

Abstract
The bacterial toxin RelE cleaves mRNA in the ribosomal A-site. Although it shares a global fold with other microbial RNases, the active site contains several positively charged residues instead of histidines and glutamates typical of ribonucleases. The pH dependences of wild-type and mutant RelE indicate it uses general acid-base catalysis, but either the general acid (proposed to be R81) or the general base must have a substantially downshifted pKa. However, which group is shifted cannot be determined using available structural and biochemical data. Here, we use a phosphorothiolate at the scissile phosphate to remove the need for a general acid. We show this modification rescues nearly all of the defect of the R81A mutation, supporting R81 as the general acid. We also find that the observed pKa of the general base is dependent on the charge of the side chain at position 81. This indicates that positive charge in the active site contributes to a general-base pKa downshifted by more than 5 units. Although this modestly reduces the effectiveness of general acid-base catalysis, it is strongly supplemented by the role of the positive charge in stabilizing the transition state for cleavage. Furthermore, we show that the ribosome is required for cleavage but not binding of mRNA by RelE. Ribosome functional groups do not directly contact the scissile phosphate, indicating that positioning and charge interactions dominate RelE catalysis. The unusual RelE active site catalyzes phosphoryl transfer at a comparable rate to similar enzymes, but in a ribosome-dependent fashion.
Keywords: RelE, ribosome, toxin, nuclease, acid-base catalysis, pKa shift
Graphical Abstract

Introduction
Phosphoryl transfer reactions, including RNA cleavage, are ubiquitous in biology1. In bacteria, several toxin-antitoxin systems act by cleaving mRNA, therefore inhibiting translation either globally or for specific transcripts2. These systems may be selfish gene elements, which cannot be removed without killing the organism due to the longer lifetime of the toxin molecules3. However, chromosomal versions respond to regulatory stimuli such as nutrient stress, suggesting they may improve the fitness of the overall population. Contrary to a proposed role in bacterial persistence by inducing a dormant state, deletion of toxin-antitoxin systems does not reduce persistence4. To repress translation, some toxins (MqsR, MazF) may bind to and cleave free mRNA, but others (RelE, YafQ, YoeB, and HigB) only cleave ribosome-bound mRNA. Toxins in both cases often have structural homology to microbial RNases5,6.
RelE is the toxin component of the RelBE system, and is ubiquitous in bacteria and archaea7–10. It has some specificity for the RNA target sequence, but an absolute requirement for ribosome-bound RNA, although no ribosome functional groups are in direct contact with the scissile phosphate11. Under single-turnover conditions, RelE cleaves RNA at more than 100 s−1, comparable to the fastest RNases12–14. This represents a 1010-fold increase over the uncatalyzed reaction in similar conditions15. Although it shares the microbial RNase fold, the active site residues are unusual (Figure 1)11. Prototypical RNases have residues with neutral pKas that can act as general acid/base partners to promote the reaction16. Instead, RelE has arginines, lysines, and a tyrosine in the active site. These residues have substantial effects on catalysis. Mutation to alanine results in defects of 102- to 106-fold on the chemical step, but their unperturbed pKas are well-removed from neutrality12.
Figure 1.
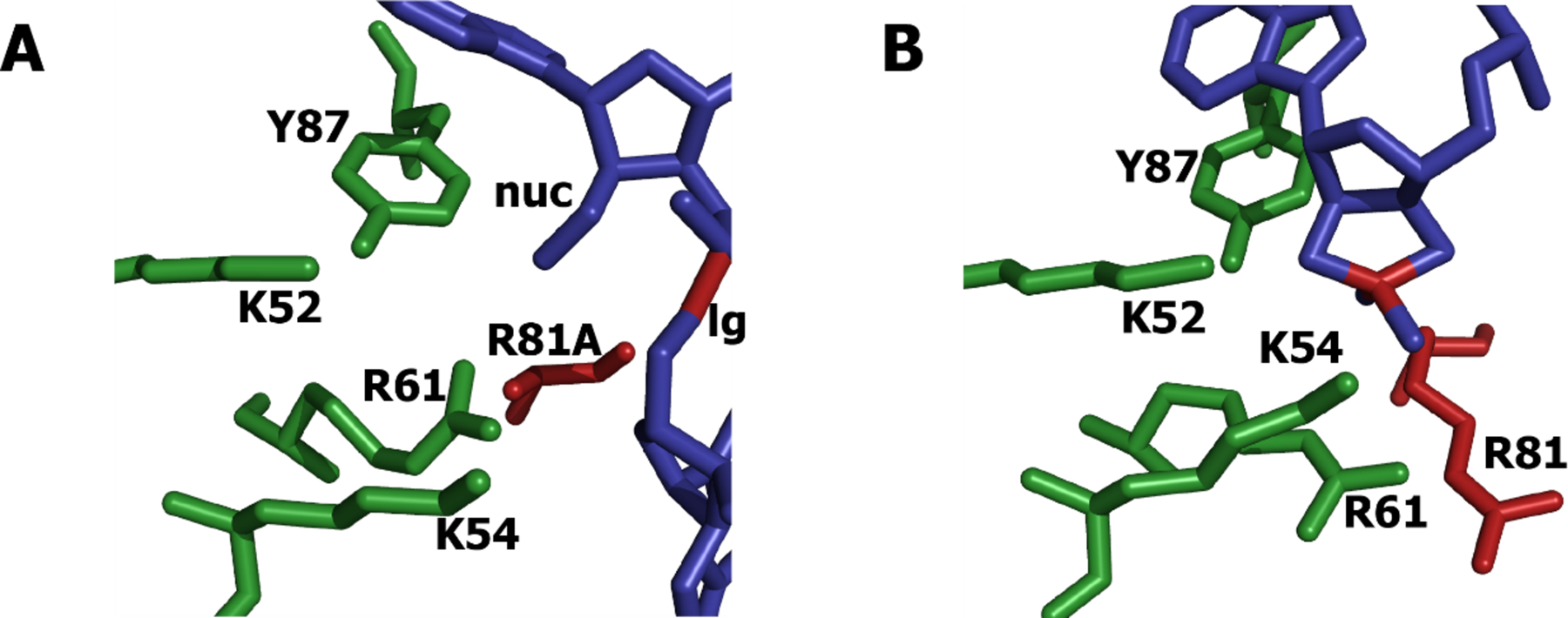
Active site of RelE. A) Precleavage structure (pdb entry 4V7J) of R81A mutant with 2’-O-methyl-modified nucleophile (nuc). 5’-oxygen leaving group is labeled (lg). R81A and scissile bond are colored red. Phosphate backbone is not in inline conformation, and catalytic residues are in the vicinity but not positioned for catalysis. B) Postcleavage structure (pdb entry 4V7K) of wild type. K52, K54, and Y87 are within hydrogen-bonding distance of the nucleophile, but the leaving group is not present. R81 and the scissile phosphorus are colored red.
To probe the role of acid-base catalysis, the pH dependences of wild-type and mutant RelE were measured17. Wild-type RelE has been shown to have no pH-rate dependence over a wide pH range (pH 5.5 to 9.0). This is consistent with acid-base catalysis only if the pKas of the general acid and general base are widely separated so that the traditional bell curve is broadened and only the wide, flat top of the curve is within the measurable range. Because the active site exclusively contains groups with high pKas in isolation (lysines and arginines) this implies that one of the acid or base must have a large pKa downshift. This could be driven by the large amount of positive charge in the active site, which will favor the deprotonated state. Similar pKa shifts have been observed in other systems, driven either by a hydrophobic or a charged environment18–21.
Several alanine mutants in the active site result in nucleases with positive pH-rate dependences. This suggests that in these contexts, there is no longer a group with a low pKa contributing to catalysis. In particular replacement of R81, the proposed general acid, by alanine results in a log-linear pH-rate relationship with a positive slope of approximately one. This suggests that the general base pKa is high in this mutant. This could be because R81 is the group with a low pKa in wild-type RelE, combining with a general base with high pKa to create the flat pH-rate dependence. Alternatively, the general base pKa in wild type is low, but the loss of positive charge at position 81 causes it to increase substantially. These two reaction mechanisms cannot be resolved using the available data. To differentiate between them, we measured the cleavage rate of wild-type and mutant RelE with phosphorothiolate-substituted RNA. We combined this with pH-rate measurements for RelE mutants with different charges in the active site. Additionally, we measured the affinity of RelE for free mRNA to determine whether ribosomes contributed to the chemical step or only to binding. Our results demonstrate the atypical catalytic strategies that RelE employs for ribosome-dependent cleavage of RNA.
Materials & Methods
Enzyme and Substrate Preparation
Wild-type and mutant enzymes were purified as previously described12.
Unmodified mRNA 5’-GGCAAGGAGGUAAAAAUGUAGAAAAACAAU-3’ and fragments were obtained from the Keck Oligonucleotide Synthesis facility at Yale University. Phosphorothiolate-modified oligos were prepared as previously described22, with the following modifications. Synthesized dinucleotide ApG (with phosphate linkage) and ApsG (with phosphorothiolate linkage) were phosphorylated using γ−32P-ATP, resulting in a radioactive phosphate on the 5’-end. The 5’-fragment 5’-GGCAAGGAGGUAAAAAUGU-3’, 3’-fragment 5’-pAAAAACAAU-3’, and DNA splint 5’-CCTCATTTTTACATCTTTTTGTTA-3’ were added in a single ligation mixture. First, annealing reactions containing 24 μM labeled ApG or ApsG dinucleotide, 110 μM 5’-fragment, 100 μM 3’-fragment, 2.5 mM EDTA, and 50 mM KCl were heated to 90 °C for 3’ and cooled 0.1 °C/s to 16 °C in a PCR machine. RNA ligase mix contained 1 part 10x RNA ligase 2 reaction buffer, 4 parts PEG 8k, and 2 parts RNA ligase 2 (New England Biolabs). Equal volumes of ligase mix and annealing reaction were mixed and incubated at 16 °C for 16 hours. Full-length 30-nucleotide mRNA was purified using 20% acrylamide TBE gels. ApG dinucleotide was converted with quantitative efficiency to full length; ApsG dinucleotide was only 0–6%. The presence of phosphorothiolate substitution was confirmed with a silver nitrate test as previously described.
mRNA with a 3’-Alexa Fluor 488 modification for fluorescence intensity and anisotropy was prepared as previously described17.
Reaction complex containing 300 nM 70S ribosomes, 400 nM tRNAfmet, and trace radiolabeled mRNA in formation buffer (50 mM Tris-HCl pH 7.5, 70 mM NH4Cl, 7 mM MgCl2, 1 mM dithiothreitol was incubated at 37 °C for 30–60 minutes. Complexes were spun at 120,000 rpm for 2 hours in a Beckman Optima Max TL centrifuge with TLA 120.1 rotor. Pellets were resuspended in MMB buffer (50 mM MES, 50 mM MOPS, 50 mM Bicine, 70 mM NH4Cl, 30 mM KCl, 2 mM MgCl2, 1 mM dithiothreitol) at a given pH.
To remove the 2’ protecting group, ribosome complexes were placed in the focus of a 100 W, 365 nm UV lamp for 5 minutes. mRNA with all phosphate linkages was cleaved at the same rate by wild-type and mutant RelE as previously reported, indicating this irradiation did not substantially affect ribosome activity (data not shown).
Kinetics
For reactions with a half-life greater than 10 seconds, enzyme and reaction complex were mixed by pipette. Trace radiolabeled reaction complex was mixed with at least 30 μM RelE at 25 °C. 2 uL aliquots were removed at defined times and added to 8 uL of quench buffer (80% formamide, 50 mM Tris-MES pH 6.5, 65 mM EDTA). Faster reactions were mixed using a KinTek RQF-3 Quench Flow instrument. Timepoints were run on 16% polyacrylamide gels and quantitated using a Storm Phosphorimager. Reactions were fit to a single exponential (equation 1). Reactions that displayed a substantial residual were fit to a double exponential (equation 2), in which case the fast rate is reported.
| (equation 1) |
| (equation 2) |
Binding Measurements by Fluorescence
Fluorescence intensity and anisotropy were measured using a PTI QuantaMaster 40. For anisotropy, increasing amounts of RelE were added to 50 nM Alexa-mRNA. All slit widths were 4mm, excitation wavelength was 485 nm, and emission was measured from 515 to 535 nm. Average anisotropy was plotted against concentration, corrected for dilution, and fit to equation 3, where A0 is the anisotropy in the absence of RelE and A∞ is the anisotropy at saturating [RelE].
| (equation 3) |
For intensity measurements, increasing amounts of unmodified mRNA were added to 100 nM RelE. The excitation wavelength was 290 nm. The area under the curve for emission from 300 to 320 nm was corrected for dilution and fit to equation 3 with [RNA] in place of [RelE].
Simulated pH-rate profiles
The dependence of rate on pH for given combinations of pKas was simulated using partition functions as previously described17. To test the contributions of linked pKas to the shape of the curve, the spreadsheet in ref 23 was used.
Calculated Contributions to Catalysis from General Acid and General Base
The fold enhancement on k1 (the intrinsic rate of the chemical step) comparing two functional groups was calculated using equation 424.
| (equation 4) |
For RelE, the values were used to give the following:
| (equation 4) |
The total enhancement on kobs, the observed rate of catalysis at a particular pH, may be attenuated by the fraction of the enzyme that is not in the correct protonation state, calculated using equation 524:
| (equation 5) |
For enzymes with pKabase = 5 and pKaacid = 10, kobs = k1. For pKabase = pKaacid = 7.5, kobs = k1 * ¼. Therefore the net rate enhancement on the observed rate for pKas near neutrality is 4.5-fold.
Results
R81A defect is rescued by phosphorothiolate modification at the scissile phosphate
Crystal structures and rate measurements of wild-type and mutant RelE have established the importance of several residues located near the scissile phosphate for catalysis11,12. In particular, R81 has been proposed to act as a general acid to protonate the leaving group oxygen. However, the pre-cleavage structure has an R81A mutation, the post-cleavage structure lacks the leaving group, and several residues have similar rate defects. Therefore, further study is required to determine whether R81 acts as a general acid.
Because the general acid is responsible for protonating the leaving group oxygen, substitution of this oxygen with a leaving group that does not need to be protonated in the rate-limiting step can rescue the rate defect of deleting the general acid. In particular, phosphorothiolate substitution, where the bridging oxygen is replaced by sulfur, has been used to identify the general acid in both protein and RNA enzymes25–29.
We made the phosphorothiolate substitution at the scissile phosphate of the mRNA substrate for RelE, and determined the rate effects on wild-type and mutant enzymes. At pH 7.5 with unsubstituted mRNA, R81A has more than a 105-fold defect compared to wild type12. However, R81A is only 10-fold slower than wild type when reacted with a phosphorothiolate mRNA (Table 1). R81A is the only enzyme tested whose rate goes up substantially with the phosphorothiolate relative to the unsubstituted mRNA. A similar uncatalyzed reaction has a 100-fold increase in reaction rate when no general acid is present30. In contrast, other RelE mutants (K54A and R61A, Table 1) catalyze cleavage of the phosphorothiolate and unsubstituted mRNAs at similar rates. The increase in reaction rate for R81A alone upon phosphorothiolate substitution, together with the available structural data, strongly supports the identification of R81 as the general acid.
Table 1.
Rates for cleavage of unmodified (PO) and phosphorothiolated (PS) mRNA by wild-type and mutant RelE at pH 7.5. Rates ± standard deviations are listed, with number of trials in parenthesis.
| PO rate (s−1)a | krel(PO) | PS rate (s−1) | krel(PS) | krel(PS)/ krel(PO) | |
|---|---|---|---|---|---|
| Wild type | 400 ± 100 | ≡ 1 | 0.8 ± 0.3 (4) | ≡ 1 | ≡ 1 |
| R81A | 0.0053 ± 0.0006 | 1.3 × 10−5 | 0.09 ± 0.02 (4) | 1 × 10−1 | 7700 |
| K54A | 0.17 ± 0.02 | 4.2 × 10−4 | 0.05 ± 0.02 (3) | 6 × 10−2 | 140 |
| R61A | 0.00032 ± 0.00008 | 8.0 × 10−7 | 0.0004 ± 0.0002 (3) | 5 × 10−4 | 630 |
data from ref 17.
Part of this rescue is due to a 500-fold decrease in the rate of wild-type cleavage of phosphorothiolate mRNA. Phosphorothiolate effects on wild-type systems have varied from a 12-fold enhancement for the Hairpin ribozyme27 to a 6-fold reduction for the Varkud Satellite (VS) ribozyme29. These effects reflect a combination of two factors. First, phosphorothiolate substitution can change the transition state of the cleavage reaction30, which can reduce the effectiveness of active site residues31. Second, by alleviating the need for a general acid, only the general base needs to be in the correct protonation state to cleave phosphorothiolate-substituted substrate. For pH values above the general acid pKa, a higher fraction of enzyme will be active for cleavage of the phosphorothiolate substrate relative to the unmodified substrate. For VS, the general acid pKa is low and phosphorothiolate substitution increases the fraction correctly protonated ribozyme by 100-fold. Because the observed effect is a 6-fold decrease, the effect on the chemical step is estimated to be a 600-fold decrease. For RelE, if the general acid pKa were low, then phosphorothiolate substitution would also increase the fraction in the correct protonation state by at least 100-fold. The true effect of the phosphorothiolate on the chemical step would then be at least 50,000-fold, far greater than the effects observed for other systems. Alternatively, the general acid has a high pKa, and the chemical step is slowed by 500-fold by phosphorothiolate substitution, similar to the value for VS.
Charge at position 81 affects pKa of general base
The flat pH-rate dependence for wild-type RelE over a wide pH range is consistent with the general acid and general base pKas being widely separated, but does not distinguish between two kinetically equivalent mechanisms. In mechanism 1, the general acid is higher than the measurable range, and the general base pKa is perturbed so it is lower than the measurable range. In mechanism 2, the pKas are reversed so that the acid is low and the base is high. Mutations of charged groups in the RelE active site cause the rate to increase with increasing pH, but it is unclear whether this change in pH dependence is due to the loss of groups with positive charge or an inability to transfer protons.
As a strategy to separate the roles of charge and proton transfer at position 81, we determined the rate and pH dependence of the R81K mutant (Figure 2a). Replacement of arginine with lysine would be expected to result in a lower pKa. The maximum reaction rate for R81K decreases 200-fold relative to wild type. This could be due to defects in proton transfer, changes in charge distribution at the active site, or structural effects. Importantly, the pH-rate dependence is different from both wild type and R81A, with a decrease in reaction rate with decreasing pH and an observed reaction pKa of 7.5. This result is consistent with a low general-base pKa in wild type (mechanism 1) that is dependent on positive charge at position 81 (Figure 2b). If K81 cannot act as a general acid, then R81A and R81K differ mainly by the charge at position 81. Introducing charge to the active site with lysine instead of alanine lowers the general-base pKa down from above 9 to 7.5.
Figure 2.

pH dependence of R81 mutants. A) WT RelE (squares) has little pH dependence. R81A (triangles) has a log-linear pH dependence with an apparent general base pKa greater than 8. R81K (diamonds), which preserves the charge but not the geometry, has an apparent pKa of 7.5. Error bars correspond to one standard deviation and are not shown when smaller than the points. B-D) Simulated curves showing the shapes of the pH-rate dependence for different combinations of acid and base pKas. Only the relative rates for each pair due to change in protonation state are shown.
If instead the general acid pKa were low (mechanism 2), replacement of arginine with lysine would drive it even lower. This would retain the flat pH-rate dependence observed in wild-type. If the general acid pKa were low in wild type and lysine were completely unable to serve as a general acid, then the reaction would have a very high pKa corresponding to the general base, as in the R81A mutant (Figure 2c). Finally, if the general acid pKa were high (mechanism 1) but decreased upon substitution with lysine, then the reaction rate should decrease at high pH (Figure 2d). None of these are observed. Instead the observed reaction pKa increases with decreasing charge at position 81.
RelE binds to mRNA in the absence of the ribosome
RelE only cleaves mRNA bound to the ribosome. This could be simply because the ribosome is required for RelE to associate with mRNA. Alternatively, the ribosome could increase the rate of the chemical step. Based on crystal structures the ribosome does not contribute functional groups to the active site, but it may participate through substrate positioning or other indirect effects11. We sought to determine whether RelE could bind to RNA in the absence of the ribosome using fluorescence anisotropy. We observed a clear, concentration-dependent increase in anisotropy upon addition of RelE (Figure 3). The calculated dissociation constant for the RelE-mRNA interaction is 21 ± 5 μM, which is seven-fold weaker than that for RelE and the ribosome-mRNA complex determined by cleavage kinetics, 3 μM12. Intrinsic fluorescence of RelE was previously used to monitor the RelE-RelB interaction32 and we used this physical property of RelE to monitor its binding to mRNA. We also saw an increase in RelE fluorescence upon addition of RNA with a similar Kd to that determined by anisotropy (data not shown). This indicates that RelE is capable of binding RNA in the absence of ribosomes, yet there is no indication that the RelE:mRNA binary complex is competent for catalysis.
Figure 3.
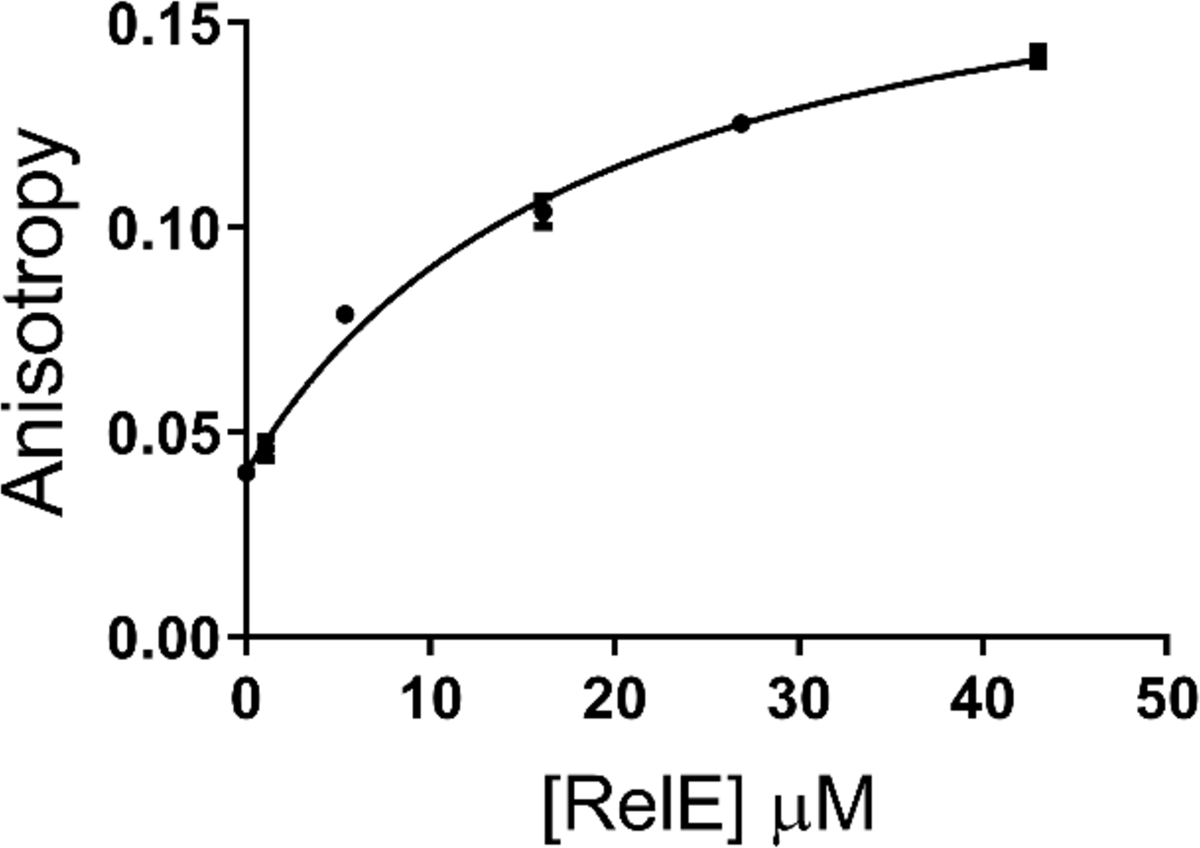
Binding of RelE to free RNA. Addition of RelE to Alexa-labeled RNA in the absence of ribosomes causes an increase in fluorescence anisotropy. Error bars correspond to one standard deviation and are not shown when smaller than the points.
Discussion
Shift in pKa of general base
Although RelE has a similar global fold to other microbial RNases, the active site only contains residues whose unperturbed pKas are well above neutrality. Previous results indicated that RelE used acid-base catalysis, but those studies could not distinguish between two distinct mechanisms17. One possibility is that the general base pKa (proposed to be K54 based on position and pH dependence12,17) is downshifted below 5.5 so that it is always deprotonated at physiological pH, and the general acid (R81) retains a pKa above 9 and is always protonated at physiological pH (mechanism 1). Alternatively, the general acid is downshifted while the general base is unperturbed (mechanism 2). Both cases would give identical, flat pH-rate profiles, consistent with the previous observations. The datasets presented here using phosphorothiolate-substituted RNA and mutant enzymes allow us to distinguish between these two models, which has interesting implications for the mechanism of catalysis.
We compared two mutations at position 81 to wild type to determine the role of charge on the pKa of the general base. When the charge is eliminated in R81A, a positive pH-rate dependence is observed with a reaction pKa at or above the observable range. In R81K the charge is nearly preserved, and the general base pKa rises only to 7.5. We conclude that R81 in wild type is essential for pushing the general base pKa below the observable range (mechanism 1).
If instead the general acid (R81) pKa were low (mechanism 2), it would have to increase upon substitution of arginine by lysine, opposite of the trend in their unperturbed pKas33. Furthermore, R81 would require a pKa shift of 8 or more units, compared with only 5 units for lysine. Similar shifts for lysine residues have been observed in other systems18,21. In one case, lysines substituted for residues in the hydrophobic core of staphylococcal nuclease had pKas as low as 5.319. In contrast, arginines were always charged, even when in the hydrophobic core34. No shifts for arginine consistent with the observed pH dependence of RelE have been reported20.
In addition to R81, other residues also yield a positive pH-rate dependence when mutated to alanine17. R61A, Y87A, and K52A have positive slopes with pKas of 7, 8.5, and greater than 9, respectively. Higher pKas indicate the loss of a more important interaction for the pKa shift of the general base. In particular, K52A has the highest pKa of any mutant tested, suggesting K52 plays a substantial role in decreasing the pKa of the general base. If K54 is the general base, then the greater effect of K52 on the general base pKa could be due to its close proximity. In acetoacetate carboxylase, an adjacent lysine drives down the pKa of the Schiff-base-forming lysine35. For this and other systems, pKa perturbation can be accomplished via charge-charge interactions or by positioning effects36. Importantly, arginines at positions 61 and 81 would be expected to retain a positive charge at all measurable pHs, consistent with their role in stabilizing the transition state charge and acting as the general acid, respectively.
Contributions to general acid and base catalysis of widely separated pKas
In textbook cases of acid/base catalysis the pKas of both groups are close to the reaction pH, yielding the classic bell curve for the pH-rate dependence. This optimizes the contribution of each group, since most groups will be in the correct state, but will also have a low barrier to proton transfer. The contribution to catalysis is related to the pKa of the candidate catalytic groups and the Brønsted coefficient24. This Brønsted coefficient reflects the extent of proton transfer in the transition state. High coefficients indicate a high degree of proton transfer and will strongly favor catalytic groups capable of transferring a proton.
Even groups with pKas two or more units removed from the reaction pH, as may be the case here, can provide substantial catalytic power24. This is because in the absence of a general acid or base, proton transfer would have to occur through water molecules. Because the pKa of water is so far from neutrality, even a titratable group with a pKa of 5 or 10 positioned for proton transfer provides significant catalytic potential. For a Brønsted value of 0.5, a 2.5-unit difference in pKa from the reaction pH produces only a 4.5-fold decrease in the observed rate (see Materials and Methods).
This mechanism involving acid-base catalysis by groups with pKas far from neutrality is most effective when the Brønsted coefficients are small; i.e. when proton transfer is relatively weak. For uncatalyzed reactions of phosphodiesters, both bond breaking and bond formation occur in the rate-limiting transition state1,37. If proton transfer from the general acid to the leaving group oxygen is weak, then this could lead to an associative transition state with high bond order to the leaving group and a buildup of negative charge on the phosphate. Mutation of positively charged residues or modifying the charge distribution of the phosphate both have significant impacts on RelE catalysis12,17. In particular, mutation of R61, which contacts the scissile phosphate, has the largest detrimental effect when mutated to alanine. This could be from decreased ability to stabilize increasing negative charge in the transition state. Additionally, the geometry of the phosphate could change during progression to the transition state, again leading to improved stabilization by R61 and other positively charged residues. These charge interactions appear to be more important than acid-base catalysis in RelE.
Binding of RelE to free and ribosome-bound mRNA
RelE binds to free mRNA with only a 7-fold worse affinity than binding to the mRNA:ribosome complex. This indicates that the additional energy of binding to the ribosome is used to drive conformational changes. Indeed, binding of RelE causes a substantial movement of the mRNA in the ribosome, such that the scissile phosphate is in an inline conformation11. It is not known whether mRNA adopts a similar conformation when bound to RelE in the absence of ribosomes. Since the ribosome does not appear to contribute functional groups to catalysis, it is likely that its role is to stabilize a catalytic conformation. In addition to electrostatic stabilization, conformational effects – in particular promoting an inline conformation – could dominate catalysis by RelE.
One possible reason that RelE’s active site differs from other RNases is the requirement for ribosome binding. However, other ribosome-dependent RNases such as YoeB and YafQ have conventional active sites including negatively charged residues and histidines (Figure 4)38,39. Alternatively, RelE offers a different solution to catalyzing phosphoryl transfer, depending to a larger extent on charge and conformational stabilization. This bears a remarkable resemblance to ribozymes, which lack an equivalent to the histidine side chain and therefore often use functional groups with either perturbed pKas or with pKas far from the reaction pH. These strategies in both RNA and protein enzymes are capable of catalyzing phosphodiester transfer at the rates required for biological function.
Figure 4.

A) Proposed mechanism of catalysis by RelE. Limits of observed pKas are shown, with unperturbed pKas in italics. The general base has not been definitively established but is shown here as K54 (red), consistent with previous biochemical data. B) Mechanism of catalysis by YoeB, which is also ribosome-dependent. The unperturbed pKas of the general acid and base are in italics.
ACKNOWLEDGMENT
We thank Meghan Griffin for advice on experimental design. This work was supported by the National Institutes of Health grants GM054839-15 and GM022778-41 (to SAS) and GM131568 (to JAP).
Funding Sources
This work was supported by the National Institutes of Health grants GM054839-15 and GM022778-41 (to SAS) and GM131568 (to JAP).
ABBREVIATIONS
- mRNA
messenger ribonucleic acid
- VS
Varkud Satellite ribozyme
- MES
2-(N-morpholino)ethanesulfonic acid
- MOPS
(3-(N-morpholino)propanesulfonic acid)
- EDTA
Ethylenediaminetetraacetic acid
- TBE
Tris borate EDTA
REFERENCES
- (1).Lassila JK; Zalatan JG; Herschlag D Biological Phosphoryl-Transfer Reactions: Understanding Mechanism and Catalysis. Annu. Rev. Biochem 2011, 80, 669–702. 10.1146/annurev-biochem-060409-092741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Yamaguchi Y; Inouye M Chapter 12 MRNA Interferases, Sequence‐Specific Endoribonucleases from the Toxin–Antitoxin Systems In Progress in Molecular Biology and Translational Science; Molecular Biology of RNA Processing and Decay in Prokaryotes; Academic Press, 2009; Vol. 85, pp 467–500. 10.1016/S0079-6603(08)00812-X. [DOI] [PubMed] [Google Scholar]
- (3).Melderen LV; Bast MSD Bacterial Toxin–Antitoxin Systems: More Than Selfish Entities? PLOS Genet. 2009, 5 (3), e1000437 10.1371/journal.pgen.1000437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Goormaghtigh F; Fraikin N; Putrinš M; Hallaert T; Hauryliuk V; Garcia-Pino A; Sjödin A; Kasvandik S; Udekwu K; Tenson T; Kaldalu N; Van Melderen L Reassessing the Role of Type II Toxin-Antitoxin Systems in Formation of Escherichia Coli Type II Persister Cells. mBio 2018, 9 (3), e00640–18. 10.1128/mBio.00640-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Takagi H; Kakuta Y; Okada T; Yao M; Tanaka I; Kimura M Crystal Structure of Archaeal Toxin-Antitoxin RelE–RelB Complex with Implications for Toxin Activity and Antitoxin Effects. Nat. Struct. Mol. Biol 2005, 12 (4), 327–331. 10.1038/nsmb911. [DOI] [PubMed] [Google Scholar]
- (6).Kamada K; Hanaoka F Conformational Change in the Catalytic Site of the Ribonuclease YoeB Toxin by YefM Antitoxin. Mol. Cell 2005, 19 (4), 497–509. 10.1016/j.molcel.2005.07.004. [DOI] [PubMed] [Google Scholar]
- (7).Gerdes K Toxin-Antitoxin Modules May Regulate Synthesis of Macromolecules during Nutritional Stress. J. Bacteriol 2000, 182 (3), 561–572. 10.1128/JB.182.3.561-572.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Christensen SK; Mikkelsen M; Pedersen K; Gerdes K RelE, a Global Inhibitor of Translation, Is Activated during Nutritional Stress. Proc. Natl. Acad. Sci. U. S. A 2001, 98 (25), 14328–14333. 10.1073/pnas.251327898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Gotfredsen M; Gerdes K The Escherichia Coli RelBE Genes Belong to a New Toxin–Antitoxin Gene Family. Mol. Microbiol 1998, 29 (4), 1065–1076. 10.1046/j.1365-2958.1998.00993.x. [DOI] [PubMed] [Google Scholar]
- (10).Pedersen K; Zavialov AV; Pavlov M. Yu.; Elf J; Gerdes K; Ehrenberg M The Bacterial Toxin RelE Displays Codon-Specific Cleavage of MRNAs in the Ribosomal A Site. Cell 2003, 112 (1), 131–140. 10.1016/S0092-8674(02)01248-5. [DOI] [PubMed] [Google Scholar]
- (11).Griffin MA; Davis JH; Strobel SA Bacterial Toxin RelE: A Highly Efficient Ribonuclease with Exquisite Substrate Specificity Using Atypical Catalytic Residues. Biochemistry 2013, 52 (48), 8633–8642. 10.1021/bi401325c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Zabinski M; Walz FG Jr Subsites and Catalytic Mechanism of Ribonuclease T1: Kinetic Studies Using GpC and GpU as Substrates. Arch. Biochem. Biophys 1976, 175 (2), 558–564. [DOI] [PubMed] [Google Scholar]
- (13).Moussaoui M; Cuchillo CM; Nogués MV A Phosphate-Binding Subsite in Bovine Pancreatic Ribonuclease A Can Be Converted into a Very Efficient Catalytic Site. Protein Sci. Publ. Protein Soc 2007, 16 (1), 99–109. 10.1110/ps.062251707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Li Y; Breaker RR Kinetics of RNA Degradation by Specific Base Catalysis of Transesterification Involving the 2’-Hydroxyl Group. J. Am. Chem. Soc 1999, 121 (23), 5364–5372. 10.1021/ja990592p. [DOI] [Google Scholar]
- (15).Neubauer C; Gao Y-G; Andersen KR; Dunham CM; Kelley AC; Hentschel J; Gerdes K; Ramakrishnan V; Brodersen DE The Structural Basis for MRNA Recognition and Cleavage by the Ribosome-Dependent Endonuclease RelE. Cell 2009, 139 (6), 1084–1095. 10.1016/j.cell.2009.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Yang W Nucleases: Diversity of Structure, Function and Mechanism. Q. Rev. Biophys 2011, 44 (1), 1–93. 10.1017/S0033583510000181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Dunican BF; Hiller DA; Strobel SA Transition State Charge Stabilization and Acid–Base Catalysis of MRNA Cleavage by the Endoribonuclease RelE. Biochemistry 2015, 54 (47), 7048–7057. 10.1021/acs.biochem.5b00866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Kokesh FC; Westheimer FH Reporter Group at the Active Site of Acetoacetate Decarboxylase. II. Ionization Constant of the Amino Group. J. Am. Chem. Soc 1971, 93 (26), 7270–7274. 10.1021/ja00755a025. [DOI] [PubMed] [Google Scholar]
- (19).Isom DG; Castañeda CA; Cannon BR; E BG-M Large Shifts in PKa Values of Lysine Residues Buried inside a Protein. Proc. Natl. Acad. Sci 2011, 108 (13), 5260–5265. 10.1073/pnas.1010750108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Harris TK; Turner GJ Structural Basis of Perturbed PKa Values of Catalytic Groups in Enzyme Active Sites. IUBMB Life 2002, 53 (2), 85–98. 10.1080/15216540211468. [DOI] [PubMed] [Google Scholar]
- (21).Barbas CF; Heine A; Zhong G; Hoffmann T; Gramatikova S; Björnestedt R; List B; Anderson J; Stura EA; Wilson IA; Lerner RA Immune Versus Natural Selection: Antibody Aldolases with Enzymic Rates But Broader Scope. Science 1997, 278 (5346), 2085–2092. 10.1126/science.278.5346.2085. [DOI] [PubMed] [Google Scholar]
- (22).Li N-S; Frederiksen JK; Koo SC; Lu J; Wilson TJ; Lilley DM; Piccirilli JA A General and Efficient Approach for the Construction of RNA Oligonucleotides Containing a 5′-Phosphorothiolate Linkage. Nucleic Acids Res. 2010, 39 (5), e31–e31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Frankel EA; Bevilacqua PC Complexity in PH-Dependent Ribozyme Kinetics: Dark PKa Shifts and Wavy Rate–PH Profiles. Biochemistry 2018, 57 (5), 483–488. 10.1021/acs.biochem.7b00784. [DOI] [PubMed] [Google Scholar]
- (24).Bevilacqua PC Mechanistic Considerations for General Acid–Base Catalysis by RNA: Revisiting the Mechanism of the Hairpin Ribozyme. Biochemistry 2003, 42 (8), 2259–2265. 10.1021/bi027273m. [DOI] [PubMed] [Google Scholar]
- (25).Das SR; Piccirilli JA General Acid Catalysis by the Hepatitis Delta Virus Ribozyme. Nat. Chem. Biol 2005, 1 (1), 45. [DOI] [PubMed] [Google Scholar]
- (26).Keenholtz RA; Mouw KW; Boocock MR; Li N-S; Piccirilli JA; Rice PA Arginine as a General Acid Catalyst in Serine Recombinase-Mediated DNA Cleavage. J. Biol. Chem 2013, jbc–M113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Kath-Schorr S; Wilson TJ; Li N-S; Lu J; Piccirilli JA; Lilley DM General Acid–Base Catalysis Mediated by Nucleobases in the Hairpin Ribozyme. J. Am. Chem. Soc 2012, 134 (40), 16717–16724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Krogh BO; Shuman S Catalytic Mechanism of DNA Topoisomerase IB. Mol. Cell 2000, 5 (6), 1035–1041. [DOI] [PubMed] [Google Scholar]
- (29).Wilson TJ; Li N-S; Lu J; Frederiksen JK; Piccirilli JA; Lilley DM Nucleobase-Mediated General Acid-Base Catalysis in the Varkud Satellite Ribozyme. Proc. Natl. Acad. Sci 2010, 107 (26), 11751–11756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Ye J-D; Barth CD; Anjaneyulu PSR; Tuschl T; Piccirilli JA Reactions of Phosphate and Phosphorothiolate Diesters with Nucleophiles: Comparison of Transition State Structures. Org. Biomol. Chem 2007, 5 (15), 2491–2497. 10.1039/B707205H. [DOI] [PubMed] [Google Scholar]
- (31).Iyer S; Hengge AC The Effects of Sulfur Substitution for the Nucleophile and Bridging Oxygen Atoms in Reactions of Hydroxyalkyl Phosphate Esters. J. Org. Chem 2008, 73 (13), 4819–4829. 10.1021/jo8002198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Overgaard M; Borch J; Gerdes K RelB and RelE of Escherichia Coli Form a Tight Complex That Represses Transcription via the Ribbon–Helix–Helix Motif in RelB. J. Mol. Biol 2009, 394 (2), 183–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Fitch CA; Platzer G; Okon M; Garcia-Moreno BE; McIntosh LP Arginine: Its PKa Value Revisited. Protein Sci. Publ. Protein Soc 2015, 24 (5), 752–761. 10.1002/pro.2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Harms MJ; Schlessman JL; Sue GR; E BG-M Arginine Residues at Internal Positions in a Protein Are Always Charged. Proc. Natl. Acad. Sci 2011, 108 (47), 18954–18959. 10.1073/pnas.1104808108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Highbarger LA; Gerlt JA; Kenyon GL Mechanism of the Reaction Catalyzed by Acetoacetate Decarboxylase. Importance of Lysine 116 in Determining the PKa of Active-Site Lysine 115. Biochemistry 1996, 35 (1), 41–46. 10.1021/bi9518306. [DOI] [PubMed] [Google Scholar]
- (36).Ho M-C; Ménétret J-F; Tsuruta H; Allen KN The Origin of the Electrostatic Perturbation in Acetoacetate Decarboxylase. Nature 2009, 459 (7245), 393–397. 10.1038/nature07938. [DOI] [PubMed] [Google Scholar]
- (37).Cassano AG; Anderson VE; Harris ME Understanding the Transition States of Phosphodiester Bond Cleavage: Insights from Heavy Atom Isotope Effects. Biopolymers 2004, 73 (1), 110–129. 10.1002/bip.10517. [DOI] [PubMed] [Google Scholar]
- (38).Feng S; Chen Y; Kamada K; Wang H; Tang K; Wang M; Gao Y-G YoeB-Ribosome Structure: A Canonical RNase That Requires the Ribosome for Its Specific Activity. Nucleic Acids Res. 2013, 41 (20), 9549–9556. 10.1093/nar/gkt742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Maehigashi T; Ruangprasert A; Miles SJ; Dunham CM Molecular Basis of Ribosome Recognition and MRNA Hydrolysis by the E. Coli YafQ Toxin. Nucleic Acids Res. 2015, 43 (16), 8002–8012. 10.1093/nar/gkv791. [DOI] [PMC free article] [PubMed] [Google Scholar]