, p = 0.00856 < 0.01) (Figure 1Jc) were significantly increased in the rLV-miR-155 group compared with those in the rLV group. Additionally, the average weight of transplanted tumors in nude mice was significantly increased in the rLV-miR-155 group compared with that in the rLV group (0.507 g ± 0.138 g versus 1.658 g ± 1.136 g, p = 0.0263 < 0.05) (Figures 2A–2C). Moreover, poorly differentiated cancer cells in transplanted tumors were significantly increased in the rLV-miR-155-Hep3B group compared with those in the rLV group (Figure 2D). Both the proliferating cell nuclear antigen (PCNA)-positive rate (45.17% ± 4.33% versus 74.27% ±± 7.94%, p < 0.01) (Figures 2Ea and 2Eb) and the Ki67-positive rate (33.57% ± 2.89% versus 70.11% ± 6.73%, p < 0.01) (Figure 2Ec) were significantly increased in the rLV-miR-155 group compared with those in the rLV group. Collectively, these results suggest that miR-155 can promote autophagy and cellular senescence, triggering the growth of liver cancer cells in vitro and in vivo.
Figure 1.

miR-155 Promotes the Growth of Liver Cancer Cell In Vitro
(A) The human hepatocarcinoma cell line Hep3B was infected with rLV-miR-155 and rLV, and the green fluorescent protein (green) was expressed. The positive cells were expanded and then imaged with a fluorescence microscope. (B) The miR-155 precursors in these two stable cell lines were detected by RT-PCR. (C) The expression of miR-155 in these two stable cell lines was detected by northern blotting. (D) qRT-PCR was used to detect mature miR-155. (E) The growth curve was determined by the CCK8 method. (F) Determination of S phase was determined by the BrdU method. OD450, optical density 450. (G) Determination of plate colony-forming ability. (a) Photograph of plate colonies. (b) Analysis of the formation of cell plate colonies. (H) Determination of cell-sphere formation ability. (a) Photograph of cell spheres. (b) Determination of cell-sphere formation rate. (I) Determination of autophagy formation ability of cells. (a) Photograph of cell autophagy. (b) Determination of cell autophagy rate. (c) Autophagy LC3 HiBiT Reporter Luc assay analysis. (J) Cellular senescence assay of cells. (a) Photograph of cell senescence. (b) Determination of cell senescence rate.
Figure 2.

Tumorigenesis Test In Vivo of Liver Cancer Cells
(A) Comparison of the tumor formation rate of liver cancer cells in nude mice. (B) In (a), Hep3B cells stably infected with rLV and rLV-miR-155 lentivirus were inoculated into the skin of BALB/c nude mice. (b) Photographs of transplanted tumors (xenografts) dissected from of BALB/c nude mice. (C) Comparison of the size (in grams) of xenografts. (D) Histopathological analysis of transplanted tumors formed in nude mice. A 4% formaldehyde-fixed, paraffin-embedded nude mouse transplanted tumor tissue section (4 μm) was subjected to hematoxylin and eosin (H&E) (original magnification, ×100). (E) In (a), immunohistochemical analysis of transplanted tumor tissue formed in nude mice. Immunohistochemical staining of anti-PCNA and anti-Ki67 (original magnification, ×100) was performed. (b) The comparison of PCNA-positive rates of xenograft. (c) The comparison of Ki67-positive rates in transplanted tumors in nude mice.
miR-155 Inhibits the Expression of Histone H3 Variant H3F3A
Given that miR-155 promotes the growth of human hepatoma cells, we will explore the effects of miR-155 on several vital signaling molecules in human hepatoma cells. RNA sequencing analysis showed that there were significant differences in the expression of several genes in the rLV-miR-155 group compared with that in the rLV group. Among them, 191 genes were upregulated and 284 genes were downregulated, including RPLP0, EIF4G2, YBX1, EEF1A1P5, EEF1A1, HNRNPA3, YWHAE, MIR30A, SMAD7, CALR, S100A6, TPT1, THBS1, and PSMB7 (downregulated expression) and HSPA5, CALM3, H3.3, and P21WAF1/Cip1 (downregulated expression) (Figure 3A). The miR-155 binds to the histone variant H3F3A mRNA 3′ untranslated region (UTR) via a 12-base seed sequence (Figure 3B). pMirtarget-H3F3A 3′ UTR-Luc luciferase reporter gene activity was significantly decreased in the rLV-miR-155 group compared with that in the rLV group (p < 0.01) (Figure 3C). Furthermore, although there was no significant change in the transcriptional capacity of H3F3A (Figure 3D), the translational capacity of H3FA was significantly attenuated in the rLV-miR-155 group compared with that in the rLV group (Figure 3E). Collectively, these results suggest that miR-155 inhibits the expression of H3F3A in liver cancer cells.
Figure 3.

Analysis of miR-155 Targeting H3F3A
(A) RNA sequencing analysis. (B) The analysis of mature miR-155 seed sequence binding to the H3F3A mRNA 3′ UTR. (C) pMirtarget-H3F3A 3′ UTR-Luc luciferase reporter gene activity was detected. (D) RT-PCR was used to detect H3F3A . β-actin was used as an internal reference gene. (E) Western blotting was used to detect the expression of H3.3. β-actin was used as an internal reference gene.
miR-155 Inhibits the Methylation Modification of Histone H3 on the 27th Lysine
Given that miR-155 inhibits the expression of histone H3F3A, we will consider whether miR-155 affects the methylation of lysine at position 27 of histone H3 by inhibiting H3F3A. Compared with the rLV-Hep3B group, the interaction between histone H3 and EZH2, SUZ12, EED, and RbAp46/48 was attenuated in the rLV-miR-155 group (Figure 4A). In particular, the interaction between EZH2 and SUZ12, EED, and RbAp46/48 was attenuated in the rLV-miR-155 group compared with that in the rLV group. However, the interaction between EZH2 and SUZ12, EED, and RbAp46/48 was not significantly changed in the rLV-miR-155+rLV-H3.3 group compared with that in the rLV group (Figures 4B and 4C). Moreover, although the interaction of histone H3 with EZH2, SUZ12, EED, and RbAp46/48 was attenuated in the rLV-miR-155 group compared with that in the rLV group, it was not significantly altered in the rLV-miR-155+rLV-H3FA-Hep3B group compared with that in the rLV group (Figure 4D). Finally, although H3K27me1, H3K27me2, and H3K27me3 were significantly reduced in the rLV-miR-155 group compared with the rLV group, they were not significantly altered in the rLV-miR-155+rLV-H3F3A-Hep3B group compared with the rLV group (Figures 4E and 4F). Collectively, these results suggest that miR-155 inhibits the methylation modification of histone H3 on the 27th lysine.
Figure 4.

miR-155 Affects Methylation Modification of Histone H3 on the 27th Lysine in Liver Cancer Cells
(A) Total proteins were extracted in two stable cell lines, rLV-Hep3B and rLV-miR-155-Hep3B, and subjected to immunoprecipitation (IP) analysis. The experimental samples were co-immunoprecipitated (coIP) with anti-histone H3, and the precipitates were analyzed by western blotting with anti-histone H3F3A. Immunoglobulin G (IgG) coIP was used as a negative control, and anti-EZH2, anti-SUZ12, anti-EED, and anti-RbAp46/48 in the samples before coprecipitation were detected by western blotting (INPUT). (B) Human hepatoma cells were infected with rLV, rLV-miR-155, and rLV-miR-155 + rLV-H3F3A, and pick up positive cells under a fluorescence microscope, and expand the fluorescence photos after culture. (C) Total proteins from the three cell lines rLV-Hep3B, rLV-miR-155-Hep3B, and rLV-miR-155+rLV-H3F3A were extracted and subjected to IP analysis. The experimental samples were coIP with anti-EZH2, and the precipitates were analyzed by western blotting with anti-SUZ12, anti-EED, and anti-RbAp46/48. IgG coIP was used as a negative control, and anti-SUZ12, anti-EED, and anti-RbAp46/48 in the samples before coprecipitation were detected by western blotting as INPUT. (D) Total proteins from the three cell lines rLV-Hep3B, rLV-miR-155-Hep3B, and rLV-miR-155+rLV- H3F3A were extracted and subjected to IP analysis. (E) Total proteins from three cell lines, including rLV-Hep3B and rLV-miR-155-Hep3B, were extracted, and H3K27me3 was detected by western blotting. Histone H3 was used as an internal reference gene. (F) The total proteins were extracted, and H3F3A and H3K27me1/2/3 were detected by western blotting. Histone H3 was used as an internal reference gene.
miR-155 Promotes the Expression of CDK2 by Inhibiting H3F3A
Given that miR-155 inhibits the expression of H3F3A, we will investigate whether miR-155 affects the expression of CDK2 dependent on H3F3A in human liver cancer cells. The ability of H3K27me1, H3K27me2, and H3K27me3 to bind to the CDK2 promoter region was significantly reduced in the rLV-miR-155 group compared with that in the rLV group (Figures 5A and 5B). pEZX-MT-CDK2 promoter-Luc luciferase reporter gene activity was significantly increased in the rLV-miR-155 group compared with that in the rLV group (26,379.94 ± 4,467.87 versus 271,502.52 ± 37,169.69, p = 0.0045388 < 0.01) (Figure 5C). Moreover, the expression of CDK2 was significantly increased in the rLV-miR-155 group compared with that in the rLV group (Figures 5D and 5E). Furthermore, although the ability of H3K27me1, H3K27me2, and H3K27me3 to bind to the CDK2 promoter region was significantly reduced in the rLV-miR-155 group compared with that in the rLV group, it was not significantly changed in the miR-155+rLV-H3F3A group compared with that in the rLV group (Figure 8F). Although the ability of H3K27me3 to bind to the CDK2 promoter cis-element probe was significantly reduced in the rLV-miR-155 group compared with that in the rLV group, it was not significantly changed in the miR-155+rLV-H3F3A group compared with the rLV group (Figure 5G). Although the pEZX-MT-CDK2 promoter-Luc luciferase reporter activity was significantly increased in the rLV-miR-155 group compared with that in the rLV group (45,050.5 ± 8,371.58 versus 244,546.37 ± 11,240.85, p = 0.00035832 < 0.01), it was not significantly altered in the miR-155+rLV-H3F3A group compared with the rLV group (45,050.5 ± 8,371.58 versus 38,966.22 ± 3,915.22, p = 0.07138 > 0.05) (Figure 5H). Although the expression of CDK2 was significantly increased in the rLV-miR-155 group compared with that in the rLV group, it was not significantly changed in the miR-155+rLV-H3F3A group compared with that in the rLV group (Figures 5I and 5J). Collectively, these results suggest that miR-155 enhances the expression of CDK2 by inhibiting H3F3A.
Figure 5.

The Expression of CDK2 on the Level of Transcription and Translation in Human Liver Cancer Cell Line Hep3B by Inhibiting H3F3A
(A) The cells were cross-linked with formaldehyde and then analyzed for chromatin IP (ChIP) using anti-H3K27me3. (B) The cells were cross-linked with formaldehyde and then analyzed for ChIP using anti-H3K27me1/2/3. (C) The pEZX-MT-CDK2-promoter-Luc luciferase reporter gene activity was tested. (D) RT-PCR was used to detect the transcription of CDK2. (E) The total protein was extracted, and CDK2 was detected by western blotting. (F) ChIP using anti-H3K9me3 was performed on three stable cell lines, rLV-Hep3B, rLV-miR-155-Hep3B, and miR-155+rLV-H3F3A-Hep3B. (G) Using biotin-labeled CDK2 promoter cis-elemental probe (Biotin-CDK2) and anti-H3K27me3, anti-Biotin, the super-electrophoretic gel mobility shift assay (super-EMSA) was performed. The activity of the pEZX-MT-CDK2-promoter-Luc luciferase reporter gene was detected. (H) The activity of the pEZX-MT-CDK2-promoter-Luc luciferase reporter gene was detected. (I) RT-PCR for CDK2 in rLV-Hep3B, rLV-miR-155-Hep3B, and miR-155+rLV-H3F3A-Hep3B. β-actin was used as an internal reference gene. (J) The total cellular proteins were extracted and detected by western blotting with anti-CDK2 in rLV-Hep3B, rLV-miR-155-Hep3B, and miR-155+rLV-H3F3A-Hep3B. β-actin was used as an internal reference gene.
Figure 8.

Excessive P21WAF1/CIP1 Abrogated the Oncogenic Effects of miR-155 in Human Liver Cancer Cells
(A) In (a), the expression of P21WAF1/CIP1 was detected by western blotting using anti-P21WAF1/CIP1 in the rLV group, rLV-miR-155 group, and rLV-miR-155+ rLV-P21WAF1/CIP1 group. (b) The mature body of miR-155 was detected by qRT-PCR. (B) The growth in vitro curves were determined by the CCK8 method. OD450, optical density 450. (C) Determination of S phase by BrdU staining. (D) Determination of colony-forming ability of liver cancer cells. (E) Determination of the ability of sphere formation. (F) In (a), determination of cellular senescence rate. (b) Enzyme chain immunoassay analysis of cytokines. (G) In (a), photographs of transplanted tumors (xenografts). (b) Comparison of the size (in grams) of transplanted tumors in nude mice. (c) Comparison of time (in days) of transplanted tumor appearance. (H) Histopathological analysis of hematoxylin and eosin (H&E) staining of transplanted tumors. (I) In (a), western blotting with anti-PCNA. (b) Quantification. (J) In (a), immunohistochemical staining of anti-PCNA was performed (original magnification, ×100). (b) Comparison of proliferating cell nuclear antigen (PCNA)-positive rates. (c) Western blotting analysis of PCNA was performed. (K) The schematic illustrates a model that miR-155 promotes growth of human liver cancer cells through activating CDK2 via targeting H3F3A.
miR-155 Inhibits Phosphorylation of CDK2
Given that miR-155-dependent H3F3A inhibits the expression of CDK2 in human liver cancer cells, we considered whether miR-155 affects the phosphorylation of CDK2. First, the ability of CTCF to bind to pre-miR-155 was significantly increased in the rLV-miR-155 group compared with that in the rLV group (Figure 6A). Thereby, the cyclized pre-miR-155 was significantly increased in the rLV-miR-155 group compared with that in the rLV group (Figures 6B–6D). Furthermore, the ability of CDK2 or cyclin E to bind to pre-miR-155 was significantly increased in the rLV-miR-155 group compared with that in the rLV group (Figure 6E). Thus, the capacity of CDK2 to bind to cyclin E was significantly increased in the rLV-miR-155 group compared with that in the rLV group (Figure 6F). Ultimately, phosphorylated CDK2 (pCDK2) was significantly increased in the rLV-miR-155 group compared with the rLV group (Figure 6G).
Figure 6.

miR-155 Promotes Phosphorylation of CDK2 in Human Liver Cancer Cells
(A) The ability of CTCF to bind to pre-miR-155 was analyzed by RNA IP (RIP) with anti-CTCF after cross-linking of formaldehyde; RNA purified from RNA coprecipitation was used as a template and pre-miR-155 primers for RT-PCR. (B) Total cellular RNA was extracted from two stable cell lines (rLV-Hep3B and rLV-miR-155-Hep3B), and circ pre-miR-155 was detected by back-to-back RT-PCR. (C) Looped pre-miR-155 was detected by back-to-back RT-PCR. (D) Circ-pre-miR-155 was detected by back-to-back RT-PCR. (E) The ability of CDK2 and cyclin E to bind to pre-miR-155 was analyzed by RIP. (F) The experimental samples were coIP with anti-Cyclin E, and (a–c) the precipitates were analyzed by western blotting with anti-CDK2. (G) Total cellular protein was extracted, and phosphorylation of CDK2 was detected by western blotting. (H) The ability of CTCF to bind to pre-miR-155 was analyzed by RIP with anti-CTCF. (I) The looped pre-miR-155 was detected by back-to-back RT-PCR in rLV-Hep3B, rLV-miR-155-Hep3B, rLV-miR-155+rLV-H3F3A-Hep3B, and rLV-miR-155+p GFP-V-RS-CTCF-Hep3B. (J) The ability of CDK2 and cyclin E to bind to pre-miR-155 was analyzed by RIP with anti-CTCF, anti-CDK2, and anti-Cyclin E. (K) The experimental samples were coIP with anti-Cyclin E, and the precipitates were analyzed by western blotting with anti-CDK2. (L) The phosphorylation of CDK2 was detected by western blotting. β-actin was used as an internal reference gene.
Furthermore, although the ability of CTCF to bind to pre-miR-155 was significantly increased in the rLV-miR-155 group compared with that in the rLV group, it was significantly reduced in the rLV-miR-155+pGFP-V-RS-CTCF group compared with that in the rLV group and was not changed in the miR-155+rLV-HF3A group (Figure 6H). Although the circular pre-miR-155 binding capacity was significantly increased in the rLV-miR-155 group, compared with that in the rLV group, it was significantly reduced in the rLV-miR-155+pGFP-V-RS-CTCF group compared with that in the rLV group and not changed in the miR-155+rLV-HF3A group (Figure 6I). Moreover, the ability of CDK2 or cyclin E to bind to pre-miR-155 was significantly increased in the rLV-miR-155 group compared with that in the rLV group; however, the ability of CDK2 or cyclin E to bind to pre-miR-155 was significantly reduced in the rLV-miR-155+pGFP-V-RS-CTCF group, and there was no significant change in the rLV-miR-155+rLV-H3.3 group compared with the rLV group (Figure 6J). In particular, the ability of CDK2 or cyclin E to bind to pre-miR-155 was significantly increased in the rLV-miR-155 group compared with that in the rLV group. However, the ability of CDK2 or cyclin E to bind to pre-miR-155 was significantly reduced in the rLV-miR-155+pGFP-V-RS-CTCF-Hep3B group, and there was no significant change in the rLV-miR-155+rLV-H3.3 group compared with the rLV group (Figure 6K). Therefore, pCDK2 was significantly increased in the rLV-miR-155 group compared with that in the rLV group. However, the phosphorylation of CDK2 was significantly reduced in the rLV-miR-155+pGFP-V-RS-CTCF group, and there was no significant change in the rLV-miR-155+rLV-H3.3 group compared with the rLV group (Figure 6L). Collectively, these results suggest that, on the one hand, miR-155-dependent CTCF loops cause the CDK2 interaction with cyclin E and that, on the other hand, miR-155 promotes the phosphorylation modification of CDK2 by inhibiting H3F3A in liver cancer cells.
miR-155 Inhibits the Expression of Tumor Suppressor Gene P21WAF1/CIP1
Given that miR-155 promotes the expression of CDK2 and its phosphorylation modification in liver cancer cells, we attempted to analyze whether miR-155 affects the expression of the tumor suppressor gene P21WAF1/CIP1 via pCDK2. Although the ability of pCDK to bind to the P21WAF1/CIP1 promoter region was significantly increased in the rLV-miR-155 group compared with that in the rLV group, it was not significantly changed in the miR-155+pGFP-V-RS-CTCF group compared with that in the rLV group (Figure 7A). Although the ability of pCDK to bind to the P21WAF1/CIP1 promoter region was significantly increased in the rLV-miR-155 group compared with that in the rLV group, it was not significantly changed in the rLV-miR-155 +rLV-H3F3A group compared with that in the rLV group (Figure 7B). The capacity of RNA polymerase II (RNA Pol II) to bind to the P21WAF1/CIP1 promoter region was significantly decreased in the rLV-miR-155 group compared with that in the rLV group. However, there was no significant change in the capacity of RNA Pol II to bind to the P21WAF1/CIP1 promoter region in the miR-155+rLV-H3F3A group compared with that in the rLV group (Figure 7C). Although the ability of RNA Pol II to bind to the P21WAF1/CIP1 promoter region was significantly reduced in the rLV-miR-155 group, compared with that in the rLV group, it was not significantly changed in the miR-155+PGFP-V-RS-CTCF group compared with that in the rLV group (Figure 7D). The ability of pCDK2 to bind to the P21WAF1/CIP1 promoter cis-element probe was significantly increased in the rLV-miR-155 group compared with that in the rLV group. However, there was no significant change in the capacity of pCDK2 to bind to the P21WAF1/CIP1 promoter region in the rLV-miR-155+ rLV-H3F3A group and the rLV-miR-155 +pGFP-V-RS-CTCF group compared with that in the rLV group (Figure 7E). Although the ability of RNA Pol II to bind to the P21WAF1/CIP1 promoter cis-element probe was significantly reduced compared with that in the rLV group, it was not significantly changed in the rLV-miR-155+rLV-H3F3A group and the miR-155+PGFP-V-RS-CTCF group compared with that in the rLV group (Figure 7F). The ability of pCDK2 to bind to the P21WAF1/CIP1 promoter-enhancer DNA loop was significantly increased in the rLV-miR-155 group compared with that in the rLV group. However, the ability of pCDK2 to bind to the P21WAF1/CIP1 promoter-enhancer loop was not significantly changed in the rLV-miR-155+ rLV-H3F3A group and the miR-155+PGFP-V-RS-CTCF group compared with that in the rLV group (Figure 7G). The ability of RNA Pol II to bind to the P21WAF1/CIP1 promoter-enhancer loop was significantly reduced in the rLV-miR-155 group compared with that in the rLV group. However, the ability of RNA Pol II to bind to the P21WAF1/CIP1 promoter-enhancer loop was not significantly changed in the rLV-miR-155+ rLV-H3F3A group or the miR-155+PGFP-V-RS-CTCF group compared with that in the rLV group (Figure 7H). The pEZX-MT-P21WAF1/CIP1 promoter-Luc luciferase reporter activity was significantly decreased in the rLV-miR-155 group compared with that in the rLV group (25,512.49 ± 4,129.56 versus 4,167.63 ± 340.89, p = 0.0054919 < 0.01). However, the luciferase reporter activity in the miR-155+rLV-H3F3A group was not significantly different from that in the rLV group (25,512.49 ± 4,129.56 versus 23,976.04 ± 3,220.07, p = 0.3233 > 0.05), and the luciferase reporter gene activity in the rLV-miR-155+pGFP-V-RS-CTCF group was not significantly different from that in the rLV group (25,512.49 ± 4,129.56 versus 27,443.78 ± 4,543.94, p = 0.22073 > 0.05) (Figure 7I). Although the expression of P21WAF1/CIP1 was significantly reduced in the rLV-miR-155 group compared with that in the rLV group, it was not significantly changed in the rLV-miR-155+rLV-H3F3A group or the rLV-miR-155+pGFP-V-RS-CTCF group compared with that in the rLV group (Figures 7J and 7K).
Figure 7.

miR-155 Inhibits the Expression of the Tumor Suppressor Gene P21WAF1/CIP1 in Liver Cancer Cells
(A) ChIP with anti-pCDK2 was performed in three stable cell lines: rLV-Hep3B, rLV-miR-155-Hep3B, and miR-155+rLV-H3F3A-Hep3B. (B) ChIP with anti-pCDK2 was performed. (C) ChIP using anti-RNA Pol II was performed. (D) ChIP using anti-RNA Pol II was performed. (E) Super-DNA-protein complex gel migration assay using biotin-labeled P21WAF1/CIP1 promoter cis-element probe (Biotin-P21WAF1/CIP1) and anti-pCDK2, anti-Biotin. (F) Super-DNA-protein complex gel migration assay using biotin-labeled P21WAF1/CIP1 promoter cis-element probe (Biotin-P21WAF1/CIP1) and anti-RNA Pol II, anti-Biotin. (G) The ability of pCDK2 to bind to the P21WAF1/CIP1 promoter-enhancer loop by chromosome configuration capture (3C)-ChIP. (H) The ability of RNA Pol II to bind to the P21WAF1/CIP1 promoter-enhancer loop was detected by the 3C-ChIP. (I) pEZX-MT-P21WAF1/CIP1-promoter-Luc luciferase reporter gene activity was detected. (J) Total RNA was detected by RT-PCR for the transcriptional capacity of P21WAF1/CIP1. (K) Western blotting was used to detect the expression of P21WAF1/CIP1. (L) qRT-PCR was used to detect miR-155 in three stable cell lines: rLV-Hep3B, rLV-miR-155-Hep3B, and miR-155+pGFP-V-RS-CAK. (a) Quantification and (b) western blot. (M) ChIP was performed in rLV-Hep3B, rLV-miR-155-Hep3B, and miR-155+pGFP-V-RS-CAK. (N) 3C-ChIP using anti-pCDK2, anti-RNA Pol II. (O) pEZX-MT-P21WAF1/CIP1-promoter luciferase reporter gene activity was examined. (P) The transcription of P21WAF1/CIP1 was detected by RT-PCR. (Q) Expression of P21WAF1/CIP1 was detected by western blotting. (R) Immunoprecipitation (IP) analysis with anti-CyclinA and anti-CDK2. (S) Western blotting with anti -E2F1.β-actin as an internal reference gene.
Furthermore, miR-155 was significantly increased in the miR-155 and rLV-miR-155+pGFP-V-RS-CAK (CDK2-activating kinase) groups compared with the rLV group (Figure 7La). pCDK2 (Thr160) was significantly increased in the rLV-miR-155 group compared with the rLV group, and pCDK2 (Thr160) and CAK were significantly reduced in the rLV-miR-155+pGFP-V-RS-CAK group compared with the rLV group (Figure 7Lb). The ability of pCDK2 to bind to the P21WAF1/CIP1 promoter region was significantly increased, and the ability of RNA Pol II to bind to the P21WAF1/CIP1 promoter region was significantly reduced in the rLV-miR-155 group compared with that in the rLV group. However, these binding abilities were not significantly changed in the rLV-miR-155+pGFP-V-RS-CAK group compared with those in the rLV group (Figure 7M). The ability of pCDK2 to bind to the P21WAF1/CIP1 promoter-enhancer loop was significantly increased, and the capacity of RNA Pol II to bind to the P21WAF1/CIP1 promoter-enhancer loop was significantly reduced in the rLV-miR-155 group compared with that in the rLV group. However, these binding abilities were not significantly changed in the rLV-miR-155+pGFP-V-RS-CAK group compared with those in the rLV group (Figure 7N). The pEZX-MT-P21WAF1/CIP1 promoter-Luc luciferase reporter gene activity was significantly decreased in the rLV-miR-155 group compared with that in the rLV group (38,502.61 ± 6,155.78 versus 5,009.03 ± 652.7, p = 0.00671 < 0.01). However, the luciferase reporter gene activity in the rLV-miR-155+pGFP-V-RS-CAK group was not significantly different from that in the rLV group (38,502.61 ± 6,155.78 versus 41,045.93 ± 2,208.55, p = 0.2442 > 0.05) (Figure 7O). The transcriptional and translational capacity of P21WAF1/CIP1 was significantly reduced in the rLV-miR-155 group compared with that in the rLV group. However, there was no significant change in the rLV-miR-155+pGFP-V-RS-CAK group compared with the rLV group (Figures 7P and 7Q). Collectively, these results suggest that miR-155 competitively inhibits the expression of P21WAF1/CIP1 in liver cancer.
Excessive P21WAF1/CIP1 Abolishes the Cancerous Functions of miR-155
In order to further investigate whether miR-155 is dependent on P21WAF1/CIP1 to promote hepatocarcinoma growth, we performed related experiments in the rLV group, the rLV-miR-155 group, and the rLV-miR-155+rLV-P21WAF1/CIP1 group. The mature miR-155 was significantly increased in the rLV-miR-155 group and the rLV-miR-155+rLV-P21WAF1/CIP1 group compared with the rLV group (Figure 8Aa). The expression of P21WAF1/CIP1 was significantly decreased in the rLV-miR-155 group and decreased in the rLV-miR-155+rLV-P21WAF1/CIP1 group compared with the rLV group (Figure 8Ab). Although the proliferation rate was significantly increased in the rLV-miR-155 group compared with that in the rLV group (p < 0.01), it was not significantly changed in the rLV-miR-155+rLV-P21WAF1/CIP1 group compared with that in the rLV group (p = 0.2236 > 0.05) (Figure 8B). The positive rate of BrdU was significantly increased in the rLV-miR-155 group compared with that in the rLV group (27.11% ± 3.61% versus 55.93% ± 8.74%, p = 0.00694 < 0.01). However, the positive rate of BrdU was not significantly altered in the rLV-miR-155+rLV-P21WAF1/CIP1 group compared with that in the rLV group. (27.11% ± 3.61% versus 25.49% ± 6.2%, p = 0.231887 > 0.05) (Figure 8C). Although the colony formation rate was significantly increased in the rLV-miR-155 group compared with that in the rLV group (37.63% ± 4.56% versus 68.59% ± 6.33%, p = 0.00778 < 0.01), the colony formation rate was not significantly altered in the rLV-miR-155+rLV-P21WAF1/CIP1 group compared with that in the rLV group (37.63% ± 4.56% versus 34.07% ± 5.93%, p = 0.2507 > 0.05) (Figure 8D). Although the sphere formation rate was significantly increased in the rLV-miR-155 group compared with that in the rLV group (8.09% ± 0.62% versus 24.87% ± 3.28%, p = 0.0087303 < 0.01), it was not significantly altered in the rLV-miR-155+rLV-P21WAF1/CIP1 group compared with that in the rLV group (8.09% ± 0.62% versus 7.28% ± 1.146%, p = 0.1885 > 0.05) (Figure 8E). Although the senescence rate was significantly increased in the rLV-miR-155 group compared with that in the rLV group (7.58% ± 0.786% versus 17.16% ± 1.776%, p = 0.0091956 < 0.01), it was not significantly altered in the rLV-miR-155+ rLV-P21WAF1/CIP1 group compared with the rLV group (7.58% ± 0.786% versus 6.71% ± 1.569%, p = 0.101169 > 0.05) (Figures 8Fa and 8Fb). The senescence factor FBLN1 was significantly increased in the rLV-miR-155 group compared with the rLV group (0.893 ± 0.065 versus 2.103 ± 0.1159, p = 0.003814 < 0.01). However, the senescence factor FBLN1 was not significantly altered in the rLV-miR-155+ rLV-P21WAF1/CIP1 group compared with the rLV group (0.893 ± 0.065 versus 0.703 ± 0.168, p = 0.1677299 > 0.05) (Figure 8Fc). Although the average weight of transplanted tumors was significantly increased in the rLV-miR-155 group compared with that in the rLV group (0.2275 g ± 0.02915 g versus 1.05375 g ± 0.1207 g, p = 0.000000097 < 0.01), it was not significantly altered in the rLV-miR-155+rLV-P21WAF1/CIP1 group compared with the rLV group (0.2275 g ± 0.02915 g versus 0.20625 g ± 0.037 g, p = 0.097268 > 0.05) (Figures 8Ga and 8Gb). The average appearance time of transplanted tumors was significantly decreased in the rLV-miR-155 group compared with that in the rLV group (9 ± 1.31 days versus 6.125 ± 1.126 days, p = 0.00175 < 0.01). However, it was not significantly altered in the rLV-miR-155+rLV-P21WAF1/CIP1 group compared with the rLV group (9 ± 1.31 days versus 9.625 ± 1.847 days, p = 0.2684 > 0.05) (Figure 8Gc). The transplanted tumor tissues contained more poorly differentiated tumor cells and fewer moderately differentiated cells in the rLV-miR-155 group than in the rLV group; however, there was no significant difference between the rLV-miR-155+rLV-P21WAF1/CIP1 group and the rLV group (Figure 8H). Although the expression of PCNA in the transplanted tumor tissues was significantly increased in the rLV-miR-155 group compared with that in the rLV group, there was no significant difference between the rLV-miR-155+rLV-P21WAF1/CIP1 group and the rLV group (Figures 8Ia and 8Ib). Although the expression of PCNA in the transplanted tumor tissues was significantly increased in the rLV-miR-155 group compared with that in the rLV group (42.59% ± 5.49% versus 85.23% ± 6.598%, p = 0.00000911 < 0.01), there was no significant difference in PCNA expression between the rLV-miR-155+ rLV-P21WAF1/CIP1 group and the rLV group (42.59% ± 5.49% versus 40.03% ± 2.75%, p = 0.130044 > 0.05) (Figure 8J). Collectively, these results suggest that excessive P21WAF1/CIP1 abrogates the oncogenic functions of miR-155 in liver cancer.
Discussion
miR-155 is a non-coding miRNA that has a certain relationship with the promotion of tumorigenesis. In this study, we indicate that miR-155 can promote the growth of liver cancer cells in vitro and in vivo and that miR-155 can play a positive role in the development of liver cancer. Moreover, we have demonstrated that miR-155 can influence a series of gene expressions through epigenetic regulation in liver cancer cells (Figure 8K). So far, a novel mechanism has emerged, particularly showing that miR-155 is associated with epigenetic modifications of histones in liver cancer.
First, our results show that miR-155 is highly expressed in human hepatocarcinoma tissues—miR-155, especially, can promote the proliferation of human hepatoma cells Hep3B in vitro and in vivo—suggesting that miR-155 has a strong function of causing liver cancer. A number of studies have reported that miR-155 can act as an accelerator to promote the development of acute.9, 10, 11 In addition to blood diseases, miR-155 is also closely related to the development of solid tumors. The study found that miR-155 plays a key role in promoting the progression of glioma and can be a prognostic factor for the survival of patients.32 These reports suggest that miR-155 is involved in the regulation of tumor development and plays a carcinogenic role. Therefore, our findings are consistent with these reports.
Second, we found that miR-155 can target H3F3A (also called H3.3) 3′ UTR and inhibit H3F3A translation and that miR-155 can inhibit H3K27me1/2/3 modification and promote CDK2 phosphorylation of p21WAF1/cip1 in liver cancer. Importantly, these functions were fully abolished after increasing the expression of H3F3A in miR-155-overexpressing liver cancer cells, indicating that, at least in liver cancer cells, miR-155 acts through H3F3A. H3F3A has been shown to be a key player in a variety of life activities.33,34 Studies have shown that the deposition of H3F3A dependent on Hira is the key to regulate cell-fate transformation. H3F3A plays a role in protecting the identity of parental cells in the early stage of cell reprogramming.35 In addition, H3F3A and its cleavage products promote transcriptional silencing and cellular senescence.36 Graber et al.37 reported overexpression of H3F3A in esophageal and lung cancer cell lines, but the function of H3F3A in tumors has rarely been studied and remains unknown.
Notably, our results show that miR-155 alters the level of histone modification in human liver cancer cells, including the following mechanisms: (1) miR-155 reduces the interaction among EZH2, SUZ12, EED, and RbAp46/48 by inhibiting H3F3A expression. (2) miR-155 inhibits the interaction of four molecules of EZH2, SUZ12, EED, and RbAp46/48 with histone H3. (3) miR-155-dependent H3F3A inhibits the level of trimethylation modification on the 27th lysine of histone H3. (4) miR-155 affects the expression of several genes through changes in the level of H3K27me3 in liver cancer cells, such as CDK2. It is well known that polycomb inhibitor complex-2 (PRC2) is a class of polycomb proteins consisting of four core proteins: SUZ12, EED, RbAp46/48, and EZH2.29 Studies have shown that EZH2 can only exert its methyltransferase activity when combined with SUZ12, EED, and RbAp46/48.38,39 The trimethylation of H3K27 is particularly dependent on the involvement of SUZ12.28 Thereby, the interaction of these four core components plays an important role in the activity of the PRC2 complex and the methylation modification of PRC2-mediated H3K27me3,24 and a decrease in the level of H3K27me3 modification leads to tumorigenesis.30,40,41 Therefore, the changes in H3K27me3 modification may be associated with liver cancer.
Strikingly, our results suggest that miR-155 is capable of forming a circular structure and functions in liver cancer cells. These results show that, on the one hand, circular miR-155 binds to cyclin E or pCDK2 and that, on the other hand, circular pre-miR-155 containing cyclin E and pCDK2 forms a topology (to be further proved) by an unknown mechanism, resulting in a direct interaction between pCDK2 and cyclin E. So far, circular RNA (circRNA) is a new class of endogenous non-coding RNAs that are widely found in eukaryotic gene transcriptomes.41, 42, 43 In addition, circRNA can also act as a central part of protein interactions. Du et al.44 have demonstrated that circFoxo3 regulates cell-cycle progression by forming a ternary complex with CDK2 and p21, but so far, there is no report on the role of miR-155 through cyclization. The mechanism for how pre-miR-155 is cyclized and plays roles remains to be further studied.
More importantly, miR-155 relies on H3F3A and H3K27me1/2/3 to alter the expression and function of cyclin in liver cancer cells, including the following mechanisms: (1) miR-155 reduces the level of methylation of histone H3K27 on the cyclin-dependent kinase (CDK)2 promoter region, which enhances the transcription and translation of CDK2; (2) miR-155 promotes phosphorylation of CDK2; (3) miR-155 enhances the binding of pCDK2 to cyclin E by cyclization and promotes the binding of pCDK2-cyclin E to the promoter region of the CDK inhibitor p21 (also known as p21WAF1/Cip1), inhibiting the expression of p21; and (4) excessive p21 abolishes oncogenic functions of miR-155. It is well known that CDK2 is regulated by cyclin E and cyclin A and is necessary for the transition of cells from the G1 phase to the S phase.45,46 Activated CDK2 is critical for the abnormal growth of cancer cells.47 In addition, Tang et al.48 reported that WTAP can promote the proliferation of renal cancer cells by regulating the stability of CDK2 mRNA and enhancing the expression of CDK2. The CDK inhibitor p21 (or p21WAF1/Cip1) is a well-known cell-cycle inhibitor that is mainly associated with inhibition of CDK2,49,50 thereby inhibiting tumor progression.51 Several reports indicate that p21WAF1/Cip1 inhibits tumorigenesis52, 53, 54 and other tumor effects. These reports are consistent with our findings. Notably, when CAK for pCDK2 (Thr160) was knocked down in miR-155-overexpressing Hep3B cells, the ability of pCDK2 or RNA Pol II to bind to the P21WAF1/CIP1 promoter region was not significantly changed, and the transcriptional and translational capacity of P21WAF1/CIP1 was also not significantly changed. Thereby, miR-155 competitively blocks the binding of RNA Pol II to the P21WAF1/CIP1 promoter by increasing the phosphorylation of CDK2, inhibiting the expression of P21WAF1/CIP1 in liver cancer.
In conclusion, we need to further explore the impact of miR-155 on the development of human liver cancer and provide a theoretical basis for the prevention and treatment of liver cancer.
Materials and Methods
Cell Lines and Lentivirus
Human hepatoma cell line Hep3B was obtained from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China). Lentivirus LV-miR and rLV-miR-155 were purchased from Wu Han Viral Therapy Technologies. pGFP-V-RS was purchased from Origene (Rockville, MD, USA).
RT-PCR
Total RNA was purified using TRIzol (Invitrogen) according to the manufacturer’s instructions.
RNA Sequencing Analysis
RNA sequencing analysis was performed according to the manufacturer’s protocol (Beijing Nuohe Zhiyuan Technology).
Co-immunoprecipitation (CoIP)
The immunoprecipitates were incubated with 30 μL protein G/A-plus agarose beads by rotation overnight at 4°C. The precipitates were washed five times with bead wash solution. Western blotting was performed with related antibodies.
Super-EMSA (Gel Shift)
Cells were washed and scraped in ice-cold PBS to prepare nuclei for electrophoretic gel mobility shift assay (EMSA) with the use of the gel shift assay system modified according to the manufacturer’s instructions (Promega).
Cell Proliferation CCK8 Assay
The cell proliferation was measured using the CCK8 Assay Kit according to the manufacturer’s instructions (Boshide, Wuhan, China).
Colony-Formation Efficiency Assay
Cell colonies on the dishes were stained with crystal violet, and the colonies were counted.
Xenograft Transplantation In Vivo
Four-week-old athymic BALB/c mice were purchased from Shi Laike (Shanghai, China) and maintained in the Tongji animal facilities approved by the China Association for Accreditation of Laboratory Animal Care. The mice were observed for 4 weeks and then sacrificed to recover the tumors.
Author Contributions
D.L. conceived the study and participated in the study design, performance, coordination, and manuscript writing. X.X., Y.L., S.X., Y.C., X.J., S.S., L.W., H.P., X.G., T.L., J.X., J.L., and S.J. performed the research. All authors have read and approved the final manuscript.
Conflicts of Interest
The authors declare no competing interests.
Acknowledgments
This study was supported by a grant from the National Natural Science Foundation of China (NCSF no. 81773158) and by a grant from the Science and Technology Commission of Shanghai Municipality Basic Research Field Project (19JC1415200).
References
- 1.Asano N., Matsuzaki J., Ichikawa M., Kawauchi J., Takizawa S., Aoki Y., Sakamoto H., Yoshida A., Kobayashi E., Tanzawa Y. A serum microRNA classifier for the diagnosis of sarcomas of various histological subtypes. Nat. Commun. 2019;10:1299. doi: 10.1038/s41467-019-09143-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bartel D.P. Metazoan MicroRNAs. Cell. 2018;173:20–51. doi: 10.1016/j.cell.2018.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lewis B.P., Burge C.B., Bartel D.P. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005;120:15–20. doi: 10.1016/j.cell.2004.12.035. [DOI] [PubMed] [Google Scholar]
- 4.Meister G., Tuschl T. Mechanisms of gene silencing by double-stranded RNA. Nature. 2004;431:343–349. doi: 10.1038/nature02873. [DOI] [PubMed] [Google Scholar]
- 5.Mello C.C., Conte D., Jr. Revealing the world of RNA interference. Nature. 2004;431:338–342. doi: 10.1038/nature02872. [DOI] [PubMed] [Google Scholar]
- 6.Tavallaie R., McCarroll J., Le Grand M., Ariotti N., Schuhmann W., Bakker E., Tilley R.D., Hibbert D.B., Kavallaris M., Gooding J.J. Nucleic acid hybridization on an electrically reconfigurable network of gold-coated magnetic nanoparticles enables microRNA detection in blood. Nat. Nanotechnol. 2018;13:1066–1071. doi: 10.1038/s41565-018-0232-x. [DOI] [PubMed] [Google Scholar]
- 7.Mann M., Mehta A., Zhao J.L., Lee K., Marinov G.K., Garcia-Flores Y., Lu L.F., Rudensky A.Y., Baltimore D. An NF-kappaB-microRNA regulatory network tunes macrophage inflammatory responses. Nat. Commun. 2017;8:851. doi: 10.1038/s41467-017-00972-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Alivernini S., Kurowska-Stolarska M., Tolusso B., Benvenuto R., Elmesmari A., Canestri S., Petricca L., Mangoni A., Fedele A.L., Di Mario C. MicroRNA-155 influences B-cell function through PU.1 in rheumatoid arthritis. Nat. Commun. 2016;7:12970. doi: 10.1038/ncomms12970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schneider E., Staffas A., Röhner L., Malmberg E.D., Ashouri A., Krowiorz K., Pochert N., Miller C., Wei S.Y., Arabanian L. Micro-ribonucleic acid-155 is a direct target of Meis1, but not a driver in acute myeloid leukemia. Haematologica. 2018;103:246–255. doi: 10.3324/haematol.2017.177485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wallace J.A., Kagele D.A., Eiring A.M., Kim C.N., Hu R., Runtsch M.C., Alexander M., Huffaker T.B., Lee S.H., Patel A.B. miR-155 promotes FLT3-ITD-induced myeloproliferative disease through inhibition of the interferon response. Blood. 2017;129:3074–3086. doi: 10.1182/blood-2016-09-740209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang T., Davidson-Moncada J.K., Mukherjee P., Furman R.R., Bhavsar E., Chen Z., Hakimpour P., Papavasiliou N., Tam W. MicroRNA-155 regulates casein kinase 1 gamma 2: a potential pathogenetic role in chronic lymphocytic leukemia. Blood Cancer J. 2017;7:e606. doi: 10.1038/bcj.2017.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Onodera Y., Teramura T., Takehara T., Obora K., Mori T., Fukuda K. miR-155 induces ROS generation through downregulation of antioxidation-related genes in mesenchymal stem cells. Aging Cell. 2017;16:1369–1380. doi: 10.1111/acel.12680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Volinia S., Calin G.A., Liu C.G., Ambs S., Cimmino A., Petrocca F., Visone R., Iorio M., Roldo C., Ferracin M. A microRNA expression signature of human solid tumors defines cancer gene targets. Proc. Natl. Acad. Sci. USA. 2006;103:2257–2261. doi: 10.1073/pnas.0510565103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kong W., He L., Coppola M., Guo J., Esposito N.N., Coppola D., Cheng J.Q. MicroRNA-155 regulates cell survival, growth, and chemosensitivity by targeting FOXO3a in breast cancer. J. Biol. Chem. 2010;285:17869–17879. doi: 10.1074/jbc.M110.101055. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 15.Fabbri M., Bottoni A., Shimizu M., Spizzo R., Nicoloso M.S., Rossi S., Barbarotto E., Cimmino A., Adair B., Wojcik S.E. Association of a microRNA/TP53 feedback circuitry with pathogenesis and outcome of B-cell chronic lymphocytic leukemia. JAMA. 2011;305:59–67. doi: 10.1001/jama.2010.1919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Blaya D., Coll M., Rodrigo-Torres D., Vila-Casadesús M., Altamirano J., Llopis M., Graupera I., Perea L., Aguilar-Bravo B., Díaz A. Integrative microRNA profiling in alcoholic hepatitis reveals a role for microRNA-182 in liver injury and inflammation. Gut. 2016;65:1535–1545. doi: 10.1136/gutjnl-2015-311314. [DOI] [PubMed] [Google Scholar]
- 17.Bala S., Csak T., Saha B., Zatsiorsky J., Kodys K., Catalano D., Satishchandran A., Szabo G. The pro-inflammatory effects of miR-155 promote liver fibrosis and alcohol-induced steatohepatitis. J. Hepatol. 2016;64:1378–1387. doi: 10.1016/j.jhep.2016.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Blaya D., Aguilar-Bravo B., Hao F., Casacuberta-Serra S., Coll M., Perea L., Vallverdú J., Graupera I., Pose E., Llovet L. Expression of microRNA-155 in inflammatory cells modulates liver injury. Hepatology. 2018;68:691–706. doi: 10.1002/hep.29833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chi P., Allis C.D., Wang G.G. Covalent histone modifications--miswritten, misinterpreted and mis-erased in human cancers. Nat. Rev. Cancer. 2010;10:457–469. doi: 10.1038/nrc2876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim J., Guermah M., McGinty R.K., Lee J.S., Tang Z., Milne T.A., Shilatifard A., Muir T.W., Roeder R.G. RAD6-Mediated transcription-coupled H2B ubiquitylation directly stimulates H3K4 methylation in human cells. Cell. 2009;137:459–471. doi: 10.1016/j.cell.2009.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cheung P., Tanner K.G., Cheung W.L., Sassone-Corsi P., Denu J.M., Allis C.D. Synergistic coupling of histone H3 phosphorylation and acetylation in response to epidermal growth factor stimulation. Mol. Cell. 2000;5:905–915. doi: 10.1016/s1097-2765(00)80256-7. [DOI] [PubMed] [Google Scholar]
- 22.Lo W.S., Trievel R.C., Rojas J.R., Duggan L., Hsu J.Y., Allis C.D., Marmorstein R., Berger S.L. Phosphorylation of serine 10 in histone H3 is functionally linked in vitro and in vivo to Gcn5-mediated acetylation at lysine 14. Mol. Cell. 2000;5:917–926. doi: 10.1016/s1097-2765(00)80257-9. [DOI] [PubMed] [Google Scholar]
- 23.Barski A., Cuddapah S., Cui K., Roh T.Y., Schones D.E., Wang Z., Wei G., Chepelev I., Zhao K. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129:823–837. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 24.Wiles E.T., Selker E.U. H3K27 methylation: a promiscuous repressive chromatin mark. Curr. Opin. Genet. Dev. 2017;43:31–37. doi: 10.1016/j.gde.2016.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ferrari K.J., Scelfo A., Jammula S., Cuomo A., Barozzi I., Stützer A., Fischle W., Bonaldi T., Pasini D. Polycomb-dependent H3K27me1 and H3K27me2 regulate active transcription and enhancer fidelity. Mol. Cell. 2014;53:49–62. doi: 10.1016/j.molcel.2013.10.030. [DOI] [PubMed] [Google Scholar]
- 26.Steiner L.A., Schulz V.P., Maksimova Y., Wong C., Gallagher P.G. Patterns of histone H3 lysine 27 monomethylation and erythroid cell type-specific gene expression. J. Biol. Chem. 2011;286:39457–39465. doi: 10.1074/jbc.M111.243006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jung H.R., Pasini D., Helin K., Jensen O.N. Quantitative mass spectrometry of histones H3.2 and H3.3 in Suz12-deficient mouse embryonic stem cells reveals distinct, dynamic post-translational modifications at Lys-27 and Lys-36. Mol. Cell. Proteomics. 2010;9:838–850. doi: 10.1074/mcp.M900489-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Højfeldt J.W., Laugesen A., Willumsen B.M., Damhofer H., Hedehus L., Tvardovskiy A., Mohammad F., Jensen O.N., Helin K. Accurate H3K27 methylation can be established de novo by SUZ12-directed PRC2. Nat. Struct. Mol. Biol. 2018;25:225–232. doi: 10.1038/s41594-018-0036-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Al-Raawi D., Jones R., Wijesinghe S., Halsall J., Petric M., Roberts S., Hotchin N.A., Kanhere A. A novel form of JARID2 is required for differentiation in lineage-committed cells. EMBO J. 2019;38:e98449. doi: 10.15252/embj.201798449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Harutyunyan A.S., Krug B., Chen H., Papillon-Cavanagh S., Zeinieh M., De Jay N., Deshmukh S., Chen C.C.L., Belle J., Mikael L.G. H3K27M induces defective chromatin spread of PRC2-mediated repressive H3K27me2/me3 and is essential for glioma tumorigenesis. Nat. Commun. 2019;10:1262. doi: 10.1038/s41467-019-09140-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang Z., Li K., Wang X., Huang W. MiR-155-5p modulates HSV-1 replication via the epigenetic regulation of SRSF2 gene expression. Epigenetics. 2019;14:494–503. doi: 10.1080/15592294.2019.1600388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wu X., Wang Y., Yu T., Nie E., Hu Q., Wu W., Zhi T., Jiang K., Wang X., Lu X. Blocking MIR155HG/miR-155 axis inhibits mesenchymal transition in glioma. Neuro Oncol. 2017;19:1195–1205. doi: 10.1093/neuonc/nox017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jang C.W., Shibata Y., Starmer J., Yee D., Magnuson T. Histone H3.3 maintains genome integrity during mammalian development. Genes Dev. 2015;29:1377–1392. doi: 10.1101/gad.264150.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Banaszynski L.A., Wen D., Dewell S., Whitcomb S.J., Lin M., Diaz N., Elsässer S.J., Chapgier A., Goldberg A.D., Canaani E. Hira-dependent histone H3.3 deposition facilitates PRC2 recruitment at developmental loci in ES cells. Cell. 2013;155:107–120. doi: 10.1016/j.cell.2013.08.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fang H.T., El Farran C.A., Xing Q.R., Zhang L.F., Li H., Lim B., Loh Y.H. Global H3.3 dynamic deposition defines its bimodal role in cell fate transition. Nat. Commun. 2018;9:1537. doi: 10.1038/s41467-018-03904-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Duarte L.F., Young A.R., Wang Z., Wu H.A., Panda T., Kou Y., Kapoor A., Hasson D., Mills N.R., Ma’ayan A. Histone H3.3 and its proteolytically processed form drive a cellular senescence programme. Nat. Commun. 2014;5:5210. doi: 10.1038/ncomms6210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Graber M.W., Schweinfest C.W., Reed C.E., Papas T.S., Baron P.L. Isolation of differentially expressed genes in carcinoma of the esophagus. Ann. Surg. Oncol. 1996;3:192–197. doi: 10.1007/BF02305800. [DOI] [PubMed] [Google Scholar]
- 38.Cao R., Zhang Y. The functions of E(Z)/EZH2-mediated methylation of lysine 27 in histone H3. Curr. Opin. Genet. Dev. 2004;14:155–164. doi: 10.1016/j.gde.2004.02.001. [DOI] [PubMed] [Google Scholar]
- 39.Czermin B., Melfi R., McCabe D., Seitz V., Imhof A., Pirrotta V. Drosophila enhancer of Zeste/ESC complexes have a histone H3 methyltransferase activity that marks chromosomal Polycomb sites. Cell. 2002;111:185–196. doi: 10.1016/s0092-8674(02)00975-3. [DOI] [PubMed] [Google Scholar]
- 40.Lee W., Teckie S., Wiesner T., Ran L., Prieto Granada C.N., Lin M., Zhu S., Cao Z., Liang Y., Sboner A. PRC2 is recurrently inactivated through EED or SUZ12 loss in malignant peripheral nerve sheath tumors. Nat. Genet. 2014;46:1227–1232. doi: 10.1038/ng.3095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hansen T.B., Jensen T.I., Clausen B.H., Bramsen J.B., Finsen B., Damgaard C.K., Kjems J. Natural RNA circles function as efficient microRNA sponges. Nature. 2013;495:384–388. doi: 10.1038/nature11993. [DOI] [PubMed] [Google Scholar]
- 42.Cherubini A., Barilani M., Rossi R.L., Jalal M.M.K., Rusconi F., Buono G., Ragni E., Cantarella G., Simpson H.A.R.W., Péault B., Lazzari L. FOXP1 circular RNA sustains mesenchymal stem cell identity via microRNA inhibition. Nucleic Acids Res. 2019;47:5325–5340. doi: 10.1093/nar/gkz199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li Z., Huang C., Bao C., Chen L., Lin M., Wang X., Zhong G., Yu B., Hu W., Dai L. Exon-intron circular RNAs regulate transcription in the nucleus. Nat. Struct. Mol. Biol. 2015;22:256–264. doi: 10.1038/nsmb.2959. [DOI] [PubMed] [Google Scholar]
- 44.Du W.W., Yang W., Liu E., Yang Z., Dhaliwal P., Yang B.B. Foxo3 circular RNA retards cell cycle progression via forming ternary complexes with p21 and CDK2. Nucleic Acids Res. 2016;44:2846–2858. doi: 10.1093/nar/gkw027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Honda R., Lowe E.D., Dubinina E., Skamnaki V., Cook A., Brown N.R., Johnson L.N. The structure of cyclin E1/CDK2: implications for CDK2 activation and CDK2-independent roles. EMBO J. 2005;24:452–463. doi: 10.1038/sj.emboj.7600554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Feng Z., Xia Y., Gao T., Xu F., Lei Q., Peng C., Yang Y., Xue Q., Hu X., Wang Q. The antipsychotic agent trifluoperazine hydrochloride suppresses triple-negative breast cancer tumor growth and brain metastasis by inducing G0/G1 arrest and apoptosis. Cell Death Dis. 2018;9:1006. doi: 10.1038/s41419-018-1046-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Peng C., Zeng W., Su J., Kuang Y., He Y., Zhao S., Zhang J., Ma W., Bode A.M., Dong Z., Chen X. Cyclin-dependent kinase 2 (CDK2) is a key mediator for EGF-induced cell transformation mediated through the ELK4/c-Fos signaling pathway. Oncogene. 2016;35:1170–1179. doi: 10.1038/onc.2015.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tang J., Wang F., Cheng G., Si S., Sun X., Han J., Yu H., Zhang W., Lv Q., Wei J.F., Yang H. Wilms’ tumor 1-associating protein promotes renal cell carcinoma proliferation by regulating CDK2 mRNA stability. J. Exp. Clin. Cancer Res. 2018;37:40. doi: 10.1186/s13046-018-0706-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Abbas T., Dutta A. p21 in cancer: intricate networks and multiple activities. Nat. Rev. Cancer. 2009;9:400–414. doi: 10.1038/nrc2657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Karimian A., Ahmadi Y., Yousefi B. Multiple functions of p21 in cell cycle, apoptosis and transcriptional regulation after DNA damage. DNA Repair (Amst.) 2016;42:63–71. doi: 10.1016/j.dnarep.2016.04.008. [DOI] [PubMed] [Google Scholar]
- 51.Luo J., Liu K., Yao Y., Sun Q., Zheng X., Zhu B., Zhang Q., Xu L., Shen Y., Ren B. DMBX1 promotes tumor proliferation and regulates cell cycle progression via repressing OTX2-mediated transcription of p21 in lung adenocarcinoma cell. Cancer Lett. 2019;453:45–56. doi: 10.1016/j.canlet.2019.03.045. [DOI] [PubMed] [Google Scholar]
- 52.Sun X., Hu Y., Wu J., Shi L., Zhu L., Xi P.-W., Wei J.-F., Ding Q. RBMS2 inhibits the proliferation by stabilizing P21 mRNA in breast cancer. J. Exp. Clin. Cancer Res. 2018;37:298. doi: 10.1186/s13046-018-0968-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li Z., Qiu R., Qiu X., Tian T. SNHG6 Promotes Tumor Growth via Repression of P21 in Colorectal Cancer. Cell. Physiol. Biochem. 2018;49:463–478. doi: 10.1159/000492986. [DOI] [PubMed] [Google Scholar]
- 54.Liu Y.W., Xia R., Lu K., Xie M., Yang F., Sun M., De W., Wang C., Ji G. LincRNAFEZF1-AS1 represses p21 expression to promote gastric cancer proliferation through LSD1-Mediated H3K4me2 demethylation. Mol. Cancer. 2017;16:39. doi: 10.1186/s12943-017-0588-9. [DOI] [PMC free article] [PubMed] [Google Scholar]